

Abstract
Background
Common bacterial blight is a devastating bacterial disease of common bean (Phaseolus vulgaris) caused by Xanthomonas citri pv. fuscans and Xanthomonas phaseoli pv. phaseoli. These phylogenetically distant strains are able to cause similar symptoms on common bean, suggesting that they have acquired common genetic determinants of adaptation to common bean. Transcription Activator-Like (TAL) effectors are bacterial type III effectors that are able to induce the expression of host genes to promote infection or resistance. Their capacity to bind to a specific host DNA sequence suggests that they are potential candidates for host adaption.
Results
To study the diversity of tal genes from Xanthomonas strains responsible for common bacterial blight of bean, whole genome sequences of 17 strains representing the diversity of X. citri pv. fuscans and X. phaseoli pv. phaseoli were obtained by single molecule real time sequencing. Analysis of these genomes revealed the existence of four tal genes named tal23A, tal20F, tal18G and tal18H, respectively. While tal20F and tal18G were chromosomic, tal23A and tal18H were carried on plasmids and shared between phylogenetically distant strains, therefore suggesting recent horizontal transfers of these genes between X. citri pv. fuscans and X. phaseoli pv. phaseoli strains. Strikingly, tal23A was present in all strains studied, suggesting that it played an important role in adaptation to common bean. In silico predictions of TAL effectors targets in the common bean genome suggested that TAL effectors shared by X. citri pv. fuscans and X. phaseoli pv. phaseoli strains target the promoters of genes of similar functions. This could be a trace of convergent evolution among TAL effectors from different phylogenetic groups, and comforts the hypothesis that TAL effectors have been implied in the adaptation to common bean.
Conclusions
Altogether, our results favour a model where plasmidic TAL effectors are able to contribute to host adaptation by being horizontally transferred between distant lineages.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4087-6) contains supplementary material, which is available to authorized users.
Keywords: Xanthomonas, Common bean, TAL effectors, Host adaptation, Horizontal gene transfer
Background
Bacterial pathogens from the genus Xanthomonas harbour singular type III effectors called transcription activator-like (TAL) effectors that are able to mimic eukaryotic transcription factors [1–3]. After being injected into host cells via the type III secretion system, TAL effectors migrate to the nucleus where they act as transcription factors thanks to an activation domain localized in the C-terminal (C-ter) region [4, 5]. The central region of the protein is composed of a variable number of quasi-identical, 34 amino acids long repeats, except for the last repeat of the central region usually corresponding to a half-repeat of 18 amino acids. The pair of residues at positions 12 and 13 of each repeat is named repeat variable diresidue (RVD) and determines the binding specificity of a repeat to a single nucleotide. Thus, the RVD sequence of a TAL effector determines its binding specificity to a target DNA sequence called Effector Binding Element (EBE) [6, 7]. A TAL code linking each RVD to their nucleotide binding affinity has been deciphered, enabling in silico prediction of EBE for a given TAL effector in the host genome [6–11]. The EBE is usually located in the promoter region of susceptibility genes, enabling TAL effectors to promote bacterial infection [12–15]. In some cases, the EBE is located upstream of resistance genes termed executor, leading to the defence reaction of the host [16, 17].
Because the function of a TAL effector requires its specific binding to DNA, each EBE is a potential source of resistance that can be exploited for developing innovative disease resistance strategies [18, 19]. For instance, any modification of an EBE can lead to loss of susceptibility because the TAL effector is not able to bind to the EBE anymore. Such strategy has been successfully used to engineer resistance of rice to X. oryzae pv. oryzae by genome editing [20, 21]. Another strategy consists in trapping TAL effectors by placing EBE of conserved TAL effectors in the promoter regions of resistance genes [22–25]. This has successfully been done in rice [26, 27]. These examples are proofs of concepts showing that TAL-based resistance engineering is possible. However, the durability of such resistances and their effectiveness against more diverse pathogens in field has still to be assessed. This requires broadening our knowledge of tal genes diversity in Xanthomonas populations. However, because of the repeated nature of their central domain, tal genes can be misassembled after whole genome sequencing based on Illumina, and/or 454 technologies [28]. Long-read sequencing strategies such as PacBio Single Molecule, Real-Time (SMRT) sequencing have been proved efficient for getting TAL effector sequences [29, 30].
Common bacterial blight of bean (CBB) is the most devastating bacterial disease on common bean. It is due to four different genetic lineages belonging to two distinct species within the genus Xanthomonas [31, 32]. The fuscous lineage (fuscans) and the non-fuscous lineages 2 (NF2) and 3 (NF3) all belong to X. citri pv. fuscans (formerly X. fuscans subsp. fuscans and X. axonopodis pv. phaseoli Rep-PCR group 9.6) while the non-fuscous lineage 1 (NF1) belongs to X. phaseoli pv. phaseoli (formerly X. axonopodis pv. phaseoli Rep-PCR group 9.4) [33–35]. Pathological convergence of strains responsible for CBB is apparently linked to large events of horizontal gene transfer (HGT) involving a hundred of genes [36]. High-quality genome sequencing of X. citri pv. fuscans strain CFBP4885 (synonym 4834-R) revealed the existence of two tal genes in this strain: Xfutal1 and Xfutal2, both located on plasmids [37]. PCR assays suggested that both Xfutal1 and Xfutal2 exist in the fuscans and NF1 lineages [36]. However, the diversity of TAL effectors in X. citri pv. fuscans and X. phaseoli pv. phaseoli has not yet been described.
In order to study the diversity of TAL effectors in Xanthomonas strains responsible for CBB, we have generated the whole genome sequence of 17 X. citri pv. fuscans and X. phaseoli pv. phaseoli strains by using PacBio SMRT sequencing. The sequence of tal genes was extracted and phylogenetic analyses including the tal gene sequences from other Xanthomonas strains were performed. The comparison of the phylogeny of tal genes with the corresponding phylogeny of strains allowed us to predict potential HGT events between the four X. citri pv. fuscans and X. phaseoli pv. phaseoli lineages, therefore suggesting a role of tal genes in host adaptation.
Results
Pathogenicity, genome quality and tal genes content
To estimate the diversity of tal genes present in strains responsible for CBB, the genome sequence of 17 strains representing the diversity of the four genetic lineages of X. citri pv. fuscans and X. phaseoli pv. phaseoli was obtained through PacBio SMRT sequencing (Table 1). All these strains were pathogenic on common bean, although with different degrees of aggressiveness (Table 1). The majority of strains (12/17) were highly aggressive on common bean while the other (5/17) were less aggressive. Overall, the average genome size and gene content (5.24 +/− 0.08 Mbp; 4596 +/− 160 genes) were quite classical for Xanthomonas strains [36–42]. Each genome sequence consisted of two to six contigs, with a maximum contig size corresponding to the expected chromosome size (5.1 Mbp) and additional circularized contigs corresponding to extrachromosomal plasmids. Plasmids A, B and C were named based on similarity with corresponding plasmids previously described in strain CFBP4885, also known as 4834-R [37] (Fig. 1).
Table 1.
Genome features of the 17 bacterial strains responsible for CBB sequenced in this study
| Pathovar (lineage) | Strain | Country (date of isolation) | Disease indexa | Contig number | Genome size (bp) | Max. contig length (bp) | % GC | Gene number |
GenBank Accession numbers |
|---|---|---|---|---|---|---|---|---|---|
| X. citri pv. fuscans (fuscans) | CFBP4885 | France (1998) | 3 | 6 | 5,173,315 | 5,012,288 | 64.66 | 4519 | CP020992 to CP020997 |
| CFBP6165 | Canada (1957) | 3 | 3 | 5,118,412 | 5,054,301 | 64.80 | 4412 | CP020998 to CP021000 | |
| CFBP6166 | South Africa (1963) | 3 | 5 | 5,200,310 | 5,025,712 | 64.67 | 4566 | CP021001 to CP021005 | |
| CFBP6167 | USA (1954) | 2 | 5 | 5,388,310 | 5,176,780 | 64.54 | 4755 | CP021018 to CP021022 | |
| CFBP6975 | France (1994) | 3 | 5 | 5,244,382 | 5,092,018 | 64.64 | 4634 | CP021006 to CP021010 | |
| CFBP7767R | Cameroon (2009) | 3 | 3 | 5,332,674 | 5,227,850 | 64.56 | 4893 | CP021012 to CP021014 | |
| X. citri pv. fuscans (NF2) | CFBP6988R | La Réunion (2000) | 3 | 2 | 5,199,272 | 5,122,265 | 64.62 | 4526 | CP020979 to CP020980 |
| CFBP6989 | La Réunion (2000) | 3 | 2 | 5,179,726 | 5,111,201 | 64.62 | 4498 | CP020981 to CP020982 | |
| CFBP6990 | La Réunion (2000) | 3 | 2 | 5,193,607 | 5,123,413 | 64.64 | 4512 | CP020983 to CP020984 | |
| CFBP6991 | La Réunion (2000) | 3 | 3 | 5,309,897 | 5,107,678 | 64.59 | 4673 | CP021015 to CP021017 | |
| X. citri pv. fuscans (NF3) | CFBP6992 | La Réunion (2000) | 1 | 2 | 5,299,787 | 5,232,548 | 64.57 | 4478 | CP020985 to CP020986 |
| CFBP6994R | Tanzania (1990) | 3 | 2 | 5,191,148 | 5,124,593 | 64.70 | 4399 | CP020987 to CP020988 | |
| CFBP6996R | La Réunion (2000) | 2 | 2 | 5,145,832 | 5,078,988 | 64.73 | 4352 | CP020989 to CP020990 | |
| X. phaseoli pv. phaseoli (NF1) | CFBP412 | USA (NA) | 2 | 3 | 5,186,811 | 5,071,497 | 64.89 | 4568 | CP020964 to CP020966 |
| CFBP6164 | Romania (1966) | 2 | 4 | 5,399,156 | 5,254,225 | 64.63 | 4833 | CP020967 to CP020970 | |
| CFBP6546R | USA (NA) | 3 | 4 | 5,258,370 | 5,081,709 | 64.78 | 4801 | CP020971 to CP020974 | |
| CFBP6982 | La Réunion (2000) | 3 | 4 | 5,291,156 | 5,100,676 | 64.75 | 4712 | CP020975 to CP020978 | |
| Average | 3 | 5,241,892 | 5,117,514 | 64.67 | 4596 |
aDisease symptoms were scored 11 days after bathing the first trifoliate leaf into bacterial suspensions at 1 × 107 CFU/mL according to the following scale: 0 = no symptoms, 1 = 1 to 50 spots per leaf, 2 = 51 to 200 spots per leaf, necrosis and sagging and 3 = more than 200 spots per leaf, necrosis, sagging or leaf death. The average of pathogenicity scores were calculated from the values of three plants per strain
Fig. 1.
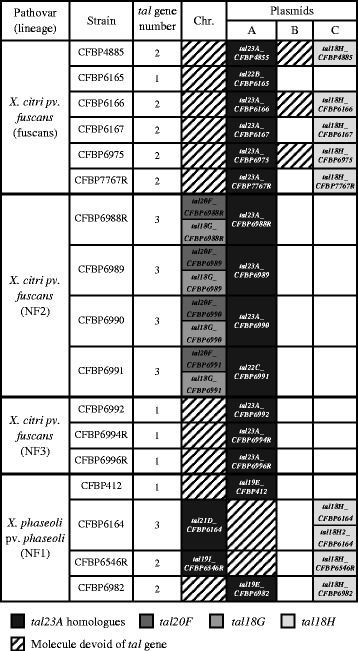
Localisation of tal genes within the genomes of 17 strains responsible for CBB. Presence of the corresponding molecule is represented by shades of grey depending on the presence of the different tal genes (see legend). Chr.: Chromosome
Following BLASTN searches in the 17 SMRT-sequenced genomes, a set of 26 complete tal genes and 14 putative tal pseudogenes were retrieved. Four pseudogenes corresponded to partial N-ter-encoding sequences, two corresponded to degenerated tal gene sequences bearing several frameshifts leading to premature stop codons, and eight corresponded to full-length tal genes containing a deletion of one nucleotide in either the N-ter- or the C-ter-encoding region (Additional file 1). Given the high rate of indels observed after SMRT sequencing [43], we sequenced the PCR products corresponding to these eight full-length tal pseudogenes with Sanger sequencing technology and all were validated as complete tal genes. This led to a total of 34 complete tal genes and six pseudogenes. Only complete tal genes were retained for further analyses (Additional file 2).
