

Abstract
Background
Tau PET ligand, [11C]PBB3, binds to a wide range of tau pathology; however, binding property of PBB3 to non-tau inclusions remains unknown. To clarify whether [11C]PBB3 binds to α-synuclein pathology, reactivity of PBB3 was assessed by in vitro fluorescence and autoradiographic labeling of brain sections from α-synucleinopathies patients.
Method
Of 10 pure Lewy body disease and 120 multiple system atrophy (MSA) cases in the Mayo Clinic brain bank, we selected 3 Lewy body disease and 4 MSA cases with a range of α-synuclein severity, based on the quantitative analysis of α-synuclein burden. PBB3 fluorescence labeling, double or single immunostaining for α-synuclein and phospho-tau, Prussian blue staining, and in vitro autoradiography with [11C]PBB3 were performed for these selected samples.
Results
PBB3 fluorescence labeled various α-synuclein lesions including Lewy bodies, Lewy neurites, spheroids, glial cytoplasmic inclusions and neuronal cytoplasmic inclusions. Meanwhile, autoradiographic labeling with [11C]PBB3 at 10 nM demonstrated no significant binding in Lewy body disease cases. In contrast, significant autoradiographic binding of [11C]PBB3 to the striatopallidal fibers was found in two MSA cases, which had high density of glial cytoplasmic inclusions without tau or iron deposits in this region.
Conclusions
Given that the maximum concentration of [11C]PBB3 in human PET scans is approximately 10 nM, the present data imply that α-synuclein pathology in Lewy body disease is undetectable by [11C]PBB3-PET, while those in a subset of MSA cases with high density of glial cytoplasmic inclusions could be captured by this radioligand.
Kew words: Lewy body disease, multiple system atrophy, α-synucleinopathy, PBB3, autoradiography
Introduction
Due to the lack of specific biomarkers, it is challenging to make a correct antemortem diagnosis of neurodegenerative disease.1–6 Since diagnoses of these diseases are confirmed at autopsy, the discrepancies between clinical diagnosis and pathologic diagnosis are inevitable. Inaccurate antemortem diagnosis of neurodegenerative diseases affects the quality of clinical studies for exploring potential biomarkers and assessing the therapeutic effect of promising drugs.7 To improve the antemortem diagnostic accuracy of neurodegenerative diseases such as tauopathies and α-synucleinopathies, it is useful to establish assay systems to visualize pathognomonic pathology for these diseases.
Alzheimer disease (AD), the most common neurodegenerative disease of dementia, has two proteins as pathologic hallmarks: amyloid and tau.8, 9 Amyloid positron emission tomography (PET) imaging using [11C]Pittsburgh Compound B (PiB) has been sucsessfully used to detect cerebral amyloid deposition in individuals with AD.10, 11 Recently, PET imaging of tau lesions has become available using several tau PET ligands, such as [11C]PBB3, [18F]AV-1451, and [18F]THK arylquinoline series.12–14 These tau ligands bind to β-pleated sheet secondary structures in tau filaments and detect tau pathology in AD as well as non-AD tauopathies.15–18 One such tau PET ligand, PBB3, has shown high-affinity binding to a wide range of tau pathologies: not only neurofibrillary tangles (NFTs) in AD, but also tufted astrocytes in progressive supranuclear palsy (PSP), astrocytic plaques in corticobasal degeneration (CBD), and coiled bodies and threads in both diseases.12 A recent study on the head-to-head comparison of [11C]PBB3 and [18F]AV-1451 has shown that PBB3 bound avidly to neuronal and glial tau lesions in PSP and CBD, whereas AV-1451 showed only vague binding.19 This indicates the high sensitivity of [11C]PBB3 for tau pathology; however, there is limited knowledge regarding the selectivity of [11C]PBB3 for other amyloidogenic proteins such as α-synuclein. Thioflavin S, a fluorescence compound binding to β-pleated sheet-rich structures, is widely used to stain amyloid and tau aggregates; 9, 20, 21 however, it also labels α-synuclein pathology exemplified by Lewy bodies and glial cytoplasmic inclusions (GCIs).22 A recent study on [18F]AV-1451-PET scan has revealed significant retention in the posterior putamen in patients with MSA.23 These findings pose the possibility that PBB3 also binds to α-synuclein pathology. Given that α-synucleinopathy is often misdiagnosed as tauopathy,5, 6 it is important to clarify whether [11C]PBB3 binds to α-synuclein pathology to avoid misinterpretation and incorrect diagnosis. To address this issue, the reactivity of PBB3 with α-synuclein inclusions was assessed by in vitro fluorescence and autoradiographic labeling of brain sections from patients with Lewy body disease (LBD) and multiple system atrophy (MSA).
Subjects and Methods
Case selection
All brain autopsies were performed after consent of the legal next-of-kin or an individual with power-of-attorney. Studies of autopsy samples were considered exempt from human subject research by Mayo Clinic Institutional Review Board. The research using autopsied human samples was approved by the Ethics Committee of National Institute of Radiological Sciences, Japan. To confirm binding specificity of PBB3 to tau, we included a neuropathologically normal individual (n = 1), AD (n = 1),8, 9 Pick’s disease (n = 1),24 and PSP (n = 1).25 To clarify whether PBB3 binds to α-synuclein pathology, we examined α-synucleinopathies that included LBD with no NFTs or amyloid β deposits (so-called pure form LBD, n = 3)26, 27 and MSA (n = 5).28 This selection of α-synucleinopathies was based on the quantitative analysis of α-synuclein burden (see below) in 10 LBD and 120 MSA cases. LBD cases were chosen to include those with the least burden (LBD-1), median burden (LBD-2), or greatest burden (LBD-3) of α-synuclein. The MSA cases were chosen to include the least burden (MSA-1), burden at the 25th percentile (MSA-2), burden at the 75th percentile (MSA-3), and greatest burden (MSA-4) of α-synuclein (see below) (Figure 1). To assess α-synuclein pathology, including neuronal cytoplasmic inclusions (NCIs), we also included an MSA case with abundant NCIs in the hippocampus (MSA-5). Demographic and pathologic characteristics of 12 cases are summarized in Table 1.
