


Abstract
The progression of genetically inherited cardiomyopathies from an altered protein structure to clinical presentation of disease is not well understood. One of the main roadblocks to mechanistic insight remains a lack of high-resolution structural information about multiprotein complexes within the cardiac sarcomere. One example is the tropomyosin (Tm) overlap region of the thin filament that is crucial for the function of the cardiac sarcomere. To address this central question, we devised coupled experimental and computational modalities to characterize the baseline function and structure of the Tm overlap, as well as the effects of mutations causing divergent patterns of ventricular remodeling on both structure and function. Because the Tm overlap contributes to the cooperativity of myofilament activation, we hypothesized that mutations that enhance the interactions between overlap proteins result in more cooperativity, and conversely, those that weaken interaction between these elements lower cooperativity. Our results suggest that the Tm overlap region is affected differentially by dilated cardiomyopathy-associated Tm D230N and hypertrophic cardiomyopathy-associated human cardiac troponin T (cTnT) R92L. The Tm D230N mutation compacts the Tm overlap region, increasing the cooperativity of the Tm filament, contributing to a dilated cardiomyopathy phenotype. The cTnT R92L mutation causes weakened interactions closer to the N-terminal end of the overlap, resulting in decreased cooperativity. These studies demonstrate that mutations with differential phenotypes exert opposite effects on the Tm–Tn overlap, and that these effects can be directly correlated to a molecular level understanding of the structure and dynamics of the component proteins.
Graphical abstract
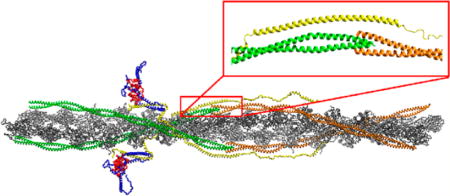
Tropomyosin (Tm) acts as the thin filament “gatekeeper” of myofilament activation.1,2 It is essential for the regulation of actin–myosin interactions. The importance of Tm in cooperative activation of the thin filament has been well established for both binding of myosin to actin3,4 and binding of calcium to troponin (Tn).5–7 The structure of the thin filament itself was confirmed to be a source of cooperativity by means of transmitting position to neighboring functional units whether via the Tm overlap or actin itself.8,9 These properties are key components of the three-state model of myofilament activation, with blocked, closed, and open myosin states,10 developed by McKillop and Geeves.11
A crucial component of cardiac myofilament cooperative activation is the noncanonical overlap structure between neighboring Tm molecules. The Tm overlap structure is composed of five α-helices: two from a C-terminal Tm dimer, two from an N-terminal Tm dimer, and one from human cardiac troponin T (cTnT).12,13 The strength of the Tm overlap interaction, determined by its structure, is responsible for the degree of positional information that is shared with neighboring functional units. The Tm components are known to be stabilized by the presence of a hydrophobic core domain, as the C-terminus of one of the Tm coiled coils “splays” to interact with the N-terminus of the next.14 Most of the previously published structures contain only the structure of the Tm portion of the overlap, omitting cTnT.15,16 There exists a high-resolution overlap structure that includes skeletal TnT, published by Murakami et al., with an antiparallel orientation against Tm.12 However, because one of the Tm N-terminal helices does not integrate with the overlap, this structure conflicts with recently published Tm-only versions.13,15,16 The interaction of cTnT with Tm at the overlap is largely via a known binding region in TNT1, one of the two proteolytic fragments of cTnT.17,18 cTnT contains significant α-helical character, which extends from the Tn core along Tm, with several prior studies reporting its antiparallel nature with respect to Tm.19–21 Additionally, the noncore regions of TnT are still not fully characterized, because they have not been crystallized,22 although the overall organization of Tn on the thin filament was recently described by Yang et al.20
Because the Tm overlap is a fundamental structure that modulates the cooperative activation of the myofilament, it follows that known human cardiomyopathic mutations within or flanking this region may cause disease by altering its structure and therefore its function. To directly assess this hypothesis, we chose two independent thin filament missense mutations known to cause highly divergent patterns of ventricular remodeling in patients, Tm D230N [dilated cardiomyopathy (DCM)] and cTnT R92L [hypertrophic cardiomyopathy (HCM)].23,24 These mutations are both located directly adjacent to the Tm overlap. We also hypothesize that these mutations may have effects on thin filament cooperativity, because previous studies demonstrated that mutations in the cardiac thin filament create structural effects that propagate to remote locations in the thin filament and lead to changes in its global flexibility.25–28
To further characterize the organization and geometry of the Tm overlap and to assess its potential role in the differential pathogenic ventricular remodeling caused by known thin filament point mutations, we utilized time-resolved Forster resonance energy transfer (TR-FRET)29 in a fully reconstituted cardiac thin filament system. To overcome some of the limitations of prior studies in which fragments of thin filament proteins were used (and the TnT N-terminus was omitted), we used full-length human cardiac isoforms of all three Tn components along with Tm and actin to create fully reconstituted thin filaments.30 Additionally, the thermodynamic stability of the fully reconstituted thin filaments was characterized via differential scanning calorimetry (DSC). These filaments were also simulated using molecular dynamics (MD) on our recently published atomistic, fully solvated model of the cardiac thin filament as shown in Figure 1.28
Figure 1.

Atomistic model of the cardiac thin filament. cTnI is colored blue, cTnC red, cTnT yellow, the C-terminal Tm green, and the N-terminal Tm orange. The subset contains the baseline structure of the Tm overlap region. The model structure for the Tm at the overlap assumes a form with orthogonal termini.
Our results demonstrate that the Tm D230N and cTnT R92L mutations differentially affect the Tm overlap as measured using all three techniques. Together, these results establish the baseline structure of the Tm overlap in terms of cooperativity of the thin filament and elucidate the differential effects of mutations that flank the Tm overlap.
MATERIALS AND METHODS
Protein Expression and Purification
Genes encoding human cardiac troponin I, human cardiac troponin T, human cardiac troponin C, and α-tropomyosin were inserted into pET3d vectors and provided by J. D. Potter. We introduced cysteines and mutations via site-directed mutagenesis using the Qiagen Quikchange II XL kit (Qiagen, Benelux, The Netherlands). The University of Arizona Genetics Core sequenced the resultant constructs that were verified using SnapGene Viewer (GSL Biotech LLC). Troponin components were expressed bacterially and purified via column chromatography as previously described.31
Tm was expressed with the N-terminal ala-ser via pET3d vector in BL21 Escherichia coli. This ensures that the Tm polymerizes and binds to actin properly.32 A 2 L culture of ZYP broth was used as the medium (1% tryptone, 0.5% yeast extract, 0.5% glycerol, 0.05% glucose, and 0.2% lactose). Bacteria were pelleted out of the medium by centrifugation at 4000g for 15 min. The pellets were resuspended in doubly distilled H2O, and then 0.1 mg/mL lysozyme was added to the solution. The resuspended pellet was incubated on ice for 1 h and then frozen at −80 °C for 10 min. The frozen solution was allowed to thaw at room temperature, with solid NaCl added to a final concentration of 1 M in the solution. The solution was sonicated three times for 3 min each, and then the lysed bacteria were centrifuged at 20000g for 45 min. The supernatant was boiled for 6 min and allowed to rest for 45 min at room temperature. The denatured lysate was centrifuged at 20000g for 20 min. The supernatant was recovered, and the pH was set to 4.6 using 1 M HCl, causing Tm to precipitate from solution. The solution was centrifuged at 20000g for 20 min. The pellet was then submerged in 1 M KCl, with the pH increased above 7.0 to resuspend the pellet. Once resuspended, the pellet was centrifuged at 20000g for 20 min. The acid/base cuts were repeated three times or until an A260/A280 ratio of <0.8 was reached.
