

Abstract
Key points
An early inflammatory response and oxidative stress are implicated in the signal transduction that alters both hepatic redox status and mitochondrial function after traumatic brain injury (TBI).
Peripheral oxidative/inflammatory responses contribute to neuronal dysfunction after TBI
Exercise training alters the profile of oxidative‐inflammatory status in liver and protects against acute hyperglycaemia and a cerebral inflammatory response after TBI.
Approaches such as exercise training, which attenuates neuronal damage after TBI, may have therapeutic potential through modulation of responses by metabolic organs.
The vulnerability of the body to oxidative/inflammatory in TBI is significantly enhanced in sedentary compared to physically active counterparts.
Abstract
Although systemic responses have been described after traumatic brain injury (TBI), little is known regarding potential interactions between brain and peripheral organs after neuronal injury. Accordingly, we aimed to investigate whether a peripheral oxidative/inflammatory response contributes to neuronal dysfunction after TBI, as well as the prophylactic role of exercise training. Animals were submitted to fluid percussion injury after 6 weeks of swimming training. Previous exercise training increased mRNA expression of X receptor alpha and ATP‐binding cassette transporter, and decreased inducible nitric oxide synthase (iNOS), cyclooxygenase‐2 (COX‐2), tumor necrosis factor (TNF)‐α and interleukin (IL)‐6 expression per se in liver. Interestingly, exercise training protected against hepatic inflammation (COX‐2, iNOS, TNF‐α and IL‐6), oxidative stress (decreases in non‐protein sulfhydryl and glutathione, as well as increases in 2′,7′‐dichlorofluorescein diacetate oxidation and protein carbonyl), which altered hepatic redox status (increases in myeloperoxidase and superoxide dismutase activity, as well as inhibition of catalase activity) mitochondrial function (decreases in methyl‐tetrazolium and Δψ, as well as inhibition of citrate synthase activity) and ion gradient homeostasis (inhibition of Na+,K+‐ATPase activity inhibition) when analysed 24 h after TBI. Previous exercise training also protected against dysglycaemia, impaired hepatic signalling (increase in phosphorylated c‐Jun NH2‐terminal kinase, phosphorylated decreases in insulin receptor substrate and phosphorylated AKT expression), high levels of circulating and neuronal cytokines, the opening of the blood–brain barrier, neutrophil infiltration and Na+,K+‐ATPase activity inhibition in the ipsilateral cortex after TBI. Moreover, the impairment of protein function, neurobehavioural (neuromotor dysfunction and spatial learning) disability and hippocampal cell damage in sedentary rats suggests that exercise training also modulates peripheral oxidative/inflammatory pathways in TBI, which corroborates the ever increasing evidence regarding health‐related outcomes with respect to a physically active lifestyle.
Keywords: cognitive dysfunction, exercise training, fluid percussion injury, inflammation, liver, neuronal damage, redox status
Key points
An early inflammatory response and oxidative stress are implicated in the signal transduction that alters both hepatic redox status and mitochondrial function after traumatic brain injury (TBI).
Peripheral oxidative/inflammatory responses contribute to neuronal dysfunction after TBI
Exercise training alters the profile of oxidative‐inflammatory status in liver and protects against acute hyperglycaemia and a cerebral inflammatory response after TBI.
Approaches such as exercise training, which attenuates neuronal damage after TBI, may have therapeutic potential through modulation of responses by metabolic organs.
The vulnerability of the body to oxidative/inflammatory in TBI is significantly enhanced in sedentary compared to physically active counterparts.
Abbreviations
- ABCA1
ATP‐binding cassette transporter
- ALT
aminotransferase
- AST
aspartate aminotransferase
- BBB
blood–brain barrier
- CAT
catalase
- COX‐2
cyclooxygenase‐2
- CT
synthase
- DCFH‐DA
2′,7′‐dichlorfluorescein diacetate
- ELISA
enzyme‐linked immunosorbent assay
- FPI
fluid percussion injury
- GSH
reduced glutathione
- H&E
haematoxylin and eosin
- HOMA2%S
homeostasis model assessment
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IRS
insulin receptor substrate
- JNK
c‐Jun NH2‐terminal kinase
- LT
lactate threshold
- LXR‐α
liver X receptor
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- ROS
reactive oxygen species
- TBI
traumatic brain injury
- TNF
tumor necrosis factor
Introduction
Traumatic brain injury (TBI) is a public health concern highly related to morbidity and mortality. This neurological injury is characterized by a combination of immediate mechanical dysfunction of the brain tissue, and secondary damage developed over a period of hours to days following the injury. Furthermore, TBI‐related injuries are not limited to the CNS because protein metabolism alterations are linked to nitrogen transfer from muscles to the liver after a TBI, which participates in the synthesis of inflammatory proteins (Mansoor et al. 1996; Mansoor et al. 1997) and leads to local dysfunctions in different tissues (Mirzayan et al. 2008).
From a metabolic perspective, injuries caused by TBI are not limited to the CNS. In this context, TBI‐induced hepatic inflammation reinforces the idea that neuronal injury signals may impact on the function of organs distant from the injury site (Moinard et al. 2008; Anthony & Couch, 2014). Indeed, the production of acute phase proteins after CNS injury leads to systemic inflammatory response syndrome, characterized by local damage as a result of the accumulation of blood inflammatory cells in different organs, such as the liver (Bao et al. 2011). Recent studies also have demonstrated that fast hepatic chemokine production after TBI amplifies focal injury (Anthony et al. 2012; Villapol et al. 2015a). However, the role of these hepatic inflammatory mediators on the neuronal dysfunction after TBI remains largely unknown.
Liver is involved in endogenous glucose production, which is regulated by insulin through insulin binding and subsequent activation of the insulin receptor substrate (IRS) (Wang et al. 2014). In this context, the unsuppressed hepatic glucose output significantly contributes to fasting hyperglycaemia as observed in patients with diabetes and/or glucose intolerance (Rizza, 2010). Furthermore, injuries (including TBI) commonly induce high glucose reactions (emergency insulin resistance) similar to high glucose and insulin levels in type‐2 diabetes (Ljungqvist et al. 2000; Tsatsoulis et al. 2013), although the mechanism of the disease is not completely understood.
The experimental model of TBI induced by fluid percussion injury (FPI) has demonstrated major decreases in liver weight and protein content, which were associated with homeostasis impairment (Moinard et al. 2005; Moinard et al. 2008). Although not as energy‐dependent as the skeletal muscle, the liver is also a highly demanding metabolic organ. In this sense, the relationships between energy metabolism, redox biochemistry and reactive oxygen species (ROS) generation at mitochondrial level are highly relevant, considering this organelle is also a primary target of uncontrolled ROS production (Ray, 2012). ROS overproduction has long been known to damage cell components in the liver, such as nucleic acids, protein and lipids (Apel & Hirt, 2004; Bergamini et al. 2004; Dong et al. 2014). Furthermore, the combination of abnormal oxygen metabolism and ATP depletion in organs such as the liver results in the collapse of the mitochondrial machinery, with the cell being destined for necrotic death (O'Connell et al. 2012).
Over past several decades, experimental and clinical evidence has validated the contribution of ROS and inflammation to TBI‐induced multidimensional secondary injury responses (Bains & Hall, 2012). However, only a few studies have addressed the role of the liver in TBI‐induced toxicity (Kamm et al. 2006; Wang & Yang, 2010; Yang et al. 2013). Furthermore, the understanding of post‐injury neuroendocrine dysregulation in TBI is essential for establishing scientific‐based rehabilitative strategies. One of these strategies may be to support exercise training from a pre‐clinical, prophylactic, point of view, considering its role in the reduction of damage onset after a TBI, which limits the secondary neuronal death outcomes and promotes neural repair and behavioural rehabilitation (Lima et al. 2009; Kim et al. 2010; Mota et al. 2012).
Regarding systemic adaptations elicited by exercise, cross‐sectional studies have suggested that regular training protects against diseases associated with chronic low‐grade systemic inflammation (Petersen & Pedersen, 2005). Aerobic exercise has been effective in reducing food intake in animals fed a fructose‐rich diet by reducing inflammatory pathways triggered through c‐Jun NH2‐terminal kinase (JNK) phosphorylation and nuclear factor kappa B activation in both the liver and skeletal muscle (Botezelli et al. 2016). Aerobic exercise training also increased liver X receptor (LXR)‐α, a member of the ligand‐activated transcription factor that regulates lipid metabolism, plasma high‐density cholesterol and inflammatory responses (Kazeminasab et al. 2012; Fu et al. 2014). This hepatic modulation of pro‐ and anti‐inflammatory responses (Abd El‐Kader et al. 2014; Motta et al. 2015), with intermittent states of rest and the exercise‐related anti‐oxidant effect, reinforces the rationale that metabolic adaptations to training are not restricted to the exercising muscles (Lima et al. 2013).
Liver plays essential roles during exercise, particularly in the energy metabolism, as well as inflammatory and oxidative responses (Barcelos et al. 2017). Although the immune modulation induced by exercise training provides a unique non‐pharmacological therapeutic approach for controlling TBI‐related secondary damage (Ang & Gomez‐Pinilla, 2007; Motta & Dutton, 2013), little information regarding the prophylactic role of exercise training in the liver is currently available. In this sense, we aimed to investigate whether: (i) acute TBI induces insulin resistance in the liver; (ii) induced resistance is associated with hepatic inflammatory responses; (iii) inflammation‐related hepatic chemokine release amplifies the neuronal injury response, as well as how these mediators contribute to cognitive dysfunction after TBI; and (iv) interference in the secondary injury development elicited by previous physical exercise breaks the progression of neuronal damage and cognitive dysfunction after TBI.
Materials and methods
Experimental design
The present study consists of two independent experiments, in which animals were randomly assigned to sedentary and exercise groups. Twenty‐four hours after the last training session, animals underwent surgery for the FPI protocol, which resulted in four groups: sedentary/sham, sedentary/TBI, exercise/sham and exercise/TBI. Then, animals were subjected to a neuromotor evaluation 24 h after FPI. Immediately afterward, animals were killed and ipsilateral cortex and liver samples were obtained for biochemical analysis (Experiment 1). Another subset of animals performed memory tests 48 h after FPI. Immediately afterward, animals were killed by decapitation and ipsilateral hippocampus samples were obtained for biochemical and histological assays (Experiment 2) (Fig. 1).
Figure 1. Schematic representation of the experimental design with the exercise training protocol.
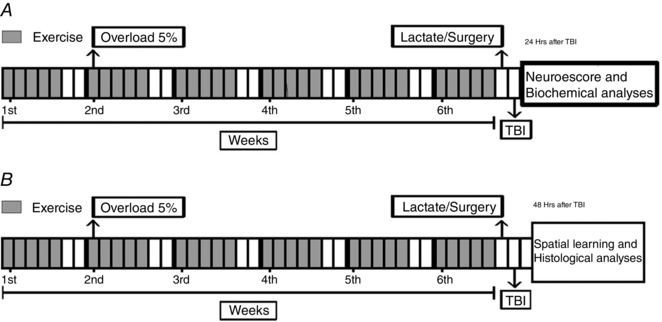
In the experiment 1 (A), animals underwent a swimming adaptation period without weights during the first week of training. After the swimming adaptation, animals trained with an extra overload equivalent to 5%/body weight during 5 weeks. One day after the last exercise session, animals were submitted to LT. Twenty‐four hours after LT, sedentary and the trained rats were subjected to lateral FPI. One day after this procedure, motor function was assessed by the neuroescore test (Experiment 1). Immediately after behavioural analysis, animals were killed for biochemical and histological analysis in ipsilateral cortex and liver. In the second experiment (B), exercise training and FPI procedures applied were the same as described in Experiment 1, except that object recognition memory test was carried out 48 h after FPI. Immediately after this behavioural test, animals were killed for histological analysis in ipsilateral hippocampus.
Animal and reagents
Male Wistar rats (250–350 g) were maintained under 12:12 h light/dark photocycle at 24 ± 1ºC and 55% relative humidity, with free access to food and water. Experimental procedures were conducted in accordance with national and international legislations (Brazilian College of Animal Experimentation and US Public Health Service Policy on Humane Care and Use of Laboratory Animals guidelines) and previously approved by the Ethics Committee on Animal Research of the Universidade Federal de Santa Maria (Protocol number 081/2014). The experiments were carried out in accordance with the principles described by Grundy (2015).
