


Abstract
DNA repair status plays a major role in mutagenesis, carcinogenesis and resistance to genotoxic agents. Because DNA repair processes involve multiple enzymatic steps, understanding cellular DNA repair status has required several assay procedures. We have developed a novel in vitro assay that allows quantitative measurement of alkylation repair via O6-methylguanine DNA methyltransferase (MGMT) and base excision repair (BER) involving methylpurine DNA glycosylase (MPG), human 8-oxoguanine DNA glycosylase (hOGG1) and yeast and human abasic endonuclease (APN1 and APE/ref-1, respectively) from a single cell extract. This approach involves preparation of cell extracts in a common buffer in which all of the DNA repair proteins are active and the use of fluorometrically labeled oligonucleotide substrates containing DNA lesions specific to each repair protein. This method enables methylation and BER capacities to be determined rapidly from a small amount of starting sample. In addition, the stability of the fluorometric oligonucleotides precludes the substrate variability caused by continual radiolabeling. In this report this technique was applied to human breast carcinoma MDA-MB231 cells overexpressing human MPG in order to assess whether up-regulation of the initial step in BER alters the activity of selected other BER (hOGG1 and APE/ref-1) or direct reversal (MGMT) repair activities.
INTRODUCTION
Two major pathways exist in mammalian cells for the removal and repair of alkylation and oxidative DNA damage (1). The most direct mechanism for repairing alkylation DNA damage is one that simply reverses the damage. An example of this type of repair mechanism is the DNA repair protein O6-methylguanine (O6-MeG)-DNA methyltransferase (MGMT or AGT), which removes alkyl groups from DNA. MGMT repairs alkylation damage through a stoichiometric reaction whereby the alkyl group is transferred from the O6 position of guanine to a cysteine residue within the protein structure (Fig. 1C). Because this reaction results in irreversible inactivation of the protein molecule, MGMT is commonly described as a ‘suicide enzyme’. If left unrepaired, O6-MeG adducts can mispair with thymine, causing a G:C→A:T transition that is associated with both mutagenesis and carcinogenesis (1).
Figure 1.

BER and MGMT repair pathways. (A) Repair of alkylated adenine (*) DNA residues initiated by MPG. (B) Repair of 8-oxoG (*) DNA residues initiated by hOGG1. (C) Repair of O6-MeG (*) by MGMT.
The DNA base excision repair (BER) pathway repairs alkylation, oxidative and ionizing radiation forms of DNA damage and provides protection against both endogenous and exogenous genotoxic agents. The first step of the BER pathway involves removal of the incorrect or damaged base by N-methylpurine DNA glycosylase (MPG or AAG) (Fig. 1A). MPG not only repairs the major cytotoxic DNA alkylation lesion, 3-methyladenine (3-meA), but also cleaves the primary product of many DNA alkylating agents, N7-methylguanine (2). MPG also recognizes and removes 1,N6-ethenoadenine, which is produced by exogenous pollutants and lipid peroxidation (3). Removal of these lesions by MPG produces an apurinic/apyrimidinic (AP), or abasic, site. Hence, the next step in the BER pathway involves cleavage of the DNA backbone at the AP site, which is mediated by AP endonuclease (APE) activity. APEs function in the recognition of AP sites, the most common form of DNA damage with about 10 000–20 000 apurinic and 500 apyrimidinic sites produced per cell each day under normal physiological conditions. The major APE in mammalian cells is APE/ref-1 (4) and in Saccharomyces cerevisiae the main APE is the functional homolog APN1. APE/ref-1 hydrolyzes the phosphodiester backbone immediately 5′ of an AP site, generating an abasic deoxyribose 5-phosphate that is released by a 5′-deoxyribophosphodiesterase (dRPase) or 5′-exonuclease. The repair process is then completed by DNA synthesis and ligation (Fig. 1A).
While MPG typifies a simple glycosylase that removes damaged bases without attacking the DNA backbone, complex glycosylases recognize damaged bases and cleave the DNA backbone on the 3′ side of the AP site through β-lyase activity (Fig. 1B). A complex glycosylase that recognizes the oxidative DNA lesion 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxoG) is 8-oxoguanine-DNA glycosylase (hOGG1). In addition, hOGG1 has also been shown to remove formamidopyrimidine lesions (5). Subsequently, the terminal ends must be modified by a 3′-phosphatase or 3′-phosphodiesterase, a reaction which has been shown to be performed by APE1, before DNA polymerase β can fill in the correct nucleotide (6). The 8-oxoG lesion is highly mutagenic in vitro and in vivo (7,8), causing G:C→T:A transversions (9,10).
DNA repair processes, including MGMT and BER, are involved in protection against various genotoxic processes. For example, MGMT is a primary mechanism of resistance to the chloroethylnitrosourea class of anticancer drugs in human tumors (11,12). BER has been shown to function in cytoprotection against alkylating agents, oxidative compounds and ionizing radiation (13,14). Because BER is a multistep process involving several repair proteins composite function of the BER pathway is important in assessing the cellular protective response. The activities of the BER enzymes should therefore be examined collectively. The method described here allows five DNA repair activities, including three components of the BER pathway, to be measured in a single cell extract. The ability to measure the activities of the enzymes that initiate the BER pathway (MPG and hOGG1) and the enzyme that subsequently either cuts the DNA backbone or ‘polishes’ the resulting termini (APE/ref-1) from a single sample in a common reaction buffer would simplify determination of the composite BER status. Furthermore, the ability to measure MGMT activity in the same buffer system also allows for direct comparisons to be made between these two important pathways for the repair of alkylation DNA damage. This method also incorporates novel fluorometric oligonucleotide substrates, which afford less variability of substrate labeling and substantial cost savings when compared to radiolabeled oligonucleotide substrates. We have employed this approach in the current study to determine whether selective overexpression of MPG in human breast carcinoma cells alters other cellular DNA repair activities, including MGMT, hOGG1 and APE/ref-1.