Actin Purification
Rabbit skeletal muscle was used to purify, prepare, and polymerize filamentous actin as previously described in the literature.33 G-Actin was then extracted from the acetone powder as described. Prior to reconstitution of the thin filaments, the G-actin was polymerized to F-actin. The G-actin was set to slowly stir at room temperature. Then, 3 M KCl was added to the actin solution slowly to a final concentration of 50 mM. Then, 1 M MgCl2 was added to the actin to a final concentration of 2 mM. The resulting actin was thoroughly mixed and polymerized at room temperature for 1 h. At the end, the viscous F-actin was pipetted using a wide-bore tip and used to reconstitute the thin filaments.
Protein Labeling
The proteins to be labeled (cysteine-substituted Tm and cTnT) were dialyzed twice for 6–8 h versus labeling buffer [3 M urea, 50 mM Tris (pH 7.4), 0.15 M NaCl, 1 mM EDTA, and 50 µM dithiothreitol (DTT)]. Prior to labeling, protein concentrations were acquired via ultraviolet–visible (UV–vis) spectroscopy. For cTnT, 5-({[(2-iodoacetyl)amino]ethyl}amino)naphthalene-1-sulfonic acid (IAEDANS) (ThermoFisher Scientific) dissolved in dimethylformamide (DMF) (Sigma-Aldrich) was added to a 10-fold molar concentration of the protein. The labeling was allowed to proceed for 24 h while the mixture was being stirred at 4 °C. For Tm, N-[4-(dimethylamino)-3,5-dinitrophenyl]maleimide (DDPM) (Setareh Biotech) was added via DMF to a 30× molar concentration of the protein. The labeling was allowed to proceed for 3 h at room temperature. Both labeling procedures were terminated by adding DTT to final concentrations of 3–5 mM. Excess label was removed by centrifugation and dialyzed out of the sample. Labeling ratios were verified via UV–vis spectrophotometry and were >0.8 in all cases.
Protein Reconstitution
cTnI, cTnC, and IAEDANS-labeled cTnT were dialyzed against a high-urea buffer for at least 8 h [6 M urea, 0.05 M Tris (pH 7.0), 0.5 M KCl, 1 mM DTT, 5 mM CaCl2, and 5 mM MgCl2]. Protein concentrations were measured using a Beckman-Colter DU-730 UV–vis spectrophotometer. The following extinction coefficients were used: ε280 = 9970 M−1 cm−1 for cTnI, ε280 = 4470 M−1 cm−1 for cTnC, and ε340 = 5900 M−1 cm−1 for cTnT[IAEDANS]. The Tn complex was reconstituted in a 1.0:1.2:1.2 cTnT:cTnI:cTnC ratio. Protein extinction coefficients were determined from the primary sequence of the protein. Extinction coefficients of labels were determined by absorption of known masses and found to be consistent with literature values.34
After Tn components had been combined, the samples underwent stepwise dialyses to remove urea, from 6 M to 4 M to 2 M to 0 M, with other solution items kept constant. Next, the KCl concentration was decreased to 0.4 M. Tm and F-actin were dialyzed versus the same solution. The following extinction coefficients were used: ε280 = 17880 M−1 cm−1 for unlabeled Tm dimer, ε430 = 3500 M−1 cm−1 for labeled Tm[DDPM], and ε340 = 5900 M−1 cm−1 for Tn[IAEDANS]. The thin filament was reconstituted at a 0.8:1.0:7.5 Tn:Tm:actin molar ratio, close to the 1:1:7 canonical ratio, similar to other in vitro experiments, to minimize free Tn. 35,36 The reconstituted thin filaments were dialyzed against working buffer [0.15 M KCl, 50 mM MOPS, 5 mM NTA, 2 mM EGTA, 5 mM MgCl2, and 1 mM DTT (pH 7.0)]. Experiments were performed without calcium and with 1 mM calcium.
Data Acquisition
Lifetime data were acquired using an ISS ChronosBH system. A 340 nm LED signal light source illuminates the sample through a 340 nm bandpass filter to remove high-wavelength artifacts. The resultant emission from the sample was passed through a 390 nm long pass filter to remove scatter from the illumination and collected via a Hamamatsu H7422 detector. The pulse repetition rate of the instrument is 10 MHz, with analog to digital conversion (ADC) at <1% of the repetition rate. The sample is maintained at 10 °C with a Julabo F-12 heating/cooling circulator. To acquire the instrument response function (IRF), the emission from a LUDOX solution was acquired through a 340 nm bandpass filter.
Data Analysis
All data were analyzed using FargoFit, a global analysis software previously developed by I. Negrashov.29 Data analysis was developed in conjunction with B. Colson. The donor only decay is represented by a sum of exponentials as shown in eq 1, which has been convolved by the IRF
| (1) |
where τDi values are the decay constants, Ai values are the amplitude terms, and t is the time, with n being the number of decay constants (three decay constants had the best χ2 fit while not increasing the complexity of the system).
The decay rate of each of these exponentials is increased in response to a nearby acceptor, with R0 being the distance at 50% of energy transfer and R the distance between the FRET pair (eq 2). The R0 for IAEDANS-DDPM was 28.04 Å for pairs using cTnT 100 IAEDANS and 28.96 Å for pairs containing cTnT 127 IAEDANS due to different probe environments. These R0 values are within range of those found in the literature.37,38 This difference in donor fluorescence spectrum due to the altered environment can be seen in Figure S1.
| (2) |
The model used to describe FRET in this system was derived by the Miki group, who previously investigated FRET between a donor on cTnT and acceptors on Tm.30 This model considers three distinct sample populations in the fluorescence decay. There is the first population of the sample in which the donor is not participating in FRET, a second population in the sample in which the donor is undergoing FRET one acceptor of the Tm dimer, assuming equidistance between the donor and either acceptor, and the third population in which the donor is interacting with both FRET acceptors of the Tm dimer. These populations are shown in eq 3
| (3) |
where FD, FDA1, and FDA2 represent the lifetime decays of each of the populations and XD, XDA1, and XDA2 represent the proportions of each population. The distances are derived from the population interacting with one acceptor of the Tm dimer (FDA1).
An example of a typical data acquisition from both a donor only and a sample with the donor and acceptor and example fits is shown in Figure 2.
Figure 2.

Time-resolved fluorescence from the cTnT 100C IAEDANS–Tm 271C DDPM FRET pair. (A) Fluorescence decay of the donor only sample (black) compared to the sample with the donor and acceptor (red). The increased rate of decay is indicative of FRET. (B) Residuals for the data shown in panel A when fit with a single discrete population undergoing FRET. (C) Residuals for the data shown in panel A when fit with two discrete populations undergoing FRET. This residual fit does not contain the periodic variance from the data that is observed in panel B.
