

Abstract
Cardioipins (CLs) are unique tetra-acylated phospholipids of mitochondria and define the bioenergetics and regulatory functions of these organelles. An unresolved paradox is the high uniformity of CL molecular species (tetra-linoleoyl-CL) in the heart, liver, and skeletal muscles—in contrast to their high diversification in the brain. Here, we combined liquid chromatography–mass-spectrometry-based phospholipidomics with genetic and nutritional manipulations to explore CLs’ biosynthetic vs postsynthetic remodeling processes in S. cerevisiae yeast cells. By applying the differential phospholipidomics analysis, we evaluated the contribution of Cld1 (CL-specific phospholipase A) and Taz1 (acyl-transferase) as the major regulatory mechanisms of the remodeling process. We further established that nutritional “pressure” by high levels of free fatty acids triggered a massive synthesis of homoacylated molecular species in all classes of phospholipids, resulting in the preponderance of the respective homoacylated CLs. We found that changes in molecular speciation of CLs induced by exogenous C18-fatty acids (C18:1 and C18:2) in wild-type (wt) cells did not occur in any of the remodeling mutant cells, including cld1Δ, taz1Δ, and cld1Δtaz1Δ. Interestingly, molecular speciation of CLs in wt and double mutant cells cld1Δtaz1Δ was markedly different. Given that the bioenergetics functions are preserved in the double mutant, this suggests that the accumulated MLCL—rather than the changed CL speciation—are the likely major contributors to the mitochondrial dysfunction in taz1Δ mutant cells (also characteristic of Barth syndrome). Biochemical studies of Cld1 specificity and computer modeling confirmed the hydrolytic selectivity of the enzyme toward C16-CL substrates and the preservation of C18:1-containing CL species.
Graphical abstract
The well-established tissue specificity of cardiolipins (CL)s1 has led to the paradigm that the molecular speciation of CLs—achieved through the process of remodeling of the nascent species—is defined by the unique mitochondrial demands of a particular tissue. This commonly accepted paradigm has been recently challenged by a clear demonstration that unremodeled CLs are functionally indistinguishable from remodeled CLs for yeast mitochondrial morphology and major bioenergetic functions, such as in oxidative phosphorylation (OxPhos),2 where it is a required structural component of the individual OxPhos complexes as well as supercomplexes.3 Recent findings have demonstrated that respiratory supercomplexes are destabilized by disturbed CL remodeling in heart mitochondria from tafazzin knockdown mice, resulting in disruption of interactions between the electron transporting complexes and the fatty acid (FA) oxidation enzymes.4
The two major reactions in the remodeling process are a phospholipase-driven production of monolyso-CL (MLCL) intermediates followed by an acyltransferase (tafazzin)-dependent “rebuilding” of the molecule that reacylates MLCL via the acyl-CoA-independent transfer of an acyl group from a donor phospholipid.2,5
Recent developments, elucidating the role of mitochondria as a platform coordinating many metabolic processes as well as life and death decisions, imply that essential functions not associated with bioenergetics may dictate the necessity for the highly diversified molecular speciation of CLs in tissues with signaling functions (e.g., in the brain). Not only has it been shown that mitochondria have functions other than bioenergetics, but CL in particular has extra-bioenergetic functions. The first demonstration of a role for CL in cell viability apart from bioenergetics was the demonstration that the crd1Δ (CL synthase) mutant has a temperature-sensitive growth defect, unlike yeast mitochondrial bioenergetics mutants, which do not exhibit temperature sensitivity.6 Perturbation of CL synthesis has been shown to affect MAPK signaling7 and vacuolar function in yeast.8 This is in sharp contrast to such tissues as cardiac and skeletal muscles in which mitochondrial energy production dictates the dominance of homoacylated tetraoleoyl-CL ((C18:1)4-CL) or tetralinoleoyl-CL ((C18:2)4-CL) species over a rich assortment of heteroacylated CLs that may be required for signaling purposes.9
CL signaling may be realized through the production of oxygenated lipid mediators,9b thus requiring CL species containing polyunsaturated fatty acids (PUFA). Therefore, the plasticity that is fine-tuned by adaptive changes in the microenvironment—rather than a rigid constitutive program—is likely to define the functions and mechanisms of CL remodeling. The machineries realizing these adaptive responses may include the availability of polyunsaturated fatty acids as well as enzymatic mechanisms involved in CL remodeling—including phospholipases A and acyl-transferases.10
Yeast cells may be ideal organisms for the exploration of these pathways because they (i) can be grown on media completely devoid of PUFA or supplemented with PUFA, (ii) rely on only one CL-specific phospholipase A, Cld1, (iii) utilize Taz1 as the major tool for reacylation of MLCL, and (iv) employ only one—Cld1/Taz1-dependent—pathway for CL remodeling. With this in mind, in the current work we employed detailed phospholipidomics analysis to explore the effects of PUFA supplementation as well as cld1Δ and taz1Δ mutants on the molecular speciation of CLs in S. cerevisiae. Herein, we report the results of differential phospholipidomics studies of S. cerevisiae that provide new insights into CL remodeling and biosynthesis. Our findings identified the roles of Cld1 and Taz1 as the major regulatory mechanisms, which are governed by the availability of PUFA esterified into phospholipids that act as donors for the Taz1-catalyzed reaction.
