

ABSTRACT
The transcriptional output of the Sonic Hedgehog morphogenic pathway is orchestrated by three Krüppel family transcription factors, Gli1 to -3, which undergo extensive posttranslational modifications, including ubiquitination and SUMOylation. Here, we report that the sentrin-specific peptidase SENP1 is the specific deSUMOylation enzyme for Gli1. We show that SUMOylation stabilizes Gli1 by competing with ubiquitination at conserved lysine residues and that SUMOylated Gli1 is enriched in the nucleus, suggesting that SUMOylation is a nuclear localization signal for Gli1. Finally, we show that small interfering RNA (siRNA)-mediated knockdown of SENP1 augments the ability of Shh to sustain the proliferation of cerebellar granule cell precursors, demonstrating the physiological significance of the negative regulation of Shh signaling by SENP1.
KEYWORDS: Sonic Hedgehog, SUMOylation, Gli1, SENP1
INTRODUCTION
Hedgehog signaling plays critical roles in specifying spatial pattern and cell fate during embryonic development and maintaining tissue homeostasis in adults (1–4). A myriad of birth defects and cancer syndromes are associated with genetic lesions in genes that transduce the Hedgehog signal (5–7). However, the precise mechanism by which Hedgehog signaling responses are regulated still remains an unresolved topic of both conceptual and practical importance (8–10).
Mammalian genomes include 3 Hedgehog genes, the major one of which is the Sonic Hedgehog (Shh) gene (11, 12). Activation of Shh signaling is initiated when Shh binds its cognate receptor, Patched 1 (Ptch1), causing it to exit the primary cilium, a microtubule-based protrusion present in interphase cells (13–16). By a still undercharacterized mechanism, ligand engagement releases Ptch1 inhibition on a membrane-bound signal transducer, Smoothened (Smo), allowing the latter to be transported into the primary cilium and turning on downstream components of the pathway (15, 17, 18). Ligand engagement also promotes endocytic turnover of Ptch1 in lysosomes by mobilizing the homology to E6AP carboxyl terminus (HECT) domain-containing E3 ligases, Smurf1 and Smurf2, which catalyze ubiquitination of Ptch1 in lipid rafts (19, 20). Downstream from the Ptch1-Smo receptor system, three Krüppel family transcription factors, Gli1, Gli2, and Gli3, orchestrate transcriptional responses of the target genes (21–25). In the absence of Shh signal, Gli2 and Gli3 undergo partial proteolysis that converts the full-length proteins into carboxyl-terminally truncated transcriptional repressors (26, 27). Production of these Gli repressors is governed by a conserved phosphorylation cascade involving protein kinase A (PKA), glycogen synthase kinase 3 (GSK3), and casein kinase I (CK1), which render the nascent Gli2 and Gli3 recognizable by the ubiquitin E3 ligase Slimb/βTRCP and send them for limited degradation in proteasomes (28–31). Activation by Smo blocks the phosphorylation and processing, causing Gli2 and Gli3 to be stabilized into full-length activators, and the induction of Gli1, which itself is an Shh transcriptional target; however, Gli1 does not undergo proteolytic processing; instead, it functions purely as an auxiliary transcription activator (22).
Small ubiquitin-related modifier (SUMO) is a ubiquitin-like protein moiety reversibly added posttranslationally to target proteins by an ATP-driven cascade of enzymes consisting of activating enzyme (E1), conjugating enzyme (E2), and ligase (E3) (32, 33). Thousands of proteins encompassing a multitude of cellular processes have been found to be SUMOylated; these processes include chromatin organization, transcription, DNA damage repair, protein trafficking, and signal transduction (34–36). Like ubiquitin, SUMO can be attached as a single entity to the ε-amino group of a lysine residue on the substrate protein or SUMO itself to form a multiunit chain. Since a common SUMOylation target consensus sequence, ΨKXE (where Ψ is a bulky aliphatic residue), recognized by the E2 SUMO-conjugating enzyme Ubc9 is also recognized by certain ubiquitin E3 ligases (37), the interplay of these two processes is often at the heart of complex mechanisms that orchestrate intricate regulation of target protein functions (38). Also, like ubiquitin, SUMO moieties on modified proteins can be removed by SUMO-specific proteases called sentrin-specific proteases (SENPs) in mammals (39, 40), further adding to the complexity of this regulation. These cysteine proteases all possess a conserved C-terminal catalytic domain that does not discriminate among different SUMO paralogues for removal; however, they do differ in their subcellular localizations and substrate preferences (41).
Recently, all three Gli proteins in mammals, as well as their Drosophila counterpart, Ci, were shown to be SUMOylated at specific sites, but the functional impacts of these modifications differ among different reports (42–44). Nevertheless, SUMOylation was shown to play important roles in regulating neural-tube development in vertebrates and somatic cyst stem cell self-renewal in the adult Drosophila testis (42, 44). We have tested all 6 mammalian SENPs for their involvement in Shh signaling and report here that SENP1 is a specific deSUMOylation enzyme for Gli1. Our data show that SENP1 negatively regulates Shh signaling and that SUMOylation likely constitutes a nuclear retention signal for Gli1.
RESULTS
SENP1 negatively modulates Shh signaling.
