

Abstract
Genomic modification with sulfur as phosphorothioate (PT) is widespread among prokaryotes, including human pathogens. Apart from its physiological functions, the redox and nucleophilic properties of PT sulfur suggest effects on bacterial fitness in stressful environments. Here we show that PTs are dynamic and labile DNA modifications that cause genomic instability during oxidative stress. Using coupled isotopic labeling-mass spectrometry, we observed sulfur replacement in PTs at a rate of ~2%/h in unstressed Escherichia coli and Salmonella enterica. While PT levels were unaffected by exposure to hydrogen peroxide (H2O2) or hypochlorous acid (HOCl), PT turnover increased to 3.8–10%/h for HOCl and was unchanged for H2O2, consistent with repair of HOCl-induced sulfur damage. PT-dependent HOCl sensitivity extended to cytotoxicity and DNA strand-breaks, which occurred at orders-of-magnitude lower doses of HOCl than H2O2. The genotoxicity of HOCl in PT-containing bacteria suggests reduced fitness in competition with HOCl-producing organisms and during human infections.
Graphical abstract
INTRODUCTION
While bacterial DNA modifications that function in restriction-modification (R-M) and epigenetics typically comprise simple nucleobase methylations,1–4 a growing variety of modification chemistries have introduced new layers of complexity in our understanding of bacterial genome protection and regulation.5–8 Here we show that phosphorothioate (PT) modifications of DNA, in which a non-bridging phosphate oxygen is replaced with sulfur, are dynamic modifications that are highly labile during oxidative stress. Although widespread among bacteria and archaea,9 including human pathogens,10,11 little is known about the fitness advantage conferred by these DNA modifications other than their role in R-M systems. PTs are synthesized by a complex of proteins (Supplementary Results, Supplementary Fig. 1), DndABCDE,9 with DndA often replaced by other cysteine desulfurases, such as IscS in Escherichia coli.12 A highly unusual feature of PTs is that, even in the presence of the cognate restriction proteins DndFGHI in those bacteria in which PTs function in R-M,13,14 only a small fraction (10–20%) of modification consensus sequences contain a PT.6 While this points to PT involvement in an atypical R-M system, many bacteria lack obvious restriction activity (DndFGHI),6,9 which suggests an alternative function for PTs, such as epigenetics.
Another unusual feature of PTs as physiological DNA modifications involves the redox and nucleophilic properties of the sulfur, which are largely unexplored in terms of biological implications. The nucleophilicity of the sulfur has been exploited to sequence RNA and DNA by alkylation to form an unstable PT triester.15 In terms of redox behavior, the PT sulfur should be readily oxidized by analogy to biological thiols,16 though there are few studies of PT oxidation under biochemically or biologically relevant conditions.6,17–19 Here we report the application of genetic and analytical tools to discover that PTs are labile DNA modifications that turn over at a significant rate in unstressed cells and that are selectively sensitive to oxidization by hypochlorous acid (HOCl), which causes genomic instability under biologically relevant conditions.
RESULTS
PT increases sensitivity of bacteria to HOCl but not H2O2
The two bacterial species chosen for study of PT effects on bacterial oxidative stress response, E. coli B7A and Salmonella enterica serovar Cerro 87, naturally possess dnd genes A–I as a R-M system and have been well characterized6–8,13,14 in terms of both quantities of PT (875 ± 68 and 620 ± 41 PT per 106 nt, respectively) and PT modifications consensus sequences (GpsAAC/GpsTTC). The choice of mutant strains lacking PTs was highly critical in light of a previous study in which S. enterica strains lacking individual dnd genes A, C, D and E were found to be more sensitive to H2O2 than wild-type S. enterica, which suggested that PTs and/or Dnd proteins were protective against oxidative stress.18 The problem with these studies is that, while deletion of individual dnd genes indeed prevents PT synthesis, the restriction activity of DndF–I proteins remains intact so that loss of PT results in significant genotoxicity and greatly reduced fitness even in the absence of oxidant stress.20 Here we used the ΔdndB–H deletion mutants of E. coli and S. enterica, which lack both PT modifications (dndB–E) and PT restriction activity (dndF–H). The choice of exposure conditions was also critical in light of the 1000-fold variation in sensitivity of wild-type E. coli to HOCl (LD50 6 μM to 6 mM) depending upon the use of phosphate-buffered saline or lysogeny broth (LB medium) for the exposure;21–24 LB is known to quench HOCl in exposure studies.22,24 To assess growth during exposures, we used M9 minimal medium25 since it sustained equivalent growth of wild-type and ΔdndB–H strains of both bacteria (Fig. 1) and caused minimal interference with oxidants during exposures.
Figure 1. The effect of PT modifications on survival and growth of bacteria following oxidant exposures.
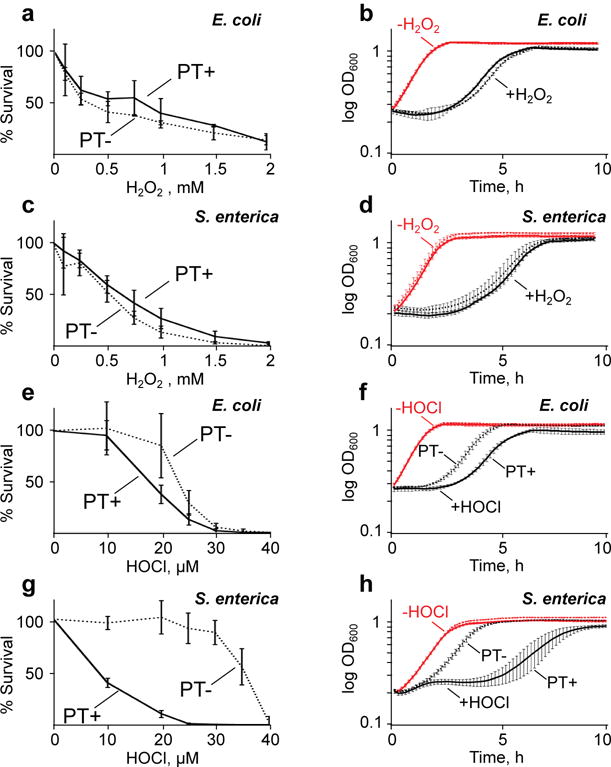
Wild-type (PT+, solid line) and ΔdndB–H (PT–, dashed line) strains of E. coli (A, B, E, F) and S. enterica (C, D, G, H) were exposed to H2O2 (A–D) or HOCl (E–H). Cytotoxicity assays (A, C, E, G) were performed with the indicated concentrations of H2O2 and HOCl. Growth curves (B, D, F, H) were then prepared using the LD80 doses of H2O2 (B, D) and HOCl (F, H). Red lines in the growth curves (B, D, F, H) indicate unexposed controls, with overlapping curves for wild-type and ΔdndB–H strains; black lines represent exposed bacteria, with data plotted in solid lines (PT+) distinguishing from dotted lines (PT–) only in panels F and H. Data represent mean ± SD for 3 biological replicates. Statistically significant differences among the data sets are discussed in the text.