RESULTS
Exogenous FAs Define the Molecular Speciation of CLs in Wild Type and Mutant S. Cerevisiae Cells
In wild-type (wt) S. cerevisiae cells grown without FA supplementation, there is an assortment of CL molecular species with different FA residues ranging from C10 to C18. Among those, only singly unsaturated CL species were present, whereas PUFA were not detected (Figure 1A, Supporting Information Figure S1). To gain insight into the mechanisms of CL remodeling and biosynthesis, we chose to expose yeast cells to FAs with different chain lengths as well as unsaturation. In particular, we employed oleic (18:1), linoleic (18:2), arachidonic (20:4), and docosahexaenoic (22:6) acids as components of the growth medium and established their effects on the phospholipidomes of S. cerevisiae cells.
Figure 1.
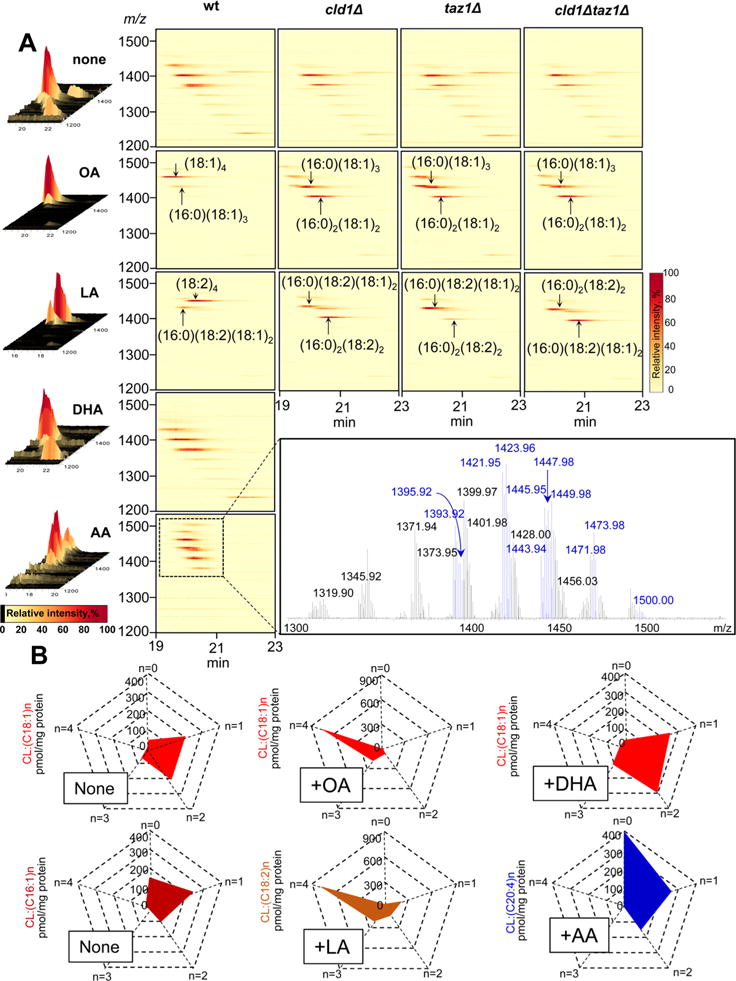
LC/MS analysis of CL in S. cerevisiae. (A) 3D-mass spectra (left panels) and MS maps (right panels) of CL obtained from wild type and cld1Δ, taz1Δ, and cld1Δtaz1Δ mutants before and after supplementation with different fatty acids. (OA, oleic acid C18:1; LA, linoleic acid C18:2, DHA, docosahexaenoic acid C22:6, AA, arachidonic acid C20:4). Inset: Mass spectrum of CL from yeast supplemented with C20:4 (AA). MS signals of C20:4 containing CL molecular species with m/z 1393.92, 1395.92, 1421.95, 1423.95, 1445.95, 1447.95, 1449.98, 1471.98, 1473.98, 1500.00 identified as C16:1_C16:1_C16:1_C20:4, C16:1_C16:1_C16:0_C20:4, C16:1_C16:1_C18:1_C20:4, C16:0_C16:1_C18:1_C20:4, C16:0_C16:1_C20:4_C20:4, C16:0_C16:0_C20:4_C20:4, C16:1_C18:1_C18:1_C20:4, C16:1_C18:1_C20:4_C20:4, C16:0_C18:1_C20:4_C20:4, and C18:1_C18:1_C20:4_C20:4, respectively, and shown in blue. (B) Quantitative assessment of hetero- and homoacylated molecular species of CLs in wt yeast cells before and after supplementation with different FAs.