Exercise training protocol and lactate threshold assay
Exercise training was carried out as previously described by Souza et al. (2009). Briefly, animals were placed in a cylindrical pool (diameter 1.05 m, height 60 cm). The swimming training protocol lasted 6 weeks with 60 min sessions five times per week. The water temperature was 32°C and swimming sessions were performed between 09.00 h and 11.00 h. Animals underwent a swimming adaptation period without weights during the first week of training. After adaptation, rats were submitted to swimming training with a workload of 5% of body weight to improve endurance. This training intensity is based on 80–90% of lactate threshold (LT) (Gobatto et al. 2001) and no animal drowned or struggled to keep the head above water. Sedentary groups were placed in a different tank with shallow water (5 cm) at 32°C for 30 min, 5 days per week, without workload. Twenty‐four hours after the last training session, LT were evaluated in sedentary (n = 6) and trained (n = 6) rats. The LT test was conducted in accordance with the protocol described previously (Marquezi et al. 2003). The test consisted on swimming sessions of 3 min (1 min of resting) with progressive workload corresponding to 4%, 5%, 6%, 7% and 8% of the body weight of each animal. During resting periods, blood samples (25 μl) were collected from the tail vein into heparinized capillary tubes to determine lactate concentration. The LT for each animal was calculated based on the point of inflection of the graph when the lactate concentration was plotted against the corresponding exercise workload.
Traumatic brain injury (TBI)
Twenty‐four hours after the LT (Experiment 1), sedentary and trained rats were subjected to lateral FPI as described by D'Ambrosio et al. (2004). Briefly, animals were anaesthetized with a single injection of xylene (10 mg kg−1)/ketamine (100 mg kg−1) mixture and placed in a rodent stereotaxic apparatus. Heart rate, temperature, tale and eyelid reflexes were monitored to verify anaesthetic depth and duration. A burr hole of 3 mm in diameter was drilled on the right convexity, 2 mm posterior to the bregma and 3 mm lateral to the midline, avoiding dura mater injury. A plastic injury cannula was placed over the craniotomy with acrylic cement. When the cement hardened, the cannula was filled with chloramphenicol and closed with a proper plastic cap. The animal was then removed from the stereotaxic device and returned to the cage. After 24 h, animals were anaesthetized with isofluorane, had the injury cannula attached to the FPI device, and were placed in a heat pad maintained at 37 ± 0.2°C. Frontoparietal TBI (2 mm posterior to the bregma and 3 mm lateral to the midline) was produced by a FPI device developed in our laboratory. A brief (10–15 ms) transient pressure fluid pulse (4.05 ± 0.17 atm) impact was applied against the exposed dura mater. Pressure pulses were measured extracranially by a transducer (Fluid Control, AmScien Instruments, Brazil) and recorded on a storage oscilloscope (Gould Ltd, Hainault, UK). During the recovery period from anaesthesia and injury, neurological function was assessed by determining the presence or absence of reflexes, including paw‐pinch withdrawal, corneal, pinna, respiratory drive and righting reflex. During the surgery and subsequent recovery, body temperature was maintained with a circulating water‐heating pad. Immediately upon responding to a paw pinch, anaesthesia was restored and the skull was sutured. Neomycin was applied on the suture and the rats were placed in a heated recovery chamber before being returned to their cages. The sham group underwent the same procedures and was coupled to the injury device, although no fluid pulse was delivered.
Assessment of neuromotor function
Twenty‐four hours after neuronal injury, the neuromotor function was tested via the neuroescore test as described by Raghupathi et al. (1998). Briefly, animals were subjected to a grid‐walk test for 1 min to allow assessment of the number of foot‐faults. Subsequently, forelimb and hindlimb functions were evaluated by suspending animals by the tail and observing how they grasped the top of the cage when lowered towards it (for the forelimbs) and with the same pattern with respect to spread and hindlimb extension during the suspension (for hindlimbs). Finally, animals were tested for both right and left resistance to lateral pulsion. Animals were scored from 0 (severely impaired) to 4 (normal) for each of the following indices: forelimb function, hindlimb function and resistance to lateral pulsion. The maximum score for each animal was 12. Evaluation of neurological motor function was conducted by an experienced researcher who was blinded to all groups.
Blood, liver and ipsilateral cortex tissue preparation
After the neuroescore test, rats were re‐anaesthetized with isofluorane and decapitated. Livers and ipsilateral cortex were rapidly dissected and stored at −80°C for further biochemical assays. Trunk blood also was collected and allowed to clot for 10 min at room temperature, centrifuged 1500 g for 15 min and stored at −80°C for further biochemical assays.
Determination of glucose and insulin resistance calculation
An Aviva Accu‐check monitor (Roche Diagnostic Corp., Indianapolis, IN, USA) was used to analyse blood glucose levels. For the insulin test, blood concentration in each group was determined using an enzyme‐linked immunosorbent assay (ELISA) kit in accordance with the manufacturer's instruction (Linco Research, St Charles, MO, USA). The Homeostasis Model Assessment (HOMA2‐%S) was calculated with a virtual platform released by the Diabetes Trials Unit, University of Oxford: HOMA2 Calculator (http://www.dtu.ox.ac.uk/homacalculator/index.php).
Cytokine, aspartate and alanine aminotransferase levels in blood
Serum interleukin (IL)‐6 and tumor necrosis factor (TNF)‐α levels were measured using a commercially available ELISA kit, in accordance with the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA). Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined with a Hitachi 7020 automatic analyser (Hitachi, Tokyo, Japan) using commercially available reagents (Shino‐Test Corporation, Tokyo, Japan).
RT‐PCR in liver samples
Hepatic total RNA was obtained by using a Trizol reagent (Life Technologies, Carlsbad, CA, USA) and quantified by spectrophotometry (NanoDrop 1000; Thermo Scientific, Waltham, MA, USA). Residual genomic DNA was removed by incubating RNA with RQ1 RNase‐free DNase (Promega, Madison, WI, USA). First‐standard cDNA was synthesized using a High‐Capacity cDNA Archive Kit (Applied Biosystems, Weiterstadt, Germany). The negative control (no transcriptase control) was performed in parallel. First‐standard cDNA was synthesized using a High‐Capacity cDNA Archive Kit (Applied Biosystems, Paisley, UK) and then amplified using TaqMan® Universal PCR Master Mix (Applied Biosystems) on a StepOnePlus™ Real‐Time PCR System (Applied Biosystems) (Filippin et al. 2011). The Taqman® probes for inducible nitric oxide synthase (iNOS) (Rn00561640 m1) and cyclooxygenase‐2 (COX‐2) (Rn01483828 m1) were acquired from Applied Biosystems. The primer sequences for LXR‐α, ATP‐binding cassette transporter (ABCA)1, iNOS, COX‐2 and β‐actin are presented in Table 1. Relative changes in gene expression levels were determined using the 2−ΔΔCT method, as described previously (García‐Mediavilla et al. 2005; Tuñón et al. 2011). The cycle number at which the transcripts were detectable (C t) was normalized to the cycle number, referred to as ΔCT.
Table 1.
Sequences and size of the primers used for quantitative PCR amplifications
| Gene | Forward primer (5′‐ to 3′) | Reverse primer (5′‐ to 3′) | Size bp |
|---|---|---|---|
| LXRs | CCTGATGTTTCTCCTGACTC | TGACTCCAACCCTATCCTTA | 147 |
| β‐actin | GGAGAAGATTTGGCACCACAC | GGATGGCTACGTACATGGCTG | 164 |
| ABCA1 | CTTGCTTCCGTTATCCAACTCCAG | GCTGTAATGTTCTCAGGACCTTGTG | 162 |
| iNOS | CTTGCAAGTCCAAGTCTTGC | GTATGTGTCTGCAGATGTGCTG | 369 |
| TNF‐α | TTCGAGTGACAAGCCTGTAGC | AGATTGACCTCAGCGCTGAGT | 390 |
| IL‐6 | CATATGAGCTGAAAGCTCTCCA | GACACAGATTCCATGGTGAAGTC | 435 |
| COX‐2 | CCCCCACAGTCAAAGACACT | AGGCAATGCGGTTCTGATAC | 348 |
| GAPDH | GTATGACTCCACTCACGGCAA | GGTCTCGCTCCTGGAAGATG | 132 |
Cytokine immunoassay
The contents of IL‐6 and TNF‐α were determined in liver homogenates. Cytokine levels were measured using a commercially available ELISA kit from R&D Systems, in accordance with the manufacturer's instructions. The concentration of cytokines was normalized to protein concentration of samples the and results are expressed as pg mg–1 protein.
Western blot analysis
Liver samples were homogenized at 4°C in 300 μl of 0.25 mm sucrose, 1 mm EDTA, 10 mm Tris and protease inhibitor cocktail (Sigma‐Aldrich, St Louis, MO, USA). The homogenate was centrifuged at 4°C for 30 min at 13 000 g. The supernatant fraction was collected and stored at −80°C in aliquots until use. Samples containing 40 μg of protein were fractionated by SDS‐PAGE and then, transferred to a polyvinylidene fluoride membrane by a Trans‐Blot TurboTM Transfer System (Bio‐Rad, Hercules, CA, USA). The membranes were incubated overnight at 4°C with the corresponding antibodies (Gobatto et al. 2001). Antibodies against iNOS (130 kDa), pIRS (130 kDa), pJNK (48 kDa) and pAkt (60 kDa) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); antibody against COX‐2 (70 kDa) was purchased from Abcam (Cambridge, UK). After washing with TBST, the membranes were incubated for 1 h at room temperature with secondary HRP conjugated antibody (dilution 1:5000; Dako, Glostrup, Denmark), and visualized using ECL detection kit (Amersham Pharmacia, Uppsala, Sweden). The blots were stripped and probed again for anti‐β‐actin (42 kDa) antibody (Sigma‐Aldrich) to confirm equal protein loading (Gobatto et al. 2001). The density of the specific bands was quantified with an imaging densitometer (Image J, version 1.46a; NIH, Bethesda, MD, USA).
Estimation of ROS production, non‐protein sulfhydryl, protein carbonyl and reduced glutathione (GSH) content
Production of ROS was estimated in liver mitochondria with the fluorescence probe 2′,7′‐dichlorfluorescein diacetate (DCFH‐DA) as described by Myhre et al. (2003). Free ‐SH groups were determined as described by Ellman & Lysko (1967). Total protein carbonyl content was determined using the method described by Yan et al. (1995) and Levine et al. (1990). For measurement of GSH levels, the method previously described by Hissin & Hilf (1976) was used.
Superoxide dismutase (SOD) and catalase (CAT) enzyme activities
The hepatic SOD activity was measured as described by Mirsra and Fridovich (1972). CAT activity was determined by following the decomposition of hydrogen peroxide in accordance with the method proposed by Aebi (1984).
Mitochondrial 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) reduction
The hepatic MTT assays were carried out with a modification (Cohen et al. 1997) of the method described by Berridge and Tan (1993) the respiration buffer was used as stock buffer.
Mitochondrial membrane potential (Δψ ) and citrate synthase (CS) activity
The hepatic mitochondrial ΔΨm determination was estimated by fluorescence changes in safranine‐O, assayed as described previously (Akerman & Wikstrom, 1976). CS activity was determined spectrophotometrically using the method of Srere & Brooks (1969), which measures the appearance of free CoA.
Na+,K+‐ATPase activity
Hepatic Na+,K+‐ATPase activity was measured as described by Silva et al. (2013a). The amount of inorganic phosphate released was quantified by the colorimetric method described by Fiske & Subbarow (1927). The Michaelis–Menten constant (K m) for ATP was then calculated using the non‐linear fit function of Prism, version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Evaluation of MPO activity as a marker of neutrophil inflitration
MPO activity in the ipsilateral cortex was assayed in accordance with the method of Suzuki et al. (1983), with some modifications. Briefly, perilesional cortex samples were homogenized with 10 volumes of 50 mm sodium acetate buffer (pH 5.4) plus 0.5% hexadecyltrimethylammonium bromide, centrifuged (11 200 g at 4°C for 20 min) and the supernatants were collected. Next, 10 μl of supernatant and 230 μl of 50 mm sodium acetate buffer (pH 5.4) containing 15% of 0.3 mm H2O2 were added in triplicate to a 96‐well plate. The reaction was initiated by the addition of 20 μl of 18.4 mm tetramethylbenzidine. The mixture was incubated for 50 min at 37 °C follwed by immersion in an ice bath. The reaction was stopped by adding 30 μl of acetic acid and absorbance was monitored at 630 nm.