Using these conditions, H2O2 did not cause PT-dependent cytotoxicity following exposure of either wild-type or ΔdndB–H strains of E. coli and S. enterica (Fig. 1a,c) in either log-growth or stationary phase (Supplementary Fig. 2). This is consistent with the conclusion that the apparent protection afforded by PT against H2O2 toxicity as reported previously18 was actually caused by heightened H2O2 sensitivity of mutant strains lacking individual dnd genes (i.e., lacking PT) as a result of unrestrained restriction enzyme activity from DndF–H. In contrast, PT-containing wild-type bacteria were more sensitive to HOCl than the ΔdndB–H strains lacking PT (Fig. 1e,g). The 1.2-fold increase in LD50 dose of HOCl for the ΔdndB–H E. coli was not statistically significant (21 ± 2.8 μM versus 17 ± 0.9 μM; mean ± SD for N=3). However, the PT-containing wild-type S. enterica was 4.8-fold more sensitive to HOCl than the ΔdndB–H mutant (LD50 6.0 ± 0.3 μM versus 29 ± 0.2 μM). That HOCl sensitivity depends on the level of PT modifications was demonstrated using the S. enterica ΔdndB strain in which loss of the DndB transcription factor causes a 2-fold increase in PT levels compared to wild-type cells (1236 ± 53 versus 620 ± 41, respectively; Supplementary Table 1).7,8 The LD50 for HOCl decreased from 6 ± 0.3 μM in the wild-type strain to 3.7 ± 1.8 μM in the ΔdndB strain (p<0.09, Student’s t-test; Supplementary Fig. 3). HOCl sensitivity differences due to genetic manipulations were ruled out by the similar HOCl sensitivities of wild-type cells and S. enterica ΔdndF–H cells lacking only the restriction activity (Supplementary Fig. 3); both strains possess identical levels of PT (Supplementary Table 1). While the presence of PT did not affect the apparent growth rate of the bacteria during LD50 exposures to either H2O2 or HOCl, PT-dependent growth effects become apparent at LD80 doses of HOCl (Fig. 1b, d, f, h). These results demonstrate that PT DNA modifications compromise bacterial fitness in the face of HOCl exposure, which raises questions about the mechanism underlying PT-dependent HOCl toxicity.
HOCl causes PT-dependent DNA damage in vitro and in vivo
As the first step in defining the mechanisms linking PT modifications with HOCl cytotoxicity, we exposed intact DNA isolated from wild-type E. coli to HOCl and quantified d(GPSA) and d(GPST) dinucleotides by LC-MS. As shown in Fig. 2a, all PTs were consumed at less than 1.6 μM HOCl, which is well below the LD50 concentration of 6–17 μM for B7A (Fig. 1) even at a 9-fold higher DNA concentration than in the in vitro experiment (50 μg/mL vs. ~6–7 μg/mL). This points to an HOCl reaction that degrades PT-containing nucleotides. However, treatment of wild-type B7A and Cerro 87 cells with LD50 doses of either HOCl or H2O2 did not cause a reduction in the steady-state level of PT (Fig. 2b). This suggests that HOCl does not access DNA in vivo, that PT is unreactive with HOCl in vivo, or that damaged PT in cells is repaired or replaced.
Figure 2. PT reaction with HOCl leads to loss of PT and strand breaks in vitro and in vivo.
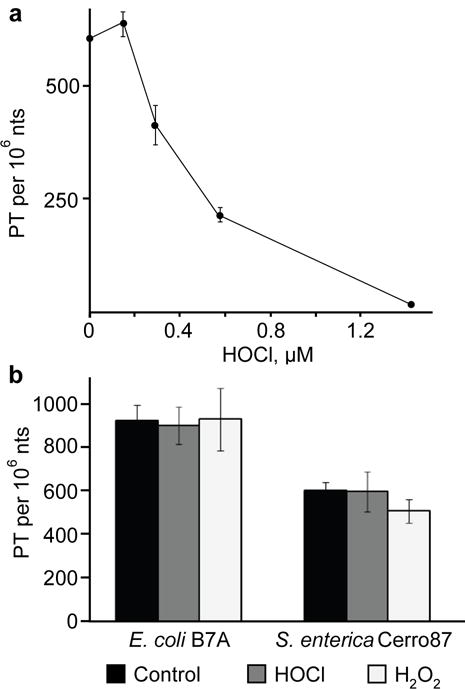
(a) Quantification of PT in wild-type E. coli B7A DNA exposed to HOCl in vitro. Data represent mean ± SD for 3 biological replicates. (b) PT-containing dinucleotides were quantified by LC-MS/MS 3 hours (E. coli wild-type) and 5 hours (S. enterica wild-type) after exposure to their respective LD50 doses of either HOCl or H2O2 (as specified in Supplementary Table 3; data represent mean ± SD for 3 biological replicates).
To begin to resolve these mechanistic possibilities, we characterized the chemical mechanisms driving PT-dependent sensitivity to HOCl exposure. The reactions are summarized in Fig. 3, which represents the most comprehensive model for PT oxidation chemistry to date. The first step was to use chromatography-coupled mass spectrometry (LC-MS) to define the products arising in reactions of d(GPSA) and d(GPST) dinucleotides with H2O2 and HOCl. Reaction of d(GPSA) with either H2O2 or HOCl resulted in a dose-dependent disappearance of the PT-containing dinucleotide and formation of desulfurated dinucleotide d(GPOA), a phosphonate-bridged dinucleotide d(GPHA), and free 2-deoxyguanosine (dG) and 2′-deoxyadenosine (dA) (Fig. 3; Supplementary Fig. 4a). The formation of desulfurated and phosphonate products in the dinucleotide reactions is consistent with previously reported observations with H2O2.18 Higher concentrations of H2O2 than HOCl were required for d(GPSA) consumption and d(GPOA) and d(GPHA) formation (Supplementary Fig. 4c), with consumption of the latter products at higher HOCl concentrations. This is consistent with the higher rate constant for HOCl reaction with thiols, such as cysteine, compared to H2O2 (2.9 vs. 3 × 10−7 M−1s−1; pH 7.4, 37°C).26,27 Our observation of complete PT consumption by four equivalents of HOCl (corresponds to 1 μM HOCl in Fig. 3) is consistent with the similar reactivity of HOCl with thiols such as cysteine.28 The heightened reactivity of HOCl compared to H2O2 is likely explained by the fact that, while both H2O2 and HOCl can be reduced to form hydroxyl radical and other reactive species (left pathway in Fig. 3), HOCl is also capable of halogenating thiols by direct reaction with the nucleophilic sulfur (right pathway in Fig. 3).29 This direct halogenation reaction is faster than HOCl reduction to release free radical species.30,31 To determine if the phosphonate in d(GPHA) is a precursor of the desulfurated phosphate in d(GPOA), HPLC-purified d(GPHA) was reacted with H2O2 and HOCl, with LC-MS analysis ruling out d(GPOA) formation and establishing d(GPOA) and d(GPHA) as independent reaction products. While these results suggest that the halogenation pathway (right side Fig. 3) will predominate at biologically relevant HOCl concentrations, DNA strand-breaks represent a common feature of both the phosphonate and halogenation pathways. Though it is highly unlikely that HOCl-induced nucleobase halogenation products (e.g., 5-chlorocytosine, 8-chloroguanine, 8-chloroadenine) influence the oxidation of PTs by HOCl, we note that no products of nucleobase halogenation were detected in MS CID scans or in UV absorbance chromatograms (in-line HPLC; 260 nm) of HOCl oxidation of PT-containing dinucleotides (Supplementary Fig. 4). Small amounts of nucleobase halogenation products are likely to have arisen in the reactions, but are apparently below the detection limit of the analyses performed for the PT oxidation products.
Figure 3. Comprehensive model for PT oxidation and alkylation in DNA.