Strain CFBP4885 was previously sequenced using a combination of Illumina, 454 and Sanger technologies [37]. This high-quality genome sequencing revealed the existence of two tal genes (Xfutal1 and Xfutal2) in this strain, therefore allowing comparison of Xfutal1 and Xfutal2 to the tal sequences obtained here by SMRT sequencing for the same strain. Unexpectedly, the distinct sequencing technologies differed in the predicted length of tal genes. The lengths of Xfutal1 and Xfutal2 in strain CFBP4885 have been previously estimated at 4935 bp and 3405 bp, respectively [37]. However, lengths of 4017 bp and 3507 bp were found for tal genes obtained after SMRT sequencing. These sequences differed only by their number of repeats, indicating that the observed differences in length were due to assembly errors in their repeat regions. To validate Xfutal1 and Xfutal2 sequence lengths, we designed PCR primers on conserved sequences corresponding to the N-ter- and C-ter-encoding regions of XfuTAL1 and XfuTAL2 proteins. PCR profiles were consistent with the lengths obtained after SMRT sequencing, and Sanger sequencing of the PCR products further confirmed that Xfutal1 and Xfutal2 sequences have been successfully sequenced and assembled in the SMRT-sequenced genomes (Additional file 3). These results corroborate previous analyses showing that SMRT sequencing led to accurate assembly of tal genes [29, 30].
The length of the 34 complete tal genes found in our 17 genome sequences ranged between 3501 bp and 4017 bp, which corresponded to TAL effectors possessing from 18 to 23 repeats. The overall RVD composition was similar to that of previously sequenced tal genes from other Xanthomonas pathovars [4, 29] with more than 97% of RVD being HD, NN, NG, NI, N* or NK (Additional file 4). The only unusual RVD was HY, retrieved in nine TAL effectors and previously reported in only six other TAL effectors from X. oryzae pv. oryzicola and Ralstonia solanacearum [29, 44]. Nine unique RVD combinations ranging from 18 to 23 RVD were identified (Fig. 2). We named these nine unique RVD combinations using their number of repeats followed by a letter corresponding to each unique RVD combination, from A to I (Fig. 2). For example, for strain CFBP4885, corrected sequences of XfuTAL1 with 23 RVD and XfuTAL2 with 18 RVD corresponded to RVD combinations TAL23A and TAL18H, respectively. Following this nomenclature, Xfutal1 and Xfutal2 genes were renamed as tal23A_CFBP4885 and tal18H_CFBP4885, respectively (Figs. 1, 2).
Fig. 2.
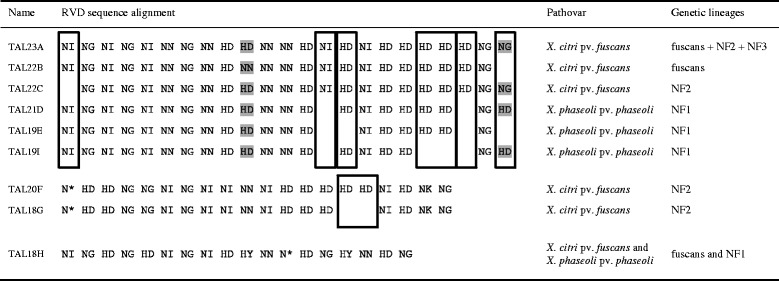
Alignment of the nine unique RVD sequences retrieved in TAL effectors from 17 strains responsible for CBB. TAL effectors that could be aligned are grouped together, indels are represented by black rectangles and putative substitutions are marked with grey colour
tal genes from X. citri pv. fuscans and X. phaseoli pv. phaseoli lineages originate from a single ancestor
To study the evolution of tal genes from strains responsible for CBB, we compared the phylogeny of tal genes to the phylogeny of X. phaseoli pv. phaseoli and X. citri pv. fuscans strains. For this, we first reconstructed the phylogeny of organisms using the whole predicted proteomes of the 17 X. phaseoli pv. phaseoli and X. citri pv. fuscans strains from our study plus 31 strains representing 10 Xanthomonas pathovars from which complete TAL effectors have previously been found, and 26 additional strains responsible for CBB sequenced previously (Additional file 5). The CVTree topology (Fig. 3a) was congruent with previously published Xanthomonas phylogenetic trees [33, 35, 45]. As previously shown, strains responsible for CBB split into four distinct genetic lineages [31, 46]. Three lineages, fuscans, NF2 and NF3 belonged to the X. citri species and one lineage, NF1, belonged to the X. phaseoli species (Fig. 3a). These groupings were confirmed by an average nucleotide identity above 99% within each genetic lineage, and under 95% between X. phaseoli pv. phaseoli and the three other lineages (Additional file 6).
Fig. 3.
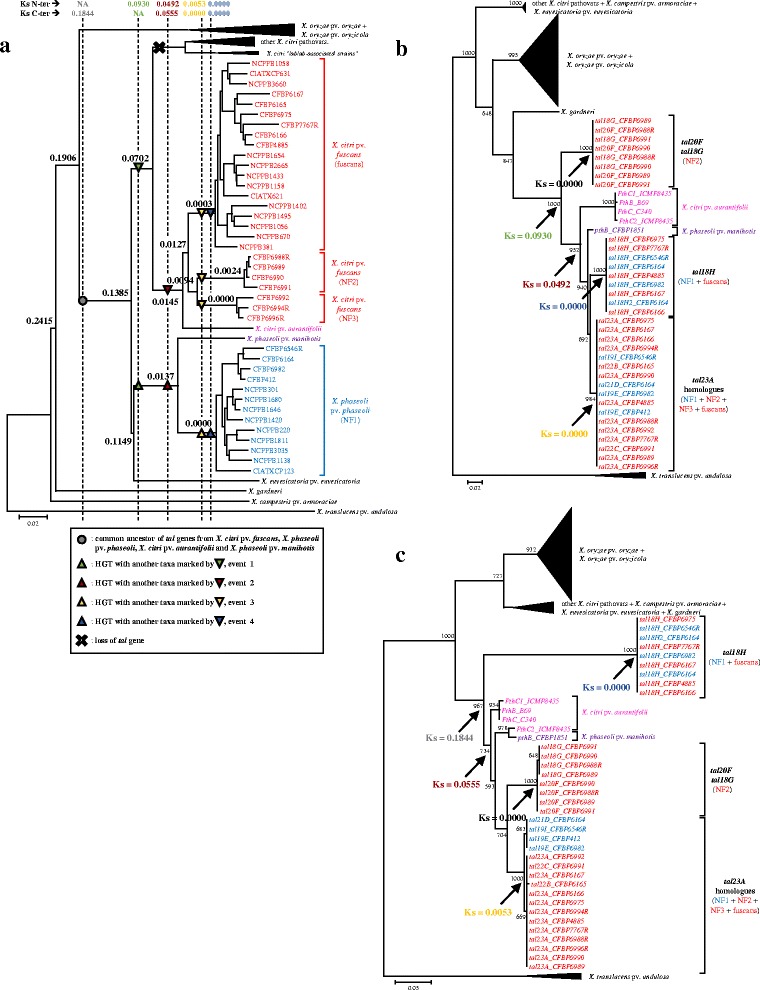
Phylogeny of Xanthomonas strains and tal genes. Strains or tal genes from the X. citri pv. fuscans genetic lineages fuscans, NF2 and NF3 are indicated in red, X. phaseoli pv. phaseoli NF1 lineage in blue, X. citri pv. aurantifolii in pink and X. phaseoli pv. manihotis in purple. Ks values corresponding to the divergence of tal genes are indicated in the different trees using the same colours. a Phylogenetic tree representing the evolution of Xanthomonas strains constructed using CVtree on the whole proteome of strains. Numbers at node correspond to mean Ks values calculated on seven housekeeping genes. Vertical dotted lines represent time of tal gene divergence estimated by Ks values written above. The most parsimonious course of events for the evolution of tal genes is highlighted according to the legend. NA: Not applicable. b ML tree constructed on the nucleotide sequences of N-ter-encoding region of tal genes. c ML tree constructed on the nucleotide sequences of C-ter-encoding region of tal genes. For (b) and (c), bootstrap values greater than 50% are shown for 1000 replicates and horizontal scale bars represent number of nucleotide substitutions per site
In evolutionary studies of tal genes, the central region is usually discarded because it is composed of a variable number of highly-conserved repeats that are able to recombine with each other, which can obscure alignments and phylogeny [29, 30]. Therefore, we reconstructed two phylogenies of tal genes using DNA sequences corresponding either to the N-ter- or to the C-ter-encoding region of complete TAL effectors (Figs. 3b, 3c). Both trees had a topology different from the organism CVTree, suggesting that recombination and/or horizontal transfers of tal genes occurred during the diversification of xanthomonads. In particular, tal genes from X. citri pv. fuscans and X. phaseoli pv. phaseoli clustered into a monophyletic group, together with tal genes from X. citri pv. aurantifolii and X. phaseoli pv. manihotis. For both trees, tal genes from X. citri pv. fuscans and X. phaseoli pv. phaseoli, were divided in three clades (Fig. 3b, c).
The CDS corresponding to RVD combinations TAL23A, TAL22B, TAL22C, TAL21D, TAL19E and TAL19I appeared to be derived from a recent common ancestor (Fig. 3b, c). Interestingly, every strain responsible for CBB possessed a single CDS corresponding to one of these six RVD combinations. These CDS were located on plasmid A for a majority of strains (15/17) except for strains CFBP6164 and CFBP6546R where it was located on the chromosome (Fig. 1). Also, all these CDS were surrounded by a conserved 17 kbp region whatever their location (see detailed explanation in the fifth section below). Together, these observations indicate that these CDS corresponded to allelic versions of a single gene, thus they were all considered as tal23A homologues. These tal23A homologues diverged by indels of complete internal repeats and/or by substitutions or recombinations affecting their RVD content (Fig. 2). Two additional genes, tal20F and tal18G, were only found in the four strains belonging to the NF2 lineage and had a chromosomic location (Fig. 1). These two tal genes were very similar to each other (Fig. 3b and c) and diverged by an indel involving two contiguous RVD (Fig. 2). Both tal genes were retrieved in every NF2 strain, indicating that they corresponded to paralogues resulting from a duplication predating the diversification of the NF2 lineage. Finally, tal18H was retrieved in most strains belonging to the fuscans (5/6) or NF1 (3/4) lineages, and was always located on plasmid C (Fig. 1). Notably, the three strains from the NF3 lineage did not present any additional tal gene other than tal23A (Fig. 1).
Partial tal sequences retrieved in previously sequenced genomes from the NF1 and fuscans lineages [36] confirmed that tal genes found in both lineages were all related to tal23A or tal18H genes (Additional file 7). Also, strains responsible for CBB isolated on lablab bean (Dolichos lablab), a legume plant phylogenetically close to common bean, have previously been described [36]. These strains clustered with X. citri pathovars other than X. citri pv. fuscans and X. citri pv. aurantifolii (Fig. 3a). Partial tal sequences were retrieved in the genomes from these lablab-associated strains. Strikingly, these tal genes did not cluster with any of the three aforementioned clades, further suggesting that TAL effectors from X. phaseoli pv. phaseoli and X. citri pv. fuscans were specific for strains having common bean as natural host.
Recombination occurred between tal genes from X. citri pv. fuscans and X. phaseoli pv. phaseoli
We observed discrepancies between the trees generated with the N-ter- or C-ter-encoding sequences. Indeed, in the tree generated using the C-ter-encoding sequence, tal18G and tal20F formed a clade external to the other tal genes from strains responsible for CBB, while in the tree generated using the N-ter-encoding sequence, tal18H clustered outside from the others (Fig. 3b, c). Tree topologies were significantly different (P < 0.05) based on the Kishino-Hasegawa-Templeton test [47]. This suggested that a recombination event occurred between the N-ter and C-ter encoding regions of these tal genes. To confirm this observation, we estimated recombination using seven nonparametric detection programs from the RDP4 package [48]. Two recombination events were retrieved on an alignment of the concatenated N-ter- and C-ter-encoding regions of tal genes from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis strains, but none were retrieved on the separated N-ter- or C-ter-encoding regions, suggesting that recombination breakpoints were located within the central repeat region of tal genes (data not shown). The first event was detected in tal18H, predicted as recombinants between the common ancestor of tal18G and tal20F, and pthB from the X. phaseoli pv. manihotis strain. The second event was detected within pthB, predicted as a recombinant between PthC2 and other tal genes from X. citri pv. aurantifolii. Both recombination events were in accordance with the differences observed between the tree topologies (Fig. 3b, c).
tal genes have undergone multiple horizontal transfers between X. citri pv. fuscans and X. phaseoli pv. phaseoli lineages
To estimate the time of divergence between tal genes, we compared the values of pairwise nucleotide synonymous substitution rates at silent sites (Ks) from the N-ter or C-ter-encoding regions of tal genes to Ks values from seven housekeeping genes (atpD, dnaK, efp, glnA1, gyrB, rpoD and suxA) [46] after removal of recombinant regions. In absence of codon usage bias, and for genes under constant evolutionary rates, Ks is an estimation of neutral evolution because it does not take into account the nonsynonymous sites that can be under selection pressure [49, 50]. Here, codon usage was constant among the different studied taxa, and no significant divergence from neutrality was retrieved (P > 0.05) after Tajima’s or Fu and Li’s relative rate tests (data not shown). Therefore, Ks could be used as an approximation of the time of divergence between genes or taxa [51], with higher Ks meaning longer time of evolution between two sequences.