Cerebral cytokine immunoassay and Na+,K+‐ATPase activity
The contents of IL‐6 and TNF‐α were determined in ipsilateral cortex homogenized in solution containing BSA (10 mg ml−1), 2 mm EGTA, 2 mm EDTA and 0.2 mm phenylmethylsulphonyl fluoride in PBS (pH 7.4). Cytokine levels were measured using a commercially available ELISA kit from R&D Systems, in accordance with the manufacturer's instructions. The concentration of cytokines was normalized to the protein concentration contained in the samples. The results are expressed as pg/mg–1 protein. The activity of Na+,K+‐ATPase was determined in the ipsilateral cortex homogenized using the method of Silva et al. (2013b).
Evaluation of blood–brain barrier (BBB) permeability
For evaluation of BBB permeability to small molecular mass compounds, 24 h after neuronal injury, a subset of animals was injected with sodium fluorescein (10 mg in 0.1 ml of sterile saline i.p.) as described by Olsen et al. (2007). In brief, animals were anaesthetized with ketamine HCl (200 mg kg−1) i.p., 45 min after sodium fluorescein injection for blood sampling. The brain was removed, weighed, homogenized (dilution 1:10) in trichloroacetic acid, centrifuged at 1250 g for 5 min and then the supernatant was stored at −70°C until biochemical analysis. Samples were analysed on fluorometer (emission 538 nm; extinction 480 nm). BBB permeability degree was measured as the percentage of sodium fluorescein in the brain per amount of sodium fluorescein in a milliliter of serum.
Barnes maze assay
To determine the effect of previous physical exercise and TBI on spatial learning memory, a subset of animals was trained to solve the Barnes maze 48 h after neuronal injury. The Barnes maze is a validated test often used for the assessment of spatial learning and memory in rodents (Barnes, 1979). The Barnes maze paradigm exploits the natural inclination of small rodents to seek escape to a darkly lit, sheltered environment when placed in an open arena under bright, aversive illumination. Our maze consists of a circular wooden table (diameter 120 cm, thickness 3.5 cm) elevated 90 cm above the floor.
Twenty holes (diameter 6 cm) were equidistantly located around the perimeter and centred 5 cm from it. The apparatus was located in a 4 × 4 m test room where four visuospatial cues made of rigid black paper (rectangle, circle, cross, triangle) were affixed to the walls but not directly over any maze hole; this increases the spatial component of the Barnes maze during training. A black wooden escape tunnel (15 × 10 × 30 cm) was placed beneath one hole, selected randomly for each rat but remained constant throughout training sessions for a given rat. The remaining 19 holes led only to a false escape box (15 × 10 × 10 cm) which, from the platform, appeared indistinguishable from an escape box but was too small to be entered. False boxes removed visual cues that might be observed through an open hole. There was a bright illumination of 300 lux over the maze.
On the first day of the experiment, rats were moved to a testing room and left undisturbed for 60 min. Following this habituation period, rats were trained to find the escape hole; they were placed in the escape box for 1 min, then into a cylindrical opaque chamber (start box) in the centre of the maze. With light on, the start box was removed and rats were allowed to explore freely and find the escape box. A maximum of 180 s to find it was allowed. Each rat was given three trials per day, over four consecutive days. In each trial, we recorded the time to reach the escape tunnel and the number of wrong holes visited. A visited hole was defined when the animal put it head into the hole. The arena, as well as the boxes, was wiped clean using distillated water both between each training session for a given rat and between each rat.
Histological procedures
To determine the effect of TBI and exercise training on neuronal damage, immediately after behavioural (spatial learning) analysis, rats were deeply anaesthetized with thiopental (125 mg kg−1; i.p.) and transcardially perfused with 600 ml of NaCl 0.9% that contained heparin (5 IU ml−1) followed by 600 mL of 4% paraformaldehyde in 0.1 m phosphate buffer (pH 7.4). Brains were removed and serial coronal sections of 50 μm (between −1.5 and – 2.1 from bregma) were cut on a vibratome (model KD400; Zhejiang Jinhua Kedi Instrumental Equipment Co., Zhejiang, China). Sections were washed in phosphate buffer and subjected to the Giemsa staining processing (Iniguez et al., 1985). The staining was performed in duplicate for each rat (i.e. two sections per animal) and a single slide contained sections for all four groups at 35 days after TBI. Sections were mounted on gelatinized glass slides and stained for 30 s in a commercially available Giemsa solution (New Prov, Pinhais, PR, Brazil), washed three times for 2 min in distilled water, dehydrated in three graded ethanol solutions (70%, 90% and 100%), cleared for 2 min in xylene and mounted with Cytoseal 60 (Thermo Scientific). The evaluation of the Giemsa‐stained slides was conducted with 10× magnification under a light microscope (DFC290; Leica Microsystems, Wetzlar, Germany) and images of the slices were taken with the Leica Application Suite, version 3.8 (Leica Microsystems). The nucleus of neurons was used as the counting unit. The number of cells within the dentate hilus was assessed at each section, and counts were averaged, resulting in a single value for each rat.
For hepatic damage, tissue blocks were placed in 10% buffered formaldehyde solution for 48 h before being embedded in paraffin. Liver histology was assessed by light microscopy (BH2; Olympus, Tokyo, Japan) of haematoxylin and eosin (H&E)‐stained 4 μm sections in a blinded fashion. Ten random fields on each slide were assessed for necrosis by standard morphological criteria (loss of architecture, vacuolization, karyolysis, increased eosinophilia) and the extent of necrosis was semiquantitatively estimated by assigning a severity score on a scale of 0–4 as described previously by Sigala et al. (2004) (0, absent; 1, mild; 2, moderate; 3, severe; 4, total necrotic destruction of the liver).
Protein determination
The protein content was colorimetrically measured using BSA (1 mg ml−1) as a standard (Bradford, 1976).
Statistical analysis
Data are expressed as the mean ± SEM or median ± interquartile range. Data analysis was conducted using one or two‐way ANOVA or non‐parametric tests, such as the Kruskal–Wallis test, depending on the experimental design. Post hoc analyses were performed using Tukey's test when appropriate. P < 0.05 was considered statistically significant.
Results
The lactate threshold test (LT)
In the present study we showed a clear stabilization of blood lactate concentration in trained rats compared to the sedentary group (F 1,16 = 7.09; P < 0.05) (Fig. 2). This finding indicates exercise training increased the aerobic resistance of the animals (Rambo et al. 2009; Souza et al. 2009; Lima et al. 2013).
Figure 2. Effect of exercise training on the lactate threshold test.
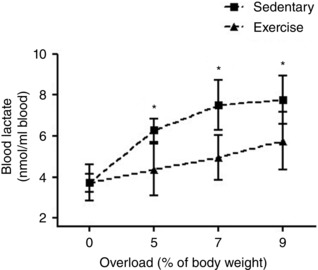
Data are expressed as the mean ± SEM for n = 6. * P < 0.05 compared to trained group (F test for simple effect).
Glycogen levels and LXR‐α and ABCA1 receptors
Our experimental data revealed that FPI did not alter glycogen levels in the liver (F 1,20 = 0.65; P > 0.05) (Fig. 3 A), although exercise training was able to increase its content (F 1,20 = 16.96; P < 0.05) (Fig. 3 A). In the present study, we showed that mRNA expression of LXR‐α (F 1,20 = 0.27; P > 0.05) (Fig. 3 B) and ABCA1 (F 1,20 = 0.11; P > 0.05) (Fig. 3 C) was not altered by the FPI injury. On the other hand, exercise training increased hepatic LXR‐α (F 1,20 = 26.96; P < 0.05) and ABCA1 (F 1,20 = 15.68; P < 0.05) mRNA expression levels in sham and TBI groups 24 h after neuronal injury.
Figure 3. The effect of exercise training and FPI on Glycogen and LXR and ABCA1 receptors.
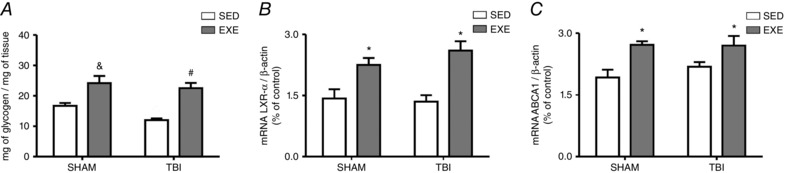
Exercise training and FPI effects on hepatic glycogen content (A), as well as mRNA expression of LXR‐α (B) and ABCA1 (B) 24 h after neuronal injury. Data are expressed as the mean ± SEM (n = 6). * P < 0.05 compared to all other groups. & P < 0.05 compared to the sedentary/sham group. # P < 0.05 compared to the sedentary/TBI group.
Inflammatory responses
To further examine the role of pro‐inflammatory enzymes in liver 24 h after neuronal injury, iNOS and COX‐2 protein expression were measured by real‐time RT‐PCR and western blotting. Previous exercise training decreased both iNOS (F 3,20 = 8.09; P < 0.05) (Fig. 4 A) and COX‐2 (F 3,20 = 36.26; P < 0.05) (Fig. 4 B) mRNA expression and protected against FPI‐induced mRNA increases of these proteins. In the same way, the immunoreactivity of iNOS (F 3,20 = 25.82; P < 0.05) (Fig. 4 C) and COX‐2 (F 3,20 = 20.71; P < 0.05) (Fig. 4 D) was reduced in the TBI‐exercised group. An immunoassay for cytokine levels also revealed that exercise training decreased hepatic levels of TNF‐α (F 1,20 = 36.12; P < 0.05) (Fig. 4 E) and protected against FPI‐induced IL‐6 increases (F 3,20 = 20.02; P < 0.05) (Fig. 4 F) and TNF‐α content (F 3,20 = 31.19; P < 0.05) (Fig. 4 E). In the present study, we revealed that pro‐inflammatory responses induced by FPI were accompanied by increased immunoreactivity of pJNK (F 3,20 = 10.50; P < 0.05) (Fig. 5 A) and decreased pIRS (F 3,20 = 13.60; P < 0.05) (Fig. 5 B) and pAKT (F 3,20 = 10.13; P < 0.05) (Fig. 5 C) content, whereas previous exercise training protected against these deleterious effect.
Figure 4. The effect of exercise training and FPI on inflammatory responses.
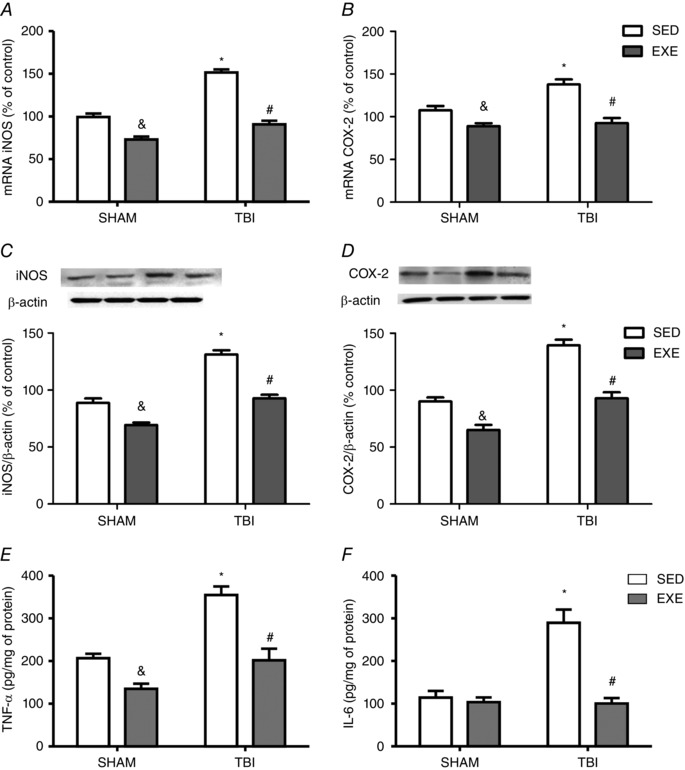
Exercise training and FPI effects on liver mRNA expression of iNOS (A) and COX‐2 (B), as well as protein content of iNOS (C) and COX‐2 (D), and also TNF‐α (E) and IL‐6 (F) levels 24 h after neuronal injury. Data are expressed as the mean ± SEM (n = 6–7). * P < 0.05 compared to all other groups. & P < 0.05 compared to the sedentary/sham group. # P < 0.05 compared to the sedentary/TBI group.
Figure 5. The effect of exercise training and FPI on hepatic pJNK, pIRS and pAKT immunoreactivity.
