

Abstract
Vascular endothelial growth factors (VEGFs) and their receptors play crucial roles in the formation of blood and lymphatic vessels during embryogenesis, and also under pathologic conditions in the adult. Despite intensive efforts over the last decades to elucidate the precise functions of VEGFs, transcriptional responses to VEGF receptor stimulation are still not fully characterized. To investigate the specific transcriptional effects of VEGFR-2 and VEGFR-3 activation, we performed a correlation analysis of previously published CAGE sequencing and microarray data of human lymphatic endothelial cells (LECs) stimulated with distinct VEGFs acting through either VEGFR-2 or VEGFR-3. We identified that specific activation of VEGFR-3 by VEGF-C156S results in the downregulation of many genes involved in immune regulation and inflammation, suggesting that VEGFR-3 stimulation has direct anti-inflammatory effects. Comparing CAGE and microarray data sets, we furthermore identified a small number of genes that showed a receptor-dependent response in LECs, demonstrating that these receptors, despite activating very similar signaling pathways, fulfill overlapping but not identical functions within the same cell type (LECs).
Subject terms: Growth factor signalling, Transcriptomics
Introduction
The vascular system, consisting of blood and lymphatic vessels, is crucially dependent on vascular endothelial growth factors (VEGFs) and their receptors (VEGFRs), which regulate the proliferation and function of endothelial cells. In humans, the VEGF family comprises five ligands, placenta growth factor (PLGF), VEGF-A, VEGF-B, VEGF-C and VEGF-D, that are structurally related and predominantly present as homodimers in their mature form1. VEGFs exert their function by binding to the three known VEGFRs, VEGFR-1, VEGFR-2 or VEGFR-3, which are expressed by blood vessel endothelial cells (BECs) in case of VEGFR-1 and -2, and lymphatic vessel endothelial cells (LECs) in case of VEGFR-2 and -3. Some expression of VEGFR-3 in angiogenic blood vessels has been described as well2. In addition, VEGFs bind to co-receptors such as neuropilins (NRP) 1 and 2 as well as heparan sulfate proteoglycans, that do not signal directly but modulate the interaction between VEGFs and VEGFRs and thereby affect VEGF signaling indirectly1.
The individual members of the VEGF family display selective binding affinities for one or several VEGFRs. PLGF and VEGF-B exclusively interact with VEGFR-1. VEGF-A, the most potent inducer of endothelial responses within the family, binds to both VEGFR-1 and VEGFR-2. Although the affinity of VEGF-A is higher for VEGFR-1 than for VEGFR-2, the latter receptor has a much higher kinase activity and is thus regarded as the most important receptor for transmitting the effects of VEGF-A. VEGF-C and VEGF-D differ from the other ligands of the family in that they are produced as immature precursor proteins, requiring proteolytic maturation in order to gain their full activity, and preferentially bind to VEGFR-3. In their fully mature form, however, both ligands also act as VEGFR-2 agonists3. Of note, a mutated form of VEGF-C, in which Cys156 is replaced by a Ser residue (VEGF-C156S), displays reduced affinity for VEGFR-2, and has thus been regarded as a selective VEGFR-3 stimulator4.
Signaling through VEGFR-2 is initiated upon ligand binding that stabilizes the receptor in a dimeric state and induces conformational changes, activating the intracellular kinase domains5. This in turn leads to the phosphorylation of specific Tyr residues in the cytoplasmic tail of the receptor, which serve as docking sites for adapter proteins needed to initiate multiple downstream signaling cascades such as the PI3K-AKT, PLC-γ and Ras-Raf-ERK signaling pathways. The biochemical processes immediately downstream of receptor activation have been investigated to considerable detail (reviewed in ref. 1). On the one hand, these signaling cascades trigger immediate, transcription independent cellular responses such as cell migration and opening of cellular junctions. On the other hand, VEGFR signaling activates several transcription factors (TFs), including AP-1, NFAT and FOXO family members, that regulate several genes involved in cell proliferation, differentiation and survival. Compared to VEGFR-2, the VEGFR-3 signaling cascade is somewhat less well understood but is regarded to activate very similar pathways as those downstream of VEGFR-2 (ref. 1). Nonetheless, the changes in gene expression downstream of the two receptors have not been fully characterized.
Using CAGE RNA sequencing6 in conjunction with the FANTOM5 project7, we recently analyzed transcriptional changes elicited by VEGF-C156S mediated VEGFR-3 stimulation in primary human lymphatic endothelial cells (LECs) and identified several TFs involved in this response. The majority of these TFs are well known ‘immediate early’ TFs that are induced upon signaling through various receptor tyrosine kinases such as EGR, FOS and JUN. Importantly, we also identified TFs with LEC-specific functions during VEGFR-3 signaling. For example, the homeobox TF HOXD10 that is constitutively present in LECs but not in BECs, is required for the induction of a second, downstream TF, namely NR4A1 (ref. 8). Another TF, MAFB, was induced and activated in LECs after VEGFR-3 and VEGFR-2 stimulation but not in BECs after VEGFR-2 stimulation, indicating that in the context of VEGF signaling, MAFB is a LEC-specific TF9. These data indicate that there are differences in TF activation downstream of VEGFRs, depending on the endothelial cell type, that are likely to result in the induction of different target genes in LECs and BECs. It is unclear, however, whether there also are receptor-specific differences in the regulation of target genes downstream of VEGFR-2 stimulation compared to VEGFR-3 stimulation, and whether these differences are cell type-dependent.