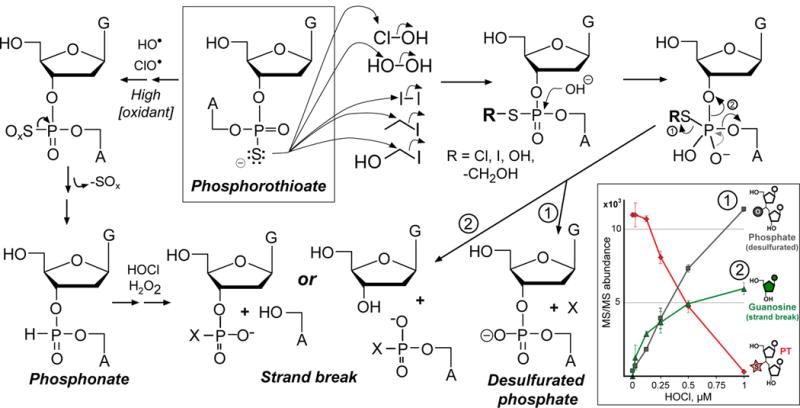
Analysis of degradation products arising in reactions of HOCl and H2O2 with PT-containing dinucleotides (inset and Supplementary Fig. 4) revealed products consistent with the reaction model shown. The sulfur in PTs can undergo two types of reactions based on nucleophilic substitution (right side) and one-electron oxidation (left side). At low concentrations of HOCl or H2O2, sulfur reacts by nucleophilic substitution to generate sulfonyl chloride (Cl-S) or sulfenic acid (HO-S), which subsequently undergo hydrolysis to eliminate the modified sulfur (pathway 1; X = sulfur-containing product) or to form a DNA strand-break (pathway 2) possibly with a modified sulfur (X). At high concentrations (left side), both H2O2 and HOCl generate radicals (hydroxyl radical, hypochoryl radical) that perform a series of one-electron oxidations leading to phosphonate formation and subsequent DNA strand-breaks containing a phosphonate (X = H), phosphate (X = O), or other product. Inset: Concentration-dependent degradation of d(GPSA) by HOCl causes both desulfuration to a phosphate-linked dinucleotide and a strand-break with release of dG; data represent mean ± SD for 3 replicates.
Two sets of studies were performed to assess the formation of DNA strand-breaks caused by PT oxidation by HOCl and H2O2 in vitro and in vivo. First, DNA isolated from wild-type and ΔdndB–H E. coli strains was exposed to the oxidants, including I2 as positive control for PT-dependent strand breaks,6 and resolved on a 0.7% agarose gel as a low-sensitivity method to detect DNA strand breaks as smearing. As shown in Fig. 4a, treatment of PT-containing DNA with I2 and micromolar concentrations of HOCl caused extensive genomic DNA degradation and smearing, which was not apparent in DNA lacking PTs. Strand-break-induced smearing is not apparent for H2O2-exposed DNA at concentrations up to 7.8 mM (Fig. 4a). The basis for the higher reactivity of H2O2 with PT in dinucleotides compared to DNA is uncertain but may relate to contamination with redox-active metals in the synthetic dinucleotides or issues of DNA secondary structure. However, similar behavior was observed in bacteria exposed for 10 min to HOCl and H2O2 concentrations 7.5- and 25-fold higher than LD50 doses (E. coli B7A wild-type and ΔdndB–H shown in Fig. 4b). The DNA damage caused by HOCl was complete within 10 min of exposure, as judged by the lack of additional DNA strand-breaks in longer incubations (Supplementary Fig. 5). The basis for the lack of DNA damage by H2O2 may relate to the presence of catalase or other H2O2 degrading activities. We conclude that HOCl, but not H2O2, produces significant amounts of PT-dependent DNA damage at biologically relevant concentrations. The evidence for HOCl-induced PT desulfuration and DNA strand-breaks coupled with unchanging levels of PT following HOCl exposure suggest a mechanism of PT damage and repair or replacement in response to the genomic instability introduced by PT.
Figure 4. PT reaction with HOCl leads to loss of PT and strand breaks in vitro and in vivo.
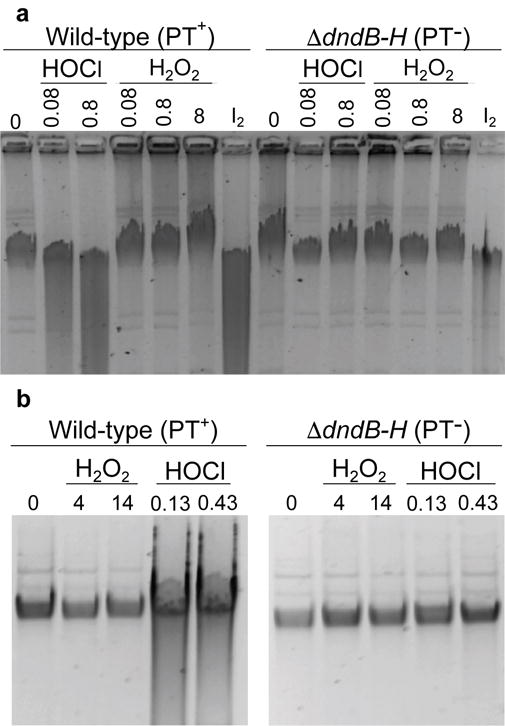
(a) Isolated DNA from wild-type (PT+) and ΔdndB–H (PT–) E. coli were exposed to 0.08–0.8 mM HOCl or 0.08–8 mM H2O2; iodine (I2) exposure serves as a positive control for PT-dependent strand breaks. HOCl-induced strand-breaks are apparent as smearing in the lane for DNA containing PT (PT+), but not for DNA lacking PT (PT–); strand-breaks are not detectable for H2O2 exposure in any case. (b) WT and ΔdndB–H E. coli cells were exposed to 7.5- and 25-times the WT LD50 concentration of H2O2 or HOCl for 10 min (4 and 14 mM for H2O2; 0.13 and 0.43 for HOCl). Again, DNA isolated from HOCl-exposed WT bacteria, but not PT– bacteria, shows strand-breaks, while no strand breaks are apparent after H2O2 treatment in either strain. Gel images are cropped for clarity (see Supplementary Fig. 9 for unprocessed images). Data for LD50 and LD80 doses are presented in Supplementary Table 3.
PTs are labile and turnover during normal growth
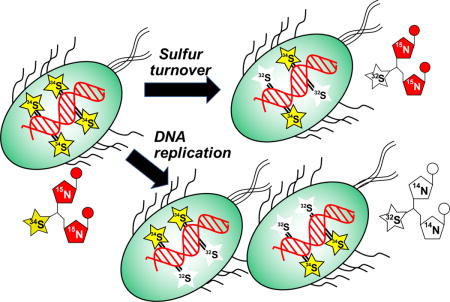
The evidence for a constant steady-state level of PTs in oxidatively stressed cells in the face of oxidative PT desulfuration and strand break formation suggests a repair-like turnover of PTs. To test this model, we developed an isotope labeling and mass spectrometry method to quantify PT dynamics in vivo, as illustrated in Figure 5a. Bacteria are first grown in a minimal medium with nitrogen and sulfur sources containing [15N] and [34S], respectively, to label DNA nucleobases with [15N] and PTs [34S].32 Experiments are initiated by changing the medium to one containing [14N]- and [32S]-labeled nutrients and then quantifying the various isotopomers of the PT dinucleotides by LC-MS/MS. Following normal DNA replication, nucleobases in the new DNA strand will be [14N]-labeled and new PTs will contain [32S] (“new PT” in Fig. 5a). If the sulfur in a PT has been removed and replaced in the original labeled DNA strand, then the PT-containing dinucleotide will have [15N]-labeled nucleobases and a [32S]-labeled PT (“turnover PT” in Fig. 5a). Finally, we also observed the presence of dinucleotides containing [34S] and [14N] (“cryptic PT” in Fig. 5a) which points to a recycling of [34S] either from residual isotopically labeled compounds in the nutrient pool or by transfer from other labeled PTs in the DNA. Mass spectra for original, new and turnover isotopomers of d(GpsT) are shown in Supplementary Fig. 6 and the LC-MS parameters for all isotopomers of d(GpsA) and d(GpsT) are shown in Supplementary Table 2. Using [13C]-labeled PT dinucleotides as internal standards for isotope-dilution mass spectrometry (Supplementary Table 2), we were able to quantify (PT per 106 nt) all PT isotopomeric dinucleotides within a single DNA sample.32
Figure 5. Quantification of PT turnover by isotope-labeling and mass spectrometry.