Supplementation of the growth medium with either 18:1 or 18:2 FAs caused dramatic changes in CL composition (Figure 1B, Supporting Information Figures S2 and S3). Only CL molecular species containing from one to four 18:1 or C18:2 were detectable, while essentially all other molecular species of CLs with shorter chains and devoid of C18-containing species were not found (Figure 1). Notably, a huge preponderance of homoacylated (18:1)4-CL and (18:2)4-CL species was observed (Figure 1B). The second largest cluster of CLs included heteroacylated tri-C18:1 and tri-C18:2 species, respectively (Figure 1). These robust effects of exogenously added C18 FAs in wt cells were much less pronounced when C20:4 was used (Figure 1). In fact, only singly and doubly 20:4-acylated CL species were detectable; no tri- or tetra-20:4-CL species were observed (Figure 1B, Supporting Information Figure S4). Upon treatment with 22:6, CLs speciation remained unchanged vs untreated wt cells (Figure 1B); 22:6-contaning CL species were not detectable. Overall, these data indicate that the remodeling and biosynthetic pathways for CLs are very specific and favor the synthesis of C18-containing molecular species. The balance of natural processes of CL hydrolysis and reacylation yields only very low concentrations of MLCLs—as intermediates of these reactions. Supplementation with FAs did not significantly affect the MLCL content in wt cells (Figure 2).
Figure 2.
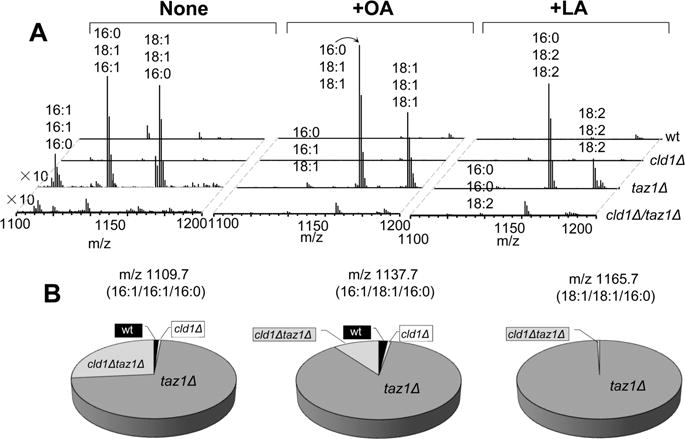
LC/MS analysis of monolyso-CL (MLCL) in S. cerevisiae. (A) Mass spectra of MLCL obtained from wt (left panel) yeast cells and cld1Δ, taz1Δ, and cld1Δtaz1Δ mutants supplemented with C18:1 (OA; middle panel) and C18:2 (LA; right panel). (B) Quantitative assessments of MLCL performed for wild type yeast cells and cld1Δ, taz1Δ, and cld1Δtaz1Δ mutants.