Our previous analysis of the CAGE RNA sequencing data only revealed upregulated but not downregulated genes after VEGF-C156S treatment of LECs9. Here, we re-analyzed the same data set using a different analysis method (DESeq2). We identified multiple downregulated genes that were frequently associated with pro-inflammatory functions. Furthermore, we performed a combined analysis of the CAGE sequencing data and our previously published microarray-based gene expression data of primary human LECs treated with VEGF-A or fully mature VEGF-C, resulting in activation of either VEGFR-2 or of both VEGFR-2 and VEGFR-3, respectively10. We found that even though the majority of target genes are comparably regulated by VEGFR-2 and VEGFR-3 activation in human LECs, a small number of distinct genes showed receptor-specific responses within the same cell type.
Results
VEGF-C156S provokes complex changes of LEC gene expression
We previously published a CAGE RNA sequencing data set of primary human dermal LECs treated with the VEGFR-3-specific ligand VEGF-C156S, analyzed at 16 different time points from 0 min (control) to 480 min (ref. 9) (Fig. 1a). In this study, we had analyzed differential gene expression by comparing each time point tn to the preceding time point tn-1 using the EdgeR method11. Since this analysis likely failed to identify genes with small, gradual changes in gene expression, we re-analyzed the data set comparing each time point tn to the baseline time point t0 using paired DESeq2 (ref. 12). Among 85774 unique CAGE peaks expressed in the data set, we identified a total of 990 upregulated (P<0.05, log2 fold change (log2FC)>0.6) and 1012 downregulated (P<0.05, log2FC<−0.6) peaks (Fig. 1b). Activation of VEGFR-3 with VEGF-C156S caused two ‘waves’ of CAGE peak upregulation, the first from 15 to 210 min, and a second one from 360 to 480 min. Downregulated CAGE peaks were most frequently detected at the 15 and 100 min time points and during the late phase of the time course (360 to 480 min), corresponding to the second wave of upregulated CAGE peaks (Fig. 1b).
Figure 1. Characterization of transcriptional changes in primary human LECs treated with VEGF-C156S by CAGE RNA sequencing.

(a) Schematic overview of the experimental procedure: Primary human dermal LECs from three individual donors were incubated with VEGF-C156S (1.5 μg/ml). RNA was extracted at 16 different time points from 0 min (baseline) to 480 min and subjected to CAGE RNA sequencing. (b) MA plots showing the log2FC compared to the expression level derived from DESeq2 analysis of differentially expressed CAGE peaks at each time point compared to the baseline. Red dots represent significantly altered CAGE peaks (P<0.05) and horizontal blue lines represent the log2FC cutoff of 0.6 resp. −0.6. The red numbers in the plots correspond to the number of significantly altered CAGE peaks above the log2FC cutoff. (c) Summary table of the number of genes corresponding to the DE CAGE peaks. (d) Heat map based on the expression levels (tpm) of all genes corresponding to DE CAGE peaks after hierarchical clustering. Colors code for log2FC values on a scale from −3 to +3. (e) Venn diagram comparing the upregulated genes between DESeq2 and previously published EdgeR analyses.
On the gene level, these differentially expressed (DE) CAGE peaks corresponded to 501 upregulated and 772 downregulated genes, according to the current CAGE peak annotation (Fig. 1c and ‘Upregulated Genes after VEGFC156S stimulation’, ‘Downregulated Genes after VEGFC156S stimulation’, Data Citation 1). Hierarchical clustering analysis of the upregulated genes over the 16 time points showed three distinct gene clusters: genes upregulated at the early time points (15 to 100 min), genes upregulated at the middle to late time points (120 to 480 min), and genes with a ‘bi-phasic’ upregulation from 60 to 100 min and from 360 to 480 min. Similarly, the downregulated genes also clustered into three groups: genes downregulated in both the middle and late phase of the time course (120 to 480 min), genes downregulated only in the late phase (360 to 480 min) and genes with a bi-phasic downregulation (15 to 100 min and 360 to-480 min) (Fig. 1d). Furthermore, we compared the 501 upregulated genes identified here with the 241 upregulated genes found in our previous analysis9. Among the 158 overlapping genes, we found several important lymphatic TFs such us KLF4, SOX18, and MAFB (Fig. 1e). Interestingly, using DESeq2, we found upregulation of further TFs such as FOXC2, ELK1, and NFATC1 which have previously been associated with vascular development and / or angiogenesis13–15. Taken together, these results indicate that selective activation of VEGFR-3 causes complex changes in gene expression in LECs, with multiple target genes showing distinct kinetics of up- or downregulation.
