

Abstract
Diffusion-based bioartificial pancreas (BAP) devices are limited by poor islet viability and functionality due to inadequate mass transfer resulting in islet hypoxia and delayed glucose-insulin kinetics. While intravascular ultrafiltration-based BAP devices possess enhanced glucose-insulin kinetics, the polymer membranes used in these devices provide inadequate ultrafiltrate flow rates and result in excessive thrombosis. Here, we report the silicon nanopore membrane (SNM), which exhibits a greater hydraulic permeability and a superior pore size selectivity compared to polymer membranes for use in BAP applications. Specifically, we demonstrate that the SNM-based intravascular BAP with ~10 and ~40 nm pore sized membranes support high islet viability (>60%) and functionality (<15 minute insulin response to glucose stimulation) at clinically relevant islet densities (5,700 and 11,400 IE/cm2) under convection in vitro. In vivo studies with ~10 nm pore sized SNM in a porcine model showed high islet viability (>85%) at clinically relevant islet density (5,700 IE/cm2), c-peptide concentration of 144 pM in the outflow ultrafiltrate, and hemocompatibility under convection. These promising findings offer insights on the development of next generation of full-scale intravascular devices to treat T1D patients in the future.
Keywords: silicon nanopore membrane, islet encapsulation, intravascular device, convection, ultrafiltration, MEMS, pigs
Graphical Abstract
We present the silicon nanopore membrane (SNM) based intravascular bioartificial pancreas (iBAP) in a porcine model that demonstrates high islet viability and functionality at clinically relevant cell density and hemocompatibility under convective transport.
Introduction
Islet encapsulation has shown great promise in treating unstable Type 1 Diabetes (T1D); however, this treatment is severely limited by the shortage of donor tissue and the need for life-long systemic immunosuppression that can be detrimental to islet cell function and result in immunosuppression-associated adverse events1. Immunoisolation via cell encapsulation may circumvent the need of immunosuppression by preventing the passage of the host’s immune factors, while allowing the exchange of glucose, insulin, nutrients and small molecules to sustain the survival of islet graft2. However, the clinical success with islet encapsulation, thus, far, has been limited3–7. Reasons for poor performance with islet micro- and macro-encapsulation include the large capsule sizes that lead to considerable diffusional gradients and implant volumes, which result in decreased islet cell viability and delayed glucose-insulin kinetics. Solutes that are critical for islet survival and function, including oxygen, nutrients, and insulin, are exchanged by diffusion across the semipermeable encapsulating membrane8. Cell necrosis occurs when islets are placed beyond the diffusion limit of tissue, which is ~150–200 μm away from the nearest blood vessel9. This distance is significantly smaller than the half-length/radial distance of most encapsulating devices10, 11. In T1D patients, the delayed glucose-insulin response due to large diffusion gradients could also endanger patients by exposing them to potential episodes of hypoglycemia and hyperglycemia5. Furthermore, transplant sites able to accommodate such large volumes of encapsulated-islets at a clinically relevant dose are restricted to the intraperitoneal space, which is poorly vascularized and therefore less favorable for cell engraftment12–14.
Our lab has developed the silicon nanopore membrane (SNM) based intravascular bioartificial pancreas (iBAP) to overcome the oxygenation and glucose-insulin kinetics challenges of diffusion-based BAP devices15–20 by convective mass transfer of nutrients and small molecules within the encased islet chamber (IC) (Fig. 1). We designed a new generation of encapsulating membranes for immunoisolation of transplanted islets based on microelectromechanical systems (MEMS) technology initially pioneered by Ferrari and colleagues21, 22 to create more uniform pore sizes at nanometer scale23. These semipermeable filtration membranes can be engineered with precise slit-shaped pore with critical widths down to 5 nm (Fig. 1,b,iv) and an uniform pore size distribution (~1%) for superior selectivity24. The slit pore microarchitecture of SNM was achieved through a series of dry oxidation of polysilicon and backside patterning with deep ion-reactive etching (DRIE)23. The DRIE process allows for fabrication of membranes with greater number of exposed nanopores per area compared to those with v-shaped sidewalls achieved by anisotropic etching25. The utilization of a sacrificial layer to define the nanopores resulted in a membrane with a straight slit-pore path that presents a shorter distance for molecules to travel compared to the previous “L” pore design26. SNM were produced with an active membrane area (6 × 6 mm) consisting of ~106 rectangular slit pores with ~7 nm in width, 300 nm in depth, and 2 μm in length (Fig. 1,b). The pore geometry optimizes the trade-off between selectivity and permeability of the membranes. The permeability – selectivity analysis for ultrafiltration showed that SNM with monodispersed slit-shaped pores exhibited greater selectivity at a given value of permeability than membranes with cylindrical pores for pore size below 100 nm27. Due to the thickness of membranes in BAP devices, convection-dominated mass transport is advantageous28, because it efficiently transports solutes such as glucose, oxygen, and insulin between the device and adjacent vascular system. In a previous study, we showed 80% reduction in cytokine passage using SNM with an average pore size of ~ 7 nm, providing immunoisolation and preserving the islet viability under convection23. While previous attempts with ultrafiltration-based devices showed some promise, they provided insufficient ultrafiltrate flow rates thereby resulting in insufficient convective mass transfer18, 19, 29–31. In contrast, the SNM produces high levels of ultrafiltrate (Table S1), which is ten times greater than the polymer membranes used in previous ultrafiltrate-based iBAP devices at physiological blood pressures24, 27, 32–35. Given advantages in the selectivity and permeability of the SNM, our long-term objective is to develop the silicon nanopore membrane (SNM) based intravascular bioartificial pancreas (iBAP), where the iBAP will be connected to arterio-venous grafts and a pressure drop between the artery and ultrafiltrate vein produces ultrafiltrate to flow through the SNM-encapsulated islet chamber (IC), carrying nutrients to the islets and insulin to the ultrafiltrate vein (Fig. S1).
Figure 1.


The assembly of intravascaular bioartificial pancreas device (iBAP). (a,i) An exploded view of the iBAP components: ultrafiltrate outlet (blue), polycarbonate backside (PC Backside), silicon nanopore membrane (SNM) (green), islet chamber (IC), and polycarbonate piece with flow path (Flow Path). (a,ii) An assembled iBAP used for in vitro and in vivo convective experiments. (b,i) An optical image of the SNM membrane. (b,ii) A top view SEM image illustrating the rectangular pore-containing regions surrounded by solid silicon regions, which provide mechanical support. (b,iii) A further magnified top view SEM image of the pore region showing individual 10 nm-wide pores. (b,iv) A cross-section SEM image showing the slit-shaped pores. (c,i) An illustration of the acrylic islet chamber (IC) where there are eight ultrafiltrate channels within the islet-agarose gel region (purple). (c,ii) A gross image of islets and agarose mixture inside the IC in which the maximum diameter surrounding each ultrafiltrate channel is 800 μm.
In this study, we tested an iBAP prototype containing SNM-encapsulated mouse islets under diffusion and convection (Fig. 1). For in vitro experiments, we investigated the SNM-encapsulated islet viability and glucose-insulin kinetics of the iBAP with varying islet densities by volume (10% vs 20%) and membrane pore sizes (~10 nm vs ~40 nm) under both diffusion and convection. Subsequently, the SNM-encapsulated islets were intravascularly implanted into Yorkshire pigs for three days. The SNM hemocompatibility, device patency, and islet viability and functionality from the porcine experiments were investigated to determine the feasibility of developing a refined SNM-based iBAP with convective transport to support a clinically relevant islet dosage to treat a T1D patient in the future.