MATERIALS AND METHODS
Materials
Oligonucleotides employed to measure MGMT activity, containing the O6-MeG lesion with or without a 5′-hexachloro-fluorescein phosphoramidite (HEX) molecule and complementary strand, were custom synthesized by Genosys Biotechnologies (The Woodlands, TX) and purified by PAGE. Oligonucleotides for measurement of MPG, hOGG1, APE1 and APN1 activities were custom synthesized by The Midland Certified Reagent Co. (Midland, TX). They contained ethanoadenine, 8-oxoG and tetrahydrofuran (THF) lesions, respectively, with or without a HEX molecule and complementary strands, and were purified by the standard manufacturer’s protocol. The fluorometric HEX molecule was incorporated into the 5′-end of the lesioned strand during synthesis. Respective oligonucleotide strands were annealed to complementary strands in 10 mM Tris–HCl, 1 mM EDTA, 33 mM NaCl, pH 8.0, by heating to 90°C for 2 min to denature and cooling to room temperature overnight, then diluted to 2 pmol/ml and stored in aliquots at –20°C. All chemical reagents were obtained from Sigma Chemical Co. (St Louis, MO), Fisher Scientific (Chicago, IL) or US Biochemical (Cleveland, OH) unless otherwise stated.
Instrumentation
Radiolabeled oligonucleotides were detected and quantitated following PAGE using a Storm 860 Phosphorimager (Molecular Dynamics, Sunnyvale, CA). Fluorometric HEX-labeled oligonucleotides were detected and quantitated using the Hitachi FMBio II Fluorescence Imaging System (Hitachi Genetic Systems, South San Francisco, CA). The HEX fluorophore is excited by a solid-state laser at 532 nm (Perkin-Elmer) and emits a fluorescent light signal at 560 nm, which is then isolated using a 585 nm filter. Fluorescence intensity units were quantitated using FMBio software (Hitachi Genetic Systems).
Cell culture
Human breast carcinoma MDA-MB231 cells were kindly provided by Dr Martin Smith (Department of Microbiology, Indiana University School of Medicine). HeLa and human leukemia K562 cells were purchased from ATCC (Rockville, MD). HeLa cells were cultured in Dulbecco’s modified essential medium supplemented with 10% bovine calf serum (Hyclone Laboratories, Logan, UT), 1% l-glutamine, 1% HEPES buffer and 2% penicillin/streptomycin (Gibco BRL Life Technologies, Rockville, MD). MDA-MD231 and K562 cells were separately cultured in RPMI-1640 medium supplemented with 10 and 5% bovine calf serum, respectively, and 1% penicillin/streptomycin (all reagents obtained from Gibco BRL Life Technologies). All cell lines were maintained in logarithmic growth phase at 37°C in a 5% CO2 atmosphere.
Construction of MPG, APE/ref-1 and hOGG1 mammalian expression vectors
The human MPG gene was cloned from human colon carcinoma HT-29 cells by RT–PCR, ligated into pAMP1 using T4 DNA ligase (Clone-AMP kit; Gibco BRL Life Technologies) and sequenced (Biochemistry Biotechnology Facility, Indiana University School of Medicine). The MPG insert was then excised with EcoRI and XhoI (New England Biolabs, Beverly, MD) and cloned into the multiple cloning site of pcDNA3.1 (Invitrogen, Carlsbad CA). Following transformation of DH5α competent Escherichia coli cells (Gibco BRL Life Technologies) and selection of ampicillin-resistant (100 µg/ml) colonies, pcDNA3.1-MPG plasmid DNA was isolated and analyzed by PCR and restriction enzyme digestion to verify MPG insertion. To construct the APE/ref-1 expression vector, pcDNA3.1 and pGEX3X-hAPE were digested with BamHI and EcoRI and fragments separated by agarose (1%) electrophoresis were purified using GENELUTE Minus EtBr spin columns (Sigma). The APE/ref-1 fragment was then inserted into pcDNA3.1. Bacterial transformation, colony selection and insert verification were preformed as described above. To construct the hOGG1 expression vector the hOGG-6 cDNA fragment was excised by BamHI and EcoRI digestion from plasmid pRSETB-hOGG-6 (generously provided by Dr Sankar Mitra, University of Texas Southwestern Medical Center, Galveston, TX) and subcloned into pBluescript SK(–) (Stratagene, La Jolla, CA). The hOGG-6 fragment was then excised by BamHI and XhoI digestion and subcloned into pcDNA3.1/Hygro (Invitrogen). Bacterial transformation, colony selection and insert verification were performed as described above.
Transfection of MPG, hOGG1 and APE/ref-1 expression vectors
MDA-MB231 cells were transfected with pcDNA3.1-MPG using the Lipofectin reagent (Gibco BRL Life Technologies) according to the manufacturer’s protocol. Briefly, Opti-MEM (Gibco BRL Life Technologies) serum-free medium was mixed with 10 µg plasmid DNA. Separately, Opti-MEM serum-free medium was mixed with the Lipofectin reagent and both mixtures were incubated at room temperature for 45 min in the dark. These solutions were then combined and incubated at room temperature for 10 min in the dark. The Lipofectin–DNA complex was added to 50–60% confluent cells and incubated under standard culture conditions overnight. After removing the Lipofectin–DNA-containing medium, normal complete medium was added and the cells were allowed to recover for 24 h before adding selective antibiotic (800 µg/ml G418) to the medium. After 10 days colonies were expanded and analyzed for gene expression by northern and western blot analyses and MPG activity assay. K562 cells were transfected first with pcDNA3.1-hAPE using DMRIE-C reagent (Gibco BRL Life Technologies) according to the manufacturer’s protocol, then following antibiotic resistance selection transfected similarly with pcDNA3.1/Hygro-hOGG-6. In each case the cells were allowed to recover for 48 h after transfection before adding selective antibiotics (800 µg/ml G418 and 200 µg/ml hygromycin, respectively) to the medium. After 10 days antibiotic selection surviving colonies were analyzed for gene expression by northern and western blot analyses and by hOGG1 and APE/ref-1 activity assays.