Figure 1.
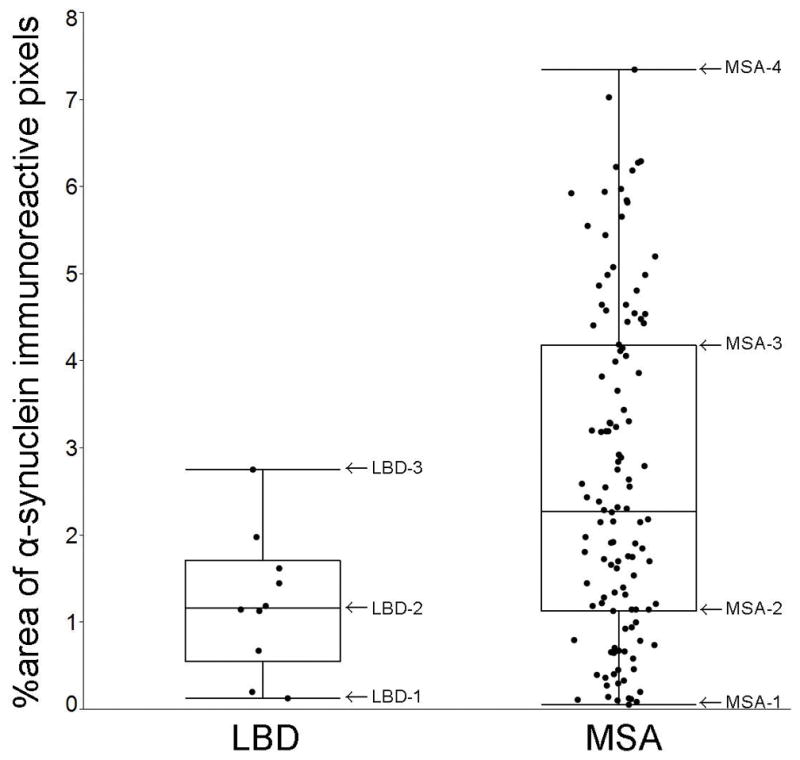
Cases with LBD and MSA are selected based on the quantitative analysis of α-synuclein burden. The graph shows %area of NACP-immunoreactive pixels to the total area of the annotated region (the amygdala in LBD and the striatopallidal fibers in MSA). Minimum, 25th percentile, Median, 75th percentile, and maximum values are 0.1% (LBD-1), 0.8%, 1.2% (LBD-2), 1.6%, and 2.7% (LBD-3) in LBD cases (n = 10) and 0.1% (MSA-1), 1.2% (MSA-2), 2.5%, 4.4% (MSA-3), and 7.3% (MSA-4) in MSA cases (n = 120).
Table 1.
Demographic and pathologic features
| Case | Pathologic diagnosis | Clinical diagnosis | Sections (FL) | Sections (ARG) | Age | Sex | Braak | Thal |
|---|---|---|---|---|---|---|---|---|
| LBD-1 | LBD | Healthy | ABG, HP, MB | ABG | 68 | F | 0 | 0 |
| LBD-2 | LBD | PDD | ABG, HP, MB | ABG | 68 | F | 0 | 0 |
| LBD-3 | LBD | PD or MSA | ABG, HP, MB | ABG | 69 | F | 0 | 0 |
| MSA-1 | MSA | Meniere disease | ABG | ABG | 82 | M | II | 1 |
| MSA-2 | MSA | MSA-P | ABG | ABG | 75 | M | II | 1 |
| MSA-3 | MSA | MSA-P | ABG | ABG | 56 | F | I | 0 |
| MSA-4 | MSA | MSA-P | ABG | ABG | 59 | M | I | 1 |
| MSA-5 | MSA | PSP or MSA-P | HP | HP | 65 | M | II | 0 |
| AD-1 | AD | AD or DLB | HP | - | 87 | F | V | 4 |
| PiD-1 | PiD | CBD | HP | - | 60 | F | 0 | 2 |
| PSP-1 | PSP | CBD | MC | - | 71 | M | III | 0 |
| HC-1 | Normal | Healthy | HP | - | 65 | F | 0 | 0 |
Abbreviations: ABG, amygdala and basal ganglia; AD, Alzheimer disease; ARG, autoradiography; CBD, corticobasal degeneration; Braak, Braak neurofibrillary tangle stage; DLB, dementia with Lewy bodies; FL, fluorescence labeling; HC, healthy control; HP, hippocampus; MB, midbrain; MC, motor cortex; LBD, Lewy body disease; MD, midbrain; MSA, multiple system atrophy; MSA-P, MSA with predominant parkinsonism; PD, Parkinson disease; PDD, Parkinson disease with dementia; PiD, Pick’s disease; PSP, progressive supranuclear palsy; Thal, Thal amyloid phase.
Neuropathologic assessment
Formalin-fixed brains underwent systematic and standardized sampling with neuropathologic evaluation by a single, experienced neuropathologist (D.W.D). Braak NFT stage and Thal amyloid phase were assigned by microscopic examinations of sections stained with thioflavin S according to published criteria.29, 30 Immunohistochemistry for α-synuclein (NACP, Mayo Clinic antibody, Jacksonville, FL)31 and phospho-tau (CP13, 1:1000; a gift from Dr. Peter Davies, Feinstein Institute for Medical Research) was conducted to establish neuropathological diagnosis as described previously.32, 33 Prussian blue stain was used to evaluate iron deposits.34 Deparaffinized slides were incubated with Prussian blue staining solution (equal volumes of 20% hydrochloric acid and 10% potassium ferrocyanide aqueous solution) for 20 minutes at room temperature. Slides were counterstained with Nuclear Fast Red Kernechtrot 0.1% (Poly Scientific R&D Corp, Bay Shore, NY) and coverslipped.
