

Abstract
Interactions between proteins frequently involve recognition sequences based on multivalent binding events. Dimeric 14‐3‐3 adapter proteins are a prominent example and typically bind partner proteins in a phosphorylation‐dependent mono‐ or bivalent manner. Herein we describe the development of a cucurbit[8]uril (Q8)‐based supramolecular system, which in conjunction with the 14‐3‐3 protein dimer acts as a binary and bivalent protein assembly platform. We fused the phenylalanine–glycine–glycine (FGG) tripeptide motif to the N‐terminus of the 14‐3‐3‐binding epitope of the estrogen receptor α (ERα) for selective binding to Q8. Q8‐induced dimerization of the ERα epitope augmented its affinity towards 14‐3‐3 through a binary bivalent binding mode. The crystal structure of the Q8‐induced ternary complex revealed molecular insight into the multiple supramolecular interactions between the protein, the peptide, and Q8.
Keywords: adapter proteins, cooperativity, cucurbiturils, host–guest systems, supramolecular chemistry
Supramolecular systems have shown great potential for the modulation of biomolecular assemblies.1, 2, 3, 4 The selective and strong recognition of peptide and protein elements in particular by synthetic supramolecular host molecules has attracted significant attention.5, 6, 7, 8, 9, 10 These prior studies have laid the foundation for supramolecularly controlled protein dimerization, functioning, and assembly orthogonal to natural recognition and switching events.3, 11, 12, 13 Notable examples include the dimerization of carbonic anhydrase by the use of a synthetic foldamer platform14, 15 and the cucurbit[8]uril (Q8)‐mediated functional reconstitution of a split luciferase enzyme.16 The study and modulation of protein–protein interactions (PPIs) is one of the most progressive areas of chemical biology and important for both fundamental research and drug discovery.17, 18 Protein assemblies frequently employ multivalent binding events, but the switching of this multivalency, for example between mono‐ and bivalent states, is not readily modulated by classical small molecules. The distinctive structural and functional features of synthetic supramolecular systems are generating new modes for modulating such protein assemblies that act in an orthogonal manner to classical small‐molecule modulation.18 Within this context, the generation of multivalent supramolecular protein assemblies and their structural elucidation requires urgent attention.
The 14‐3‐3 adapter proteins are an especially interesting protein class because of their interaction with several hundred protein partners, many of which are involved in human disease,19, 20 including the breast‐cancer target, estrogen receptor α (ERα).21 They are dimerized proteins that typically bind their partner proteins through short, phosphorylated motifs22 either at two single‐motif binding sites or at a tandem binding site, which greatly enhances the binding affinity.23 Whereas PPIs with the 14‐3‐3 monomer can be modulated by natural products,23 peptide derivatives,22, 24, 25 synthetic molecules,26 or supramolecular ligands,3 molecular approaches to control the valency of 14‐3‐3–protein complexes are currently absent. Therefore, we rationally designed a binary bivalent supramolecular assembly platform based on cucurbit[8]uril and dimeric adapter protein 14‐3‐3. The resulting dual‐inducible bivalent system is based on an ERα phosphopeptide,21 which, in addition to the 14‐3‐3 binding motif at its C‐terminus, harbors a phenylalanine–glycine–glycine (FGG) motif at its N‐terminus. The supramolecular platform Q8 reversibly binds two FGG motifs in an antiparallel fashion5 and thus enables reversible chemical “switching” of the 14‐3‐3‐binding peptide between the mono‐ and bivalent states (Figure 1). The dimeric 14‐3‐3 and Q8 platforms resulted in strong enhancement of complex assembly and the first structural elucidation of a Q8–protein architecture.
Figure 1.
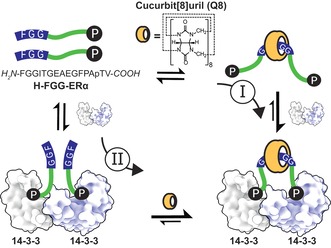
Multicomponent supramolecular protein assembly directed synergistically by the Q8 host and 14‐3‐3 protein dimer. Independent of the supramolecular assembly pathway (I or II), the two distinct binding epitopes of the two FGG‐ERα phosphopeptides steer the formation of the binary bivalent assembly.