Results
iBAP testing in vitro
The iBAP comprising a ~10 nm pore sized SNM with 10% (5,700 IE/cm2) or 20% (11,400 IE/cm2) mouse islet densities was investigated for glucose-stimulated insulin response and viability of the encapsulated islets after three days. Under convection, the iBAP with 10% mouse islet density and ~10 nm pore sized SNM showed an increase in insulin secretion within 10 minutes of high glucose exposure (Fig. 2.a,i), which was consistent with normal islet function of biphasic insulin release36 (i.e. the first insulin phase appeared within 5–10 minutes followed by a second sustained phase that is slower and delayed as times goes longer). Furthermore, during the period of 63 to 78 minutes in which the glucose concentration decreased, the corresponding stimulated-insulin secretion also dropped. However, when the cell density increased from 10% to 20% in the iBAP with ~10 nm pore sized SNM under convection, no significant change in glucose-stimulated insulin level was observed within the first few minutes of high glucose exposure (Fig. 2,a,ii). The stimulation index (SI), the ratio of stimulated to basal insulin secretion normalized by the insulin content, was calculated as 4.4 ± 0.6 and 1.1 ± 0.1 for the iBAP of ~10 nm pore sized SNM with 10% and 20% mouse islet densities, respectively (Table 1). It is well-recognized that delay of insulin secretion in response to glucose (>15 min) has been a common problem encountered in the early extravascular hollow-fiber systems37. Our iBAP with ~10 nm pore size under convection supported the normal insulin function at 10% islet density with no significant delay in glucose-stimulated insulin response. However, the glucose stimulated-insulin response at 20% islet density under convection with ~10 nm pore sized SNM showed abnormal insulin-functioning behavior, indicating that encased cells were likely not in optimal health in that environment.
Figure 2.



In vitro testing of the intravascular bioartificial pancreas device (iBAP) with 10% or 20% islet density encapsulated with 10 nm-pore size SNM. (a) Glucose-insulin kinetics of the SNM-encapsulated iBAP with 10% (i) or 20% (ii) islet densities under convection was measured from exposing them to a series of low, high, and low glucose conditions. (b) The SNM-encapsulated iBAP with 10% islet density under convection (10% convection) showed significantly higher viability compared to that of 10% islet density under diffusion (10% diffusion), and 20% islet density under both diffusion (20% diffusion) and convection (20% convection) after 3 days. (n > 3, *p < 0.05). Viabilities of islets that were immediately encapsulated in agarose and dispensed into the islet chamber (IC) without further testing were evaluated as the in vitro positive control. (c) Viable (green) and dead (red) cells were stained for in vitro positive control (i), 10% islet density under diffusion (ii), 10% islet density under convection (iii), 20% islet density under diffusion (iv), and 20% islet density under convection (v) (scale bar = 50 μm). The SNM-encapsulated iBAP with 10% islet density under convection (iii) showed higher viability than that of 10% islet density under diffusion (ii), and 20% islet density under both diffusion (iv) and convection (v). The 10% islet density under diffusion (ii), and 20% islet density under both diffusion (iv) and convection (v) showed similar viability with significant amount of cell death.
Table 1.
A list of SI and viability information for in vitro experimental cases
| Experimental Condition | Mode of Transport | Viability (%) | SI | |
|---|---|---|---|---|
| 10 nm | 10% cell density | Convection | 40 ± 11 | 4.4 ± 0.6 |
| Diffusion | 4 ± 1 | - | ||
| 20% cell density | Convection | 17 ± 11 | 1.1 ± 0.1 | |
| Diffusion | 11 ± 5.8 | - | ||
| 20 nm | 10% cell density | Convection | 66 ± 4.8 | 3.2 ± 1.3 |
| Diffusion | 24 ± 6.8 | - | ||
| 20% cell density | Convection | 61 ± 3.0 | 9.1 ± 1.2 | |
| Diffusion | 5 ± 1 | - | ||
The viability study of ~10 nm pore sized SNM in the iBAP demonstrated that 10% mouse islet density under convection (40 ± 11%) showed a higher viability compared to that under diffusion (4 ± 1%) (Fig. 2,b&c) (Table 1). Furthermore, as the islet density increased to 20% within the islet chamber, the viability significantly decreased under diffusion (11 ± 5.8%) and convection (17 ± 11%) (Fig. 2,b&c) (Table 1). In summary, the ~10 nm pore sized SNM under convection is sufficient to support the viability and glucose-insulin response of 10% (but not 20%) mouse islet density.
To verify whether the pore size was the limiting factor in causing cell death at the higher density, the ~40 nm pore sized SNM with 10% or 20% mouse islet densities was studied in the iBAP under diffusion and convection. The glucose-stimulated insulin study showed that both 10% (Fig. 3,a,i) and 20% (Fig. 3.a,ii) demonstrated the characteristic insulin biphasic release curves. The iBAP at 10% and 20% mouse islet densities with ~40 nm pore sized SNM indicated that the first spike in glucose-stimulated insulin production occurred within 10 minutes of high glucose exposure. Both conditions showed insulin shut down as glucose concentration decreased. The SI at 10% and 20% mouse islet densities with ~40 nm pore sized SNM were 3.2 ± 1.3 and 9.1 ± 1.2, respectively (Table 1). Although the absolute amount of insulin secreted did not double when cell density increased from 10% to 20%, the latter showed a 1.9-fold increase in SI factor, indicating the magnitude of insulin stimulated from basal to high glucose level almost doubled. The viability study demonstrated that 10% mouse islet density under convection (66 ± 4.8%) showed a higher viability compared to that under diffusion (24 ± 6.8%) (Fig. 3,b&c) (Table 1). Furthermore, as the islet density increased to 20% within the islet chamber, the viability of islets under convection (61 ± 3.0%) exhibited a significant increase in viability compared with that under diffusion (5 ± 1%) (Fig. 3,b&c) (Table 1). Overall, the iBAP using ~40 nm pore sized SNM under convection supported the viability and glucose-insulin response at both 10% and 20% mouse islet densities.
Figure 3.



In vitro testing of the intravascular bioartificial pancreas device (iBAP) with 10% or 20% islet density encapsulated with 40 nm-pore size SNM. (a) Glucose-insulin kinetics of the SNM-encapsulated iBAP with 10% (i) or 20% (ii) islet densities under convection was measured from exposing them to a series of low, high, and low glucose conditions. (b) The SNM-encapsulated iBAP with 10% and 20% islet density under convection (10% & 20% convection) showed significantly higher viability compared to that of 10% and 20% islet density under diffusion (10% & 20% diffusion) after 3 days (n > 3, *p < 0.05). Viabilities of islets that were immediately encapsulated in agarose and dispensed into the islet chamber (IC) without further testing were evaluated as the in vitro positive control. (c) Viable (green) and dead (red) cells were stained for in vitro positive control (i), 10% islet density under diffusion (ii), 10% islet density under convection (iii), 20% islet density under diffusion (iv), and 20% islet density under convection (v) (scale bar = 50 μm). The SNM-encapsulated iBAP with 10% and 20% islet density under convection (iii & v) showed higher viability than those under diffusion (ii & iv). In particular, the 20% islet density under diffusion (iv) showed significant amount of cell death.
Compared to the previous in vitro experiments using ~10 nm pore sized SNM, we observed that the ~40 nm pore sized SNM enhanced the viability at 10% and 20% mouse islet densities to 66 ± 4.8% and 61 ± 3.0% as compared to 40 ± 11% and 17 ± 11% for the ~10 nm pore sized SNM under convection, respectively (Fig. 2,b & 3,b) (Table 1). Islet viability correlates positively with an increase in pore size dimension under convection. The greater amount of ultrafiltrate produced by ~40 nm pore size under convection enhanced the viability and functionality of encapsulated islets. In contrast, diffusion provides inadequate mass transfer to support a greater islet density. Although an increase in pore size improved islet viability at a lower cell density (10%) under diffusion from ~10 nm (4 ± 1%) to ~40 nm pore size (24 ± 6.8%), the islet viability at a higher cell density (20%) showed no significant difference under diffusion ((11 ± 5.8%) vs. (5 ± 1%)) (Table 1). These data show that nutrients and oxygen remained severely depleted under diffusion even when the pore size was increased to ~40 nm. Diffusive mass transport has been widely reported for porous materials with nanometer-sized pores38, 39, as one study showed that the diffusion of 45 nm nanoparticles was slowed down by a factor of 2 in 300 nm cylindrical pores due to hydrodynamic friction40. Therefore, given the greater cell density, the large diffusion distance, and the restriction of nanoscaled pores under diffusion, insufficient transfer of nutrients and oxygen would likely result in cell necrosis and hypoxia. In summary, our in vitro testing of the iBAP demonstrated that convection is the key to supporting 10% or 20% mouse islet density with either ~10 nm or ~40 nm pore sized SNM with higher islet viability and providing appropriate glucose-stimulated insulin response (Fig. 2,a&3,a) (Table 1).