Gene ontology analysis reveals downregulation of immune response-related genes after VEGFR-3 stimulation
To characterize the transcriptional changes elicited by VEGFR-3 activation in LECs functionally, we grouped the differentially expressed genes into 3 groups, roughly corresponding to the clusters observed before: early phase genes (15 to 120 min), middle phase genes (150 to 240 min) and late phase genes (300 to 480 min) (Fig. 2a). Then, we performed a gene ontology (GO) analysis of the up- and downregulated genes in each of these phases, considering the ‘biologic process’ GO terms (Fig. 2b–d and ‘GO_BP analysis’, Data Citation 1). Strikingly, we found that downregulated genes were enriched for GO terms related to immune regulation and cytokine (particularly interferon) responses, indicating that VEGFR-3 stimulation has a direct, anti-inflammatory effect on LECs. Downregulated genes included adhesion molecules (SELE, VCAM1), cytokines and chemokines (CXCL10, CXCL11) and several INF-γ related genes (IRF2, IRF3, IFIH1, IFI16, IFI35, IFIT3). In line with our previous analysis9, the upregulated genes were enriched for GO terms related to chromatin organization, mRNA expression, and protein translation and localization (Fig. 2b–d). We found an enrichment of RNA splicing-associated genes among the early downregulated genes, whereas the upregulated genes were enriched for cell differentiation-associated genes (Fig. 2b). Similar results were obtained when ‘molecular function’ GO terms were used (‘GO_MF analysis’, Data Citation 1). Taken together, these results indicate that VEGFR-3 stimulation results in the upregulation of genes associated with gene expression and protein translation, whereas genes involved in immune regulation and cytokine (interferon) responses are downregulated.
Figure 2. Gene ontology analysis and clustering of up- and downregulated genes.

(a) DE genes were grouped into three distinct phases of the time course experiment: early (15 to 120 min), middle (150 to 240 min), and late phase (300 to 360 min). (b–d) Gene ontology (GO) analysis and clustering of the 25 most highly enriched GO biological process terms into related topics for the early (b), middle (c) and late (d) phase of the time course. GO analysis of downregulated genes is shown in the left side panels, GO analysis of upregulated genes is shown in the right side panels.
Comparison of VEGF-A and VEGF-C156S induced gene expression reveals a small set of receptor-specific target genes
VEGF-C156S is a selective agonist of VEGFR-3 (ref. 4), whereas VEGF-A mainly acts through VEGFR-2, and mature wildtype VEGF-C triggers both receptors (Fig. 3a). We previously published microarray-based (ABI Human Genome Survey V2.0) gene expression data of primary dermal LECs stimulated with VEGF-A or mature wildtype VEGF-C (Data Citation 2)10, 60 min, 240 min, 480 min and 24 h after stimulation. We decided to use the VEGF-A data to identify genes regulated by either VEGFR-2 or VEGFR-3 stimulation, comparing them to the VEGF-C156S stimulation time course at the three overlapping time points 60 min, 240 min, and 480 min (Fig. 3b). To this end, we correlated the log2FC values of each gene after VEGF-A or VEGF-C156S stimulation with each other (Fig. 3c). The correlation was relatively low, especially at the two later time points, probably due to technical biases between CAGE RNA sequencing and the microarrays. Correspondingly, we observed a similarly low correlation when we compared VEGF-C156S with wildtype VEGF-C induced gene expression, but a much higher correlation when we compared VEGF-A with wildtype VEGF-C induced gene expression (Supplementary Fig. 1). Nonetheless, at the 60 min time point, a positive correlation between the VEGF-C156S and VEGF-A data sets was observed, indicating that the majority of target genes are similarly regulated by VEGFR-2 and VEGFR-3 stimulation. However, we also noted several ‘outlier’ genes, which appeared to be regulated much more (or even exclusively) by either VEGF-C156S or by VEGF-A, and could thus represent receptor-specific target genes.
Figure 3. Differential gene regulation by VEGFR-2 and VEGFR-3 in LECs.

(a) Schematic representation of the binding abilities of VEGF ligands to VEGFR-2 and -3. VEGF-A binds to VEGFR-2 exclusively, whereas fully mature, wildtype VEGF-C can act on both VEGFR-2 and VEGFR-3. VEGF-C with a Cys156=>Ser mutation acts as a selective VEGFR-3 agonist. (b) Schematic illustration of the three time course gene expression data sets used for the analysis. CAGE RNA sequencing of LECs stimulated with VEGF-C156S was performed at 16 different time points over a course of 480 min. The 60, 240 and 480 min time points were compared with previously published microarray based gene expression data of LECs stimulated with VEGF-A or mature wildtype VEGF-C. (c) Correlation of log2FC of all matchable genes induced by VEGF-A (y-axes) and VEGF-C156S (x-axes) in LECs at the 60, 240 and 480 min time point. Linear regression and Pearson correlation coefficients are indicated in red. The most differentially regulated genes (up to 5) are indicated in dark red (genes up- or downregulated by VEGF-A but not by VEGF-C156S) and green (genes up- or downregulated by VEGF-C156S but not by VEGF-A). (d) Genes were considered as differentially regulated by VEGF-C156S and VEGF-A if they were strongly regulated by at least one of the ligands (P<0.05, log2FC>1 or <−1), and if the difference in log2FC values after VEGF-A and VEGF-C156S stimulation was at least four-fold. The heat maps show the log2FC values of all differentially regulated genes after VEGF-C156S, wildtype VEGF-C and VEGF-A stimulation.