To assess α-synuclein burden quantitatively, sections of the amygdala and basal ganglia from LBD and MSA cases were scanned on the ScanScopeXT (Aperio Technologies, Vista, CA) The amygdala in LBD cases and striatopallidal fibers in MSA cases were annotated using ImageScope-11.2 (Aperio Technologies) because these regions had the greatest density of Lewy bodies in LBD 35, 36 and GCIs in MSA31. Annotated regions were analyzed in Spectrum-11.2 using a custom-designed color deconvolution algorithm to detect only NACP-positive pathology. A burden of α-synuclein pathology was expressed as a percent ratio of the area of immunoreactive pixels to the total area of the annotated region (Supplementary Figure 1).
PBB3 fluorescence labeling and immunofluorescence doublestaining
Following deparaffinization, antigen retrieval was performed by steaming slides in distilled water for 30 minutes. Then, slides were incubated in 32.3 μM of PBB3 with 50% ethanol for 30 minutes at room temperature. Subsequently, the slides were immersed in 50% ethanol for several dips for decolorization, and in deionized water for 5 minutes. The slides were coverslipped with Vectashield mounting media (Vector Laboratories, Burlingame, CA). PBB3 fluorescence images were captured by BX50 fluorescent microscope (Olympus Co. Ltd., Tokyo, Japan) using U-MWBV2 blue-violet excitation fluorescence filter (Olympus Co. Ltd., excitation wavelength: 400–440 nm, emission wavelength: 475 nm). After capturing images, coverglass was removed. Sections were pretreated with 95% formic acid for 30 minutes, steamed in distilled water for 30 minutes, blocked with Protein Block plus Serum Free (Dako) for 1 hour, and then, incubated with anti-NACP (1:2000) and anti-CP13 (1:500) primary antibodies diluted in with Antibody Diluent with Background-Reducing Components (Dako) overnight at 4°C. Sections were washed three times with 1xPBS (Sigma-Aldrich) at room temperature, and then incubated with secondary antibodies Alexa Fluor 488 and Alexa Fluor 568 (1:500, Thermo Fisher Scientific Inc) and diluted with Antibody Diluent for 1.5 hours at room temperature in a dark chamber. Sections were washed three times with 1xPBS and mounted with Vectashield mounting media. Representative images were taken with the BX50 fluorescent microscope.
Thioflavin S fluorescence and immunofluorescence doublestaining
Following deparaffinization, slides were incubated in thioflavin S solution for 7 minutes at room temperature. Subsequently, the slides were immersed in 70% ethanol for several dips for decolorization, and in deionized water for 1 minute. The slides were coverslipped with Vectashield mounting media (Vector Laboratories). Subsequent procedures were the same as PBB3 fluorescence labeling and immunofluorescence doublestaining.
Radiosynthesis
[11C]PBB3 was radiosynthesized using its desmethyl precursor as described previously in detail.12, 37 Specific activity of [11C]PBB3 at the end of synthesis was 133.0 GBq/μmol, and [11C]PBB3 maintained its radioactive purity exceeding 90% for over 3 hours after formulation.
In vitro autoradiography
In vitro autoradiography was performed using 6-μm-thick deparaffinized sections derived from LBD and MSA brains. For labeling with [11C]PBB3, sections were pre-incubated in 50 mM Tris-HCl buffer, pH 7.4, containing 20% ethanol at room temperature for 30 minutes, and incubated in 50 mM Tris-HCl buffer, pH 7.4, containing 20% ethanol and a radioligand (10 nM) at room temperature for 60 minutes. The samples were then rinsed with ice-cold Tris-HCl buffer containing 20% ethanol twice for 2 minutes and dipped into ice-cold water for 10 seconds. The sections labeled with [11C]PBB3 were subsequently dried by treating with warm air, and exposed to an imaging plate (BAS-MS2025; Fuji Film). The imaging plate was scanned with BAS-5000 system (Fuji Film) to acquire autoradiograms. Excess concentration (100 μM) of PBB5 was added to the reaction to determine nonspecific radiotracer binding. All autoradiography procedures with [11C]PBB3 were performed without exposure to fluorescence lights to circumvent photoisomerization of this compound.37
Results
Fluorescence labeling with PBB3
To confirm binding of PBB3 to tau aggregates, we examined fluorescence labeling and immunofluorescence doublestaining with anti-NACP and anti-CP13 antibodies in tauopathy cases. As expected, PBB3 labeled a wide range of tau pathology, including NFTs and threads in AD, Pick bodies in Pick’s disease, and coiled bodies and tufted astrocytes in PSP, which were positive for CP13 and negative for NACP (Figure 2A–C). Next, we applied the same analysis to α-synucleinopathies. In LBD, PBB3 labeled brainstem type Lewy bodies in the substantia nigra (Figure 2D), basal nucleus of Meynert, and hypothalamus; cortical type Lewy bodies in the CA4 sector and entorhinal cortex; abundant dystrophic Lewy neurites in the CA2/CA3 sectors (Figure 2E); and spheroids in the substantia nigra and basal nucleus of Meynert. These PBB3-positive lesions were also positive for NACP and negative for CP13 (Figure 2D–E). In MSA cases, PBB3 labeled abundant GCIs in the putamen, especially in striatopallidal fibers, globus pallidus (Figure 2F), hypothalamus, amygdala, and internal capsule of all MSA cases. Many NCIs in the pyramidal layer of CA1/subiculum (Figure 2G) and dentate gyrus of MSA-5 were labeled by PBB3. Immunofluorescence doublestaining confirmed that these GCIs and NCIs were positive for NACP and negative for CP13 (Figure 2F–G). In a neurologically normal control subject with no NFTs, amyloid β, or α-synuclein pathology, PBB3 did not label any significant pathology, although lipofuscin autofluorescence was present (Supplementary Figure 2). To further support PBB3 binding to both tau and α-synuclein pathologies with fibrillar β-pleated sheet structures, thioflavin S staining was performed. As with PBB3, thioflavin S labeled tau pathology (NFTs, threads, Pick bodies, coiled bodies, and tufted astrocytes) as well as α-synuclein pathology (Lewy bodies, Lewy neurites, GCIs, and NCIs (Supplementary Figure 3)). It is noteworthy that binding of both PBB3 (Supplementary Figure 2) and thioflavin S to Lewy-related pathology, GCIs and NCIs, disappeared after formic acid treatment, which disrupts β-pleated sheet structure.21, 38
Figure 2.