iBAP implantation in pigs
As a first step to study the device and membrane patency, the diffusion-based iBAP with ~10 nm pore sized SNM and a 5% mouse islet density (2,850 IE/cm2) was intravascularly grafted in the porcine model for three days. The angiogram showed no thrombosis formation and obstruction in the blood flow path of the device during explant (Fig. 4,a,i) (Video S1). This data matched with previous studies in which the iBAP device was intravascularly implanted into Class A dogs where the device was patent throughout the experiment, possessed no thrombus formation, and generated 27.5 ml of ultrafiltrate based on a SNM pore size of 5.6 nm after explanted at 8 days32. A cytokine panel indicated an expected increase in the pro-inflammatory response from pig immediately after the surgery (Fig. S2). SEM images of blood-contacting SNM displayed some non-catastrophic attachment and aggregations of cells and adhesive proteins (Fig. 4,a,ii). In particular, we observed red blood cells, white blood cells, and platelets deposited on the membrane surface. Subsequent immunohistochemical analysis showed that while platelet adhesion mostly in the porous regions of SNM (which contain the nanopores) as indicated by the green CD41 marker, there was minimal platelet activation observed as stained by the red CD62p marker (Fig. 4,b). The viability study demonstrated that the diffusion-based iBAP in the pig supported the viability at 5% mouse islet density with ~10 nm pore size (88 ± 4.9%) which was comparable with the in vitro conditions (89 ± 2.1%) (Fig. 4,c) (Table 2). To avoid hypoxia and necrosis of cells located at the center of diffusion-based devices, islet density of the macrocapsules has been suggested to be 5–10% of the volume fraction in order to ensure the proper exchange of nutrients and waste of islets5. Our iBAP with ~10 nm pore sized SNM demonstrated sufficient mass transfer to support the viability of 5% islet density under diffusion (Fig. 4,d) (Table 2). Unfortunately, the concentration of mouse insulin and c-peptide in porcine systemic circulation was below the detection limit.
Figure 4.


In vivo testing of the intravascular bioartificial pancreas device (iBAP) with 5% islet density encapsulated with 10 nm-pore size SNM for 3 days. (a) An image of the explanted diffusion-based iBAP (i). An SEM image of the implanted membrane showing attachment of red blood cells and platelets (ii) (scale bar = 10 μm). (b) Immunofluorescence staining of platelet adhesion CD41 marker (green) and platelet activation CD62p marker (red). The rectangular pore-containing regions surrounded by solid silicon regions were shown in the bright field image (i). The platelet adhesion (green) mostly occurred in the window regions where pores reside, whereas minimal platelet activation (red) was detected (ii) (scale bar = 20 μm). (c) The SNM-encapsulated iBAP with 5% islet density under diffusion both in vitro (in vitro 5% diffusion) and in vivo (in vivo 5% diffusion) showed significantly higher viability compared to the in vitro negative control (n > 3, *p < 0.05). The in vitro negative control was those islets that were assembled in the iBAP with no medium circulation for 3 days. Viabilities of islets that were immediately encapsulated in agarose and dispensed into the islet chamber (IC) without further testing were evaluated as the in vitro positive control. (d) Viable (green) and dead (red) cells were stained for in vitro positive control (i), in vitro negative control (ii), in vitro 5% islet density under diffusion (iii), in vivo 5% islet density under diffusion (iv) (scale bar = 50 μm). The SNM-encapsulated iBAP with 5% islet density under convection (iii & v) showed similar viability to the in vitro positive control.
Table 2.
A list of viability information for in vivo experimental conditions with respect to in vitro controls that were conducted together
| Experimental Condition | Mode of Transport | Viability (%) | ||
|---|---|---|---|---|
| 10 nm | 5% cell density | in vivo | Diffusion | 88 ± 4.9 |
| in vitro | 89 ± 2.1 | |||
| 10% cell density | in vivo | Convection | 85 ± 4.4 | |
| Diffusion | 73 ± 4.1 | |||
| in vitro | Convection | 40 ± 11 | ||
| Diffusion | 2 ± 1 | |||
Next, we evaluated the performance of the iBAP with SNM-encapsulated mouse islets under diffusion and convection at a higher islet density to demonstrate the effectiveness of convective mass transfer for supporting islet viability and functionality. Specifically, the iBAP with ~10 nm pore size SNM and a 10% mouse islet density (5,700 IE/cm2) with convective and diffusive mechanism was grafted to the carotid artery and vein of a pig for three days. A pro-inflammatory response was also observed for this pig immediately after the surgery (Fig. S3). No ultrafiltration was generated for the diffusive side, whereas ultrafiltrate production was observed on the convective side. The ultrafiltrate was directly drained into the interstitial space of the animal. After three days, we observed no significant change in device blood flow rate and the ultrafiltrate appeared to be clear, indicating the membranes were intact during the in vivo experiment (Fig. 5,a,i) (Video S2,a). The angiogram also showed no thrombus formation and obstruction in the blood flow path of the device during explant (Video S2,b). Gross inspection of the blood-contacting membrane surfaces showed minimal cellular adhesion for both diffusive and convective conditions; however, the back side of the SNM under convection exhibited a white layer of proteinaceous materials (Fig. 5,b,iii). Our previous study showed that a pore size of ~7 nm SNM can prevent the passage of large molecules such as bovine serum albumin (66.5 kDa)23. Given the pore size (~10 nm) used in this convective transport study, we speculated that some blood-rich proteins such as albumin or fibrinogen passed through and deposited on the back side of the membrane. SEM images of blood-contacting SNM displayed minimal cellular attachment for the diffusive case, whereas there appeared to be more cellular deposition were present for the convective condition (Fig. 5,a,ii). Subsequent immunohistochemical analysis showed that the convection resulted in more platelet adhesion and activation on the blood-exposed SNM surface compared with diffusion (Fig. 5,b). More importantly, the viability of 10% mouse islet density with ~10 nm pore size was higher in the convective condition (85 ± 4.4%) compared to the diffusive scenario (73 ± 4.1%) (Table 2). Interestingly, the in vivo viability at 10% mouse islet density with ~10 nm pore size under diffusion (73 ± 4.1%) was greater than the in vitro viability of those under diffusion (2 ± 1%) and convection (40 ± 11%) (Table 2). The difference in viability between in vivo and in vitro testing under diffusion (Table 1 vs. Table 2) could potentially be due to that blood serves as a better growth medium with essential nutrients for the islets. We also speculate that the higher in vivo cell viability was a result of the ability of hemoglobin from the blood flow path to scavenge nitric oxide (NO), which could be produced by islets under hypoxia41. Previous in vitro studies showed that encapsulating hemoglobin with islets improved islet viability and function as compared to islet encapsulation without hemoglobin42, 43. Furthermore, the ultrafiltrate generated directly from the islet chamber on the convective side indicated a mouse c-peptide concentration of 144 pM (or 12 pg/min/IE (islet equivalent) insulin production rate), exhibiting the functionality of the encapsulated islets. These data demonstrate that SNM encapsulation under convection preserved islet viability and functionality of the encapsulated cells at a cell density for macroencapsulation.
Figure 5.