The mean Ks for tal genes from X. citri pv. fuscans, X. phaseoli pv. phaseoli, X. citri pv. aurantifolii and X. phaseoli pv. manihotis strains were 0.0930 and 0.1844 for the N-ter- and C-ter-encoding regions, respectively (Fig. 3b, c). Thus, Ks value for the C-ter-encoding region was approximately twice as high as the Ks value for the N-ter-encoding region, indicating that the C-ter-encoding region has accumulated more synonymous substitutions at silent sites than the N-ter-encoding region did. This suggested that the C-ter-encoding region had a more ancient origin than the N-ter-encoding region in these strains. The Ks value for the C-ter-encoding region (0.1844) was in the same range as the Ks value calculated for housekeeping genes corresponding to the split between the X. oryzae species and the X. axonopodis species complex (0.1906), suggesting that the C-ter-encoding region has been transmitted vertically from the ancestor of X. axonopodis to the present days (Fig. 3a). In contrast, Ks value for the N-ter-encoding region (0.0930) was lower than the Ks value for housekeeping genes corresponding to the split between X. euvesicatoria and X. phaseoli species (Ks = 0.1149) and higher than the Ks value corresponding to the emergence of the X. citri species (Ks = 0.0702). This suggested that the N-ter-encoding region of tal genes from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis diverged after the split between X. euvesicatoria and X. phaseoli species and before the emergence of the X. citri species. This can be explained by a HGT between the ancestors of the X. phaseoli and X. citri species (event 1 on Fig. 3). Then, absence of orthologous tal genes in other X. citri pathovars would be due to a tal gene loss (Fig. 3a).
For both N-ter- and C-ter-encoding regions, tal genes from X. citri pv. aurantifolii, X. citri pv. fuscans, X. phaseoli pv. manihotis and X. phaseoli pv. phaseoli strains were grouped in a clade having tal18G and tal20F or tal18H as direct outgroups, respectively (Fig. 3b and c). In absence of HGT, the time of divergence for this clade should reflect the time of divergence between the X. citri and X. phaseoli species (Ks = 0.1385). However, the N-ter- and C-ter-encoding regions diverged at Ks values of 0.0492 and 0.0555, respectively (Fig. 3b and c). These Ks values were similar to each other, and lower than the Ks at the split between X. citri pathovars aurantifolii plus fuscans and the other X. citri pathovars (0.0702), but higher than Ks at the split between X. citri pv. aurantifolii and X. citri pv. fuscans (0.0145) and at the split between X. phaseoli pv. manihotis and X. phaseoli pv. phaseoli (0.0137; Fig. 3a). This suggested that a HGT of tal genes occurred between the ancestor of X. citri pv. aurantifolii and X. citri pv. fuscans, and the ancestor of X. phaseoli pv. manihotis and X. phaseoli pv. phaseoli (event 2 on Fig. 3a).
Presence of tal23A homologues in the phylogenetically distant X. citri pv. fuscans and X. phaseoli pv. phaseoli strains suggested that these tal genes were horizontally transferred between the ancestor of the NF1 lineage and the ancestor of one of the three X. citri pv. fuscans lineages (event 3 on Fig. 3a). Ks values for tal23A homologues between X. citri pv. fuscans and X. phaseoli pv. phaseoli lineages were zero for the N-ter-encoding region and 0.0053 for the C-ter-encoding region, indicating that tal23A homologues accumulated very few synonymous substitutions between X. citri pv. fuscans and X. phaseoli pv. phaseoli lineages, and indicating that this HGT was very recent. These low Ks values were similar to the values found within each lineage for housekeeping genes (NF1: 0.0000; NF2: 0.0024; NF3: 0.0000; fuscans: 0.0003), further confirming that this HGT occurred in the same time frame as the divergence of strains within each lineage. Moreover, most tal23A homologues were located on plasmid A. This plasmid was present in all X. citri pv. fuscans and X. phaseoli pv. phaseoli strains but not in X. citri pv. aurantifolii and X. phaseoli pv. manihotis strains [52, 53] (Fig. 1). Plasmid A was highly conserved with, on average, more than 97% nucleotide identity on more than 36% of the total plasmid length between the strains from the three X. citri pv. fuscans lineages and the strains from the NF1 lineage (Additional file 8, Additional file 9). This suggested that the event responsible for the horizontal transfer of tal23A homologues corresponded in fact to the horizontal transfer of a large portion of plasmid A. The largest conserved portion of plasmid A between X. citri pv. fuscans and X. phaseoli pv. phaseoli strains corresponded to 76% of plasmid A from strain CFBP6546R (NF1 lineage) and 77% of plasmid A from strain CFBP6988R (NF2 lineage) (Additional file 9). Thus, the most probable event would correspond to a HGT between the NF2 and NF1 lineages. The region between strains CFBP6988R and CFBP6546R corresponded to 39 kbp comprising 38 predicted protein-encoding genes (Additional file 10a, Additional file 11). These predicted proteins included 11 hypothetical proteins and 27 proteins with predicted functions including seven proteins involved in the type IV conjugal transfer system, three transposases, and two DNA topoisomerases (Additional file 11). In strain CFBP6546R, the tal23A homologue was located on the chromosome. Therefore, it was not retrieved in the 39 kbp region shared by both strains. In strain CFBP6988R, this tal23A homologue was located contiguous to the 39kbp region for a total of ~44 kbp (Additional file 10a). This suggests that a region of ~44 kbp from plasmid A was horizontally transferred between the NF2 and NF1 lineages, followed by the integration of tal23A homologues in the chromosome of some strains belonging to the NF1 lineage (this event is explained with more details in the next section).
Finally, tal18H was common to most strains from the NF1 and fuscans lineages but absent in the other lineages, suggesting that tal18H has been horizontally transferred between the ancestors of NF1 and fuscans strains (event 4 on Fig. 3a). In accordance with this, Ks values between NF1 and fuscans lineages were zero for both N-ter and C-ter-encoding regions, indicating that tal18H did not accumulate any nonsynonymous substitutions between the NF1 and fuscans lineages. Moreover, tal18H was located on plasmid C, and its presence was directly correlated with the presence of plasmid C (Fig. 1). Plasmid C was highly conserved with, on average, more than 99% nucleotide identity on more than 59% of the total plasmid length between NF1 and fuscans strains (Additional file 8, Additional file 12), suggesting that tal18H has been horizontally transferred along with a large portion of plasmid C. The largest conserved portion between X. citri pv. fuscans (fuscans) and X. phaseoli pv. phaseoli (NF1) strains corresponded to 68% of plasmid C from strain CFBP6982 (NF1 lineage) and 76% of plasmid C from strain CFBP6166 (fuscans lineage) (Additional file 12). The horizontally transferred region corresponded to ~30 kbp that were split in different smaller regions and/or duplicated depending on the strain, suggesting that recombination and/or local duplications occurred after the HGT event (Additional file 10b, Additional file 13). In addition to the tal18H gene, these regions comprised 26 predicted protein-encoding genes including eight hypothetical proteins and 18 proteins with predicted functions including the type III effector XopC1, two proteins involved in the type IV conjugal transfer system, three ribonucleases and two type II-like restriction endonucleases (Additional file 13). Strains CFBP412 and CFBP6165 were the only strains from the NF1 and fuscans lineages not possessing plasmid C, suggesting that they recently lost this plasmid, together with the tal18H gene (Fig. 1).
Recent tal gene duplications and movements are associated with the presence of insertion sequences (IS) and transposons
Presence of IS and/or Tn3-like transposons has been proved as a hallmark of tal gene movements and duplication [54]. Here, all tal genes from X. citri pv. fuscans and X. phaseoli pv. phaseoli were associated with IS3 and/or Tn3-like transposons. To estimate if these IS3 and/or Tn3-like transposons were responsible for tal gene movements or duplications, we analysed the genomic environment of relocated or duplicated tal genes.
As described above, our analyses indicated that tal23A homologues have been transmitted by a HGT between X. citri pv. fuscans and X. phaseoli pv. phaseoli strains involving plasmid A. The majority of tal23A homologues (15/17) was located on plasmid A except for two X. phaseoli pv. phaseoli strains, CFBP6164 and CFBP6546R, where tal21D_CFBP6164 and tal19I_CFBP6546R had a chromosomic location (Fig. 1). Strains CFBP6164 and CFBP6546R were closer to each other than any other strains (Fig. 3a). This suggested that the tal gene moved from the plasmid to the chromosome in the common ancestor of strains CFBP6164 and CFBP6546R. Analysis of the genomic environment indicated that this movement was associated with the relocation of a 17 kbp region surrounding this tal gene (Additional file 14a). Presence of intact IS3 sequences framing the receiving region of the chromosome of CFBP6164 and CFBP6546R strains, and presence of an IS3 remnant at the donor site in plasmid A in these strains suggested that recombination between IS3 sequences was involved in this movement, although we could not define the breakpoints precisely (Additional file 14a).
In each NF2 strain, tal genes tal20F and tal18G had nearly-identical sequences. These tal genes were not retrieved outside from the NF2 lineage, and the Ks value between these two copies was zero for both N-ter- and C-ter-encoding regions, indicating that these two genes resulted from a duplication in the direct ancestor of the NF2 lineage (Figs. 1 and 3). These tal genes were oriented back to back on the chromosome, and separated by around 720 kb. Both tal genes were surrounded by two Tn3-like transposons that were intact except for the region downstream tal18G (Additional file 14b). This suggested that this duplication involved a recombination between Tn3-like transposons, and indicated that tal18G was a copy of tal20F.
In plasmid C, tal18H was present as a single copy except for the CFBP6164 strain where two copies were found on plasmid C (tal18H_CFBP6164 and tal18H2_CFBP6164). This duplication corresponded to the tandem duplication of a seven kbp region including the tal gene and flanked by two IS3 sequences, suggesting that this duplication was the consequence of an unequal crossing-over between these two IS3 (Additional file 14c). Interestingly, the N-ter-encoding region of tal18H2_CFBP6164 comprised the insertion of a cytosine 88 bp after the start codon. This led to a frameshift resulting in the relocation of the start codon 27 aminoacids upstream of the classical N-ter-encoding region and modifying the first 30 aminoacids of the TAL18H2_CFBP6164 effector.
In silico predictions suggest that tal18H and tal23A homologues were involved in X. citri pv. fuscans and X. phaseoli pv. phaseoli adaptation to common bean
The repeat region of TAL effectors is important because it is involved in the binding specificity of TAL effectors to the promoters of targeted host plant genes [6, 7]. Previous studies suggested that recombination, duplication and deletion events within repeats were involved in tal gene evolution [54–56]. To study the evolution of the repeat regions of tal genes from X. citri pv. fuscans and X. phaseoli pv. phaseoli, we used the DisTAL program that is dedicated to the analysis of the evolution of TAL repeats excluding the RVD [57]. TAL23A homologues were distributed according to their genetic lineages (Fig. 4a). An important diversification was observed for the NF1 lineage that comprised genes encoding RVD combinations TAL19E, TAL19I and TAL21D (Figs. 2, 4a). In fuscans, NF2 and NF3 lineages, repeats were less diversified and TAL23A RVD combination was retrieved in 11 strains out of 13. An interesting point is that genes encoding TAL23A RVD combination presented a lineage-dependant diversification of their repeats outside of the RVD, while keeping the same RVD combination. Indeed, repeats encoding TAL23A in the NF2 and NF3 lineages were divergent from those found in the fuscans lineage (Fig. 4a). Also, within genes encoding TAL23A RVD combination in the fuscans lineage, tal23A_CFBP6975 presented a repeat region with a slight divergence from its orthologues in other strains from the fuscans lineage. Similarly, diversification occurred in the repeats of tal18H, leading to two different repeat profiles in the NF1 lineage and three in the fuscans lineage (Figs. 2, 4a). Thus, for TAL23A and TAL18H RVD combinations, RVD were retained despite repeat diversification, suggesting that these RVD combinations were functionally important for CBB causal agents.
Fig. 4.

Distance trees of repeat regions or RVD sequences of TAL effectors. TAL effectors from the X. citri pv. fuscans genetic lineages fuscans, NF2 and NF3 are indicated in red, X. phaseoli pv. phaseoli NF1 lineage in blue, X. citri pv. aurantifolii in pink and X. phaseoli pv. manihotis in purple. a Phylogenetic classification of the repeat regions of TAL effectors constructed with the DisTAL program (Pérez-Quintero et al., 2015). Analysis was performed on amino acid sequences of the repeat region (excluding RVD) of TAL effectors from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis, using eight TAL effectors from X. translucens pv. undulosa XT4699 as outgroups. b Neighbour Joining tree representing the distance between RVD sequences, constructed using the FuncTAL program. Analysis was performed on the RVD sequences of TAL effectors from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis
We performed a comparative analysis of the 772 individual repeats from the 39 TAL effectors retrieved in X. citri pv. fuscans, X. phaseoli pv. phaseoli, X. citri pv. aurantifolii and X. phaseoli pv. manihotis stains after removal of the RVD. These repeats were distributed in 48 unique repeat sequences (Additional file 15). All repeats comprised 34 amino-acids, or 18 amino-acids for the last half-repeat, and no repeat of unusual length was retrieved [58]. One predominant repeat was found 259 times (in red in Additional file 15, Additional file 16), while 14 repeats were retrieved only once. Interestingly, the predominant repeat was conserved in all TAL effectors whatever the associated RVD. Also, this repeat differed by at least one amino acid from any other repeat found in other pathovars described so far (data not shown), further highlighting that tal genes from X. citri pv. fuscans, X. phaseoli pv. phaseoli, X. citri pv. aurantifolii and X. phaseoli pv. manihotis strains share a common evolutionary history different from the evolutionary history of tal genes found in other Xanthomonas strains. A second repeat found 33 times was shared by all TAL effectors whatever the associated RVD, except for X. phaseoli pv. manihotis (in green in Additional file 15, Additional file 16). A third repeat found 31 times was shared among TAL23A homologues, TAL20F, TAL18G, and PthC from X. citri pv. aurantifolii strain (in purple in Additional file 15, Additional file 16). Two additional repeats were specific for TAL effectors from X. citri pv. aurantifolii and X. phaseoli pv. manihotis strains (in blue and yellow in Additional file 15, Additional file 16). This is in agreement with the DisTAL tree general topology (Fig. 4a) and suggested that TAL effectors of X. citri pv. aurantifolii and X. phaseoli pv. manihotis strains had a specific evolutionary history compared to X. citri pv. fuscans and X. phaseoli pv. phaseoli strains.