PBB3 fluorescence labeling and immunofluorescence doublestaining for α-synuclein (NACP, 1:2000) and phospho-tau (CP13, 1:500). PBB3 labels tau pathology; neurofibrillary tangles in AD (A), Pick body in Pick’s disease (B), and coiled body in PSP (C), which are positive for CP13 but negative for NACP. PBB3 also labels α-synuclein pathology; brainstem type Lewy body (D) and Lewy neurites (E) in LBD, and glial cytoplasmic inclusion (F) and neuronal cytoplasmic inclusions (G) in MSA, which are positive for NACP but negative for CP13. Bar = 20 μm. All images are the same magnification.
Autoradiographic labeling with [11C]PBB3
To assess binding of [11C]PBB3 to α-synuclein pathology at a low concentration (10 nM), we performed autoradiography using sections of the amygdala and basal ganglia from LBD and MSA cases. Total binding of [11C]PBB3 was almost completely abolished after adding excessive nonradioactive PBB5, indicating the specificity of the signal. The three LBD cases showed no [11C]PBB3 binding in the amygdala, basal forebrain, or basal ganglia, areas with significant α-synuclein immunoreactivity on the adjacent sections (Figure 3A). In contrast, MSA cases showed specific binding of [11C]PBB3 according to the burden of α-synuclein pathology. Two cases with mild pathology (MSA-1 and -2) had no binding of [11C]PBB3 to the putamen, striatopallidal fibers, or globus pallidus (Figure 3B). MSA-3, which had severe α-synuclein pathology, showed weak [11C]PBB3 radiosignals in striatopallidal fibers that had many GCIs (Figure 3B). MSA-4, the case with the greatest burden of α-synuclein, showed strong radioligand binding to the putamen, especially to the striatopallidal fibers that corresponded to an area with extremely high density of GCIs (Figure 1). Immunohistochemistry for phospho-tau confirmed the absence of tau pathology in this region. Taken together, results of autoradiography in MSA most likely represent the binding of [11C]PBB3 to GCIs, not tau pathology. In all images, minor binding was seen in the anterior commissure and optic tract. This signal was considered to be due to nonspecific binding to unknown molecular structures as reported in amyloid β-targeting tracers.39, 40 To clarify whether [11C]PBB3 also binds to NCIs, we examined autoradiography using a section of the hippocampus of MSA-5; however, it showed no [11C]PBB3 binding in this region (data not shown). Although MSA-5 had abundant NCIs in the hippocampus, the density of NCIs was much lower than that of GCIs: <0.1% in the CA1 sector (mainly NCIs) and 2.1% in the hippocampal alveus (mainly GCIs).
Figure 3.

Autoradiographic labeling of sections including the amygdala and basal ganglia derive from patients with LBD (A), and MSA (B) with 10 nM of [11C]PBB3 in the absence (top rows) and presence (bottom rows) of 100 μM of nonradioactive PBB5. Small boxes in top rows indicate the area of inset images, which show immunohistochemistry for NACP and CP13 (A–B, inset). Large boxes in top rows indicate the area of higher magnification images of autoradiography in selected cases (B, inset). A: Three pure form LBD cases with different α-synuclein burden show no [11C]PBB3 binding in the amygdala. B: No specific binding of [11C]PBB3 are detected in two MSA cases (MSA-1 and MSA-2), but weak binding of [11C]PBB3 to the striatopallidal fibers is seen in MSA-3 (top left inset) and strong binding to this region is observed in MSA-4 (top left inset). These bindings correspond to severe α-synuclein pathology (top right, inset). Immunohistochemistry for CP13 confirms the absent of tau deposits in this region (bottom right inset). Less-intense signal is seen in the anterior commissure (ac) and optic tract (opt) due to nonspecific binding in all cases (A–B).
Assessment of iron deposits
The result of the recent study on [18F]AV-1451-PET scan in MSA raises the possibility that the binding of [18F]AV-1451 to the posterior putamen in MSA patients is due to its off-target binding to iron deposits.23 Therefore, to clarify whether autoradiographic binding of [11C]PBB3 to the striatopallidal fibers was due to the presence of iron deposits, we performed Prussian blue staining in 3 LBD and 4 MSA cases. Variable degree of iron deposits was detected throughout the sections, especially in the globus pallidus and putamen in all cases (dense staining was marked with arrows in Supplementary Figure 4). Although LBD-1 and LBD-3 showed abundant iron deposits within the striatopallidal fibers (Supplemental Figure 4, LBD-1 and LBD-3), no autoradiographic binding of [11C]PBB3 was observed. Similarly, MSA-1, which had no binding of [11C]PBB3 to the striatopallidal fibers, showed striking accumulation of iron (Supplemental Figure 4, MSA-1). In contrast, MSA-3, weak binding of [11C]PBB3 to this region, had almost no iron deposits and MSA-4, strong binding of [11C]PBB3, showed only slight iron deposits (Supplemental Figure 4, MSA-3 and MSA-4). Taken together, we conclude that the binding of [11C]PBB3 was unrelated to the burden of iron deposits.
Discussion
The present study had two major findings. First, tau PET ligand PBB3 at a high concentration (32.3 μM) can bind to not only tau, but also α-synuclein pathology, including Lewy bodies, Lewy neurites, spheroids, GCIs, and NCIs, since fluorescence labeling of these lesions with PBB3 was observed in all cases we examined. Second, autoradiographic evaluation of [11C]PBB3 at a low concentration (10 nM) showed no binding to α-synuclein pathology in LBD cases, but MSA cases with a high burden of α-synuclein pathology (MSA-3 and MSA-4) had significant radioligand binding to the striatopallidal fibers, which corresponded to abundant GCIs in this region.
