

Abstract
The aging process and several age-related neurodegenerative disorders have been linked to elevated levels of DNA damage induced by ROS and deficiency in DNA repair mechanisms. DNA damage induced by ROS is a byproduct of cellular respiration and accumulation of damage over time, is a fundamental aspect of a main theory of aging. Mitochondria have a pivotal role in generating cellular oxidative stress, and mitochondrial dysfunction has been associated with several diseases. DNA base excision repair is considered the major pathway for repair of oxidized bases in DNA both in the nuclei and in mitochondria, and in neurons this mechanism is particularly important because non-diving cells have limited back-up DNA repair mechanisms. An association between elevated oxidative stress and a decrease in BER is strongly related to the aging process and has special relevance in age-related neurodegenerative diseases. Here, we review the role of DNA repair in aging, focusing on the implications of the DNA base excision repair pathways and how alterations in expression of these DNA repair proteins are related to the aging process and to age-related neurodegenerative diseases.
Keywords: Aging, DNA repair, oxidative damage, oxidative stress, neurodegeneration, Alzheimers disease
1) Introduction
The aging process can be described as a progressive impairment of organismal function with increasing vulnerability to environmental changes and heightened risk of disease and mortality (Kirkwood and Austad 2000, Kirkwood 2005). Significant effort has been undertaken to understanding the molecular basis of aging. However, many key aspects of the process remain unclear. Considerable circumstantial evidence show that genomic instability has a central role in age-related decline, potentially due to the accumulation of somatic DNA damage in aged cells and this evidence will be further discussed in this review.
Genomic instability occurs upon exposure to exogenous and endogenous DNA damaging agents that can generate chemical modifications in DNA (Pages and Fuchs 2002). An important factor related to the age-dependent exacerbation of genomic instability is chronic, elevated oxidative stress. Oxidative stress is a result of the loss of equilibrium between reactive oxygen species (ROS) formation and the antioxidant defense mechanisms within the cell.
Several mechanisms of protection are available to cells subjected to the deleterious action of ROS depending on the nature of ROS and its subcellular site of generation. The cell can elicit specific antioxidants, in the form of enzymes that sequester ROS before the free radicals can damage other molecules (Lu and Holmgren 2014). In mammalian cells, the initial defense against ROS is mediated by antioxidants which include the thioredoxin (NADPH, thioredoxin reductase and thioredoxin), and glutathione-glutaredoxin system, (NADPH, glutathione reductase, GSH, Glutaredoxin) which make up the two major thiol-dependent antioxidant systems in the cell (Lu and Holmgren 2014). Cells that are not able to maintain this homeostatic ROS equilibrium are susceptible to deleterious effects of DNA damage induced by ROS which can include a multitude of DNA modifications including base oxidation and DNA strand breaks (Dizdaroglu 1991). There are more than 100 base modifications caused by DNA oxidation (Dizdaroglu 1991, Croteau and Bohr 1997) which have the potential to cause mutagenic and cytotoxic lesions (Lindahl 1993).
Mechanisms to repair DNA damage are numerous and overlapping. In mammalian cells these DNA repair pathways include: direct repair, base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination repair (HR) and non-homologous end-joining repair (NHEJ) (Bjelland and Seeberg 2003, Sancar, Lindsey-Boltz et al. 2004). The failure of these DNA repair mechanisms has been associated with several human diseases. Table 1 shows central nervous system diseases associated with dysfunctional DNA repair. The following is a discussion of the role of these DNA repair mechanisms in age-related diseases.
Table 1.
Diseases of the CNS associated with DNA instability
| Diseases of the CNS | Defective gene | Pathway |
|---|---|---|
| Alzheimer’s disease | MRN complex/NHEJ/BER/mtDNA mutations | |
| Huntington disease | FEN1 | BER*/mtDNA deletion |
| Amyotrophic lateral sclerosis | BER*/mtDNA deletion | |
| Parkinson disease | BER/mtDNA deletion | |
| Hereditary spastic paraplegia | mtDNA deletion | |
| Friedreich ataxia | mtDNA deletion | |
| Ataxia with oculomotor apraxia 1 | Aprataxin | BER/SSBR |
| Spinocerebellar ataxia with axonal neuropathy | TDP1 | BER/SSBR |
| Xeroderma pigmentosum | XP | NER |
| Trichothiodystrophy | XP | NER |
| Cockayne syndrome | CS | BER/NER |
| Ataxia telangectasia | ATM/MRE1 | Double strand break recognition |
| Nijmegan breakage syndrome | NBS1 | Double strand break recognition |
| Fanconi anemia | FA | |
| Werner syndrome | WRN | Helicase |
| Bloom syndrome | BLM | Helicase |
| Ataxia with oculomotor apraxia2 | Senataxin | Helicase |
There are two major direct repair mechanisms, the repair of UV-induced pyrimidine dimers mediated by photolyase and repair of O6-methylguanine lesions by methylguanine DNA methyltransferase (MGMT). MGMT is present in human cells (Sancar, Lindsey-Boltz et al. 2004) and is a suicide protein that recognizes the damage and acts by transferring the methyl group to an active cysteine site in an irreversible reaction (Pegg 1990, Daniels and Tainer 2000). It was observed that a decrease in MGMT activity occurs during the aging process (Anisimov 2001). Further, the overexpression of this protein correlates with an increased life span in mice (Qin, Zhang et al. 2000).
There is extensive evidence supporting that MMR has importance in the aging progress and that levels of MMR proteins decrease with age (Ben Yehuda, Globerson et al. 2000, Krichevsky, Pawelec et al. 2004, Neri, Gardini et al. 2005). This DNA repair pathway plays a role in removing unpaired bases resulting from replication error (Jiricny 2006), recombination imperfections and deamination of 5-methyl-cytosine. It has been suggested that MMR plays a role in oxidized DNA damage repair but the mechanisms involved are not yet clear (Skinner and Turker 2005). It has been reported that the induction of base-pair substitutions caused by oxidative stress may further oxidized DNA promoting mutagenesis (Diaz-Llera, Podlutsky et al. 2000). Thus, decline or deficiency in MMR can lead to heightened susceptibility to disease.
NER is a DNA repair pathway that involves multiple protein complexes. The specific role of the pathway is to remove bulky DNA lesions. This damage includes chemical compounds and covalent linkages between adjacent pyrimidines caused by exposure to UV light (Hanawalt 2002). The role of NER in the aging process has been extensively studied and there is significant evidence that supports the function of this pathway in longevity (Gorbunova, Seluanov et al. 2007). The importance of the pathway is seen from mutations in NER genes leading to rare human diseases such as Cockayne’s Syndrome (CS) and Xeroderma Pigmentosum (XP) (see Table 1). These diseases have premature aging features including extensive neurological symptoms (Menck and Munford 2014). Despite these diseases having progeria symptoms, there is not a consensus whether NER capacity decreases in normal human aging. For example, it was reported that cyclobutane pyrimidine dimers (CPD) induced by UV radiation were reduced in senescent skin fibroblast but not in young cells when cultured in media with serum. However, under culture conditions where cells are serum-starved there is a reduction in CPD removal in young cells (Boyle, Kill et al. 2005), thus the authors suggested that the results are related to age-related decline in NER capacity in proliferative cells. However, other studied have shown that aged human fibroblasts and trabecular osteoblasts also treated with UV radiation were as efficient as young cells in the removal of CPD lesions (Christiansen, Stevnsner et al. 2000). These conflicting results may be explained by different cell culture conditions and cell cycle distribution along the treatment (Al-Baker, Oshin et al. 2005). Most studies seem to converge on the notion that NER declines with age.
Double strand breaks (DSB) are lethal DNA lesions in a replicating cell and are repaired by HR or NHEJ (Sancar, Lindsey-Boltz et al. 2004). When DSBs are not repaired, chromosome segments can become displaced leading to chromosome rearrangements and a drastic reduction in cell survival (Gorbunova, Seluanov et al. 2007). The role of DSB repair in the aging process has also been extensively studied and disruption of genes involved in this pathway leads to premature aging phenotypes such as those observed in Werner syndrome and Ataxia telangiectasia (Gorbunova and Seluanov 2005) (see Table 1). Werner syndrome is caused by a mutation in a RecQ DNA helicase/nuclease (WRN). Werner patients show increased risk for atherosclerosis, osteoporosis, diabetes, cancer, and also present with early characteristics of the normal aging process including loss of hair and cataracts (Epstein, Martin et al. 1966, Goto 1997). WRN interacts with many common components of DSB repair (Bohr 2002, Croteau, Popuri et al. 2014) and also is a substrate for DNA-PK, a protein that has a central role in NHEJ (Yannone, Roy et al. 2001, Karmakar, Piotrowski et al. 2002). Several reports show that dysfunction in DSB repair pathways during aging correlates with an increase in genomic rearrangements (Dolle, Giese et al. 1997, Vijg and Dolle 2002). In support, knockout of NHEJ genes causes accelerated aging as observed in DNA-PK deficient mice that have shorter lifespan, loss of bone density and early onset lymphomas (Espejel, Martin et al. 2004).