As mentioned before, RVD diversification of TAL effectors from CBB causal agents led to nine different RVD combinations (Fig. 2). To analyse the functional relationships between these TAL effectors, we used the FuncTAL program that compares DNA binding specificities using RVD sequences [57]. Despite the diversification of RVD sequences observed among TAL23A homologues, or between TAL20F and TAL18G, the FuncTAL tree showed that TAL effectors coming from a same clade clustered together (Fig. 4b). Interestingly, TAL23A homologues were closer to TAL18H than to TAL20F and TAL18G, while their repeats were more distant from TAL18H according to the DisTAL tree. This could be a trace of functional convergence between TAL23A homologues and TAL18H.
To search for putative common targets of TAL effectors within and between the three phylogenetic groups of tal genes, we used the TALVEZ EBE prediction tool on the promoterome (3 kb-promoter regions) of the common bean genome sequence [11, 59]. For each single RVD combination found in TAL effectors from X. phaseoli pv. phaseoli and X. citri pv. fuscans strains, the top 10 predicted targets according to the scores calculated by TALVEZ did not correspond to any other previously described target of TAL effectors from other Xanthomonas pathovars [12, 18, 60–67] (Additional file 17). However, several top 10 predicted targets were shared by TAL effectors coming from a same phylogenetic group. Indeed, eight predicted targets were shared by at least two of the six TAL23A homologues, including two targets predicted for four RVD combinations (Additional file 17). This was the case for a peptidase (Phvul.006G032900) predicted for TAL23A, TAL22B, TAL19E and TAL19I, and a 40S ribosomal subunit (Phvul.002G052200) predicted for TAL23A, TAL22C, TAL21D and TAL19I. In addition, two different genes encoding nucleolar ribonucleoprotein subunits (Phvul.008G022975 and Phvul.003G201700) were predicted as targets for TAL23A, TAL22B and TAL21D (Additional file 17). Also, four out of the first 10 predicted targets were shared by TAL20F and TAL18G. This included a protein of unknown function (Phvul.005G097300), a carboxyl methyltransferase (Phvul.006G048600), a nucleic acid deaminase (Phvul.003G292800) and an oligopeptide transporter-related protein (Phvul.010G009400). Interestingly, two genes encoding UDP-glycosyl transferases (Phvul.003G097300 and Phvul.009G148800) were predicted as targets for TAL23A, TAL22C and TAL21D as well as for TAL18H, further suggesting that a functional convergence exist between TAL23A homologues and TAL18H (Additional file 17).
Discussion
To study the diversity of TAL effectors from Xanthomonas strains responsible for CBB, we generated whole genome sequences for 17 strains representing the diversity of the four X. citri pv. fuscans and X. phaseoli pv. phaseoli genetic lineages. PacBio SMRT sequencing assemblies allowed us to retrieve 40 sequences of complete tal genes and pseudogenes. Previously, only two tal genes, Xfutal1 and Xfutal2 from CFBP4885 strain were reported for CBB agents [37]. Here, we show that Xfutal1 and Xfutal2 repeat regions contained assembly errors despite having been sequenced using a combination of Sanger, Illumina and 454 sequencing strategies. We thus confirmed that SMRT sequencing is a good methodology to obtain the sequence of TAL effector repeats [29, 30, 68, 69], which is essential for good prediction of their potential targets in host genomes [6, 7, 11]. However, corrections had to be made for deletions observed in the N-ter- and/or C-ter-encoding regions. These deletions were observed only in G- or C-rich stretches, which could correspond to error hotspots for SMRT sequencing.
Recombination and HGT are important driving forces of ecological adaptation and diversification in bacteria such as xanthomonads [45, 70–72]. Here, phylogenetic analyses revealed that the overall topology of tal genes differed from the topology of strains, indicating that the evolution of tal genes has been impacted by different events of recombination and/or HGT (Fig. 3). It was particularly interesting to observe that whether for the repeats, the N-ter-, or the C-ter-encoding regions, tal genes from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis strains clustered in a single clade (Figs. 3b, c, 4a). This strongly supports a scenario where tal genes from these four pathovars share a common ancestor different from tal genes found in other Xanthomonas species. Ks values for the N-ter- and C-ter-encoding regions suggested that two HGT involving tal genes occurred between the ancestors of X. citri and X. phaseoli species, and between the ancestors of X. citri pv. aurantifolii and X. citri pv. fuscans strains and the ancestor of X. phaseoli pv. manihotis and X. phaseoli pv. phaseoli strains (events 1 and 2 on Fig. 3a). Absence of orthologues in X. citri pathovars other than X. citri pv. aurantifolii and X. citri pv. fuscans certainly should be a result of a specific gene loss in these other X. citri pathovars. In particular, X. citri strains that were isolated on lablab and able to cause CBB on common bean [36, 73] did not possess any tal gene orthologous to the tal genes found in strains isolated on common bean. This further strengthens that the tal genes described here are specific to Xanthomonas strains having common bean as a natural host. Also, it showed that these lablab-associated strains were able to cause CBB symptoms on common bean without any contribution of the TAL effectors found in the genuine common bean pathogens, suggesting that these TAL effectors were not fully needed for pathogenicity and would rather contribute to aggressiveness. It would be interesting to obtain the RVD combination of TAL effectors from lablab-associated strains to analyse the contribution of these TAL effectors to the aggressiveness of Xanthomonas strains on common bean.
In strains responsible for CBB, tal gene diversification led to three distinct clades of tal genes. Subsequent to this diversification, we observed tal gene duplications and movements associated with IS and Tn3-like transposons, in accordance with previous observations in X. citri subsp. citri and other xanthomonads [54]. IS and transposons appear to have facilitated recombination, thus contributing to tal gene duplications and movements in strains responsible for CBB. It is tempting to speculate that the transfer of tal23A homologues that was observed from the plasmid A to the chromosome in strains CFBP6164 and CFBP6546 (Additional file 14) would allow a stable maintenance of these tal genes in the genome, and thus would underline the primary importance of tal23A homologues in adaptation of Xanthomonas to common bean.
Combination of phylogeny and Ks analyses unveiled events of recombination and HGT that shaped the evolution of tal genes in strains responsible for CBB. First, these analyses showed that the different X. citri pv. fuscans and X. phaseoli pv. phaseoli lineages bear different combinations of tal genes, suggesting that these lineages evolved in different ways for manipulating the common bean transcriptome. Strains from X. citri pv. fuscans NF3 lineage contained tal23A homologues only. Strains from X. citri pv. fuscans NF2 lineage also contained tal23A homologues, and were the only strains carrying tal20F and tal18G, suggesting that these genes emerged specifically in the X. citri pv. fuscans NF2 lineage. Finally, most strains from the X. citri pv. fuscans fuscans and X. phaseoli pv. phaseoli NF1 lineages, despite being phylogenetically distant, contained tal23A homologues and tal18H which is highly suggestive of HGT (Fig. 1). Ks analyses confirmed that tal18H has been horizontally transferred between these two lineages and that tal23A homologues have been transmitted by HGT between the NF1 lineage and the NF2 lineage. Moreover, plasmid conservation among phylogenetically distant lineages showed that HGT involving both tal18H and tal23A homologues were not restricted to these tal genes alone, and highlight the role of plasmids for these two HGT. These results are reminiscent of studies showing that numerous genes including type three effectors have been horizontally transferred between X. citri pv. fuscans and X. phaseoli pv. phaseoli lineages [36, 74]. This also suggests that tal18H and tal23A homologues have been involved in Xanthomonas adaptation to common bean. In particular, tal23A homologues were conserved in all strains responsible for CBB, suggesting that they could play a crucial role in X. citri pv. fuscans and X. phaseoli pv. phaseoli pathogenicity.
The study of repeats and RVD evolution shed new light on the importance of tal genes for the interaction between Xanthomonas strains and common bean. The DisTAL tree revealed that TAL23A and TAL18H presented variations in their repeats for aminoacids other than the RVD, resulting in three variants of TAL23A and five variants of TAL18H (Fig. 4a). As RVD sequence is responsible for DNA-binding specificity, these observations suggested that selection pressure acted against RVD diversification in both TAL23A and TAL18H, highlighting the importance of TAL23A and TAL18H functional maintenance in different X. citri pv. fuscans and X. phaseoli pv. phaseoli lineages. In particular, TAL18H RVD combination was completely conserved in both NF1 and fuscans lineages, which is in line with TAL18H being involved in Xanthomonas adaptation to common bean. To our knowledge, this is the first case of RVD sequence conservation across distant Xanthomonas lineages described so far.
FuncTAL analysis and EBE predictions showed that the function of TAL effectors appeared relatively conserved within each tal clade (Fig. 4b, Additional file 17). Another interesting observation was that predicted targets of TAL23A homologues and TAL18H both comprised genes encoding an UDP-glycosyltransferase. So far, UDP-glycosyltransferases have not been described as TAL effectors targets [12, 18, 60–67]. UDP-glycosyltransferase transcription has previously been associated with plant resistance to fungi and bacteria [75–77]. Further analyses including mutant construction, phenotyping and transcriptomics are underway to functionally validate the targets of TAL effectors of strains responsible for CBB.
The origins of the strains used for this study gave additional information on tal gene evolution in the different X. citri pv. fuscans and X. phaseoli pv. phaseoli lineages (Table 1). Indeed, all four lineages have been found in La Réunion Island in 2000 (http://catalogue-cfbp.inra.fr/recherche.php), indicating that sympatry exists among all these lineages in this place, rendering possible further HGT events between strains from these four lineages. Also, in contrast to the NF2 and NF3 lineages, fuscans and NF1 strains have been collected across very distant geographical areas spanning American, European, and African continents. This widespread distribution could have been facilitated by transport of contaminated seeds [78, 79], in frame with agricultural intensification and expansion [46]. Finally, two fuscans strains collected in 1954 and 2009 and two NF1 strains collected in 1966 and 2000 all contained tal18H and tal23A homologues, indicating that these tal genes have been maintained in both lineages for dozens of years. This further highlights the importance of these TAL effectors in these two lineages.
Our finding that plasmidic localisation has played a role in tal gene dissemination across phylogenetically distant Xanthomonas lineages adapted to common bean shed new light in tal genes potential of evolution. Indeed, it appears that two different ways of evolution exist for tal genes in Xanthomonas. First, it is striking that X. oryzae pv. oryzae and X. oryzae pv. oryzicola pathovars rely on numerous (up to 28) chromosomal tal genes [29, 68, 80–82]. Also, several cases of convergent targeting of SWEET family genes by X. oryzae pv. oryzae and X. oryzae pv. oryzicola TAL effectors have been described [12, 58, 61, 83, 84]. Moreover, polymorphism in EBE, or localisation of EBE upstream of executor genes led to the resistance of several rice accession [15, 22–25]. Together, these observations in rice suggested that consecutive events of coevolution occurred between rice and X. oryzae pv. oryzae / X. oryzae pv. oryzicola TAL effectors. An interpretation to this would be that presence of numerous tal genes localised in the chromosome would be an evidence of long-termed co-evolution between TAL effectors and the host plant genome. In sharp contrast with what was observed in X. oryzae pv. oryzae and X. oryzae pv. oryzicola, other pathovars rely solely on few tal genes localised on plasmids. For example, X. euvesicatoria contains only two tal genes, AvrBs3 and AvrBs4, both encoded on plasmids [85], while X. citri subsp. citri bears up to four pthA homologues on two different plasmids [86]. In X. phaseoli pv. phaseoli and X. citri pv. fuscans, we observed only one to three tal genes per strain and the majority was plasmidic. The tal genes with chromosomic localisation were the result of a recent duplication in the NF2 lineage, which could be a trace of recent coevolution with common bean. On the other hand, plasmidic TAL effectors were conserved in different lineages and some of them bore the exact same RVD sequence, arguing for a recent horizontal transfer of these tal genes. It is tempting to speculate that RVD conservation in horizontally-transferred tal genes reflects the fact that the host plant did not have enough time to evolve any resistance against these TAL effectors. An interpretation to this would be that a short number of tal genes with plasmidic localisation would be an evidence of short-termed evolution without coevolution between the TAL effector and the host genome. As such, horizontal transfers of tal genes can lead to the rapid modification of TAL repertoires, and could participate to the acquisition of novel traits for host adaptation.