To identify deSUMOylation enzymes that regulate Shh signaling, we resorted to using small interfering RNAs (siRNAs) to test the effect of silencing each of the 6 mammalian SENPs on Shh-induced Gli1 transcription in a stable line of NIH 3T3 cells that contain the genomically integrated 8× GliBS-luc reporter (45, 46). The efficacies of these siRNAs were tested in the same NIH cells (see Fig. S1 in the supplemental material), which exhibited a robust transcriptional response to the treatment (24 h) by ShhN-conditioned medium (ShhN-CM; see Materials and Methods for a definition), and that response was substantially enhanced when the cells received siSENP1 delivered by transient transfection (Fig. 1A). Surprisingly, siSENP2 actually downregulated the 8× GliBS-luc response, while siRNAs against other SENPs showed no effect (Fig. 1A). Western analyses of the endogenous Gli1 levels showed effects similar to those in the luciferase reporter assay on Shh signaling (Fig. 1B). To further ascertain the roles of SENP1 and SENP2 in Shh signaling, we analyzed the induction of Gli1 and Ptch1, another well-known Shh target, in SENP1−/− and SENP2−/− murine embryonic fibroblasts (MEFs) (47, 48). Compared to that in the wild-type (WT) control MEFs, ShhN-CM treatment induced severalfold-higher activation of the two genes in SENP1−/− MEFs, but the induction of both genes was dramatically curtailed in SENP2−/− MEFs (Fig. 1C to F). Likewise, Western analyses of Gli1 expression in these mutant SENP1−/− and SENP2−/− MEFs corroborated the Gli1 mRNA measurements (Fig. 1G), indicating the observed effects of SENP1 and -2 on Shh signaling were at the level of transcription. Of note, the basal levels of Gli1 in these mutant MEFs remained comparable to that in the normal control MEFs, suggesting that the Shh pathway was not activated and that the roles of SENP1 and SENP2 are likely secondary to pathway activation. The fact that loss of SENP1 and SENP2 exhibited opposing effects despite the close similarity in their protein sequences and that these two enzymes are known to regulate distinct cellular processes (49, 50) strongly argues that they likely control different aspects of Shh signaling. For this investigation, we deliberately chose SENP1 for further analysis of its potential role in deSUMOylating Gli1, the major transcriptional activator of Shh target genes. When reintroduced into SENP1−/− MEFs, wild-type SENP1 strongly inhibited the induction of both Gli1 mRNA and protein expression, but inhibition by a mutant SENP1m that lacks the deSUMOylase activity was much weaker (Fig. 1H and I). Finally, overexpressing wild-type SENP1 but not the mutant SENP1m in Ptch1−/− MEFs drastically repressed the constitutive expression of Gli1 mRNA and protein (Fig. 1J and K), demonstrating the ability of SENP1 to negatively modulate Shh signaling.
FIG 1.

Identification of SENP1 as a negative regulator of Shh signaling. (A and B) Luciferase assays for Shh induction of 8× GliBS-luc reporter (A) and Western analyses of Gli1 protein (B) in NIH 3T3 cells that were transfected with nonspecific control (NC) or SENP-specific siRNAs as indicated. IB, immunoblotting. (C to F) RT-qPCR quantification of Shh-induced Gli1 and Ptch1 mRNA expression in SENP1−/− and SENP2−/− mutant MEFs. (G) Western analysis of Shh-induced Gli1 protein expression in SENP1−/− and SENP2−/− mutant MEFs. (H and I) Western analysis (H) and RT-qPCR quantification thereof (I) showing that reexpression of SENP1 but not the deSUMOylase-deficient SENP1m restored the normal induced level of Gli1 expression in SENP1−/− MEFs by Shh. (J and K) Western analysis (J) and RT-qPCR quantification thereof (K) showing that reexpression of SENP1 but not SENP1m suppressed the elevated Gli1 expression in Ptch−/− MEFs. ShhN-CM treatment was for 24 h, and the knockdown efficiency of each siRNA is shown in Fig. S1 in the supplemental material. The data are presented as means and standard deviations (SD) of the results of three independent experiments. Student's t tests were used for statistical analysis. *, P < 0.05; **, P < 0.01.
SENP1 is the deSUMOylation enzyme of Gli1.
Previously, two SUMOylation receptor sites with the consensus sequence motif ΨKXE encompassing K180 and K815, respectively, were reported in human Gli1 (42). A careful search using a software GPS-SUMO algorithm revealed a third putative acceptor site surrounding K415 (Fig. 2A). Since SENP1 was found to negatively modulate Shh signaling, we speculated that it might act by deSUMOylating Gli1. To test this hypothesis, we expressed FLAG-tagged SUMO1 (FLAG-SUMO1) and hemagglutinin (HA)-tagged human Gli1 (HA-Gli1) in NIH 3T3 cells and then precipitated Gli1 from the cell lysates, which was followed by Western analysis for SUMOylated Gli1 with anti-HA antibody. The results showed that Gli1 was indeed SUMOylated, as evidenced by a FLAG-tagged band appearing above the 170-kDa molecular mass marker (Fig. 2B). Moreover, ShhN-CM treatment of transfected NIH 3T3 cells enhanced the SUMOylation (Fig. 2B). We were also able to detect endogenous SUMOylated Gli1 in SENP1−/− MEFs, but only when the cells were treated with ShhN-CM, which induced not only the production of total Gli1 but also the accumulation of SUMOylated Gli1 (Fig. 2C). This result corroborated the observation noted above that regulation of Gli1 by SUMO is likely secondary to Shh pathway activation. SUMOylation of exogenous HA-Gli1 was also demonstrated in transfected HEK293 cells by Western analysis following immunoprecipitation of cotransfected FLAG-SUMO1 with anti-FLAG, but simultaneously replacing K180, K415, and K815 with arginine completely abolished SUMOylation of the HA-Gli1-3KR mutant (Fig. 2D), suggesting that all three lysine residues are potential SUMO acceptor sites. Also, in SENP1−/− MEFs, proximity ligation assay (PLA) indicated that mutating these lysine residues in any binary combination reduced the PLA signal of HA-tagged Gli1 mutants and SUMO1, while mutating all three completely abolished it (see Fig. S2 in the supplemental material), once again suggesting that any of the three lysines can serve as the SUMOylation acceptor. Finally, coexpressing SENP1 along with wild-type Gli1 and SUMO1 in HEK293T cells completely blocked the SUMOylation of Gli1, but the catalytically inert SENP1m failed to alter the SUMOylation status of Gli1 (Fig. 2E), thus confirming the direct role of SENP1 in removing SUMO modification from Gli1. In contrast, coexpressing both SENP2 and the catalytically inert SENP2m moderately reduced the level of SUMOylation of HA-Gli1 in HEK293T cells (Fig. 2F), implying a nonspecific effect. Taken together, these results indicate that SENP1 is the specific deSUMOylating enzyme of Gli1.
FIG 2.