Standard DNA repair assays
MGMT assay. The assay was performed as described previously (15,16) with minor modifications. Cells (10 × 106) were harvested by trypsinization and resuspended in serum-containing culture medium, then centrifuged at 1500 g for 5 min. The cells were washed with 5–10 ml of PBS and recentrifuged, then resuspended in 0.5 ml assay buffer (50 mM Tris–HCl, 1 mM DTT, 0.5 mM EDTA, 5% glycerol, pH 8.0) and kept on ice. The cells were then pulse-sonicated on ice five times for 5 s each, then centrifuged at 14 000 g at 4°C for 30 min. Protein concentration was quantitated by measuring absorbance at 595 nm using the Coomasie Blue Protein Reagent (Pierce, Rockford, IL). The 18 bp oligonucleotide substrate contains a single O6-MeG residue nested within a PvuII restriction site. The radiolabeled substrate was produced by filling in the recessed 3′-end of the complementary 16 bp strand with [α-32P]TTP such that cleavage by PvuII produced a radiolabeled 8mer fragment. The HEX-labeled substrate incorporated a single HEX molecule into the 5′-end of the 18 bp complementary strand and PvuII cleavage produced a HEX-labeled 10mer fragment. To measure MGMT activity 0.2 pmol of the 32P- or HEX-labeled oligo (2 pmol/ml) was first incubated with 10–100 µg cell protein in a total volume of 150 µl of assay buffer at 37°C for 2 h. Following two cycles of phenol/chloroform/isoamyl alcohol (25:24:1) extraction and one chloroform/isoamyl alcohol (24:1) extraction the oligonucleotide was precipitated using sodium acetate and ethanol with tRNA (10 µg) as a carrier. The purified oligo was then digested with PvuII (Roche Molecular, Indianapolis, IN) in a total reaction volume of 20 µl at 37°C for 2 h. Then formamide loading buffer (10 µl), without dyes when the HEX-labeled oligo was employed, was added to each sample and the samples were denatured by heating at 95°C for 5 min. Sample solutions (24 µl) were applied to a 20% polyacrylamide gel (30 × 40 cm) containing 8 M urea in 1× Tris–borate EDTA (TBE) buffer and run at 26 mA for 75 min at room temperature.
MPG and hOGG1 assays. Cells (10 × 106) were harvested as described above and resuspended in 0.5 ml of harvesting buffer (PBS containing 2 mM DTT), then kept on ice. The cells were then pulse-sonicated as described above and centrifuged at 14 000 g at 4°C for 5 min. Protein concentration was also quantitated as described above. The 26 bp MPG and hOGG1 oligonucleotide substrates contained either a single ethanoadenine or 8-oxoG residue, respectively, in the middle, yielding either a 32P- or HEX-labeled 13mer fragment upon repair. To measure MPG activity 0.2 pmol of the 32P- or HEX-labeled oligo (2 pmol/ml) was first incubated with 5–20 µg cell protein in a total volume of 20 µl of assay buffer (25 mM HEPES–KOH, 0.5 mM DTT, 0.5 mM EDTA, 150 mM KCl, 1% glycerol, pH 7.8) (17) at 37°C for 1 h. Similarly, to measure hOGG1 activity 0.2 pmol of the 32P- or HEX-labeled oligo (2 pmol/ml) was first incubated with 5–50 µg cellular protein in a total volume of 20 µl of assay buffer (70 mM HEPES–KOH, 0.1 M KCl, 10 mM EDTA, pH 7.6) (18) at 37°C for 1 h. For both assays reactions were terminated by adding formamide loading buffer (10 µl), without dyes when the HEX-labeled oligo was employed, to each sample and heating each sample at 95°C for 5 min to denature the oligos. Electrophoresis was carried out as described above except that medium sized gels (15 × 17 cm) were used for 32P-labeled samples and large (30 × 40 cm) gels for HEX-labeled samples.
APE/ref-1 and APN1 assays. Cells (10 × 106) were harvested as described above and resuspended in 0.5 ml of harvesting buffer (PBS containing 2 mM DTT), then kept on ice. The cells were pulse-sonicated as described above, then centrifuged at 14 000 g at 4°C for 10 min. Protein concentration was also quantitated as described above. The 26 bp oligonucleotide substrate contained a single THF residue in the middle, yielding either a 32P- or HEX-labeled 13mer fragment upon repair. The 32P-labeled oligo was obtained by polynucleotide kinase incorporation of [32P]ATP into the lesioned strand, followed by Sephadex column purification. The radiolabeled strand was then annealed to the complementary strand in 10 mM Tris–HCl, 10 mM MgCl2, 1 mM EDTA, pH 7.5, in a 90°C water bath and cooled to room temperature overnight. The radiolabeled annealed oligo was then purified by non-denaturing SDS–PAGE in 1× TBE at 150 V for 1 h, excised from the gel and purified using a Centrex MF microfilter containing 300 µl of 35 mM HEPES, pH 7.5. Following ethanol precipitation the 32P-labeled oligo was resuspended in TE buffer (2.5 pmol/µl). APE/ref-1 or APN1 activity was measured by first incubating 2.5 pmol 32P-labeled or 0.2 pmol HEX-labeled (2 pmol/ml) excess oligo substrate with 0.5–5 µg cellular protein in a total volume of 20 µl of assay buffer (50 mM HEPES, 50 mM KCl, 10 mM MgCl2, 1% BSA, 0.05% Triton X-100, pH 7.5) (19) at 37°C for 15 min. APE/ref-1 is Mg2+ dependent and inactive in EDTA-containing buffer, whereas APN1 is Mg2+ independent. Thus, when APN1 activity was being measured the reaction mixture was supplemented with EDTA (10 mM final concentration) (20). Reactions were terminated by adding formamide loading buffer (10 µl), without dyes when the HEX-labeled oligo was employed, to each sample and heating each sample at 95°C for 5 min to denature the oligos. Electrophoresis was carried out as described above, with medium sized gels (15 × 17 cm) employed for 32P-labeled samples and large (30 × 40 cm) gels for HEX-labeled samples.
DNA repair assays using common buffer
Cells (10 × 106) were harvested by trypsinization and resuspended in serum-containing culture medium, then centrifuged at 1500 g for 5 min. After the cells were washed with 5–10 ml of PBS and recentrifuged, they were resuspended in 0.5 ml of common buffer (25 mM HEPES–KOH, 0.5 mM DTT, 0.5 mM EDTA, 150 mM KCl, 1% glycerol, pH 7.8) (17) and kept on ice. The cells were then pulse-sonicated on ice five times for 5 s, then centrifuged at 14 000 g at 4°C for 30 min. Protein concentration was quantitated as described above. MGMT activity was measured by first incubating 0.2 pmol HEX-labeled oligo (2 pmol/ml) with 10–100 µg cellular protein in a total volume of 150 µl of common buffer at 37°C for 2 h. The oligonucleotide substrate was next phenol/chloroform extracted and PvuII digested as described above. Samples were then prepared and electrophoresed as described above. In the same cellular extract MPG and hOGG1 activity were separately measured by first incubating 0.2 pmol of the respective HEX-labeled oligo (2 pmol/ml) with 5–50 µg cell protein in a total volume of 20 µl of assay buffer at 37°C for 1 h. The reactions were terminated and samples electrophoresed as described above. In the same cell extract APN1 and APE/ref-1 activities were separately measured by first incubating 0.2 pmol HEX-labeled oligo (2 pmol/ml) with 0.1–1.0 µg cell protein in a total volume of 20 µl of assay buffer. APE/ref-1 reactions were supplemented with MgCl2 (stock solution of 100 mM to a final concentration of 10 mM). The samples were then incubated at 37°C for 15 min, the reactions were terminated and samples were electrophoresed as described above using 30 × 40 cm gels.