To analyze differences between the VEGF-A induced and VEGF-C156S induced target genes in an unbiased way, we selected candidate genes that were significantly and strongly regulated by at least one of the ligands (P<0.05, log2FC>1 or <−1), and that showed at least a 4-fold difference in their log2FC induced by VEGF-A and VEGF-C156S. We identified 108 differentially regulated (DR) genes at the 60 min time point, 232 at the 240 min time point, and 194 at the 480 min time point (Fig. 3d and ‘Genes differentially regulated by VEGF-A and VEGF-C156S’, Data Citation 1). The vast majority of these were specifically regulated by VEGF-A / VEGFR-2 (the 5 most specific genes are highlighted in red in Fig. 3c) and included well-known VEGF-A target genes such as ESM1, ANGPT2 and RCAN1 (refs 16–19). We also identified a small group of genes specifically regulated by VEGF-C156S (highlighted in green in Fig. 3c). Interestingly, these genes included two important TFs in LEC differentiation and biology, SOX18 and KLF4 (refs 9,20,21).
RCAN1, ANGPT2, and ESM1 are selectively induced by VEGF-A/VEGFR-2 signaling
To confirm the selective upregulation of RCAN1, ANGPT2, and ESM1 by VEGF-A/VEGF-R2 signaling, we quantified the gene expression of these genes after VEGF-A (20 ngml−1), wildtype VEGF-C (500 ngml−1) and VEGF-C156S (1.5 μgml−1) incubation at 60, 240 and 480 min, using qPCR. Consistent with our in silico results, RCAN1 was selectively induced at the 60 min time point (Fig. 4a), and ESM1 and ANGPT2 at the 480 min time point, after VEGF-A stimulation (Fig. 4b,c).
Figure 4. RCAN1, ANGPT2 and ESM1 are selectively regulated by VEGF-A/VEGFR-2 signaling.
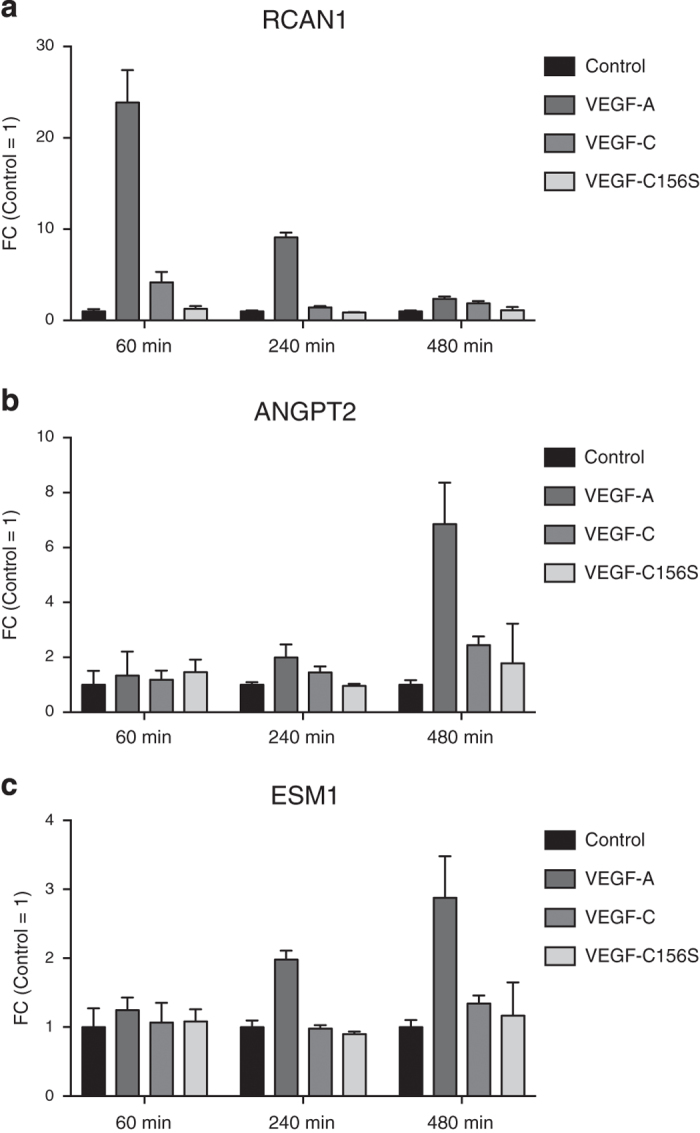
(a–c) Validation of the selective upregulation of (a) RCAN1, (b) ANGPT2 and (c) ESM1 by VEGF-A (20 ngml−1) but not by mature wildtype VEGF-C (500 ngml−1) or VEGF-C156S (1.5 μgml−1) using qPCR (one representative experiment is shown).