While each of the pathways described above have commanding roles in the repair of a wide range of lesions, it is DNA damage induced by ROS that is most commonly attributed to the advancement of aging pathology. Base excision repair (BER) is considered the dominant mechanism responsible for averting deleterious effects of DNA damage modifications caused by ROS in both mitochondrial and nuclear DNA (nDNA) (Mitra, Boldogh et al. 2001, Wallace 2002). This review will focus on the role of BER age-related neurodegenerative diseases.
2) Base Excision Repair (BER)
BER is an indispensable mechanism for repair of oxidized DNA bases. All aerobic organisms are continually exposed to endogenous ROS formation as a by-product of respiration hence proteins of the BER pathway are highly conserved. BER preferentially repairs non-bulky, non-helix distorting lesions, removing and repairing damage sustained by deamination, alkylation or the oxidation process (Sancar, Lindsey-Boltz et al. 2004). The BER mechanism can be broken into general steps that include: lesion recognition and excision by a DNA glycosylase forming an intermediate abasic site; subsequent incision of the resulting abasic site by an AP-endonuclease (APE1) or AP-lyase action; and phosphodiesterase activity to convert the 5′-dRP fragment; gap filling by a DNA polymerase and finally a DNA ligase seals the nick and restores the continuity of the phosphodiester backbone (Dianov, Bischoff et al. 1998, Wilson and Kunkel 2000, Kim and Wilson 2012).
There are two major BER sub-pathways in mammalian cells, long and short patch BER (LP-BER, SP-BER respectively) (Figure 1). Short-patch BER employs a single mechanism of nucleotide replacement using the core proteins: APE1, DNA Polymerase β (Pol β), DNA ligase III (LIG3) and X-ray repair cross-complementing protein 1 (XRCC1). In contrast, LP-BER uses APE1 to make a 5′ incision at the AP-site and a combination of DNA Polymerases β/δ/ε, proliferating cell nuclear antigen (PCNA) and flap endonuclease (FEN1) displaces the strand 3′ to the nick producing a flap of 2–10 nucleotides, and then FEN1 endonuclease is responsible for cutting the junction of the single to double strand transition. Pol δ/ε in conjunction with PCNA synthetizes the oligonucleotide and ligation is performed by Ligase I (LIG1) (Frosina, Fortini et al. 1996, Klungland and Lindahl 1997, Sancar, Lindsey-Boltz et al. 2004). Interestingly, Pol β can also have an essential role in LP-BER, since in the absence of replicative polymerases it becomes responsible for strand displacement synthesis (Dianov, Prasad et al. 1999), in combination with the nuclease FEN1 (Prasad, Dianov et al. 2000, Podlutsky, Dianova et al. 2001, Liu, Beard et al. 2005). In post-mitotic tissue, LP-BER utilizes a PCNA independent sub-pathway catalyzed by Pol β/FEN1 that occurs via a “hit and run” mechanism (Liu, Beard et al. 2005). The PCNA-independent LP-BER is important in neurons. The 5′-blocked termini may be refractory to Pol β dRP lyase activity and requires the action of FEN1. Hence, the lack of replicative proteins in post-mitotic cells may result in a higher proportion of alternative PCNA-independent LP-BER repair events (Wei and Englander 2008). It was observed that neurons rely heavily on this PCNA independent pathway for repair of oxidized DNA damage with damaged termini, however the lesion processing is comparatively slow (Wei and Englander 2008). Single strand break repair (SSBR) is considered a BER sub-pathway, an attenuated version of SP-BER. The pathways include specialized termini “clean-up” proteins many of which have been associated with neurodegenerative diseases. SSBR and SP-BER represents the major BER pathways in the brains however residual LP-BER activity is present in neuronal cells (Wei and Englander 2008, Akbari, Pena-Diaz et al. 2009).
Figure 1. Base Excision Repair Pathway.

BER is responsible for the repair of cellular oxidative DNA damage. Depending on the lesion, the process begins with the action of one of many glycosylases (listed left not comprehensive), creating an intermediate AP site. This site is processed by the major human AP endonuclease, APE1. BER then branches into two major sub-pathways, Short and Long patch BER (SP-BER and LP-BER respectively). SP-BER comprises of four core BER proteins; APE1, Polβ (incorporation)and XRCC1/Ligase III (ligation). Knock down of any of the core SP-BER genes results in embryonic lethality. Alternatively, replicating cells have access to a second robust LP-BER pathway. In comparison to SP-BER, the proteins involved in LP-BER are primarily attributed to replication but are able to carry out repair function giving the cells alternate overlapping repair response.
When comparing differentiated and undifferentiated cells, Montecucco et al. observed a decrease in DNA ligase I mRNA levels in non-proliferating cells relative to proliferative cells in a variety of cell types including human lymphocytes, rat neurons and mouse fibroblasts (Montecucco, Biamonti et al. 1992). In another study it was observed that mouse differentiated myotubes showed increases in oxidative damage markers and a decrease in BER activity compared to proliferating myoblast (Narciso, Fortini et al. 2007). Akibari et al. demonstrated that only proliferating lymphocytes (stimulated by phytohemaglutin A) had capacity for LP-BER compared with human non-proliferating lymphocytes (Akbari, Pena-Diaz et al. 2009). They also saw differences in BER pathways for keratinocyte cell lines (HaCaT cells) in proliferating versus non-proliferating conditions. These results and others establish that LP-BER is associated with replicative synthesis, this aspect has direct ramifications for post mitotic, highly differentiated cells such as neurons.
Sykora et al. used mitotic and post-mitotic human neuroblastoma cells to clarify the changes in BER pathways in neurons. Their study showed that the modulation of BER depends on the differentiation status of the cell (Figure 2). It was observed that post-mitotic neurons were more susceptible to oxidative stress than mitotic cells despite no difference when cells were treated with an alkylating agent. A decrease in LP-BER proteins, FEN1, PCNA, Polε and LIG1, was observed resulting in an overall reduced LP-BER capacity after neuronal differentiation (Sykora, Yang et al. 2013). Therefore, oxidative stress-induced DNA damage may be better tolerated in replicative cells than in non-proliferative cells due to a more robust LP-BER activity able to modulate repair of substrates more efficiently.
Figure 2. BER is dependent on cellular proliferative status.
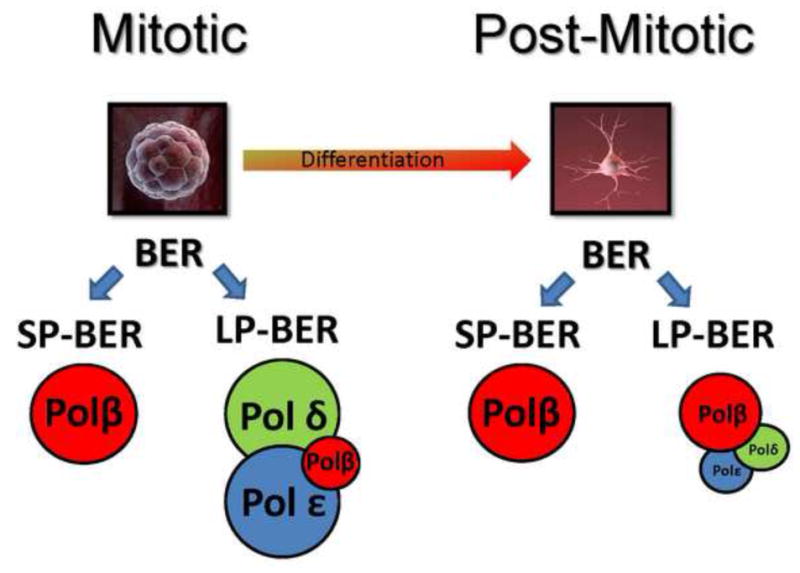
Mitotic/undifferentiated cells have a BER system that uses three different DNA polymerases providing a robust repair response with substrate overlap. As an example, a neuroblast goes through the process of differentiation to become a terminally differentiate neuron. The neuron has a significantly changed BER system compared to the neuroblast that relies heavily on DNA polymerase β with replicative polymerase δ/ε strongly attenuated. This may leave the neuron more vulnerable to certain forms of DNA damage preferentially repaired by LP-BER in replicating systems. Also see Sykora et.al. 2013, Neurobiology of aging.