SENP1 is a Gli1-specific deSUMOylase. (A) Schematic representation of human Gli1 amino acid sequence showing relative positions of 3 consensus SUMOylation sites. NES, nuclear export signal. (B) Western analysis of SUMO1-modified Gli1 (arrow). FLAG-SUMO1 and HA-Gli1 were cotransfected into NIH 3T3 cells, and the SUMO1-modified proteins were isolated by anti-FLAG immunoprecipitation (IP), eluted, and then analyzed by Western blotting. (C) IP-Western analysis showing the accumulation of endogenous SUMOylated Gli1 in SENP1−/− MEFs in response to ShhN-CM treatment. (D) IP-Western analysis showing that replacing all 3 lysine residues of the consensus SUMOylation sites blocked SUMO1 modification of Gli1. HA-Gli1 or HA-Gli1-3KR was transfected alone or with FLAG-SUMO1 into HEK293T cells. The lysate was immunoprecipitated with anti-FLAG and then blotted by anti-HA in Western analysis. (E) Western analysis of SUMO1-modified Gli1 showing that SENP1, but not SENP1m, promotes the deSUMOylation of Gli1 in transfected HEK293 cells. (F) Western analysis showing nonspecific effects of SENP2 and SENP2m on SUMOylation of Gli1.
SUMOylation stabilizes Gli1 by competitively inhibiting ubiquitination.
One of the mechanisms by which SUMOylation is known to modulate activities of transcriptional factors is through competitive inhibition of ubiquitination, which marks proteins for destruction in proteasomes (34). In light of the elevated induction of Gli1 by Shh in SENP1−/− MEFs and the ability of SENP1 to deSUMOylate Gli1, we sought to determine if SENP1 regulates Gli1 stability and if it does so through competing with ubiquitination. Using cycloheximide to block protein synthesis, we quantified the turnover rates of Gli1 in SENP1−/− and its matching wild-type control MEFs. The results showed that the half-life of Gli1 increased from approximately 11 h in the control MEFs to slightly over 24 h in SENP1−/− MEFs (Fig. 3A and B). We also determined the half-life of exogenously expressed HA-Gli1 in normal MEFs to be about 25 h, but in comparison, the half-life of SUMOylation acceptor site mutant HA-Gli1-3KR was, surprisingly, over 48 h (Fig. 3C and D), indicating that the mutant was much more stable. Since the consensus SUMOylation sites are also shared by ubiquitination, our interpretation of this observation was that ubiquitination has a dominant effect on Gli1 stability over SUMOylation so that mutating these acceptor sites also compromises its ability to be modified by ubiquitination. Surprisingly, when assayed under comparable conditions, the level of ubiquitin modification of the HA-Gli1-3KR mutant was only moderately reduced compared to that of wild-type HA-Gli1 (see Fig. S3 in the supplemental material) rather than completely blocked, as was SUMOylation (Fig. 2D). Given the notorious promiscuity of ubiquitination in selecting acceptor sites, these results nevertheless showed that the three SUMO acceptor sites do influence Gli1 ubiquitination and, more prominently, its stability. To demonstrate that ubiquitination and SUMOylation of Gli1 are actually competing, we cotransfected a fixed amount of HA-ubiquitin with increasing amounts of His-SUMO1, along with FLAG-Gli1, into HEK293T cells and used MG132 to block protein degradation. The results showed that at high concentrations of SUMO1, the level of ubiquitinated Gli1 was effectively reduced (Fig. 3E and F). Furthermore, coexpressing SENP1, which removes SUMO1, markedly enhanced the level of ubiquitinated Gli1 in transfected HEK293T cells (Fig. 3G and H). Taken together, the above-mentioned data indicate that competitive inhibition of ubiquitination-mediated degradation by SUMOylation is at least one of the mechanisms through which SENP1 controls Gli1 stability.
FIG 3.

SENP1 promotes Gli1 turnover by enhancing ubiquitination at the same consensus sites. (A and B) Western analysis (A) and quantification (B) of endogenous Gli1 turnover in SENP1−/− and control MEFs. The cells were first treated with ShhN-CM for 24 h, and thereafter, cycloheximide (CHX) (20 μM) was added to the culture medium for the durations indicated. (C and D) Western analysis (C) and quantification (D) of exogenously expressed HA-Gli1 and HA-Gli1-3KR turnover in NIH 3T3 cells. The experiments were performed as for panel A. (E and F) Western analysis (E) and quantification (F) of ubiquitin-modified FLAG-Gli1 showing competition between SUMOylation and ubiquitination. HEK293T cells were transfected with FLAG-Gli1, HA-ubiquitin, and increasing amounts of HIS-SUMO1. The cells were treated with MG132 (20 μM) 6 h prior to harvesting for immunoprecipitation using anti-FLAG M2 beads. (G and H) Western analysis (G) and quantification (H) of ubiquitin-modified FLAG-Gli1 as for panel E, showing that SENP1 promotes ubiquitination of Gli1. In the above-described experiments, β-actin was used as a loading control. The arrows indicated ubiquitin-modified Gli1. Student's t tests were used for statistical analysis. *, P < 0.05; ***, P < 0.001. The error bars indicate SD.
SENP1 is required for the nuclear export of Gli1.