Conclusion
Together, our observations favour a model where plasmidic tal genes are able to contribute to host adaptation by being horizontally transferred between distant lineages. Such horizontal gene transfer can take place either on a host plant or on another asymptomatic plant where a broader Xanthomonas diversity is expected to live [45]. Because we are not completely aware of this diversity, we may still underestimate the evolutionary potential of TAL effector to circumvent TAL-based resistance engineering strategies. To anticipate such potential and elaborate durable resistances against diseases caused by xanthomonads, it is important to make use of third-generation sequencing strategies on a much broader diversity of Xanthomonas strains than what has been made so far, and to explore more in-depth the whole TAL repertoire of xanthomonads.
Methods
Bacterial strains, plant material and growing conditions
Xanthomonas strains used and sequenced for this study are listed in Table 1. Strains were grown 48 h at 28 °C on TSA medium (17.0 g/L pancreatic digest of casein; 3.0 g/L enzymatic digest of soya bean; 5.0 g/L NaCl; 2.5 g/L K2HPO4; 2.5 g/L glucose; 15 g/L agar), then 24 h at 28 °C on a 1/10 dilution of TSA medium to obtain fresh grown bacteria. When needed, rifamycin was added at a final concentration of 50 mg/L.
The seeds from common bean cultivar Flavert used in this study were kindly provided by Vilmorin (La Ménitré, France) and were considered free of X. citri pv. fuscans and X. phaseoli pv. phaseoli following the analysis of approximately 100,000 seed per lot with standard tests (International Seed Testing Association 2007). Bean plants were grown in plastic square pots (8 cm) containing compost (Neuhaus humin substrat S NF 44–551) in a controlled climatic room under 95% relative humidity with an alternation of 16 h of light at 23 °C and 8 h of darkness at 20 °C until the first trifoliate leaf was fully expanded. Plants were watered every 2 days and supplemented with N-P-K (18:14:18) at 0.3 g/L once a week.
Pathogenicity assays
The day before inoculation, and for the duration of pathogenicity assays, plants were incubated under 95% relative humidity with an alternation of 16 h of light at 28 °C and 8 h of darkness at 25 °C. Pathogenicity assays were performed with bacterial suspensions calibrated at 1 × 107 CFU/mL in sterile distilled H2O. Pathogenicity assays were performed by bathing the first trifoliate leave for 30 s into bacterial suspensions. One trifoliate per plant and three plants per strain were inoculated. Symptoms were scored on the 11th day following inoculation according to the following scale: 0 = no symptoms, 1 = 1 to 50 spots per leaf, 2 = 51 to 200 spots per leaf, necrosis and sagging and 3 = more than 200 spots per leaf, necrosis, sagging or leaf death. Average scores were calculated from the values of three plants per strain. Pathogenicity assays were carried out under quarantine at UMR1345 IRHS, Centre INRA, Beaucouzé, France.
Genomic DNA extraction, genome sequencing, assembly and annotation
Bacterial cells were scraped from agar plates, suspended in sterile distilled H2O and collected by centrifugation. Genomic DNA was extracted with The Wizard® Genomic DNA Purification Kit (Promega) according to manufacturer’s recommendations. Whole genomes were sequenced using the PacBio SMRT technology [87] at Icahn School of Medicine at Mount Sinai (New York, NY). One SMRT cell was used per strain to achieve ~100× coverage. De novo assembling was performed using HGAP assembler [88] version 3.0 (Pacific Biosciences, Menlo Park, CA). Annotation of whole genome assemblies was performed with EuGene-PP version 1.2 automated pipeline [89], using SWISS-PROT as protein and training protein databases (http://www.uniprot.org/). The sequences of tal genes were manually retrieved using BLASTN search with the N-ter- and C-ter-encoding regions of Xfutal1 and Xfutal2 as queries [37]. In case of indels leading to tal pseudogenes, sequence verifications were performed by PCR amplifications using specific primers on genomic DNA, followed by Sanger sequencing of PCR products (Additional file 18).
Phylogenetic analyses and detection of recombination
The phylogeny of organisms was performed using CVTree version 4.2.1 [90] on the whole predicted proteome of strains listed in Table 1, plus 31 proteomes available in public database that belong to Xanthomonas pathovars where tal genes have previously been described (Additional file 5).
For tal genes phylogeny, we used the tal genes from strains listed in Table 1, plus 151 tal genes encoding unique RVD sequences, available in public databases and sequenced by SMRT sequencing or functionally validated (Additional file 5). The amino acid sequences of the N-ter and C-ter regions of these TAL effectors were aligned with MAFFT version 7 using the L-ins-I strategy [91], then this alignment was used as template for generating a codon-based nucleotide alignment with RevTrans 2.0 [92]. For both alignments, the best model of evolution determined using ModelTest 3.7 in Paup was the TVM + I + G model. Under this model, Maximum Likelihood (ML) phylogenetic trees were generated using PhyML 3.0 version 20,110,919 [93] and bootstraps analyses were done with 1000 iterations. Trees were visualised and manually edited with Mega 7.0.14 [94].
For detecting recombination, two additional trees constructed using nucleotide alignments of the N-ter- or C-ter-encoding regions of tal genes from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli, X. phaseoli pv. manihotis strains and a tal gene from X. translucens pv. undulosa as outgroup were produced using the aforementioned method. For the N-ter -encoding region tree, the model of evolution was K81uf + I and for the C-ter-encoding region tree, the model of evolution was TVM + G (Additional file 19). The Kishino-Hasegawa-Templeton test [47] implemented in the DNAML program from PHYLIP version 3.69 [95] was used to test the congruence of tree topologies. Also, nucleotide alignments from both N-ter- and C-ter-encoding regions of tal genes from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis strains were concatenated. Detection of potential recombination events were searched for on the three nucleotide alignments (separated N-ter- and C-ter-encoding regions, and concatenated) using a set of seven nonparametric detection programs implemented in RDP version 4 beta 69 [48]: RDP [96]; Geneconv [97]; Bootscan [98]; MaxChi [99]; Chimera [100]; SiScan [101] and 3Seq [102]. Default parameter settings were used for each method except as follows: RDP (internal reference sequence); Bootscan (window size = 150, neighbour joining trees, 200 bootstrap replicates, 95% cutoff, Jin and Nei model, variation coefficient = 2). For general settings, the maximum P value for accepting recombination was set at 0.001 after a Bonferroni correction. Only recombination events retrieved by at least three programs with a P value <0.001 were considered.
Average nucleotide identity calculations
Pairwise comparisons of X. citri pv. fuscans and X. phaseoli pv. phaseoli genome and plasmids nucleotide identities were performed by calculating the Average Nucleotide Identity by using BLASTn with JSpecies using default parameters [103] (Additional file 6, Additional file 8, Additional file 9).
Ks calculations
Ks was estimated on nucleotide alignments of N-ter- and C-ter-encoding regions of tal genes from X. citri pv. aurantifolii, X. citri pv. fuscans, X. phaseoli pv. manihotis and X. phaseoli pv. phaseoli strains, as well as on a concatenated alignment of seven previously described housekeeping genes from X. oryzae, X. citri, X. phaseoli, and X. euvesicatoria species [46]. Alignments and recombination analyses were done as described in the “Phylogenetic analyses and detection of recombination” section. After removal of recombinant regions, Ks was determined using DNAsp version 5 with default parameters [104]. On the same alignment, codon usage bias was calculated using DNAsp version 5 and no major difference was observed between the different Xanthomonas species (data not shown). Also Tajima’s and Fu and Li’s relative rate test were performed with DNAsp version 5 and no significant divergence from neutrality was retrieved (P > 0.05).
Repeats and RVD analyses
Amino acid sequences of every repeats from TAL effectors of X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis strains were manually extracted and aligned using MAFFT version 7 [91], after RVD exclusion. Then, unique repeats were searched, and repeats found multiple times were counted using Geneious version 9.1.3 [105]. For Fig. 1, amino acid RVD sequence of TAL effectors from X. citri pv. fuscans and X. phaseoli pv. phaseoli strains were extracted and an alignment was performed with MAFFT version 7 [91].
The evolution of TAL effector repeat regions and RVD sequences were analysed using the QueTAL suite [57]. First, amino acid sequences of repeat regions from TAL effectors of X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli, X. phaseoli pv. manihotis strains were extracted and aligned manually according to the QueTAL’s instructions, using TAL effectors from X. translucens pv. undulosa as outgroup for the DisTAL analysis and no outgroup for the FuncTAL analysis. Then, these alignments were used to construct a distance tree of TAL effector repeat regions using DisTAL version 1.1 and a distance tree of RVD sequences using FuncTAL version 1.1 [57]. DisTAL program considers repeat sequences without RVD whereas FuncTAL program only considers RVD sequences.
With the nine unique RVD combinations, predictions of EBE were performed using TALVEZ version 3.2 with correction matrix starting from the 14th RVD [11] on the common bean promoterome consisting of the 3 kb region upstream of the transcriptional start of each transcript annotated on the genome of common bean genotype G19833 version 2.1 [59] available on Phytozome 12 (https://phytozome.jgi.doe.gov/).