Although various types of α-synuclein pathology were captured by PBB3 fluorescence labeling, it should be noted that structures labeled by PBB3 fluorescence were not necessarily detected by autoradiography. The high sensitivity of fluorescence labeling is consistent with literature on another tau ligand, [18F]AV-1451. Autoradiographic studies showed that [18F]AV-1451 preferentially bound to paired helical filaments (PHFs), rather than straight filaments.41, 42 Marquie and colleagues reported, however, that the AV-1451 fluorescent analogue also labeled non-PHF-tau lesions, amyloid plaque cores, and amyloid-laden vessels at high concentration.41 In the present study, the concentration of PBB3 for fluorescence labeling (32.3 μM) experiment is much higher than that for autoradiography (10 nM). The higher concentration may attribute to greater sensitivity and less specificity of fluorescence labeling, as the compound could cross-react with low-affinity, high-capacity binding sites in non-tau fibers.
The current autoradiographic evaluations indicated distinct detectabilities of α-synuclein inclusions in LBD and MSA by [11C]PBB3. We did not find any positive radiosignals in LBD, even in a case with the most severe α-synuclein deposition (LBD-3). Given the relatively low density of α-synuclein pathology within the amygdala(compared to the density of those within the striatopallidal fibers of MSA, see Figure 1), binding of [11C]PBB3 to this region would be negligible. This is supported by our pilot binding assays using LBD brain homogenates; dissociation constant (Kd) value for binding of [11C]PBB3 to α-synuclein deposits is 10–50 times higher than Kd for binding to tau deposits in AD and PSP brain samples, indicating that [11C]PBB3 has a low affinity for α-synuclein (Ono et al., unpublished data). Taken together, our results indicate that α-synuclein pathology in LBD would be undetectable by [11C]PBB3-PET. It is worth noting that LBD cases with SNCA duplication/triplication might be exceptions because severe α-synuclein pathology often coexists with tau pathology, which would hamper interpretation of autoradiographic labeling.43
In contrast to LBD, autoradiographic evaluations showed significant binding of [11C]PBB3 to the striatopallidal fibers in two MSA cases, despite low affinity of [11C]PBB3 for α-synuclein fibrils. Immunohistochemistry confirmed abundant GCIs and absence of tau pathology in this region. Furthermore, results of Prussian blue staining showed that the autoradiographic binding of [11C]PBB3 was unrelated to burden of iron deposits. These findings indicate that the positive signals in autoradiography are attributable to binding of [11C]PBB3 to GCIs but not to tau pathology or iron deposits. Compared to the strong binding of [11C]PBB3 in MSA-4, the striatopallidal fibers were weakly labeled by [11C]PBB3 in MSA-3. Assuming that the α-synuclein burden of MSA-3 (4.4%, at the 75th percentile) is at the lower limit of detection by [11C]PBB3, 25% of MSA cases would show positive striatal binding. Given that the maximum concentration of [11C]PBB3 in human PET scans is approximately 10 nM, the present data imply that α-synuclein pathology in a subset of MSA cases with the high density of GCIs might be captured by this radioligand. It should be noted, however, that the dynamic range of PET signals is determined by binding properties of [11C]PBB3 (i.e. reactivity for the target versus off-target molecules) as well as the uptake of [11C]PBB3 in the brain, clearance of free radioligands from the brain, and entry of radiometabolites into the brain.
It should also be noted that several non-tau structures have been reported as off-target binding components for tau PET ligands.19, 41, 42, 44–46 Autoradiographic evaluations of [18F]AV-1451 demonstrated the off-target binding in neuromelanin-containing cells in substantia nigra of PSP cases as well as basal ganglia regardless of disease type.19, 41, 42 In contrast, a recent study by Ono et al. reported that only minimal binding of [11C]PBB3 in the basal ganglia of healthy control subjects was observed.19 This finding suggests that off-target binding of [11C]PBB3 does not apparently influence in vitro autoradiography in these areas. The off-target binding of monoamine oxidase (MAO)-A and MAO-B has also been reported.45–47 Although we do not have the direct evidence that PBB3 binds MAOs (i.e. MAO-A and MAO-B) yet, the autoradiographic signals in the striatopallidal fibers are unlikely due to the off-target binding in MAOs because both MAOs are found in neurons and astroglia, but not in oligodendrocytes.48 To determine the binding of [11C]PBB3 to MAOs, in vivo PET studies with MAOs inhibitors are needed.
As with tauopathy, α-synucleinopathy is one of the major categories of neurodegenerative disease. The fact that clinical presentations of α-synucleinopathies such as dementia and parkinsonism often mimic the clinical phenotype of tauopathies makes accurate antemortem diagnosis difficult.5, 6 Imaging modalities to differentiate tauopathy and α-synucleinopathy will solve this clinical challenge. Our present study raises the possibility that derivative compounds of [11C]PBB3 might be potential candidates as α-synuclein PET ligands. The fact that PBB3 potentially binds to α-synuclein pathology can be explained by the fact that PBB3 binds to β-pleated sheet structures and α-synuclein filaments have a β-pleated sheet conformation that is similar to the structure described for amyloid fibrils.49, 50 Similar to our findings, [11C]BF-227, known as a PET tracer for in vivo detection of β-amyloid deposits, also bound to synthetic α-synuclein as well as β-amyloid aggregates in vitro.51 In addition, Kikuchi et al reported that [11C]BF-227-PET demonstrated significant retention in the subcortical white matter, putamen, and other regions, typically affected in MSA.52 These findings suggest that [11C]PBB3-PET imaging might also be positive in patients with MSA. Although the binding affinity of PBB3 to α-synuclein pathology is lower than its affinity to tau pathology, screening derivative compounds of [11C]PBB3 to develop novel ligands that have higher affinity to α-synuclein pathology is much anticipated. Although the distribution and the burden of Lewy pathology are quite limited in the early stages of Parkinson disease,53 it would be possible to detect Lewy pathology if the α-synuclein PET ligand has high enough binding affinity, at least 10–50 times higher than that of PBB3. The threshold of signal detection can be confirmed by direct comparison between in vivo PET imaging and post-mortem pathology in the same subjects. As Murray et al. clarified the cut-off standardized uptake value of [11C]PiB-PET which was approximately equivalent to Thal amyloid phase of 1–2,54 this type of study will determine whether the α-synuclein PET ligand can detect the α-synuclein pathology even in the early stages of Parkinson disease patients.