CL remodeling from MLCL in yeast cells occurs via a single metabolic reaction catalyzed by Taz1:11
where PL-2FA is a donor diacylated phospholipid, MLCL-3FA is an acceptor MLCL, LPL is lyso-phospholipid formed, and CL-4FA is a remodeled CL. For CL reacylation, the enzyme utilizes donor phospholipids, particularly PC and PE. Therefore, we also studied the effects of C18:1 and C18:2 supplementations on the phospholipidome of these cells. We found no changes in the total content of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) in supplemented cells. However, supplementation with 18:1 resulted in the accumulation of PE and PC species containing 18:1 FA residues (Figure 3, Supporting Information Figure S5). Similarly, addition to the medium of 18:2 was associated with higher levels of 18:2-containing PE and PC (Figure 3, Supporting Information Figure S5). The enrichment with C18 FA residues was at the expense of the decreased contents of PE and PC species containing 16:0 and 16:1 (Figure 3). Further, we found that supplementation of cells with either 18:1 or 18:2 resulted in the accumulation of homoacylated PC and PE molecular species (containing either 18:1 or 18:2 in both sn-1 and sn-2 positions). Subsequently, we detected an increased content of 18:1-lyso-PC(LPC)/lyso-PE(LPE) and 18:2-LPC/LPE after supplementation of cells with these FAs (Supporting Information Figure S6). This suggests that homoacylated species of PC and PE were effectively utilized by Taz1 as preferred substrates for the reacylation of MLCL. It is also possible that these FAs were readily incorporated via de novo biosynthesis (presumably of phosphatidic acid (PA)). Direct experimental assessment of the contribution of these alternative pathways can be deduced from our lipidomics data obtained from wt and mutant cells supplemented with FA, as shown below.
Figure 3.
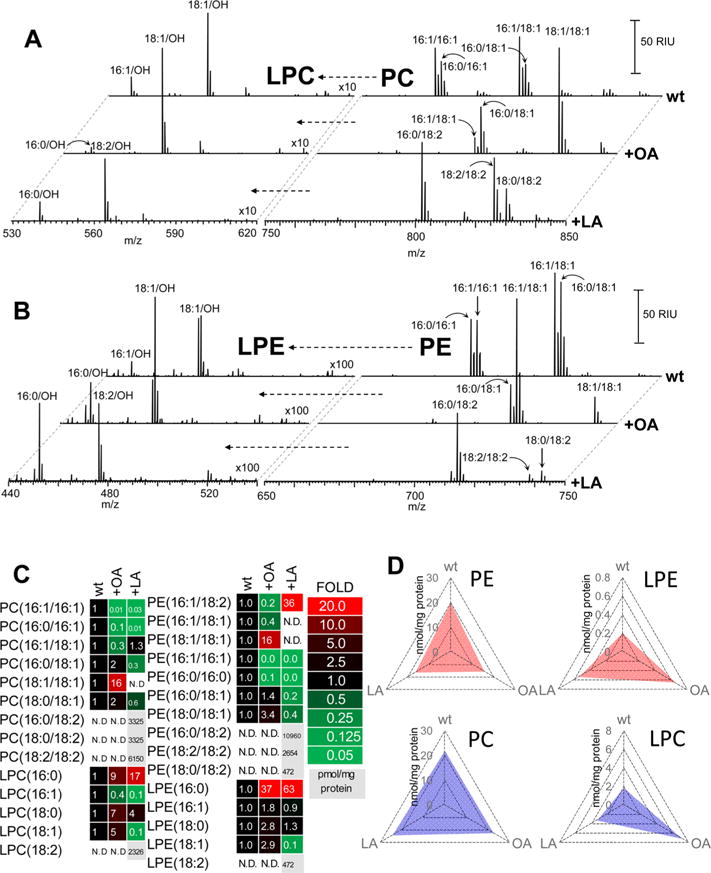
LC/MS analysis and quantitative assessment of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) as well as their hydrolysis products—lyso-PC (LPC) and lyso-PE (LPE)—in S. cerevisiae. (A) Mass spectra of PC (left panels) and LPC (right panels) for wild type yeast cells before and after supplementation with either C18:1 (OA) or C18:2 (LA). (B) Mass spectra of PE (left panels) and LPE (right panels) for wild type yeast cells before and after supplementation with either C18:1 (OA) or C18:2 (LA). (C,D) Heat maps of PC, PE, LPC and LPE for wild type yeast cells before and after supplementation with either C18:1 (OA) or C18:2 (LA). Values in heat maps pixels show the fold change in phospholipid content in supplemented yeast cells compared to nonsupplemented wild type cells. Red pixels without numbers inside designate lipid species produced only after LA supplementation (but not detectable in non-C18:2-supplemented cells).