Discussion
Activation of VEGFR-3 results in various cellular responses in lymphatic endothelial cells, including cell migration, proliferation and changes in the intercellular junctions, which affects monolayer permeability22–24. Some of these cellular effects occur within minutes after receptor stimulation and are therefore considered to be independent of de novo gene expression, whereas others clearly require a transcriptional response. We recently characterized this transcriptional response of primary human LECs during the first 8 h after treatment with the VEGFR-3 specific ligand VEGF-C156S using CAGE RNA sequencing, which allowed us to identify several TFs involved in the induction of downstream genes8,9. However, we initially did not identify any downregulated genes. Changing the in silico analysis approach as described here, we now discovered a large number of downregulated genes based on the same CAGE RNA sequencing data set. Likely, the observed differences in the analysis results are due to 3 main reasons: First, we used a different analysis algorithm, namely DESeq2 (ref. 12), as compared to EdgeR11 used previously. Both DESeq2 and EdgeR are commonly employed for the analysis of RNA sequencing data but differ in various crucial aspects, including the normalization method and the way how the data dispersion is estimated25. Consequently, side-by-side comparison of EdgeR and DESeq2 resulted in overlapping, but not identical sets of DE genes in previous studies25–27. As of now, there is no consensus which of the two methods should be used preferentially. However, one study found that DESeq2 is slightly more conservative than EdgeR, meaning that the rate of true positive results, but also the rate of type I errors (false positives) are lower26. This suggests that the increased number of DE genes identified here is not simply due to false positives. Secondly, we used a different scheme to calculate the contrasts, comparing each time point tn of the stimulation with the baseline t0 (0 min), whereas previously, tn was compared to tn-1, which probably resulted in type II errors (false negatives) in case of genes with small and/or gradual changes in expression. Finally, using a multi-factorial design, we now adjusted for differences between the 3 individual LEC donors, which conceivably increased the statistical power of our analysis.
In agreement with our previous analysis9, the new analysis confirmed that the genes upregulated during the first hours after VEGFR-3 stimulation are to a large extent associated with transcription-related processes, including chromatin organization, RNA synthesis and stability. In addition to our previous analysis, we identified further TFs induced by VEGF-C156S, including FOXC2, ELK1, and NFATC1, which have been reported to regulate vascular development and angiogenesis before13–15. Importantly, our data also indicate that VEGFR-3 activation exerts direct anti-inflammatory effects on LEC, by downregulation of several immune- and cytokine-response associated genes. This is in line with the reported anti-inflammatory effects of VEGF-C on macrophages28,29 and indicates that VEGF-C, that has previously been found to reduce inflammation when applied in vivo30–32, not only acts via an increase of the lymphatic drainage, but also by inhibiting inflammatory signaling in LEC. Interestingly, these anti-inflammatory effects of VEGF-C are reminiscent of the effects of VEGF-A signaling in BEC33,34.
VEGFR-2 and VEGFR-3 are structurally related and considered to activate similar intracellular signaling cascades1. Nonetheless, VEGF-A and VEGF-C have been described to elicit different effects on lymphatic vessels in vivo, although the literature is not entirely consistent in this regard. For example, adenoviral delivery of VEGF-A into the ear of mice has been reported to induce massive dilation of lymphatic vessels35, whereas others found lymphatic dilation and formation of new lymphatic vessels only after delivery of VEGF-C, but not of VEGF-A36–38. This prompted us to attempt to identify genes differentially regulated by VEGFR-2 and VEGFR-3 within the same cell type, namely primary human LECs, by comparing our CAGE RNA sequencing data set of VEGF-C156S stimulated LECs with a microarray study of VEGF-A and wildtype VEGF-C stimulated LECs previously published by our group10. Naturally, cross-platform comparison of gene expression is problematic, due to inherent technical biases. Many studies have described a relatively good correlation between conventional RNA sequencing and microarray results obtained from the same starting material39–41. Similarly, CAGE RNA sequencing, in which only short (ca. 20 nt) 5’ fragments of mature, capped RNAs are analyzed, has been found to correlate well with conventional RNA sequencing42. Nonetheless, CAGE RNA sequencing is different from microarray and conventional RNA sequencing methods in that it does not assess the abundance of entire transcripts, but rather measures the activity of transcriptional start sites. Perhaps not surprisingly, a study comparing CAGE RNA sequencing directly with microarray and qPCR data found a rather low correlation between the data obtained by these techniques43. In our case, we additionally performed a cross-study comparison that likely introduced further bias due to biological differences in the starting material as well as experimental procedures and handling. Additionally, the data sets differed regarding the dosage of the growth factors applied: 1.5 μgml−1 in case of VEGF-C156S, 500 ngml−1 of wildtype VEGF-C, and 20 ng ml−1 VEGF-A. However, these doses reflect the differences in affinity and activity of the VEGFs, and are well within the range of what is commonly used by our lab and others23,44,45.
Despite these challenges, we observed the expected upregulation of an overlapping gene set after VEGFR-2 and VEGFR-3 stimulation with our approach, at least early after stimulation (60 min) when the induction of many downstream genes peaked. Furthermore, we identified several genes that were selectively regulated by stimulation of VEGFR-2 or VEGFR-3 only. These differentially regulated genes included the selective VEGF-A target genes RCAN1, ESM1 and ANGPT2. The specific induction of these genes by VEGF-A could also be validated experimentally, supporting the overall validity of our analysis. Selective upregulation of genes by VEGFR-2 might be explained simply by a difference in signal ‘strength’, e.g. due to higher ligand-receptor affinity, or higher kinase activity of the receptor, which are hard to control or manipulate experimentally. However, the fact that we also found a few genes regulated selectively by VEGFR-3 suggests that the quality of the signal downstream of the two receptors differs to some extent, e.g. by activating different kinases which in turn activate different TFs. Interestingly, stimulation with wildtype VEGF-C resulted in a ‘mixed’ response, with some genes being regulated as by VEGF-C156S and others as by VEGF-A.