3) Mitochondrial DNA damage, repair, and aging
Mitochondria are membrane-enclosed organelles that are essential in cellular homeostasis. Due to their central role in energy metabolism, they are also the most abundant source of endogenous ROS (Kowaltowski, de Souza-Pinto et al. 2009). Mitochondria have their own genome, an artifact from the organelles proteo-bacteria origins (Lang, Burger et al. 1997, Gray, Burger et al. 2001). ATP generation in mitochondria depends on the transport of high-energy electrons using the electron transport chain in the inner mitochondrial membrane. Electron leak from this process can react with O2 generating ROS. The proximity of the ROS formation exposes mitochondrial DNA (mtDNA) to higher levels of oxidative stress. DNA damage induced by ROS in mtDNA can cause dysfunction in mitochondrial oxidative phosphorylation (OXPHOS), leading to an induction of ROS, and provoking more mtDNA damage, beginning a vicious cycle (Sanz, Caro et al. 2006) (Figure 3, Point 4). The levels of damage induced by ROS in mtDNA may be higher (Richter, Park et al. 1988, Hudson, Hogue et al. 1998), more persistent and more extensive (Yakes and Van Houten 1997) than in nDNA in human cells.
Figure 3. Mitochondrial dysfunction can trigger a positive feedback loop.

The mitochondrial theory of aging states that oxidation can damage DNA leading to less efficient oxidative phosphorylation thus creating more ROS, further damaging DNA. (1) ROS is not only a damaging agent but an important signaling molecule. Even under normal mitochondrial function ROS is released but anti-oxidant enzymes are able to sequester the free radicals before DNA damage occurs. (2) Mitochondrial dysfunction can occur when the amount of ROS produced in the mitochondria exceeds the anti-oxidant capacity of the cell. The proximity of mtDNA to ROS production means that it is more likely to be damaged (3) With persistent elevation of ROS the nDNA can also come under oxidative attack. Since the majority of mitochondrial genes are nuclear encoded, this can accelerate mitochondrial deterioration leading to the (4) positive feedback cycle which is the foundation of the mitochondrial theory of aging, ROS formation exposes mtDNA to increased oxidative stress and the subsequently oxidized bases in mtDNA causes dysfunction in mitochondrial oxidative phosphorylation that induces ROS formation leading to more mtDNA damage.
In 1956, Harman suggested an association between free radical-dependent cellular damage accumulation and the aging process by introducing the mitochondrial free radical theory of aging (Harman 1956, Harman 1981). This theory assumes a connection between mitochondrial OXPHOS, oxygen free radical induction, and cellular damage, leading to the age-related deterioration of organisms (Sohal and Brunk 1992). It has received significant support and is now cited as a major theory of aging (Harman 1981). However, some research has questioned Harman’s theory (Stuart, Maddalena et al. 2014). For example, it has been showed that simply decreasing ROS production (Labinskyy, Csiszar et al. 2006, Lambert, Boysen et al. 2007, Brown, McClelland et al. 2009), or increasing antioxidant defense (Salway, Page et al. 2011) is not enough to increase an organism’s life-span. These results are suggestive of ROS playing a crucial role in alternate metabolic and/or signaling pathways and suggest caution when labeling ROS solely as a damaging agent. Thus, further elucidation of this theory and its contribution to the aging process is required (Stuart, Maddalena et al. 2014).
Considering the importance of nDNA repair it is perhaps not surprising that the mitochondria can also repair mtDNA using similar mechanisms to those observed in nDNA. However, there are considerable differences between DNA repair pathways in mitochondria and in the nucleus. mtDNA relies on MMR (de Souza-Pinto, Mason et al. 2009) and BER (mtBER). BER is the best characterized DNA repair process among mtDNA repair mechanisms and was the first to be identified in mitochondria (Muftuoglu, Mori et al. 2014). mtBER proteins are not encoded in the mitochondria but are found as isoforms or splice variants of the nuclear BER (nBER) proteins (Nilsen, Otterlei et al. 1997, Nishioka, Ohtsubo et al. 1999, Chattopadhyay, Wiederhold et al. 2006). Several DNA glycosylases have a mitochondrial isoform, including oxoguanine glycosylase (OGG1) and uracil-DNA glycosylase (UDG) (Svilar, Goellner et al. 2011). After excision of the damaged DNA base, APE1 is responsible for the incision of the abasic site but it is unclear whether the same form of this protein operates in both nBER and mtBER (Tell, Damante et al. 2005). DNA polymerase γ (Pol γ) is an exclusively mitochondrial polymerase responsible for repair and replication events in mitochondria (Kaguni 2004). For the nick sealing in mitochondria, an alternative LIG3 isoform is the sole enzyme and, unlike in the nucleus, the mitochondrial form is not dependent on XRCC1 (Lakshmipathy and Campbell 1999). Until recently LP-BER was believed to be present only in the nucleus, however recently LP-BER components were found in mitochondria (Akbari, Visnes et al. 2008, Liu, Qian et al. 2008, Szczesny, Tann et al. 2008). For example, FEN1 was found in mammalian mitochondrial extracts (Liu, Qian et al. 2008, Szczesny, Tann et al. 2008). Despite the obvious similarities between nBER and mtBER, mtBER does have distinct proteins. Transcription factor A-mitochondrial (TFAM) is an essential component of mitochondrial nucleoids, and participates in mtBER (Canugovi, Maynard et al. 2010). TFAM can modulate mtBER by binding DNA. TFAM preferentially binds to DNA containing 8-oxo Guanine (8-oxoG) in vitro, and lysates from TFAM knockdown cells had higher 8-oxoG incision activity but increased mtDNA damage accumulation compared to the control cells (Canugovi, Maynard et al. 2010). An age-dependent decline of mtBER activities was observed in rat cerebral cortices as well as a decrease in expression of OGG1 and Pol γ enzymes (Chen, Cao et al. 2002), and it was also reported that there was an age-dependent decrease of mtDNA glycosylases activities in five different mouse brain regions (Imam, Karahalil et al. 2006).
Mitochondrial dysfunction may also have a commanding role in Down’s syndrome (DS) or trisomy 21. This disorder has a complex phenotype characterized by developmental delay (Roizen and Patterson 2003) and symptoms of accelerated aging (Tiano and Busciglio 2011). One particularly important feature of DS is an early onset of Alzheimer’s disease (AD) by middle age (Coyle, Oster-Granite et al. 1986, Mann 1988). Interestingly, DS fibroblasts were impaired in mitochondrial DNA repair after induction of damage by the damaging oxidative agent menadione (Druzhyna, Nair et al. 1998). And, similar to other diseases associated with DNA repair deficiency, DS is also associated with elevated levels of oxidative stress and mitochondrial abnormalities including reduced mitochondrial membrane potential, reduced ATP production and decreased oxido-reductase activity (Brooksbank and Balazs 1984, Arbuzova, Hutchin et al. 2002). In DS the accelerated aging process may be related to oxidative stress and mitochondrial dysfunction as can occur in some senile neuropathies and also in the normal aging process (Tiano and Busciglio 2011). The premature aging syndrome CS is characterized by progressive neurological dysfunction, leukodystrophy, cachexia and growth retardation (Nance and Berry 1992, Koob, Laugel et al. 2010, Natale 2011). It is caused by mutations in the cockayne syndrome group B (CSB) protein (80% of the cases) or the group A protein (CSA) (20% of the cases) that participate in multiple pathways involved in genomic integrity including transcription coupled NER (Anindya, Mari et al. 2010), transcription (Le May, Mota-Fernandes et al. 2010) and BER (Stevnsner, Muftuoglu et al. 2008). CSB may also have a direct role in mtDNA maintenance (Aamann, Sorensen et al. 2010, Kamenisch, Fousteri et al. 2010, Scheibye-Knudsen, Ramamoorthy et al. 2012). In support, CSB can exert a role as an accessory factor to BER enzymes APE1 (Wong, Muftuoglu et al. 2007) and neil-endonuclease VIII-like 1 (NEIL1) (Muftuoglu, de Souza-Pinto et al. 2009), suggesting a possible additional role for CSB in mtBER. CSB deficient cells show deficient mitophagy, causing accumulation of damage in mitochondria, which can be reversed by the addition of lithium chloride and rapamycin to cell culture medium (Scheibye-Knudsen, Ramamoorthy et al. 2012).
4) BER and neurodegeneration
Aging is the strongest risk factor for most common neurodegenerative diseases including AD and Parkinson’s (PD). Age-related neurodegeneration can lead to functional or structural neuronal loss culminating in a decline in the quantity of neurons, a consequence of elevated apoptotic activation (Jeppesen, Bohr et al. 2011) but may also include autophagy and necrosis. (Yuan, Lipinski et al. 2003). Since BER is considered the major DNA repair pathway in neurons (Wei and Englander 2008), BER deficiencies have been highlighted as an important factor in neurodegenerative disorders.