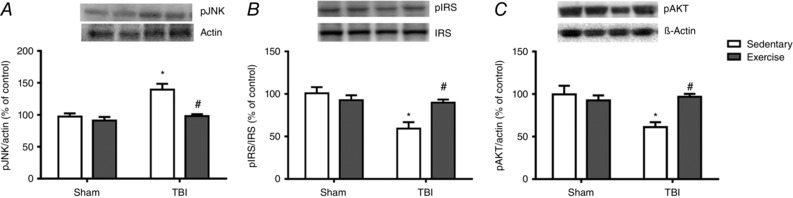
Exercise training and FPI effects on liver protein content of pJNK (A), pIRS/IRS ratio (B) and pAKT (C) 24 h after neuronal injury. Data are expressed as the mean ± SEM (n = 6–7). * P < 0.05 compared to all other groups. # P < 0.05 compared to the sedentary/TBI group.
Oxidative stress and redox status markers
The results obtained in the present study reveal that training attenuated DCFH‐DA oxidation (F 1,22 = 26.40; P < 0.05) (Fig. 6 A) and decreased protein carbonyl content (F 1,18 = 8.67; P < 0.05) (Fig. 6 B). In the same line, increases in free –SH groups (F 1,20 = 19.13; P < 0.05) (Fig. 6 C) and GSH levels (F 1,16 = 25.46; P < 0.05) (Fig. 6 D) were seen after exercise training. Following FPI‐induced injury, exercise training prevented DCFH‐DA oxidation (F 3,22 = 26.40; P < 0.05) (Fig. 6 A) and protein carbonylation increases (F 3,18 = 26.17; P < 0.05) (Fig. 6 B), whereas free ‐SH groups (F 3,20 = 20.05) (P < 0.05) (Fig. 6 C) and GSH levels (F 3,16 = 18.90; P < 0.05) (Fig. 6 D) were increased compared to TBI‐sedentary rats. Statistical analysis also revealed that previous exercise training protected against FPI‐induced SOD activity increases (F 3,20 = 9.26; P < 0.05) (Fig. 6 F) and CAT inhibition (F 3,20 = 6.62; P < 0.001) (Fig. 6 E) when analysed 24 h after neuronal injury.
Figure 6. The effect of exercise training and FPI on hepatic oxidative and redox status.
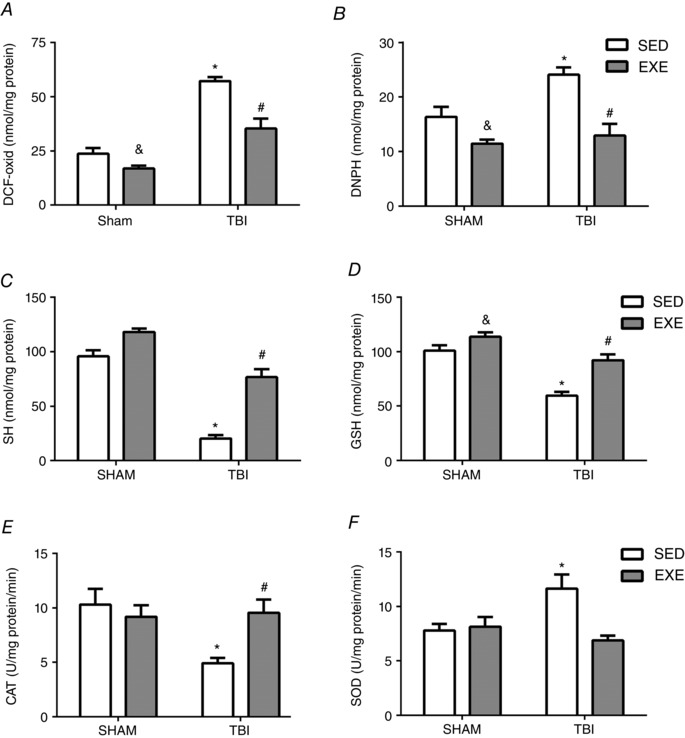
Exercise training and FPI effects on liver DCFH‐DA oxidation (A), DNPH (B), free‐SH (C) and GSH levels (D), as well as CAT (E) and SOD (F) activities 24 h after neuronal injury. Data are expressed as the mean ± SEM (n = 6–7). * P < 0.05 compared to all other groups. & P < 0.05 compared to the sedentary/sham group. # P < 0.05 compared to the sedentary/TBI group.
Mitochondrial assays
Exercise training was effective against FPI‐induced liver mitochondria dysfunction, as characterized by increases for both dehydrogenases, measured as MTT reduction (F 3,24 = 6.41; P < 0.05) (Fig. 7 A), and CS activity (F 3,20 = 5.01; P < 0.05) (Fig. 7 B) 24 h after neuronal injury. Moreover, Δψ decreased after FPI (F 3,20 = 7.51 P < 0.05) (Fig. 7 C), whereas exercise training reverted this effect back to baseline values.
Figure 7. The effect of exercise training and FPI on hepatic mitochondrial function.
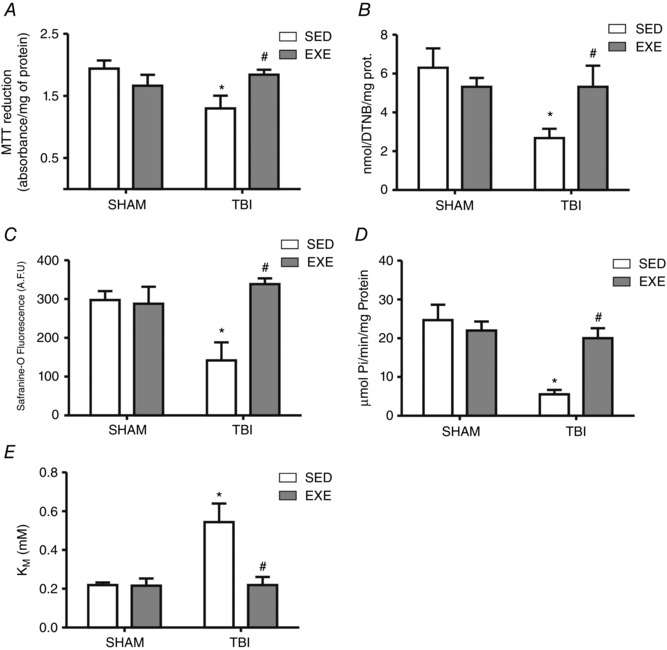
Exercise training and FPI effects on liver mitochondrial MTT reduction (A), CS (B), Δψ (C), Na+, K+‐ATPase activity (D) and K m for ATP (E) 24 h after TBI. Data are expressed as the mean ± SEM (n = 5–8). * P < 0.05 compared to all other groups. # P < 0.05 compared to the sedentary/TBI group.
Na+,K+‐ATPase and ATP K m
Our experimental data showed that FPI decreased Na+,K+‐ATPase activity (F 1,20 = 10.99; P < 0.05) (Fig. 7 D). Additionally, functional analysis of Na+,K+‐ATPase dependence for ATP revealed that ATP K m was also altered with FPI (F 1,20 = 8.19; P < 0.05) (Fig. 7 E). However, exercise training prevented both a FPI‐induced Na+,K+‐ATPase decrease (F 3,20 = 8.58 P < 0.05) (Fig. 7 D) and an ATP K m increase (F 3,20 = 5.51 P < 0.05) (Fig. 7 E) 24 h after neuronal injury.
Change of metabolic characteristics and serum cytokine levels after TBI
Statistical analyses revealed that FPI induced blood insulin (F 1,20 = 17.32; P < 0.05) (Fig. 9 A) and glucose (F 1,20 = 8.90; P < 0.05) (Fig. 9 B) increases; previous exercise training protected against hyperglycaemia. Calculation of HOMA‐2% indicated that insulin sensitivity was low in FPI‐sedentary compared to the FPI‐trained group 24 h after neuronal injury (F 1,20 = 7.38; P < 0.05) (Fig. 9 C). As shown in Table 2, previous exercise training protected against serum IL‐6 (F 3,20 = 14.15 P < 0.05) and TNF‐α (F 3,20 = 16.89; P < 0.05) increases 24 h after FPI.
Figure 9. The effect of exercise training and FPI on metabolic profile.
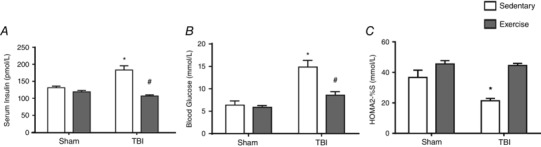
Exercise training and FPI effects on plasma insulin (A), glucose (B) and HOMA2 %S (C) 24 h after TBI. Data are expressed as the mean ± SEM (n = 6–7). * P < 0.05 compared to all other groups. # P < 0.05 compared to the sedentary/TBI group.
Table 2.
Metabolic profile of sham‐operated and TBI‐rats
| Day 1 post TBI | |||
|---|---|---|---|
| Parameter | Baseline (n = 6–8) | Sedentary group (n = 6–8) | Trained group (n = 6–8) |
| Body weight (g) | |||
| Sham | 319.9 ± 2.7 | 316.9 ± 2.9 | 322.5 ± 3.0 |
| TBI | 314.7 ± 3.1 | 316.8 ± 3.6 | 320.6 ± 2.1 |
| Liver weight (g) | |||
| Sham | 9.9 ± 0.5 | 10.1 ± 0.3 | |
| TBI | 10.0 ± 1.6 | 9.7 ± 0.4 | |
| Serum IL‐6 (pg ml–1) | |||
| Sham | 104.24 ± 31.0 | 98.09 ± 28.5 | |
| TBI | 225.70 ± 69.10* | 106.18 ± 31.4# | |
| Serum TNF‐α (pg ml–1) | |||
| Sham | 160.84 ± 42.1 | 159.39 ± 38.5 | |
| TBI | 338.89 ± 89.99* | 198.18 ± 59.4# | |
| Serum ALT (IU mL–1) | |||
| Sham | 18.13 ± 4.1 | 24.19 ± 7.5 | |
| TBI | 22.33 ± 6.1 | 23.13 ± 5.6 | |
| Serum AST (IU mL–1) | |||
| Sham | 102.4 ± 7.98 | 103.39 ± 8.50 | |
| TBI | 106.3 ± 12.09 | 108.18 ± 11.10 | |
Data are given as the mean ± SEM. * P < 0.05 sham/group. #P < 0.05 TBI/group.
Liver damage markers
Statistical analyses revealed that neither FPI, nor exercise training altered liver and body weights, as well as serum ALT (F 1,20 = 1.09; P > 0.05) (Table 2) and AST (F 1,20 = 0.58; P > 0.05) (Table 2) concentrations, compared to the respective control groups. Equally, H&E staining did not show tissue damage in hepatic cells and histological assessment revealed that livers from the different groups displayed a well‐preserved architecture 24 h after FPI injury (Fig. 8).
Figure 8. Exercise training and FPI effects on hepatocellular morphology 24 h after TBI.
Histological analysis in the liver displays as: sham/sedentary group (A), TBI/sedentary (B), sham/trained (C) and TBI/trained (D) animals demonstrated by H&E staining. [Color figure can be viewed at wileyonlinelibrary.com]
Opening of the blood‐brain barrier (BBB) followed by neurtrophils infiltration and cerebral inflammation, contributes to failure of Na+,K+‐ATPase activity
In the present study, we showed that previous exercise training significantly attenuated an FPI‐induced increase of plasma fluorescein extravasation (indicator of BBB breakdown) (F 3,20 = 8,50; P < 0.05) (Fig. 10 A) and protected against MPO activity increases (F 3,20 = 7,04; P < 0.05) (Fig. 10 B) 24 h after neuronal injury. Exercise training was also effective against FPI‐induced IL‐6 (F 3,20 = 10.15) (P < 0.05) (Fig. 10 C) and TNF‐α content (F 3,20 = 18.06; P < 0.05) (Fig. 10 D) increases, as well as Na+,K+‐ATPase activity inhibition (F 3,20 = 14,74; P < 0.05;Fig. 10 E).
Figure 10. The effect of exercise training on neuronal oxidative inflammatory‐related cascades after FPI.