Of note, the genes selectively induced by VEGFR-3 stimulation included two TFs, SOX18 and KLF4, that both play important roles in LEC function and differentiation. SOX18 is induced in LEC progenitors early during embryogenesis and is required for establishment and maintenance of the LEC phenotype20. KLF4 is a direct transcriptional target of MAFB, a TF activated by VEGFR-3 signaling9, and may be involved in LEC differentiation by regulating the ‘master’ TF of LEC, PROX1 (ref. 21). Thus, in line with our previous study9, the results of our meta-analysis presented here suggest that VEGFR-3, which is predominantly expressed in LECs, exerts a specific function in LEC differentiation, whereas more general processes shared between LECs and BECs such as cell migration and proliferation are regulated by both VEGFR-2 and VEGFR-3 alike.
Methods
Gene expression analysis of VEGF-C156S stimulated LEC
CAGE RNA sequencing data of primary human LECs stimulated with VEGF-C156S have been published previously9,46. In brief, dermal LECs isolated from the foreskin of 3 individual donors47 were starved in EBM medium (Lonza) + 0.2% bovine serum albumine (BSA) over night and were subsequently incubated with 1.5 μgml−1 VEGF-C156S. RNA was extracted at 16 different timepoints (0 min, 15 min, 30 min, 45 min, 60 min, 80 min, 100 min, 120 min, 150 min, 180 min, 210 min, 240 min, 300 min, 360 min, 420 min, 480 min) using TRIzol (Thermo Fisher) and subjected to CAGE RNA sequencing by the Fantom5 consortium at the RIKEN Institute (Yokohama, Japan) as described9. Raw counts were downloaded from the freely available Fantom5 data repository (http://fantom.gsc.riken.jp/5). CAGE peak expression analysis over the entire time course was done in R, using Bioconductor’s DESeq2 package (version: 1.12.4 (ref. 12)). A multi-factorial model design was used to compare time point 0 min (control) to the other 15 time points after VEGFC156S stimulation, pairing the samples from each individual donor. Lowly expressed CAGE peaks were removed if their raw count sum was=<1. CAGE peaks with a P-value<0.05 and a log2FC>0.6 or <−0.6 were considered as differentially expressed (DE). MA plots showing the log2FC compared to the average expression of the time point-specific CAGE peaks were done using the DESeq2 package. The corresponding genes (501 upregulated, 772 downregulated) are provided in ‘Upregulated Genes after VEGFC156S stimulation’, ‘Downregulated Genes after VEGFC156S stimulation’, Data Citation 1. Heat maps and hierarchical clustering of the up- and downregulated genes were performed using Genesis (Release 1.76, TU Graz, Austria).
Gene ontology analysis
The DE genes were grouped into early (0 to 120 min), middle (150 to 240 min) and late (300 to 480 min) phase genes. Gene ontology (GO) enrichment analysis for each of these phases was performed using the Gene Ontology Consortium Database (http://geneontology.org, release 22.8.2016). GO biological process (BP) terms and molecular function (MF) terms with an adjusted P-value<0.01 were considered significantly enriched. The top 25 enriched GO terms (by fold enrichment) from each category were clustered manually into biologically related themes and are listed in ‘GO_BP analysis’ and ‘GO_MF analysis’, Data Citation 1.
Gene expression analysis of VEGF-A and VEGF-C stimulated LECs
Generation of gene expression data of primary human LECs treated with VEGF-A and wildtype VEGF-C has been published before10. In brief, dermal LECs were starved in EBM + 0.2% BSA over night and were subsequently incubated with 20 ngml−1 VEGF-A or 500 ngml−1 mature wildtype VEGF-C (both from R&D Systems). RNA was extracted after 0 min (control), 60 min, 240 min, 480 min and 24 h, and subjected to microarray-based gene expression analysis using the Human Genome Survey V2.0 (Applied Biosystems) as described10. This data set has been deposited in the gene expression omnibus (GEO) repository under the accession number GSE11228 (Data Citation 2). Normalized data were retrieved and analyzed using the GEOquery package in Bioconductor, comparing the gene expression at the time points 60, 240 and 480 min to the 0 min (control) time point.
Comparison of VEGF-C156S, wildtype VEGF-C and VEGF-A regulated genes in LECs
Comparison of the VEGF-C156S stimulation time course and the VEGF-A and VEGF-C stimulation time courses was done based on the log2FC values at the matching time points 60, 240 and 480 min. To this end, we first removed all ambiguous CAGE peaks and probe sets. In the case of multiple peaks or probe sets associated with one gene, only the peak or probe set with the highest average expression was considered. Next, we used Entrez Gene IDs derived from corresponding annotation tables (Fantom5 annotation table available at http://fantom.gsc.riken.jp/5/, and GPL2986 from the GEO repository) to match the genes in the three data sets. Genes without available Entrez Gene ID in the corresponding annotation tables were additionally annotated using Bioconductor and the org.Hs.eg.db package (version 3.2.3) based on their gene symbol. The final matching of genes in the three data sets based on the Entrez Gene ID was done using R. From 16639 genes represented on the microarray, 10952 (at 60 min), 10951 (at 240 min), and 10989 (at 480 min) could be matched to the CAGE RNA sequencing data in this way. Log2FC values of all matched genes were plotted, and linear regression and correlation coefficients (Pearson) were calculated using GraphPad Prism V5. For the 3 time points, we selected genes differentially regulated by VEGF-C156S and VEGF-A if they were strongly regulated by either VEGF-A or VEGF-C156S (P<0.05 and log2FC>1 or <−1) and if there was at least a four-fold difference in the log2FC value after VEGF-C156S stimulation compared to the log2FC after VEGF-A stimulation (‘Genes differentially regulated by VEGF-A and VEGF-C156S’, Data Citation 1).