Reduced DNA repair was observed in tissue of old (24-month) compared to young (4-month) mice measuring in vitro BER activity. The authors tested brain, liver, spleen and testes and observed a significantly reduced capacity of BER (50–75%) in the 24 month group (Cabelof, Raffoul et al. 2002). Moreover, the observed decline in BER activity correlates with a decrease in levels of enzymatic activity of Pol β, as well a decrease in transcript and protein levels of this DNA polymerase (Cabelof, Raffoul et al. 2002). A decrease in DNA repair activity was also observed in extracts from neurons taken from old rats (Rao, Annapurna et al. 2000, Rao, Annapurna et al. 2001), and proof-of-principle was shown by supplementing Pol β to recover this activity. Further, the ability to repair DNA damage was also measured in young (3 month) compared to old (30 month) old mice when exposed to isobaric hyperoxia, a condition that elevates ROS levels in the brain and increases apoptosis. Six hours after the exposure, an induction of APE1 was observed in the hippocampus and basal forebrain of young but not old mice (Edwards, Rassin et al. 1998). The importance of BER proteins is also noted in results from knock-out studies. The knock-out of many core BER genes results in severe defects in embryonic development resulting in early gestational lethality in mice. Human mutations in two peripheral SSBR proteins ataxia-oculomotor apraxia-1 (AOA1) and tyrosyl-DNA phosphodiesterase 1 (TDP-1) clearly show the importance of BER in human neuronal maintenance.
Ataxia oculomotor apraxia-1 (AOA1) is an autosomal recessive neurodegenerative syndrome associated with progressive cerebellar atrophy caused by a severe loss of Purkinje cells, late axonal peripheral motor neuropathy, ataxia, oculomotor apraxia and variable age of onset (Date, Onodera et al. 2001, Moreira, Barbot et al. 2001) Moreover, patients show cognitive impairments, hypoalbuminaemia, and hypercholesterolaemia (Madabhushi, Pan et al. 2014). The disease is associated with mutations in the APTX gene. The APTX gene product, aprataxin, is involved in the resolution of 5′-AMP ligation intermediates. It was observed that AOA1 cells were sensitive to hydrogen peroxide (H2O2) and methyl methanesulfonate (MMS) that can induce strand breaks (Clements, Breslin et al. 2004, Gueven, Becherel et al. 2004). However, aprataxin might have an important role in the repair of both single strand breaks (SSBs) and double strand breaks (DSBs) (Ahel, Rass et al. 2006, Rass, Ahel et al. 2008), as suggested by the interaction of aprataxin with phosphorylated XRCC1 and XRCC4 (Clements, Breslin et al. 2004, Gueven, Becherel et al. 2004). It remains unclear what the specific role of aprataxin is in DSB remediation.
Spinocerebellar ataxia with axonal neuropathy-1 (SCAN1)is another rare autosomal recessive neurodegenerative disease characterized by progressive cerebellar atrophy and peripheral neuropathy (Takashima, Boerkoel et al. 2002). SCAN1 patients show no cognitive impairment (Takashima, Boerkoel et al. 2002). In SCAN1, the mutated gene encodes TDP1, an enzyme responsible for hydrolysis of a phosphotyrosyl protein linkage at the 3′ ends of DNA SSBs and DSBs (Yang, Burgin et al. 1996). Phosphotyrosyl linkage results from abortive activity of the topoisomerase (TOP1) that catalyzes the relaxation of DNA supercoils formed ahead of polymerases (Pourquier, Waltman et al. 2001). SCAN1 cell lines are also defective in the repair of SSBs caused by the treatment with H2O2, ionizing radiation and camptothecin (El-Khamisy, Saifi et al. 2005, Katyal, el-Khamisy et al. 2007).
It remains unclear why mutations in these two relatively obscure SSBR genes result in largely neurological symptoms. Further, unlike mutations in other DNA repair related genes these patients do not appear to present with elevated cancer incidence. It has been suggested that proliferating cells are more capable of repairing SSBs because they can be converted to DSBs and then repaired by HR in S/G2 phase, whereas neurons, as post-mitotic cells, do not have this option (Rulten and Caldecott 2013). Alternatively, back-up DNA repair pathways for the specific lesion thought to accumulate in the diseases may be absent in post-mitotic cells. For AOA1 this DNA repair pathway is most likely to be LP-BER using the action of the replication dependent enzyme FEN1 to clear the damaged termini, this enzyme has severely reduced levels in post-mitotic neurons (Sykora, Yang et al. 2013).
5-Age related neurodegenerative diseases
Age-associated neurodegenerative disorders such as AD and PD have also been linked to higher DNA damage induced by ROS and decreased DNA repair capacity. These conditions are distinct in pathology, yet common features between them are oxidative stress and mitochondrial dysfunction (Canugovi, Misiak et al. 2013). PD is a neurodegenerative disorder associated with loss of substantia nigra cells in the mid brain normally responsible for producing dopamine (Dauer and Przedborski 2003, Jankovic 2008), leading to motor dysfunction and dementia. The PD neurons show accumulation of Lewy bodies that are proportional to the severity of the symptoms (Schulz-Schaeffer 2010). The importance of oxidative stress and genomic instability in PD was observed in several studies (Fukae, Takanashi et al. 2005, Arai, Fukae et al. 2006, Bender, Krishnan et al. 2006). Increased levels of mtDNA deletions were detected in PD individuals (Bender, Krishnan et al. 2006). These deletions may be related to respiratory chain deficiency and have been identified as an important factor for neuronal loss (Bender, Krishnan et al. 2006). In addition, immunohistochemistry and biochemical studies on brain tissue from patients with PD show up-regulation of the human MutY homolog (hMUTYH) which removes mis-incorporated adenine opposite 8-oxoG in DNA (Arai, Fukae et al. 2006). Therefore, oxidative stress and genomic instability plays a role in PD disease progression.
Alzheimer’s disease is a chronic, senile neurodegenerative disease considered the most common elderly form of dementia and the 6th leading cause of death in the US. According to the Policy Brief for Heads of Government: The Global Impact of Dementia 2013–2050, the number of people living with dementia worldwide in 2013 is estimated at 44.35 million, reaching 75.62 million in 2030 and 135.46 million in 2050. Considering the daunting incidence of AD incidents, we are yet to find a definite way to prevent, pause or cure AD progression. In AD, the disease histological hallmarks are extracellular deposits of oligomerized amyloid-β precursor protein (AβPP) caused by abnormal processing and intracellular tau tangles (Tiraboschi, Hansen et al. 2004). Other characteristics in AD include loss of neuronal cells, mitochondrial dysfunction, aberrant calcium regulation and inflammation (Hirai, Aliev et al. 2001, Wyss-Coray and Mucke 2002, Caspersen, Wang et al. 2005, Isaacs, Senn et al. 2006). In addition, deregulation of DNA repair mechanisms have been reported in AD, including elevated levels of DNA breaks, reduction of DSB repair proteins and a decline in BER activity (Mullaart, Boerrigter et al. 1990, Adamec, Vonsattel et al. 1999, Hirai, Aliev et al. 2001, Wyss-Coray and Mucke 2002, Jacobsen, Beach et al. 2004, Caspersen, Wang et al. 2005, Isaacs, Senn et al. 2006, Shackelford 2006)
Another important characteristic of AD is the accumulation of oxidized DNA bases that is considered one of the earliest sign of AD (Mecocci, MacGarvey et al. 1994, Gabbita, Lovell et al. 1998, Wang, Xiong et al. 2005, Wang, Markesbery et al. 2006, Lovell, Soman et al. 2011). Several studies have shown an increase in oxidized DNA bases in AD brains, both in nDNA and mtDNA (Mecocci, MacGarvey et al. 1994, Gabbita, Lovell et al. 1998, Wang, Xiong et al. 2005). In Mild Cognitive Impairment (MCI) patients, considered a transitional zone between normal aging and dementia, especially AD (Wolf, Hensel et al. 2004), elevated levels of oxidized DNA marker 8-oxoG were also detected both in mtDNA and nDNA. (Wang, Markesbery et al. 2006). The authors suggest that oxidized DNA bases appears early in AD pathology and it can be an important factor in neurodegeneration.