The design of our system was based on inspection of a previously reported 14‐3‐3/ERα crystal structure.21 The two binding sites for the phosphorylated threonine (pT) of the ERα peptide are located 25 Å from one another in the 14‐3‐3 dimer. Consequently, we designed a flexible 15‐mer peptide of sufficient length to adequately span half of this distance, composed of the wild‐type (wt) ERα peptide epitope and an N‐terminal FGG motif for dimerization through Q8 binding (Figure 1). The affinity of this FGG‐ERα peptide towards the 14‐3‐3 dimer was measured in competitive fluorescence polarization (FP) assays (Figure 2 A), in which increasing concentrations of the FGG‐ERα peptide competed with a constant concentration of fluorescently labeled bivalent ExoS peptide. The latter peptide was designed, in analogy with previous examples,23, 27 by connecting two more weakly binding monovalent ExoS motifs with a flexible linker, which resulted in over 100‐fold affinity enhancement (see Figure 2 in the Supporting Information). In the absence of Q8, the FGG‐ERα peptide was observed to displace the ExoS peptide with an IC50 value of 1.0 μm (Figure 2 B, black line). Upon the addition of Q8 at increasing concentrations, the apparent affinity of the FGG‐ERα peptide for 14‐3‐3 increased circa 14‐fold, culminating in an IC50 value of 0.07 μm FGG‐ERα. The enhancement effect of Q8 appeared to saturate at approximately 10 μm of Q8, with no evidence of combinatorial inhibition at high Q8 concentrations, as testimony to the high cooperativity of this multicomponent Q8:14‐3‐3:FGG‐ERα complex.28, 29 By contrast, no such difference in binding affinity was observed with the N‐terminal acetylated peptide (Ac‐FGG‐ERα) upon the addition of Q8 (Figure 2 C). Furthermore, the analogous cucurbit[7]uril, Q7, which binds only one FGG moiety,30 did not enhance the apparent affinity of FGG‐ERα for the 14‐3‐3 protein (Figure 2 E). Combined, these reference experiments provide experimental evidence that the enhancement of the apparent affinity between the FGG‐ERα peptide cargo and 14‐3‐3 protein by Q8 relies exclusively on its postulated bivalent FGG recognition. Nonspecific effects of the supramolecular host are absent in the protein‐binding event (see also Figures 5–8 in the Supporting Information).
Figure 2.
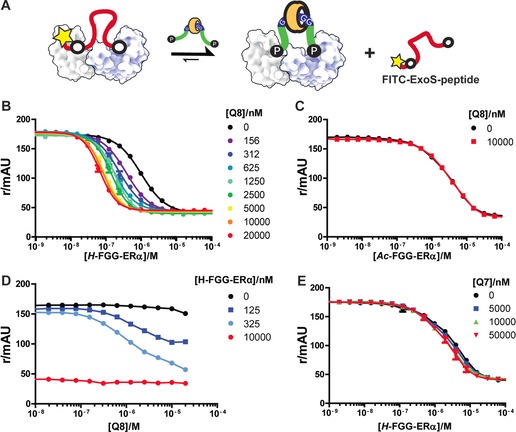
A) Q8‐enhanced bivalent binding of FGG‐ERα peptide to 14‐3‐3, as determined by competitive fluorescence polarization assays: FITC‐BIEXOS27 (10 nm) and 14‐3‐3β (40 nm) in FP‐Buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 0.01 % TWEEN20, 1 mg mL−1 BSA). B) Variation of the FGG‐ERα concentration at a constant Q8 concentration. C) As in (B), but with Ac‐FGG‐ERα instead of FGG‐ERα. D) Variation of the Q8 concentration at a constant FGG‐ERα concentration. E) As in (B), but with Q7 instead of Q8. Data points are the average of three measurements; error bars represent the standard deviation.