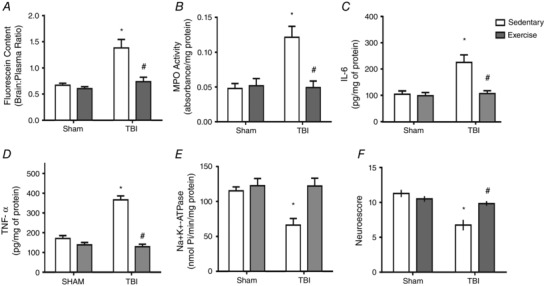
Exercise training and FPI effects on plasma fluorescein extravasation (indicator of BBB breakdown) (A), neuronal MPO activity (B), IL‐6 (C), TNF‐α levels (D), Na+,K+‐ATPase activity (E) and neuroscore (F) 24 h after TBI. Data are expressed as the mean ± SEM (n = 6–7). * P < 0.05 compared to all other groups. # P < 0.05 compared to the sedentary/TBI group.
Exercise training protects against TBI‐induced motor impairment and spatial learning dysfunction
Motor impairment is a common TBI disability (Mota et al. 2012; da Silva Fiorin et al. 2016). Using the composite neuroscore, we demonstrated that FPI‐induced motor impairment (F 1,20 = 10.58; P < 0.05) (Fig. 10 F) and previous exercise training protected against neurological impairment 24 h after neuronal injury. Results showed that 6 weeks of swimming training enhanced spatial learning per se characterized by escape latency (F 1,42 = 5.10 P < 0.05) (Fig. 11 B and C) and decreased number of errors (F(1,42 = 20.21 P < 0.05) (Fig. 12 B–D) in the naive group compared to the sedentary animals. Exercise training also protected against FPI‐induced increases on escape latency (F 2,42 = 7.03 P < 0.05) (Fig. 11 B and C) and number of errors (F 2,42 = 6.23 P < 0.05) (Fig. 12 B–D). The open field test revealed that neither swimming training, nor TBI altered crossing numbers (F 2,42 = 0.10; P > 0.05) or rearing responses (F 2,42 = 0.35; P > 0.05), indicating that FPI‐related impaired performance in the Barnes maze was unrelated to motor disabilities (supplementary data).
Figure 11. Previous physical training protects against FPI‐induced spatial learning dysfunction.
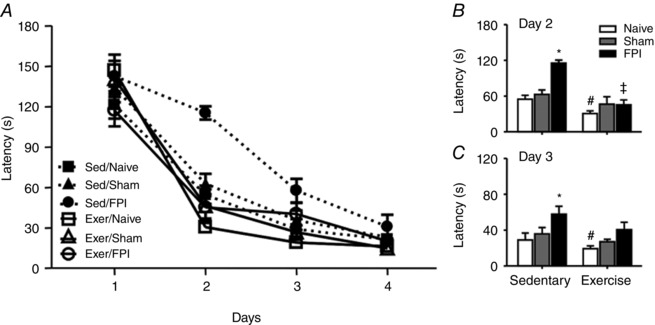
Exercise training and FPI effects on escape latency in Barnes maze test (A). B and C, previous swimming training decreased the latency for escape per se and protected against latency for escape increase induced by FPI. Data are the mean ± SEM (n = 7–9 in each group). * P < 0.05 compared to sham and naive sedentary group. # P < 0.05 compared to the naïve‐sedentary group.
Figure 12. Previous physical training protects against the number of errors increase after FPI.
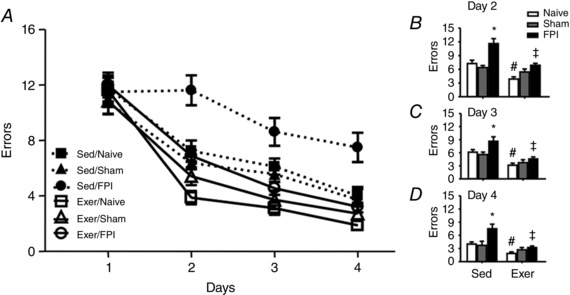
Exercise training and FPI effects on the number of errors in Barnes maze test (A). B–D, previous swimming training decreased the number of errors per se and protected against the number of errors increase induced by FPI. Data are the mean ± SEM (n = 7–9 in each group). * P < 0.05 compared to the sham and naive sedentary group. # P < 0.05 compared with naïve‐sedentary group.
Exercise training protects against hippocampal cell loss after FPI
Considering secondary injury processes lead to necrotic, apoptotic and autophagic neuronal cell death and synaptic loss (Bigford et al. 2009), we performed histological analyses in the hippocampus. H&E staining demonstrated previous exercise training protected against cell loss in the dentate gyrus hilus after the Barnes maze test (F 3,21 = 13.25; P < 0.05) (Fig. 13 A).
Figure 13. Previous physical training protects hippocampal cell loss after FPI.
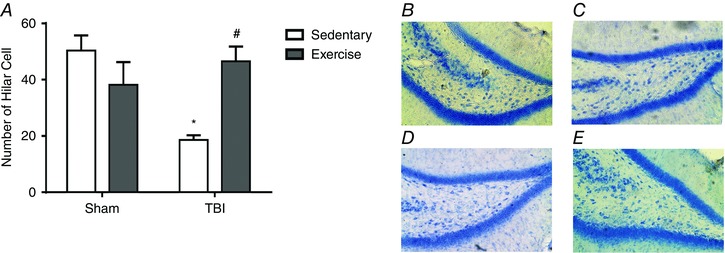
Histological analysis (H&E staining) demonstrated previous exercise training protected against cell loss in the dentate gyrus hilus induced by FPI (A). Histological analysis in the displays: sham/sedentary group (B), sham/trained (C), TBI/sedentary (D) and TBI/trained (E) animals. Data are expressed as the mean ± SEM (n = 6–7). * P < 0.05 compared to all other groups. # P < 0.05 compared to the sedentary/TBI group. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
The results reported in the present study confirm and extend previous findings indicating that a single FPI episode in the rat parietal cortex induces a major peripheral inflammatory response (Moinard et al. 2008). From a metabolic point of view, our experimental data suggest that an early inflammatory response and oxidative stress are implicated in the signal transduction that alters redox status, mitochondrial function and insulin signalling in the liver. By contrast, the stress response in this TBI model did not induce early hepatocellular damage, although the ion gradient collapse provides an explanation for the crucial role that the liver may play in secondary damage after a neuronal injury (Anthony et al. 2012).
Recently, considerable evidence has demonstrated that hyperglycaemia (both peak glucose and persistent hyperglycaemia) is one of the most common secondary complications of severe TBI (Shi et al. 2016). In line with this view, the present study shows that blood glucose, insulin and HOMA‐2%S were significantly elevated following acute neuronal injury in sedentary rats. The development of hyperglycaemia after neuronal injury was accompanied by hepatic immunoreactivity of a pJNK increase and pIRS and pAKT decreases. Our data mimicked the findings of previous studies in experimental haemorrhage and burn models and further highlight the importance of local hepatic inflammation and ROS generation in the pathogenesis of glucose metabolic abnormalities in acute illnesses (Xu et al. 2008; Wang et al. 2011). Our experimental data also reinforce that the idea that TBI‐related injuries are not limited to the CNS. In this sense, the hepatic acute‐phase response leads to the mobilization of leukocytes, cytokines and chemokines (Villapol et al. 2015b). These signalling molecules enter the bloodstream and reach the cerebral circulation to affect the inflammatory status of the brain.
In the present study, we have demonstrated that high levels of circulating cytokines elicited by FPI affect the brain response to injury and contribute to BBB opening, neurtrophil infiltration and Na+,K+‐ATPase activity inhibition. These results are in line with previous findings from our group suggesting that a single episode of FPI affects the inflammatory status of the brain (Mota et al. 2012), impairs performance in spatial learning and induces cell loss in the dentate gyrus hilus (Lima et al. 2008; da Silva Fiorin et al. 2016). However, the understanding of secondary damage post‐injury is essential for establishing scientific‐based rehabilitation following TBI. One of the possible strategies may be the inclusion of exercise training as a prophylactic means for TBI sequelae management. Evidence accumulated over the years has demonstrated that exercise training exerts a broad range of beneficial effects on the body, including improvements in cardiovascular function and a potential reduction in the incidence of several neurological diseases (Radak et al. 2008). In the present study, a clear stabilization of blood lactate concentration in the trained group during LT confirms the assumption that training increases aerobic resistance (Gobatto et al. 2001; Lima et al. 2009; Rambo et al. 2009; Souza et al. 2009; Lima et al. 2013). In addition, the results show that hepatic LXR‐α and ABCA1 mRNA expression is higher in trained animals. LXRs are ligand‐activated transcription factors that play an important role in the hepatic metabolism regulation of lipids and carbohydrates.
Apart from being glucose sensors (Mitro et al. 2007), LXRs also induce genes that inhibit the expression of inflammatory mediators, such as the ABCA1 (Spillmann et al. 2014). Because glucose is an endogenous LXR ligand and ABCA1 inhibits the expression of inflammatory mediators (Spillmann et al. 2014), it is plausible to propose that the anti‐inflammatory effects elicited by exercise training involve hepatic glycogen metabolism (Hoene & Weigert, 2010) followed by LXR‐α and ABCA1 mRNA increases (Ghanbari‐Niaki et al. 2007; Kazeminasab et al. 2013). Noteworthy, inflammatory modulation may differ among TBI acute and delayed phases (Ziebell & Morganti‐Kossmann, 2010). Although physiological nitric oxide (NO) production has beneficial anti‐microbial and anti‐tumor effects, excessive NO produced by iNOS mediates inflammatory signalling, causing ROS‐mediated cell damage via peroxynitrite (Huang et al. 2012). Furthermore, lipid peroxidation initiated by peroxynitrite releases arachidonic acid from the cell membrane, which in turn activates COX‐2 (Salvemini et al. 1995; Huang et al. 2012). Considering that prostaglandin E2, a product of COX‐2, is one of the strongest inflammatory mediators, it is plausible to propose the protection elicited by exercise training may be associated to prostaglandin E2 blockade through COX‐2 downregulation. Western blot assays have revealed that exercise training also protected against FPI‐induced increases in IL‐6 and TNF‐α content in the liver. Because the hepatic pro‐inflammatory cytokines TNF‐α and IL‐6 are relevant mediators for the stimulation of PGs synthesis by COX‐2, our experimental data suggest that this exercise‐mediated cytokine inhibition may account for COX‐2 downregulation (Anthony et al. 2012). These data are consistent with the hypothesis that favourable changes in the hepatic anti‐inflammatory status elicited by previous exercise training may exert a prophylactic effect on the early inflammatory response after severe TBI.
Liver is the main organ involved in endogenous glucose production, which is regulated by insulin though IRS and AKT pathways (Bugianesi et al. 2005). In this sense, the increase of blood glucose, insulin and HOMA‐2%S after neuronal injury in the sedentary FPI group corroborates data from obese animal and human studies indicating that hepatic cytokine expression (TNF‐α) activates intracellular JNK and phosphorylate insulin receptor substrate (IRS‐1) at serine residues (Cai et al. 2005; Moschen & Tilg, 2008). Our experimental data also suggest that acute stress, such as TBI, may lead to hyperglycaemia, in the absence of diabetes, and also that strategies such as exercise training can enhance glucose control and reduce damage as a result of hyperglycaemia to several cells (Tsuzuki et al. 2015; Shi et al. 2016). Indeed, our experimental protocol of exercise training protected against blood glucose, insulin and HOMA‐2%S increases, as well as hepatic immunoreactivity of pJNK increase, and also pIRS and pAKT decreases after neuronal injury.
Regarding the effects of TBI on liver function, it is important to consider that neutrophils produce superoxide anions (O2 .−) via NADPH oxidases: O2 .− is further dismutated by SOD into hydrogen peroxide (H2O2) and molecular oxygen. In the presence of CAT, H2O2 is converted into H2O and oxygen; otherwise, it can be used as a substrate by MPO to produce hypochlorous acid (HOCl), a highly reactive ROS that is associated with the production of protein carbonyls (Yan et al. 1996; Handelman et al. 1998). The increases in MPO and SOD activity combined with dampened CAT in the FPI sedentary group may indicate that higher levels of H2O2 formed by the SOD‐mediated O2 .− dismutation persist, increasing H2O2 production. The DCFH‐oxidation data corroborate this rationale, considering that this is a H2O2‐based assay (LeBel et al. 1992). Furthermore, the compromised CAT activity, a H2O2 decomposing enzyme, is usually a result of excessive H2O2, as well as 4‐HNE, which forms aldehyde‐protein adducts inactivating its active site (Kostyuk et al. 2010; Lubrano et al. 2015). Under these circumstances, the development of compensatory responses elicited by exercise training may be effective in attenuating FPI‐induced redox status disruption, characterized in the present study by a decrease in GSH and ‐SH groups combined with increased protein carbonylation and DCFH‐DA in the liver (Salo et al. 1991; Viguie et al. 1993; Leeuwenburgh & Heinecke, 2001). As such, our experimental data agree with the assumption that exercise training reinforces the liver anti‐oxidant system (Boveris & Navarro, 2008; Sun et al. 2010).