Peripheral tissues of AD individuals also showed increased levels of ROS induced DNA damage, as was demonstrated in peripheral leukocytes of AD and MCI patients (Migliore, Fontana et al. 2005). Furthermore, it was observed that human fibroblasts treated with H2O2 or menadione to stimulate oxidative stress showed a gene expression pattern similar to AD-patient-derived fibroblasts (Ramamoorthy, Sykora et al. 2012). Several studies have also observed gene expression changes in DNA repair genes in AD tissue. A study of OGG1 activity, an enzyme responsible for excision of 8-oxoG, demonstrated a decrease in enzymatic activity in four different AD brain regions, suggesting impairment in the capability of repair of 8-oxoG in AD (Lovell, Xie et al. 2000). UDG was found down-regulated in AD patients when compared to age-matched individuals (Weissman, Jo et al. 2007). MTH1, an enzyme with important roles in processing oxidized DNA bases in the dNTP precursor pools, was found to be decreased in hippocampus of AD brains compared with controls (Furuta, Iida et al. 2001). Alterations in Pol β and APE1 regulation were also observed in AD tissues (Tan, Sun et al. 1998, Copani, Hoozemans et al. 2006).
A study of BER capacity in brain tissue from sporadic AD patients and normal age-matched controls indicated an impairment of BER function in AD that is related to a reduction in Pol β activity as well as a decrease in glycosylase UDG (Weissman, Jo et al. 2007) (Figure 4). MCI brains also showed deregulation in DNA repair whereas BER activity was inversely correlated to severity of the disease (Weissman, Jo et al. 2007). Moreover, BER impairment was not restricted to the regions damaged in AD, it was observed in affected and non-affected regions, suggesting defective BER as a general feature of AD brain pathology. BER activity was also measured in mitochondrial extracts of post-mortem brain inferior parietal regions, sampled from AD and normal aging individuals (Figure 4). The results showed that 5-hydroxyuracil incision and ligase activities were lower in AD brains, however uracil incision, abasic site cleavage and deoxyribonucleotide triphosphate incorporation activities were normal (Canugovi, Shamanna et al. 2014). Further, Gredilla et al. measured repair of oxidized DNA base in mitochondria extracts derived from synaptosomes of mouse brain using normal aging mice models and 3xTgAD mice that carry PS1M146V, APPswe, and TauP301L transgenes (Oddo, Caccamo et al. 2003). They observed a decline in BER capacity during the normal aging process comparing animals with 5 weeks, 5 months and 23 months, which was associated with a decrease in the level of BER proteins, however, there was no difference between pre-symptomatic and symptomatic 3xTgAD mice (Gredilla, Weissman et al. 2012). The results suggest that BER dysfunction is associated with synaptic dysfunction during the aging process. In humans, Silva et al. compared hippocampi of patients with clinical and pathological AD and also individuals with AD neuropathology but no evidence of cognitive impairment, to normal aging individuals. They observed reduced levels of DNA repair and cell-cycle inhibition markers, as well as elevated levels of cell cycle progression markers and cell death in post-mitotic neurons of clinical and pathological AD patients compared to age matched controls. Patients with just AD neuropathology presented an expression profile similar to normal aging individuals (Silva, Santos et al. 2014). These experiments provide preliminary evidence that cognitive impairment may be associated with deregulation in DNA damage repair and cell cycle maintenance.
Figure 4. Patients with Alzheimer’s (AD) or mild cognitive impairment (MCI) have reduced repair capacity.
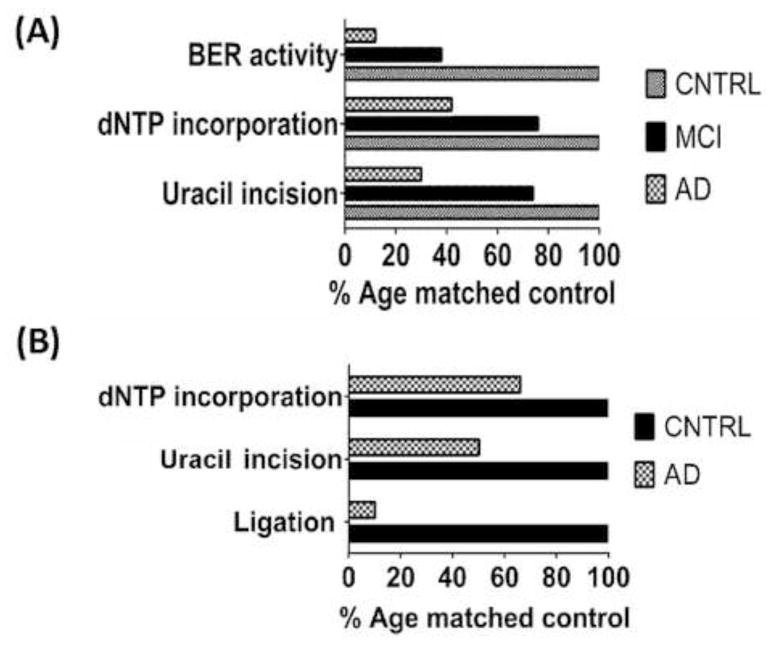
(A) Short post-mortem interval brain tissue was taken from 10 AD, 9 MCI and 10 age matched patients and measured for repair capacity. Using oligonucleotide substrates we found that uracil incision, polymerase dNTP incorporation and overall BER repair capacity was reduced in MCI samples and further reduced in samples from AD patients. This reduction in repair capacity correlated with the level of cognitive impairment. Adapted from Weissman et.al. NAR, Vol.35, No.6, 2007. Graph shows median results as a percentage of control activity set at 100%. (B) Mitochondrial BER was also reduced in AD patient brain extracts. Polymerase activity, Uracil incision and total ligation activity were reduced in AD post mortem brain samples compared to age matched controls. Adapted from Canugovi, NAB, Vol. 35, 2014. Graph shows mean results as a percentage of control activity.
7) Final considerations
The research described in this review highlights the reliance of an organism on DNA repair to thwart, not only normal aging, but also age-related neurodegenerative diseases. We consider the fundamental role of mitochondria in ROS formation and the importance of these organelles, with dysfunction clearly linked to onset and progression of neurological disorders. The evidence highlights that further research is required to understand the implication of BER deficiencies in aging and neurodegenerative process. Moreover, it is important to determine the influence of the BER mechanisms in the aging brain, only then we can more completely address the neurodegenerative problems common to the aging population.
Highlights.
DNA repair can protect against aging and age-related neurodegenerative disorders.
Mutations of key DNA repair proteins can cause rare ataxias.
Alzheimer’s disease has elevated oxidized bases and DNA repair decline.