In addition to stability, SUMOylation is also known to control protein distribution within a cell (32). As shown by immunohistochemistry (IHC) staining, the level of resting-state Gli1 was low, and it was distributed in the cytoplasm in normal MEFs until the cells were activated by ShhN-CM, which induced the nuclear enrichment of Gli1 (Fig. 4A and B). In SENP1−/− MEFs, however, ShhN-CM treatment not only drastically increased the expression level of Gli1, the localization of Gli1 appeared to become exclusively nuclear (Fig. 4A and B). Both measures imply a hypersensitized Shh pathway. Interestingly, even in the resting state, Gli1 was already highly enriched in the nuclei of SENP1−/− MEFs, although the percentage of cells with Gli1-enriched nuclei was low (Fig. 4A and B). Nuclear and cytoplasmic fractionation analysis reciprocated the IHC experiment (Fig. 4C and D). In light of previous quantification of Gli1/Ptch1 mRNA and protein (Fig. 1C to G), our results showed that loss of SENP1 is not sufficient to cause Shh pathway activation. Exogenous HA-Gli1 delivered by transient transfection also behaved similarly in that in normal control MEFs, ShhN-CM induced nuclear accumulation of HA-Gli1 in 23% of cells within 24 h of treatment, but in SENP1−/− MEFs, this treatment essentially drove nuclear accumulation of HA-Gli1 in the entire cell population (Fig. 4E; see Fig. S4 in the supplemental material). Although the increased nuclear enrichment could have resulted from the intrinsic attribute of elevated Gli1 in the absence of SENP1, we found that little, if any, HA-Gli1-3KR entered the nucleus regardless of ShhN-CM treatment or the cell type tested (Fig. 4F; see Fig. S4), reaffirming the essential role of SUMOylTaken together, our data indicateation in the nuclear distribution of Gli1.
FIG 4.

SENP1 promotes the nuclear export of Gli1. (A) IHC staining of endogenous Gli1 in SENP1−/− and SENP1WT control MEFs. ShhN-CM treatment was given for 24 h. (B) Quantification of nuclear-cytoplasmic distribution of Gli1 as for panel A. Fifty cells were counted for each bar graph data point. (C) Biochemical fractionation of SENP1−/− and SENP1WT control MEFs, followed by Western analysis of endogenous Gli1. ShhN-CM treatment was given for 24 h, and lamin B (LMNB) and tubulin were used as the nuclear and cytoplasmic markers, respectively. (D) quantification of nuclear Gli1 as for panel C. (E and F) Nuclear-cytoplasmic distribution of exogenously expressed HA-Gli1 (E) and HA-Gli1-3KR (F) in SENP1−/− and SENP1WT control MEFs. The Gli proteins were visualized by anti-HA immunofluorescence, and the percentage of cells with mostly nuclear Gli1 staining was calculated from at least 50 cells. ShhN-CM treatment was given for the durations indicated. The regression curves were done in GraphPad Prism 5, and statistical significance between the two ShhN-CM-treated groups is indicated. Immunofluorescence images of panels E and F are shown in Fig. S4 in the supplemental material. (G) GliBS-luc (8×) luciferase assay of Gli1 transcriptional activity in HEK293 cells. (H and I) PLA (H) and quantification thereof (I) showing the enrichment of SUMO1-modified Gli1 in the nucleus in SENP1−/− and SENP1WT control MEFs. Following transfection with HA-Gli1 and FLAG-SUMO1, the cells were treated with ShhN-CM for 24 h. For quantification, 50 cells were counted for each bar graph data point. Student t tests were used for statistical analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The red PLA signal indicates where the SUMO1-modified Gli1 localized. The error bars indicate SD.
Since blocking SUMOylation by removing all three consensus acceptor sites precluded HA-Gli1-3KR from entering the nucleus, one would expect that the 3KR mutation would abolish Gli1 transcriptional activity and that nuclear Gli1 has to be SUMOylated. Indeed, when tested in the GliBS-luc reporter assay in HEK293 cells, the 3KR replacement showed complete blockage of Gli1 transcriptional activity, whereas various combinations of 1KR or 2KR mutations moderately reduced it (Fig. 4G). To test the latter possibility, we resorted to the proximity ligation assay to determine the subcellular localization of SUMOylated Gli1. In this assay, we transiently coexpressed HA-Gli1 and FLAG-SUMO1 in SENP1WT and SENP1−/− MEFs and detected the two proteins using forward and reverse primer-conjugated secondary antibodies. Only when Gli1 and SUMO were located within 40 nm of each other did the fluorescence signal become detectable after PCR amplification employing the antibody-linked primers. Using this assay, we detected little signal in normal control MEFs, possibly due to the robust deSUMOylation activity of SENP1; however, in SENP1−/− MEFs, we observed strong punctate signals within the nucleus, which were further enhanced by the ShhN-CM treatment (Fig. 4H and I). Thus, our data indicate that SENP1-mediated deSUMOylation is required for the nuclear export of Gli1.
SUMOylation is a nuclear retention signal for Gli1.
The exclusive nuclear localization of SUMOylated Gli1 suggests that SUMOylation may serve as a nuclear retention signal. To demonstrate this point, we mutated the nuclear localization signal (NLS) of Gli1 (HA-Gli1-mNLS) (Fig. 5A) and found that it was as good a substrate for SUMOylation as the normal Gli1 (Fig. 5B). However, HA-Gli1-mNLS was incapable of translocating into the nucleus regardless of the ligand treatment (Fig. 5C), even under SENP1-deficient conditions, which are conducive to nuclear accumulation of Gli1 (Fig. 4A and B). A proximity ligation assay confirmed that HA-Gli1-mNLS retained in the cytoplasm was SUMOylated in SENP1−/− cells (Fig. 5D). Once again, immunofluorescence (IF) experiments indicated that removing one or two consensus ΨKXE sites reduced but did not abolish nuclear accumulation of HA-Gli1 until all three sites were removed (Fig. 5E; see Fig. S5 in the supplemental material). However, when Crm1-mediated nuclear export was blocked with leptomycin B (LMB), all Gli mutants, including 3KR, were sequestered in the nucleus (Fig. 5F; see Fig. S1 in the supplemental material). Furthermore, IF staining experiments indicated that endogenous SENP1 was colocalized with HA-Gli1 but not the 3KR mutant in the nucleus in normal control MEFs and that ShhN-CM treatment enhanced that colocalization (Fig. 5G). Taken together, our data indicate that SUMOylation of Gli1, which occurs in the cytoplasm, serves as a nuclear retention signal whose removal by SENP1 in the nucleus sets up the export of Gli1.
FIG 5.