Additional files
Correction of sequences from tal pseudogenes containing indels in the SMRT sequence. (XLSX 10 kb)
Amino acid sequences of TAL effector from the 17 strains responsible for CBB obtained in this study. (DOCX 19 kb)
Gel photograph showing the sizes obtained for tal23A_CFBP4885 (Xfutal1) and tal18H_CFBP4885 (Xfutal2) after PCR amplification. Amplification was performed using XapF2 and XapR primers on plasmidic DNA from the CFBP4885 strain (Additional file 18: Table S11). Expected PCR product sizes for tal23A_CFBP4885 and tal18H_CFBP4885 were 4604 bp and 3074 bp for Sanger sequencing of the entire genome, respectively, and 3686 bp and 3176 bp for SMRT sequencing of the entire genome, respectively. (DOCX 2788 kb)
RVD frequencies in TAL effectors from the 17 X. citri pv. fuscans and X. phaseoli pv. phaseoli strains used in this study. (DOCX 20 kb)
References for previously published tal genes and whole genome sequences from Xanthomonas spp. strains used in this study. (XLSX 19 kb)
Average nucleotide identity and percentage of alignment between whole genome sequences from 17 X. citri pv. fuscans and X. phaseoli pv. phaseoli strains. BLAST calculation of average nucleotide identity (ANIb) and alignment percentages were estimated using Jspecies (Richter and Rossello-Mora, 2009). (XLSX 15 kb)
Phylogenetic trees of N-ter and C-ter-encoding regions of tal genes with sequences published by Aritua et al. (2015). Bootstrap values greater than 50% (100 replicates) are shown and horizontal scale bars represent the number of nucleotide substitutions per site. The tal genes from the X. citri pv. fuscans genetic lineages fuscans, NF2 and NF3 are indicated in red, X. phaseoli pv. phaseoli NF1 lineage in blue, X. citri pv. aurantifolii in pink and X. phaseoli pv. manihotis in purple. The sequences published by Aritua et al. (2015) are in bold. Both ML trees were constructed using tal genes from X. translucens pv. undulosa XT4699 as outgroups. a ML tree constructed on a nucleotide alignment of the N-ter-encoding region of tal genes. b ML tree constructed on a nucleotide alignment of the C-ter-encoding region of tal genes. (PPTX 137 kb)
Circular representation of plasmids A and C. Genomic sequences were compared and converted in a graphical map using CGView (Grant et al., 2012) with strain CFBP4885 as reference. Colours differ according to identity percentage (see legend). Strains order, ANIb and alignment percentages are indicated in the center of each graphical map. Localisation of tal genes are highlighted by green zones. Absence of tal genes in plasmid A from strains CFBP6164 and CFBP6546R is indicated by a black cross. (PDF 444 kb)
Average nucleotide identity and percentage of alignment between sequences of plasmid A from 17 X. citri pv. fuscans and X. phaseoli pv. phaseoli strains. BLAST calculation of average nucleotide identity (ANIb) and alignment percentages were estimated using Jspecies (Richter and Rossello-Mora, 2009). (XLSX 14 kb)
Circular representation of the regions shared by phylogenetically distant lineages in plasmids A and C. Genomic sequences were compared and converted in a graphical map using CGView (Grant et al., 2012). Colours differ according to identity percentage (see legend). Regions shared by both strains are surrounded by a black line. Numbers represent genes conserved in both strains (see Additional files 11 and 13 for more details). a Plasmids A from strains CFBP6988R (outer circle = reference) and CFBP6546R (inner circle). b Plasmids C from strains CFBP6166 (outer circle = reference) and CFBP6982 (inner circle). (PDF 168 kb)
Genes localized in Plasmid A shared by strains CFBP6988R and CFBP6546R. Please refer to Additional file 10a for graphic representation of the data. (XLSX 15 kb)
Average nucleotide identity and percentage of alignment between sequences of plasmid C from 8 X. citri pv. fuscans fuscans and X. phaseoli pv. phaseoli NF1 strains. BLAST calculation of average nucleotide identity (ANIb) and alignment percentages were estimated using Jspecies (Richter and Rossello-Mora, 2009). (XLSX 11 kb)
Genes localized in Plasmid C shared by strains CFBP6982 and CFBP6166. Please refer to Additional file 10b for graphic representation of the data. (XLSX 15 kb)
Duplications and movements of tal genes associated with IS3 and Tn3-like transposons. Arrows represent tal genes (yellow), IS3 sequences (orange) or Tn3-like transposons (dark blue). Arrowheads correspond to truncated IS3 (orange) or Tn3-like transposon (dark blue). Striped areas correspond to conserved regions. Dashed lines represent the borders of duplication events between different regions of a single molecule. a Movement of tal23A homologues from the plasmid A to the chromosome associated with IS3 sequences. b Duplication of tal20F and tal18G associated with Tn3-like transposons. c. Duplication of tal18H_CFBP6164 associated with IS3 sequences in strain CFBP6164. (PPTX 74 kb)
Repeats found within all TAL effectors from X. citri pv. fuscans, X. phaseoli pv. phaseoli, X. citri pv. aurantifolii and X. phaseoli pv. manihotis strains. Sequences were compared after removal of RVD. Xcf = X. citri pv. fuscans; Xpp = X. phaseoli pv. phaseoli; Xca = X. citri pv. aurantifolii; Xpm = X. phaseoli pv. manihotis. A coloured line indicates that the repeat is shared between TAL effectors from different groups, one colour being associated with a single repeat and corresponding to the colour of repeats in Additional file 16: Figure S7. (XLSX 12 kb)
Repeats conserved and shared between different TAL effectors from X. citri pv. fuscans, X. phaseoli pv. phaseoli, X. citri pv. aurantifolii and X. phaseoli pv. manihotis strains. Repeats conserved between TAL effectors from different groups are indicated by the same colour (red, purple, green, blue or yellow). Half-coloured boxes indicate that polymorphism exist among TAL effectors homologues from different strains. Please see Additional file 15: Table S9 for more detailed information. (PPTX 69 kb)
Top 10 predicted targets for the nine TAL effectors with unique RVD sequences retrieved in 17 X. citri pv. fuscans and X. phaseoli pv. phaseoli strains. Target names correspond to locus tags from the P. vulgaris genome available at https://phytozome.jgi.doe.gov/. Similar targets predicted for different TAL effectors are highlighted by colours corresponding to the predicted function of the target (see the legend below). (XLSX 17 kb)
Primers used in this study. (DOCX 19 kb)
Phylogenetic trees of N-ter- and C-ter-encoding regions of tal genes used for the Kishino-Hasegawa-Templeton test. Bootstrap values greater than 50% are shown for 1000 replicates and horizontal scale bars represent the number of nucleotide substitutions per site. a ML tree constructed on a nucleotide alignment of the N-ter-encoding region of tal genes from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis strains using a tal gene from X. translucens pv. undulosa XT4699 as outgroup. b ML tree constructed on a nucleotide alignment of the C-ter-encoding region of tal genes from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis strains using a tal gene from X. translucens pv. undulosa XT4699 as outgroup. (PPTX 56 kb)
Acknowledgements
The authors thank Alvaro L. Pérez-Quintero for helping in TAL effector target predictions, Chrystelle Brin and Armelle Darrasse for sharing preliminary data on the diversity of TAL effectors from X. citri pv. fuscans and X. phaseoli pv. phaseoli and Matthieu Barret for critical review of the manuscript. The authors also thank the French Network on Xanthomonads (FNX) (https://www.reseau-xantho.org/) and particularly Laurent D. Noël, Boris Szurek, Lionel Gagnevin and Ralf Koebnik for recurrent scientific exchanges on Xanthomonas within the framework of the ANR project CROpTAL led by Laurent D. Noël. PacBio sequencing was performed at Icahn School of Medicine at Mount Sinai (NY, USA) under the supervision of Robert Sebra and preliminary quality control of DNA was performed at the ANAN platform from the SFR 4207 QUASAV (Angers, France). We thank the CIRM-CFBP (Angers, France) for strain preservation and supply and the members of the EmerSys and INEM teams (IRHS, Angers) for their participation in pathogenicity assays.
Funding
This study was supported by the French National Research Agency (ANR-14-CE19–0002-04 CROpTAL project to NWGC. MR is funded by a PhD grant (X-HOT project) from Angers-Loire Metropole, France. The funding bodies did not participate in any of the study design, data collection and analysis, or writing the manuscript.
Availability of data and materials
The datasets (genome sequences) generated through various projects and used in this study have been deposited in GenBank under accession numbers listed in Table 1.
Abbreviations
- ANI
Average nucleotide identity
- CBB
Common bacterial blight
- CFBP
Collection Française de Bactéries associées aux Plantes
- CFU
Colony forming unit
- EBE
Effector binding element
- HGT
Horizontal gene transfer
- IS
Insertion sequence
- ML
Maximum likelihood
- NF
Non-fuscous
- PCR
Polymerase chain reaction
- RVD
Repeat variable diresidue
- SMRT
Single molecule, real-time
- TAL
Transcription activator-like
Authors’ contributions
MR, MAJ and NWGC designed the study and wrote the manuscript. MR, MB, SB and NWGC contributed to data analyses. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4087-6) contains supplementary material, which is available to authorized users.
Contributor Information
Mylène Ruh, Email: mylene.ruh@agrocampus-ouest.fr.
Martial Briand, Email: martial.briand@inra.fr.
Sophie Bonneau, Email: sophie.bonneau@inra.fr.
Marie-Agnès Jacques, Email: marie-agnes.jacques@inra.fr.
Nicolas W.G. Chen, Email: nicolas.chen@agrocampus-ouest.fr
References
- 1.Bonas U, Stall RE, Staskawicz B. Genetic and structural characterization of the avirulence gene avrBs3 from Xanthomonas campestris pv. vesicatoria. Mol Gen Genet. 1989;218:127–136. doi: 10.1007/BF00330575. [DOI] [PubMed] [Google Scholar]
- 2.de d LO, Wolf C, Dietze J, Elsaesser J, Morbitzer R, Lahaye T. Programmable DNA-binding proteins from Burkholderia provide a fresh perspective on the TALE-like repeat domain. Nucleic Acids Res. 2014;42:7436–7449. doi: 10.1093/nar/gku329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heuer H, Yin YN, Xue QY, Smalla K, Guo JH. Repeat domain diversity of avrBs3-like genes in Ralstonia solanacearum strains and association with host preferences in the field. Appl Environ Microbiol. 2007;73:4379–4384. doi: 10.1128/AEM.00367-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boch J, Bonas U. Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol. 2010;48:419–436. doi: 10.1146/annurev-phyto-080508-081936. [DOI] [PubMed] [Google Scholar]
- 5.Mak ANS, Bradley P, Bogdanove AJ, Stoddard BL. TAL effectors: Function, structure, engineering and applications. Curr Opin Struct Biol. 2013;23:93–99. doi: 10.1016/j.sbi.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science (80- ) 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 7.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science (80-. ) 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Zhang Y, Yuan P, Zhou Y, Cai C, Ren Q, et al. Complete decoding of TAL effectors for DNA recognition. Cell Res. 2014;24:628–631. doi: 10.1038/cr.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40:117–122. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grau J, Wolf A, Reschke M, Bonas U, Posch S, Boch J. Computational Predictions Provide Insights into the Biology of TAL Effector Target Sites. PLoS Comput Biol. 2013;9 [DOI] [PMC free article] [PubMed]
- 11.Pérez-Quintero AL, Rodriguez-R LM, Dereeper A, López C, Koebnik R, Szurek B, et al. An Improved Method for TAL Effectors DNA-Binding Sites Prediction Reveals Functional Convergence in TAL Repertoires of Xanthomonas oryzae Strains. PLoS One. 2013;8:e68464. doi: 10.1371/journal.pone.0068464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang B, Sugio A, White FF. Os8N3 is a host disease-susceptibility gene for bacterial blight of rice. Proc Natl Acad Sci U S A. 2006;103:10503–10508. doi: 10.1073/pnas.0604088103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kay S, Hahn S, Marois E, Hause G, Bonas U. A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science (80-. ) 2007;318:648–651. doi: 10.1126/science.1144956. [DOI] [PubMed] [Google Scholar]
- 14.Chen L-Q, Hou B-H, Lalonde S, Takanaga H, Hartung ML, Qu X-Q, et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature. 2010;468:527–532. doi: 10.1038/nature09606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Römer P, Recht S, Strauss T, Elsaesser J, Schornack S, Boch J, et al. Promoter elements of rice susceptibility genes are bound and activated by specific TAL effectors from the bacterial blight pathogen, Xanthomonas oryzae pv. oryzae. New Phytol. 2010;187:1048–1057. doi: 10.1111/j.1469-8137.2010.03217.x. [DOI] [PubMed] [Google Scholar]
- 16.Gu K, Yang B, Tian D, Wu L, Wang D, Sreekala C, et al. R gene expression induced by a type-III effector triggers disease resistance in rice. Nature. 2005;435:1122–1125. doi: 10.1038/nature03630. [DOI] [PubMed] [Google Scholar]
- 17.Römer P, Hahn S, Jordan T, Strauss T, Bonas U, Lahaye T. Plant pathogen recognition mediated by promoter activation of the pepper Bs3 resistance gene. Science (80-. ) 2007;318:645–648. doi: 10.1126/science.1144958. [DOI] [PubMed] [Google Scholar]
- 18.Boch J, Bonas U, Lahaye T. TAL effectors - pathogen strategies and plant resistance engineering. New Phytol. 2014;204:823–832. doi: 10.1111/nph.13015. [DOI] [PubMed] [Google Scholar]
- 19.Schornack S, Moscou MJ, Ward ER, Horvath DM. Engineering Plant Disease Resistance Based on TAL Effectors. Annu Rev Phytopathol. 2013;51:383–406. doi: 10.1146/annurev-phyto-082712-102255. [DOI] [PubMed] [Google Scholar]