Differential Scanning Calorimetry
Actin, Tm, and Tn were dialyzed against a reducing solution containing 2-mercaptoethanol as described previously.39 Actin, Tm, and Tn were reconstituted against each other in a 5:3:3 molar ratio in accordance with DSC experiments previously performed on fully reconstituted thin filaments by Kremneva et al.39 Samples were heated from 25 to 75 °C at a rate of 0.5 °C/min using the NanoDSC instrument (TA Instruments). The resultant calorimetric heating profile was analyzed using NanoAnalyze software provided by TA Instruments. Gaussian distributions were fit to the heating profile, based upon prior work in which DSC measurements of Tn–Tm–actin had been characterized.39,40
ATPase Assay
The ATPase assay using the label-conjugated version of the Cys variant proteins used in FRET demonstrated proper calcium-based Tn regulation of the thin filament (data not shown).41 The assays were performed at 25 °C in 10 mM KCl, 4 mM MgCl2, and 20 mM Tris-HCl (pH 7.6), with 0.05 mM CaCl2 for samples with calcium and 1 mM EGTA for samples without calcium. The protein concentrations were 4 µM F-actin, 0.7 µM Tm, 0.8 µM Tn, and 1 µM S1. The reaction was coupled with a NADH regeneration system described by Matsuo et al. in which the time course of NADH absorption at 340 nm is followed over 10 min.42
Molecular Dynamics
MD simulations of the fully atomistic thin filament model were performed according to published methods.28 As we have described, a full model of the entire thin filament is necessary to obtain biologically relevant computational predictions, especially when the questions of interest involve the relative placement of one component of the thin filament (Tm) with respect to another (cTnT). To briefly summarize, the model was generated using NAMD version 2.9 using CHARMM27 force field parameters. The models were minimized for 5000 steps. The cell size of the original box was maintained, with periodic boundaries. The particle mesh Ewald (PME) method was used to calculate long-range electrostatic interactions, and the van der Waals interactions were cut off at 12 Å. The PME grid spacing was 1.0 Å, and the tolerance was 10−6. The SHAKE algorithm constrained hydrogen length with a tolerance of 1.0−8 Å.
The models were heated at a rate of 1 K/ps to 300 K, followed by 10 ps with rescaling at 300 K to ensure the model reached 300 K. The model was then equilibrated for 690 ps in an isothermal–isobaric ensemble, with a Langevin piston used to maintain a constant pressure at 1 atm, and a Langevin temperature control maintaining a constant temperature of 300 K. As a check, we have compared results with equilibrations run for 20 ns, and the overall results are unchanged. MD production runs of 10 ns were simulated under isothermal– isobaric conditions. The average structures of the MD production runs allow for the visualization of conformational trends of the models. The resultant fully solvated model contains roughly 5 million atoms and was simulated on the University of Arizona high-performance computing facility utilizing GPU-equipped nodes.
Statistical Analysis
Tukey’s multiple-comparison test was used to determine statistical significance on the sample using GraphPad Prism 7. A p value of <0.05 was considered statistically significant.
RESULTS AND DISCUSSION
In Vitro Characterization of the Structure of the Human Tropomyosin Overlap Domain
To determine whether cTnT is antiparallel with respect to the orientation of Tm at the overlap, we utilized FRET. As mentioned in Materials and Methods, the ATPase assay using the label-conjugated version of the Cys variant proteins used for FRET demonstrated proper calcium-based Tn regulation of the thin filament. The measured distances without calcium are listed in Table S1 and with calcium in Table S2. With the FRET donor IAEDANS at cTnT residue 100 and the FRET acceptor DDPM at both C-terminal Tm sites 265, the distance between the sites is measured to be 30.95 Å in the absence of calcium. This distance then decreases as the acceptor probes are moved along the C-terminus, specifically to Tm 271 (27.80 Å) and then Tm 279 (24.80 Å). The distance between cTnT 100C and the N-terminal Tm 13C was 27.17 Å. This indicates that cTnT site 100 is close to the middle of the overlap. This fully agrees with the placement in the model, as seen by the labeled sites in Figure 3.
Figure 3.

Locations of labeled sites. On cTnT, IAEDANS is attached to either site 100 or site 127 as a FRET donor. On Tm, site 13, 265, 271, or 279 is used to link the FRET acceptor DDPM.
With the FRET donor covalently linked to residue 127 of cTnT, the C-terminal Tm FRET acceptor sites surround this residue in the wild-type state. In the WT thin filament in the absence of calcium, cTnT 127 is closer to Tm 271 (28.38 Å) than either Tm 265 (28.87 Å) or Tm 279 (30.30 Å). Additionally, there was no discernible FRET between cTnT 127 and Tm 13. Because cTnT 127 is far from site 13C of Tm, this localizes cTnT 127 farther from the overlap along the C-terminal Tm. Thus, the overall finding from the FRET pairs used to query cTnT 127 suggests that this location is proximal to the C-terminal region of the Tm overlap, but out of FRET range of the N-terminus. The distances were >40 Å compared to the R0 of 28.96 Å.
Calcium Affects Only Residues Distal from the Overlap Center
The addition of calcium induced significant changes only in the cTnT 127–Tm 265 FRET pair. The overall result of adding calcium to a system was an increase in the distance between this pair. The impact of calcium on distances is illustrated in Figure 4 as well as in Tables S1 and S2. Significance and variance were observed at similar levels seen in thin filament systems utilizing the same FRET probes.43,44 We were expecting relatively small changes in distance at the overlap due to calcium or mutations, as large changes would be expected to disrupt the structure in a manner that would likely impact overall viability. The rest of the FRET pairs did not reveal statistically significant effects of calcium on distance. An overall lack of calcium-induced movement may indicate two phenomena: the role of the overlap itself as a binding region or the fact that there are no large changes in the absence of myosin.17 These results are consistent with previous findings from other groups, e.g., a lack of calcium-induced movement between cTnT and Tm.30
Figure 4.

Adding calcium to a thin filament increases the distance between Tm and cTnT in the region adjacent to the C-terminal portion of the overlap. The magnitude of this change is around 2 Å. This is small in comparison with the larger-scale calcium-induced movements in the Tn core. We observe an overall lack of calcium-induced movement at the overlap itself.
Previous studies utilizing FRET and the thin filament have found a minimal effect of calcium on the relative position of thin filament proteins.45–48 However, there have been some larger-scale calcium-induced changes observed near the Tn core with respect to Tm.30 Kimura-Sakiyama et al. did not observe any significant movement of Tm compared to the TnT fragment at the overlap,30 while they did note calcium-induced changes between the two proteins near the Tn core region. There is a slight decrease in distance that is induced by calcium at the sites farthest from the Tm overlap and closest to the Tn core, around 1–2 Å. However, at the center of the Tm overlap, it is likely that any calcium-induced changes are small enough that they cannot be resolved using FRET. The increased calcium effects distal from the overlap compared to the overlap center provide structural insight. The C-terminal portion of the overlap is likely the location of the Tm–Tn binding region,49 and thus, the proteins are closely associated with one another in a unique configuration regardless of calcium status. As one examines Tm–Tn interactions while changing perspective from the Tm overlap toward the Tn core, the nature of the relationship between these proteins changes. There is no longer a binding region between the proteins until the Tn core itself, and this may lead to more calcium-induced changes between the proteins. Thus, we may be seeing a transition between two types of interactions of the cTnT and Tm system, such that the calcium-induced C-terminal shift may be indicative of displacement of the Tn core in response to calcium.