Western blot analysis
To analyze MDA-MB231 cells overexpressing nuclear MPG (231+nMPG) cells were collected from a confluent plate, pelleted, washed with Dulbecco’s phosphate-buffered saline (Gibco BRL Life Technologies) and lysed with protein loading dye (0.0313 M Tris, 0.1 M SDS, 3.6 mM bromophenol blue, 2.75 mM β-mercaptoethanol, 50% glycerol, final concentration 1×). The samples were boiled to denature the proteins and quantitated using the Bradford Protein Assay (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein (20 µg) were separated on a 12% SDS–polyacrylamide gel and transferred to a nitrocellulose filter. The Roche Chemiluminescence kit (Indianapolis, IN) was used for the blocking and detection reagents. Primary MPG polyclonal antibody, kindly provided by Dr Tim O’Connor (Department of Biology, Beckman Research Institute, CA), was added at a dilution of 1:1000 and rotated overnight at 4°C. Following primary antibody incubation the filter was washed and the rabbit secondary antibody was added for 45–60 min at room temperature. Again the filter was washed and the detection solutions were added. The Roche kit contained a horseradish peroxidase-labeled secondary antibody and a luminol substrate solution for the detection of bands. After these solutions were added to the filter it was exposed to high performance autoradiography film (Amersham Pharmacia Biotech, Little Chalfont, UK).
Statistics
The statistical software SAS (SAS Institute, Cary, NC) was employed for linear regression analysis using zero intercepts of the repair activity curves, expressed as percent oligonucleotide cleaved as a function of cellular extract protein. Specific activities were determined from the slopes of the respective linear portion of the curves normalized to time. The slopes were also employed for comparison between two groups, including 32P- versus HEX-labeled oligonucleotides, standard versus common buffers and wild-type versus MPG-overexpressing MDA-MB231 cells. Results were considered to be significantly different for P < 0.05.
RESULTS
HEX- versus 32P-labeled oligonucleotides
The amounts of the 32P- and HEX-labeled oligonucleotides employed in the assays were within the linear range of quantitative detection by phosphorimaging and fluorescence intensity, respectively (Fig. 2). In both cases 200 fmol starting substrate was used per sample; however, only half of each sample was loaded for electrophoresis when the 32P-labeled substrates were used, while the entire sample was loaded for the HEX-labeled substrates. Hence, the total amounts of substrate that were quantitatively analyzed for the 32P- and HEX-labeled oligonucleotides were 100 and 200 fmol, respectively. As shown in Figure 2, the respective detection limits permitted the same level of sensitivity to be attained for both substrates with respect to the assay, 1% cleavage of the oligonucleotide. To compare the abilities of the 32P- and HEX-labeled oligonucleotides to serve as substrates for the DNA repair proteins, MGMT, MPG, hOGG1, APE/ref-1 and APN1 activities were determined separately for both substrates under the respective standard assay conditions. The oligonucleotide substrates employed for each repair protein are described in Table 1 and representative results are shown in Figure 3. In all cases no cleavage product was detected using the HEX-labeled substrates in the absence of cell extract (data not shown), indicating a lack of incomplete synthesis products or non-specific oligonucleotide degradation. In HeLa cells MGMT activity increased linearly with increasing protein concentration using either the 32P-labeled (r2 = 0.984) or HEX-labeled (r2 = 0.968) substrate, as shown in Figure 3A. The slopes of these curves (µg protein extract versus fmol substrate cleaved) yielded respective MGMT specific activities of 3.0 and 2.9 fmol O6-MeG removed/µg protein. Hence, the HEX-labeled oligonucleotide exhibited similar MGMT reactivity to the 32P-labeled oligonucleotide and there was no significant difference in the MGMT specific activities when either oligonucleotide was used.
Figure 2.


Sensitivity and linearity of quantitative analysis of 32P- and HEX-labeled oligonucleotides. (A) Quantitative analysis of various amounts (0–140 fmol) of 32P-labeled oligonucleotide by phosphorimaging. (B) Quantitative analysis of various amounts (0–250 fmol) of HEX-labeled oligonucleotide by fluorescence intensity imaging.
Table 1. Labeled substrates and products for DNA repair activity assays.
| Protein |
Substrate |
Size (bp) |
Cleavage product |
Size |
| MGMT | 5′-HEX-GCCCGGCCA GaCTGCAGTT | 18 | 5′-HEX-GCCCGGCCAG | 10mer |
| CGGGCCGGT C GACGTCAA | ||||
| MPG | 5′-HEX-AATTCACCGGTACC AbCCTAGAATTCG | 26 | 5′-HEX-AATTCACCGGTAC | 13mer |
| TTAAGTGGCCATGG T GGATCTTAAGC | ||||
| hOGG1 | 5′-HEX-AATTCACCGGTACC GcTCTAGAATTCG | 26 | 5′-HEX-AATTCACCGGTAC | 13mer |
| TTAAGTGGCCATGG C AGATCTTAAGC | ||||
| APE/ref-1 | 5′-HEX-AATTCACCGGTACC TdTCTAGAATTCG | 26 | 5′-HEX-AATTCACCGGTAC | 13mer |
| TTAAGTGGCCATGG A AGATCTTAAGC | ||||
| APN1 | 5′-HEX-AATTCACCGGTACC TdTCTAGAATTCG | 26 | 5′-HEX-AATTCACCGGTAC | 13mer |
| TTAAGTGGCCATGG A AGATCTTAAGC |
aG, O6-methylguanine.
bA, ethanoadenine.
cG, 8-oxoguanine.
dT, tetrahydrofuran.
Figure 3.