In the present study, previous exercise training was effective against FPI‐induced liver mitochondria dysfunction. It is worth noting that ROS generation is a necessary and unavoidable consequence of aerobic metabolism, and the rate of ROS generation in biological tissue is closely related to oxygen consumption (Toldy et al. 2005). Consequently, mitochondria are probably both a source and target of oxidant agents (Boveris & Navarro, 2008). Furthermore, the development of compensatory responses to oxidative stress elicited by exercise training (Salo et al. 1991; Viguie et al. 1993; Leeuwenburgh & Heinecke, 2001) is a reflection of decreased markers of ROS generation, which translates into the maintenance of mitochondrial homeostasis in FPI‐trained rats. This hypothesis has been upheld in animal studies, which indicate that a significant adaptive response to exercise training involves a greatly increased endurance capacity, facilitated by increases of O2 consumption and mitochondrial biogenesis (Boveris & Navarro, 2008; Packer et al. 2008). In the present study, exercise training prevented the downregulation of relevant mitochondrial functioning parameters, such as the Δψ, as well as CS and dehydrogenases activities.
Any cellular constituent may be a ROS target, although selected targets are more prone to induce toxicity (Lima et al. 2009). The Na+,K+‐ATPase appears to be particularly sensitive to ROS‐induced damage, considering that its inhibition has been associated with alterations in membrane lipid composition, ‐SH groups redox and amino acid residues induced by ROS (Jamme et al. 1995; Siems et al. 1996). Therefore, it is tempting to propose that the presently reported FPI‐induced ROS generation and Na+,K+‐ATPase activity decrease in liver are causally related. Although the stress response of liver did not induce early hepatocellular damage, the collapse of ion gradient homeostasis provides an explanation for the crucial role played by the liver in secondary damage after neuronal injury (Anthony et al. 2012). Indeed, the increase in the Michaelis–Menten constant (i.e. the K m) for the main Na+,K+‐ATPase substrate in the FPI‐sedentary group suggests that ATP affinity is reduced in the liver of these animals. Importantly, the reduced affinity of hepatic Na+,K+‐ATPase for ATP could be exacerbated by mitochondrial energy metabolism dysfunction. This finding is particularly important, considering the exercise training protocol induced compensatory responses to oxidative stress and protected against FPI‐induced Na+,K+‐ATPase activity diminution. Thus, the findings of the present study support the assumption that exercise training modulates oxidative‐inflammatory functions in the liver (Barcelos et al. 2017) and thus delays or prevents secondary cascades that lead to several disabilities after a TBI (Griesbach et al. 2004; Gomez‐Pinilla & Kostenkova, 2008).
Furthermore, favourable changes in the hepatic oxidative‐inflammatory status elicited by previous exercise training may exert prophylactic effects on acute hyperglycaemia and the cerebral inflammatory response induced by severe TBI. It is important to note that the influx of leukocytes to an inflammatory site is orchestrated by a sequential upregulation of adhesion molecules on vascular endothelium, leading post‐traumatic oedema and BBB breakdown (Baskaya et al. 1997; Unterberg et al. 1997). Moreover, an imbalance in the ratio of harmful ROS/RNS ratio and BBB dysfunction after TBI causes delayed neuronal dysfunction and death through secondary processes involving increased excitatory amino acids levels and ionic equilibrium loss (Lenzlinger et al. 2004; Shlosberg et al. 2010). Our experimental data agree with this assumption when TBI induced plasma fluorescein extravasation (an indicator of BBB breakdown) and cerebral MPO activity increase, whereas previous exercise training attenuated this neuronal dysfunction. Exercise training was also effective against an increase in IL‐6 and TNF‐α content, and Na+,K+‐ATPase activity inhibition. Considering that alterations in the redox state of regulatory ‐SH groups in selected targets, such as Na+, K+‐ATPase (Morel et al. 1998), increases cellular excitability after TBI (Rao et al. 2008), our data reinforce the idea that ROS‐induced Na+,K+‐ATPase inhibition may contribute to FPI‐induced neurological dysfunction characterized by early neuromotor dysfunction and spatial learning impairment (Lima et al. 2008; Mota et al. 2012; da Silva Fiorin et al. 2016).
Recently, a considerable body of evidence has demonstrated that oxidative inflammatory‐related cascades resulting from TBI have been implicated in altered signal transduction in peripheral organs and the CNS (Hall et al. 2004; Bayir et al. 2007; Lima et al. 2013). Although a pre‐injury regimen for humans may not be the most effective treatment because the time of injury cannot be predicted (Vaynman & Gomez‐Pinilla, 2005), the effective protection exerted by exercise training in this model of TBI is of particular interest because it supports the assumption that metabolic chances elicited by exercise delay or prevent secondary cascades which lead to long‐term cell damage after TBI. Indeed, our experimental data revealed that previous exercise training protected from cell loss induced by TBI. Considering that a loss in number of interneurons after experimental TBI (Lowenstein et al. 1992; Santhakumar et al. 2000; Grady et al. 2003; Hall et al. 2005) can be associated with a reduction in granule cells synaptic inhibition (Hunt et al. 2011; Pavlov et al. 2011; Gupta et al. 2012), we suggest that exercise‐induced reduction of the initial damage limits the long‐term secondary degeneration and supports neural repair or behavioural compensation after neuronal injury (Wannamethee & Shaper, 1992; Wang et al. 2001; Klein et al. 2003). However, it is worth noting that simultaneous hippocampal cell loss and spatial memory dysfunction in this TBI model do not necessary imply a cause–effect relationship between these events. It is important to note that hippocampal cells are a mixed population of mostly inhibitory interneurons and the assay used does not allow the identification of these cells. Consequently, our experimental data are speculative and additional studies are necessary to clarify the involvement of these pathways in the prophylactic effect of exercise training after TBI.
In conclusion, the present study describes TBI induction of inflammation and oxidative stress of rats, which disrupt redox status and mitochondrial function in the liver. These responses originate not from early hepatocellular damage, but from a collapse of the ion gradient homeostasis followed by dysglycaemia and impaired hepatic signalling observed in sedentary rats after neuronal injury, which suggests that neuronal injury signals may impact on organs away from the injury site. High levels of circulating cytokines after TBI also affected brain function, characterized in the present study by BBB opening, neutrophil infiltration and oxidative‐inflammatory increases. Neuronal inflammation and oxidative stress leads to a marked impairment of protein function, neurobehavioural disability and long‐term cell damage after TBI. Although a pre‐injury prophylactic strategy may not be the most effective treatment for humans because the occurrence of injury cannot be predicted, the effect of exercise on the expression/activity of specific endogenous proteins is particularly interesting. The findings reported in the present study support the idea that exercise training modulates oxidative‐inflammatory functions (Barcelos et al. 2017) and thus delays or prevents secondary cascades that lead to several disabilities after a TBI (Griesbach et al. 2004; Gomez‐Pinilla & Kostenkova, 2008). Therefore, approaches aiming to counteract or attenuate secondary damage developed over a period of hours to days after a TBI, such as exercise training, may have preventive and/or therapeutic potential through the modulation of responses by metabolic‐related organs, such as the liver and brain.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author's contributions
LFFR, MRTC, GB and JGG conceived and designed the experiments. MRTC, APOF, GLB, LRHS, MEPJ, FF, RPB, MJC and GA performed the assays. LFFR, MRTC, MRF and MECL analysed the data. LFFR, MRTC, MECL, MRF, GB and JGG drafted the article. All authors approved the final version of the manuscript submitted for publication.
Funding
The present study was supported by CNPq/FAPERGS (grants: Pronem 11/2082‐4). M. R. Fighera and L. F. F. Royes are recipients of CNPq fellowships (CNPq: 306309/2013‐0). CIBERehd is funded by Instituto de Salud Carlos III, Spain.
Acknowledgements
We thank Dr Mauro Schneider Oliveira, Dra Ana Flavia Furian and Dr Felix Soares for reviewing the study. We are also indebted to Dr Adair Roberto Soares dos Santos for his contribution to the inflammatory measurements.
This is an Editor's Choice article from the 1 September 2017 issue.
References
- Abd El‐Kader SM, Al‐Jiffri OH & Al‐Shreef FM (2014). Markers of liver function and inflammatory cytokines modulation by aerobic versus resisted exercise training for nonalcoholic steatohepatitis patients. Afr Health Sci 14, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebi H (1984). Catalase in vitro. Methods Enzymol 105, 121–126. [DOI] [PubMed] [Google Scholar]
- Akerman KE & Wikstrom MK (1976). Safranine as a probe of the mitochondrial membrane potential. FEBS Lett 68, 191–197. [DOI] [PubMed] [Google Scholar]
- Ang ET & Gomez‐Pinilla F (2007). Potential therapeutic effects of exercise to the brain. Curr Med Chem 14, 2564–2571. [DOI] [PubMed] [Google Scholar]
- Anthony DC & Couch Y (2014). The systemic response to CNS injury. Exp Neurol 258, 105–111. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Couch Y, Losey P & Evans MC (2012). The systemic response to brain injury and disease. Brain Behav Immun 26, 534–540. [DOI] [PubMed] [Google Scholar]
- Apel K & Hirt H (2004). Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55, 373–399. [DOI] [PubMed] [Google Scholar]
- Bains M & Hall ED (2012). Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta 1822, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F, Brown A, Dekaban GA, Omana V & Weaver LC (2011). CD11d integrin blockade reduces the systemic inflammatory response syndrome after spinal cord injury. Exp Neurol 231, 272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcelos RP, Royes LF, Gonzalez‐Gallego J & Bresciani G (2017). Oxidative stress and inflammation: liver responses and adaptations to acute and regular exercise. Free Radic Res 1–32. [DOI] [PubMed] [Google Scholar]
- Barnes CA (1979). Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93, 74–104. [DOI] [PubMed] [Google Scholar]
- Baskaya MK, Rao AM, Dogan A, Donaldson D & Dempsey RJ (1997). The biphasic opening of the blood‐brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci Lett 226, 33–36. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Clark RS, Janesko‐Feldman K, Rafikov R, Huang Z, Zhang X, Vagni V, Billiar TR & Kochanek PM (2007). Neuronal NOS‐mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J Neurochem 101, 168–181. [DOI] [PubMed] [Google Scholar]
- Bergamini CM, Gambetti S, Dondi A & Cervellati C (2004). Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des 10, 1611–1626. [DOI] [PubMed] [Google Scholar]
- Berridge MV & Tan AS (1993). Characterization of the cellular reduction of 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys 303, 474–482. [DOI] [PubMed] [Google Scholar]