Acknowledgments
This work was supported by funds from the intramural program of the National Institute on Aging, National Institutes of Health, US. Giovana S Leandro was supported by Fundacao de Amparo à Pesquisa do Estado de Sao Paulo-FAPESP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aamann MD, Sorensen MM, Hvitby C, Berquist BR, Muftuoglu M, Tian J, de Souza-Pinto NC, Scheibye-Knudsen M, Wilson DM, 3rd, Stevnsner T, Bohr VA. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. Faseb j. 2010;24(7):2334–2346. doi: 10.1096/fj.09-147991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamec E, Vonsattel JP, Nixon RA. DNA strand breaks in Alzheimer’s disease. Brain Res. 1999;849(1–2):67–77. doi: 10.1016/s0006-8993(99)02004-1. [DOI] [PubMed] [Google Scholar]
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443(7112):713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- Akbari M, Pena-Diaz J, Andersen S, Liabakk NB, Otterlei M, Krokan HE. Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair (Amst) 2009;8(7):834–843. doi: 10.1016/j.dnarep.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Akbari M, Visnes T, Krokan HE, Otterlei M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair (Amst) 2008;7(4):605–616. doi: 10.1016/j.dnarep.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Al-Baker EA, Oshin M, Hutchison CJ, Kill IR. Analysis of UV-induced damage and repair in young and senescent human dermal fibroblasts using the comet assay. Mech Ageing Dev. 2005;126(6–7):664–672. doi: 10.1016/j.mad.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Anindya R, Mari PO, Kristensen U, Kool H, Giglia-Mari G, Mullenders LH, Fousteri M, Vermeulen W, Egly JM, Svejstrup JQ. A ubiquitin-binding domain in Cockayne syndrome B required for transcription-coupled nucleotide excision repair. Mol Cell. 2010;38(5):637–648. doi: 10.1016/j.molcel.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimov VN. Mutant and genetically modified mice as models for studying the relationship between aging and carcinogenesis. Mech Ageing Dev. 2001;122(12):1221–1255. doi: 10.1016/s0047-6374(01)00262-7. [DOI] [PubMed] [Google Scholar]
- Arai T, Fukae J, Hatano T, Kubo S, Ohtsubo T, Nakabeppu Y, Mori H, Mizuno Y, Hattori N. Up-regulation of hMUTYH, a DNA repair enzyme, in the mitochondria of substantia nigra in Parkinson’s disease. Acta Neuropathol. 2006;112(2):139–145. doi: 10.1007/s00401-006-0081-9. [DOI] [PubMed] [Google Scholar]
- Arbuzova S, Hutchin T, Cuckle H. Mitochondrial dysfunction and Down’s syndrome. Bioessays. 2002;24(8):681–684. doi: 10.1002/bies.10138. [DOI] [PubMed] [Google Scholar]
- Ben Yehuda A, Globerson A, Krichevsky S, Bar On H, Kidron M, Friedlander Y, Friedman G, Ben Yehuda D. Ageing and the mismatch repair system. Mech Ageing Dev. 2000;121(1–3):173–179. doi: 10.1016/s0047-6374(00)00208-6. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38(5):515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res. 2003;531(1–2):37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Bohr VA. Human premature aging syndromes and genomic instability. Mech Ageing Dev. 2002;123(8):987–993. doi: 10.1016/s0047-6374(02)00039-8. [DOI] [PubMed] [Google Scholar]
- Boyle J, I, Kill R, Parris CN. Heterogeneity of dimer excision in young and senescent human dermal fibroblasts. Aging Cell. 2005;4(5):247–255. doi: 10.1111/j.1474-9726.2005.00167.x. [DOI] [PubMed] [Google Scholar]
- Brooksbank BW, Balazs R. Superoxide dismutase, glutathione peroxidase and lipoperoxidation in Down’s syndrome fetal brain. Brain Res. 1984;318(1):37–44. doi: 10.1016/0165-3806(84)90060-9. [DOI] [PubMed] [Google Scholar]
- Brown JC, McClelland GB, Faure PA, Klaiman JM, Staples JF. Examining the mechanisms responsible for lower ROS release rates in liver mitochondria from the long-lived house sparrow (Passer domesticus) and big brown bat (Eptesicus fuscus) compared to the short-lived mouse (Mus musculus) Mech Ageing Dev. 2009;130(8):467–476. doi: 10.1016/j.mad.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Cabelof DC, Raffoul JJ, Yanamadala S, Ganir C, Guo Z, Heydari AR. Attenuation of DNA polymerase beta-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat Res. 2002;500(1–2):135–145. doi: 10.1016/s0027-5107(02)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canugovi C, Maynard S, Bayne AC, Sykora P, Tian J, de Souza-Pinto NC, Croteau DL, Bohr VA. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst) 2010;9(10):1080–1089. doi: 10.1016/j.dnarep.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canugovi C, Misiak M, Ferrarelli LK, Croteau DL, Bohr VA. The role of DNA repair in brain related disease pathology. DNA Repair (Amst) 2013;12(8):578–587. doi: 10.1016/j.dnarep.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canugovi C, Shamanna RA, Croteau DL, Bohr VA. Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiol Aging. 2014;35(6):1293–1300. doi: 10.1016/j.neurobiolaging.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial A beta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. Faseb j. 2005;19(14):2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, Mitra S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34(7):2067–2076. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Cao G, Hastings T, Feng Y, Pei W, O’Horo C, Chen J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J Neurochem. 2002;81(6):1273–1284. doi: 10.1046/j.1471-4159.2002.00916.x. [DOI] [PubMed] [Google Scholar]
- Christiansen M, Stevnsner T, Bohr VA, Clark BF, Rattan SI. Gene-specific DNA repair of pyrimidine dimers does not decline during cellular aging in vitro. Exp Cell Res. 2000;256(1):308–314. doi: 10.1006/excr.2000.4826. [DOI] [PubMed] [Google Scholar]
- Clements PM, Breslin C, Deeks ED, Byrd PJ, Ju L, Bieganowski P, Brenner C, Moreira MC, Taylor AM, Caldecott KW. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst) 2004;3(11):1493–1502. doi: 10.1016/j.dnarep.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Copani A, Hoozemans JJ, Caraci F, Calafiore M, Van Haastert ES, Veerhuis R, Rozemuller AJ, Aronica E, Sortino MA, Nicoletti F. DNA polymerase-beta is expressed early in neurons of Alzheimer’s disease brain and is loaded into DNA replication forks in neurons challenged with beta-amyloid. J Neurosci. 2006;26(43):10949–10957. doi: 10.1523/JNEUROSCI.2793-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Oster-Granite ML, Gearhart JD. The neurobiologic consequences of Down syndrome. Brain Res Bull. 1986;16(6):773–787. doi: 10.1016/0361-9230(86)90074-2. [DOI] [PubMed] [Google Scholar]
- Croteau DL, V, Bohr A. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem. 1997;272(41):25409–25412. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–552. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DS, Tainer JA. Conserved structural motifs governing the stoichiometric repair of alkylated DNA by O(6)-alkylguanine-DNA alkyltransferase. Mutat Res. 2000;460(3–4):151–163. doi: 10.1016/s0921-8777(00)00024-0. [DOI] [PubMed] [Google Scholar]
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, Sakai T, Takahashi T, Nagatomo H, Sekijima Y, Kawachi I, Takiyama Y, Nishizawa M, Fukuhara N, Saito K, Sugano S, Tsuji S. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29(2):184–188. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- de Souza-Pinto NC, Mason PA, Hashiguchi K, Weissman L, Tian J, Guay D, Lebel M, Stevnsner TV, Rasmussen LJ, Bohr VA. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair (Amst) 2009;8(6):704–719. doi: 10.1016/j.dnarep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov G, Bischoff C, Piotrowski J, Bohr VA. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J Biol Chem. 1998;273(50):33811–33816. doi: 10.1074/jbc.273.50.33811. [DOI] [PubMed] [Google Scholar]
- Dianov GL, Prasad R, Wilson SH, Bohr VA. Role of DNA polymerase beta in the excision step of long patch mammalian base excision repair. J Biol Chem. 1999;274(20):13741–13743. doi: 10.1074/jbc.274.20.13741. [DOI] [PubMed] [Google Scholar]
- Diaz-Llera S, Podlutsky A, Osterholm AM, Hou SM, Lambert B. Hydrogen peroxide induced mutations at the HPRT locus in primary human T-lymphocytes. Mutat Res. 2000;469(1):51–61. doi: 10.1016/s1383-5718(00)00058-9. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M. Chemical determination of free radical-induced damage to DNA. Free Radic Biol Med. 1991;10(3–4):225–242. doi: 10.1016/0891-5849(91)90080-m. [DOI] [PubMed] [Google Scholar]
- Dolle ME, Giese H, Hopkins CL, Martus HJ, Hausdorff JM, Vijg J. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat Genet. 1997;17(4):431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- Druzhyna N, Nair RG, LeDoux SP, Wilson GL. Defective repair of oxidative damage in mitochondrial DNA in Down’s syndrome. Mutat Res. 1998;409(2):81–89. doi: 10.1016/s0921-8777(98)00042-1. [DOI] [PubMed] [Google Scholar]
- Edwards M, Rassin DK, Izumi T, Mitra S, Perez-Polo JR. APE/Ref-1 responses to oxidative stress in aged rats. J Neurosci Res. 1998;54(5):635–638. doi: 10.1002/(SICI)1097-4547(19981201)54:5<635::AID-JNR8>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434(7029):108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966;45(3):177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- Espejel S, Martin M, Klatt P, Martin-Caballero J, Flores JM, Blasco MA. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice. EMBO Rep. 2004;5(5):503–509. doi: 10.1038/sj.embor.7400127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271(16):9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- Fukae J, Takanashi M, Kubo S, Nishioka K, Nakabeppu Y, Mori H, Mizuno Y, Hattori N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005;109(3):256–262. doi: 10.1007/s00401-004-0937-9. [DOI] [PubMed] [Google Scholar]