Validation of selected genes by qPCR
24 h before stimulation, 70000 primary LECs were seeded on collagen type-I coated 12-well plates. Next, LECs were starved over night in EBM + 0.2% BSA and were subsequently treated for 60, 240 or 480 min with 20 ngml−1 VEGF-A (Cell Sciences), 500 ngml−1 mature wildtype VEGF-C (R&D Systems) or 1.5 μgml−1 recombinant VEGF-C156S (kindly provided by Dr. Kari Alitalo, Wihuri Research Center, Helsinki, Finland). Total RNA was isolated using Genezol reagent (Geneaid) and extracted following the manufacturer’s protocol. The concentration of RNA was measured using a NanoDrop. Equal amounts of RNA were reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression of selected genes was quantified by qPCR using FastStart Universal SYBR Green Master (ROX) on a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems). RPLP0 was used as housekeeping control to normalize Ct values. Relative expression was calculated according to the comparative Ct method. Primer sequences are listed in Table 1.
Table 1. List of qPCR primers.
| Gene | Forward | Reverse |
|---|---|---|
| RPLP0 | CAGATTGGCTACCCAACTGGT | GGGAAGGTGTAATCCGTCTCC |
| RCAN1 | CAGAATAAACTTCAGCAACCC | CCTATGTGTAAGGTCTGAGC |
| ANGPT2 | CAATTATTCAGCGACGTGAG | AAGGGTTACCAAATCCCAC |
| ESM1 | GTATCTGCAAAGACTGTCCCT | TGTCACAGATGCCTGACTG |
Additional Information
How to cite this article: Dieterich, L. C. et al. Distinct transcriptional responses of lymphatic endothelial cells to VEGFR-3 and VEGFR-2 stimulation. Sci. Data 4:170106 doi: 10.1038/sdata.2017.106 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The authors would like to thank Sarah Klein (ETH Zurich, Zurich), Anthony Mathelier (University of British Columbia, Vancouver) and the entire FANTOM5 consortium for their crucial contributions to the generation and evaluation of the CAGE RNA sequencing data set used in this meta-analysis.
Data Citations
- Dieterich L. C., Ducoli L., Shin J., Detmar M. 2017. figshare. https://doi.org/10.6084/m9.figshare.c.3581339
- Shin J., Huggenberger R., Detmar M. 2008. Gene expression Omnibus. GSE11228
References
- Koch S., Tugues S., Li X., Gualandi L. & Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J 437, 169–183 (2011). [DOI] [PubMed] [Google Scholar]
- Valtola R. et al. VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in breast cancer. Am J Pathol 154, 1381–1390 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V. et al. Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J 16, 3898–3911 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V. et al. A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation, and vascular permeability activities. J Biol Chem 273, 6599–6602 (1998). [DOI] [PubMed] [Google Scholar]
- Manni S. et al. Structural and functional characterization of alternative transmembrane domain conformations in VEGF receptor 2 activation. Structure 22, 1077–1089 (2014). [DOI] [PubMed] [Google Scholar]
- Shiraki T. et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc Natl Acad Sci USA 100, 15776–15781 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner E. et al. Gene regulation. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S. et al. DeepCAGE transcriptomics identify HOXD10 as a transcription factor regulating lymphatic endothelial responses to VEGF-C. J Cell Sci 129, 2573–2585 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich L. C. et al. DeepCAGE Transcriptomics Reveal an Important Role of the Transcription Factor MAFB in the Lymphatic Endothelium. Cell Rep 13, 1493–1504 (2015). [DOI] [PubMed] [Google Scholar]
- Shin J. W., Huggenberger R. & Detmar M. Transcriptional profiling of VEGF-A and VEGF-C target genes in lymphatic endothelium reveals endothelial-specific molecule-1 as a novel mediator of lymphangiogenesis. Blood 112, 2318–2326 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. & Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I., Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov K. I. et al. Phosphorylation Regulates FOXC2-Mediated Transcription in Lymphatic Endothelial Cells. Mol Cell Biol 33, 3749–3761 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro-Jorganes A. et al. VEGF-Induced Expression of miR-17-92 Cluster in Endothelial Cells Is Mediated by ERK/ELK1 Activation and Regulates Angiogenesis. Circ Res 118, 38–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suehiro J. et al. Genome-wide Approaches Reveal Functional Vascular Endothelial Growth Factor (VEGF)-inducible Nuclear Factor of Activated T Cells (NFAT) c1 Binding to Angiogenesis-related Genes in the Endothelium. J Biol Chem 289, 29044–29059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes K., Chapman E., See V. & Cross M. J. VEGF stimulates RCAN1.4 expression in endothelial cells via a pathway requiring Ca2+/calcineurin and protein kinase C-delta. PLoS One 5, e11435 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich L. C. et al. Transcriptional profiling of human glioblastoma vessels indicates a key role of VEGF-A and TGFbeta2 in vascular abnormalization. J Pathol 228, 378–390 (2012). [DOI] [PubMed] [Google Scholar]
- Rennel E. et al. Endocan is a VEGF-A and PI3K regulated gene with increased expression in human renal cancer. Exp Cell Res 313, 1285–1294 (2007). [DOI] [PubMed] [Google Scholar]