In vivo testing of the intravascular bioartificial pancreas device (iBAP) with 10% islet density encapsulated with 10 nm-pore size SNM under either diffusion or convection for 3 days. (a) An image of the explanted iBAP with diffusion (back) and convection (front) of the device (i). An SEM image of the diffusion-side implanted membrane showed a patent surface (ii, top) (scale bar = 100 μm) and an SEM image of the convection-side membrane presented coverage of proteins and cells on the surface (ii) (scale bar = 10 μm). (b) Immunofluorescence staining of platelet adhesion CD41 marker (green) and platelet activation CD62p marker (red). The rectangular pore-containing regions surrounded by solid silicon regions were shown in the bright field image for diffusion-side membrane (i) and convection-side membrane (iii). The platelet adhesion (green) was minimal on the diffusion-side membrane (ii), whereas more platelet adhesion (green) and activation (red) was detected on the convection-side membrane (iv) (scale bar = 20 μm). (c) The SNM-encapsulated iBAP with 10% islet density under convection both in vitro (in vitro 10% convection) and in vivo (in vivo 10% convection) showed higher cell viability compared to that under diffusion in vitro (in vitro 10% diffusion) and in vivo (in vivo 10% diffusion). (n > 3, *p < 0.05). The in vitro negative control was those islets that were assembled in the iBAP with no medium circulation for 3 days. Viabilities of islets that were immediately encapsulated in agarose and dispensed into the islet chamber (IC) without further testing were evaluated as the in vitro positive control. (d) Viable (green) and dead (red) cells were stained for in vitro positive control (i), in vitro negative control (ii), in vitro 10% islet density under diffusion (iii), in vitro 10% islet density under convection (iv), in vivo 10% islet density under diffusion (v), in vivo 10% islet density under convection (vi) (scale bar = 50 μm). The SNM-encapsulated iBAP with 10% islet density under convection in vivo (vi) showed similar viability to the in vitro positive control.
To summarize, the in vitro testing of the iBAP demonstrated that SNM with ~10 nm pore size showed an improved viability at 10% mouse islet density (5,700 IE/cm2) under convection, and SNM with ~40 nm pore size demonstrated an increase in viability at 10% (5,700 IE/cm2) and 20% (11,400 IE/cm2) mouse islet densities compared to those tested under diffusion. Furthermore, the glucose-insulin kinetics experiments showed physiological glucose-insulin response and a clinically relevant absolute insulin production rate. Furthermore, porcine studies demonstrated both device and membrane patency under convection and diffusion, a higher islet viability at 10% mouse islet density with convection, and a clinically relevant mouse insulin production rate on a per IE basis. Overall, these studies suggest the feasibility of designing a full-scale SNM-based iBAP to achieve long-term blood flow patency, improved islet viability with convection in comparison to diffusion at clinically relevant densities, and sustained clinically relevant insulin secretion on a per IE basis.
Discussion
A biocompatible, retrievable, and immunoisolating bioartificial pancreas (BAP) is a promising approach to expand islet transplantation to additional T1D patients. Native islets are highly perfused with arterial blood at high pO2 (~100 mmHg) levels and possess short diffusion distances between capillaries and cells (<50 μm) resulting in high mass transfer rates of nutrients, wastes, and insulin16, 44. Cell necrosis occurs when islets are placed beyond the diffusion limit of tissue, which is ~150–200 μm away from the nearest blood vessels9. The high mass transfer rates meet the islets’ high metabolic demands and ensure rapid glycemic control19. Consequently, a successful BAP must possess sufficient mass transfer to achieve physiologic glycemic control. While diffusion-based macrocapsule BAP devices have demonstrated successful alloimmunoisolation and efficacy in small animal models45–47, previous BAP in large animal models have demonstrated poor islet viability and insufficient glycemic control due to inadequate mass transfer. More specifically, inadequate mass transfer resulted from diffusive mass transport over large distances (>500 μm), which created large diffusion gradients of glucose, oxygen, and insulin. The unfavorable gradient reduced islet viability and resulted in poor glucose-insulin kinetics16. In addition, many of these devices were implanted extravascularly resulting in low pO2 (10–50 mmHg) and fibrous tissue formation at the device surface severely limiting oxygen delivery48. Recently, the Viacyte VC-01 device demonstrated successful alloimmunoisolation, efficacy in small animal models, and promising early clinical data on cell viability and differentiation, and device biocompatibility and vascularization45. However, the low oxygen concentration and fibrous tissue at the device surface as well as the large diffusion distances could significantly reduce oxygen, glucose, and insulin mass transfer ultimately resulting in a prohibitively large device for implantation and poor clinical outcomes.
Recognizing the serious oxygenation barrier to BAP development, recent BAP devices have attempted to enhance islet oxygenation. Beta-O2 Technologies (Rosh-Haayin, Israel) has implemented exogenous oxygen supplementation by daily oxygen injection through transcutaneous ports15, 49–52, while Giner (Newton, MA) and their collaborators have been developing an implantable electrochemical oxygen generator17, 53. Both devices enable continuous and long-term supply of oxygen to implanted islets. While these devices have been designed and optimized for enhanced BAP oxygenation and demonstrated superior islet viability and pre-clinical functionality, early clinical translation of the Beta-O2 device yielded poor graft function with a clinically relevant islet dose (160,000 IE)15, 46, 54. While clinical testing of the Beta-O2 device may have achieved poor glycemic control for various reasons, the large diffusion distance (>600 um) (>10x normal islet physiology) for glucose into the device and especially insulin out of the device is likely a significant factor. The large diffusion distance requires a significantly higher insulin production rate within the device to achieve the same systemic insulin delivery rate. The large diffusion distance may also cause high insulin concentrations within the islet local microenvironment, consequently inhibiting insulin production55. In addition to the Giner device being susceptible to the Beta-O2 device limitations, the Giner device’s small surface area (3–9 cm2) may also significantly reduce insulin release, since the rate of insulin release is directly dependent on device surface area55. Consequently both the Beta-O2 and Giner devices may yield a significant delay in insulin release and consequently poor glucose-insulin kinetics. Proper glucose-insulin kinetics is essential to create a functional BAP and even small delays of insulin release may result in hyperinsulinemia and hypoglycemia19, 55. In summary, while these devices supply adequate oxygen, they are not optimized for glucose-insulin kinetics, and may ultimately yield poor glycemic control in the patient.
The use of vascular perfusion devices have been studied to avoid the primary limitations, namely hypoxia and necrosis of cells and the delay of insulin secretion in response to glucose, associated with diffusive transport in extravascular macroencapsulation. Chick and co-workers developed an intravascular hollow fiber device consisting of neonatal islets where the unidirectional blood flow causes the pressure of first part of the fiber lumen to be greater than the pressure in the periphery of the islet compartment, resulting in ultrafiltrate crosses from the bloodstream to the islet graft56. However, blood-contacting hollow-fiber shaped devices with islets at the outside of the artificial capillaries failed due to excess blood clotting in the lumen of those small-diameter capillaries57. Maki and co-workers first demonstrated the long-term use of vascular perfusing BAP in large animals58–61. The hybrid pancreas device contained an acrylic housing with islets separated from the common iliac artery and vein in dogs through the semipermeable membrane (80 kDa). Although no gross fibrosis detected throughout the membrane, clotting occurred at either the anastomosis sites or the junction of the graft. Layers of fibrin-like materials were adhered to the luminal surface of the membrane. Implanted hybrid BAP devices in three dogs failed immediately due to excessive clotting and thrombosis, collapse of membrane and vascular connection, and loss of islet function related to device patency. Although these studies demonstrated the feasibility and clinical applicability of the intravascular hybrid BAP devices, it did not move to the clinical stage due to potential risks associated with thrombosis and hemorrhage. Vascular perfusion devices are advantageous in creating high oxygen tension of the arterial blood exposed to islets, however, risks associated with the surgery required for creating arterio-venous (AV) or arterio-arterio (AA) shunts, vascular thrombosis, and potentials risks in diversion of large volume of blood from the distal extremity limited the development and application of this approach5.
There are three major areas to be considered for making the successful design of next generation of intravascular macroencapsulation devices: (1) sustaining cell viability by minimizing hypoxia and necrosis, (2) demonstrating proper glucose-insulin kinetics, and (3) exhibiting device patency with no blood clotting and thrombosis with minimal systematic anticoagulants. As mentioned above, although intravascular ultrafiltration devices performed better than diffusion-based devices, the low hydraulic permeability of the polymer membranes used in vascular perfusion devices severely limited their performance. The polymer membranes produced inadequate levels of ultrafiltrate resulting in insufficient convective mass transport; the hydraulic permeability of the polymer membranes was about an order of magnitude too low to achieve ideal glucose-insulin kinetics (~10x too low)18, 19, 29, 30. Additionally, the low hydraulic permeability also severely restricted convective oxygen transport, which adversely affected islet viability and function. Consequently, a membrane with a significantly greater hydraulic permeability is needed to provide the convective mass transport rates necessary to ensure proper long-term performance of the promising ultrafiltrate-based intravascular bioartificial pancreas device. Conventional polymer membranes used in previous ultrafiltration-based intravascular BAP devices exhibited hydraulic permeabilities of ~0.40 × 102 ml/hr/m2/mmHg19. Our SNM provides an extremely high hydraulic permeability (3.8 and 230-fold greater for 10 and 40 nm pores, respectively; Table S1) and molecular selectivity with ~1% pore size variation at nanometer scale23 to enhance immunoisolation in comparison to polymer membranes which possess large pore size distributions (~30%)62.