Tm D230N
Tm D230N decreases the distance across the center of the Tm overlap. In particular, upon measurement between cTnT 100 and Tm 271, the distance decreased as a result of this mutation when compared with that of WT Tm, by 0.90 Å in the presence of calcium. Tm D230N increases the distance between site cTnT 127 and two locations of Tm, Tm 271 and Tm 279. The distances were increased by 2.46 and 1.06 Å, respectively, in the samples with calcium. These increases in distance are located farther from the overlap. There were no significant changes in distance induced by Tm D230N observed at residue 13 of Tm. Notably, this residue is at the N-terminus of Tm, and thus, if measurements at this site were affected by the Tm D230N mutation, it would have been either propagated over a distance much longer than those of the other Tm acceptor sites or affected via interactions across the overlap. Overall, the effects of Tm D230N impact the overlap structure on the portion proximal to the mutation and are represented in Figure 5.
Figure 5.

Effect of the Tm D230N mutation and the cTnT R92L mutation on the FRET distances at the overlap shown as arrows, with a black contour representing the overall changes at the overlap in the presence of calcium. Tm D230N (red arrows) decreases the distance between cTnT and Tm at the center of the overlap (between cTnT 100 and Tm 271). cTnT R92L (green arrows) increases the distance between cTnT and Tm at the center of the overlap region. Both mutations result in an opposing compensatory change in distance between cTnT 127 and Tm 279. The change in overlap distance along with the compensation is represented by the black contour on cTnT.
cTnT R92L
cTnT R92L increased the distance between cTnT 100 and Tm 271. There was also a slightly decreased distance between cTnT 100 and Tm 279, distal to the N-terminal overlap region. There was only one significant difference involving cTnT 127, at Tm 279, which was most proximal to cTnT R92L. These increases in distance between cTnT and Tm were more pronounced in the presence of calcium. Overall, the R92L mutant effects appear to be more concentrated in the N-terminal portion of Tm, proximal to the mutation, as shown in Figure 5. These differential effects on Tm–TnT structure at the overlap region likely affect global Tm flexibility and thus cooperativity.
Opposing Effects of Tm D230N and cTnT R92L between Two FRET Pairs at the Overlap
Tm D230N and cTnT R92L resulted in opposite effects when compared to the wild type for the FRET pairs from cTnT 127 to Tm 279 and cTnT 100 to Tm 271. These opposing effects were amplified and statistically significant in the experiments with calcium. We see changes in distance at the center of the Tm overlap between cTnT 100 and Tm 271, which could explain changes in cooperativity, in addition to an opposing compensatory change in distance between cTnT 127 and Tm 279. The sums of these effects are represented by the black contours in Figure 5. The overall distances from the FRET experiments are shown in Figures 6 and 7.
Figure 6.

Distances ± SE for the wild type (WT), Tm D230N, and cTnT R92L containing thin filaments using cTnT site 100 as the FRET donor site. *p < 0.05; **p < 0.01.
Figure 7.

Distances ± SE for the wild type (WT), Tm D230N, and cTnT R92L containing thin filaments using cTnT site 127 as the FRET donor site. *p < 0.05; **p < 0.01; ****p < 0.0001; +p < 0.05; + +p < 0.01; +++p < 0.001.
DSC
The interactions of fully reconstituted thin filaments were examined via DSC. There is a highly cooperative transition associated with the Tm–actin interaction at roughly 47 °C.39 This transition is surrounded by a dashed outline in Figure 8. The melt transition temperature of the Tm–actin interaction was significantly affected by the presence of the Tm D230N mutation but showed only modest effects in the presence of the cTnT R92L mutation. The melt temperature for the filament containing Tm D230N was 47.9 ± 0.21 °C when that of the wild-type thin filament was 47.2 ± 0.14 °C. This increased thermal stability is indicative of an enhanced interaction between the Tm and actin in the presence of Tm D230N. The melt temperature for Tn–Tm–actin interaction for the thin filament with cTnT R92L was 47.3 ± 0.06 °C, similar to that of the wild type.
Figure 8.

DSC results for fully reconstituted thin filaments of (A) wild-type, (B) Tm D230N, and (C) cTnT R92L variants. The fwhm of the actin–Tm binding interaction is represented by the sharp peak near 47 °C and represents the cooperativity of the interaction. This interaction is outlined in the wild-type data with a dashed line. The wild-type complex peak (0.993 ± 0.01 °C) is wider than that of the peak from the Tm D230N complex (0.883 ± 0.01 °C) but narrower than that of the cTnT R92L complex (1.38 ± 0.14 °C). The standard deviation resulted from two or three independent experiments.
The full width at half-maximum (fwhm) of the unfolding process of Tn–Tm–actin interaction narrowed from 0.993 ± 0.01 to 0.883 ± 0.01 °C in the presence of Tm D230N. The decreased fwhm of this unwinding is consistent with a highly ordered or cooperative system. When the Tn–Tm complex and actin begin to dissociate, the process occurs more rapidly. This increased cooperativity is consistent with an enhanced interaction across the Tm overlap region. In combination with the increased unfolding temperature, we observe two metrics that indicate enhanced overlap interactions. The cTnT R92L mutant thin filament led to an increased fwhm of the dissociation of the Tn–Tm complex from actin, from 0.998 ± 0.01 to 1.38 ± 0.14 °C, contrasting with the effect of the Tm D230N mutation on this measurement. The increased fwhm of the Tm–actin interaction suggests weakened interactions between adjacent Tm units, and a less cooperative Tm filament.
Molecular Dynamics
The structure and dynamics of the cardiac thin filament were studied using our all-atom model of the cardiac thin filament for the wild type and Tm D230N and cTnT R92L mutants. Because the thin filament is a cooperative, highly allosteric multisubunit “machine”, it is crucial to study the intact structure, to maximize functional and dynamic insight. It is only by understanding how mutations in one part of the complex transmit structural and dynamic information to another that we can make useful theoretical predictions about the nature of mutational effects.
The results showed overall effects that were consistent with the in vitro studies. Compared to a wild-type thin filament, inclusion of the Tm D230N mutation decreased the average distance between residues 94–133 of cTnT and the center of the Tm molecule from 19.867 to 18.464 Å. Conversely, including the cTnT R92L residue increased the distance between these same residues from 19.867 to 21.157 Å. The overall effects are displayed in Figure 9. The modulation in overlap distances by the mutations reflects the altered protein– protein interactions.
Figure 9.