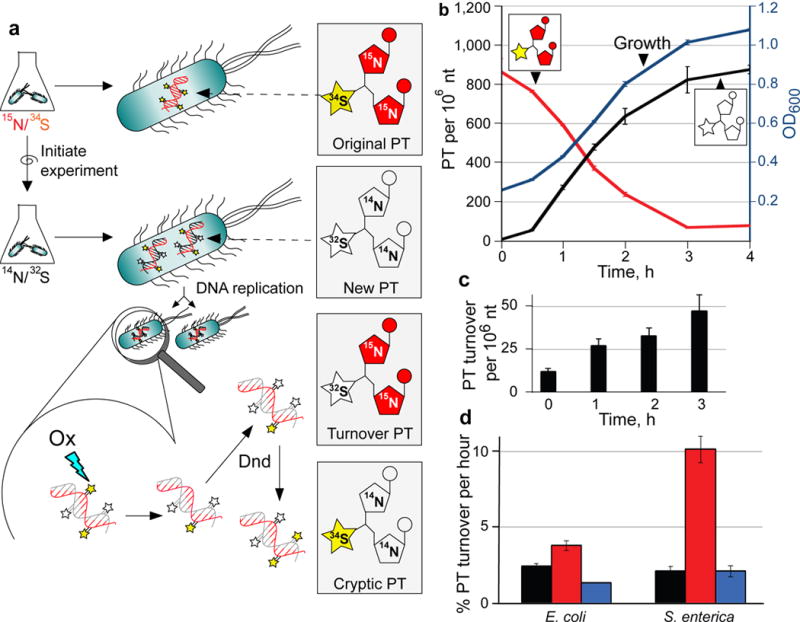
(a) Principle of PT turnover assay. Bacteria were grown in minimal medium containing [15N]/[34S]-labeled nutrients, with resulting bacterial DNA containing [15N]-labeled nucleobases with [34S]-labeled PTs (Original PT). To initiate an experiment, cells are transferred to minimal medium with [14N]/[32S]-labeled constituents and subjected to stresses. Newly synthesized PT-linked dinucleotides (New PT) are labeled with [14N] and [32S]. The [34S] in the original PTs was found to undergo replacement with [32S] (Turnover PT) in both untreated and oxidant-treated cells. We also observed low levels of PT dinucleotides labeled with [34S] and [14N] (Cryptic PT) resulting from reuse of [34S] from nutrient pools or other PT modification sites. In all cases, isolated DNA was hydrolyzed to canonical mononucleotides and PT-containing dinucleotides, and quantities of various isotope-labeled dinucleotides were determined by LC-MS/MS analysis. (b) Validation of PT turnover method. Cultures of wild-type E. coli B7A were grown with [15N]/[34S]-containing medium, and then cells were transferred to [14N]/[32S]-containing medium. At various times, genomic DNA was isolated and subjected to LC-MS/MS analysis of PT-containing dinucleotides. The plot shows cell growth (OD600; blue line), original PT (red line) and new PT (black line). (c) The level of [15N]/[33S]-labeled, PT-containing dinucleotides (“turnover” PT) were determined in wild-type, untreated E. coli B7A as a function of time. The level of this turnover PT as a function of total PT was used to calculate the %PT turnover. (d) Estimates of the rate of PT turnover per hour were calculated by performing linear regression analysis of plots of %PT turnover as a function of time in unstressed E. coli B7A and S. enterica Cerro 87 (black) and for bacteria exposed to HOCl (red) and H2O2 (blue), with regression analysis illustrated in Supplementary Figure 7 for wild-type, untreated Cerro 87. Data represent mean ± SD for 3 biological replicates and values for HOCl-treated samples are significantly different from untreated and H2)2-treated by Student’s t-test at P < 0.05.
The isotope labeling method was validated in log-growing, unstressed E. coli. As shown in Fig. 5b, the quantity of heavy isotope-labeled PTs in [15N]/[34S]-d(GpsA) and [15N]/[34S]-d(GpsT) at the start of the experiment amounted to 865 ± 67 PTs per 106 nt. Growing the bacteria in unlabeled medium resulted in a decrease in the level of the original, heavy isotope-labeled PTs (red line in Fig. 5b) as DNA replication occurred. As expected, the level of unlabeled PTs ([14N]/[32S]-d(GpsA) and [14N]/[32S]-d(GpsT)) increased from zero to a maximal steady-state level of 875 ± 21 PT per 106 nt (black line in Fig. 5b). The rate of increase of unlabeled PTs paralleled the OD600 metric for bacterial growth (blue line in Fig. 5b), which is consistent with incorporation of PT during or after DNA replication. The level of heavy isotope-labeled PTs ([15N]/[34S]-d(GpsA) and [15N]/[34S]-d(GpsT)) in the parental labeled DNA strand, originally at 865 ± 67 PTs per 106 nt, remained at nearly this level for many cell divisions as expected given conservation of the parental DNA strand in progeny bacteria. However, as shown in Fig. 5c, we observed a consistent conversion of [34S] to [32S] in PT-containing dinucleotides derived from the original [15N]/[34S]-labeled DNA (“turnover” PT in Fig. 5c). This shift demonstrates that PT modifications are labile and that there is a natural loss of PTs and a replacement or repair by Dnd proteins during normal metabolism in log-growing cells. Analysis of the PT replacement (i.e., turnover) over time revealed a nearly linear relationship (Supplementary Fig. 7) consistent with a roughly constant rate of PT turnover events at 2% hr−1 in S. enterica. These results demonstrate the dynamic nature of PT modifications in DNA.
HOCl causes PT turnover at biologically relevant doses
If PTs are more reactive with HOCl than H2O2, as suggested by the cytotoxicity and DNA damage results, then we would expect HOCl exposure to cause a higher rate of PT turnover reflecting increased damage and subsequent repair by Dnd proteins. We tested this hypothesis by applying the PT turnover assay following exposure of wild-type E. coli and S. enterica to biologically relevant (LD50) doses of HOCl and H2O2. Oxidant-induced changes in the percentage of replaced PTs as a function of time are shown in Supplementary Fig. 8 and the PT turnover rates derived from linear regression of these data are plotted in Fig. 5d. Exposure of E. coli and S. enterica to H2O2 did not significantly (p=0.97) affect PT turnover, with untreated and H2O2-treated S. enterica showing turnover rates of 2.1% (± 0.3%) hr−1 and 2.1% (± 0.4%) hr−1, respectively. Similarly, H2O2 exposure did not change PT turnover in E. coli B7A: 2.5% (± 0.2%) hr−1 and 1.4% (± 0.04%) hr−1 for untreated and H2O2-treated cells, respectively. These results are consistent with lack of PT-dependent H2O2 toxicity in wild-type and ΔdndB–H strains of both E. coli and S. enterica (Fig. 1). However, HOCl treatment significantly increased the PT turnover to 3.8% (± 0.3%; p<0.005) hr−1 in E. coli and 10% (± 0.8%; p<0.001) hr−1 for S. enterica. This increase can be explained by desulfuration of PT to form phosphate17 or by repair of HOCl-induced strand breaks, followed by PT reinsertion with [32S] by the Dnd enzymes. The results are also consistent with the greater HOCl sensitivity of the wild-type, PT-containing bacteria compared to ΔdndB–H strains in the survival (Fig. 1) and DNA damage (Fig. 2 and Fig. 4) assays.