SUMOylation constitutes a nuclear retention signal of Gli1. (A) Schematic representation of Gli1 sequence encompassing the NLS. The amino acid residue substitutions used for creating the NLS mutant, HA-Gli1-mNLS, are shown in red. (B) Western analysis of SUMO1-modified HA-Gli1 and the NLS mutant HA-Gli1-mNLS in HEK293T cells. (C) Immunofluorescence staining and quantification of HA-Gli1 and HA-Gli1-mNLS in SENP1−/− MEFs. (D) PLA analysis of SUMO1-modified HA-Gli1 and HA-Gli1-mNLS in SENP1−/− MEFs. ShhN-CM treatment was given for 24 h. (E and F) Nuclear-cytoplasmic distribution of various SUMOylation site mutants of Gli1 in SENP1−/− MEFs (E) and in SENP1−/− MEFs (F) treated with LMB for 6 h to block nuclear export. Each bar graph data point was calculated based on data from at least 50 cells. (G) Immunofluorescence staining of endogenous SENP1 and exogenously expressed HA-Gli1 or HA-Gli1-3KR in normal MEFs. Note the colocalization of HA-Gli1 but not HA-Gli1-3KR with SUMO1 in the nucleus under ShhN-CM treatment. Student's t tests were used for statistical analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars indicate SD.
SENP1 attenuates the Shh-dependent proliferation of cerebellar granule cell precursors.
To assess the physiological significance of SENP1-mediated deSUMOylation in Shh signaling, we took advantage of cerebellar granule cell precursors (GCPs), whose proliferation is absolutely dependent upon Shh signaling (20). To this end, we isolated GCPs from 7-day-old mouse pups and cultured them in vitro in the presence of a fluorescence-labeled thymidine analogue, EdU, which can be conveniently used as a surrogate measure of cell proliferation because it is incorporated into the chromosomes during DNA synthesis. These primary cells underwent apoptosis once put in culture and died out within 48 h unless their proliferation was sustained with ShhN-CM (Fig. 6A). Knockdown of SENP1 with siRNA further increased the level of proliferation of GCPs (Fig. 6A). On the other hand, forced expression of SENP1 but not the catalytically mutant SENP1m markedly reduced the level of Shh-dependent growth of GCPs in culture (Fig. 6B). These results suggest that SENP1 normally exerts a restraint on the physiological function of Shh signaling in sustaining the proliferation of GCPs.
FIG 6.
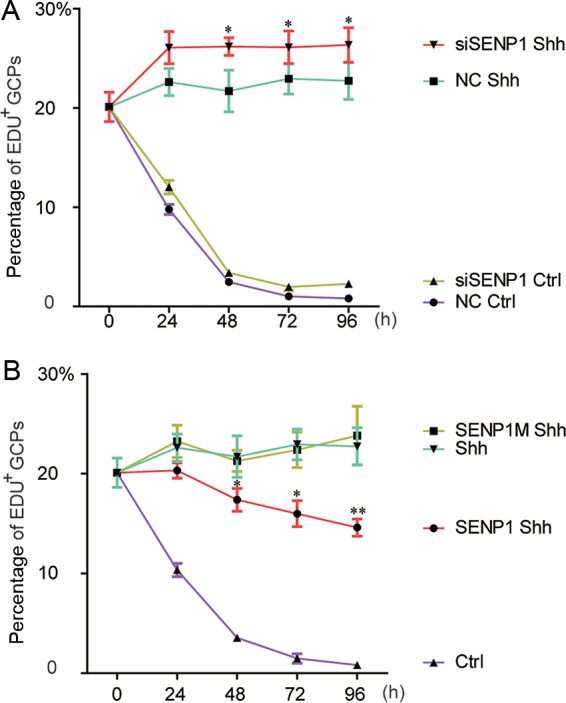
SENP1 attenuates Shh signaling required to sustain the proliferation of cerebellar granule cell precursors. (A) Quantification of EdU incorporation in freshly isolated cerebellar granule cell precursors in which SENP1 expression was knocked down by an siRNA specific for SENP1. (B) Quantification of EdU incorporation in freshly isolated cerebellar granule cell precursors that received exogenously expressed SENP1 or SENP1m. Each data point represents cell counts from 5 separate fields, and the error bars indicate SD. NC, nonspecific control siRNA. The statistical significance between siRNA versus SENP1 and NC (A) or SENP1 Shh and Shh samples (B) is shown. Student t tests were used for statistical analysis. *, P < 0.05; **, P < 0.01.
DISCUSSION
Precise control of its signaling output is essential to the morphogenic roles of the Shh pathway (51), and yet, how this is achieved still remains to be fully determined. Here, we present evidence that shows SENP1 is the specific deSUMOylation enzyme of Gli1, and we demonstrate that SUMOylation stabilizes Gli1 by competing with ubiquitination and serves as a nuclear retention signal for Gli1. Thus, SENP1 negatively regulates Shh signaling by at least two mechanisms, namely, promoting Gli1 turnover and nuclear export. Our data further reveal that regulation of Gli1 activities by SENP1 is of physiological significance in sustaining the proliferation of cerebellar granule cell precursors, adding to the already complex mechanisms that orchestrate Shh signaling responses.
Several recent studies showing SUMOylation of Gli proteins have been reported (42–44), revealing that all 3 Gli proteins are modified at several phylogenetically conserved sites encompassing the consensus sequence ΨKXE, but these studies diverge on the role of this modification in Shh signaling. In one report, forced expression of the putative Gli SUMOylation E3 ligase PIAS1 in chicken embryos was shown to increase Gli activity and cause ectopic expression of a Gli target gene in the neural tube (42). This positive role was corroborated in a separate study that showed that mutating 10 lysine residues of possible SUMOylation sites in Ci abolished the transcriptional activity of the mutant Ci in the Drosophila testis (44). However, another report showed that mutating Gli2 SUMOylation sites significantly increased Gli2 transcriptional activity both in a cell-based assay and in mice, possibly because these sites were also shown to be required for recruiting the histone modifier HDAC5, which represses transcription (43). Currently, there is no clear explanation given in the literature for these discrepancies; however, it is possible that they could arise from the use of lysine mutants to block SUMOylation, which recognizes the same substrates as ubiquitination. Indeed, our experiments showed that SUMOylation competes with ubiquitination for modification of Gli1 and thereby increases its stability (Fig. 3). Thus, depending on the particular assay and experimental procedure, one type of modification could display a dominant effect over the other. Alternatively, SUMO moieties on these Gli proteins could be removed by different SENPs, which could play opposite roles under different circumstances, as we demonstrated for SENP1 and SENP2 in our assays for Shh signaling activities (Fig. 1A and B). In this regard, approaches that genetically alter the activities of modifying enzymes, as taken in our investigation here, may prove to be methods of choice to ascertain the function of SUMOylation in Shh signaling.