Both Cld1 and Taz1 Modify the Cardiolipinome
Cld1 is a critical participant of the CL remodeling process, as it catalyzes phospholipid hydrolysis with a significant selectivity toward 16:0- and 16:1-containing molecular species.5a This suggests that disruption of this process in the cld1Δ mutant should result in an altered molecular speciation of CLs, particularly with regard to longer-chain (C16 and C18) species. However, according to a recently reported shotgun lipidomics study, the lipid profile of cld1Δ was similar to that of wt, whereas taz1Δ had a reduction of CL and an accumulation of MLCL.2 To resolve this apparent conundrum, we performed detailed LC-MS-based characterization of cardiolipinomes in wt and mutant S. cerevisiae cells. We found that the composition of CL from wt cells was different from that detected in either cld1Δ, taz1Δ, or cld1Δtaz1Δ mutants (Supporting Information Figure S1, Tables S1 and S2). In addition, cld1Δ contained slightly (but significantly) higher levels of CLs compared to wt cells (data not shown). In line with published data, the changes in the total content of CL and MLCL found in taz1Δ mutants were no longer detectable in cld1Δtaz1Δ mutants. Indeed, a dramatic accumulation of MLCL was detected in taz1Δ but not in cld1Δ or cld1Δtaz1Δ mutants (Figure 2). However, molecular speciation of CLs in wt cells vs double mutant cells was markedly different. Similar results were obtained with wt cells and mutants supplemented with OA or LA (Figure 2, Supporting Information Figures S3 and S4). These data are compatible with the role of Cld1 in “preparing” CLs for remodeling via hydrolysis/reacylation and suggest that elimination of the hydrolytic pathway by deletion of CLD1 would lead to “conservation” of CLs. Consistent with this prediction, cld1Δ mutant cells were essentially unable to produce remodeled tetra- and tri-18:1 species of CLs (Figure 1B, Supporting Information Table S2). Instead, their CLs were enriched with 16:0 and 16:1 containing molecular species (Figure 1B, Supporting Information Table S1).
Assuming that Cld1 is a participant in the only remodeling process in S. cerevisiae cells, one can distinguish between the contribution of de novo biosynthesis versus remodeling processes in the overall diversification of CLs. In wt cells, both the biosynthesis de novo and remodeling reactions contribute to the overall diversity and content of CLs. Deletion of CLD1 would eliminate the remodeling process and reveal the nascent CL species generated via its de novo biosynthesis. Thus, two categories of CLs can be determined by comparison of LC/MS spectra of CLs from wt and cld1Δ cells (Figure 4), including (i) those produced by the biosynthetic pathway, revealed in the cld1Δ mutant and (ii) those generated as products (increased) or utilized as substrates by the remodeling pathway (decreased). These latter respective categories of CLs can be identified in the LC/MS spectra of CLs in ± (wt minus cld1Δ) by the differential analysis.
Figure 4.