A major limitation of this study is the lack of in vivo [11C]PBB3-PET imaging in patients with α-synucleinopathy, which prevents direct comparisons between in vivo and post-mortem neuropathology in the same subjects. Since results of in vitro autoradiography are not always directly reflective of the in vivo PET scans, it is imperative to conduct an in vivo [11C]PBB3-PET imaging study on patients with α-synucleinopathy to validate the results of present study. Another limitation is that we used phosphorylation-dependent tau antibody (CP13) to assess tau pathology in MSA cases. Since the presence of unphosphorylated tau in GCI was reported,55 we might fail to evaluate the presence of this type of tau in GCI. We assume, however, that the contribution of the unphosphorylated tau might be minimal because these tau proteins are not the principal constituents of the fibrillary structures of GCIs.56 A notable strength of our study is that we selected cases based on quantitative analysis of α-synuclein burden. We performed autoradiography using cases with a wide range of α-synuclein burden, including an MSA case with the highest α-synuclein burden in our brain bank. Although recent studies have shown that [18F]AV-1451 do not seem to bind α-synuclein pathology,41, 42 the burden of α-synuclein in cases studied was not described. It is important to compare a wide range of α-synuclein burden in each α-synucleinopathy to assess binding properties of PBB3 and its derivatives. Another advantage of this study is that we examined pure LBD cases rather than LBD cases with varying amounts of coexisting Alzheimer-type pathology. If these LBD cases were included in this study, it would be hard to interpret whether observed binding of PBB3 was to α-synuclein, tau or Aβ.
In conclusion, the present results suggest that α-synuclein pathology in LBD would not be detectable by [11C]PBB3-PET, while a subset of severe MSA cases with high density of α-synuclein pathology might be positive. Further studies including direct comparison between in vivo PET images and post-mortem neuropathology in the same subjects are necessary to demonstrate the usefulness of [11C]PBB3-PET to differentiate tauopathies from α-synucleinopathies.
Supplementary Material
Supplementary Figure 1: Quantitative assessment of α-synuclein burden for case selection. A: Immunohistochemistry for α-synuclein (NACP, 1:3000) in LBD (top) and MSA (bottom) cases. Amygdala in LBD and striatopallidal fibers in MSA are annotated for quantitative analysis. Boxes indicate the area of high magnification images shown in B and C. Bars = 2 mm. B: High magnification images of amygdala (top) and the striatopallidal fibers (Bottom) C: The images showing the custom-designed color deconvolution algorithm to highlight α-synuclein deposits (shown in red). Lewy bodies, Lewy neurites, and Lewy dots in LBD (top) and glial cytoplasmic inclusions in MSA (bottom) are observed and quantified. Bar = 50 μm.
Supplementary Figure 2: PBB3 fluorescence labels tau (A) and α-synuclein (B) pathology, formic acid treatment disappear the signals. Lipofuscin autofluorescence is still visible after formic acid treatment (C). Bars = 20 μm. Abbreviations: GCIs, glial cytoplasmic inclusions; FA, formic acid.
Supplementary Figure 3: Thioflavin S fluorescence and immunofluorescence doublestaining. Thioflavin S labels tau pathology; neurofibrillary tangles in AD (A), Pick body in Pick’s disease (B), and coiled body in PSP (C), which are positive for CP13 but negative for NACP. Thioflavin S also labels α-synuclein pathology; Lewy body (D) and Lewy neurites (E) in LBD, and glial cytoplasmic inclusion (F) and neuronal cytoplasmic inclusions (G) in MSA, which are positive for NACP but negative for CP13. Bar = 20 μm. All images are the same magnification.
Supplementary Figure 4: Prussian blue staining in the striatopallidal fibers in LBD (top) and MSA (bottom) cases. Various degree of iron deposits (blue) is observed especially in the perivascular regions. Arrows indicate representative iron deposits. Bar = 100 μm. All images are the same magnification.
Acknowledgments
We would like to thank the patients and their families who donated brains to help further the scientific understanding of neurodegeneration. The authors would also like to acknowledge Dr. Peter Davies (Feinstein Institute for Medical Research, LIJ-North Shore Health System, NY) for the monoclonal anti-tau antibodies CP13, Linda Rousseau (Mayo Clinic, Jacksonville, FL) and Virginia Phillips (Mayo Clinic) for histologic support, Monica Castanedes-Casey (Mayo Clinic) for immunohistochemistry support, and Lewis-Tuffin, Laura J. (Mayo Clinic) for fluorescence microscopy support.
Footnotes
Author Contributions:
Shunsuke Koga: Execution of the project; Analysis and interpretation of data; Writing of the first draft.
Maiko Ono: Execution of the project; Analysis and interpretation of data;
Naruhiko Sahara: Conception of the project; Interpretation of data; Review and critique.
Makoto Higuchi: Conception of the project; Interpretation of data; Review and critique.
Dennis W Dickson: Conception and organization of the project; Interpretation of data; Review and critique.
Relevant conflicts of interest/financial disclosures
S.K. receives support from the Postdoctoral Fellowship for Research Abroad, Japan Society for the Promotion of Science.
M.O. receives grants from the Takeda Science Foundation.