(A) Average structures of molecular dynamics simulations for wild-type, D230N, and R92L complexes. The Tm’s are shown as the wild type, while the positioning of the cTnT of the mutants is shown with the Tm’s aligned with the wild-type structures. The C-terminal end of Tm is colored green, the N-terminal end of Tm orange, wild-type cTnT yellow, D230N blue, and R92L red. (B) Closer view of the N-terminus of the cTnT. The cTnT D89 and R92 side chains that create the salt bridge that is eliminated by the R92L mutation are shown in ball-and-stick representation. Cyan spheres are carbon atoms, white spheres hydrogen atoms, blue spheres nitrogen atoms, and red spheres oxygen atoms.
Additionally, the shape of cTnT along Tm was affected by the cTnT R92L mutant. While the Tm D230N mutant maintained the curvature of the TNT1 of the cTnT region along the Tm that the wild type exhibited, the cTnT R92L TNT1 region is straight throughout the span of the Tm, which is more pronounced at the N-terminal end of the TNT1 α-helix. The removal of the curvature increased the average distances between cTnT and Tm. This is also demonstrated in Figure 9. We note that for both the Tm D230N and cTnT R92L results, the overall changes in geometry recapitulate the curves shown in Figure 5.
For decades, it has been known that the unique head-to-tail array of Tm overlap contributes to both the strength and flexibility of the thin filament. The overlap structure also plays a central functional role in modulating the cooperativity of myofilament activation from calcium and myosin. This supports an important and highly conserved function of this domain. The origin of cooperativity in the thin filament has been attributed to transmission of structure either via the head-to-tail overlap or via actin conformational changes.8,9 Previous reports have estimated that in addition to many Tm mutations, >65% of all cTnT mutations occur within regions near or flanking the Tm overlap region.50 More recently, independent mutations predicted to affect the overlap have been shown to cause divergent patterns of ventricular remodeling, a clinically relevant and complex observation.23,24 Despite extensive high-resolution studies, including molecular dynamics simulations, X-ray crystallography, cryo-EM, and FRET, mechanistic insight regarding how single-amino acid substitutions within the same region could cause divergent phenotypes is lacking.15,16,30,51 To directly address this, we combined DSC, FRET, and MD simulations. We have characterized the Tm overlap structure at baseline and in the presence of the two mutations, Tm D230N and cTnT R92L. We find that the overall divergent effects of the two mutations on the baseline structure and properties of the overlap highlight its importance in thin filament cooperativity. Our findings suggest that there is a basis for altered cooperativity that arises from Tm–Tm head-to-tail interactions. Although we have elucidated the primary effect of these mutations on thin filament structure, the link between these biophysical effects and the eventual remodeling that results in HCM or DCM over time still warrants further investigation.
The DCM-associated Tm D230N mutation has been well characterized in two large multigenerational families in a clinical study, as well as in molecular studies.52,53 Because of the proximity of the Tm D230N mutation to the overlap, we predicted that this mutation may lead to opposite structural effects on the Tm overlap when compared to the HCM causing the cTnT R92L mutation.24 The overlap would be predicted to be less flexible, thus leading to increased cooperativity of the Tm filament, reflecting a stronger Tm–Tm connection. This hypothesis agrees with the results of Gupte et al., who showed that the D230N mutation increased the cooperativity of binding under low-salt conditions.53
The cTnT R92L mutation is located at a mutational hot spot, located adjacent to the Tm overlap.24 The effect of the TNT1 region of cTnT on the cooperativity of calcium activation has been previously demonstrated by our group.31,54,55 There was no evidence of haploinsufficiency leading to the cTnT R92L effects, and thus, the mechanism of disease was believed to be biophysical alterations of the TnT itself. cTnT R92L led to a weakened interaction between the cTnT and Tm proteins. The prior work also showed that cTnT R92L was less cooperative than the wild-type complex. This decrease in cooperativity was correlated to an increase in the flexibility of the TNT1 region, specifically the hinge region near sites 104–110. The R92 mutations have also recently been found to have effects that propagate along the molecule to the Tn core, where R92L and R92W differentially affect the rate of dissociation of calcium from cTnC,28 coupled with differential downstream calcium handling shown by other studies.56,57 In another study, Sewenan et al. showed HCM causing mutants Tm D175N and Tm E180G led to less cooperative inhibition between regulatory units of the thin filament.58 On the basis of the prior results, we hypothesized that HCM-associated cTnT R92L would result in a more flexible TNT1. The Tm–actin unfolding cooperativity decreased in the presence of cTnT R92L. MD results showed the cTnT R92L mutation led to an increased Tn–Tm distance along the whole cTnT N-terminal region. As a whole, the results from these independent approaches show that cTnT R92L increases interprotein distances at the overlap and thus decreases the cooperativity of the Tm filament. The proximity of the R92 hot spot and its predicted effects on the cTnT hinge region59 are in agreement with the results of our current study. A possible mechanism for primary effects from the cTnT R92L mutation is the elimination of a local interaction, the salt bridge created between the side chains of R92 and D89 of the cTnT, as seen in MD (Figure 9B). Without the salt bridge between R92 and D89, the N-terminal end of the TNT1 α-helix is straight because of the increased degree of freedom that occurs without the salt bridge pushing the D89 farther away.
Previous studies that focused on the Tm overlap region have provided insights into its structure. For example, Hill et al. and Jin et al. have shown its importance as a cTnT binding region.17,60 There is ample evidence of a Tm–cTnT binding site at the overlap.17,61 This is one of two Tm–Tn binding sites, and thus a site for Tn to modulate Tm structure and dynamics. Greenfield et al. and Li et al. have shown the orientation of the interdigitating N- to C-terminal Tms at the overlap.13,15 These structures highlight the relationship between the ends of adjacent molecules, and thus their role in thin filament regulation. Li et al. demonstrated that the flexibility across the overlap structure is small, implying that positional information can be translated to neighboring Tm.13 Additionally, it has been found that the stiffness of the overlap can be modulated, for example, with phosphorylation of Tm S283.62 Tm S283 phosphorylation has been demonstrated to increase the stiffness of the Tm overlap region.62 Additionally, Tm S283 phosphorylation has been shown to extend the cooperativity of myosin binding to the thin filament.63 Thus, if effects from phosphorylation and these mutations (cTnT R92L and Tm D230N) were additive, we would observe a further increase in the effects of Tm D230N on the overlap (further enhanced cooperativity), and a decrease in the effects of cTnT R92L (an increase in cooperativity from Tm phosphorylation countering the decrease in cooperativity from cTnT R92L). Studies to address these questions are to be pursued.
Of note, β-Tm is known to be expressed in the fetal heart, and neonatal expression is associated with heart failure.64 One study found that β-Tm significantly reduced the cooperativity of myofilament activation upon being phosphorylated.65 One of the amino acids at the Tm overlap that differs between α and β is residue 276, which is a His (positive charge) in α-Tm and an Asn (polar) in β-Tm. This substitution was postulated as the cause of the reduced level of binding between β-Tm and Tn, specifically between Tm and TNT166 at the Tm overlap. Thus, the presence of β-Tm may amplify the effects of cTnT R92L and alleviate effects from Tm D230N. This does not take into account unique interactions that may occur between the mutated thin filament proteins and the β-Tm.