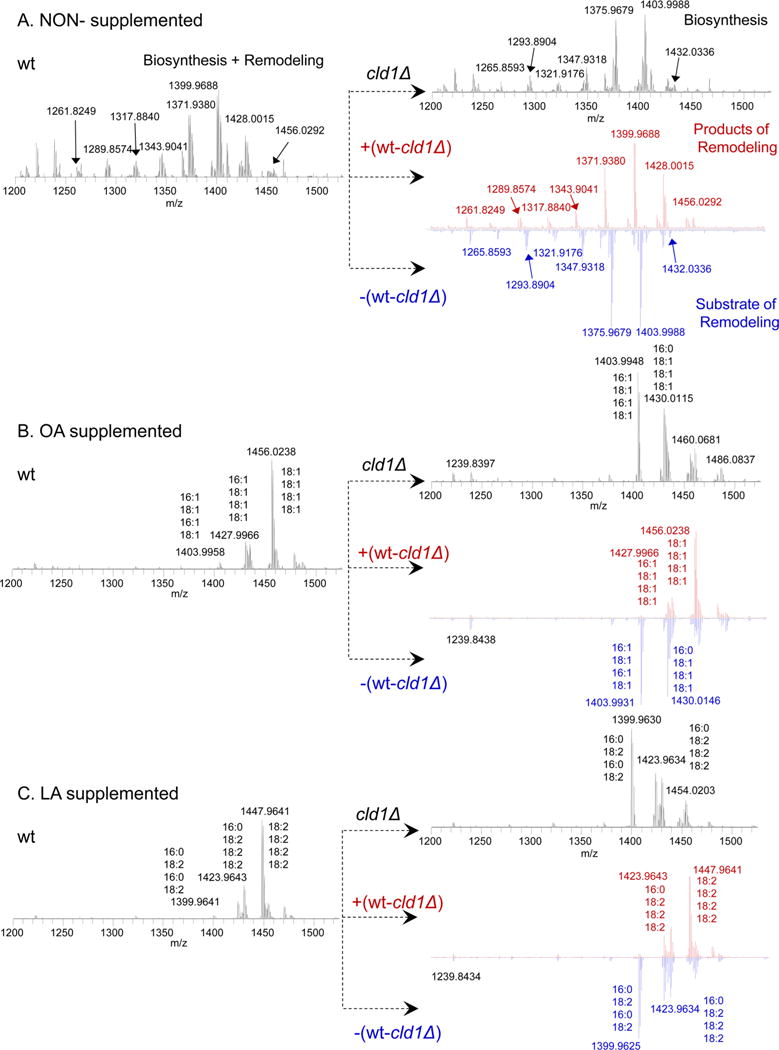
Contribution of de novo biosynthesis versus remodeling processes to the diversification of CLs in S. cerevisiae. Mass spectra of CL obtained from wild type yeast cells and cld1Δ mutant cells with or without oleic/linoleic acid (OA/LA) supplementation. The resulting differential mass spectra of CL species produced and consumed by the remodeling process were obtained, as shown in the positive and negative spectra of wt minus cld1Δ mutants, respectively. (A) Mass spectra of CL obtained from nonsupplemented wild type yeast cells and cld1Δ mutant. CL molecular species were identified as C10:0_C16:1_C16:1_C16:1 (m/z 1261.8249); C10:0_C16:0_C16:0_C16:1 (m/z 1265.8593); C10:0_C16:1_C16:1_C18:1 (m/z 1289.8574); C12:0_C16:0_C16:0_C16:1 (m/z1293.8904); C12:0_C16:1_C16:1_C18:1 (m/z 1317.8840); C12:0_C16:0_C16:1_C16:1 (m/z 1321.9176); C16:1_C16:1_C16:1_C16:1 (m/z 1343.9041); C12:0_C16:1_C18:1_C18:1 (m/z 1345.9184); C12:0_C16:0_C16:1_C18:0 (m/z 1347.9318); C16:1_C16:1_C16:1_C18:1 (m/z 1371.9380); C16:0_C16:1_C16:0_C18:1 (m/z 1375.9669); C16:1_C18:1_C16:1_C18:1 (m/z 1399.9688); C16:0_C18:1_C16:0_C18:1 (m/z 1403.9988); C16:1_C18:1_C18:1_C18:1 (m/z 1428.0015); C16:0_C18:1_C18:0_C18:1 (m/z 1432.0336); and C18:1/C18:1/C18:1/C18:1 (m/z 1456.0292). (B) Mass spectra of CL obtained from OA supplemented wt yeast cells and cld1Δ mutant. Major CL species are annotated on mass spectra. (C) Mass spectra of CL obtained from LA supplemented wt yeast cells and cld1Δ mutant. Major CL species are annotated on mass spectra.