With the decades of profound technological advances and improved clinical experience with prosthetic vascular grafts used in arterial bypass and dialysis access to develop a safe and effective intravascular device, the development of long-term vascular perfusion devices is now more achievable. Our current in vitro and in vivo porcine studies of the SNM-encapsulated iBAP demonstrated the advantages of using convective transport to support a high islet viability at a density of 10% and 20% by volume, providing fast glucose-insulin kinetics, and maintaining device and membrane patency with only aspirin and Plavix given postoperatively. Specifically, the SNM-based iBAP feasibility was demonstrated by: (1) in vitro experiments in which ~10 nm and ~40 nm pore sized SNM enabled high islet viability (>60%) and functionality (<15 minute insulin response to glucose stimulation) at clinically relevant islet densities (5,700 and 11,400 IE/cm2 or 10% and 20% islet densities by volume) under convection; and (2) porcine studies in which ~10 nm pore sized SNM enabled high islet viability (>85%) at clinically relevant islet density (5,700 IE/cm2 or 10% islet density), c-peptide concentration of 144 pM (or 12 pg/min/IE insulin) and hemocompatibility under convection.
Outlook
The iBAP with SNM-encapsulated mouse islets under convection provides ultrafiltrate rates at physiologic blood pressure supporting functional islets at clinically relevant islet densities (5,700 and 11,400 IE/cm2). A full-scale SNM-encapsulated iBAP must next be developed to accommodate a clinically relevant number of islets (50,000–300,000 IE)46, 63 and investigated for the long-term hemocompatibility, ultrafiltrate production, and cell viability and functionality in large diabetic animals and then T1D patients in the future.
Materials and Methods
Silicon Nanopore Membranes (SNM) architecture and fabrication
Silicon nanopore membranes (SNM) have been prototyped from silicon substrates by MEMS technology as previously reported23. Briefly, the process used the growth of a thin SiO2 (oxide) layer on 400 μm-thick double side polished (DSP) silicon wafers followed by a low pressure chemical vapor deposition (LPCVD) of polysilicon (~500 nm). The wafers were then specifically patterned, dry oxidized, wet etched, deposited with a second polysilicon layer, and finally blanket-etched until 400 nm of polysilicon remained and the underlying vertical oxide layer was exposed. The vertical sacrificial oxide layer defined the critical nanoscale pore size of the membranes. The low temperature oxide (LTO) (~1 μm) was deposited onto polysilicon of the wafers to serve as the hard mask for membrane protection. Deep reactive ion etching (DRIE) removed the backside of each window until membranes were disclosed. Eventually, the sacrificial oxide was etched away in 49% hydrofluoric acid (HF) during the final step of the fabrication process to leave behind open nanoscale slit pores. The wafers were subsequently cut into 1×1 cm chips with an effective area of 6×6 mm2 containing 1500 windows each, with a total of 106 pores per membrane. Each rectangular pore was 300 nm in depth and 2 μm in length. The SNM with an average pore size width of ~10 nm and ~40 nm were used in this study. All membranes were cleaned using a conventional “piranha” clean procedure, which involved a 20 min-immersion in 3:1 sulfuric acid (H2SO4)/hydrogen peroxide (H2O2) mixture, followed by thorough rinses in deionized (DI) water. Images of SNM were obtained using scanning electron microscope (SEM) (Leo 1550) (Fig. 1,b).
Surface modification of SNM with poly(ethylene glycol) (PEG)
SNM were covalently modified with PEG using a previously reported protocol to prevent protein fouling on the membrane surface23. The technique used for PEG attachment involved a single reaction step which covalently couples silicon surface silanol group (Si-OH) to a chain of PEG polymer through a trimethoxysilane group forming a Si-O-Si-PEG sequence. Briefly, SNM were immersed in a solution of 3 mM 2-[methoxy(polyethyleneoxy)propyl]trimethoxysilane (PEG-silane) (Gelest: SIM6492.7) in toluene for 2 hr at 70 °C. A series of extensive washing steps involving toluene, ethanol, and DI water was used to remove unbounded PEG residue.
Hydraulic permeability for SNM pore size characterization
An automated mass and pressure measurement system was utilized for characterizing liquid flow through the SNM under a tangential-flow filtration operation23. The pore size of the SNM can be related to filtration flow parameters using
| (Eq. 1) |
where h is pore width, μ is the viscosity, l is the membrane thickness, Q is the volumetric flow rate, n is the number of pores per membrane, w is the pore length, and ΔP is the transmembrane pressure. To assemble the overall system for SNM pore size characterization, air was applied through a syringe pump (Sigma: Z675709) into a water reservoir. Water was circulated by a peristaltic pump (Masterflex: 07551-00) through a differential pressure transducer (Omega: PX429 015GI), a flow cell with enclosed membrane, and returned to the original water reservoir. The flow cell was assembled with the SNM submerged under water to remove air bubbles from all compartments. Specifically, a membrane was positioned with the polysilicon interface facing down with a customized silicone gasket positioned on top of the membrane, followed by the final placement of a filtrate chamber on top of the gasket. All sections were fastened together and secured to the base with hand-tightened hex bolts until the gasket was visibly compressed. The ultrafiltrate permeated through the membrane and was routed to a liquid collection container that rested on a precision mass balance (Mettler Toledo: XS205). Measurements from the differential pressure transducer and the mass balance were automatically collected with a data acquisition laptop. A typical membrane hydraulic permeability test consisted of 5 ml/min flow rate and 4 pressure cycles (5, 1, 5, and 1 psi) for durations of 150 s each. Using the specifications for pore length, membrane thickness, and total number of pores provided based on individual wafer designs, the average pore size of SNM was calculated using Equation 1. All SNM membranes in this study were surface-modified with PEG and exhibited an average pore size of ~10 nm and ~40 nm.
Development of the islet chamber (IC)
In this study, IC possessed a thickness of 1000 μm and high islet densities of 10% (5,700 IE/cm2) and 20% (11,400 IE/cm2). Islet density by percentage was calculated as the ratio of total islet volume expressed in islet equivalents and the IC volume. Islet density by surface area was calculated by dividing the total number of islet equivalents (IE) by the device membrane surface area. A biocompatible acrylic sheet (McMaster: 8589K11) was first laser-cut to create ~2.4 mm × ~2.4 mm × ~1 mm thick void region which was inserted with eight 100 μm diameter polytetrafluoroethlene (PTFE) coated wires (McMaster:1749T11). A 2% agarose-islet mixture was then poured into this void region of acrylic sheets. After the agarose-islet mixture was cured, all wires were removed (Fig. 1,c,i). Using this process, a hexagonal arrangement of eight 800-μm cylindrical agarose-islet regions (dotted red circle) with a central 100 μm cylindrical channel (solid red circles) was obtained for the IC (Fig. 1,c,ii). This configuration created a diffusion distance ≤ 400 μm between the islets and ultrafiltrate. After IC construction, it was assembled in the iBAP as described in Figure 1,a with gaskets between the various iBAP components.