- Furuta A, Iida T, Nakabeppu Y, Iwaki T. Expression of hMTH1 in the hippocampi of control and Alzheimer’s disease. Neuroreport. 2001;12(13):2895–2899. doi: 10.1097/00001756-200109170-00028. [DOI] [PubMed] [Google Scholar]
- Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J Neurochem. 1998;71(5):2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A. Making ends meet in old age: DSB repair and aging. Mech Ageing Dev. 2005;126(6–7):621–628. doi: 10.1016/j.mad.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A, Mao Z, Hine C. Changes in DNA repair during aging. Nucleic Acids Res. 2007;35(22):7466–7474. doi: 10.1093/nar/gkm756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M. Hierarchical deterioration of body systems in Werner’s syndrome: implications for normal ageing. Mech Ageing Dev. 1997;98(3):239–254. doi: 10.1016/s0047-6374(97)00111-5. [DOI] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF. The origin and early evolution of mitochondria. Genome Biol. 2001;2(6) doi: 10.1186/gb-2001-2-6-reviews1018. Reviews 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gredilla R, Weissman L, Yang JL, Bohr VA, Stevnsner T. Mitochondrial base excision repair in mouse synaptosomes during normal aging and in a model of Alzheimer’s disease. Neurobiol Aging. 2012;33(4):694–707. doi: 10.1016/j.neurobiolaging.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueven N, Becherel OJ, Kijas AW, Chen P, Howe O, Rudolph JH, Gatti R, Date H, Onodera O, Taucher-Scholz G, Lavin MF. Aprataxin, a novel protein that protects against genotoxic stress. Hum Mol Genet. 2004;13(10):1081–1093. doi: 10.1093/hmg/ddh122. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC. Subpathways of nucleotide excision repair and their regulation. Oncogene. 2002;21(58):8949–8956. doi: 10.1038/sj.onc.1206096. [DOI] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Harman D. The aging process. Proc Natl Acad Sci U S A. 1981;78(11):7124–7128. doi: 10.1073/pnas.78.11.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21(9):3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson EK, Hogue BA, Souza-Pinto NC, Croteau DL, Anson RM, Bohr VA, Hansford RG. Age-associated change in mitochondrial DNA damage. Free Radic Res. 1998;29(6):573–579. doi: 10.1080/10715769800300611. [DOI] [PubMed] [Google Scholar]
- Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27(8):1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Isaacs AM, Senn DB, Yuan M, Shine JP, Yankner BA. Acceleration of amyloid beta-peptide aggregation by physiological concentrations of calcium. J Biol Chem. 2006;281(38):27916–27923. doi: 10.1074/jbc.M602061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen E, Beach T, Shen Y, Li R, Chang Y. Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Brain Res Mol Brain Res. 2004;128(1):1–7. doi: 10.1016/j.molbrainres.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Jeppesen DK, V, Bohr A, Stevnsner T. DNA repair deficiency in neurodegeneration. Prog Neurobiol. 2011;94(2):166–200. doi: 10.1016/j.pneurobio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7(5):335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- Kamenisch Y, Fousteri M, Knoch J, von Thaler AK, Fehrenbacher B, Kato H, Becker T, Dolle ME, Kuiper R, Majora M, Schaller M, van der Horst GT, van Steeg H, Rocken M, Rapaport D, Krutmann J, Mullenders LH, Berneburg M. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J Exp Med. 2010;207(2):379–390. doi: 10.1084/jem.20091834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar P, Piotrowski J, Brosh RM, Jr, Sommers JA, Miller SP, Cheng WH, Snowden CM, Ramsden DA, Bohr VA. Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J Biol Chem. 2002;277(21):18291–18302. doi: 10.1074/jbc.M111523200. [DOI] [PubMed] [Google Scholar]
- Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. Embo j. 2007;26(22):4720–4731. doi: 10.1038/sj.emboj.7601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Wilson DM., 3rd Overview of base excision repair biochemistry. Curr Mol Pharmacol. 2012;5(1):3–13. doi: 10.2174/1874467211205010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120(4):437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408(6809):233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16(11):3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob M, Laugel V, Durand M, Fothergill H, Dalloz C, Sauvanaud F, Dollfus H, Namer IJ, Dietemann JL. Neuroimaging in Cockayne syndrome. AJNR Am J Neuroradiol. 2010;31(9):1623–1630. doi: 10.3174/ajnr.A2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009;47(4):333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Krichevsky S, Pawelec G, Gural A, Effros RB, Globerson A, Yehuda DB, Yehuda AB. Age related microsatellite instability in T cells from healthy individuals. Exp Gerontol. 2004;39(4):507–515. doi: 10.1016/j.exger.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Labinskyy N, Csiszar A, Orosz Z, Smith K, Rivera A, Buffenstein R, Ungvari Z. Comparison of endothelial function, O2-* and H2O2 production, and vascular oxidative stress resistance between the longest-living rodent, the naked mole rat, and mice. Am J Physiol Heart Circ Physiol. 2006;291(6):H2698–2704. doi: 10.1152/ajpheart.00534.2006. [DOI] [PubMed] [Google Scholar]
- Lakshmipathy U, Campbell C. Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 1999;27(4):1198–1204. doi: 10.1093/nar/27.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AJ, Boysen HM, Buckingham JA, Yang T, Podlutsky A, Austad SN, Kunz TH, Buffenstein R, Brand MD. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell. 2007;6(5):607–618. doi: 10.1111/j.1474-9726.2007.00312.x. [DOI] [PubMed] [Google Scholar]
- Lang BF, Burger G, O’Kelly CJ, Cedergren R, Golding GB, Lemieux C, Sankoff D, Turmel M, Gray MW. An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature. 1997;387(6632):493–497. doi: 10.1038/387493a0. [DOI] [PubMed] [Google Scholar]
- Le May N, Mota-Fernandes D, Velez-Cruz R, Iltis I, Biard D, Egly JM. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell. 2010;38(1):54–66. doi: 10.1016/j.molcel.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362(6422):709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Liu P, Qian L, Sung JS, de Souza-Pinto NC, Zheng L, Bogenhagen DF, Bohr VA, Wilson DM, 3rd, Shen B, Demple B. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mo Cell Biol. 2008;28(16):4975–4987. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 2005;280(5):3665–3674. doi: 10.1074/jbc.M412922200. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Soman S, Bradley MA. Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech Ageing Dev. 2011;132(8–9):443–448. doi: 10.1016/j.mad.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell MA, Xie C, Markesbery WR. Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 2000;855(1):116–123. doi: 10.1016/s0006-8993(99)02335-5. [DOI] [PubMed] [Google Scholar]
- Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron. 2014;83(2):266–282. doi: 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM. Alzheimer’s disease and Down’s syndrome. Histopathology. 1988;13(2):125–137. doi: 10.1111/j.1365-2559.1988.tb02018.x. [DOI] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36(5):747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- Menck CF, Munford V. DNA repair diseases: What do they tell us about cancer and aging? Genet Mol Biol. 2014;37(1 Suppl):220–233. doi: 10.1590/s1415-47572014000200008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore L, Fontana I, Trippi F, Colognato R, Coppede F, Tognoni G, Nucciarone B, Siciliano G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol Aging. 2005;26(5):567–573. doi: 10.1016/j.neurobiolaging.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Mitra S, Boldogh I, Izumi T, Hazra TK. Complexities of the DNA base excision repair pathway for repair of oxidative DNA damage. Environ Mol Mutagen. 2001;38(2–3):180–190. doi: 10.1002/em.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco A, Biamonti G, Savini E, Focher F, Spadari S, Ciarrocchi G. DNA ligase I gene expression during differentiation and cell proliferation. Nucleic Acids Res. 1992;20(23):6209–6214. doi: 10.1093/nar/20.23.6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29(2):189–193. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- Muftuoglu M, de Souza-Pinto NC, Dogan A, Aamann M, Stevnsner T, Rybanska I, Kirkali G, Dizdaroglu M, Bohr VA. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J Biol Chem. 2009;284(14):9270–9279. doi: 10.1074/jbc.M807006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M, Mori MP, Souza-Pinto NC. Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion. 2014;17C:164–181. doi: 10.1016/j.mito.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Mullaart E, Boerrigter ME, Ravid R, Swaab DF, Vijg J. Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol Aging. 1990;11(3):169–173. doi: 10.1016/0197-4580(90)90542-8. [DOI] [PubMed] [Google Scholar]
- Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet. 1992;42(1):68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- Narciso L, Fortini P, Pajalunga D, Franchitto A, Liu P, Degan P, Frechet M, Demple B, Crescenzi M, Dogliotti E. Terminally differentiated muscle cells are defective in base excision DNA repair and hypersensitive to oxygen injury. Proc Natl Acad Sci U S A. 2007;104(43):17010–17015. doi: 10.1073/pnas.0701743104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale V. A comprehensive description of the severity groups in Cockayne syndrome. Am J Med Genet A. 2011;155a(5):1081–1095. doi: 10.1002/ajmg.a.33933. [DOI] [PubMed] [Google Scholar]