We also performed the competitive FP assay in an alternative format, by titrating Q8 at increasing concentrations against the FGG‐ERα peptide at a constant concentration (Figure 2 D). At very low and very high FGG‐ERα peptide concentrations, the effect of Q8 was negligible, since at very low peptide concentrations insufficient peptide is available for 14‐3‐3 binding, and full saturation is reached at very high concentrations. Revealingly, a clear dose‐response curve was observed at intermediate peptide concentrations—in line with the concentration‐dependent effects observed in Figure 2 B—thus resulting in IC50 values for Q8 of around 1 μm under these assay conditions. These results highlight the binary nature of our supramolecular bivalent platforms and show that the same equilibrium is achieved whichever route is taken (Figure 1). Asymmetric flow field‐flow fractionation experiments confirmed the formation of a compact particle with RH≈3.0 nm by the Q8:14‐3‐3:FGG‐ERα complex (see Figures 9 and 10 in the Supporting Information).
A complementary pathway to assemble the multicomponent system is to use 14‐3‐3β as the bivalent platform that facilitates the binding of two FGG‐ERα peptides to Q8 (II, Figure 1). This binding can be measured in a fluorimetric assay on the basis of the displacement of acridine orange (AO) from the cavity of Q8 (Figure 3 A).31 Thus, the FGG‐ERα was titrated into a solution of Q8 precomplexed with AO in the presence of varying concentrations of 14‐3‐3β (Figure 3 B). In the absence of 14‐3‐3β (Figure 3 B, black line), high concentrations of the FGG‐ERα peptide (IC50=62 μm) were required to displace the AO, in line with previous observations regarding the relatively weak competitive binding of FGG peptides.16 In the presence of 14‐3‐3β, however, much lower concentrations of FGG‐ERα sufficed to displace AO. At intermediate 14‐3‐3β concentrations, a mixed behavior reflecting the substoichiometric concentration of 14‐3‐3β as compared to Q8 and FGG‐ERα became evident in the titration curves. In the presence of excess 14‐3‐3β, the IC50 value of the FGG‐ERα peptide decreased to 2.5 μm, thus representing a 25‐fold enhancement. The two orthogonal assay types each address the concentration‐dependent role of one of the two bivalent platforms, Q8 or 14‐3‐3β, and show that both assembly pathways (Figure 1) converge to the same end point through the double bivalency mechanism.32, 33
Figure 3.
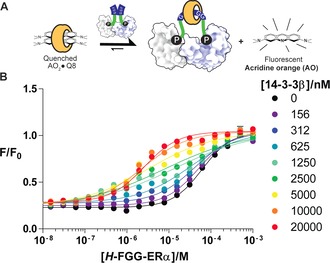
A) 14‐3‐3‐enhanced bivalent binding of ERα phosphopeptides to Q8, as determined by a fluorimetric displacement assay. B) Titration of FGG‐ERα peptide into a solution of acridine orange (1 μm) and Q8 (1 μm) in FP‐Buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 0.01 % TWEEN20, 1 mg mL−1 BSA) at various set concentrations of 14‐3‐3β. Data points are the average of three measurements; error bars represent the standard deviation.
The cocrystal structure of the 14‐3‐3β/FGG‐ERα/Q8 complex was solved at 1.67 Å resolution (PDB code: 5N10). For this purpose, a truncated 14‐3‐3βΔC variant was used to enable efficient crystallization23 (see the Supporting Information). In the asymmetric unit one 14‐3‐3β dimer can be found, with the electron density allowing the building of one Q8 molecule; one entire FGG‐ERα peptide, bound to one 14‐3‐3 monomer and Q8; the phosphorylated C‐terminus of a second FGG‐ERα peptide bound to a second 14‐3‐3 monomer; and the FGG motif of a second FGG‐ERα peptide (Figure 4 A; see also Figures 1 and 2 in the Supporting Information). The phosphorylated epitope region of both ERα peptides is bound to 14‐3‐3 as previously described (see Figure 3 in the Supporting Information),21 with the most important contacts established between residues pT594, R58, R129, Y130, and K49 from 14‐3‐3β (Figure 4 B).