- 20.Blanvillain-Baufumé S, Reschke M, Solé M, Auguy F, Doucoure H, Szurek B, et al. Targeted promoter editing for rice resistance to Xanthomonas oryzae pv. oryzae reveals differential activities for SWEET14-inducing TAL effectors. Plant Biotechnol J. 2017;15:306–317. doi: 10.1111/pbi.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Wei J, Lin Y, Chen H. Gene silencing using the recessive rice bacterial blight resistance gene xa13 as a new paradigm in plant breeding. Plant Cell Rep. 2012;31:851–862. doi: 10.1007/s00299-011-1206-8. [DOI] [PubMed] [Google Scholar]
- 22.Hummel AW, Wilkins KE, Wang L, Cernadas RA, Bogdanove AJ. A transcription activator-like effector from Xanthomonas oryzae pv. oryzicola elicits dose-dependent resistance in rice. Mol. Plant Pathol. 2016;18:55–56. doi: 10.1111/mpp.12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutin M, Césari S, Chalvon V, Michel C, Tran TT, Boch J, et al. Ectopic activation of the rice NLR heteropair RGA4/RGA5 confers resistance to bacterial blight and bacterial leaf streak diseases. Plant J. 2016;88:43–55. doi: 10.1111/tpj.13231. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Zhang X, Fan Y, Gao Y, Zhu Q, Zheng C, et al. XA23 Is an executor r protein and confers broad-spectrum disease resistance in rice. Mol Plant. 2015;8:290–302. doi: 10.1016/j.molp.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Yin Z, White F. TAL effectors and the executor R genes. Front Plant Sci. 2015;6:641. doi: 10.3389/fpls.2015.00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hummel AW, Doyle EL, Bogdanove AJ. Addition of transcription activator-like effector binding sites to a pathogen strain-specific rice bacterial blight resistance gene makes it effective against additional strains and against bacterial leaf streak. New Phytol. 2012;195:883–893. doi: 10.1111/j.1469-8137.2012.04216.x. [DOI] [PubMed] [Google Scholar]
- 27.Zeng X, Tian D, Gu K, Zhou Z, Yang X, Luo Y, et al. Genetic engineering of the Xa10 promoter for broad-spectrum and durable resistance to Xanthomonas oryzae pv. oryzae. Plant Biotechnol. J. 2015;13:993–1001. doi: 10.1111/pbi.12342. [DOI] [PubMed] [Google Scholar]
- 28.Gardiner DM, Upadhyaya NM, Stiller J, Ellis JG, Dodds PN, Kazan K, et al. Genomic analysis of Xanthomonas translucens pathogenic on wheat and barley reveals cross-kingdom gene transfer events and diverse protein delivery systems. PLoS One. 2014;9 [DOI] [PMC free article] [PubMed]
- 29.Wilkins KE, Booher NJ, Wang L, Bogdanove AJ. TAL effectors and activation of predicted host targets distinguish Asian from African strains of the rice pathogen Xanthomonas oryzae pv. oryzicola while strict conservation suggests universal importance of five TAL effectors. Front Plant Sci. 2015;6:1–15. doi: 10.3389/fpls.2015.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng Z, Hu Y, Xie J, Potnis N, Akhunova A, Jones J, et al. Long read and single molecule DNA sequencing simplifies genome assembly and TAL effector gene analysis of Xanthomonas translucens. BMC Genomics. 2016;17:21. doi: 10.1186/s12864-015-2348-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alavi SM, Sanjari S, Durand F, Brin C, Manceau C, Poussier S. Assessment of the genetic diversity of Xanthomonas axonopodis pv. phaseoli and Xanthomonas fuscans subsp. fuscans as a basis to identify putative pathogenicity genes and a type III secretion system of the SPI-1 family by multiple suppression subtractive. Appl. Environ. Microbiol. 2008;74:3295–3301. doi: 10.1128/AEM.02507-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.EFSA Panel on Plant Health Scientific Opinion on the pest categorisation of Xanthomonas axonopodis pv. phaseoli and Xanthomonas fuscans subsp. fuscans. EFSA J. 2014;12:3856. doi: 10.2903/j.efsa.2014.3856. [DOI] [Google Scholar]
- 33.Constantin EC, Cleenwerck I, Maes M, Baeyen S, Van Malderghem C, De Vos P, et al. Genetic characterization of strains named as Xanthomonas axonopodis pv. dieffenbachiae leads to a taxonomic revision of the X. axonopodis species complex. Plant Pathol. 2016;65:792–806. doi: 10.1111/ppa.12461. [DOI] [Google Scholar]
- 34.Vauterin L, Hoste B, Kersters K, Swings J. Reclassification of Xanthomonas. Int J Syst Bacteriol. 1995;45:472–489. doi: 10.1099/00207713-45-3-472. [DOI] [Google Scholar]
- 35.Rademaker JLW, Louws FJ, Schultz MH, Rossbach U, Vauterin L, Swings J, et al. A comprehensive species to strain taxonomic framework for Xanthomonas. Phytopathology. 2005;95:1098–1111. doi: 10.1094/PHYTO-95-1098. [DOI] [PubMed] [Google Scholar]
- 36.Aritua V, Harrison J, Sapp M, Buruchara R, Smith J, Studholme DJ. Genome sequencing reveals a new lineage associated with lablab bean and genetic exchange between Xanthomonas axonopodis pv. phaseoli and Xanthomonas fuscans subsp. fuscans. Front. Microbiol. 2015;6:1–18. doi: 10.3389/fmicb.2015.01080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darrasse A, Carrère S, Barbe V, Boureau T, Arrieta-Ortiz ML, Bonneau S, et al. Genome sequence of Xanthomonas fuscans subsp. fuscans strain 4834-R reveals that flagellar motility is not a general feature of xanthomonads. BMC Genomics. 2013;14:761. doi: 10.1186/1471-2164-14-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Indiana A, Briand M, Arlat M, Gagnevin L, Koebnik R, Noël L, et al. Draft genome sequence of the flagellated Xanthomonas fuscans subsp. fuscans strain CFBP 4884. Genome Announc. 2014;2:e00966–e00914. doi: 10.1128/genomeA.00966-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jalan N, Kumar D, Yu F, Jones JB, Graham JH, Wang N. Complete Genome Sequence of Xanthomonas citri subsp. citri Strain Aw12879, a Restricted-Host-Range Citrus Canker-Causing Bacterium. Genome Announc. 2013;1:e00235–e00213. doi: 10.1128/genomeA.00235-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolot S, Guy E, Carrere S, Barbe V, Arlat M, Noël LD. Xanthomonas campestris pv. campestris Strain Xca5. Genome Announc. 2013;1:e00032–e00012. doi: 10.1128/genomeA.00032-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cesbron S, Briand M, Essakhi S, Gironde S, Boureau T, Manceau C, et al. Comparative Genomics of Pathogenic and Nonpathogenic Strains of Xanthomonas arboricola Unveil Molecular and Evolutionary Events Linked to Pathoadaptation. Front Plant Sci. 2015;6:1126. doi: 10.3389/fpls.2015.01126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gagnevin L, Bolot S, Gordon JL, Pruvost O, Vernière C, Robène I, et al. Draft Genome Sequence of Xanthomonas axonopodis pv. allii Strain CFBP 6369. Genome Announc. 2014;2:e00727–e00714. doi: 10.1128/genomeA.00727-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quail M, Smith ME, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Lange O, Schreiber T, Schandry N, Radeck J, Braun KH, Koszinowski J, et al. Breaking the DNA-binding code of Ralstonia solanacearum TAL effectors provides new possibilities to generate plant resistance genes against bacterial wilt disease. New Phytol. 2013;199:773–786. doi: 10.1111/nph.12324. [DOI] [PubMed] [Google Scholar]
- 45.Jacques M-A, Arlat M, Boulanger A, Boureau T, Carrère S, Cesbron S, et al. Using Ecology, Physiology, and Genomics to Understand Host Specificity in Xanthomonas : French Network on Xanthomonads (FNX) Annu Rev Phytopathol. 2016;54:163–187. doi: 10.1146/annurev-phyto-080615-100147. [DOI] [PubMed] [Google Scholar]
- 46.Mhedbi-Hajri N, Hajri A, Boureau T, Darrasse A, Durand K, Brin C, et al. Evolutionary History of the Plant Pathogenic Bacterium Xanthomonas axonopodis. PLoS One. 2013;8:e58474. doi: 10.1371/journal.pone.0058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kishino H, Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol. 1989;29:170–179. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- 48.Martin DP, Williamson C, Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinforma Appl Note. 2005;21:260–26210. doi: 10.1093/bioinformatics/bth490. [DOI] [PubMed] [Google Scholar]
- 49.Ochman H, Elwyn S, Moran NA. Calibrating bacterial evolution. Proc Natl Acad Sci U S A. 1999;96:12638–12643. doi: 10.1073/pnas.96.22.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuo C-H, Ochman H. Inferring clocks when lacking rocks: the variable rates of molecular evolution in bacteria. Biol Direct. 2009;4:35. doi: 10.1186/1745-6150-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schuenzel EL, Scally M, Stouthamer R, Nunney L. A Multigene Phylogenetic Study of Clonal Diversity and Divergence in North American Strains of the Plant Pathogen Xylella fastidiosa A Multigene Phylogenetic Study of Clonal Diversity and Divergence in North American Strains of the Plant Pathogen Xylella. Appl Environ Microbiol. 2005;71:3832–3839. doi: 10.1128/AEM.71.7.3832-3839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreira LM, Almeida NF, Potnis N, Digiampietri L a, Adi SS, Bortolossi JC, et al. Novel insights into the genomic basis of citrus canker based on the genome sequences of two strains of Xanthomonas fuscans subsp. aurantifolii. BMC Genomics. 2010;11:238. doi: 10.1186/1471-2164-11-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bart R, Cohn M, Kassen A, McCallum EJ, Shybut M, Petriello A, et al. High-throughput genomic sequencing of cassava bacterial blight strains identifies conserved effectors to target for durable resistance. Proc Natl Acad Sci U S A. 2012;109:E1972–E1979. doi: 10.1073/pnas.1208003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferreira RM, de Oliveira ACP, Moreira LM, Belasque JJ, Gourbeyre E, Siguier P, et al. A TALE of Transposition: Tn3-like transposons play a major role in the spread of pathogenicity determinants of xanthomonas citri and other xanthomonads. MBio. 2015;6:1–12. doi: 10.3391/mbi.2015.6.1.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vera Cruz CM, Bai J, Ona I, Leung H, Nelson RJ, Mew TW, et al. Predicting durability of a disease resistance gene based on an assessment of the fitness loss and epidemiological consequences of avirulence gene mutation. Proc Natl Acad Sci U S A. 2000;97:13500–13505. doi: 10.1073/pnas.250271997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bogdanove AJ, Koebnik R, Lu H, Furutani A, Angiuoli SV, Patil PB, et al. Two new complete genome sequences offer insight into host and tissue specificity of plant pathogenic Xanthomonas spp. J. Bacteriol. 2011;193:5450–5464. doi: 10.1128/JB.05262-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pérez-Quintero AL, Lamy L, Gordon JL, Escalon A, Cunnac S, Szurek B, et al. QueTAL: a suite of tools to classify and compare TAL effectors functionally and phylogenetically. Front Plant Sci. 2015;6:545. doi: 10.3389/fpls.2015.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Richter A, Streubel J, Blücher C, Szurek B, Reschke M, Grau J, et al. A TAL effector repeat architecture for frameshift binding. Nat Commun. 2014;5:3447. doi: 10.1038/ncomms4447. [DOI] [PubMed] [Google Scholar]
- 59.Schmutz J, McClean PE, Mamidi S, Wu GA, Cannon SB, Grimwood J, et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet. 2014;46:707–713. doi: 10.1038/ng.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sugio A, Yang B, Zhu T, White FF. Two type III effector genes of Xanthomonas oryzae pv. oryzae control the induction of the host genes OsTFIIAgamma1 and OsTFX1 during bacterial blight of rice. Proc Natl Acad Sci U S A. 2007;104:10720–10725. doi: 10.1073/pnas.0701742104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Streubel J, Pesce C, Hutin M, Koebnik R, Boch J, Szurek B. Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae. New Phytol. 2013;200:808–819. doi: 10.1111/nph.12411. [DOI] [PubMed] [Google Scholar]
- 62.Cernadas RA, Doyle EL, Niño-Liu DO, Wilkins KE, Bancroft T, Wang L, et al. Code-Assisted Discovery of TAL Effector Targets in Bacterial Leaf Streak of Rice Reveals Contrast with Bacterial Blight and a Novel Susceptibility Gene. PLoS Pathog. 2014;10:e1003972. doi: 10.1371/journal.ppat.1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu Y, Zhang J, Jia H, Sosso D, Li T, Frommer WB, et al. Lateral organ boundaries 1 is a disease susceptibility gene for citrus bacterial canker disease. Proc Natl Acad Sci U S A. 2014;111:E521–E529. doi: 10.1073/pnas.1313271111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cohn M, Bart RS, Shybut M, Dahlbeck D, Gomez M, Morbitzer R, et al. Xanthomonas axonopodis virulence is promoted by a transcription activator-like effector-mediated induction of a SWEET sugar transporter in cassava. Mol Plant-Microbe Interact. 2014;27:1186–1198. doi: 10.1094/MPMI-06-14-0161-R. [DOI] [PubMed] [Google Scholar]
- 65.Zhou J, Peng Z, Long J, Sosso D, Liu B, Eom JS, et al. Gene targeting by the TAL effector PthXo2 reveals cryptic resistance gene for bacterial blight of rice. Plant J. 2015;82:632–643. doi: 10.1111/tpj.12838. [DOI] [PubMed] [Google Scholar]
- 66.Schwartz AR, Morbitzer R, Lahaye T, Staskawicz BJ. TALE-induced bHLH transcription factors that activate a pectate lyase contribute to water soaking in bacterial spot of tomato. Proc Natl Acad Sci U S A. 2017;114:E897–E903. doi: 10.1073/pnas.1620407114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cox KL, Meng F, Wilkins KE, Li F, Wang P, Booher NJ, et al. TAL effector driven induction of a SWEET gene confers susceptibility to bacterial blight of cotton. Nat Commun. 2017;8:1–14. doi: 10.1038/s41467-016-0009-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huguet-Tapia JC, Peng Z, Yang B, Yin Z, Liu S, White FF. Complete Genome Sequence of the African Strain AXO1947 of Xanthomonas oryzae pv. oryzae. Genome Announc. 2016;4:10–11. doi: 10.1128/genomeA.01730-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Booher NJ, Carpenter SCD, Sebra RP, Wang L, Salzberg SL, Leach JE, et al. Single molecule real-time sequencing of Xanthomonas oryzae genomes reveals a dynamic structure and complex TAL (transcription activator-like) effector gene relationships. Microb Genomics. 2015;1:1–22. doi: 10.1099/mgen.0.000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wiedenbeck J, Cohan FM. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol Rev. 2011;35:957–976. doi: 10.1111/j.1574-6976.2011.00292.x. [DOI] [PubMed] [Google Scholar]