- Roudnicky F. et al. Endocan is upregulated on tumor vessels in invasive bladder cancer where it mediates VEGF-A-induced angiogenesis. Cancer Res 73, 1097–1106 (2013). [DOI] [PubMed] [Google Scholar]
- François M. et al. Sox18 induces development of the lymphatic vasculature in mice. Nature 456, 643–647 (2008). [DOI] [PubMed] [Google Scholar]
- Park D. Y. et al. Lymphatic regulator PROX1 determines Schlemm’s canal integrity and identity. J Clin Invest 124, 3960–3974 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin J. W., Yuan S. Y. & Wu M. H. VEGF-C alters barrier function of cultured lymphatic endothelial cells through a VEGFR-3-dependent mechanism. Lymphat Res Biol 5, 105–113 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkinen T. et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J 20, 4762–4773 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacconi C. et al. Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology 148, 1438–1451.e1438 (2015). [DOI] [PubMed] [Google Scholar]
- Rapaport F. et al. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol 14, R95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C. & Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics 14, 91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yendrek C. R., Ainsworth E. A. & Thimmapuram J. The bench scientist’s guide to statistical analysis of RNA-Seq data. BMC Res Notes 5, 506 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio S. et al. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J Clin Invest 124, 3863–3878 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. Activation of vascular endothelial growth factor receptor-3 in macrophages restrains TLR4-NF-kappaB signaling and protects against endotoxin shock. Immunity 40, 501–514 (2014). [DOI] [PubMed] [Google Scholar]
- Huggenberger R. et al. An important role of lymphatic vessel activation in limiting acute inflammation. Blood 117, 4667–4678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggenberger R. et al. Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J Exp Med 207, 2255–2269 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q. et al. Vascular endothelial growth factor C attenuates joint damage in chronic inflammatory arthritis by accelerating local lymphatic drainage in mice. Arthritis Rheum 63, 2318–2328 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB J 29, 227–238 (2015). [DOI] [PubMed] [Google Scholar]
- Dirkx A. E. et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res 63, 2322–2329 (2003). [PubMed] [Google Scholar]
- Nagy J. A. et al. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J Exp Med 196, 1497–1506 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. et al. Comparative evaluation of FGF-2-, VEGF-A-, and VEGF-C-induced angiogenesis, lymphangiogenesis, vascular fenestrations, and permeability. Circ Res 94, 664–670 (2004). [DOI] [PubMed] [Google Scholar]
- Rissanen T. T. et al. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res 92, 1098–1106 (2003). [DOI] [PubMed] [Google Scholar]
- Enholm B. et al. Adenoviral expression of vascular endothelial growth factor-C induces lymphangiogenesis in the skin. Circ Res 88, 623–629 (2001). [DOI] [PubMed] [Google Scholar]
- Xu X. et al. Parallel comparison of Illumina RNA-Seq and Affymetrix microarray platforms on transcriptomic profiles generated from 5-aza-deoxy-cytidine treated HT-29 colon cancer cells and simulated datasets. BMC Bioinformatics 14(Suppl 9): S1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni J. C., Mason C. E., Mane S. M., Stephens M. & Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18, 1509–1517 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu A., Kerr G., Crane M. & Ruskin H. J. RNA-Seq vs dual- and single-channel microarray data: sensitivity analysis for differential expression and clustering. PLoS One 7, e50986 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaji H. et al. Comparison of CAGE and RNA-seq transcriptome profiling using clonally amplified and single-molecule next-generation sequencing. Genome Res 24, 708–717 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita A. et al. Comparison of gene expression profiles produced by CAGE, illumina microarray and real time RT-PCR. Genome Inform 24, 56–68 (2010). [PubMed] [Google Scholar]
- Kajiya K., Hirakawa S., Ma B., Drinnenberg I. & Detmar M. Hepatocyte growth factor promotes lymphatic vessel formation and function. EMBO J 24, 2885–2895 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veikkola T. et al. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J 20, 1223–1231 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi S. et al. FANTOM5 CAGE profiles of human and mouse samples. Sci. Data 4, 170–112 doi: 10.1038/sdata.2017.112 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S. et al. Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am J Pathol 162, 575–586 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Dieterich L. C., Ducoli L., Shin J., Detmar M. 2017. figshare. https://doi.org/10.6084/m9.figshare.c.3581339
- Shin J., Huggenberger R., Detmar M. 2008. Gene expression Omnibus. GSE11228