Assembly of an intravascular bioartificial pancreas device (iBAP) for islet encapsulation
The intravascular bioartificial pancreas device (iBAP) is shown in an exploded view in Figure 1,a: the polycarbonate flow path component containing the blood flow path, two SNM sandwiching the islet chamber (IC) containing the agarose (Sigma: A2576)-seeded mouse islets, the polycarbonate backside (PC Backside), and the ultrafiltrate port (Ultrafiltrate Outlet). The parallel-plate blood flow path was modeled with SolidWorks and computational fluid dynamics (CFD) to create ideal flow characteristics to minimize thrombosis34. The iBAP was symmetrical on both sides and could be assembled with one IC on each side. The iBAP can possess up to 0.72 cm2 of SNM area. In operation, fluid flows through the Flow Path component at an elevated pressure creating a transmembrane pressure (TMP) of ~80 mmHg between the blood flow path and the Ultrafiltrate Outlet resulting in ultrafiltrate flow through the SNM, IC, PC Backside, and Ultrafiltrate Outlet, which was collected in a tube in vitro or drained into interstitial tissue space in vivo. Under the diffusive condition, the PC Backside was capped-off resulting in no ultrafiltrate flow through the system. All device components were individually sterilized either by autoclave or Nolvasan for both in vitro and in vivo experiments.
Testing of the intravascular bioartificial pancreas device (iBAP) in vitro
All procedures involving isolation of mouse islets were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Francisco (UCSF). Mouse islets were isolated from 8 to 10-week-old male B6 mice (Jackson Laboratories) based on previously described protocols. Harvested islets were maintained in suspension culture with RPMI 1640 with L-glutamine and 11.1 mM glucose (Gibco: 11875-093), 10% fetal bovine serum (FBS) (Gibco: 16000), and 1% penicillin-streptomycin (P/S) (UCSF Cell Culture Facility: CCFGK003). A 2% agarose gel mixed with appropriate amount of mouse islets was dispensed into the previously described IC to create high islet densities of 10% or 20% by volume, respectively. The SNM with ~10 nm and ~40 nm pore sizes were chosen to encapsulate the IC with 10% and 20% islet density. For in vitro viability tests, a mock-loop circuit was set up with a peristaltic pump flowing culture medium through the iBAP at TMP of ~80 mmHg to generate ultrafiltrate for convective condition (Fig. 1,a,ii), whereas no ultrafiltrate was produced for the diffusive condition. The viability experiments studied both ~10 nm and ~40 nm SNM encapsulating both 10% and 20% islet densities. After 3 days of culture, the devices were disassembled and the islets were assessed for viability. Using this same mock-loop circuit, glucose-insulin kinetics was explored in iBAPs containing either 10% or 20% islet density and either ~10 nm or ~40 nm pore sized SNM. SNM-encapsulated mouse islets in the iBAP were exposed to a low, high, and low glucose (Gibco: 11879) challenge on day 0. Ultrafiltrate directly produced from the IC under convection was collected for insulin measurements. Insulin content was analyzed with mouse insulin enzyme-linked immunosorbent assay (ELISA) kits (Mercodia: 10-1247-01) with accounted dilutions.
Implantation of the intravascular bioartificial pancreas device (iBAP) in pigs
A preliminary proof-of-concept study was designed with a swine model because of the comparably sized vasculature and hematologic similarities with humans. The study was approved by the IACUC review committee at PMI Preclinical CRO, San Carlos, CA. All pigs used in this study were non-diabetic. Pigs were given heparin perioperatively and intraoperatively, while only aspirin and Plavix were given postoperatively.
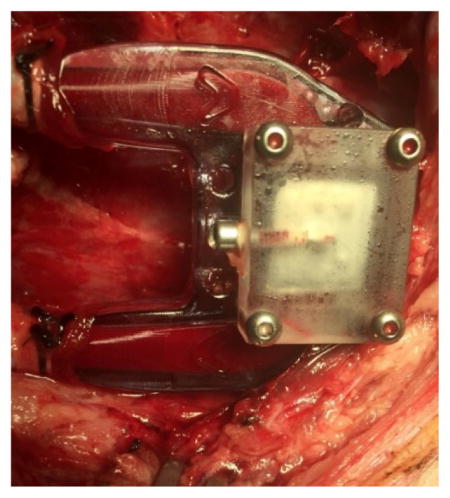
For pig #1, the device was assembled as previously described with each chamber containing a 5% islet equivalents (IE) density by volume of mouse islets suspended in agarose gel. Oral aspirin (81mg) and clopidogrel (75mg) were given to a 75kg female Yorkshire pig for 3 days preoperatively and then daily thereafter. After general anesthesia was induced, a vertical incision was made to the left of midline to expose the left external jugular vein. A 15Fr double-lumen tunneled catheter (NextStep®, Teleflex, Morrisville, NC) was placed in the left external jugular vein for blood sampling. The right carotid artery and right external jugular vein were then exposed via a similar vertical incision on the right side of the neck. Once the vessels were exposed a subcutaneous pocket was created caudally for eventual device placement. Heparin was given intraoperatively targeting an activated clotting time (ACT) of greater than 200 seconds. The 6mm externally-supported polytetrafluoroethylene (PTFE) grafts were then anastomosed end-to-side to the internal carotid artery for inflow and the external jugular vein for outflow. The device was then placed in the subcutaneous pocket and anchored to surrounding soft tissue. The inflow and outflow grafts were then connected to the device and clamps were removed to allow blood flow through the device, which was visually confirmed (Fig. 4). The overlying soft tissue and skin were then closed in layers and then animal was extubated and allowed to recover. Meloxicam and buprenorphine were administered as needed for post-operative pain.
For pig #2, the device was assembled as previously described with 10% density by volume for each chamber. One chamber had a channel in communication with the islets that was open to atmosphere, allowing for ultrafiltrate flow through the chamber (Fig. 1,a,ii). The ultrafiltrate then passed through the channel and was freely deposited into the surrounding tissue and subcutaneous pocket (Fig. 5,a). The other chamber’s ultrafiltrate outlet was capped resulting in a diffusive chamber. The technical aspects of the implant procedure were identical to Pig #1. Blood flow through the device and ultrafiltrate deposition into the surrounding tissue was visually confirmed prior to closure of the incision.
Assessment of islet function in vivo
Islet function was assessed using standard intravenous glucose tolerance tests (IVGTT) with administration of glucose (0.5g/kg in 40% solution) via the tunneled venous catheter. Blood was drawn to measure serum glucose using a standard glucometer (Accu-Chek Compact Plus: 1002–5021) at time 0, 5, 10, 15, 30, 60 and 90 minutes. The IVGTT was administered on post-operative day (POD) 1 and 2 prior to the animals eating their morning meals. On POD 3 the test was performed intra-operatively prior to planned explant of the device and islet retrieval. For pig #1, blood sampling for the intra-operative IVGTT was performed via direct cannulation of the external jugular venous outflow tract immediately distal to the anastomosis. All samples from systematic circulation and directly collected from ultrafiltration port were stored on ice prior to testing for mouse insulin enzyme-linked immunosorbent assay (ELISA) (Mercodia: 10-1247-01) and c-peptide ELISA kits (EMD Millipore: EZRMCP2-21K).
Patency assessment and device explant with islet retrieval
On POD 3 both animals were taken back to the operating room for assessment of patency and device retrieval. Once the animal was intubated and sedated the incision was re-opened and the device was delivered into the superficial tissue for visual assessment and confirmation of maintained patency. A final IVGTT was administered. As mentioned, for pig #1, blood was sampled directly from the outflow vein via direct cannulation of the external jugular vein distal to the anastomosis. For pig #2, blood was sampled from the tunneled catheter. Once the IVGTT was completed, the carotid was cannulated proximal to the anastomosis with a 5Fr catheter. Radiopaque contrast media was then injected (Visipaque™, GE Healthcare, Little Chalfont, United Kingdom) to fluoroscopically confirm flow through the device (Fig. 4,a & 5,a). The inflow and outflow grafts were then clamped and the device was then explanted and subsequently flushed with culture media prior to disassembly and retrieval of the islets from the chamber.
Islet viability
Islet viability was assessed by double staining with the Live/Dead Cell Imaging Kit (488/570) (Life Technology: R37601). Live cells are distinguished by the presence of intensely fluorescent calcein (green) which is well-retained within live cells, whereas dead cells are stained with red. Briefly, agarose-encapsulated mouse islets were incubated in the mixture of live (green) and dead (red) kit components for 15 min and extensively washed in phosphate buffered saline (PBS) to remove excess staining. Images of mouse islets were obtained using laser scanning Nikon Spectral C1si confocal microscope (Nikon Instruments). The percentage of viability was calculated based on the ratio of non-dead or the green area over the entire area of that islet.