DISCUSSION
Here we have shown that PTs are labile DNA modifications that compromise bacterial fitness in environments that risk oxidative stress and exposure to hypohalous acids. Given the widespread horizontal transfer of dnd genes among bacteria and archaea, including human pathogens,10,11 PTs clearly confer benefits to bacteria in terms of restriction-modification activity that protects against pathogenic DNA invasion or by virtue of controlling gene expression in the sizable portion of organisms that lack restriction enzymes.9 In spite of these benefits, however, we have demonstrated a direct chemical link between PTs and lethal genomic instability caused by exposure to HOCl, but not H2O2, at levels encountered in many biologically relevant environments. One of the most well defined environments for HOCl exposure involves granulocytes of the human innate immune system, in which activated neutrophils produce up to 10–15 μM H2O233 and 30–40 μM HOCl.34 This level of HOCl is well above the LD50 values we determined for wild-type E. coli (17 μM) and S. enterica (6 μM). Eosinophil peroxidease similarly produces large quantities of HOBr,35 as well as NO2 radical and hypothiocyanous acid (HOSCN), all of which are potentially capable of oxidizing PTs. This raises questions about the fitness cost of PTs in terms of virulence for human pathogens possessing dnd genes. These include the 50% of clinical isolates of Mycobacterium abscessus10 and the 4% of clinical isolates of Pseudomonas aeruginosa11 that possess PTs. HOCl, HOBr and HOI are also frequently encountered in biofilms, soil and marine environments. Haloperoxidases are widely distributed in bacteria and fungi, and produce biocidal levels of hypohalous acids in competition for resources.36–38 For bacteria without the PT modifications, the extensive production of extracellular HOX would be a clear advantage.
Even without exogenous exposure to oxidants, PT modifications in the bacterial strains studied here turn over relatively frequently, with ~2% of PTs replaced every hour, the time of one cell division in minimal media. There is no known PT removal enzyme, so it is likely that PT turnover results from endogenous oxidants, metals, or alkylating agents modifying the PT sulfur, with this damage leading to desulfuration, phosphonate formation, or DNA strand-break chemistry that we observed in vitro (Fig. 3). Another possible scenario to explain PT turnover is that PTs move from one location to another, though maintaining a constant steady-state level. While this scenario is plausible for bacteria lacking PT-dependent restriction activity, which represent a significant portion of PT-containing bacteria,6 this would seem unlikely when sequence-specific restriction activity is present as in the E. coli and S. enterica strains studied here. Unlike chemically stable methyl modifications of DNA, the PT modifications thus present the first case of a physiological DNA modification that is subject to damage and repair.
In parallel with this PT damage and repair activity, PT oxidation produces significant levels of DNA damage, which almost certainly contributes to the fitness cost of bearing PT modifications. Exposure to HOCl caused high levels of DNA strand-breaks that were detectable with low-sensitivity agarose gel analysis, while H2O2 exposure did not produce detectable damage (Fig. 4). Though our characterization of PT oxidation chemistry is the most comprehensive to date (Fig. 3), the presence of undefined chemical structures at some of the 5′- and 3′-ends of the strand-breaks could alter DNA repair efficiency. Notably, the formation of phosphonates in the intact DNA backbone is completely unexplored in terms of effects on protein binding, the existence of repair mechanisms, and their genetic toxicology. The few reports that exist for methylphosphonates (H replaced with CH3, Fig. 3) point to significant changes in DNA structure, with bending and deviations from B-like structure near the phosphonate, which could contribute to toxic disruption of DNA replication and transcription.39,40 Nonetheless, while we do not know the chemistry of the PT-dependent strand-break ends, it is possible that these modified ends could be removed by known 5′- and 3′-trimming DNA repair activities, such as endonuclease IV or exonuclease III for 3′-termini clean-up and DNA pol I for both 3′- and 5′-termini clean-up.41 The PT oxidation products may thus present novel challenges to the DNA repair machinery in bacteria, with damage occurring even under unstressed conditions as a result of PT reaction with endogenous oxidants and alkylating agents. Finally, HOCl is known to produce other types of PT-independent DNA damage, such as halogenated and oxidized nucleobases (e.g., 5-chlorocytosine, 8-chloroguanine, 8-oxoguanine, 8-chloroadenine). For several reasons, it is highly unlikely that these nucleobase damage products play a role in the differences in HOCl toxicity or DNA strand-break formation in wild-type bacteria compared to bacteria lacking PT modification genes. First, repair of halogenated nucleobases as a cause of the DNA strand-breaks observed in HOCl-treated cells is unlikely given the low level of nucleobase damage expected in the bacteria. Previous studies demonstrated that treating mammalian cells with HOCl at 500 μM led to levels of halogenated nucleobases ranging over 1–10 lesions per 105 nt, with damage in RNA only 50% higher.42 This is too low to account for the high levels of DNA strand-breaks observed at lower HOCl concentrations on the insensitive agarose gels in Figure 4. More importantly, the HOCl-induced DNA strand-breaks were entirely PT-dependent, as was the increase in cytotoxicity in the PT-containing bacterial strains.
We have demonstrated that PTs are highly dynamic DNA modifications that, while conferring benefits to host bacteria, compromise bacterial fitness due to lethal genomic instability under commonly encountered chemical stresses. How the lability of PTs affects the ecological niche of bacteria remains a mystery awaiting quantitative analysis. Along with questions about the evolutionary adaptation of PT-containing bacteria to hostile environments,43 our observations have implications for the virulence of bacterial pathogens during human infections and for bacterial fitness in other environments.
ONLINE Methods
Media and strains
Modified M9 medium was prepared by mixing individual sterile-filtered solutions of 100 mM MgCl2, 100 mM Na2SO4, 100 mM CaCl2 and 20% glucose, 100 mM Na2[34S]O4, 20% [13C6]-glucose. 10× M9 medium (68 g/L Na2HPO4, 50 g/L KH2PO4, 2.5 g/L NaCl and 10 g/L NH4Cl) and 10× [15N]-M9 medium (68 g/L Na2HPO4, 50 g/L KH2PO4, 2.5 g/L NaCl and 10 g/L [15N]-NH4Cl) were prepared and sterilized by autoclaving. These M9 media were mixed in varying proportions to achieve the noted isotopic combinations as a 1× M9 medium mixture (Final concentrations: 1× M9 salts, 2 mM MgCl2, 2 mM Na2SO4, 0.1 mM CaCl2 and 0.4% glucose). All salts were purchased from Sigma-Aldrich-Isotec. LB-agar plates were prepared by autoclaving 25 g/L Difco LB (Becton Dickinson) together with 20 g/L agar (Becton Dickinson) and pouring into 100×15mm plastic plates (VWR). Plates were stored at 4 °C until use. Supplementary Table 1 lists all strains of bacteria, their genotypes and their sources.