With respect to Tm and actin interactions, there are two hypotheses that focus on either the importance of site specific interactions or Tm global properties. Singh et al. proposed a mechanism of binding of Tm to actin based on specific interaction sites. They have linked alterations that destabilize Tm to a reduced level of actin binding, by means of changing the flexibility of the filament, and thus changing interprotein contacts.67 In contrast, Holmes and Lehman emphasized the shape itself, rather than the flexibility allowing for specific binding, as being more crucial in determining the binding of Tm to actin.1 We did not examine residue specific interactions between Tm and actin and thus cannot comment on specific actin–Tm interaction sites. Our results directly support the perspective of the polymeric Tm filament taken as a whole, as the properties of the overlap had a significant effect on protein interactions, as measured by DSC.
Tm can shift in position relative to cTnT in positions that are defined by the α-helical repeats. Some differences between our MD and FRET results at the residue specific level may be due to the dynamic nature of the overlap. The results do not agree on a residue specific basis. These differences are demonstrated in Figure 10, particularly with cTnT 127. Although the residues near cTnT 100 have good agreement with the model, cTnT 127 should appear closest to Tm 265 via our FRET. Despite this inconsistency, the model does provide overall structural insight into possible mechanisms of the cTnT R92L mutation. The residue specific differences are now informing the hunt for an alternative free energy minimum structure that fits all new data.
Figure 10.

Predicted positions of the labeling sites using the all-atom model of the thin filament (top). Schematic of the relative orientations of the labeling sites as revealed from FRET (bottom). cTnT 100 is in the same relative position in both models. cTnT 127 appears roughly equidistant in the FRET model between the C-terminal sites of Tm, compared to the model in which it is more distant from site 279 than from site 265.
We have defined the baseline structure of the Tm overlap in the presence of cTnT and established the biophysical basis for the effects of HCM- and DCM-causing mutations on the Tm overlap. Of note, these mutations have differential effects clinically, and thus, defining a differential effect in a crucial functioning region of the protein is another step toward understanding the primary biophysical impairment that leads to disease, a crucial step toward the development of modern therapeutics. Finally, our results set the framework for developing a novel approach to predicting the pathogenicity of de novo thin filament mutations in patients with HCM and DCM.
Supplementary Material
Acknowledgments
The authors thank Dr. Wenji Dong (Washington State University, Pullman, WA) and Dr. Brett Colson (University of Arizona, Tucson, AZ) for technical and analytical advice regarding FRET.
Funding
This work was supported by National Institutes of Health Grants R01HL107046 (J.C.T. and S.D.S.) and R01HL075619 (J.C.T.). M.M. was supported by National Institutes of Health Grant T32HL007955. M.L.L. was supported by National Institutes of Health Grant T32HL07249 and American Heart Association Grant 16PRE27260116. J.C.T. acknowledges the support of the Gootter Foundation for the Prevention of Sudden Cardiac Death.
ABBREVIATIONS
- Tm
tropomyosin
- Tn
troponin
- cTnT
human cardiac troponin T
- cTnI
human cardiac troponin I
- cTnC
human cardiac troponin C
- DSC
differential scanning calorimetry
- MD
molecular dynamics
- FRET
fluorescence resonance energy transfer
- fwhm
full-width at half-maximum
- HCM
hypertrophic cardiomyopathy
- DCM
dilated cardiomyopathy
- PME
particle mesh Ewald
- SE
standard error
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00266.
Steady-state emission spectra of IAEDANS conjugated to fully reconstituted thin filament samples and tables of FRET distances (PDF)
Author Contributions
M.M. conducted most of the in vitro experiments. L.T.G. designed and tested all vectors. M.R.W. performed the in silico modeling under the supervision of S.D.S. M.L.L. set up the DSC technique and assisted with analysis. B.A.S. and O.Z.F. optimized protein expression. J.C.T. and L.T.G. conceived the project, and J.C.T. supervised all experimental work. M.M., M.R.W., S.D.S., and J.C.T. wrote the manuscript.
The authors declare no competing financial interest.
References
- 1.Holmes KC, Lehman W. Gestalt-binding of tropomyosin to actin filaments. J. Muscle Res. Cell Motil. 2008;29:213–219. doi: 10.1007/s10974-008-9157-6. [DOI] [PubMed] [Google Scholar]
- 2.Tardiff JC. Tropomyosin and dilated cardiomyopathy: revenge of the actinomyosin ″gatekeeper″. J. Am. Coll. Cardiol. 2010;55:330–332. doi: 10.1016/j.jacc.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 3.Bremel RD, Weber A. Cooperation within actin filament in vertebrate skeletal muscle. Nat. New Biol. 1972;238:97–101. doi: 10.1038/newbio238097a0. [DOI] [PubMed] [Google Scholar]
- 4.Weber A, Murray JM. Molecular control mechanisms in muscle contraction. Physiol. Rev. 1973;53:612–673. doi: 10.1152/physrev.1973.53.3.612. [DOI] [PubMed] [Google Scholar]
- 5.Tobacman LS, Sawyer D. Calcium binds cooperatively to the regulatory sites of the cardiac thin filament. J. Biol. Chem. 1990;265:931–939. [PubMed] [Google Scholar]
- 6.Sun YB, Lou F, Irving M. Calcium- and myosindependent changes in troponin structure during activation of heart muscle. J. Physiol. 2009;587:155–163. doi: 10.1113/jphysiol.2008.164707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell SG, Lionetti FV, Campbell KS, McCulloch AD. Coupling of adjacent tropomyosins enhances cross-bridge-mediated cooperative activation in a markov model of the cardiac thin filament. Biophys. J. 2010;98:2254–2264. doi: 10.1016/j.bpj.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tobacman LS, Butters CA. A new model of cooperative myosin-thin filament binding. J. Biol. Chem. 2000;275:27587–27593. doi: 10.1074/jbc.M003648200. [DOI] [PubMed] [Google Scholar]
- 9.Smith DA, Maytum R, Geeves MA. Cooperative regulation of myosin-actin interactions by a continuous flexible chain I: actin-tropomyosin systems. Biophys. J. 2003;84:3155–3167. doi: 10.1016/S0006-3495(03)70040-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sousa DR, Stagg SM, Stroupe ME. Cryo-EM structures of the actin:tropomyosin filament reveal the mechanism for the transition from C- to M-state. J. Mol. Biol. 2013;425:4544–4555. doi: 10.1016/j.jmb.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murakami K, Stewart M, Nozawa K, Tomii K, Kudou N, Igarashi N, Shirakihara Y, Wakatsuki S, Yasunaga T, Wakabayashi T. Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7200–7205. doi: 10.1073/pnas.0801950105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li XE, Orzechowski M, Lehman W, Fischer S. Structure and flexibility of the tropomyosin overlap junction. Biochem. Biophys. Res. Commun. 2014;446:304–308. doi: 10.1016/j.bbrc.2014.02.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orzechowski M, Li XE, Fischer S, Lehman W. An atomic model of the tropomyosin cable on F-actin. Biophys. J. 2014;107:694–699. doi: 10.1016/j.bpj.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenfield NJ, Huang YJ, Swapna GV, Bhattacharya A, Rapp B, Singh A, Montelione GT, Hitchcock-DeGregori SE. Solution NMR structure of the junction between tropomyosin molecules: implications for actin binding and regulation. J. Mol. Biol. 2006;364:80–96. doi: 10.1016/j.jmb.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 16.Frye J, Klenchin VA, Rayment I. Structure of the tropomyosin overlap complex from chicken smooth muscle: insight into the diversity of N-terminus recognition. Biochemistry. 2010;49:4908–4920. doi: 10.1021/bi100349a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin JP, Chong SM. Localization of the two tropomyosin-binding sites of troponin T. Arch. Biochem. Biophys. 2010;500:144–150. doi: 10.1016/j.abb.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biesiadecki BJ, Chong SM, Nosek TM, Jin JP. Troponin T core structure and the regulatory NH(2)-terminal variable region. Biochemistry. 2007;46:1368–1379. doi: 10.1021/bi061949m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harada K, Potter JD. Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J. Biol. Chem. 2004;279:14488–14495. doi: 10.1074/jbc.M309355200. [DOI] [PubMed] [Google Scholar]
- 20.Yang S, Barbu-Tudoran L, Orzechowski M, Craig R, Trinick J, White H, Lehman W. Three-dimensional organization of troponin on cardiac muscle thin filaments in the relaxed state. Biophys. J. 2014;106:855–864. doi: 10.1016/j.bpj.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White SP, Cohen C, Phillips GN., Jr Structure of co-crystals of tropomyosin and troponin. Nature. 1987;325:826–828. doi: 10.1038/325826a0. [DOI] [PubMed] [Google Scholar]
- 22.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 23.Lakdawala NK, Dellefave L, Redwood CS, Sparks E, Cirino AL, Depalma S, Colan SD, Funke B, Zimmerman RS, Robinson P, Watkins H, Seidman CE, Seidman JG, McNally EM, Ho CY. Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J. Am. Coll. Cardiol. 2010;55:320–329. doi: 10.1016/j.jacc.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forissier J-Fo, Carrier L, Farza H, Bonne Gl, Bercovici J, Richard P, Hainque B, Townsend PJ, Yacoub MH, Fauré S, Dubourg O, Millaire A, Hagége AA, Desnos M, Komajda M, Schwartz K. Codon 102 of the cardiac troponin T gene is a putative hot spot for mutations in familial hypertrophic cardiomyopathy. Circulation. 1996;94:3069–3073. doi: 10.1161/01.cir.94.12.3069. [DOI] [PubMed] [Google Scholar]
- 25.Yar S, Chowdhury SA, Davis RT, 3rd, Kobayashi M, Monasky MM, Rajan S, Wolska BM, Gaponenko V, Kobayashi T, Wieczorek DF, Solaro RJ. Conserved Asp-137 is important for both structure and regulatory functions of cardiac alphatropomyosin (alpha-TM) in a novel transgenic mouse model expressing alpha-TM-D137L. J. Biol. Chem. 2013;288:16235–16246. doi: 10.1074/jbc.M113.458695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sumida JP, Wu E, Lehrer SS. Conserved Asp-137 imparts flexibility to tropomyosin and affects function. J. Biol. Chem. 2008;283:6728–6734. doi: 10.1074/jbc.M707485200. [DOI] [PubMed] [Google Scholar]
- 27.Li XE, Suphamungmee W, Janco M, Geeves MA, Marston SB, Fischer S, Lehman W. The flexibility of two tropomyosin mutants, D175N and E180G, that cause hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2012;424:493–496. doi: 10.1016/j.bbrc.2012.06.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams MR, Lehman SJ, Tardiff JC, Schwartz SD. Atomic resolution probe for allostery in the regulatory thin filament. Proc. Natl. Acad. Sci. U. S. A. 2016;113:3257–3262. doi: 10.1073/pnas.1519541113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kast D, Espinoza-Fonseca LM, Yi C, Thomas DD. Phosphorylation-induced structural changes in smooth muscle myosin regulatory light chain. Proc. Natl. Acad. Sci. U. S. A. 2010;107:8207–8212. doi: 10.1073/pnas.1001941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimura-Sakiyama C, Ueno Y, Wakabayashi K, Miki M. Fluorescence resonance energy transfer between residues on troponin and tropomyosin in the reconstituted thin filament: modeling the troponin-tropomyosin complex. J. Mol. Biol. 2008;376:80–91. doi: 10.1016/j.jmb.2007.10.078. [DOI] [PubMed] [Google Scholar]
- 31.Manning EP, Guinto PJ, Tardiff JC. Correlation of molecular and functional effects of mutations in cardiac troponin T linked to familial hypertrophic cardiomyopathy: an integrative in silico/in vitro approach. J. Biol. Chem. 2012;287:14515–14523. doi: 10.1074/jbc.M111.257436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monteiro PB, Lataro RC, Ferro JA, Reinach FdC. Functional alpha-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J. Biol. Chem. 1994;269:10461–10466. [PubMed] [Google Scholar]
- 33.Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 34.Dedova IV, Nikolaeva OP, Safer D, De La Cruz EM, dos Remedios CG. Thymosin beta4 induces a conformational change in actin monomers. Biophys. J. 2006;90:985–992. doi: 10.1529/biophysj.105.063081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landis C, Back N, Homsher E, Tobacman LS. Effects of tropomyosin internal deletions on thin filament function. J. Biol. Chem. 1999;274:31279–31285. doi: 10.1074/jbc.274.44.31279. [DOI] [PubMed] [Google Scholar]
- 36.Gordon AM, LaMadrid MA, Chen Y, Luo Z, Chase PB. Calcium regulation of skeletal muscle thin filament motility in vitro. Biophys. J. 1997;72:1295–1307. doi: 10.1016/S0006-3495(97)78776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xing J, Jayasundar JJ, Ouyang Y, Dong WJ. Forster resonance energy transfer structural kinetic studies of cardiac thin filament deactivation. J. Biol. Chem. 2009;284:16432–16441. doi: 10.1074/jbc.M808075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Remedios CGd. Fluorescence Resonance Energy Transfer. John Wiley & Sons, Ltd; New York: 2001. [Google Scholar]
- 39.Kremneva EV, Nikolaeva OP, Gusev NB, Levitsky DI. Effects of troponin on thermal unfolding of actin-bound tropomyosin. Biochemistry (Moscow) 2003;68:802–809. doi: 10.1023/a:1025043202615. [DOI] [PubMed] [Google Scholar]
- 40.Matyushenko AM, Shchepkin DV, Kopylova GV, Popruga KE, Artemova NV, Pivovarova AV, Bershitsky SY, Levitsky DI. Structural and functional effects of cardiomyopathy-causing mutations in the troponin T-binding region of cardiac tropomyosin. Biochemistry. 2017;56:250–259. doi: 10.1021/acs.biochem.6b00994. [DOI] [PubMed] [Google Scholar]