Figure 4.
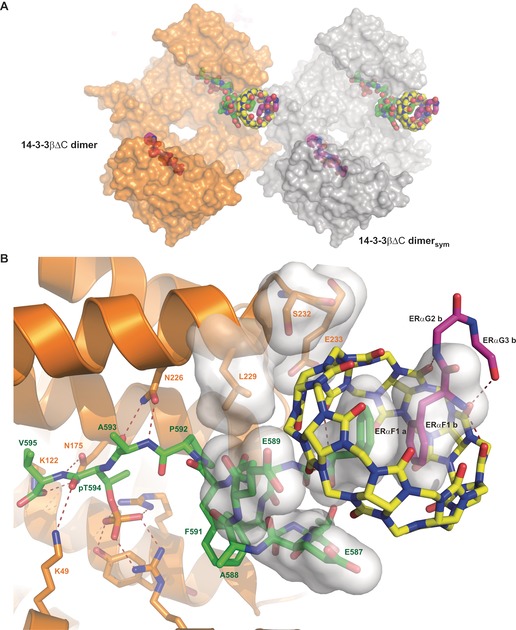
Crystal structure of the 14‐3‐3β/FGG‐ERα/Q8 complex (PDB code: 5N10). A) Overview of the crystal structure. One 14‐3‐3 dimer (orange and white surfaces) binds two FGG‐ERα peptides (green and magenta spheres) and one Q8 molecule (yellow spheres). B) Details of the interface between 14‐3‐3β (orange), FGG‐ERα (green, magenta), and Q8 (yellow). Polar contacts are depicted as dotted lines, and residues contributing interaction surfaces are shown as semitransparent white surfaces.
Interestingly, Q8 is located at the interface between two symmetry‐related 14‐3‐3 dimers (Figure 4 A, orange and white surfaces) and bound to two N‐terminal FGG motifs (Figure 4, green and magenta sticks) with the N‐terminal phenylalanine residues stacked within Q8 in the expected5 antiparallel mode (Figure 4 B). Within the asymmetric unit, Q8 makes additional polar contacts with both FGG‐ERα peptides and hydrophobic contacts with the hydrocarbon part of side chains from both the FGG‐ERα peptide (E589, E587) and 14‐3‐3β protein (E233, S232, L229; Figure 4 B). Five residues (N69sym–R73sym) from the symmetry‐related 14‐3‐3β dimer (14‐3‐3βsym) also contribute to the accommodation of Q8, and participate in establishing a water network, together with R224, W230, and E233, which helps to lock Q8 in place (see Figure 1 in the Supporting Information). In this context, essential roles are also played by the N‐terminal phenylalanine residues of the two FGG‐ERα peptides as well as E587 and E589 from the FGG‐ERα peptide accommodated within monomer A of 14‐3‐3β (Figure 4 B).
A short study on the intramolecular distances between FGG‐ERα peptides in symmetry‐related 14‐3‐3 dimers (white, 22 Å) and within the same 14‐3‐3 dimer (orange, 44 Å) concluded that the nine nonvisible amino acids of the second FGG‐ERα peptide (magenta) can only bridge symmetry‐related 14‐3‐3 dimers (see Figure 4 in the Supporting Information). In solution, one Q8 molecule dimerizes two FGG‐ERα peptides, which simultaneously bind to the two monomers in a 14‐3‐3 dimer, thus rationalizing the mutual increase in apparent affinity between 14‐3‐3 and FGG‐ERα (in solution). In this configuration, however, the peptides would remain flexible and thus not visible in the electron density of an X‐ray crystallography experiment. We therefore believe that the constellation observed in the solid state probably results from the geometry of the lattice, and is also imposed by the high concentrations of the molecular components, with the stabilization of Q8 by a symmetry‐related 14‐3‐3 dimer being a crystallographic prerequisite enabling the elucidation of its structure.