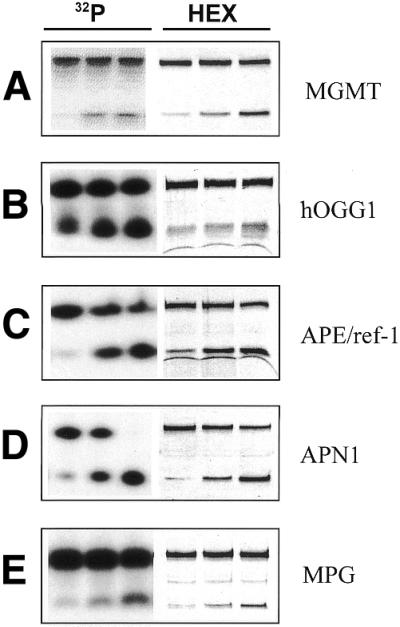
Comparison of 32P- versus HEX-labeled oligonucleotides using standard buffers. Activities using either 32P- (lanes 1–3) or HEX-labeled (lanes 4–6) oligonucleotides are shown with increasing amounts of cell extract from left to right, respectively. (A) MGMT activity was measured in HeLa cells (10, 20 and 50 µg). In lanes 1–3 (32P-labeled) the upper and lower bands represent the 18 bp substrate and 8 bp cleavage product, respectively; in lanes 4–6 (HEX-labeled) the upper and lower bands represent the 18 bp substrate and 10 bp cleavage product, respectively. (B) hOGG1 activity was measured in K562-OGG/APE cells (5, 10 and 20 µg). (C) APE/ref-1 activity was measured in K562-OGG/APE cells (0.1, 0.25 and 0.5 µg). (D) APN1 activity was measured in K562-APN1 cells (0.1, 0.5 and 1.0 µg). (E) MPG activity was measured in MDA-MB231 cells (5, 10 and 20 µg). In (B)–(E) all of the upper and lower bands represent the 26 bp substrates and 13 bp cleavage products, respectively. Data shown are of a representative experiment out of at least three independent experiments.
Human leukemia K562 cells overexpressing both hOGG1 and APE/ref-1 (K562-OGG/APE) were used to compare the 32P- and HEX-labeled substrates for both of these proteins. To measure hOGG1 activity varying amounts of cell extract were incubated separately with each substrate. As shown in Figure 3B, hOGG1 activity increased linearly with increasing protein concentration whether the 32P-labeled (r2 = 0.878) or the HEX-labeled (r2 = 0.928) oligonucleotide was used. The slopes of the curves yielded specific hOGG1 activities of 82 and 75 fmol 8-oxoG repaired/mg protein/min for the 32P- and HEX-labeled substrates, respectively. Hence, no significant difference in hOGG1 specific activity was found between the two oligonucleotide substrates. In the same cells APE/ref-1 activity was compared using markedly less cell extract (0.1–1 µg protein) due to the high expression of APE/ref-1 in most mammalian cells. The APE/ref-1 activity increased linearly with protein concentration using either the 32P- or HEX-labeled substrate (r2 = 0.983 and 0.963, respectively), as shown in Figure 3C. The specific APE/ref-1 activities using the 32P- and HEX-labeled substrates were 15.6 and 14.3 pmol THF lesions repaired/mg protein/min, respectively, and no difference in APE/ref-1 specific activity was observed between the substrates. The arced lines below the 13 bp cleavage fragments of the HEX-labeled hOGG1 and APE/ref-1 substrates (Fig. 3B and C) are apparent buffer effects under the electrophoresis conditions.
The activity of the yeast repair protein APN1 using the 32P- and HEX-labeled substrates was compared in K562 cells overexpressing APN1. As shown in Figure 3D, APN1 activity increased linearly with respect to cell extract protein using either the 32P- and HEX-labeled substrate (r2 = 0.984 and 0.959, respectively). APN1 specific activities were 5.7 and 6.7 pmol THF lesions repaired/mg protein/min for the 32P- and HEX-labeled substrates, respectively. The increase in APN1 specific activity when the HEX-labeled oligonucleotide was used was slight, but statistically significant (P = 0.031).
To compare MPG activity using the 32P- and HEX-labeled substrates varying amounts of MDA-MB231 cell extract were separately incubated with each oligonucleotide. With either substrate MPG activity was undetectable at the lowest protein concentration and a maximum of ∼20% of the substrate was cleaved at the highest concentration (Fig. 3E). MPG activity increased linearly with respect to cell protein in both cases (r2 = 0.960 and 0.980 for the 32P- and HEX-labeled substrates, respectively). The slopes of the curves yielded specific MPG activities of 31 and 27 fmol methylpurine repaired/mg protein/min for the 32P- and HEX-labeled substrates, respectively, and no significant difference was detected between MPG specific activities using either oligonucleotide.
DNA repair activity assays using the HEX-labeled oligonucleotide substrates in a common buffer
To enable MGMT, MPG, hOGG1, APE/ref-1 and APN1 activities to be rapidly measured from a single cell extract a common buffer in which each of the proteins exhibited similar activity to the respective standard assay buffers was developed. Because the MGMT and MPG standard assay buffers contained the greatest number of unique components and because of the apparent similarities between them (Table 2), the repair activities were first measured using the MGMT standard buffer and then compared to the respective standard assay buffers for each protein. In K562-OGG/APE cells the activities of hOGG1 and APE/ref-1 were both higher in the MGMT assay buffer than in the respective standard buffers (data not shown). However, MPG specific activity in 231-MPG cells was markedly lower using the MGMT buffer than the standard buffer (data not shown). The repair activities were subsequently assayed using the MPG standard buffer and compared to the respective standard assay buffers. In HeLa cells, MGMT activity when MPG buffer was used was similar to that obtained with the standard buffer, with MGMT specific activity being 2.5 fmol O6-MeG removed/µg protein with MPG buffer (Fig. 4A). hOGG1 activity in K562-OGG/APE cells was significantly increased in MPG buffer compared to standard hOGG1 buffer (Fig. 4B), resulting in an ∼5-fold increase in hOGG1 specific activity. Similarly, APE/ref-1 activity was significantly higher at all of the protein concentrations when MPG buffer was used (Fig. 4C) and the specific activity was similarly increased by ∼5-fold. Also, the arcs observed below the 13 bp HEX-labeled cleavage products using standard hOGG1 and APE/ref-1 buffers (Fig. 3B and C) were not detected when the common buffer was used. APN1 specific activity in K562-APN1 cells when MPG buffer was used was slightly increased to 8.4 THF lesions repaired/mg protein/min (Fig. 4D), although the increase was not statistically significant. In summary, MPG buffer enhanced hOGG1 and APE/ref-1 activities compared to the respective standard buffers (P = 0.013 and 0.029, respectively), while APN1 and MGMT activities were not significantly different between the MPG and respective standard buffers. Therefore, MPG standard buffer functioned effectively as a common buffer in which MPG, MGMT, hOGG1, APE/ref-1 and APN1 activities could be measured and was employed as a common buffer in further studies.