The observation that loss of SENP1 drastically sensitized cellular response to Shh and caused Gli1 to be enriched in the nucleus is intriguing. First, it suggests that SENP1 may also play an important role in controlling the nuclear-cytoplasmic distribution of transcriptional factors, such as Gli1, putting SENP1 in the same functional category as SENP2, which has been shown to interact with components of the nuclear pore complexes. Second, given the result where the NLS mutant Gli1 was nevertheless still capable of being modified by SUMO, nuclear accumulation of Gli1 in the absence of SENP1 implies that SUMOylation and deSUMOylation are likely compartmentalized in the cytoplasm and the nucleus, respectively. Third, since mutating the lysine residues of all 3 SUMOylation sites in Gli1 blocked the nuclear localization of the mutant Gli1, SUMOylation likely constitutes a nuclear retention signal for Gli1. In keeping with this notion, we observed that Shh signaling promotes the SUMOylation of Gli1 (Fig. 2B and C), thus enhancing its nuclear function. Although the precise timing of SUMO modification relative to subcellular movements of Gli1, as well as chromatin binding, is not known, SENP1 likely plays an important role in attenuating Shh signaling by triggering nuclear exit of Gli1 via deSUMOylation (Fig. 7). Future studies are required to ascertain if attenuation by SENP1 contributes to defining the boundaries of Shh signaling domains.
FIG 7.
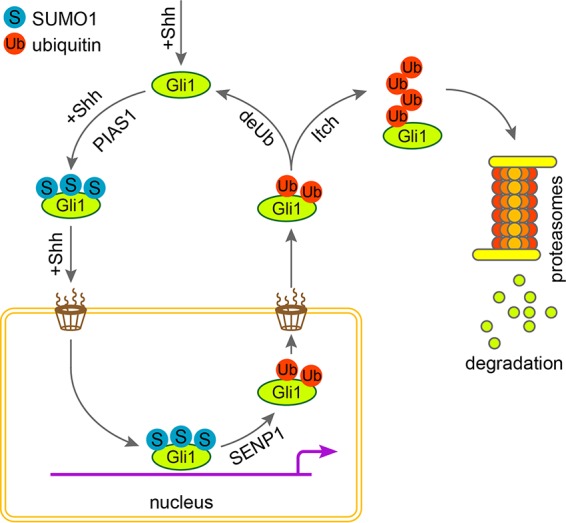
Model for the role of SENP1 in promoting the nuclear export of Gli1. While Shh signaling drives the transport of Gli1 into the nucleus, SUMOylation, likely catalyzed by PIAS1 in the cytoplasm, enables Gli1 to fulfill its transcriptional-activator function by serving as a nuclear retention signal. In tissues or cells where Shh is actively suppressed, SENP1 releases Gli1 to be exported into the cytoplasm by removing SUMO modification and thereby attenuates Shh signaling.
MATERIALS AND METHODS
Cell lines, plasmids, and siRNAs.
Mutant SENP1−/− and SENP2−/− and their matching WT control MEFs, as well as SENP1, SENP2, FLAG-SUMO1, HA-SUMO1, and HIS-SUMO1 cDNA expression constructs, were generous gifts from Jinke Cheng's laboratory, Shanghai Jiaotong University. The cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Full-length human Gli1 cDNA was obtained from the ATCC; the HA and FLAG tags were added by PCR, and the resultant cDNA expression constructs were sequence verified and subcloned into the pRK5 vector. The SUMOylation site KR mutants and the NLS mutant of Gli1 were generated with Mut Express and MultiS fast mutagenesis kits (Vazyme; C215-01/02), respectively. siRNAs were purchased from GenePharma (Shanghai, China), and their sequences are shown in Table S1 in the supplemental material. Plasmid transfection was carried out using Lipofectamine Transfection Plus reagent (Invitrogen), and siRNA transfection was done using Lipofectamine 2000 transfection reagents (Invitrogen). The ShhN-conditioned medium was produced from HEK293 cells that were transiently transfected with a plasmid vector expressing the N-terminal fragment of Shh and was used at 1:16 dilution.
Real-time PCR and quantitative real-time PCR (qRT-PCR).
Total RNAs were isolated from cultured cells with RNAiso Plus reagent (TaKaRa) and reverse transcribed using Vazyme HiScript II Q RTSuper mix (Vazyme). Quantitative real-time PCR (qPCR) was carried out using FastStart essential DNA green master (Roche). Each measurement was repeated three times, and each sample was analyzed in triplicate with hypoxanthine phosphoribosyltransferase (HPRT) as an internal control. The PCR and qPCR primers are listed in Table S2 in the supplemental material.
Immunoprecipitation and Western analysis.
Cultured cells were lysed for 30 min at 4°C in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, pH 8.0, 1% NP-40, 0.5% sodium deoxycholate, and 1× Roche Complete protease inhibitor cocktail [Roche]). The lysate was clarified by centrifugation for 20 min at 20,000 × g and 4°C. Total protein was quantified using a Thermo Pierce bicinchoninic acid (BCA) kit (Thermo), and 400 μg of total protein was used for each precipitation by incubating at 4°C overnight with anti-FLAG M2 beads (Sigma; 1:1,000). The beads were then washed three times with washing buffer (50 mM Tris-HCl, pH 7.4, 300 mM NaCl, 1 mM EDTA, pH 8.0, 0.5% NP-40, 10% glycerol) for Western analysis.
Antibodies and immunofluorescence microscopy.