Bacterial growth assays
Single colonies of the respective bacterial strain were grown in 5 mL isotopically unlabeled M9 medium overnight at 37 °C. Cells (2.7 mL) were pelleted in 50 mL falcon tubes at 3000×g for 8 min at ambient temperature (unless indicated otherwise) and resuspended with fresh M9 medium to a starting optical density at 600 nm (OD600) of 0.2, followed by growth at 37°C, 230 rpm for 60 min. H2O2 and HOCl solutions were prepared from chemical stocks (Sigma-Aldrich) maintained at 4 °C in the dark, with dilution of the stocks in sterile water immediately before use. The concentrations of stock solutions were determined before use by measuring the absorbance of HOCl as sodium hypochlorite in 0.01 M NaOH at 292 nm (ε = 350 M−1cm−1)44 and H2O2 in water at 240 nm (ε = 43.6 M−1cm−1)45. The stocks were diluted with sterile water immediately before the experiment and distributed in flat-bottom 96 well-plates. Cell stocks were added to each well containing the oxidant, mixed rapidly, and growth was monitored in 10 min increments at 600 nm in an ELX808iu plate-reader (BIO-TEK Instrument Inc.) set for 37 °C. The OD of each biological replicate was averaged (N=3) and plotted with the standard deviation.
Survival assay and LD50 calculation
Cells were prepared as described in the growth curve assay. After 60 min oxidant exposure, cells were removed and diluted 1:105 with fresh M9 and 70 μL plated using 4 mm glass-beads (VWR 26396-456) on LB-agar plates. Plates were incubated over-night at 37 °C and colonies counted using a FluorChem 8900 (Alpha Innotech). Colony counts were plotted over dose range and the LD50 dose was calculated from the logarithmic regression equation for each biological replicate (N=3), with data plotted as mean ± standard deviation.
HOCl exposure of PT-containing dinucleotides
For detection of reaction products, 104 pmol PT-bridged dinucleotides d(GPSA) and d(GPST) with the natural Rp configuration (IBA Lifesciences) were treated with 20-fold excess HOCl and reaction products were identified by LC-MS/MS using MS2Scan mode (see section HPLC-MS). d(GPOT) and d(GPOA) dinucleotides were commercially available from IBA Lifesciences. A dose response on PT dinucleotide consumption was established by incubation of 2.6 pmol d(GPSA) with 0, 2.6, 5.2, 7.8 and 10.4 pmol HOCl (corresponding to 0–4 reaction equivalents) in 10 μL water for 60 min at 37 °C. The reaction was stopped by addition of 100 pmol H2O2 (OCl− + H2O2 → Cl− + H2O + O2) to protect the mass spectrometer from HOCl induced damage. To exclude possible synergistic effects of the H2O2/HOCl reaction,46–49 several tests without addition of H2O2 as stopping reagent were performed. No difference in PT reactivity was observed with or without addition of H2O2. The formation of previously established reaction products dG and d(GPOA) were monitored by LC-MS/MS.
Preparation of [13C]-labeled PT dinucleotides for absolute quantification of PT
Wild-type E. coli B7A was grown overnight in 5 mL [13C6]-glucose M9 medium and 1 mL of this culture was used to inoculate 100 mL [13C6]-glucose M9 medium for growth to OD600 ~1.4 at 37 °C and 230 rpm shaking. Bacteria were then centrifuged (3500×g) at 4 °C for 10 min. DNA was isolated with the Qiagen genomic tip 500G kit, according to manufacturer’s instructions. Isolated DNA (~1 mg) was digested with 10u Nuclease P1 (Sigma-Aldrich) in 30 mM NH4OAc pH 5.3 and 20 μM ZnCl2 at 55 °C for 5 h. Calf intestine alkaline phosphatase (34 units; Sigma-Aldrich) and 1/10 volume of 1 M Tris pH 8 were added, with incubation for an additional 4 h at 37 °C to release 2′-deoxynucleosides and PT dinucleotides. Enzymes were removed by centrifugation of the digest through a 10 kDa MWCO column at 12,000×g for 10 min. A 100 μL volume of the digest was injected onto a Hydro-RP HPLC column (4 μm particle size, 80 Å pore size, 250 mm length, 4.6 mm inner diameter; Phenomenex, Torrance, CA) with an Agilent 1200 HPLC with binary pump, degasser, autoinjector, DAD detector, and fraction collector. Ammonium acetate (5 mM, pH 5.3) was used as solvent A at 1 mL/min flow rate and 35 °C column temperature. Elution started with 3% acetonitrile (ACN; solvent B) for 5 min, increasing to 10% ACN after 30 min, followed by a 2 min column rinsing with 20% ACN and then back to 3% ACN at 34 min. Eluting 2′-deoxyribonucleosides and PT dinucleotides were monitored at 260 nm and collected using a fraction collector. Under these conditions, d(GPSA) eluted at 25.8 min and d(GPST) at 28.0 min. Fractions containing each dinucleotide were combined and lyophilized, and the concentration of PT was adjusted to roughly 100 fmol/μL; 10 mM theophylline was added as an external standard. The resulting internal standard mix is considered a 10× SILIS (stable isotope-labeled internal standard) that results in a final concentration of 10 fmol/μL in the sample.
LC-MS/MS analysis of PT-containing dinucleotides
Synthetic PT dinucleotides or nuclease P1 hydrolyzed DNA (1–10 μg depending on the growth state noted in each experiment) was analyzed by LC-MS/MS on an Agilent 1290 series HPLC system equipped with a Synergy Fusion RP HPLC column (2.5 μm particle size, 100 Å pore size, 100 mm length, 2 mm inner diameter) with a guard column (Phenomenex) and a diode array detector (DAD). The HPLC was coupled to an Agilent 6430 triple quadrupole mass spectrometer (MS). The column was eluted at 0.35 mL/min at 35 °C with a linear gradient of 3–8% acetonitrile in 97% solvent A (5 mM ammonium acetate buffer, pH 5.3) over 11 min. The column is rinsed with 40% acetonitrile in A for 1 min and initial conditions were regenerated by rinsing with 97% solvent A for 2 min. Canonical ribonucleosides eluting from the column were quantified by 260 nm UV absorbance using the DAD. PT-containing dinucleotides were identified and quantified by tandem quadrupole mass spectrometry, with electrospray ionization (ESI) operated with the following parameters: N2 temperature 350 °C, N2 flow 10 L/min, nebulizer pressure 40 psi, 3500 V capillary voltage and 2 V cell accelerator voltage. For product identification the MS was operated in positive ion by scanning a 200–1000 Da range or product ion scan mode, respectively. For quantification and PT metabolism assays, the MS was operated mode by monitoring multiple fragmentation reactions (MRM mode) under previously optimized MS conditions. The transitions and retention times used for identification of dinucleotides and nucleosides can be found in Supplementary Table 2.