Table 2. Standard buffer components for DNA repair assays and common buffer components.
| MGMT buffer | 50 mM Tris–HCl, 0.5 mM EDTA, 1.0 mM DTT, 5% glycerol, pH 8.0 |
| hOGG1 buffer | 70 mM HEPES–KOH, 100 mM KCl, 10 mM EDTA, pH 7.6 |
| APE/ref-1 and APN1 buffer | 50 mM HEPES, 50 mM KCl, (10 mM MgCl2), 1% BSA, 0.05% Triton X-100, pH 7.5 |
| MPG buffer/common buffer | 25 mM HEPES–KOH, 0.5 mM EDTA, 0.5 mM DTT, 150 mM KCl, 1% glycerol, pH 7.8 |
Figure 4.

Comparison of activities using standard versus common buffers. Percent cleavage of HEX-labeled oligonucleotide substrates is shown as a function of cell extract using either the respective standard buffer (open circles) or the common buffer (filled circles). (A) MGMT activity was measured in K562-OGG/APE cells. (B) hOGG1 activity was measured in K562-OGG/APE cells. (C) APE/ref-1 activity was measured in K562-APN cells. (D) APN1 activity was measured in HeLa cells. All data are represented as the means of at least three independent experiments. Error bars represent the standard deviation (SD); where error bars are absent the SD is smaller than the symbol.
DNA repair status in MDA-MB231 cells overexpressing MPG
To determine whether overexpression of MPG in human breast carcinoma MDA-MB231 cells produced alterations in other DNA repair proteins the activities of MGMT, hOGG1 and APE/ref-1 were compared in single cell extracts of wild-type MDA-MB231 (wt-231) and MPG-overexpressing MDA-MB231 (231-MPG) cells, respectively, prepared in the common buffer described above. 231-MPG cells expressed dramatically higher levels of MPG protein than wt-231 cells, as shown by western blot analysis (Fig. 5), and exhibited a dramatic increase in MPG activity (P < 0.0001) (Fig. 6A). hOGG1 specific activities were 18.5 and 16.9 fmol 8-oxoG repaired/mg protein/min in wt-231 and 231-MPG cells, respectively (Fig. 6B). Hence, MPG overexpression did not significantly alter hOGG1 activity in these cells. APE/ref-1 specific activities in wt-231 and 231-MPG cells were 24.3 and 22.7 pmol THF lesions repaired/mg protein/min (Fig. 6C) and no significant difference in APE/ref-1 activity was found between wt-231 and 231-MPG cells. Interestingly, MGMT activity was undetectable in both wt-231 and 231-MPG cells at up to 100 µg cell extract (Fig. 6D), indicating that MDA-MB231 cells are methylation repair-deficient (MGMT–). MGMT protein was not detected in wt-231 cells by western blot analysis while APE/ref-1 protein was present (data not shown), confirming the results obtained by the activity assay.
Figure 5.
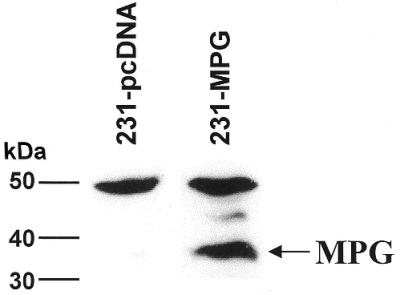
Western blot analysis of MPG expression. MPG expression was determined in MDA-MB231 cells transfected with the pcDNA vector (231-pcDNA) or with vector containing MPG cDNA (231-MPG) using a polyclonal antibody. The arrow indicates the MPG protein, ∼35 kDa in size. The 50 kDa band represents non-specific binding to an unknown protein, which serves as an internal control.
Figure 6.
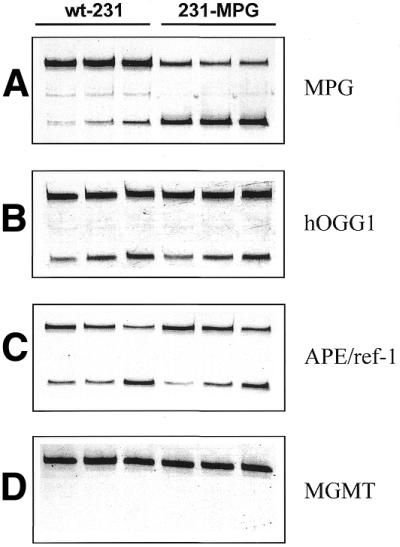
Comparison of DNA repair activities in wild-type MDA-MB231 and MDA-MB231 cells overexpressing MPG. Repair activities using HEX-labeled oligonucleotides in the common buffer are shown in wild-type MDA-MB231 cells (wt-231) and MDA-MB231 cells overexpressing MPG (231-MPG) by increasing amounts of cell extract from left to right, respectively. (A) MPG activity (5, 10 and 20 µg). (B) hOGG1 activity (10, 20 and 50 µg). (C) APE/ref-1 activity (0.1, 0.25 and 0.5 µg). (D) MGMT activity (25, 50 and 100 µg). Data shown are of a representative experiment out of at least three independent experiments.