- Neri S, Gardini A, Facchini A, Olivieri F, Franceschi C, Ravaglia G, Mariani E. Mismatch repair system and aging: microsatellite instability in peripheral blood cells from differently aged participants. J Gerontol A Biol Sci Med Sci. 2005;60(3):285–292. doi: 10.1093/gerona/60.3.285. [DOI] [PubMed] [Google Scholar]
- Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan HE. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997;25(4):750–755. doi: 10.1093/nar/25.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Ohtsubo T, Oda H, Fujiwara T, Kang D, Sugimachi K, Nakabeppu Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol Biol Cell. 1999;10(5):1637–1652. doi: 10.1091/mbc.10.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Pages V, Fuchs RP. How DNA lesions are turned into mutations within cells? Oncogene. 2002;21(58):8957–8966. doi: 10.1038/sj.onc.1206006. [DOI] [PubMed] [Google Scholar]
- Pegg AE. Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990;50(19):6119–6129. [PubMed] [Google Scholar]
- Podlutsky AJ, Dianova, Podust VN, Bohr VA, Dianov GL. Human DNA polymerase beta initiates DNA synthesis during long-patch repair of reduced AP sites in DNA. EMBO J. 2001;20(6):1477–1482. doi: 10.1093/emboj/20.6.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourquier P, Waltman JL, Urasaki Y, Loktionova NA, Pegg AE, Nitiss JL, Pommier Y. Topoisomerase I-mediated cytotoxicity of N-methyl-N′-nitro-N-nitrosoguanidine: trapping of topoisomerase I by the O6-methylguanine. Cancer Res. 2001;61(1):53–58. [PubMed] [Google Scholar]
- Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase beta mediates an excision step in mammalian long patch base excision repair. J Biol Chem. 2000;275(6):4460–4466. doi: 10.1074/jbc.275.6.4460. [DOI] [PubMed] [Google Scholar]
- Qin X, Zhang S, Matsukuma S, Zarkovic M, Shimizu S, Ishikawa T, Nakatsuru Y. Protection against malignant progression of spontaneously developing liver tumors in transgenic mice expressing O(6)-methylguanine-DNA methyltransferase. Jpn J Cancer Res. 2000;91(11):1085–1089. doi: 10.1111/j.1349-7006.2000.tb00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy M, Sykora P, Scheibye-Knudsen M, Dunn C, Kasmer C, Zhang Y, Becker KG, Croteau DL, Bohr VA. Sporadic Alzheimer disease fibroblasts display an oxidative stress phenotype. Free Radic Biol Med. 2012;53(6):1371–1380. doi: 10.1016/j.freeradbiomed.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao KS, Annapurna VV, Raji NS. DNA polymerase-beta may be the main player for defective DNA repair in aging rat neurons. Ann N Y Acad Sci. 2001;928:113–120. doi: 10.1111/j.1749-6632.2001.tb05641.x. [DOI] [PubMed] [Google Scholar]
- Rao KS, V, Annapurna V, Raji NS, Harikrishna T. Loss of base excision repair in aging rat neurons and its restoration by DNA polymerase beta. Brain Res Mol Brain Res. 2000;85(1–2):251–259. doi: 10.1016/s0169-328x(00)00266-7. [DOI] [PubMed] [Google Scholar]
- Rass U, Ahel I, West SC. Molecular mechanism of DNA deadenylation by the neurological disease protein aprataxin. J Biol Chem. 2008;283(49):33994–34001. doi: 10.1074/jbc.M807124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988;85(17):6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizen NJ, Patterson D. Down’s syndrome. Lancet. 2003;361(9365):1281–1289. doi: 10.1016/S0140-6736(03)12987-X. [DOI] [PubMed] [Google Scholar]
- Rulten SL, Caldecott KW. DNA strand break repair and neurodegeneration. DNA Repair (Amst) 2013;12(8):558–567. doi: 10.1016/j.dnarep.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Salway KD, Page MM, Faure PA, Burness G, Stuart JA. Enhanced protein repair and recycling are not correlated with longevity in 15 vertebrate endotherm species. Age (Dordr) 2011;33(1):33–47. doi: 10.1007/s11357-010-9157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Sanz A, Caro P, Gomez J, Barja G. Testing the vicious cycle theory of mitochondrial ROS production: effects of H2O2 and cumene hydroperoxide treatment on heart mitochondria. J Bioenerg Biomembr. 2006;38(2):121–127. doi: 10.1007/s10863-006-9011-8. [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Ramamoorthy M, Sykora P, Maynard S, Lin PC, Minor RK, Wilson DM, 3rd, Cooper M, Spencer R, de Cabo R, Croteau DL, Bohr VA. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med. 2012;209(4):855–869. doi: 10.1084/jem.20111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010;120(2):131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DA. DNA end joining activity is reduced in Alzheimer’s disease. Neurobiol Aging. 2006;27(4):596–605. doi: 10.1016/j.neurobiolaging.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Silva AR, Santos AC, Farfel JM, Grinberg LT, Ferretti RE, Campos AH, Cunha IW, Begnami MD, Rocha RM, Carraro DM, de Braganca Pereira CA, Jacob-Filho W, Brentani H. Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS One. 2014;9(6):e99897. doi: 10.1371/journal.pone.0099897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner AM, Turker MS. Oxidative mutagenesis, mismatch repair, and aging. Sci Aging Knowledge Environ. 2005;2005(9):re3. doi: 10.1126/sageke.2005.9.re3. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Brunk UT. Mitochondrial production of pro-oxidants and cellular senescence. Mutat Res. 1992;275(3–6):295–304. doi: 10.1016/0921-8734(92)90033-l. [DOI] [PubMed] [Google Scholar]
- Stevnsner T, Muftuoglu M, Aamann MD, Bohr VA. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech Ageing Dev. 2008;129(7–8):441–448. doi: 10.1016/j.mad.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JA, Maddalena LA, Merilovich M, Robb EL. A midlife crisis for the mitochondrial free radical theory of aging. Longev Healthspan. 2014;3(1):4. doi: 10.1186/2046-2395-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svilar D, Goellner EM, Almeida KH, Sobol RW. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Signal. 2011;14(12):2491–2507. doi: 10.1089/ars.2010.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykora P, Yang JL, Ferrarelli LK, Tian J, Tadokoro T, Kulkarni A, Weissman L, Keijzers G, Wilson DM, 3rd, Mattson MP, Bohr VA. Modulation of DNA base excision repair during neuronal differentiation. Neurobiol Aging. 2013;34(7):1717–1727. doi: 10.1016/j.neurobiolaging.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Tann AW, Longley MJ, Copeland WC, Mitra S. Long patch base excision repair in mammalian mitochondrial genomes. J Biol Chem. 2008;283(39):26349–26356. doi: 10.1074/jbc.M803491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32(2):267–272. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- Tan Z, Sun N, Schreiber SS. Immunohistochemical localization of redox factor-1 (Ref-1) in Alzheimer’s hippocampus. Neuroreport. 1998;9(12):2749–2752. doi: 10.1097/00001756-199808240-00012. [DOI] [PubMed] [Google Scholar]
- Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7(3–4):367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- Tiano L, Busciglio J. Mitochondrial dysfunction and Down’s syndrome: is there a role for coenzyme Q(10) ? Biofactors. 2011;37(5):386–392. doi: 10.1002/biof.184. [DOI] [PubMed] [Google Scholar]
- Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62(11):1984–1989. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- Vijg J, Dolle ME. Large genome rearrangements as a primary cause of aging. Mech Ageing Dev. 2002;123(8):907–915. doi: 10.1016/s0047-6374(02)00028-3. [DOI] [PubMed] [Google Scholar]
- Wallace SS. Biological consequences of free radical-damaged DNA bases. Free Radic Biol Med. 2002;33(1):1–14. doi: 10.1016/s0891-5849(02)00827-4. [DOI] [PubMed] [Google Scholar]
- Wang J, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem. 2006;96(3):825–832. doi: 10.1111/j.1471-4159.2005.03615.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem. 2005;93(4):953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- Wei W, Englander EW. DNA polymerase beta-catalyzed-PCNA independent long patch base excision repair synthesis: a mechanism for repair of oxidatively damaged DNA ends in post-mitotic brain. J Neurochem. 2008;107(3):734–744. doi: 10.1111/j.1471-4159.2008.05644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35(16):5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nat Struct Biol. 2000;7(3):176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- Wolf H, Hensel A, Kruggel F, Riedel-Heller SG, Arendt T, Wahlund LO, Gertz HJ. Structural correlates of mild cognitive impairment. Neurobiol Aging. 2004;25(7):913–924. doi: 10.1016/j.neurobiolaging.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Wong HK, Muftuoglu M, Beck G, Imam SZ, Bohr VA, Wilson DM., 3rd Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007;35(12):4103–4113. doi: 10.1093/nar/gkm404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron. 2002;35(3):419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94(2):514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JL, Weissman L, Bohr VA, Mattson MP. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair (Amst) 2008;7(7):1110–1120. doi: 10.1016/j.dnarep.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SW, Burgin AB, Jr, Huizenga BN, Robertson CA, Yao KC, Nash HA. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci U S A. 1996;93(21):11534–11539. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannone SM, Roy S, Chan DW, Murphy MB, Huang S, Campisi J, Chen DJ. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J Biol Chem. 2001;276(41):38242–38248. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40(2):401–413. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]