HOCl exposure of isolated DNA
Genomic DNA (1 μg) from wild-type and ΔdndB–H E. coli were incubated with 0–60 pmol of HOCl in 20 μL total for 60 min at 37°C. The reaction was stopped by addition of 1/10 volume (2 μL) of 5 M NH4OAC and 10 volumes (22 μL) of isopropanol. The precipitated DNA was pelleted by centrifugation at 16,000×g for 15 min at 4 °C, washed with isopropanol, and dried under vacuum. Following resuspension in water, DNA was enzymatically hydrolyzed for PT quantification by LC-MS/MS according to the method below. For analysis by gel electrophoresis, DNA (10 μg) was incubated with 360 pmol HOCl as described above, precipitated, washed, resuspended in water with gel loading buffer, and resolved on a 0.7% agarose gel. As a positive control, the same amount of DNA was incubated with a final concentration of 3 mM iodine (Fluka, NY) and 50 mM Na2HPO4 buffer for 5 min at 65 °C to cause strand-breaks at PT sites, as described elsewhere.6
Analysis of DNA strand-breaks in HOCl- and H2O2-exposed bacteria
S. enterica wild-type, ΔdndB and ΔdndB–H, or E. coli B7A wild-type and ΔdndB–H were prepared as described for the survival assay. After 60 min of growth, cells were placed on ice and exposed to various doses of HOCl or H2O2. HOCl dependent strand-breaks were first observed at 7.5-fold×LD50 for each wild-type strain (see Supplementary Table 3), and these doses were then used for each corresponding ΔB–H strain. Effects caused by the genetic background of the strains and by the resulting difference in LD50 doses were ruled out by exposure of all bacterial strains to higher doses (25-fold×LD50 see Supplementary Table 3). Cells (in 1 mL LB) were pelleted at 8000×g for 2 min at 4 °C. Using reagents from the EZNA Bacterial DNA Isolation Kit (Omega Biotek), DNA was isolated by adding 2 μL lysozyme and 20 μL BTL digestion buffer for 10 min at 37 °C, followed by 20 μL proteinase K for 40 min at 55 °C and then 2 μL RNaseA for 2 min at 24 °C. The digestion mix was filtered through a 0.2 μm spin filter at 10,000×g for 10 min to remove insoluble cellular debris. A 40 μL aliquot was mixed with 5 μL of loading dye (6× dye from New England Biolabs) and loaded on a 0.7% agarose gel. A 1kb extended size marker from NEB was used. DNA was stained with ethidium bromide and visualized with FluorChem 8900 (Alpha Innotech).
PT metabolism assay
Cells from a single-colony were grown in 6 mL of [34S]- and [15N]-labeled M9 at 37 °C overnight. Cells in a 2.7 mL volume were pelleted in 50 mL polypropylene tubes at 3000×g for 8 min at ambient temperature and resuspended to a starting OD600 of 0.2 with fresh unlabeled M9 medium. Cells were then incubated at 37 °C with shaking (230 rpm) for 5 h. Aliquots (4 mL) of untreated cells were removed at 30, 60, 120, 180, 240 and 300 min and immediately pelleted and frozen at –80 °C. After 60 min of incubation, HOCl or H2O2 was added to an LD50 concentration in 24 mL of culture and 4 mL aliquots were then removed 60, 120, 180 and 240 min after adding the oxidants; aliquots were also removed for CFU-based analysis of bacterial survival after 60 min of exposure. 4 mL aliquots were pelleted at 10,000×g for 2 min at 4 °C and genomic DNA was isolated using PureLink Pro 96 Genomic DNA Purification Kits according to the manufacturer’s protocol (Thermo Fisher Scientific). The DNA was further purified by addition of 1/10 volume 5 M NH4OAc and 2 volumes isopropanol and precipitated 10 minutes at 3500×g. DNA pellets were resuspended in 30 μL of a digestion mix consisting of 0.3 units Nuclease P1 (Sigma-Aldrich), 30 mM NH4OAc pH 5.3, 20 μM ZnCl2. After 2 h at 55 °C, 2 units of calf intestine alkaline phosphatase (Sigma-Aldrich) and 1/10 volume of 1 M Tris (pH 8) were added, followed by incubation for 60 min at 37°C. Enzymes were removed by centrifugal-filtration of the digest through a 10 kDa MWCO column (VWR) at 12000×g for 10 min. A 3 μL volume of [13C6]-d(GpsA) or [13C6]-d(GpsT) SILIS was added to 27 μL of each sample digest and PT levels were quantified using LC-MS analysis.
Statistical analyses
Significance testing was performed using an unpaired, two-tailed Student’s t-test based on data from at least three replicate analyses with technical duplicates.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge funding from the Deutsche Forschungsgemeinschaft (SK), the US National Science Foundation (CHE-1019990), the US National Institute of Environmental Health Science (ES002109), the US National Institute of Allergy and Infectious Disease (AI112711), the National Natural Science Foundation of China (31630002), and the Singapore-MIT Alliance for Research and Technology sponsored by the National Research Foundation of Singapore.
Footnotes
AUTHOR CONTRIBUTIONS
SK, MSD, BR, BC and PCD planned the experiments. SK and MSD either directly performed or supervised all experiments. CPC performed gel experiments in Fig. 4B, Suppl. Fig. 5B, and Suppl. Fig. 6. BR performed experiments for Fig. 1. BC performed experiments involved in Suppl. Fig. 3. DY developed the bacterial strains used in all experiments. SK, MSD, BR, BC and PCD performed data analysis. SK, MSD, BR, BC, DY and PCD and wrote the manuscript.
ADDITIONAL INFORMATION
Supplementary Information is available. Correspondence and requests for materials should be addressed to PCD.
COMPETING INTERESTS
The authors declare no competing financial interest.
DATA AVAILABILITY
All data generated or analyzed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author on reasonable request.
References
- 1.Bayliss CD. Determinants of phase variation rate and the fitness implications of differing rates for bacterial pathogens and commensals. FEMS Microbiol Rev. 2009;33:504–520. doi: 10.1111/j.1574-6976.2009.00162.x. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez-Romero MA, Cota I, Casadesus J. DNA methylation in bacteria: from the methyl group to the methylome. Curr Opin Microbiol. 2015;25:9–16. doi: 10.1016/j.mib.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Tock MR, Dryden DT. The biology of restriction and anti-restriction. Curr Opin Microbiol. 2005;8:466–472. doi: 10.1016/j.mib.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Wilson GG, Murray NE. Restriction and modification systems. Ann Rev Genetics. 1991;25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
- 5.Thiaville JJ, et al. Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc Natl Acad Sci USA. 2016;113(11):E1452–E1459. doi: 10.1073/pnas.1518570113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao B, et al. Genomic mapping of phosphorothioates reveals partial modification of short consensus sequences. Nat Commun. 2014;5:3951. doi: 10.1038/ncomms4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, et al. Phosphorothioation of DNA in bacteria by dnd genes. Nat Chem Biol. 2007;3:709–710. doi: 10.1038/nchembio.2007.39. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, et al. DNA phosphorothioation is widespread and quantized in bacterial genomes. Proc Natl Acad Sci USA. 2011;108:2963–2968. doi: 10.1073/pnas.1017261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, Chen S, Deng Z. In: DNA Replication - Current Advances. Seligmann H, editor. Intech; 2011. [Google Scholar]
- 10.Howard ST, et al. Insertion site and distribution of a genomic island conferring DNA phosphorothioation in the Mycobacterium abscessus complex. Microbiology. 2013;159:2323–2332. doi: 10.1099/mic.0.070318-0. [DOI] [PubMed] [Google Scholar]