The primary antibodies were mouse anti-β-actin (Santa Cruz Biotechnology; 1:1,000), rabbit anti-Gli1 (Cell Signaling Technology; 1:1,000 for Western analysis and 1:200 for IHC), mouse anti-FLAG (Sigma 1:1,000), rat anti-HA (Roche; 1:1,000 for Western analysis and 1:300 for IF), rabbit anti-SENP1 (Sigma; 1:1,000 for Western analysis and 1:100 for IF), mouse anti-HA (Protein-Tech; 1:100 in PLA), rabbit anti-SUMO1 (Cell Signaling Technology; 1:1,000 for Western analysis and 1:100 for PLA), and rabbit antiubiquitin (Santa Cruz Biotechnology; 1:1,000). The secondary antibodies were donkey anti-mouse (Santa Cruz Biotechnology; 1:5,000 for Western analysis), goat anti-rabbit (Santa Cruz Biotechnology; 1:5,000 for Western analysis), goat anti-rat (Santa Cruz Biotechnology; 1:5,000 for Western analysis), and Alexa Fluor-conjugated (Invitrogen; 1:200 for IF) antibodies. For immunofluorescence microscopy, cells were fixed with 4% paraformaldehyde (PFA) for 10 min at 4°C, permeabilized, and blocked with 0.3% NP-40–3% BSA in phosphate-buffered saline (PBS) for 30 min at room temperature.
PLA.
SENP1−/− and matching control MEFs were transfected with pRK5-HA-Gli1 for 24 h and then stimulated with ShhN-CM for another 24 h. The cells were fixed in 4% formaldehyde and permeabilized with 0.3% NP-40. PLA was carried out using Duolink in situ detection reagents red (Sigma). Microscopic images were taken using a confocal microscope (LSM710; Zeiss).
GCP isolation and proliferation assay.
Mouse cerebellar GCPs were isolated from 7-day-old pups as described previously (52). Plasmid and siRNA transfection was carried out using FugeneHD Transfection Reagent (Promega). The rate of GCP proliferation was measured using Click-iT EdU cell proliferation assays (Life Technology). EdU was added to the cell culture 12 h prior to the end of each ShhN-CM treatment time point and then analyzed according to the manufacturer's instructions. Images were acquired using an inverted fluorescence microscope (DMI 300B; Leica), and the EdU-positive GCPs were quantified with ImageJ software.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jinke Cheng for providing various reagents.
This work was supported by grants from the Chinese National Science Foundation (81272238 and 81672748 to S.Y.C.; 81602431 to T.-T.Y.) and the National Basic Research Program of China (973 Program; 2012CB945003 to S.Y.C.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00579-16.
REFERENCES
- 1.Ryan KE, Chiang C. 2012. Hedgehog secretion and signal transduction in vertebrates. J Biol Chem 287:17905–17913. doi: 10.1074/jbc.R112.356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robbins DJ, Fei DL, Riobo NA. 2012. The Hedgehog signal transduction network. Sci Signal 5:re6. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang J, Hui CC. 2008. Hedgehog signaling in development and cancer. Dev Cell 15:801–812. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrova R, Joyner AL. 2014. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 141:3445–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alman BA. 2015. The role of hedgehog signalling in skeletal health and disease. Nat Rev Rheumatol 11:552–560. doi: 10.1038/nrrheum.2015.84. [DOI] [PubMed] [Google Scholar]
- 6.Briscoe J, Therond PP. 2013. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 7.Hui CC, Angers S. 2011. Gli proteins in development and disease. Annu Rev Cell Dev Biol 27:513–537. doi: 10.1146/annurev-cellbio-092910-154048. [DOI] [PubMed] [Google Scholar]
- 8.Amakye D, Jagani Z, Dorsch M. 2013. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med 19:1410–1422. doi: 10.1038/nm.3389. [DOI] [PubMed] [Google Scholar]
- 9.Scales SJ, de Sauvage FJ. 2009. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci 30:303–312. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 10.Yao E, Chuang PT. 2015. Hedgehog signaling: from basic research to clinical applications. J Formos Med Assoc 114:569–576. doi: 10.1016/j.jfma.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Ingham PW, McMahon AP. 2001. Hedgehog signaling in animal development: paradigms and principles. Genes Dev 15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 12.Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. 1993. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 13.Taipale J, Cooper MK, Maiti T, Beachy PA. 2002. Patched acts catalytically to suppress the activity of Smoothened. Nature 418:892–897. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- 14.Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. 2003. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 15.Rohatgi R, Milenkovic L, Scott MP. 2007. Patched1 regulates hedgehog signaling at the primary cilium. Science 317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 16.Rohatgi R, Scott MP. 2007. Patching the gaps in Hedgehog signalling. Nat Cell Biol 9:1005–1009. doi: 10.1038/ncb435. [DOI] [PubMed] [Google Scholar]
- 17.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. 2005. Vertebrate Smoothened functions at the primary cilium. Nature 437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 18.Kovacs JJ, Whalen EJ, Liu R, Xiao K, Kim J, Chen M, Wang J, Chen W, Lefkowitz RJ. 2008. Beta-arrestin-mediated localization of smoothened to the primary cilium. Science 320:1777–1781. doi: 10.1126/science.1157983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang S, Zhang Z, Zhang C, Lv X, Zheng X, Chen Z, Sun L, Wang H, Zhu Y, Zhang J, Yang S, Lu Y, Sun Q, Tao Y, Liu F, Zhao Y, Chen D. 2013. Activation of Smurf E3 ligase promoted by smoothened regulates hedgehog signaling through targeting patched turnover. PLoS Biol 11:e1001721. doi: 10.1371/journal.pbio.1001721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yue S, Tang LY, Tang Y, Shen QH, Ding J, Chen Y, Zhang Z, Yu TT, Zhang YE, Cheng SY. 12 June 2014 Requirement of Smurf-mediated endocytosis of Patched1 in sonic hedgehog signal reception. eLife 3. doi: 10.7554/eLife.02555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding Q, Motoyama J, Gasca S, Mo R, Sasaki H, Rossant J, Hui CC. 1998. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125:2533–2543. [DOI] [PubMed] [Google Scholar]