Explanted membrane analysis
For observation, SNM were fixed in a solution containing 3% glutaraldehyde (Sigma: G7651), 1 M sodium cacodylate (Polysciences) and 0.1 M sucrose (Sigma). After 2 days, the substrates were washed with distilled water. Dehydration was achieved by placing these scaffolds in an increasing concentration of ethanol (50–100%). Dehydrated samples were then mounted on aluminum stubs, sputter-coated with gold–palladium, and examined with scanning electron microscopy (SEM) (Ultra 55, Carl Zeiss).
Blood platelet adhesion and activation
The SNM were fixed with 4% paraformaldehyde followed by PBS washes and incubated in blocking solution (PBS, 1% bovine serum albumin (BSA)) for 30 min. Samples were then incubated with CD41 antibody (Biorbyt: orb181793) for platelet adhesion (green) and CD62p antibody (Bioss: bs-0561R-Cy3) for platelet activation (red) at a dilution of 1:300 for 4 h and repeatedly washed with PBS to remove residues. Images were obtained using 6D High Throughput Perfect Focus System (Nikon Instruments).
Statistical analysis
Sample pairs were analyzed using Student’s t-test. Multiple samples were evaluated with one-way or two-way analysis of variance (ANOVA) followed by Bonferroni and multiple comparison using Graphpad Prism software (San Diego, CA). A p value of <0.05 was accepted as statistically significant for all analyses.
Supplementary Material
Acknowledgments
This research was supported by National Science Foundation (NSF) Graduate Research Fellowship (SS), Harry Wm. & Diana V. Hind Distinguished Professorship in Pharmaceutical Sciences II (SR), and the Juvenile Diabetes Research Foundation (JDRF). We would like to thank colleagues Gregory Szot and Vi Dang for their help with mouse islet isolation, and Eun Jung Kim for SEM images of explanted SNM.
Footnotes
Conflict of interest statement. Dr. Shuvo Roy and Dr. William Fissell are founders of Silicon Kidney, LLC. Charles Blaha and Nathan Wright have worked as employees of Silicon Kidney, LLC.
Author contribution. S.S., C.B., and S.R. designed research; N.W. provided flow cell drawing and device components; S.S. and C.B. performed in vitro and in vivo device preparation and assembly; S.S. analyzed in vitro and in vivo cell viability, immunohistochemistry, and SEM staining; C.B helped with assay testing; J.P. provided unreleased membranes; S.S, C.B, W.M. and S.V. helped with in vivo implantation; S.S. and C.B. wrote the paper; and W.M. J.P., N.W., J.G., W.F, A.M.P., S.V., and S.R. all participated in the project and manuscript discussion.
References
- 1.Gibly RF, Graham JG, Luo X, Lowe WL, Jr, Hering BJ, Shea LD. Diabetologia. 2011;54:2494–2505. doi: 10.1007/s00125-011-2243-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang TM. Science. 1964;146:524–525. doi: 10.1126/science.146.3643.524. [DOI] [PubMed] [Google Scholar]
- 3.Monaco AP, Maki T, Ozato H, Carretta M, Sullivan SJ, Borland KM, Mahoney MD, Chick WL, Muller TE, Wolfrum J, Solomon B. Ann Surg. 1991;214:339–362. doi: 10.1097/00000658-199109000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Sullivan ES, Vegas A, Anderson DG, Weir GC. Endocrine reviews. 2011;32:827–844. doi: 10.1210/er.2010-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song S, Roy S. Biotechnology and bioengineering. 2016;113:1381–1402. doi: 10.1002/bit.25895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sullivan SJ, Maki T, Borland KM, Mahoney MD, Solomon BA, Muller TE, Monaco AP, Chick WL. Science. 1991;252:718–721. doi: 10.1126/science.2024124. [DOI] [PubMed] [Google Scholar]
- 7.Tuch BE, Keogh GW, Williams LJ, Wu W, Foster JL, Vaithilingam V, Philips R. Diabetes care. 2009;32:1887–1889. doi: 10.2337/dc09-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colton CK. Cell Transplant. 1995;4:415–436. doi: 10.1177/096368979500400413. [DOI] [PubMed] [Google Scholar]
- 9.Thomlinson RH, Gray LH. British journal of cancer. 1955;9:539–549. doi: 10.1038/bjc.1955.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchwald P. Theoretical biology & medical modelling. 2011;8:20. doi: 10.1186/1742-4682-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams SJ, Huang HH, Kover K, Moore W, Berkland C, Singh M, Smirnova IV, MacGregor R, Stehno-Bittel L. Organogenesis. 2010;6:115–124. doi: 10.4161/org.6.2.10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calafiore R. Diabetes care. 1997;20:889–896. doi: 10.2337/diacare.20.5.889. [DOI] [PubMed] [Google Scholar]
- 13.Colton CK, Avgoustiniatos ES. Journal of biomechanical engineering. 1991;113:152–170. doi: 10.1115/1.2891229. [DOI] [PubMed] [Google Scholar]
- 14.Elliott RB, Escobar L, Tan PL, Garkavenko O, Calafiore R, Basta P, Vasconcellos AV, Emerich DF, Thanos C, Bambra C. Transplantation proceedings. 2005;37:3505–3508. doi: 10.1016/j.transproceed.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 15.Ludwig B, Reichel A, Steffen A, Zimerman B, Schally AV, Block NL, Colton CK, Ludwig S, Kersting S, Bonifacio E, Solimena M, Gendler Z, Rotem A, Barkai U, Bornstein SR. P Natl Acad Sci USA. 2013;110:19054–19058. doi: 10.1073/pnas.1317561110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mikos AG, Papadaki MG, Kouvroukoglou S, Ishaug SL, Thomson RC. Biotechnology and bioengineering. 1994;43:673–677. doi: 10.1002/bit.260430717. [DOI] [PubMed] [Google Scholar]
- 17.Papas KK, Steyn L, Davies M, Loudovaris T, Limesand S, Tempelman L, Avgoustiniatos ES. Tissue Eng Pt A. 2014;20:S123–S123. [Google Scholar]
- 18.Reach G, Jaffrin MY. Comput Meth Prog Bio. 1990;32:277–285. doi: 10.1016/0169-2607(90)90110-u. [DOI] [PubMed] [Google Scholar]
- 19.Reach G, Jaffrin MY, Desjeux JF. Diabetes. 1984;33:752–761. doi: 10.2337/diab.33.8.752. [DOI] [PubMed] [Google Scholar]
- 20.Thijssen DHJ, Dawson EA, Black MA, Hopman MTE, Cable NT, Green DJ. Med Sci Sport Exer. 2009;41:1072–1079. doi: 10.1249/MSS.0b013e3181923957. [DOI] [PubMed] [Google Scholar]
- 21.Chu W, Huen T, Tu J, Ferrari M. P Soc Photo-Opt Ins. 1997;2978:111–122. [Google Scholar]
- 22.Desai TA, Chu WH, Tu JK, Beattie GM, Hayek A, Ferrari M. Biotechnology and bioengineering. 1998;57:118–120. doi: 10.1002/(sici)1097-0290(19980105)57:1<118::aid-bit14>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 23.Song S, Faleo G, Yeung R, Kant R, Posselt AM, Desai TA, Tang Q, Roy S. Scientific reports. 2016;6:23679. doi: 10.1038/srep23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fissell WH, Dubnisheva A, Eldridge AN, Fleischman AJ, Zydney AL, Roy S. J Membrane Sci. 2009;326:58–63. doi: 10.1016/j.memsci.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desai TA, Hansford DJ, Ferrari M. Biomolecular engineering. 2000;17:23–36. doi: 10.1016/s1389-0344(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 26.Sabek OM, Ferrati S, Fraga DW, Sih J, Zabre EV, Fine DH, Ferrari M, Gaber AO, Grattoni A. Lab on a chip. 2013;13:3675–3688. doi: 10.1039/c3lc50601k. [DOI] [PubMed] [Google Scholar]