DISCUSSION
This study demonstrates that fluorometric HEX-labeled oligonucleotides can be effectively employed to measure the activities of several DNA repair enzymes, specifically MGMT, MPG, hOGG1, APE/ref-1 and APN1, in cell extracts in vitro. The major benefits of the fluorometric assay are decreased substrate variability and improved experimental reproducibility, elimination of radioactivity and a substantial cost reduction when compared to conventional radiolabeled substrates. We recently reported use of a HEX-labeled oligonucleotide substrate to measure MGMT activity in several normal murine tissues and in human glioma SF767 tumor xenografts (21). In addition, a fluorometric FITC-labeled oligonucleotide substrate has been employed to measure hOGG1 activity in extracts of normal and neoplastic human colorectal tissues (22). However, the current study is the first to demonstrate the use of fluorometric oligonucleotide substrates for MPG, APE/ref-1 and APN1. Fluorometric oligonucleotide substrates could potentially be used to measure the activities of other DNA repair proteins, such as uracil-DNA glycosylase. Furthermore, in our laboratory, using the Hitachi FMBio II Fluorescence Imaging System (Hitachi Genetic Systems), HEX-labeled oligonucleotides exhibited greater fluorescence intensity on a femtomolar basis than FITC-labeled oligonucleotides, resulting in enhanced sensitivity of the assay, especially at low levels of repair activity (E.L.Kreklau, C.Kurpad and L.C.Erickson, unpublished observations).
We have also identified a common buffer system in which all five of these repair proteins can be assayed from a single starting sample. Because DNA repair encompasses several processes, measurement of multiple repair activities provides a composite view of cellular DNA repair function. Furthermore, BER and methylation repair participate in protection against various genotoxic agents, including ionizing radiation and anticancer therapeutics. The method described here permits measurement of MGMT and several BER proteins from a limited starting sample such as that obtained from a tumor biopsy, which may offer a useful clinical assessment of tumor chemosensitivity. Based upon our studies of murine tissues, including liver, lung, kidney, brain, spleen and bone marrow, as well as several human tumor cell lines and primary human bone marrow samples, ∼1–2 × 106 cells is sufficient to employ the assay described herein for most tissues, even those with relatively low repair activity. The common buffer did not alter the specific activities of MGMT or APN1, while the activities of hOGG-1 and APE/ref-1 were enhanced, compared to the respective standard buffers. The reasons for the enhanced specific activities of hOGG1 and APE/ref-1 in the common buffer are unknown. However, APE/ref-1 is known to have important redox properties that function in its regulation of transcription factor binding to DNA (23) and to be inducible by oxidative stress (24,25). In addition, we have shown recently that APE/ref-1 activity is augmented by the reducing agent dithiothreitol (DTT) (26). Thus the presence of DTT in the common buffer may be a contributing factor in the observed enhancement of APE/ref-1 activity. The role of redox state in modulating hOGG1 activity is unknown but may be involved in its increased activity in association with the common buffer. Alternatively, other factors, such as stabilization by the presence of glycerol or the increase in pH of the common buffer, may mediate the enhancement of hOGG1 activity. It has recently been shown that APE/ref-1 stimulates hOGG1 activity (27), suggesting that the enhanced APE/ref-1 activity may continue to the enhancement of hOGG1 activity in the common buffer.
Modulation of MGMT and BER have been employed in attempts to alter cellular sensitivity to various genotoxic agents. For example, inhibition of MGMT significantly reverses tumor resistance to chloroethylnitrosourea in cell culture and in xenografts (15,28). In the case of BER, however, modulation of one of the repair enzymes may produce an imbalance in the pathway. Hence, an important question that arises in studies that specifically modulate the activity of DNA repair proteins is whether other proteins, particularly those in the same pathway, are concomitantly altered. For example, a change in the activity of one BER protein may elicit a compensatory response elsewhere within the pathway. Because the cellular protective response is determined by the function of the pathway as a whole, the activities of all of the BER proteins should be examined when one step in the pathway is altered. In the current study selective overexpression of the simple glycosylase MPG in human breast carcinoma cells did not alter the activities of a complex glycosylase, hOGG1, or of the protein immediately downstream in the BER pathway, APE/ref-1. Interestingly, the MDA-MB231 cell line appears to be deficient in methylation repair, as indicated by a lack of MGMT activity. Taken altogether, the MPG overexpressing cells would thus be expected to generate more abasic sites in response to MPG-responsive genotoxic damage. Indeed, overexpression of MPG has been found to cause chromosomal aberrations and increased sensitivity to alkylating agents as a result of accumulation of cytotoxic AP sites (6,29). In another case overexpression of APE/ref-1 did not increase cellular resistance to alkylation or radiation (30), perhaps suggesting that endogenous APE/ref-1 activity is not limiting for these repair processes. The rate limiting step in the BER pathway, however, remains controversial. These studies demonstrate the importance of measuring DNA repair activities collectively when assessing cellular protective capacity against genotoxic injury, as well as when developing strategies to modulate cellular responses to genotoxic agents. The method presented herein offers an effective and simplified approach to measure MGMT and BER functions from a single experimental sample.
Acknowledgments
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the expert statistical consultation and analyses performed by Dr Sin-ho Jung (Division of Biostatistics, Indiana University Cancer Center). This research was supported by National Cancer Institute grants R01-CA45628 (L.C.E.) and F32-CA86405 (E.L.K.) and M.R.K. is supported in part by National Institutes of Health grants R01-CA76643, NS38506, R43-CA83507, P30-DK49218 and P01-CA75426, DOD/CDMRP grant OC990085 and the Riley Memorial Association.