This study highlights the potential of bivalent Q8 to act in conjunction with the bivalent 14‐3‐3 protein as a binary bivalent platform to control supramolecular protein assembly. The interplay of the synthetic supramolecular concept with PPIs yields complementary platforms and provides orthogonal control over the protein assembly process. The interplay of the different molecular elements culminated in the first structural elucidation of a Q8–protein complex. We are currently exploring ways to integrate the switchable behavior of our binary platform concept to modulate complex, multivalent PPIs in a controlled manner.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was funded by the Netherlands Organization for Scientific Research (NWO) through Gravity program 024.001.035 and VICI grant 016.150.366 and by the Deutsche Forschungsgemeinschaft (DFG) through Collaborative Research Centre 1093. We thank David Williams for help with asymmetric flow field‐flow fractionation experiments.
P. J. de Vink, J. M. Briels, T. Schrader, L.-G. Milroy, L. Brunsveld, C. Ottmann, Angew. Chem. Int. Ed. 2017, 56, 8998.
Contributor Information
Prof. Luc Brunsveld, Email: l.brunsveld@tue.nl.
Prof. Christian Ottmann, Email: c.ottmann@tue.nl.
References
- 1. Zhang L., Wu Y., Brunsveld L., Angew. Chem. Int. Ed. 2007, 46, 1798–1802; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1830–1834. [Google Scholar]
- 2. Sakamoto S., Kudo K., J. Am. Chem. Soc. 2008, 130, 9574–9582. [DOI] [PubMed] [Google Scholar]
- 3. Bier D., Rose R., Bravo-Rodriguez K., Bartel M., Ramirez-Anguita J. M., Dutt S., Wilch C., Klärner F.-G., Sanchez-Garcia E., Schrader T., Ottmann C., Nat. Chem. 2013, 5, 234–239. [DOI] [PubMed] [Google Scholar]
- 4. Zimmerman S. C., Beilstein J. Org. Chem. 2016, 12, 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heitmann L. M., Taylor A. B., Hart P. J., Urbach A. R., J. Am. Chem. Soc. 2006, 128, 12574–12581. [DOI] [PubMed] [Google Scholar]
- 6. Nguyen H. D., Dang D. T., van Dongen J. L. J., Brunsveld L., Angew. Chem. Int. Ed. 2010, 49, 895–898; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 907–910. [Google Scholar]
- 7. Uhlenheuer D. A., Petkau K., Brunsveld L., Chem. Soc. Rev. 2010, 39, 2817. [DOI] [PubMed] [Google Scholar]
- 8. McGovern R. E., Fernandes H., Khan A. R., Power N. P., Crowley P. B., Nat. Chem. 2012, 4, 527–533. [DOI] [PubMed] [Google Scholar]
- 9. Sonzini S., Marcozzi A., Gubeli R. J., van der Walle C. F., Ravn P., Herrmann A., Scherman O. A., Angew. Chem. Int. Ed. 2016, 55, 14000–14004; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14206–14210. [Google Scholar]
- 10. Hewitt S. H., Wilson A. J., Chem. Commun. 2016, 52, 9745–9756. [DOI] [PubMed] [Google Scholar]
- 11. Dang D. T., Nguyen H. D., Merkx M., Brunsveld L., Angew. Chem. Int. Ed. 2013, 52, 2915–2919; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2987–2991. [Google Scholar]
- 12. McGovern R. E., McCarthy A. A., Crowley P. B., Chem. Commun. 2014, 50, 10412. [DOI] [PubMed] [Google Scholar]
- 13. Sankaran S., Kiren M. C., Jonkheijm P., ACS Nano 2015, 9, 3579–3586. [DOI] [PubMed] [Google Scholar]
- 14. Buratto J., Colombo C., Stupfel M., Dawson S. J., Dolain C., Langlois d'Estaintot B., Fischer L., Granier T., Laguerre M., Gallois B., Huc I., Angew. Chem. Int. Ed. 2014, 53, 883–887; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 902–906. [Google Scholar]