- Bigford GE, Alonso OF, Dietrich D & Keane RW (2009). A novel protein complex in membrane rafts linking the NR2B glutamate receptor and autophagy is disrupted following traumatic brain injury. J Neurotrauma 26, 703–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botezelli JD, Coope A, Ghezzi AC, Cambri LT, Moura LP, Scariot PP, Gaspar RS, Mekary RA, Ropelle ER & Pauli JR (2016). Strength training prevents hyperinsulinemia, insulin resistance, and inflammation independent of weight loss in fructose‐fed animals. Sci Rep 6, 31106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A & Navarro A (2008). Systemic and mitochondrial adaptive responses to moderate exercise in rodents. Free Radic Biol Med 44, 224–229. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Bradley PP, Christensen RD & Rothstein G (1982). Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood 60, 618–622. [PubMed] [Google Scholar]
- Bugianesi E, McCullough AJ & Marchesini G (2005). Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology 42, 987–1000. [DOI] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J & Shoelson SE (2005). Local and systemic insulin resistance resulting from hepatic activation of IKK‐beta and NF‐kappaB. Nat Med 11, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen G, Farooqui R & Kesler N (1997). Parkinson disease: a new link between monoamine oxidase and mitochondrial electron flow. Proc Natl Acad Sci USA 94, 4890–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle DL & Miller JW (2004). Post‐traumatic epilepsy following fluid percussion injury in the rat. Brain: A Journal of Neurology 127, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Fiorin F, de Oliveira Ferreira AP, Ribeiro LR, Silva LF, de Castro MR, da Silva LR, da Silveira ME, Jr. , Zemolin AP, Dobrachinski F, Marchesan de Oliveira S, Franco JL, Soares FA, Furian AF, Oliveira MS, Fighera MR & Freire Royes LF (2016). The impact of previous physical training on redox signaling after traumatic brain injury in rats: a behavioral and neurochemical approach. J Neurotrauma 33, 1317–1330. [DOI] [PubMed] [Google Scholar]
- Dong GZ, Lee JH, Ki SH, Yang JH, Cho IJ, Kang SH, Zhao RJ, Kim SC & Kim YW (2014). AMPK activation by isorhamnetin protects hepatocytes against oxidative stress and mitochondrial dysfunction. Eur J Pharmacol 740, 634–640. [DOI] [PubMed] [Google Scholar]
- Ellman GL & Lysko H (1967). Disulfide and sulfhydryl compounds in TCA extracts of human blood and plasma. J Lab Clin Med 70, 518–527. [PubMed] [Google Scholar]
- Filippin LI, Cuevas MJ, Lima E, Marroni NP, Gonzalez‐Gallego J & Xavier RM (2011). The role of nitric oxide during healing of trauma to the skeletal muscle. Inflamm Res 60, 347–356. [DOI] [PubMed] [Google Scholar]
- Fiske CH & Subbarow Y (1927). The nature of the "inorganic phosphate" in voluntary muscle. Science 65, 401–403. [DOI] [PubMed] [Google Scholar]
- Fu Y, Tian Y, Wei Z, Liu H, Song X, Liu W, Zhang W, Wang W, Cao Y & Zhang N (2014). Liver X receptor agonist prevents LPS‐induced mastitis in mice. Int Immunopharmacol 22, 379–383. [DOI] [PubMed] [Google Scholar]
- García‐Mediavilla MV, Sánchez‐Campos S, González‐Pérez P, Gómez‐Gonzalo M, Majano PL, López‐Cabrera M, Clemente G, García‐Monzón C & González‐Gallego J (2005). Differential contribution of hepatitis C virus NS5A and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte‐derived cells. J Hepatol 43, 606–613. [DOI] [PubMed] [Google Scholar]
- Ghanbari‐Niaki A, Khabazian BM, Hossaini‐Kakhak SA, Rahbarizadeh F & Hedayati M (2007). Treadmill exercise enhances ABCA1 expression in rat liver. Biochem Biophys Res Commun 361, 841–846. [DOI] [PubMed] [Google Scholar]
- Gobatto CA, de Mello MA, Sibuya CY, de Azevedo JR, dos Santos LA & Kokubun E (2001). Maximal lactate steady state in rats submitted to swimming exercise. Comp Biochem Physiol A Mol Integr Physiol 130, 21–27. [DOI] [PubMed] [Google Scholar]
- Gomez‐Pinilla F & Kostenkova K (2008). The influence of diet and physical activity on brain repair and neurosurgical outcome. Surg Neurol 70, 333–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady MS, Charleston JS, Maris D, Witgen BM & Lifshitz J (2003). Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: analysis by stereological estimation. J Neurotrauma 20, 929–941. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Molteni R, Wu A & Gomez‐Pinilla F (2004). Voluntary exercise following traumatic brain injury: brain‐derived neurotrophic factor upregulation and recovery of function. Neuroscience 125, 129–139. [DOI] [PubMed] [Google Scholar]
- Gupta A, Elgammal FS, Proddutur A, Shah S & Santhakumar V (2012). Decrease in tonic inhibition contributes to increase in dentate semilunar granule cell excitability after brain injury. J Neurosci 32, 2523–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K & Kupina NC (2004). Peroxynitrite‐mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma 21, 9–20. [DOI] [PubMed] [Google Scholar]
- Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM & Scheff SW (2005). Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma 22, 252–265. [DOI] [PubMed] [Google Scholar]
- Handelman GJ, Nightingale ZD, Dolnikowski GG & Blumberg JB (1998). Formation of carbonyls during attack on insulin by submolar amounts of hypochlorite. Anal Biochem 258, 339–348. [DOI] [PubMed] [Google Scholar]
- Hissin PJ & Hilf R (1976). A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem 74, 214–226. [DOI] [PubMed] [Google Scholar]
- Hoene M & Weigert C (2010). The stress response of the liver to physical exercise. Exerc Immunol Rev 16, 163–183. [PubMed] [Google Scholar]
- Huang GJ, Bhaskar Reddy MV, Kuo PC, Huang CH, Shih HC, Lee EJ, Yang ML, Leu YL & Wu TS (2012). A concise synthesis of viscolin, and its anti‐inflammatory effects through the suppression of iNOS, COX‐2, ERK phosphorylation and proinflammatory cytokines expressions. Eur J Med Chem 48, 371–378. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW & Smith BN (2011). Synaptic reorganization of inhibitory hilar interneuron circuitry after traumatic brain injury in mice. J Neurosci 31, 6880–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iniguez C, Gayoso MJ & Carreres J (1985). A versatile and simple method for staining nervous tissue using Giemsa dye. J Neurosci Methods. 13, 77–86. [DOI] [PubMed] [Google Scholar]
- Jamme I, Petit E, Divoux D, Gerbi A, Maixent JM & Nouvelot A (1995). Modulation of mouse cerebral Na+,K(+)‐ATPase activity by oxygen free radicals. Neuroreport 7, 333–337. [PubMed] [Google Scholar]
- Kamm K, Vanderkolk W, Lawrence C, Jonker M & Davis AT (2006). The effect of traumatic brain injury upon the concentration and expression of interleukin‐1beta and interleukin‐10 in the rat. J Trauma 60, 152–157. [DOI] [PubMed] [Google Scholar]
- Kazeminasab F, Marandi M, Ghaedi K, Esfarjani F & Moshtaghian J (2013). Endurance training enhances LXRalpha gene expression in Wistar male rats. Eur J Appl Physiol 113, 2285–2290. [DOI] [PubMed] [Google Scholar]
- Kazeminasab S, Esmaeilzadeh‐Gharehdaghi E, Oladnabi M, Ohadi M, Mirabzadeh A & Hosseinkhani S (2012). Aberrant expression of activating transcription factor 6 (ATF6) in major psychiatric disorders. Psychiatry Res 200, 1086–1087. [DOI] [PubMed] [Google Scholar]
- Kim DH, Ko IG, Kim BK, Kim TW, Kim SE, Shin MS, Kim CJ, Kim H, Kim KM & Baek SS (2010). Treadmill exercise inhibits traumatic brain injury‐induced hippocampal apoptosis. Physiol Behav 101, 660–665. [DOI] [PubMed] [Google Scholar]
- Klein E, Caspi Y & Gil S (2003). The relation between memory of the traumatic event and PTSD: evidence from studies of traumatic brain injury. Can J Psychiatry 48, 28–33. [DOI] [PubMed] [Google Scholar]
- Kostyuk VA, Potapovich AI, Cesareo E, Brescia S, Guerra L, Valacchi G, Pecorelli A, Deeva IB, Raskovic D, De Luca C, Pastore S & Korkina LG (2010). Dysfunction of glutathione S‐transferase leads to excess 4‐hydroxy‐2‐nonenal and H(2)O(2) and impaired cytokine pattern in cultured keratinocytes and blood of vitiligo patients. Antioxid Redox Signal 13, 607–620. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Ischiropoulos H & Bondy SC (1992). Evaluation of the probe 2′,7′‐dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5, 227–231. [DOI] [PubMed] [Google Scholar]
- Leeuwenburgh C & Heinecke JW (2001). Oxidative stress and antioxidants in exercise. Curr Med Chem 8, 829–838. [DOI] [PubMed] [Google Scholar]
- Lenzlinger PM, Saatman KE, Hoover RC, Cheney JA, Bareyre FM, Raghupathi R, Arnold LD & McIntosh TK (2004). Inhibition of vascular endothelial growth factor receptor (VEGFR) signaling by BSF476921 attenuates regional cerebral edema following traumatic brain injury in rats. Restor Neurol Neurosci 22, 73–79. [PubMed] [Google Scholar]
- Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S & Stadtman ER (1990). Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 186, 464–478. [DOI] [PubMed] [Google Scholar]
- Lima FD, Oliveira MS, Furian AF, Souza MA, Rambo LM, Ribeiro LR, Silva LF, Retamoso LT, Hoffmann MS, Magni DV, Pereira L, Fighera MR, Mello CF & Royes LF (2009). Adaptation to oxidative challenge induced by chronic physical exercise prevents Na+,K+‐ATPase activity inhibition after traumatic brain injury. Brain Res 1279, 147–155. [DOI] [PubMed] [Google Scholar]
- Lima FD, Souza MA, Furian AF, Rambo LM, Ribeiro LR, Martignoni FV, Hoffmann MS, Fighera MR, Royes LF, Oliveira MS & de Mello CF (2008). Na+,K+‐ATPase activity impairment after experimental traumatic brain injury: relationship to spatial learning deficits and oxidative stress. Behav Brain Res 193, 306–310. [DOI] [PubMed] [Google Scholar]
- Lima FD, Stamm DN, Della‐Pace ID, Dobrachinski F, de Carvalho NR, Royes LF, Soares FA, Rocha JB, Gonzalez‐Gallego J & Bresciani G (2013). Swimming training induces liver mitochondrial adaptations to oxidative stress in rats submitted to repeated exhaustive swimming bouts. PLoS ONE 8, e55668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungqvist O, Nygren J & Thorell A (2000). Insulin resistance and elective surgery. Surgery 128, 757–760. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH & McIntosh TK (1992). Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci 12, 4846–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubrano C, Valacchi G, Specchia P, Gnessi L, Rubanenko EP, Shuginina EA, Trukhanov AI, Korkina LG & De Luca C (2015). Integrated haematological profiles of redox status, lipid, and inflammatory protein biomarkers in benign obesity and unhealthy obesity with metabolic syndrome. Oxid Med Cell Longev 2015, 490613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoor O, Beaufrere B, Boirie Y, Ralliere C, Taillandier D, Aurousseau E, Schoeffler P, Arnal M & Attaix D (1996). Increased mRNA levels for components of the lysosomal, Ca2+‐activated, and ATP‐ubiquitin‐dependent proteolytic pathways in skeletal muscle from head trauma patients. Proc Natl Acad Sci USA 93, 2714–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoor O, Cayol M, Gachon P, Boirie Y, Schoeffler P, Obled C & Beaufrere B (1997). Albumin and fibrinogen syntheses increase while muscle protein synthesis decreases in head‐injured patients. Am J Physiol Endocrinol Metab 273, E898–E902. [DOI] [PubMed] [Google Scholar]
- Marquezi ML, Roschel HA, dos Santa Costa A, Sawada LA & Lancha AH, Jr (2003). Effect of aspartate and asparagine supplementation on fatigue determinants in intense exercise. Int J Sport Nutr Exerc Metab 13, 65–75. [DOI] [PubMed] [Google Scholar]
- Mirzayan MJ, Probst C, Krettek C, Samii M, Pape HC, van Griensven M & Samii A (2008). Systemic effects of isolated brain injury: an experimental animal study. Neurol Res 30, 457–460. [DOI] [PubMed] [Google Scholar]
- Misra HP & Fridovich I (1972). The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem 247, 3170–3175. [PubMed] [Google Scholar]
- Mitro N, Mak PA, Vargas L, Godio C, Hampton E, Molteni V, Kreusch A & Saez E (2007). The nuclear receptor LXR is a glucose sensor. Nature 445, 219–223. [DOI] [PubMed] [Google Scholar]
- Moinard C, Gupta S, Besson V, Morio B, Marchand‐Leroux C, Chaumeil JC, Cynober L & Charrueau C (2008). Evidence for impairment of hepatic energy homeostasis in head‐injured rat. J Neurotrauma 25, 124–129. [DOI] [PubMed] [Google Scholar]