References
- 1.Hansen W.K. and Kelley,M.R. (2000) Review of mammalian DNA repair and translational implications. J. Pharmacol. Exp. Ther., 295, 1–9. [PubMed] [Google Scholar]
- 2.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 3.Singer B., Antoccia,A., Basu,A.K., Dosanjh,M.K., Fraenkel-Conrat,H., Kusmierek,J.T., Qiu,Z.H. and Rydberg,B. (1992) Both purified human 1,N6-ethenoadenine-binding protein and purified human 3-methyladenine-DNA glycosylase act on 1,N6-ethenoadenine and 3-methyladenine. Proc. Natl Acad. Sci. USA, 89, 9386–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans A.R., Limp-Foster,M. and Kelley,M.R. (2000) Going APE over ref-1. Mutat. Res., 461, 83–108. [DOI] [PubMed] [Google Scholar]
- 5.Bjoras M., Luna,L., Johnsen,B., Hoff,E., Haug,T., Rognes,T. and Seeberg,E. (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J., 16, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Limp-Foster M. and Kelley,M.R. (2000) DNA repair and gene therapy: implications for translational uses. Environ. Mol. Mutagen., 35, 71–81. [DOI] [PubMed] [Google Scholar]
- 7.Kasai H., Chung,M.H., Jones,D.S., Inoue,H., Ishikawa,H., Kamiya,H., Ohtsuka,E. and Nishimura,S. (1991) 8-Hydroxyguanine, a DNA adduct formed by oxygen radicals: its implication on oxygen radical-involved mutagenesis/carcinogenesis. J. Toxicol. Sci., 16 (suppl. 1), 95–105. [DOI] [PubMed] [Google Scholar]
- 8.Michaels M.L., Cruz,C., Grollman,A.P. and Miller,J.H. (1992) Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl Acad. Sci. USA, 89, 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tchou J., Kasai,H., Shibutani,S., Chung,M.H., Laval,J., Grollman,A.P. and Nishimura,S. (1991) 8-Oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc. Natl Acad. Sci. USA, 88, 4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng K.C., Cahill,D.S., Kasai,H., Nishimura,S. and Loeb,L.A. (1992) 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G→T and A→C substitutions. J. Biol. Chem., 267, 166–172. [PubMed] [Google Scholar]
- 11.Robins P., Harris,A.L., Goldsmith,I. and Lindahl,T. (1983) Cross-linking of DNA induced by chloroethylnitrosourea is prevented by O6-methylguanine DNA methyltransferase. Nucleic Acids Res., 11, 7743–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolan M.E., Young,G. and Pegg,A. (1986) Effect of O6-alkylguanine pretreatment on the sensitivity of human colon tumor cells to the cytotoxic effects of chloroethylating agents. Cancer Res., 46, 4500–4504. [PubMed] [Google Scholar]
- 13.Cunningham R.P. (1997) DNA glycosylases. Mutat. Res., 383, 189–196. [DOI] [PubMed] [Google Scholar]
- 14.Wood R.D. (1996) DNA repair in eukaryotes. Annu. Rev. Biochem., 65, 135–167. [DOI] [PubMed] [Google Scholar]
- 15.Futscher B.W., Micetich,K.C., Barnes,D.M., Fisher,R.I. and Erickson,L.C. (1989) Inhibition of specific DNA repair system and nitrosourea cytotoxicity in resistant human cancer cells. Cancer Commun., 1, 65–73. [DOI] [PubMed] [Google Scholar]
- 16.Wu R., Hurst-Calderone,S. and Kohn,K. (1987) Measurement of O-6 alkylguanine DNA alykyltransferase activity in human cells and tumor tissues by restriction endonuclease inhibition. Cancer Res., 47, 6229–6235. [PubMed] [Google Scholar]
- 17.Miao F., Bouziane,M. and O’Connor,T.R. (1998) Interaction of the recombinant human methylpurine-DNA glycosylase (MPG protein) with oligodeoxyribonucleotides containing either hypoxanthine or abasic sites. Nucleic Acids Res., 26, 4034–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Kemp P.A., Thomas,D., Barbey,R., de Oliveira,R. and Boiteux,S. (1996) Cloning and expression in Escherichiacoli of the OGG1 gene of Saccharomycescerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N- methylformamidopyrimidine. Proc. Natl Acad. Sci. USA, 93, 5197–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson D.M., Takeshita,M., Grollman,A.P. and Demple,B. (1995) Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem., 270, 16002–16007. [DOI] [PubMed] [Google Scholar]
- 20.Levin J.D. and Demple,B. (1990) Analysis of class II (hydrolytic) and class I (β-lyase) apurinic/apyrimidinic endonucleases with a synthetic DNA substrate. Nucleic Acids Res., 18, 5069–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreklau E.L., Liu,N., Li,Z., Cornetta,K., Williams,D.A. and Erickson,L.C. (2001) Comparison of single- versus double-bolus treatments of O6-benzylguanine for depletion of O6-methylguanine DNA methyltransferase (MGMT) activity in vivo: development of a novel fluorometric oligonucleotide assay for measurement of MGMT activity. J. Pharmacol. Exp. Ther., 297, 524–530. [PubMed] [Google Scholar]
- 22.Kondo S., Toyokuni,S., Tanaka T., Hiai,H., Onodera H., Kasai,H. and Imamura,M. (2000) Overexpression of the hOGG1 gene and high 8-hydroxy-2′-deoxyguanosine (8-OHdG) lyase activity in human colorectal carcinoma: regulation mechanism of the 8-OHdG level in DNA. Clin. Cancer Res., 6, 1394–1400. [PubMed] [Google Scholar]
- 23.Barzilay G., Walker,L.J., Rothwell,D.G. and Hickson,I.D. (1996) Role of the HAP1 protein in repair of oxidative DNA damage and regulation of transcription factors. Br. J. Cancer, 74, S145–S150. [PMC free article] [PubMed] [Google Scholar]
- 24.Grosch S., Fritz,G. and Kaina,B. (1998) Apurinic endonuclease (Ref-1) is induced in mammalian cells by oxidative stress and involved in clastogenic adaptation. Cancer Res., 58, 4410–4416. [PubMed] [Google Scholar]
- 25.Ramana C.V., Boldogh,I., Izumi,T. and Mitra,S. (1998) Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc. Natl Acad. Sci. USA, 95, 5061–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelley M.R. and Parsons,S.H. (2001) Reduction-oxidation (redox) regulation of the DNA repair function of the human AP endonuclease Ape1/ref-1. Antioxidants Redox Signaling, Vol. 3. [DOI] [PubMed] [Google Scholar]
- 27.Hill J.W., Hazra,T.K., Izumi,T. and Mitra,S. (2001) Stimulation of human 8-oxoguanine DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res., 29, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dolan M.E., Moschel,R. and Pegg,A. (1990) Depletion of O6-alkylguanine DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl Acad. Sci. USA, 87, 5368–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coquerelle T., Dosch,J. and Kaina,B. (1995) Overexpression of N-methylpurine-DNA glycosylase in Chinese hamster ovary cells renders them more sensitive to the production of chromosomal aberrations by methylating agents—a case of imbalanced DNA repair. Mutat. Res., 336, 9–17. [DOI] [PubMed] [Google Scholar]
- 30.Herring C.J., Deans,B., Elder R.H., Rafferty,J.A., MacKinnon, J, Barzilay,G., Hickson,I.D., Hendry,J.H. and Margison,G.P. (1999) Expression levels of the DNA repair enzyme HAP1 do not correlate with the radiosensitivities of human or HAP1-transfected rat cell lines. Br. J. Cancer, 80, 940–945. [DOI] [PMC free article] [PubMed] [Google Scholar]