N.S. receives Grants-in-Aid for Scientific Research on Innovation Area (“Brain Protein Aging”, 26117001) and Scientific Research (C) (15K06793) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
M.H. receives Grants-in-Aid for Core Research for Evolutional Science and Technology (14533254), the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS; 15653129) and Research and Development Grants for Dementia (16768966) from the Japan Agency for Medical Research and Development, and the Japan Advanced Molecular Imaging Program and Scientific Research on Innovative Areas (“Brain Environment” 23111009) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. M. H. hold a patent on compounds related to the present report (JP 5422782/EP 12 884 742.3).
D.W.D. receives support from the NIH (P50-NS072187). D.W.D. is an editorial board member of Acta Neuropathologica, Annals of Neurology, Brain, Brain Pathology, and Neuropathology, and he is editor in chief of American Journal of Neurodegenerative Disease, and International Journal of Clinical and Experimental Pathology.
References
- 1.Adler CH, Beach TG, Hentz JG, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology. 2014;83(5):406–412. doi: 10.1212/WNL.0000000000000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koga S, Aoki N, Uitti RJ, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology. 2015;85(5):404–412. doi: 10.1212/WNL.0000000000001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55(3):181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koga S, Josephs KA, Ogaki K, et al. Cerebellar ataxia in progressive supranuclear palsy: An autopsy study of PSP-C. Mov Disord. 2016;31(5):653–662. doi: 10.1002/mds.26499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125(Pt 4):861–870. doi: 10.1093/brain/awf080. [DOI] [PubMed] [Google Scholar]
- 6.Josephs KA, Dickson DW. Diagnostic accuracy of progressive supranuclear palsy in the Society for Progressive Supranuclear Palsy brain bank. Mov Disord. 2003;18(9):1018–1026. doi: 10.1002/mds.10488. [DOI] [PubMed] [Google Scholar]
- 7.Ubhi K, Low P, Masliah E. Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci. 2011;34(11):581–590. doi: 10.1016/j.tins.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 11.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79(6):1094–1108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34(2):457–468. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 14.Okamura N, Furumoto S, Harada R, et al. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med. 2013;54(8):1420–1427. doi: 10.2967/jnumed.112.117341. [DOI] [PubMed] [Google Scholar]
- 15.Whitwell JL, Lowe VJ, Tosakulwong N, et al. [18 F]AV-1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord. 2016 doi: 10.1002/mds.26834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammes J, Bischof GN, Giehl K, et al. Elevated in vivo [18F]-AV-1451 uptake in a patient with progressive supranuclear palsy. Mov Disord. 2016 doi: 10.1002/mds.26727. [DOI] [PubMed] [Google Scholar]
- 17.Harada R, Okamura N, Furumoto S, et al. [(18)F]THK-5117 PET for assessing neurofibrillary pathology in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2015;42(7):1052–1061. doi: 10.1007/s00259-015-3035-4. [DOI] [PubMed] [Google Scholar]
- 18.Harada R, Okamura N, Furumoto S, et al. Characteristics of Tau and Its Ligands in PET Imaging. Biomolecules. 2016;6(1):7. doi: 10.3390/biom6010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ono M, Sahara N, Kumata K, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain. 2017 doi: 10.1093/brain/aww339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bussiere T, Bard F, Barbour R, et al. Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am J Pathol. 2004;165(3):987–995. doi: 10.1016/s0002-9440(10)63360-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun A, Nguyen XV, Bing G. Comparative analysis of an improved thioflavin-s stain, Gallyas silver stain, and immunohistochemistry for neurofibrillary tangle demonstration on the same sections. J Histochem Cytochem. 2002;50(4):463–472. doi: 10.1177/002215540205000403. [DOI] [PubMed] [Google Scholar]
- 22.Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ. Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem. 2002;277(7):5411–5417. doi: 10.1074/jbc.M105326200. [DOI] [PubMed] [Google Scholar]
- 23.Cho H, Choi JY, Lee SH, Ryu YH, Lee MS, Lyoo CH. 18F-AV-1451 binds to putamen in multiple system atrophy. Mov Disord. 2016 doi: 10.1002/mds.26857. [DOI] [PubMed] [Google Scholar]
- 24.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology. 1994;44(11):2015–2019. doi: 10.1212/wnl.44.11.2015. [DOI] [PubMed] [Google Scholar]
- 26.Kosaka K. Diffuse Lewy body disease in Japan. J Neurol. 1990;237(3):197–204. doi: 10.1007/BF00314594. [DOI] [PubMed] [Google Scholar]
- 27.Kosaka K, Yoshimura M, Ikeda K, Budka H. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree--a new disease? Clin Neuropathol. 1984;3(5):185–192. [PubMed] [Google Scholar]
- 28.Trojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. 2007;33(6):615–620. doi: 10.1111/j.1365-2990.2007.00907.x. [DOI] [PubMed] [Google Scholar]
- 29.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 30.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 31.Dickson DW, Liu W, Hardy J, et al. Widespread alterations of alpha-synuclein in multiple system atrophy. Am J Pathol. 1999;155(4):1241–1251. doi: 10.1016/s0002-9440(10)65226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koga S, Dickson DW, Bieniek KF. Chronic Traumatic Encephalopathy Pathology in Multiple System Atrophy. J Neuropathol Exp Neurol. 2016;75(10):963–970. doi: 10.1093/jnen/nlw073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koga S, Parks A, Uitti RJ, et al. Profile of cognitive impairment and underlying pathology in multiple system atrophy. Mov Disord. 2016 doi: 10.1002/mds.26874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bancroft JD, Gamble M. Theory and practice of histological techniques. 5. London: Churchill Livingstone; 2002. [Google Scholar]