- 15. Jewginski M., Fischer L., Colombo C., Huc I., Mackereth C. D., ChemBioChem 2016, 17, 727–736. [DOI] [PubMed] [Google Scholar]
- 16. Bosmans R. P. G., Briels J. M., Milroy L.-G., de Greef T. F. A., Merkx M., Brunsveld L., Angew. Chem. Int. Ed. 2016, 55, 8899–8903; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9045–9049. [Google Scholar]
- 17. Arkin M. R., Tang Y., Wells J. A., Chem. Biol. 2014, 21, 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Milroy L.-G., Grossmann T. N., Hennig S., Brunsveld L., Ottmann C., Chem. Rev. 2014, 114, 4695–4748. [DOI] [PubMed] [Google Scholar]
- 19. Milroy L.-G., Brunsveld L., Ottmann C., ACS Chem. Biol. 2013, 8, 27–35. [DOI] [PubMed] [Google Scholar]
- 20. Aghazadeh Y., Papadopoulos V., Drug Discovery Today 2016, 21, 278–287. [DOI] [PubMed] [Google Scholar]
- 21. De Vries-van Leeuwen I. J., da Costa Pereira D., Flach K. D., Piersma S. R., Haase C., Bier D., Yalcin Z., Michalides R., Feenstra K. A., Jiménez C. R., de Greef T. F. A., Brunsveld L., Ottmann C., Zwart W., de Boer A. H., Proc. Natl. Acad. Sci. USA 2013, 110, 8894–8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson C., Crowther S., Stafford M. J., Campbell D. G., Toth R., MacKintosh C., Biochem. J. 2010, 427, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stevers L. M., Lam C. V., Leysen S. F. R., Meijer F. A., van Scheppingen D. S., de Vries R. M. J. M., Carlile G. W., Milroy L. G., Thomas D. Y., Brunsveld L., Ottmann C., Proc. Natl. Acad. Sci. USA 2016, 113, E1152–E1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Glas A., Bier D., Hahne G., Rademacher C., Ottmann C., Grossmann T. N., Angew. Chem. Int. Ed. 2014, 53, 2489–2493; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2522–2526. [Google Scholar]
- 25. Milroy L.-G., Bartel M., Henen M. A., Leysen S., Adriaans J. M. C., Brunsveld L., Landrieu I., Ottmann C., Angew. Chem. Int. Ed. 2015, 54, 15720–15724; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15946–15950. [Google Scholar]
- 26. Zhao J., Du Y., Horton J. R., Upadhyay A. K., Lou B., Bai Y., Zhang X., Du L., Li M., Wang B., Zhang L., Barbieri J. T., Khuri F. R., Cheng X., Fu H., Proc. Natl. Acad. Sci. USA 2011, 108, 16212–16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ottmann C., Yasmin L., Weyand M., Veesenmeyer J. L., Diaz M. H., Palmer R. H., Francis M. S., Hauser A. R., Wittinghofer A., Hallberg B., EMBO J. 2007, 26, 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levchenko A., Bruck J., Sternberg P. W., Proc. Natl. Acad. Sci. USA 2000, 97, 5818–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. den Hamer A., Lemmens L. J. M., Nijenhuis M. A. D., Ottmann C., Merkx M., de Greef T. F. A., Brunsveld L., ChemBioChem 2017, 18, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chinai J. M., Taylor A. B., Ryno L. M., Hargreaves N. D., Morris C. A., Hart P. J., Urbach A. R., J. Am. Chem. Soc. 2011, 133, 8810–8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Montes-Navajas P., Corma A., Garcia H., ChemPhysChem 2008, 9, 713–720. [DOI] [PubMed] [Google Scholar]
- 32. Fasting C., Schalley C. A., Weber M., Seitz O., Hecht S., Koksch B., Dernedde J., Graf C., Knapp E.-W., Haag R., Angew. Chem. Int. Ed. 2012, 51, 10472–10498; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10622–10650. [Google Scholar]
- 33. He Z., Jiang W., Schalley C. A., Chem. Soc. Rev. 2015, 44, 779–789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary