


Abstract
Hammerhead ribozymes were expressed under the control of similar tRNA promoters, localizing transcripts either in the cytoplasm or the nucleus. The tRNAVal-driven ribozyme (tRNA-Rz; tRNA with extra sequences at the 3′ end) that has been used in our ribozyme studies was exported efficiently into the cytoplasm and ribozyme activity was detected only in the cytoplasmic fraction. Both ends of the transported tRNA-Rz were characterized comprehensively and the results confirmed that tRNA-Rz had unprocessed 5′ and 3′ ends. Furthermore, it was also demonstrated that the activity of the exported ribozyme was significantly higher than that of the ribozyme which remained in the nucleus. We suggest that it is possible to engineer tRNA-Rz, which can be exported to the cytoplasm based on an understanding of secondary structures, and then tRNA-driven ribozymes may be co-localized with their target mRNAs in the cytoplasm of mammalian cells.
INTRODUCTION
Hammerhead ribozymes are small and versatile catalytic RNA molecules that cleave RNAs at specific sites (1–16). In studies to investigate the applications of these potentially useful tools in vivo (17–24), a tRNA-based system for expression of ribozymes was established that resulted in both high-level expression and intracellular stability (25–37). In the RNA polymerase III (pol III) transcription system, the promoter is located within the tRNA sequence being transcribed. For this reason, it is inevitable that a portion of the tRNA becomes incorporated into the ribozyme. Thus, in our expression system, the ribozymes are linked downstream of the partially modified human tRNAVal through a linker (Fig. 1).
Figure 1.

Secondary structures of mature tRNAs and tRNA-driven ribozymes. (A) Comparison of pre-tRNAVal with mature tRNAVal. The wild-type tRNA is synthesized as a precursor that undergoes a series of maturation steps. (B) Secondary structures of the various tRNA-Rz that were efficiently exported to the cytoplasm. Artificial sequences in the linker region are indicated by lowercase letters. The ribozyme sequence is shown in red and the substrate-binding sites in orange. (C) Secondary structure of tBR-Rz and (D) Rz-N, which accumulated in the nucleus. The sequence underlined in purple is the sequence complementary to the sequences of a probe used for in situ hybridization in Figure 3A.
In eukaryotic cells, RNAs are transcribed initially as precursors that are processed to yield mature molecules in the nucleus. It is generally accepted that only tRNAs with mature 5′ and 3′ ends can be exported to the cytoplasm (Fig. 1A) (38–49). The exchange of macromolecules between the nucleus and the cytoplasm proceeds through nuclear pore complexes (NPCs). In general, such an exchange is mediated by transport receptors. These receptors recognize signals on cargo molecules, which interact directly with NPCs and shuttle continuously between the nucleus and the cytoplasm. Several pathways mediated by transport receptors have been identified. The export of tRNAs is rapid and is mediated by a saturable carrier. Newly transcribed pre-tRNAs undergo a series of maturation steps in the nucleus that involve trimming of the 5′ and 3′ ends, base modifications and, in some cases, removal of small introns. It was proposed recently that exportin-t (Xpo-t) (40,44), a transport receptor, recognizes the 3′ ends and TψC loops of tRNAs (41,47). It has also been suggested that, in Xenopus oocytes, the aminoacylation of a tRNA prior to transport serves as a proofreading step and prevents pre-tRNAs from leaving the nucleus (43,45).
In this study, we investigated the localization of tRNAVal-driven ribozymes (tRNA-Rz), in mammalian cells. From northern blotting, in situ hybridization and microinjection analyses, it was shown that the nucleocytoplasmic transport of the tRNA-Rz was effective. In addition, specific cleavage of the substrate RNA was detected when it was mixed with the cytoplasmic RNAs extracted from HeLa cells that had been transfected by an expression vector for tRNA-driven ribozymes, whereas no such activity was detected from the nuclear RNA extracts. Therefore, the exported tRNA-Rz maintained the extra (ribozyme) sequence at their 3′ end. Furthermore, the exact sequence of the 5′ end of the exported tRNA-Rz was determined. From these analyses, we confirmed that, in mammalian cells, tRNA-driven ribozymes could be exported, without being processed at 5′ and 3′ ends, from the nucleus to the cytoplasm where they came into contact with their target mRNAs. Importantly, intracellular activity of the transportable tRNA-Rz was revealed to be higher than that of nuclear localizing ribozymes.
MATERIALS AND METHODS
Cell culture and the transfection
HeLa S3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco BRL, Gaithersburg, MD) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco BRL). Transfection was carried out using Lipofectin™ reagent (Gibco BRL) according to the manufacturer’s protocol.
Northern blotting analysis
Cells were grown to ∼80% confluence (1 × 107 cells) and transfected with a tRNA-Rz expression vector using the Lipofectin™ reagent (Gibco BRL). Following transfection, cells were incubated for 36 h before being harvested. To prepare the cytoplasmic fraction, collected cells were washed 2× with PBS and then resuspended in digitonin lysis buffer (50 mM HEPES–KOH pH 7.5, 50 mM potassium acetate, 8 mM MgCl2, 2 mM EGTA and 50 µg/ml digitonin) on ice for 10 min. The lysate was centrifuged at 1000 g for 5 min and the supernatant collected as the cytoplasmic fraction. The pellets were resuspended in NP-40 lysis buffer (20 mM Tris–HCl pH 7.5, 50 mM KCl, 10 mM NaCl, 1 mM EDTA and 0.5% NP-40) and incubated on ice for 10 min, then the resultant lysate was used as the nuclear fraction. Cytoplasmic RNA and nuclear RNA were extracted and purified from the cytoplasmic fraction and the nuclear fraction, respectively, with ISOGEN reagent (Wako, Osaka, Japan). Thirty micrograms total RNA per lane was loaded on a 3.0% NuSieveTM (3:1) agarose gel (FMC Inc., Rockland, ME). After electrophoresis, RNA was transferred to a Hybond-NTM nylon membrane (Amersham Co., Buckinghamshire, UK). The membrane was probed with synthetic oligonucleotides complementary to the sequences of the ribozymes being investigated (35). A synthetic probe complementary to the sequence of each respective ribozyme was used and all probes were labeled with 32P by T4 polynucleotide kinase (Takara Shuzo Co., Kyoto, Japan).
In situ hybridization
HeLa cells that had been transfected in advance with one of the plasmids and cultured on a coverslip, were washed in fresh PBS and fixed in fix/permeabilization buffer (50 mM HEPES–KOH pH 7.5, 50 mM potassium acetate, 8 mM MgCl2, 2 mM EGTA, 2% paraformaldehyde, 0.1% NP-40, 0.02% SDS) for 15 min at room temperature. A reaction mixture consisting of 70 µg Cy3-labeled oligodeoxynucleotide probe complementary to the underlined sequence in Figure 1 (Tm >90°C) and 20 µg tRNA from Escherichia coli MRE 600 (Boehringer Mannheim, Mannheim, Germany) dissolved in 10 µl deionized formamide was denatured by heating at 70°C for 10 min. Twenty microliters of hybridization solution, containing 20% dextran sulfate and 2% BSA in 4× SSC, with denatured probe were placed on the coverslip and the coverslip was inverted on a glass slide, sealed with rubber cement and incubated for 16 h at 37°C. Cells were rinsed in 2× SSC/50% formamide then 2× SSC at room temperature for 20 min each. The coverslip was mounted with Vectashield (Vector Laboratories, Burlingame, CA) on a glass slide and cells were analyzed using a confocal laser scanning microscope (LSM 510; Carl Zeiss, Jena, Germany).
Microinjection of tRNA-Rz into HeLa cells
Microinjection analysis was performed as previously described (50,51). FITC-labeled tRNA-Rz (tRNA-HIV Rz2) was prepared using the AmpliScribeTM T7 transcription kit (Epicentre Technologies, Madison, WI) in combination with ChromaTideTM BODIPY FL-14-rUTP (Molecular Probes, Eugene, OR). The molar ratio of FL-14-rUTP to UTP in the reaction was ∼1:5. Human tRNAVal was used in the unmodified form. For injection of FITC-labeled tRNA-Rz in combination with excess amount of wild-type tRNAVal, non-labeled tRNAVal was mixed with tRNA-Rz at final concentrations of 2.4 µg/µl and 20 ng/µl, respectively. Before injection, HeLa cells were grown in 35 mm glass base dishes. RNA at a final concentration of ∼20 ng/µl in PBS was injected into the nucleus of HeLa cells using a micromanipulation system (micromanipulator 5171 and transjector 5246; Eppendorf, Hamburg, Germany). Simultaneously, FluoroLinkTM Cy3-labeled goat anti-rabbit IgG (Amersham Pharmacia Biotech, Piscataway, NJ), which cannot pass through nuclear membranes, was injected into nuclei as an internal control. Approximately 5 min after injection, the location of the injected RNA in cells was monitored with LSM 510.
Sequencing analysis of the isolated tRNA-ribozymes from HeLa cells
We isolated tRNA-ribozymes from HeLa cells as described above. The prepared RNAs were linked, by T4 RNA ligase (Invitrogen, Carlsbad, CA), to a DNA adaptor that had 3′ amino ends to prevent self-ligation of the adaptors. After reverse transcription of the RNA moieties, the ligated constructs were amplified by PCR with a 5′-side primer that included the linker sequence and a 3′-side primer that was complementary to the adaptor sequence. The amplified DNA fragments were cloned into pCR vector using the TA Cloning Kit (Invitrogen) and the sequence was analyzed with 377 DNA Sequencer (PE Applied Biosystems, Foster City, CA).
Cleavage activities of RNAs extracted from HeLa cells
Cytoplasmic RNAs and nucleic RNAs were prepared as described above, from extracts of HeLa cells that had been transfected with tRNA-Rz expression vectors. Thirty micrograms of cytoplasmic and nuclear RNAs were incubated with 2 nM 32P-labeled S11 in 50 mM Tris–HCl pH 7.5, 10 mM MgCl2 at 37°C. The substrate and the products of the reaction were separated by electrophoresis on a 20% polyacrylamide–7 M urea denaturing gel and were detected by autoradiography. The extent of cleavage was determined by quantitation of radioactivity in the bands of substrate and product with a Bio-Image Analyzer (BA2000; Fuji Film, Tokyo).
In vitro cleavage activity of tRNA-Rz
tRNA-Rz (tRNAVal-HIV Rz2 and tBR-Rz) were prepared by transcription in vitro. Ribozyme expression plasmids [the expression cassette for each ribozyme (tRNAVal-HIV Rz2 or tBR-Rz) that was subcloned into pUC 19 plasmid] were used as DNA templates for PCR to make DNA templates for the in vitro transcription. Primers were synthesized for each template and the sense strand contained the T7 promoter. PCR products were gel-purified. T7 transcription in vitro and purification were performed as described (33). Reaction rates were measured in 10 mM MgCl2 and 50 mM Tris–HCl pH 8.0, under the ribozyme-saturating (single-turnover) conditions at 37°C. The reactions were initiated by the addition of MgCl2 to a buffered solution containing the ribozyme and its substrate. The substrate RNA had the sequence 5′-CAGAACA(GUC)AGACUCAUC-3′. Both ribozymes were designed to cleave the U5 region of HIV-1 RNA, at 568 nt of the pNL432 infectious molecular clone of HIV-1 (52). The 5′-terminus of the substrate was labeled with [γ-32P]ATP by T4 polynucleotide kinase (New England Biolabs, Beverly, MA). Ribozymes were used at 50 nM, 500 nM or 1 µM with <5 nM substrate. Aliquots from the reaction mixture were removed at appropriate time intervals and mixed with an equivalent volume of 200 mM EDTA, 0.1% xylene cyanol, 0.1% bromophenol blue and 20% glycerol on ice to terminate the reaction. The substrate and the product of the reaction were separated by electrophoresis on a 12% polyacrylamide–7 M urea denaturing gel and were detected by autoradiography. The extent of cleavage was determined by quantitation of radioactivity in the bands of substrate and products with a Bio-Image Analyzer (Molecular Dynamics).
Luciferase assay
HeLa cells were plated at 80% confluence in 12 well plates and incubated at 37°C in a CO2 incubator. The cells were washed twice with PBS before (co)-transfection. A reaction mix containing 3 µg each ribozyme expression plasmid, 500 ng target gene-expressing plasmid pGV-V1, which encoded the chimeric HIV-1 LTR sequence luciferase gene and 4 µl Lipofectin reagent in 400 µl serum-reduced medium (OPTI-MEM I; Gibco BRL) was prepared and incubated for 30 min at room temperature. The mixture was then gently added to cells. After 12 h, the medium was replaced by DMEM supplemented with 10% FBS and cells were cultured for a further 24 h.
Luciferase activity was measured with a PicaGene kit (Toyo-inki, Tokyo, Japan) as described (33,35). In order to normalize the efficiency of transfection by reference to β-galactosidase activity, cells were co-transfected with pSV-β-galactosidase control vector (Promega, Madison, WI) and then the chemiluminescent signal due to β-galactosidase was determined with a luminescent β-galactosidase Genetic Reporter System (Clontech, Palo Alto, CA) as described (35).
RESULTS
In our strategy for expression of tRNAVal-driven ribozymes (Fig. 1B), we removed the last seven bases of the wild-type mature tRNAVal in order to block 3′ end processing of the transcript (35,38). We replaced these bases by a linker (indicated by lowercase letters in Fig. 1B) followed by a ribozyme (indicated by red capital letters). The freedom or availability of the substrate-recognition arms of the ribozyme was controlled by the linker sequence via formation of stable stem structures in combination with the sequence of tRNAVal, which accounted for about two-thirds of the whole sequence (Fig. 1B). Thus, it is relatively easy to predict, by computer folding, the secondary structure of the entire tRNAVal transcript and the accessibility of each recognition arm.
Co-localization of a ribozyme with its target is clearly an important determinant of the ribozyme’s efficiency (28–36,53). Figure 2A shows the results of analysis by northern blotting of the subcellular localization of various kinds of tRNA-Rz. HeLa cells were grown to ∼80% confluence (1 × 107 cells) and transfected with the vector that encoded the tRNAVal-driven ribozyme. Thirty-six hours after transfection, total RNA was extracted as nuclear and cytoplasmic fractions. The conditions for the separation of cytoplasmic and nuclear fractions were optimized using western blotting and northern blotting (data not shown). Since there was a possibility that treatment with digitonin, which we used for the separation of the cytoplasmic fractions, might not only have disrupted cell membranes but might also have damaged nuclear membranes, we carefully set the conditions for digitonin treatment such that only cell membranes were removed, without any damage to nuclear membranes. The successful separation of the cytoplasmic fraction was monitored by mixing the digitonin-treated suspension of cells with 148 kDa fluorescent dextran (Fig. 2B). If nuclear membranes had remained intact, this large fluorescent dextran molecule should have been excluded from nuclei. The confocal image indicated that the nuclear membranes had indeed remained intact and that the separation of the cytoplasmic fraction had been successful (Fig. 2B).
Figure 2.

The intracellular localization of pol III transcripts. (A) Steady-state levels of expression and intracellular localization of tRNA-Rz. Northern blotting analysis was performed with total RNA from intracellular fractions (N, nuclear; C, cytoplasmic). (B) Cell images after treatment for separation of the cytoplasmic fraction. We confirmed that, under our conditions for treatment with digitonin, only cell membranes were disrupted and nuclear membranes remained intact. (C) Northern blots showing the kinetics of the export of tRNA-Rz at various times after transfection. As a control, intracellular U6 snRNA, which remains in the nucleus, was also analyzed.
Having established these conditions, we purified cytoplasmic RNA and nuclear RNA 36 h after transfection from the separated cytoplasmic and nuclear fractions. Transcripts of ∼130 nt in length, which corresponded in size to tRNA-Rz, were detected with a ribozyme-specific probe (Fig. 2A, upper panels) and we confirmed that the tRNA-Rz transcripts [tRNAVal CPP Rz and tRNAVal HIV Rz2 (Fig. 1B)] had been exported to the cytoplasm. The predicted secondary structures of the various tRNA-Rz are shown in Figure 1B. We also investigated the kinetics of the export of tRNA-Rz. Total RNA from HeLa cells that had been transfected with various plasmids was extracted 6, 12, 18, 24, 30 and 36 h after transfection. These samples of total RNA were also separated into nuclear and cytoplasmic fractions. Even initially, tRNA-Rz was found in the cytoplasmic fraction and none was detected in the nuclear fraction (Fig. 2C, upper panels), confirming the high efficacy, for the 5′- and 3′-extended version of tRNAs, of the transport system in mammalian cells. The transported tRNA-based ribozymes included cloverleaf structures in their predicted secondary structures.
In contrast, other transcripts, whose secondary structures did not include such a cloverleaf, remained predominantly in the nucleus. A tRNAiMet-linked ribozyme is known to accumulate in the nucleus (29), although it is also transcribed under the control of a tRNA promoter. The predicted secondary structure of this specific transcript is shown in Figure 1C [tRNAiMet-BR Rz (tBR-Rz)]. We investigated the subcellular localization of this transcript (tBR-Rz) under the conditions described above. As shown in Figure 2A and C, tBR-Rz (∼130 nt in length) accumulated in the nucleus without being transported into the cytoplasm. In addition, a tRNAVal-driven ribozyme without a cloverleaf motif in the secondary structure (Rz-N in Fig. 1D) remained in the nucleus in HeLa cells (Fig. 2A). The predicted secondary structure of Rz-N is quite different from that of tRNAVal-HIV Rz2 even though the sequences corresponding to the A and B boxes in the promoter elements and all of the remaining nucleotides within the tRNAVal segment are identical in transcripts tRNAVal-HIV Rz2 and Rz-N (Fig. 2).
It should be emphasized that endogenous U6 RNA, which is significantly smaller (∼100 nt) than that of tRNA-Rz (130–150 nt), remained in the nucleus without being transported into the cytoplasm. Next, under more natural conditions, we attempted to detect the tRNA-Rz transcript directly in mammalian cells by in situ hybridization (Fig. 3A). HeLa cells that had been transfected with the vector that encoded the tRNAVal-HIV Rz2 were fixed and allowed to hybridize with Cy3-labeled oligodeoxynucleotide probe. Because the probe was specific for the ribozyme sequence, endogenous tRNAs were not detected by the probe. As seen in Figure 3A, tRNA-Rz was detected only in the cytoplasm.
Figure 3.
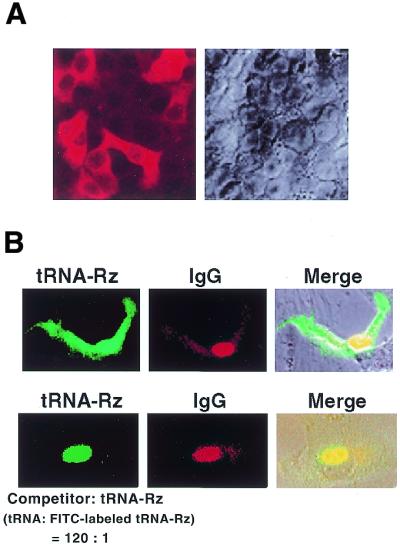
The efficient transport of the tRNA-Rz in mammalian cells. (A) Analysis by in situ hybridization of tRNA-Rz in HeLa S3 cells. (B) Microinjection analysis of tRNA-Rz in HeLa S3 cells. FITC-labeled tRNA-Rz was injected into nuclei of HeLa cells with excess amount of wild-type tRNAVal (bottom).
In order to further strengthen our conclusion, we examined the transport of tRNA-Rz in mammalian cells by microinjection analysis. We prepared FITC internally labeled tRNA-Rz and then injected the RNA into the nuclei of HeLa cells. Simultaneously, Cy3-labeled IgG, which should not pass through the nuclear membrane, was injected into the nucleus as an internal control. Figure 3B shows the typical microscopic images that demonstrate the export of RNAs from the nuclei of HeLa cells 5 min after injection. The tRNA-Rz was efficiently exported into the cytoplasm within 5 min (Fig. 3B, upper panels). Importantly, when an excess amount of the unlabeled wild-type tRNAVal was co-injected simultaneously into HeLa cells, the wild-type tRNAVal apparently competed with the FITC-labeled tRNA-Rz for the nucleocytoplasmic transport and significantly inhibited the export of the FITC-labeled tRNA-Rz (Fig. 3B, lower panels), indicating that tRNA-Rz indeed shares the same transport pathway with wild-type tRNAVal. These results seem to confirm that tRNA-Rz was exported effectively to the cytoplasm in HeLa cells via the same transport pathway as mature tRNAs.
These results contradict the concept that only tRNAs with mature 5′ and 3′ ends can be exported to the cytoplasm (38–49), as our tRNA-Rz might be classified as immature tRNAs that have extremely extended 3′ ends (including the ribozyme sequence). Although we confirmed that the tRNA-Rz isolated from the cytoplasmic fraction were of the expected size by northern blotting analysis, we could not completely exclude the possibility of trimming and addition of a CCA end. Therefore, we examined tRNA-Rz that had been exported to the cytoplasm in mammalian cells in terms of processing at the 5′ and 3′ ends.
We first confirmed that the extra sequence at the 3′ end (ribozyme portion) had not been cleaved in mammalian cells. Using the method described above, we collected cytoplasmic RNAs from HeLa cells that had been transfected with vectors that encoded tRNA-Rz. In order to confirm that the cytoplasmic tRNA-Rz contained the intact ribozyme sequence, we examined each ribozyme’s cleavage activity by incubating the collected cytoplasmic RNA with a 32P-labeled substrate RNA in vitro (Fig. 4A). The S11 RNA is a specific substrate for the R32 ribozyme that is used in this analysis (12,33). In the presence of 10 mM MgCl2, cleavage of S11 occurred only during incubation with the cytoplasmic RNA that had been isolated from cells transfected with the vector that encoded tRNA-R32 (Fig. 4A). The cytoplasmic fractions including other tRNA-Rz and the nuclear fractions had no such cleavage activity. Thus, it is clear that the exported tRNA-Rz had a catalytically active ribozyme sequence at its 3′ end.
Figure 4.

Demonstration of the integrity of exported tRNA-Rz isolated from the cytoplasm. (A) Cleavage activity of the ribozyme (tRNAVal-R32) extracted from the cytoplasmic fraction. The 32P-labeled substrate RNA (S11) was cleaved only by RNA extracted from the cytoplasmic fraction of cells that had been transfected with the plasmid encoding the ribozyme that cleaved S11. Other ribozymes (tBR-Rz and tRNA CPP Rz) were not designed to cleave S11. (B) Determination of the 5′ end of tRNA-Rz that had been exported to the cytoplasm. Reverse transcription was performed with cytoplasmic RNAs and the 5′-32P-labeled ribozyme-specific primer (RT). As size markers (M), tRNA-Rz RNAs that had correctly processed 5′ ends and 5′-extended ends were prepared by T7 transcription.
Next, to investigate the 5′ ends of tRNA-Rz, we performed reverse transcription with the isolated cytoplasmic RNAs and 5′-32P-labeled ribozyme-specific primers. As size markers, we prepared two kinds of T7-transcribed RNA: one kind that had the correctly processed 5′ end (RNAs that start at +1 in Fig. 1B) and one kind that had the unprocessed 5′-extended end (RNAs that start at –7 in Fig. 1B). From the mobility of each exported tRNA-Rz, as shown in Figure 4B, it was clear that each exported tRNA-Rz had an unprocessed 5′ end.
To further demonstrate that the transported tRNA-Rz had indeed maintained the expected structure without being trimmed at the 3′ end, we performed sequencing analysis. We isolated tRNA-Rz from HeLa cells and linked them to a DNA adaptor that had 3′ amino ends to prevent self-ligation of the adaptors. After reverse transcription of the RNA moieties, the ligated constructs were amplified by PCR with a 5′-side primer that included the linker sequence and a 3′-side primer that was complementary to the adaptor sequence (Fig. 5A). The results of sequencing the products are summarized in Figure 5B. The sequenced cDNA of the tRNA-Rz had various numbers of thymidines at their 3′ ends (terminator), suggesting termination of transcription at several locations. However, all the transported tRNA-Rz obviously included the complete ribozyme sequence at their 3′ ends. The sequencing analysis of 5′ end of tRNA-Rz was also performed using a modified DNA adaptor. Basically, the sequence of the 5′ end was the same as that shown in Figure 1B and all tRNA-Rz had unprocessed 5′ ends (data not shown). From these results, we confirmed that tRNA-Rz that contained a significant extra sequence at both 5′ and 3′ ends (3′ ends extended by a ribozyme sequence of >40 nt) had been exported efficiently into the cytoplasm in an intact form (without being trimmed) in mammalian cells.
Figure 5.

Determination of the sequences of the 3′ ends of various tRNA-driven ribozyme transcripts. (A) Schematic representation of the experimental procedure for cloning each tRNA-Rz. We isolated tRNA-Rz from HeLa cells and ligated them to a DNA adaptor that had a 3′ amino end to avoid self-ligation of adaptors. The ligated products were amplified by RT–PCR with 5′ and 3′ primers. (B) Sequences of the cDNAs that corresponded to the 3′ ends of various tRNA-Rz. Dotted lines indicate deletions. Underlining indicates mutations. The sequences of the 3′ portions of genes for ribozymes on the expression plasmids were as follows: Rz2, 5′-TCGGAAACGGTTTTTTTCTATCGCGTC-3′; Rz-BR, 5′-TTCGGTCCG CTTTTTTTGGCTGCAGCG-3′; Rz-N, 5′-ACTCGAGCGCTTTTTTTCTATCGCGTC-3′. Continual Ts are those of the terminator sequence. Bold letters in the table correspond to those terminator Ts in figure legend. Some T residues attach to transcribed ribozymes as shown in (B).
Our proposal that all the exported tRNA-Rz had unprocessed 5′ and 3′ ends was confirmed. We then evaluated for comparison the catalytic activity of the cytoplasmic localizing ribozyme and of the nuclear localizing ribozyme. At first, we measured cleavage activities of tRNAVal-HIV Rz2 and tBR-Rz in vitro. Both tRNAVal-HIV Rz2 and tBR-Rz had an identical ribozyme sequence (except for the stem–loop II region) targeting to the identical site on HIV-1 LTR mRNA. However, their predicted secondary structures were quite different (compare Fig. 1B and C), because of the different linker sequence that was inserted between the tRNA sequence and the ribozyme sequence, as shown in Figure 1. Typical gel images of the cleavage reactions mediated by either tRNAVal-HIV Rz2 or tBR-Rz are shown in Figure 6A. Calculated apparent rate constants (kobs) for both ribozymes were very similar, indicating that both tRNA-Rz had almost the same cleavage activities in vitro.
Figure 6.

Comparison of in vitro and intracellular activities of the nuclear- or cytoplasmic-localizing ribozyme in HeLa cells. (A) Activities of the tRNAVal HIV Rz2 and tBR-Rz in vitro. The typical gel image of time courses of the cleavage reactions and the calculated kobs values for both ribozymes are shown. (B) Activities of the tRNAVal-HIV Rz2 and tBR-Rz in vivo. Although the tRNAVal HIV Rz2 was efficiently transported into the cytoplasm, the tBR-Rz remained in the nucleus. The results shown are the averages of results from four sets of experiments.
We then compared the activities of these ribozymes in mammalian cells. We used each tRNA-ribozyme expression plasmid and a target gene-expressing plasmid, which encoded a chimeric target HIV-1 LTR sequence-luciferase gene (pGV-V1), to co-transfect HeLa cells. After transient expression of both genes in each cell lysate, we estimated the intracellular activity of each tRNA-driven ribozyme by measuring the luciferase activity. We also confirmed by northern blotting analysis, using the respective probes described in Figure 2, that the level of expression of each ribozyme was nearly identical for both tRNAVal-HIV Rz2-expressing and tBR-Rz-expressing cells (data not shown). The luciferase activity recorded when we used the pGV-V1 and pUC 19 was taken as 100% (Fig. 6B). Figure 6B shows intercellular activities of tRNAVal-HIV Rz2 and tBR-Rz. Importantly, the intercellular activity of the ribozyme that was transported into the cytoplasm (tRNAVal-HIV Rz2) was significantly higher than that of the ribozyme that remained in the nucleus (Fig. 6B).
DISCUSSION
While some tRNA-based ribozymes do not work well in cells, some tRNA-based hammerhead ribozymes are extremely active in vivo (25–27,29–37). Many successful attempts at using tRNA-based ribozymes for the suppression of gene expression have been reported. However, the efficacy of ribozymes in vitro does not necessarily correlate with their functional activity in vivo (35). When a ribozyme is generated within a cell by transcription from an expression vector, various factors, such as the transcription rate, the stability and localization of the transcript and the cleavage activity, are likely to influence the functional activity of the ribozyme in vivo. Since such factors depend, to a large part, on the activities of cellular proteins, it is important that ribozyme’s characteristics should be appropriate for optimal functioning in the intracellular environment (54–58).
Our recent data (32–37) suggest that, among several parameters that determine the intracellular activity of tRNA-based ribozymes, the stability and localization in vivo of the transcribed ribozyme are more important than their intrinsic chemical cleavage activity in vitro. As long as we depend on the tRNA-based expression system, it is relatively easy to predict, by computer folding, the secondary structure of the entire tRNAVal transcript and the accessibility of each recognition arm and it became clear that the difference between an effective and an ineffective ribozyme in cells depended on secondary structure, at least to some extent. Effective ribozymes maintained the cloverleaf structure, whereas less effective ribozymes did not (32–37).
These observations suggest that the transporter of tRNA-Rz might recognize the higher-order structure of the tRNA rather than any specific sequence. We examined the kinetics of export of tRNA-Rz by microinjecting fluorescently labeled tRNA-Rz into the nucleus of HeLa cells. A significant fraction of the tRNA-Rz that had been injected into the nucleus was exported to the cytoplasm within a few minutes (Fig. 3B). Furthermore, when an excess amount of the unlabeled wild-type tRNAVal was injected simultaneously into HeLa cells, the wild-type tRNAVal apparently competed with the FITC-labeled tRNA-Rz for the nucleocytoplasmic transport and significantly inhibited the export of the FITC-labeled tRNA-Rz, indicating that tRNA-Rz indeed share the same transport pathway with wild-type tRNAVal (Fig. 3B).
We confirmed that the tRNA-Rz isolated from the cytoplasmic fraction were of the expected size by northern blotting analysis, by the ribozyme activity of the cytoplasmic RNA extracts and also by sequencing of the cytoplasmic tRNA-Rz. Therefore, there is no doubt that various tRNA-Rz can be exported to the cytoplasm in mammalian cells, without being processed at the 5′ and 3′ ends, in their immature forms (with Rz attached) rather than as trimmed and matured forms. This transport of tRNA-Rz is important in maintaining the activity of tRNA-driven ribozymes in vivo (Fig. 6B).
Another important parameter that determines the intracellular activity of tRNA-Rz is its stability in vivo, which can be estimated by measuring its steady-state levels by northern blotting analysis (33–36). When we examined the steady-state levels of tBR-Rz, which tends to accumulate in the nucleus, and of tRNAVal-HIV Rz2, which is transported efficiently to the cytoplasm, they had similar levels of transcripts, indicating that their intracellular stability is similar (35). Nevertheless, tRNAVal-HIV Rz2 was significantly more active than tBR-Rz in vivo (Fig. 6B).
Therefore, as demonstrated in this report, depending on the localization, the activity of the ribozyme changed significantly, indicating the importance of co-localization of a ribozyme with its target mRNA in the cytoplasm. This conclusion is also valid for other known ribozymes, such as hairpin and HDV ribozymes (57). Taken together, our results suggest that mature mRNAs in the cytoplasm are more accessible to ribozymes than pre-mRNAs in the nucleus. Therefore, it should be very useful to be able to design transportable tRNA-derivatives that have high stability and high levels of expression in mammalian cells. The co-localization in the cytoplasm is an important determinant of high-level ribozyme activity not only in cultured cells (28,32–36) but also in animal models (37).
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Professor David Engelke of the Department of Biological Chemistry, University of Michigan, for providing the protocol for in situ hybridization. This research was supported by various grants from the Ministry of Economy, Trade and Industry (METI) of Japan and also by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. T.K. and M.W. are recipients of research fellowships for young scientists from the Japan Society for the Promotion of Science.
References
- 1.Uhlenbeck O.C. (1987) A small catalytic oligoribonucleotide. Nature, 328, 596–600. [DOI] [PubMed] [Google Scholar]
- 2.Haseloff J. and Gerlach,W.L. (1988) Simple RNA enzymes with new and highly specific endoribonuclease activities. Nature, 334, 585–591. [DOI] [PubMed] [Google Scholar]
- 3.Symons R.H. (1989) Self-cleavage of RNA in the replication of small pathogens of plants and animals. Trends Biochem. Sci., 14, 445–450. [DOI] [PubMed] [Google Scholar]
- 4.Pyle A.M. (1993) Ribozymes: a distinct class of metalloenzymes. Science, 261, 709–714. [DOI] [PubMed] [Google Scholar]
- 5.Sawata S., Komiyama,M. and Taira,K. (1995) Kinetic evidence based on solvent isotope effects for the nonexistence of a proton-transfer process in reactions catalyzed by a hammerhead ribozyme: implications to the double-metal-ion mechanism of catalysis. J. Am. Chem. Soc., 117, 2357–2358. [Google Scholar]
- 6.Amontov S.V. and Taira,K. (1996) Hammerhead minizymes with high cleavage activities: a dimeric structure as the active conformation of minizymes. J. Am. Chem. Soc., 118, 1624–1628. [Google Scholar]
- 7.Zhou D.-M., Usman,N., Wincott,F.E., Matulic-Adamic,J., Orita,M., Zhang,L.-H., Komiyama,M.. Kumar,P.K.R. and Taira,K. (1996) Evidence for the rate-limiting departure of the 5′-oxygen in nonenzymatic and hammerhead ribozyme-catalyzed reactions. J. Am. Chem. Soc., 118, 5862–5866. [Google Scholar]
- 8.Zhou D.-M., Kumar,P.K.R., Zhang,L.-H. and Taira,K. (1996) Ribozyme mechanism revisited: evidence against direct coordination of a Mg2+ ion with the pro-R oxygen of the scissile phosphate in the transition state of a hammerhead ribozyme-catalyzed reaction. J. Am. Chem. Soc., 118, 8969–8970. [Google Scholar]
- 9.Birikh K.R., Heaton,P.A. and Eckstein,F. (1997) The structure, function and application of the hammerhead ribozyme. Eur. J. Biochem., 245, 1–16. [DOI] [PubMed] [Google Scholar]
- 10.Warashina M., Takagi,Y., Sawata,S., Zhou,D.-M., Kuwabara,T. and Taira,K. (1997) Entropically driven enhancement of cleavage activity of a DNA-armed hammerhead ribozyme: mechanism of action of hammerhead ribozymes. J. Org. Chem., 62, 9138– 9147. [Google Scholar]
- 11.Zhou D.-M. and Taira,K. (1998) The hydrolysis of RNA: from theoretical calculations to the hammerhead ribozyme-mediated cleavage of RNA. Chem. Rev., 98, 991–1026. [DOI] [PubMed] [Google Scholar]
- 12.Lilley D.M.J. (1999) Structure, folding and catalysis of the small nucleolytic ribozymes. Curr. Opin. Struct. Biol., 9, 330–338. [DOI] [PubMed] [Google Scholar]
- 13.Wang S., Karbstein,K., Peracchi,A., Beigelman,L. and Herschlag,D. (1999) Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate. Biochemistry, 38, 14363–14378. [DOI] [PubMed] [Google Scholar]
- 14.Nakamatsu Y., Warashina,M., Kuwabara,T., Tanaka,Y., Yoshinari,K. and Taira,K. (2000) Significant activity of a modified ribozyme with N7-deazaguanine at G10.1: implication to the double-metal-ion mechanism of catalysis in reactions catalyzed by hammerhead ribozymes. Genes Cells, 5, 603–612. [DOI] [PubMed] [Google Scholar]
- 15.Yoshinari K. and Taira,K. (2000) A further investigation and reappraisal of the thio effect in the cleavage reaction catalyzed by a hammerhead ribozyme. Nucleic Acids Res., 28, 1730–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takagi Y., Warashina,M., Stec,W.J., Yoshinari,K. and Taira,K. (2001) Recent advances in the elucidation of the mechanisms of action of ribozymes. Nucleic Acids Res., 29, 1815–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossi J.J. and Sarver,N. (1990) RNA enzymes (ribozymes) as antiviral therapeutic agents. Trends Biotechnol., 8, 179–183. [DOI] [PubMed] [Google Scholar]
- 18.Sarver N., Cantin,E.M., Chang,P.S., Zaida,J.A., Ladne,P.A., Stephenes,D.A. and Rossi,J.J. (1990) Ribozymes as potential anti-HIV-1 therapeutic agents. Science, 247, 1222–1225. [DOI] [PubMed] [Google Scholar]
- 19.Erickson R.P. and Izant,J. (1992) Gene Regulation: Biology of Antisense RNA and DNA. Raven Press, New York, NY.
- 20.Murray J.A.H. (1992) Antisense RNA and DNA. Wiley-Liss Inc., New York, NY.
- 21.Rossi J.J. (1995) Controlled, targeted, intracellular expression of ribozymes: progress and problems. Trends Biotechnol., 13, 301–306. [DOI] [PubMed] [Google Scholar]
- 22.Eckstein F. and Lilley,D.M.J. (1996) Nucleic Acids and Molecular Biology: Catalytic RNA, vol. 10. Springer-Verlag, Berlin, Germany.
- 23.Turner P.C. (1997) Methods in Molecular Biology: Ribozyme Protocols, 74. Humana Press, Totowa, NJ.
- 24.Krupp G. and Gaur,R.K. (2000) Ribozyme, Biochemistry and Biotechnology. Eaton Publishing, MA.
- 25.Cotten M. and Birnstiel,M. (1989) Ribozyme mediated destruction of RNA in vivo. EMBO J., 8, 3861–3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu M., Ojwang,J.O., Yamada,O., Hampel,A., Rappaport,J., Looney,D. and Wong-Staal,F. (1993) A hairpin ribozyme inhibits expression of diverse strains of human immunodeficiency virus type 1. Proc. Natl Acad. Sci. USA, 90, 6340–6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson J.D., Ayers,D.F., Malmstorm,T.A., Mackenzie,T.L., Ganousis,L., Chowrira,B.M., Couture,L. and Stinchcomb,D.T. (1995) Improved accumulation and activity of ribozymes expressed from a tRNA-based RNA polymerase III promoter. Nucleic Acids Res., 23, 2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawasaki H., Ohkawa,J., Tanishige,N., Yoshinari,K., Murata,T., Yokoyama,K.K. and Taira,K. (1996) Selection of the best target site for ribozyme-mediated cleavage within a fusion gene for adenovirus E1A-associated 300 kDa protein (p300) and luciferase. Nucleic Acids Res., 24, 3010–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertrand E., Castanotto,D., Zhou,C., Carbonnelle,C., Lee,N.S., Good,P., Chatterjee,S., Grange,T., Pictet,R., Kohn,D., Engelke,D. and Rossi,J.J. (1997) The expression cassette determines the functional activity of ribozymes in mammalian cells by controlling their intracellular localization. RNA, 3, 75–88. [PMC free article] [PubMed] [Google Scholar]
- 30.Good P.D., Krikos,A.J., Li,S.X., Lee,N.S., Gilver,L., Ellington,A., Zaia,J.A., Rossi,J.J. and Engelke,D.R. (1997) Expression of small, therapeutic RNAs in human cell nuclei. Gene Ther., 4, 45–54. [DOI] [PubMed] [Google Scholar]
- 31.Du Z., Ricord,C., Inverardi,L., Podack,E. and Pastori,R.L. (1998) Efficient ex vivo inhibition of perforin and Fas ligand expression by chimeric tRNA-hammerhead ribozymes. Hum. Gene Ther., 9, 1551–1560. [DOI] [PubMed] [Google Scholar]
- 32.Kawasaki H., Ecker,R., Yao,T.-P., Taira,K., Chiu,R., Livingston,D.M. and Yokoyama,K.K. (1998) Distinct roles of the co-activators p300 and CBP in retinoic- acid-induced F9-cell differentiation. Nature, 393, 284–289. [DOI] [PubMed] [Google Scholar]
- 33.Kuwabara T., Warashina,M., Orita,M., Koseki,S., Ohkawa,J. and Taira,K. (1998) Formation in vitro and in cells of a catalytically active dimer by tRNAVal-driven short ribozymes. Nat. Biotechnol., 16, 961–965. [DOI] [PubMed] [Google Scholar]
- 34.Kuwabara T., Warashina,M., Tanabe,T., Tani,K., Asano,S. and Taira,K. (1998) A novel allosterically trans-activated ribozyme, the maxizyme, with exceptional specificity in vitro and in vivo. Mol. Cell, 2, 617–627. [DOI] [PubMed] [Google Scholar]
- 35.Koseki S., Tanabe,T., Tani,K., Asano,S., Shioda,T., Nagai,Y., Shimada,T., Ohkawa,J. and Taira,K. (1999) Factors governing the activity in vivo of ribozymes transcribed by RNA polymerase III. J. Virol., 73, 1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuwabara T., Warashina,M., Nakayama,A., Ohkawa,J. and Taira,K. (1999) Novel tRNAVal-heterodimeric maxizymes with high potential as gene-inactivating agents: simultaneous cleavage at two sites in HIV-1 tat mRNA in cultured cells. Proc. Natl Acad. Sci. USA, 96, 1886–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanabe T., Kuwabara,T., Warashina,M., Tani,K., Taira,K. and Asano,S. (2000) Oncogene inactivation in a mouse model: tissue invasion by leukaemic cells is stalled by loading them with a designer ribozyme. Nature, 406, 473–474. [DOI] [PubMed] [Google Scholar]
- 38.Adeniyi-Jones S., Romeo,P.H. and Zasloff,M. (1984) Generation of long read through transcripts in vivo and in vitro by deletion of 3′ termination and processing sequences in the human tRNAiMet gene. Nucleic Acids Res., 12, 1101–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boelens W., Palacios,I. and Mattaj,I.W. (1995) Nuclear retention of RNA as a mechanism for localization. RNA, 1, 273–283. [PMC free article] [PubMed] [Google Scholar]
- 40.Arts G.-J., Fornerod,M. and Mattaj,I.W. (1998) Identification of a nuclear export receptor for tRNA. Curr. Biol., 8, 305–314. [DOI] [PubMed] [Google Scholar]
- 41.Arts G.-J., Kuersten,S., Romby,P., Ehresmann,B. and Mattaj,I.W. (1998) The role of exportin-t in selective nuclear export of mature tRNAs. EMBO J., 17, 7430–7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hellmuth K., Lau,D.M., Bischoff,F.R., Künzler,M., Hurt,E.D. and Simos,G. (1998) Yeast Los1p has properties of an exportin-like nucleocytoplasmic transport factor for tRNA. Mol. Cell. Biol., 18, 6374–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hopper A.K. (1998) Nuclear functions charge ahead. Science, 282, 2003–2004. [DOI] [PubMed] [Google Scholar]
- 44.Kutay U., Lipowsky,G., Izaurralde,E., Bischoff,F.R., Schwarzmaier,P., Hartmann,E. and Görlich,D. (1998) Identification of a tRNA-specific nuclear export receptor. Mol. Cell, 1, 359–369. [DOI] [PubMed] [Google Scholar]
- 45.Lund E. and Dahlberg,J.E. (1998) Proofreading and aminoacylation of tRNAs before export from the nucleus. Science, 282, 2082–2085. [DOI] [PubMed] [Google Scholar]
- 46.Sarkar S. and Hopper,A.K. (1998) tRNA nuclear export in Saccharomyces cerevisiae: in situ hybridization analysis. Mol. Biol. Cell, 9, 3041–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lipowsky G., Bischoff,F.R., Izaurralde,E., Kutay,U., Scharfer,S., Gross,H.J., Beier,H. and Görlich,D. (1999) Coordination of tRNA nuclear export with processing of tRNA. RNA, 5, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simos G. and Hurt,E. (1999) Transfer RNA biogenesis: a visa to leave the nucleus? Curr. Biol., 9, 238–241. [DOI] [PubMed] [Google Scholar]
- 49.Wolin S.L. and Matera,A.G. (1999) The trials and travels of tRNA. Genes Dev., 13, 1–10. [DOI] [PubMed] [Google Scholar]
- 50.Li J., Tang,H., Mullen,T.-M., Westberg,S., Reddy,T., Rose,D. and Wong-Staal,F. (1999) A role for RNA helicase A in post-transcriptional regulation of HIV type A. Proc. Natl Acad. Sci. USA, 96, 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang Y. and Cullen,B.R. (1999) The human Tap protein is a nuclear mRNA export factor that contains novel RNA-binding and nucleocytoplasmic transport sequences. Genes Dev., 13, 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adachi A., Gendelman,H.E., Koening,A., Folks,T., Willey,R., Rabson,A. and Martin,M.A. (1986) Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol., 59, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sullenger B.A. and Cech,T.R. (1993) Tethering ribozymes to a retroviral packaging signal for destruction of viral RNA. Science, 262, 1566–1569. [DOI] [PubMed] [Google Scholar]
- 54.Kuwabara T., Warashina,M. and Taira,K. (2000) Allosterically controllable ribozymes with biosensor functions. Curr. Opin. Chem. Biol., 4, 669–677. [DOI] [PubMed] [Google Scholar]
- 55.Kuwabara T., Warashina,M. and Taira,K. (2000) Allosterically controllable maxizymes cleave mRNA with high efficiency and specificity. Trends Biotechnol., 18, 462–468. [DOI] [PubMed] [Google Scholar]
- 56.Warashina M., Kuwabara,T. and Taira,K. (2000) Working at the cutting edge: the creation of allosteric ribozymes. Structure, 8, 207–212. [DOI] [PubMed] [Google Scholar]
- 57.Kato Y., Kuwabara,T., Warashina,M., Toda,H. and Taira,K. (2001) Relationships between the activities in vitro and in vivo of various kinds of ribozyme and their intracellular localization in mammalian cells. J. Biol. Chem., 276, 15378–15385. [DOI] [PubMed] [Google Scholar]
- 58.Warashina M., Kuwabara,T., Kato,Y., Sano,M. and Taira,K. (2001) RNA–protein hybrid ribozymes that efficiently cleave any mRNA independently of the structure of the target RNA. Proc. Natl Acad. Sci. USA, 98, 5572–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]