- Moinard C, Neveux N, Royo N, Genthon C, Marchand‐Verrecchia C, Plotkine M & Cynober L (2005). Characterization of the alteration of nutritional state in brain injury induced by fluid percussion in rats. Intensive Care Med 31, 281–288. [DOI] [PubMed] [Google Scholar]
- Morel P, Tallineau C, Pontcharraud R, Piriou A & Huguet F (1998). Effects of 4‐hydroxynonenal, a lipid peroxidation product, on dopamine transport and Na+/K+ ATPase in rat striatal synaptosomes. Neurochem Int 33, 531–540. [DOI] [PubMed] [Google Scholar]
- Moschen AR & Tilg H (2008). Nutrition in pathophysiology and treatment of nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care 11, 620–625. [DOI] [PubMed] [Google Scholar]
- Mota BC, Pereira L, Souza MA, Silva LF, Magni DV, Ferreira AP, Oliveira MS, Furian AF, Mazzardo‐Martins L, Silva MD, Santos AR, Ferreira J, Fighera MR & Royes LF (2012). Exercise pre‐conditioning reduces brain inflammation and protects against toxicity induced by traumatic brain injury: behavioral and neurochemical approach. Neurotox Res 21, 175–184. [DOI] [PubMed] [Google Scholar]
- Motta L & Dutton E (2013). Suspected exercise‐induced seizures in a young dog. J Small Anim Pract 54, 213–218. [DOI] [PubMed] [Google Scholar]
- Motta VF, Aguila MB & Mandarim‐De‐Lacerda CA (2015). High‐intensity interval training (swimming) significantly improves the adverse metabolism and comorbidities in diet‐induced obese mice. J Sports Med Phys Fitness 56, 655–663. [PubMed] [Google Scholar]
- Myhre O, Andersen JM, Aarnes H & Fonnum F (2003). Evaluation of the probes 2′,7′‐dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochem Pharmacol 65, 1575–1582. [DOI] [PubMed] [Google Scholar]
- O'Connell KM, Littleton‐Kearney MT, Bridges E & Bibb SC (2012). Evaluating the Joint Theater Trauma Registry as a data source to benchmark casualty care. Mil Med 177, 546–552. [DOI] [PubMed] [Google Scholar]
- Olsen AL, Morrey JD, Smee DF & Sidwell RW (2007). Correlation between breakdown of the blood‐brain barrier and disease outcome of viral encephalitis in mice. Antiviral Res 75, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer L, Cadenas E & Davies KJ (2008). Free radicals and exercise: an introduction. Free Radic Biol Med 44, 123–125. [DOI] [PubMed] [Google Scholar]
- Pavlov I, Huusko N, Drexel M, Kirchmair E, Sperk G, Pitkanen A & Walker MC (2011). Progressive loss of phasic, but not tonic, GABAA receptor‐mediated inhibition in dentate granule cells in a model of post‐traumatic epilepsy in rats. Neuroscience 194, 208–219. [DOI] [PubMed] [Google Scholar]
- Petersen AM & Pedersen BK (2005). The anti‐inflammatory effect of exercise. J Appl Physiol 98, 1154–1162. [DOI] [PubMed] [Google Scholar]
- Radak Z, Chung HY, Koltai E, Taylor AW & Goto S (2008). Exercise, oxidative stress and hormesis. Ageing Res Rev 7, 34–42. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, Fernandez SC, Murai H, Trusko SP, Scott RW, Nishioka WK & McIntosh TK (1998). BCL‐2 overexpression attenuates cortical cell loss after traumatic brain injury in transgenic mice. J Cereb Blood Flow Metab 18, 1259–1269. [DOI] [PubMed] [Google Scholar]
- Rambo LM, Ribeiro LR, Oliveira MS, Furian AF, Lima FD, Souza MA, Silva LF, Retamoso LT, Corte CL, Puntel GO, de Avila DS, Soares FA, Fighera MR, Mello CF & Royes LF (2009). Additive anticonvulsant effects of creatine supplementation and physical exercise against pentylenetetrazol‐induced seizures. Neurochem Int 55, 333–340. [DOI] [PubMed] [Google Scholar]
- Rao V, Spiro JR, Handel S & Onyike CU (2008). Clinical correlates of personality changes associated with traumatic brain injury. J Neuropsychiatry Clin Neurosci 20, 118–119. [DOI] [PubMed] [Google Scholar]
- Ray K (2012). Liver: Activation of NF‐kappaB signaling in hepatocytes induces liver fibrosis. Nat Rev Gastroenterol Hepatol 9, 244. [DOI] [PubMed] [Google Scholar]
- Rizza RA (2010). Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: implications for therapy. Diabetes 59, 2697–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salo DC, Donovan CM & Davies KJ (1991). HSP70 and other possible heat shock or oxidative stress proteins are induced in skeletal muscle, heart, and liver during exercise. Free Radic Biol Med 11, 239–246. [DOI] [PubMed] [Google Scholar]
- Salvemini D, Seibert K, Masferrer JL, Settle SL, Currie MG & Needleman P (1995). Nitric oxide activates the cyclooxygenase pathway in inflammation. Am J Ther 2, 616–619. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Bender R, Frotscher M, Ross ST, Hollrigel GS, Toth Z & Soltesz I (2000). Granule cell hyperexcitability in the early post‐traumatic rat dentate gyrus: the ‘irritable mossy cell’ hypothesis. J Physiol 524, 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Dong B, Mao Y, Guan W, Cao J, Zhu R & Wang S (2016). Review: Traumatic brain injury and hyperglycemia, a potentially modifiable risk factor. Oncotarget. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlosberg D, Benifla M, Kaufer D & Friedman A. Blood‐brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 6, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlosberg D, Benifla M, Kaufer D & Friedman A (2010). Blood‐brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 6, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siems WG, Hapner SJ & van Kuijk FJ (1996). 4‐hydroxynonenal inhibits Na(+)‐K(+)‐ATPase. Free Radic Biol Med 20, 215–223. [DOI] [PubMed] [Google Scholar]
- Sigala F, Kostopanagiotou G, Andreadou I, Kavatzas N, Felekouras E, Sigalas P, Bastounis E & Papalambros E (2004). Histological and lipid peroxidation changes after administration of 2‐acetylaminofluorene in a rat liver injury model following selective periportal and pericentral damage. Toxicology 196, 155–163. [DOI] [PubMed] [Google Scholar]
- Silva LF, Hoffmann MS, Gerbatin R, Fiorin F, Dobrachinski F, Mota BC, Wouters AT, Soares FA, Pavarini SP, Fighera MR & Royes LF (2013a). Treadmill exercise protects against pentylenetetrazol‐induced seizures and oxidative stress after traumatic brain injury. J Neurotrauma 30, 1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva VS, Oliveira L & Goncalves PP (2013b). Alteration of aluminium inhibition of synaptosomal (Na(+)/K(+))ATPase by colestipol administration. J Inorg Biochem 128, 208–214. [DOI] [PubMed] [Google Scholar]
- Souza MA, Oliveira MS, Furian AF, Rambo LM, Ribeiro LR, Lima FD, Dalla Corte LC, Silva LF, Retamoso LT, Dalla Corte CL, Puntel GO, de Avila DS, Soares FA, Fighera MR, de Mello CF & Royes LF (2009). Swimming training prevents pentylenetetrazol‐induced inhibition of Na+, K+‐ATPase activity, seizures, and oxidative stress. Epilepsia 50, 811–823. [DOI] [PubMed] [Google Scholar]
- Spillmann F, Van Linthout S, Miteva K, Lorenz M, Stangl V, Schultheiss HP & Tschope C (2014). LXR agonism improves TNF‐alpha‐induced endothelial dysfunction in the absence of its cholesterol‐modulating effects. Atherosclerosis 232, 1–9. [DOI] [PubMed] [Google Scholar]
- Srere PA & Brooks GC (1969). The circular dichroism of glucagon solutions. Arch Biochem Biophys 129, 708–710. [DOI] [PubMed] [Google Scholar]
- Sun L, Shen W, Liu Z, Guan S, Liu J & Ding S (2010). Endurance exercise causes mitochondrial and oxidative stress in rat liver: effects of a combination of mitochondrial targeting nutrients. Life Sci 86, 39–44. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ota H, Sasagawa S, Sakatani T & Fujikura T (1983). Assay method for myeloperoxidase in human polymorphonuclear leukocytes. Anal Biochem 132, 345–352. [DOI] [PubMed] [Google Scholar]
- Toldy A, Stadler K, Sasvari M, Jakus J, Jung KJ, Chung HY, Berkes I, Nyakas C & Radak Z (2005). The effect of exercise and nettle supplementation on oxidative stress markers in the rat brain. Brain Res Bull 65, 487–493. [DOI] [PubMed] [Google Scholar]
- Tsatsoulis A, Mantzaris MD, Bellou S & Andrikoula M (2013). Insulin resistance: an adaptive mechanism becomes maladaptive in the current environment ‐ an evolutionary perspective. Metabolism 62, 622–633. [DOI] [PubMed] [Google Scholar]
- Tsuzuki T, Shinozaki S, Nakamoto H, Kaneki M, Goto S, Shimokado K, Kobayashi H & Naito H (2015). Voluntary exercise can ameliorate insulin resistance by reducing iNOS‐mediated S‐nitrosylation of akt in the liver in obese rats. PLoS ONE 10, e0132029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuñón MJ, San Miguel B, Crespo I, Jorquera F, Santamaría E, Alvarez M, Prieto J & González‐Gallego J (2011). Melatonin attenuates apoptotic liver damage in fulminant hepatic failure induced by the rabbit hemorrhagic disease virus. J Pineal Res 50, 38–45. [DOI] [PubMed] [Google Scholar]
- Unterberg AW, Stroop R, Thomale UW, Kiening KL, Pauser S & Vollmann W (1997). Characterisation of brain edema following ‘controlled cortical impact injury’ in rats. Acta Neurochir Suppl 70, 106–108. [DOI] [PubMed] [Google Scholar]
- Vaynman S & Gomez‐Pinilla F (2005). License to run: exercise impacts functional plasticity in the intact and injured central nervous system by using neurotrophins. Neurorehabil Neural Repair 19, 283–295. [DOI] [PubMed] [Google Scholar]
- Viguie CA, Frei B, Shigenaga MK, Ames BN, Packer L & Brooks GA (1993). Antioxidant status and indexes of oxidative stress during consecutive days of exercise. J Appl Physiol 75, 566–572. [DOI] [PubMed] [Google Scholar]
- Villapol S, Balarezo MG, Affram K, Saavedra JM & Symes AJ (2015a). Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 138, 3299–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Kryndushkin D, Balarezo MG, Campbell AM, Saavedra JM, Shewmaker FP & Symes AJ (2015b). Hepatic expression of serum amyloid A1 is induced by traumatic brain injury and modulated by telmisartan. Am J Pathol 185, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GX, Li GR, Wang YD, Yang TS & Ouyang YB (2001). Characterization of neuronal cell death in normal and diabetic rats following exprimental focal cerebral ischemia. Life Sci 69, 2801–2810. [DOI] [PubMed] [Google Scholar]
- Wang XC, Zhan XR, Li XY, Yu JJ & Liu XM (2014). MicroRNA‐185 regulates expression of lipid metabolism genes and improves insulin sensitivity in mice with non‐alcoholic fatty liver disease. World J Gastroenterol 20, 17914–17923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YB & Yang ZX (2010). Effect of oxygen free radicals on bacterial translocation in rats with traumatic brain injury. Zhonghua Yi Xue Za Zhi 90, 1716–1718. [PubMed] [Google Scholar]
- Wang YD, Chen WD, Yu D, Forman BM & Huang W (2011). The G‐protein‐coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light‐chain enhancer of activated B cells (NF‐kappaB) in mice. Hepatology 54, 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannamethee G & Shaper AG (1992). Physical activity and stroke in British middle aged men. BMJ 304, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kim HT, Ma Y, Zhao L, Zhai L, Kokorina N, Wang P & Messina JL (2008). Trauma and hemorrhage‐induced acute hepatic insulin resistance: dominant role of tumor necrosis factor‐alpha. Endocrinology 149, 2369–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LJ, Traber MG, Kobuchi H, Matsugo S, Tritschler HJ & Packer L (1996). Efficacy of hypochlorous acid scavengers in the prevention of protein carbonyl formation. Arch Biochem Biophys 327, 330–334. [DOI] [PubMed] [Google Scholar]
- Yan LJ, Traber MG & Packer L (1995). Spectrophotometric method for determination of carbonyls in oxidatively modified apolipoprotein B of human low‐density lipoproteins. Anal Biochem 228, 349–351. [DOI] [PubMed] [Google Scholar]
- Yang S, Ma Y, Liu Y, Que H, Zhu C & Liu S (2013). Elevated serum haptoglobin after traumatic brain injury is synthesized mainly in liver. J Neurosci Res 91, 230–239. [DOI] [PubMed] [Google Scholar]
- Ziebell JM & Morganti‐Kossmann MC (2010). Involvement of pro‐ and anti‐inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]