- 11.Romling U, Tummler B. Achieving 100% typeability of Pseudomonas aeruginosa by pulsed-field gel electrophoresis. J Clin Microbiol. 2000;38:464–465. doi: 10.1128/jcm.38.1.464-465.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.An X, et al. A novel target of IscS in Escherichia coli: participating in DNA phosphorothioation. PLoS One. 2012;7:e51265. doi: 10.1371/journal.pone.0051265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu T, Yao F, Zhou X, Deng Z, You D. A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Res. 2010;38:7133–7141. doi: 10.1093/nar/gkq610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gan R, et al. DNA phosphorothioate modifications influence the global transcriptional response and protect DNA from double-stranded breaks. Sci Rep. 2014;4:6642. doi: 10.1038/srep06642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gish G, Eckstein F. DNA and RNA sequence determination based on phosphorothioate chemistry. Science. 1988;240:1520–1522. doi: 10.1126/science.2453926. [DOI] [PubMed] [Google Scholar]
- 16.Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Laird DA. Abiotic transformation of chlorpyrifos to chlorpyrifos oxon in chlorinated water. Environ Toxicol Chem. 2003;22:261–264. [PubMed] [Google Scholar]
- 18.Xie X, et al. Phosphorothioate DNA as an antioxidant in bacteria. Nucleic Acids Res. 2012;40:9115–9124. doi: 10.1093/nar/gks650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schatz D, Leberman R, Eckstein F. Interaction of Escherichia coli tRNA(Ser) with its cognate aminoacyl-tRNA synthetase as determined by footprinting with phosphorothioate-containing tRNA transcripts. Proc Natl Acad Sci USA. 1991;88:6132–6136. doi: 10.1073/pnas.88.14.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao B, et al. Pathological phenotypes and in vivo DNA cleavage by unrestrained activity of a phosphorothioate-based restriction system in Salmonella. Molec Microbiol. 2014;93:776–785. doi: 10.1111/mmi.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drazic A, et al. Role of cysteines in the stability and DNA-binding activity of the hypochlorite-specific transcription factor HypT. PLoS One. 2013;8:e75683. doi: 10.1371/journal.pone.0075683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drazic A, Kutzner E, Winter J, Eisenreich W. Metabolic Response of Escherichia coli upon Treatment with Hypochlorite at Sub-Lethal Concentrations. PLoS One. 2015;10:e0125823. doi: 10.1371/journal.pone.0125823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gundlach J, Winter J. Evolution of Escherichia coli for maximum HOCl resistance through constitutive expression of the OxyR regulon. Microbiology. 2014;160:1690–1704. doi: 10.1099/mic.0.074815-0. [DOI] [PubMed] [Google Scholar]
- 24.Winter J, Ilbert M, Graf PC, Ozcelik D, Jakob U. Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell. 2008;135:691–701. doi: 10.1016/j.cell.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.M9 minimal medium (standard) Cold Spring Harbor Prot. 2010;2010 pdb.rec12295. [Google Scholar]
- 26.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 27.Pattison DI, Davies MJ. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem Res Toxicol. 2001;14:1453–1464. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 28.Winterbourn CC. Comparative reactivities of various biological compounds with myeloperoxidase-hydrogen peroxide-chloride, and similarity of the oxidant to hypochlorite. Biochim Biophys Acta. 1985;840:204–210. doi: 10.1016/0304-4165(85)90120-5. [DOI] [PubMed] [Google Scholar]
- 29.Armesto XL, Canle LM, Fernández MI, García MV, Santaballa JA. First Steps in the Oxidation of Sulfur-Containing Amino Acids by Hypohalogenation: Very Fast Generation of Intermediate Sulfenyl Halides and Halosulfonium Cations. Tetrahedron. 2000;56:1103–1109. [Google Scholar]
- 30.Folkes LK, Candeias LP, Wardman P. Kinetics and mechanisms of hypochlorous acid reactions. Arch Biochem Biophys. 1995;323:120–126. doi: 10.1006/abbi.1995.0017. [DOI] [PubMed] [Google Scholar]
- 31.Prutz WA. Hypochlorous acid interactions with thiols, nucleotides, DNA, and other biological substrates. Arch Biochem Biophys. 1996;332:110–120. doi: 10.1006/abbi.1996.0322. [DOI] [PubMed] [Google Scholar]
- 32.Kellner S, et al. Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res. 2014;42:e142. doi: 10.1093/nar/gku733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Test ST, Weiss SJ. Quantitative and temporal characterization of the extracellular H2O2 pool generated by human neutrophils. J Biol Chem. 1984;259:399–405. [PubMed] [Google Scholar]
- 34.Bergt C, et al. Human neutrophils employ the myeloperoxidase/hydrogen peroxide/chloride system to oxidatively damage apolipoprotein A–I. Eur J Biochem. 2001;268:3523–3531. doi: 10.1046/j.1432-1327.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Slungaard A. Role of eosinophil peroxidase in host defense and disease pathology. Arch Biochem Biophys. 2006;445:256–260. doi: 10.1016/j.abb.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Bengtson P, Bastviken D, de Boer W, Oberg G. Possible role of reactive chlorine in microbial antagonism and organic matter chlorination in terrestrial environments. Environ Microbiol. 2009;11:1330–1339. doi: 10.1111/j.1462-2920.2009.01915.x. [DOI] [PubMed] [Google Scholar]
- 37.Zamocky M, Gasselhuber B, Furtmuller PG, Obinger C. Turning points in the evolution of peroxidase-catalase superfamily: molecular phylogeny of hybrid heme peroxidases. Cell Mol Life Sci. 2014;71:4681–4696. doi: 10.1007/s00018-014-1643-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofrichter M, Ullrich R. Heme-thiolate haloperoxidases: versatile biocatalysts with biotechnological and environmental significance. Appl Microbiol Biotechnol. 2006;71:276–288. doi: 10.1007/s00253-006-0417-3. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, et al. Stereospecific effects determine the structure of a four-way DNA junction. Chem Biol. 2005;12:217–228. doi: 10.1016/j.chembiol.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 40.Soliva R, et al. Solution structure of a DNA duplex with a chiral alkyl phosphonate moiety. Nucleic Acids Res. 2001;29:2973–2985. doi: 10.1093/nar/29.14.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krwawicz J, Arczewska KD, Speina E, Maciejewska A, Grzesiuk E. Bacterial DNA repair genes and their eukaryotic homologues: 1. Mutations in genes involved in base excision repair (BER) and DNA-end processors and their implication in mutagenesis and human disease. Acta Biochim Pol. 2007;54:413–434. [PubMed] [Google Scholar]
- 42.Badouard C, et al. Detection of chlorinated DNA and RNA nucleosides by HPLC coupled to tandem mass spectrometry as potential biomarkers of inflammation. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;827:26–31. doi: 10.1016/j.jchromb.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 43.van der Veen S, Tang CM. The BER necessities: the repair of DNA damage in human-adapted bacterial pathogens. Nat Rev Microbiol. 2015;13:83–94. doi: 10.1038/nrmicro3391. [DOI] [PubMed] [Google Scholar]
ONLINE METHODS REFERENCES
- 44.Perez-Cruz F, et al. Use of pyrogallol red and pyranine as probes to evaluate antioxidant capacities towards hypochlorite. Molecules. 2013;18:1638–1652. doi: 10.3390/molecules18021638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beers RF, Jr, Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- 46.Held AM, Halko DJ, Hurst JK. Mechanisms of chlorine oxidation of hydrogen peroxide. J Am Chem Soc. 1978;100:5732–5740. [Google Scholar]
- 47.Evans DF, Upton MW. Studies on singlet oxygen in aqueous solution. Part 1. Formation of singlet oxygen from hydrogen peroxide with two-electron oxidants. J Chem Soc Dalton Trans. 1985:1141–1145. [Google Scholar]
- 48.Shams El Din AM, Mohammed RA. Kinetics of the reaction between hydrogen peroxide and hypochlorite. Desalination. 1998;115:145–153. [Google Scholar]
- 49.Lontsi Djimeli C, et al. Mixture of Sodium Hypochlorite and Hydrogen Peroxide on Adhered Aeromonas hydrophila to Solid Substrate in Water: Impact of Concentration and Assessment of the Synergistic Effect. Int J Bacteriol. 2014;2014:121367. doi: 10.1155/2014/121367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.