- 41.Ueda K, Kimura-Sakiyama C, Aihara T, Miki M, Arata T. Interaction sites of tropomyosin in muscle thin filament as identified by site-directed spin-labeling. Biophys. J. 2011;100:2432–2439. doi: 10.1016/j.bpj.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsuo N, Nagata Y, Nakamura J, Yamamoto T. Coupling of calcium transport with ATP hydrolysis in scallop sarcoplasmic reticulum. J. Biochem. 2002;131:375–381. doi: 10.1093/oxfordjournals.jbchem.a003112. [DOI] [PubMed] [Google Scholar]
- 43.Schlecht W, Zhou Z, Li KL, Rieck D, Ouyang Y, Dong WJ. FRET study of the structural and kinetic effects of PKC phosphomimetic cardiac troponin T mutants on thin filament regulation. Arch. Biochem. Biophys. 2014;550–551:1–11. doi: 10.1016/j.abb.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li KL, Rieck D, Solaro RJ, Dong W. In situ time-resolved FRET reveals effects of sarcomere length on cardiac thin-filament activation. Biophys. J. 2014;107:682–693. doi: 10.1016/j.bpj.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tao T, Lamkin M, Lehrer SS, Morris EP. Excitation energy transfer studies of the proximity between tropomyosin and actin in reconstituted skeletal muscle thin filaments. Computer simulation study on the extent of energy transfer from a single Tm-bound donor to multiple actin-bound acceptors. Biochemistry. 1983;22:3059–3066. doi: 10.1021/bi00282a006. [DOI] [PubMed] [Google Scholar]
- 46.Miki M, Miura T, Sano K, Kimura H, Kondo H, Ishida H, Maeda Y. Fluorescence resonance energy transfer between points on tropomyosin and actin in skeletal muscle thin filaments: does tropomyosin move? J. Biochem. 1998;123:1104–1111. doi: 10.1093/oxfordjournals.jbchem.a022049. [DOI] [PubMed] [Google Scholar]
- 47.Bacchiocchi C, Lehrer SS. Ca2+-induced movement of tropomyosin in skeletal muscle thin filaments observed by multi-site FRET. Biophys. J. 2002;82:1524–1536. doi: 10.1016/S0006-3495(02)75505-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miki M, Hai H, Saeki K, Shitaka Y, Sano K, Maeda Y, Wakabayashi T. Fluorescence resonance energy transfer between points on actin and the C-terminal region of tropomyosin in skeletal muscle thin filaments. J. Biochem. 2004;136:39–47. doi: 10.1093/jb/mvh090. [DOI] [PubMed] [Google Scholar]
- 49.Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ. Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys. J. 2001;81:2827–2837. doi: 10.1016/S0006-3495(01)75924-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circ. Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.von der Ecken J, Muller M, Lehman W, Manstein DJ, Penczek PA, Raunser S. Structure of the F-actintropomyosin complex. Nature. 2015;519:114–117. doi: 10.1038/nature14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Memo M, Leung MC, Ward DG, dos Remedios C, Morimoto S, Zhang L, Ravenscroft G, McNamara E, Nowak KJ, Marston SB, Messer AE. Familial dilated cardiomyopathy mutations uncouple troponin I phosphorylation from changes in myofibrillar Ca(2)(+) sensitivity. Cardiovasc. Res. 2013;99:65–73. doi: 10.1093/cvr/cvt071. [DOI] [PubMed] [Google Scholar]
- 53.Gupte TM, Haque F, Gangadharan B, Sunitha MS, Mukherjee S, Anandhan S, Rani DS, Mukundan N, Jambekar A, Thangaraj K, Sowdhamini R, Sommese RF, Nag S, Spudich JA, Mercer JA. Mechanistic heterogeneity in contractile properties of alpha-tropomyosin (TPM1) mutants associated with inherited cardiomyopathies. J. Biol. Chem. 2015;290:7003–7015. doi: 10.1074/jbc.M114.596676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manning EP, Tardiff JC, Schwartz SD. A model of calcium activation of the cardiac thin filament. Biochemistry. 2011;50:7405–7413. doi: 10.1021/bi200506k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guinto PJ, Manning EP, Schwartz SD, Tardiff JC. Computational characterization of mutations in cardiac troponin T known to cause familial hypertrophic cardiomyopathy. J. Theor. Comput. Chem. 2007;6:413. doi: 10.1142/S0219633607003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sommese RF, Nag S, Sutton S, Miller SM, Spudich JA, Ruppel KM. Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human beta-cardiac myosin. PLoS One. 2013;8:e83403. doi: 10.1371/journal.pone.0083403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tikunova SB, Liu B, Swindle N, Little SC, Gomes AV, Swartz DR, Davis JP. Effect of calcium-sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry. 2010;49:1975–1984. doi: 10.1021/bi901867s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sewanan LR, Moore JR, Lehman W, Campbell SG. Predicting effects of tropomyosin mutations on cardiac muscle contraction through myofilament modeling. Front. Physiol. 2016;7:473. doi: 10.3389/fphys.2016.00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ertz-Berger BR, He H, Dowell C, Factor SM, Haim TE, Nunez S, Schwartz SD, Ingwall JS, Tardiff JC. Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 2005;102:18219–18224. doi: 10.1073/pnas.0509181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hill LE, Mehegan JP, Butters CA, Tobacman LS. Analysis of troponintropomyosin binding to actin. Troponin does not promote interactions between tropomyosin molecules. J. Biol. Chem. 1992;267:16106–16113. [PubMed] [Google Scholar]
- 61.Willadsen KA, Butters CA, Hill LE, Tobacman LS. Effects of the amino-terminal regions of tropomyosin and troponin T on thin filament assembly. J. Biol. Chem. 1992;267:23746–23752. [PubMed] [Google Scholar]
- 62.Lehman W, Medlock G, Li XE, Suphamungmee W, Tu AY, Schmidtmann A, Ujfalusi Z, Fischer S, Moore JR, Geeves MA, Regnier M. Phosphorylation of Ser283 enhances the stiffness of the tropomyosin head-to-tail overlap domain. Arch. Biochem. Biophys. 2015;571:10–15. doi: 10.1016/j.abb.2015.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rao VS, Marongelli EN, Guilford WH. Phosphorylation of tropomyosin extends cooperative binding of myosin beyond a single regulatory unit. Cell Motil. Cytoskeleton. 2009;66:10–23. doi: 10.1002/cm.20321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muthuchamy M, Boivin GP, Grupp IL, Wieczorek DF. Beta-tropomyosin overexpression induces severe cardiac abnormalities. J. Mol. Cell. Cardiol. 1998;30:1545–1557. doi: 10.1006/jmcc.1998.0720. [DOI] [PubMed] [Google Scholar]
- 65.Lu X, Heeley DH, Smillie LB, Kawai M. The role of tropomyosin isoforms and phosphorylation in force generation in thin-filament reconstituted bovine cardiac muscle fibres. J. Muscle Res. Cell Motil. 2010;31:93–109. doi: 10.1007/s10974-010-9213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pearlstone JR, Smillie LB. Binding of troponin-T fragments to several types of tropomyosin. Sensitivity to Ca2+ in the presence of troponin-C. J. Biol. Chem. 1982;257:10587–10592. [PubMed] [Google Scholar]
- 67.Singh A, Hitchcock-DeGregori SE. Local destabilization of the tropomyosin coiled coil gives the molecular flexibility required for actin binding. Biochemistry. 2003;42:14114–14121. doi: 10.1021/bi0348462. [DOI] [PubMed] [Google Scholar]
- 68.Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 2014;106:1236–1249. doi: 10.1016/j.bpj.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.