- 35.Rezaie P, Cairns NJ, Chadwick A, Lantos PL. Lewy bodies are located preferentially in limbic areas in diffuse Lewy body disease. Neurosci Lett. 1996;212(2):111–114. doi: 10.1016/0304-3940(96)12775-0. [DOI] [PubMed] [Google Scholar]
- 36.Dickson DW. Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol. 2009;3(1):1–23. [PMC free article] [PubMed] [Google Scholar]
- 37.Hashimoto H, Kawamura K, Igarashi N, et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J Nucl Med. 2014;55(9):1532–1538. doi: 10.2967/jnumed.114.139550. [DOI] [PubMed] [Google Scholar]
- 38.Kitamoto T, Ogomori K, Tateishi J, Prusiner SB. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest. 1987;57(2):230–236. [PubMed] [Google Scholar]
- 39.Forsberg A, Jureus A, Cselenyi Z, et al. Low background and high contrast PET imaging of amyloid-beta with [11C]AZD2995 and [11C]AZD2184 in Alzheimer’s disease patients. Eur J Nucl Med Mol Imaging. 2013;40(4):580–593. doi: 10.1007/s00259-012-2322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cantone MC, Hoeschen C. Radiation physics for nuclear medicine. Berlin; London: Springer; 2011. [Google Scholar]
- 41.Marquie M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015;78(5):787–800. doi: 10.1002/ana.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowe VJ, Curran G, Fang P, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun. 2016;4(1):58. doi: 10.1186/s40478-016-0315-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujishiro H, Imamura AY, Lin WL, et al. Diversity of pathological features other than Lewy bodies in familial Parkinson’s disease due to SNCA mutations. Am J Neurodegener Dis. 2013;2(4):266–275. [PMC free article] [PubMed] [Google Scholar]
- 44.Hansen AK, Knudsen K, Lillethorup TP, et al. In vivo imaging of neuromelanin in Parkinson’s disease using 18F-AV-1451 PET. Brain. 2016;139(Pt 7):2039–2049. doi: 10.1093/brain/aww098. [DOI] [PubMed] [Google Scholar]
- 45.Xia CF, Arteaga J, Chen G, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement. 2013;9(6):666–676. doi: 10.1016/j.jalz.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 46.Ng KP, Massarweh G, Soucy JP, et al. Selegiline reduces brain [18F]THK5351 binding. 11th Human Amyloid Imaging; 2017; Miami Beach. 2017. [Google Scholar]
- 47.Hostetler ED, Walji AM, Zeng Z, et al. Preclinical Characterization of 18F-MK-6240, a Promising PET Tracer for In Vivo Quantification of Human Neurofibrillary Tangles. J Nucl Med. 2016;57(10):1599–1606. doi: 10.2967/jnumed.115.171678. [DOI] [PubMed] [Google Scholar]
- 48.Riederer P, Konradi C, Schay V, et al. Localization of MAO-A and MAO-B in human brain: a step in understanding the therapeutic action of L-deprenyl. Adv Neurol. 1987;45:111–118. [PubMed] [Google Scholar]
- 49.Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97(9):4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000;39(10):2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 51.Fodero-Tavoletti MT, Mulligan RS, Okamura N, et al. In vitro characterisation of BF227 binding to alpha-synuclein/Lewy bodies. Eur J Pharmacol. 2009;617(1–3):54–58. doi: 10.1016/j.ejphar.2009.06.042. [DOI] [PubMed] [Google Scholar]
- 52.Kikuchi A, Takeda A, Okamura N, et al. In vivo visualization of alpha-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy]benzoxazole positron emission tomography in multiple system atrophy. Brain. 2010;133(Pt 6):1772–1778. doi: 10.1093/brain/awq091. [DOI] [PubMed] [Google Scholar]
- 53.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 54.Murray ME, Lowe VJ, Graff-Radford NR, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain. 2015;138(Pt 5):1370–1381. doi: 10.1093/brain/awv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cairns NJ, Atkinson PF, Hanger DP, Anderton BH, Daniel SE, Lantos PL. Tau protein in the glial cytoplasmic inclusions of multiple system atrophy can be distinguished from abnormal tau in Alzheimer’s disease. Neurosci Lett. 1997;230(1):49–52. doi: 10.1016/s0304-3940(97)00474-6. [DOI] [PubMed] [Google Scholar]
- 56.Shibuya K, Uchihara T, Nakamura A, et al. Reversible conformational change of tau2 epitope on exposure to detergent in glial cytoplasmic inclusions of multiple system atrophy. Acta Neuropathol. 2003;105(5):508–514. doi: 10.1007/s00401-003-0675-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Quantitative assessment of α-synuclein burden for case selection. A: Immunohistochemistry for α-synuclein (NACP, 1:3000) in LBD (top) and MSA (bottom) cases. Amygdala in LBD and striatopallidal fibers in MSA are annotated for quantitative analysis. Boxes indicate the area of high magnification images shown in B and C. Bars = 2 mm. B: High magnification images of amygdala (top) and the striatopallidal fibers (Bottom) C: The images showing the custom-designed color deconvolution algorithm to highlight α-synuclein deposits (shown in red). Lewy bodies, Lewy neurites, and Lewy dots in LBD (top) and glial cytoplasmic inclusions in MSA (bottom) are observed and quantified. Bar = 50 μm.
Supplementary Figure 2: PBB3 fluorescence labels tau (A) and α-synuclein (B) pathology, formic acid treatment disappear the signals. Lipofuscin autofluorescence is still visible after formic acid treatment (C). Bars = 20 μm. Abbreviations: GCIs, glial cytoplasmic inclusions; FA, formic acid.
Supplementary Figure 3: Thioflavin S fluorescence and immunofluorescence doublestaining. Thioflavin S labels tau pathology; neurofibrillary tangles in AD (A), Pick body in Pick’s disease (B), and coiled body in PSP (C), which are positive for CP13 but negative for NACP. Thioflavin S also labels α-synuclein pathology; Lewy body (D) and Lewy neurites (E) in LBD, and glial cytoplasmic inclusion (F) and neuronal cytoplasmic inclusions (G) in MSA, which are positive for NACP but negative for CP13. Bar = 20 μm. All images are the same magnification.
Supplementary Figure 4: Prussian blue staining in the striatopallidal fibers in LBD (top) and MSA (bottom) cases. Various degree of iron deposits (blue) is observed especially in the perivascular regions. Arrows indicate representative iron deposits. Bar = 100 μm. All images are the same magnification.