- 71.Darmon E, Leach DRF. Bacterial genome instability. Microbiol Mol Biol Rev. 2014;78:1–39. doi: 10.1128/MMBR.00035-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.El Yacoubi B, Brunings AM, Yuan Q, Shankar S, Gabriel DW. In planta horizontal transfer of a major pathogenicity effector gene. Appl Environ Microbiol. 2007;73:1612–1621. doi: 10.1128/AEM.00261-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sabet KA. Studies in the bacterial diseases of Sudan crops: III. On th occurrence, host range and taxonomiy of the bacteria causing leaf blight diseases of certain leguminous plants. Ann Appl Biol. 1959;47:318–331. doi: 10.1111/j.1744-7348.1959.tb02547.x. [DOI] [Google Scholar]
- 74.Hajri A, Brin C, Hunault G, Lardeux F, Lemaire C, Manceau C, et al. A «repertoire for repertoire» hypothesis: Repertoires of type three effectors are candidate determinants of host specificity in Xanthomonas. PLoS One. 2009;4:e6632. doi: 10.1371/journal.pone.0006632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le Roy J, Huss B, Creach A, Hawkins S, Neutelings G. Glycosylation Is a Major Regulator of Phenylpropanoid Availability and Biological Activity in Plants. Front Plant Sci. 2016;7:735. doi: 10.3389/fpls.2016.00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Langlois-Meurinne M, Gachon CMM, Saindrenan P. Pathogen-responsive expression of glycosyltransferase genes UGT73B3 and UGT73B5 is necessary for resistance to Pseudomonas syringae pv tomato in Arabidopsis. Plant Physiol. 2005;139:1890–1901. doi: 10.1104/pp.105.067223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simon C, Langlois-Meurinne M, Didierlaurent L, Chaouch S, Bellvert F, Massoud K, et al. The secondary metabolism glycosyltransferases UGT73B3 and UGT73B5 are components of redox status in resistance of Arabidopsis to Pseudomonas syringae pv. tomato. Plant Cell Environ. 2014;37:1114–1129. doi: 10.1111/pce.12221. [DOI] [PubMed] [Google Scholar]
- 78.Darrasse A, Darsonval A, Boureau T, Brisset MN, Durand K, Jacques MA. Transmission of plant-pathogenic bacteria by nonhost seeds without induction of an associated defense reaction at emergence. Appl Environ Microbiol. 2010;76:6787–6796. doi: 10.1128/AEM.01098-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Akhavan A, Bahar M, Askarian H, Lak MR, Nazemi A, Zamani Z. Bean common bacterial blight: pathogen epiphytic life and effect of irrigation practices. Spring. 2013;2:41. doi: 10.1186/2193-1801-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee BM, Park YJ, Park DS, Kang HW, Kim JG, Song ES, et al. The genome sequence of Xanthomonas oryzae pathovar oryzae KACC10331, the bacterial blight pathogen of rice. Nucleic Acids Res. 2005;33:577–586. doi: 10.1093/nar/gki206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ochiai H, Inoue Y, Takeya M, Sasaki A, Kaku H. Genome sequence of Xanthomonas oryzae pv. oryzae suggests contribution of large numbers of effector genes and insertion sequences to its race diversity. Japan Agric Res Q. 2005;39:275–287. doi: 10.6090/jarq.39.275. [DOI] [Google Scholar]
- 82.Salzberg SL, Sommer DD, Schatz MC, Phillippy AM, Rabinowicz PD, Tsuge S, et al. Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. oryzae PXO99A. BMC Genomics. 2008;9:204. doi: 10.1186/1471-2164-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Antony G, Zhou J, Huang S, Li T, Liu B, White F, et al. Rice xa13 recessive resistance to bacterial blight is defeated by induction of the disease susceptibility gene Os-11N3. Plant Cell. 2010;22:3864–3876. doi: 10.1105/tpc.110.078964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu Y, Streubel J, Balzergue S, Champion A, Boch J, Koebnik R, et al. Colonization of rice leaf blades by an african strain of Xanthomonas oryzae pv. oryzae depends on a new TAL effector that induces the rice nodulin-3 Os11N3 gene. Mol Plant-Microbe Interact. 2011;24:1102–1113. doi: 10.1094/MPMI-11-10-0254. [DOI] [PubMed] [Google Scholar]
- 85.Barak JD, Vancheva T, Lefeuvre P, Jones JB, Timilsina S, Minsavage G V, et al. Whole-Genome Sequences of Xanthomonas euvesicatoria Strains Clarify Taxonomy and Reveal a Stepwise Erosion of Type 3 Effectors. Front Plant Sci. 2016;7:1805. [DOI] [PMC free article] [PubMed]
- 86.da Silva ACR, Ferro JA, Reinach FC, Farah CS, Furlan LR, Quaggio RB, et al. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature. 2002;417:459–463. doi: 10.1038/417459a. [DOI] [PubMed] [Google Scholar]
- 87.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Single Polymerase Molecules. Science (80-. ) 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 88.Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 89.Sallet E, Gouzy J, Schiex T. EuGene-PP: A next-generation automated annotation pipeline for prokaryotic genomes. Bioinformatics. 2014;30:2659–2661. doi: 10.1093/bioinformatics/btu366. [DOI] [PubMed] [Google Scholar]
- 90.Qi J, Luo H, Hao B. CVTree: A phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res. 2004;32:45–47. doi: 10.1093/nar/gkh362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wernersson R, Pedersen AG. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003;31:3537–3539. doi: 10.1093/nar/gkg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 94.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:msw054. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Felsenstein J. PHYLIP - Phylogeny inference package - v3.2. Cladistics. 1989. pp. 164–166. [Google Scholar]
- 96.Martin D, Rybicki E. RDP: detection of recombination amongst aligned sequences. Bioinformatics. 2000;16:562–563. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- 97.Padidam M, Sawyer S, Fauquet CM. Possible emergence of new geminiviruses by frequent recombination. Virology. 1999;265:218–225. doi: 10.1006/viro.1999.0056. [DOI] [PubMed] [Google Scholar]
- 98.Martin DP, Posada D, Crandall K a, Williamson C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retrovir. 2005;21:98–102. doi: 10.1089/aid.2005.21.98. [DOI] [PubMed] [Google Scholar]
- 99.Maynard-Smith J. Analyzing the mosaic structure of genes. J Mol Evol. 1992;34:126–129. doi: 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- 100.Posada D, Crandall KA. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc Natl Acad Sci. 2001;98:13757–13762. doi: 10.1073/pnas.241370698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gibbs MJ, Armstrong JS, Gibbs a J. Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics. 2000;16:573–582. doi: 10.1093/bioinformatics/16.7.573. [DOI] [PubMed] [Google Scholar]
- 102.Boni MF, Posada D, Feldman MW. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics. 2007;176:1035–1047. doi: 10.1534/genetics.106.068874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 105.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Correction of sequences from tal pseudogenes containing indels in the SMRT sequence. (XLSX 10 kb)
Amino acid sequences of TAL effector from the 17 strains responsible for CBB obtained in this study. (DOCX 19 kb)
Gel photograph showing the sizes obtained for tal23A_CFBP4885 (Xfutal1) and tal18H_CFBP4885 (Xfutal2) after PCR amplification. Amplification was performed using XapF2 and XapR primers on plasmidic DNA from the CFBP4885 strain (Additional file 18: Table S11). Expected PCR product sizes for tal23A_CFBP4885 and tal18H_CFBP4885 were 4604 bp and 3074 bp for Sanger sequencing of the entire genome, respectively, and 3686 bp and 3176 bp for SMRT sequencing of the entire genome, respectively. (DOCX 2788 kb)
RVD frequencies in TAL effectors from the 17 X. citri pv. fuscans and X. phaseoli pv. phaseoli strains used in this study. (DOCX 20 kb)
References for previously published tal genes and whole genome sequences from Xanthomonas spp. strains used in this study. (XLSX 19 kb)
Average nucleotide identity and percentage of alignment between whole genome sequences from 17 X. citri pv. fuscans and X. phaseoli pv. phaseoli strains. BLAST calculation of average nucleotide identity (ANIb) and alignment percentages were estimated using Jspecies (Richter and Rossello-Mora, 2009). (XLSX 15 kb)
Phylogenetic trees of N-ter and C-ter-encoding regions of tal genes with sequences published by Aritua et al. (2015). Bootstrap values greater than 50% (100 replicates) are shown and horizontal scale bars represent the number of nucleotide substitutions per site. The tal genes from the X. citri pv. fuscans genetic lineages fuscans, NF2 and NF3 are indicated in red, X. phaseoli pv. phaseoli NF1 lineage in blue, X. citri pv. aurantifolii in pink and X. phaseoli pv. manihotis in purple. The sequences published by Aritua et al. (2015) are in bold. Both ML trees were constructed using tal genes from X. translucens pv. undulosa XT4699 as outgroups. a ML tree constructed on a nucleotide alignment of the N-ter-encoding region of tal genes. b ML tree constructed on a nucleotide alignment of the C-ter-encoding region of tal genes. (PPTX 137 kb)
Circular representation of plasmids A and C. Genomic sequences were compared and converted in a graphical map using CGView (Grant et al., 2012) with strain CFBP4885 as reference. Colours differ according to identity percentage (see legend). Strains order, ANIb and alignment percentages are indicated in the center of each graphical map. Localisation of tal genes are highlighted by green zones. Absence of tal genes in plasmid A from strains CFBP6164 and CFBP6546R is indicated by a black cross. (PDF 444 kb)
Average nucleotide identity and percentage of alignment between sequences of plasmid A from 17 X. citri pv. fuscans and X. phaseoli pv. phaseoli strains. BLAST calculation of average nucleotide identity (ANIb) and alignment percentages were estimated using Jspecies (Richter and Rossello-Mora, 2009). (XLSX 14 kb)
Circular representation of the regions shared by phylogenetically distant lineages in plasmids A and C. Genomic sequences were compared and converted in a graphical map using CGView (Grant et al., 2012). Colours differ according to identity percentage (see legend). Regions shared by both strains are surrounded by a black line. Numbers represent genes conserved in both strains (see Additional files 11 and 13 for more details). a Plasmids A from strains CFBP6988R (outer circle = reference) and CFBP6546R (inner circle). b Plasmids C from strains CFBP6166 (outer circle = reference) and CFBP6982 (inner circle). (PDF 168 kb)
Genes localized in Plasmid A shared by strains CFBP6988R and CFBP6546R. Please refer to Additional file 10a for graphic representation of the data. (XLSX 15 kb)
Average nucleotide identity and percentage of alignment between sequences of plasmid C from 8 X. citri pv. fuscans fuscans and X. phaseoli pv. phaseoli NF1 strains. BLAST calculation of average nucleotide identity (ANIb) and alignment percentages were estimated using Jspecies (Richter and Rossello-Mora, 2009). (XLSX 11 kb)
Genes localized in Plasmid C shared by strains CFBP6982 and CFBP6166. Please refer to Additional file 10b for graphic representation of the data. (XLSX 15 kb)
Duplications and movements of tal genes associated with IS3 and Tn3-like transposons. Arrows represent tal genes (yellow), IS3 sequences (orange) or Tn3-like transposons (dark blue). Arrowheads correspond to truncated IS3 (orange) or Tn3-like transposon (dark blue). Striped areas correspond to conserved regions. Dashed lines represent the borders of duplication events between different regions of a single molecule. a Movement of tal23A homologues from the plasmid A to the chromosome associated with IS3 sequences. b Duplication of tal20F and tal18G associated with Tn3-like transposons. c. Duplication of tal18H_CFBP6164 associated with IS3 sequences in strain CFBP6164. (PPTX 74 kb)
Repeats found within all TAL effectors from X. citri pv. fuscans, X. phaseoli pv. phaseoli, X. citri pv. aurantifolii and X. phaseoli pv. manihotis strains. Sequences were compared after removal of RVD. Xcf = X. citri pv. fuscans; Xpp = X. phaseoli pv. phaseoli; Xca = X. citri pv. aurantifolii; Xpm = X. phaseoli pv. manihotis. A coloured line indicates that the repeat is shared between TAL effectors from different groups, one colour being associated with a single repeat and corresponding to the colour of repeats in Additional file 16: Figure S7. (XLSX 12 kb)
Repeats conserved and shared between different TAL effectors from X. citri pv. fuscans, X. phaseoli pv. phaseoli, X. citri pv. aurantifolii and X. phaseoli pv. manihotis strains. Repeats conserved between TAL effectors from different groups are indicated by the same colour (red, purple, green, blue or yellow). Half-coloured boxes indicate that polymorphism exist among TAL effectors homologues from different strains. Please see Additional file 15: Table S9 for more detailed information. (PPTX 69 kb)
Top 10 predicted targets for the nine TAL effectors with unique RVD sequences retrieved in 17 X. citri pv. fuscans and X. phaseoli pv. phaseoli strains. Target names correspond to locus tags from the P. vulgaris genome available at https://phytozome.jgi.doe.gov/. Similar targets predicted for different TAL effectors are highlighted by colours corresponding to the predicted function of the target (see the legend below). (XLSX 17 kb)
Primers used in this study. (DOCX 19 kb)
Phylogenetic trees of N-ter- and C-ter-encoding regions of tal genes used for the Kishino-Hasegawa-Templeton test. Bootstrap values greater than 50% are shown for 1000 replicates and horizontal scale bars represent the number of nucleotide substitutions per site. a ML tree constructed on a nucleotide alignment of the N-ter-encoding region of tal genes from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis strains using a tal gene from X. translucens pv. undulosa XT4699 as outgroup. b ML tree constructed on a nucleotide alignment of the C-ter-encoding region of tal genes from X. citri pv. fuscans, X. citri pv. aurantifolii, X. phaseoli pv. phaseoli and X. phaseoli pv. manihotis strains using a tal gene from X. translucens pv. undulosa XT4699 as outgroup. (PPTX 56 kb)
Data Availability Statement
The datasets (genome sequences) generated through various projects and used in this study have been deposited in GenBank under accession numbers listed in Table 1.