- 27.Kanani DM, Fissell WH, Roy S, Dubnisheva A, Fleischman A, Zydney AL. J Membrane Sci. 2010;349:405–410. doi: 10.1016/j.memsci.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maher JF. Replacement of Renal Function by Dialysis: A Text Book of Dialysis. Springer Science & Business Media; 1989. [Google Scholar]
- 29.Jaffrin MY, Reach G, Notelet D. Journal of biomechanical engineering. 1988;110:1–10. doi: 10.1115/1.3108400. [DOI] [PubMed] [Google Scholar]
- 30.Moussy F, Rouchette J, Reach G, Cannon R, Jaffrin MY. Artificial organs. 1989;13:109–115. doi: 10.1111/j.1525-1594.1989.tb02845.x. [DOI] [PubMed] [Google Scholar]
- 31.Reach G. The International journal of artificial organs. 1990;13:329–336. [PubMed] [Google Scholar]
- 32.Kensinger C, Karp S, Kant R, Chui BW, Goldman K, Yeager T, Gould ER, Buck A, Laneve DC, Groszek JJ, Roy S, Fissell WH. ASAIO J. 2016;62:491–495. doi: 10.1097/MAT.0000000000000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muthusubramaniam L, Lowe R, Fissell WH, Li L, Marchant RE, Desai TA, Roy S. Annals of biomedical engineering. 2011;39:1296–1305. doi: 10.1007/s10439-011-0256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melvin ME, Fissell WH, Roy S, Brown DL. ASAIO J. 2010;56:344–348. doi: 10.1097/MAT.0b013e3181d98cf8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conlisk AT, Datta S, Fissell WH, Roy S. Annals of biomedical engineering. 2009;37:722–736. doi: 10.1007/s10439-009-9642-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hedeskov CJ. Physiological reviews. 1980;60:442–509. doi: 10.1152/physrev.1980.60.2.442. [DOI] [PubMed] [Google Scholar]
- 37.Sparks RE, Mason NS, Finley TC, Scharp DW. Transactions - American Society for Artificial Internal Organs. 1982;28:229–231. [PubMed] [Google Scholar]
- 38.Nagumo R, Takaba H, Nakao SI. Chem Phys Lett. 2008;458:281–284. [Google Scholar]
- 39.Jiang XQ, Mishra N, Turner JN, Spencer MG. Microfluid Nanofluid. 2008;5:695–701. [Google Scholar]
- 40.Chen F, Neupane B, Li P, Su W, Wang G. Electrophoresis. 2016;37:2129–2138. doi: 10.1002/elps.201600158. [DOI] [PubMed] [Google Scholar]
- 41.Lanza R, Langer R, Vacanti J. Principles of Tissue Engineering, 3rd Edition. 2007:Xxxi-Xxxi. doi: 10.1016/B978-012370615-7/50002-0. [DOI] [Google Scholar]
- 42.Chae SY, Lee M, Kim SW, Bae YH. Biomaterials. 2004;25:843–850. doi: 10.1016/s0142-9612(03)00605-7. [DOI] [PubMed] [Google Scholar]
- 43.Nadithe V, Mishra D, Bae YH. Biotechnology and bioengineering. 2012;109:2392–2401. doi: 10.1002/bit.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim A, Miller K, Jo J, Kilimnik G, Wojcik P, Hara M. Islets. 2009;1:129–136. doi: 10.4161/isl.1.2.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agulnick AD, Ambruzs DM, Moorman MA, Bhoumik A, Cesario RM, Payne JK, Kelly JR, Haakmeester C, Srijemac R, Wilson AZ, Kerr J, Frazier MA, Kroon EJ, D’Amour KA. Stem cells translational medicine. 2015;4:1214–1222. doi: 10.5966/sctm.2015-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colton CK. Advanced drug delivery reviews. 2014;67–68:93–110. doi: 10.1016/j.addr.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 47.Kumagai-Braesch M, Jacobson S, Mori H, Jia X, Takahashi T, Wernerson A, Flodstrom-Tullberg M, Tibell A. Cell Transplant. 2013;22:1137–1146. doi: 10.3727/096368912X657486. [DOI] [PubMed] [Google Scholar]
- 48.Thrash M. ASAIO J. 2010;56:338–343. doi: 10.1097/MAT.0b013e3181e3f874. [DOI] [PubMed] [Google Scholar]
- 49.Ludwig B, Ludwig S. Langenbeck’s archives of surgery. 2015;400:531–540. doi: 10.1007/s00423-015-1314-y. [DOI] [PubMed] [Google Scholar]
- 50.Barkai U, Weir GC, Colton CK, Ludwig B, Bornstein SR, Brendel MD, Neufeld T, Bremer C, Leon A, Evron Y, Yavriyants K, Azarov D, Zimermann B, Maimon S, Shabtay N, Balyura M, Rozenshtein T, Vardi P, Bloch K, de Vos P, Rotem A. Cell Transplant. 2013;22:1463–1476. doi: 10.3727/096368912X657341. [DOI] [PubMed] [Google Scholar]
- 51.Neufeld T, Ludwig B, Barkai U, Weir GC, Colton CK, Evron Y, Balyura M, Yavriyants K, Zimermann B, Azarov D, Maimon S, Shabtay N, Rozenshtein T, Lorber D, Steffen A, Willenz U, Bloch K, Vardi P, Taube R, de Vos P, Lewis EC, Bornstein SR, Rotem A. PloS one. 2013;8:e70150. doi: 10.1371/journal.pone.0070150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ludwig B, Zimerman B, Steffen A, Yavriants K, Azarov D, Reichel A, Vardi P, German T, Shabtay N, Rotem A, Evron Y, Neufeld T, Mimon S, Ludwig S, Brendel MD, Bornstein SR, Barkai U. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2010;42:918–922. doi: 10.1055/s-0030-1267916. [DOI] [PubMed] [Google Scholar]
- 53.Papas KK, Mueller KR, Penrod LV, Davis MA, Kitzmann JP, Gruessner SG, Suszynski TM, Jie T, Tempelman L, Limesand SW, Avgoustiniatos ES. Transplantation. 2013;96:S71–S71. [Google Scholar]
- 54.Warnock GL, Rajotte RV. Diabetes. 1988;37:467–470. doi: 10.2337/diab.37.4.467. [DOI] [PubMed] [Google Scholar]
- 55.Scharp DW, Marchetti P. Advanced drug delivery reviews. 2014;67–68:35–73. doi: 10.1016/j.addr.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 56.Chick WL, Perna JJ, Lauris V, Low D, Galletti PM, Panol G, Whittemore AD, Like AA, Colton CK, Lysaght MJ. Science. 1977;197:780–782. doi: 10.1126/science.407649. [DOI] [PubMed] [Google Scholar]
- 57.Colton CK. Cell Transplant. 1995;4:415–436. doi: 10.1177/096368979500400413. [DOI] [PubMed] [Google Scholar]
- 58.Maki T, Lodge JP, Carretta M, Ohzato H, Borland KM, Sullivan SJ, Staruk J, Muller TE, Solomon BA, Chick WL, et al. Transplantation. 1993;55:713–717. doi: 10.1097/00007890-199304000-00005. discussion 717–718. [DOI] [PubMed] [Google Scholar]
- 59.Maki T, Otsu I, ONeil JJ, Dunleavy K, Mullon CJP, Solomon BA, Monaco AP. Diabetes. 1996;45:342–347. doi: 10.2337/diab.45.3.342. [DOI] [PubMed] [Google Scholar]
- 60.Maki T, Ubhi CS, Sanchez-Farpon H, Sullivan SJ, Borland K, Muller TE, Solomon BA, Chick WL, Monaco AP. Transplantation. 1991;51:43–51. doi: 10.1097/00007890-199101000-00006. [DOI] [PubMed] [Google Scholar]
- 61.Maki T, Ubhi CS, Sanchez-Farpon H, Sullivan SJ, Borland K, Muller TE, Solomon BA, Chick WL, Monaco AP. Transplantation proceedings. 1991;23:754–755. [PubMed] [Google Scholar]
- 62.Dunleavy M. Medical device technology. 1996;7:14–16. 18–21. [PubMed] [Google Scholar]
- 63.Fujita Y, Takita M, Shimoda M, Itoh T, Sugimoto K, Noguchi H, Naziruddin B, Levy MF, Matsumoto S. Islets. 2011;3:1–5. doi: 10.4161/isl.3.1.14131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.