- 22.Falkenstein KN, Vokes SA. 2014. Transcriptional regulation of graded Hedgehog signaling. Semin Cell Dev Biol 33:73–80. doi: 10.1016/j.semcdb.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hui CC, Joyner AL. 1993. A mouse model of Greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet 3:241–246. doi: 10.1038/ng0393-241. [DOI] [PubMed] [Google Scholar]
- 24.Kinzler KW, Vogelstein B. 1990. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol Cell Biol 10:634–642. doi: 10.1128/MCB.10.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, Nakashima M, Joyner AL. 2000. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 127:1593–1605. [DOI] [PubMed] [Google Scholar]
- 26.Aza-Blanc P, Ramirez-Weber FA, Laget MP, Schwartz C, Kornberg TB. 1997. Proteolysis that is inhibited by hedgehog targets cubitus interruptus protein to the nucleus and converts it to a repressor. Cell 89:1043–1053. doi: 10.1016/S0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- 27.Wang B, Fallon JF, Beachy PA. 2000. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100:423–434. doi: 10.1016/S0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- 28.Jia J, Zhang L, Zhang Q, Tong C, Wang B, Hou F, Amanai K, Jiang J. 2005. Phosphorylation by double-time/CKIepsilon and CKIalpha targets cubitus interruptus for Slimb/beta-TRCP-mediated proteolytic processing. Dev Cell 9:819–830. doi: 10.1016/j.devcel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Niewiadomski P, Kong JH, Ahrends R, Ma Y, Humke EW, Khan S, Teruel MN, Novitch BG, Rohatgi R. 2014. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep 6:168–181. doi: 10.1016/j.celrep.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan Y, Wang C, Wang B. 2009. Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev Biol 326:177–189. doi: 10.1016/j.ydbio.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price MA, Kalderon D. 2002. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by glycogen synthase kinase 3 and casein kinase 1. Cell 108:823–835. doi: 10.1016/S0092-8674(02)00664-5. [DOI] [PubMed] [Google Scholar]
- 32.Flotho A, Melchior F. 2013. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem 82:357–385. doi: 10.1146/annurev-biochem-061909-093311. [DOI] [PubMed] [Google Scholar]
- 33.Matunis MJ, Coutavas E, Blobel G. 1996. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J Cell Biol 135:1457–1470. doi: 10.1083/jcb.135.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gareau JR, Lima CD. 2010. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11:861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hendriks IA, Lyon D, Young C, Jensen LJ, Vertegaal AC, Nielsen ML. 2017. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat Struct Mol Biol 24:325–336. doi: 10.1038/nsmb.3366. [DOI] [PubMed] [Google Scholar]
- 36.Hendriks IA, Vertegaal AC. 2016. A comprehensive compilation of SUMO proteomics. Nat Rev Mol Cell Biol 17:581–595. doi: 10.1038/nrm.2016.81. [DOI] [PubMed] [Google Scholar]
- 37.Matic I, Schimmel J, Hendriks IA, van Santen MA, van de Rijke F, van Dam H, Gnad F, Mann M, Vertegaal AC. 2010. Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Mol Cell 39:641–652. doi: 10.1016/j.molcel.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 38.Geiss-Friedlander R, Melchior F. 2007. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 39.Hickey CM, Wilson NR, Hochstrasser M. 2012. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol 13:755–766. doi: 10.1038/nrm3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nayak A, Muller S. 2014. SUMO-specific proteases/isopeptidases: SENPs and beyond. Genome Biol 15:422. doi: 10.1186/s13059-014-0422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim JH, Baek SH. 2009. Emerging roles of desumoylating enzymes. Biochim Biophys Acta 1792:155–162. doi: 10.1016/j.bbadis.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 42.Cox B, Briscoe J, Ulloa F. 2010. SUMOylation by Pias1 regulates the activity of the Hedgehog dependent Gli transcription factors. PLoS One 5:e11996. doi: 10.1371/journal.pone.0011996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han L, Pan Y, Wang B. 2012. Small ubiquitin-like modifier (SUMO) modification inhibits GLI2 protein transcriptional activity in vitro and in vivo. J Biol Chem 287:20483–20489. doi: 10.1074/jbc.M112.359299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lv X, Pan C, Zhang Z, Xia Y, Chen H, Zhang S, Guo T, Han H, Song H, Zhang L, Zhao Y. 2016. SUMO regulates somatic cyst stem cell maintenance and directly targets the Hedgehog pathway in adult Drosophila testis. Development 143:1655–1662. doi: 10.1242/dev.130773. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Yue S, Xie L, Pu XH, Jin T, Cheng SY. 2011. Dual phosphorylation of suppressor of fused (Sufu) by PKA and GSK3beta regulates its stability and localization in the primary cilium. J Biol Chem 286:13502–13511. doi: 10.1074/jbc.M110.217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sasaki H, Hui C, Nakafuku M, Kondoh H. 1997. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 124:1313–1322. [DOI] [PubMed] [Google Scholar]
- 47.Qi Y, Zuo Y, Yeh ET, Cheng J. 2014. An essential role of small ubiquitin-like modifier (SUMO)-specific protease 2 in myostatin expression and myogenesis. J Biol Chem 289:3288–3293. doi: 10.1074/jbc.M113.518282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Nguyen T, Angkasekwinai P, Dou H, Lin FM, Lu LS, Cheng J, Chin YE, Dong C, Yeh ET. 2012. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol Cell 45:210–221. doi: 10.1016/j.molcel.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hang J, Dasso M. 2002. Association of the human SUMO-1 protease SENP2 with the nuclear pore. J Biol Chem 277:19961–19966. doi: 10.1074/jbc.M201799200. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H, Saitoh H, Matunis MJ. 2002. Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Mol Cell Biol 22:6498–6508. doi: 10.1128/MCB.22.18.6498-6508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ingham PW, Nakano Y, Seger C. 2011. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet 12:393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- 52.Gallo V, Ciotti MT, Coletti A, Aloisi F, Levi G. 1982. Selective release of glutamate from cerebellar granule cells differentiating in culture. Proc Natl Acad Sci U S A 79:7919–7923. doi: 10.1073/pnas.79.24.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.