By applying these differential (subtraction) protocols [please note that the specific feature of the software utilized for the subtraction disallows plotting of the negative spectra; therefore −(WT − cld1Δ) plots were used as explained in detail in the Methods], we found significant differences in molecular speciation of CLs in wt and mutant cells. In cld1Δ cells (lacking the remodeling pathway), CLs formed in the reaction catalyzed by CL synthase were mainly represented by molecular species containing 16:0, 16:1, and 18:1 FA residues (Figure 4, Supporting Information Figure S7). This was supported by LC/MS analysis of the CL biosynthesis precursor, phosphatidylglycerol (PG). We found that PGs were predominantly represented by 16:1/18:1, 16:1/18:1, and 16:0/16:1 molecular species in wt cells (Figure 5). On the other hand, during the remodeling, esterified 16:0 was substituted with the 16:1 fatty acid residue (Figure 5). These data suggest that in non-FA-supplemented wt cells, Cld1 predominantly hydrolyzed 16:0-containing CLs that were then remodeled with 16:1 by Taz1-catalyzed reacylation.
Figure 5.
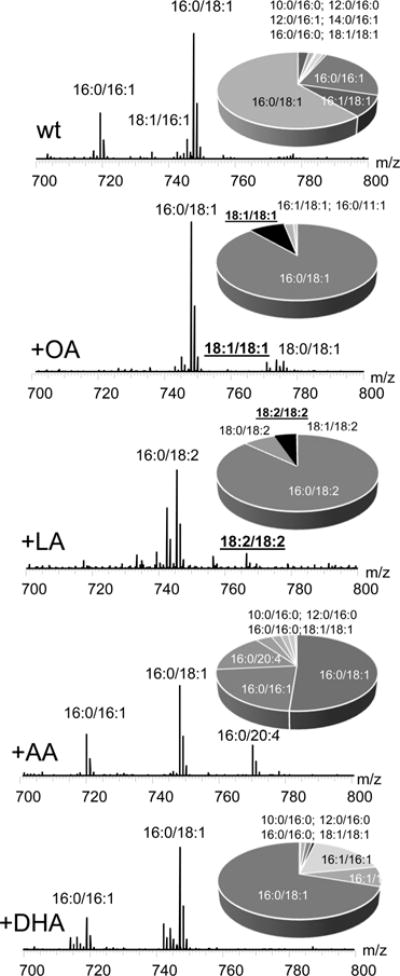
LC/MS analysis of phosphatidylglycerol (PG) in wild type S. cerevisae before and after supplementation with different fatty acids. Mass spectra and quantitative assessments (insets) of PG obtained from control wild type yeast cells and wild type yeast cells supplemented with C18:1 (OA), C18:2 (LA), C20:4 (AA), and C22:6 (DHA).
We further applied the same approach to distinguish between the CL species formed through either biosynthetic or remodeling pathways in wt cells supplemented with either 18:1 or 18:2. This analysis revealed that the remodeling route was entirely accountable for the replacement of fatty acid residues in CLs yielding predominantly homoacylated tetra-18:1 or tetra-18:2 molecular species of CLs, respectively (Figures 1B, 4B,C, Supporting Information Figure S7). On the basis of these newly developed protocols for the assessment of biosynthetic versus remodeling origins of the CL species, we compared the relative contributions of these two pathways to all CLs in determining the pool of homoacylated species. We found that biosynthesis was the major (wt cells, 18:2-treated cells) or significant (C18:1-treated cells) contributor to the entire pool of CLs. Interestingly, the remodeling process was, by far, the most important contributor to the pool of homoacylated CLs. Specifically, we found that CL(18:1/18:1/18:1/18:1) was the major product (Figure 4B, wt-cld1Δ), while CL(16:1_18:1_16:1_18:1) and CL(16:0_18:1_18:1_18:1) were the major substrates of the remodeling process (Figure 4B, -(wt-cld1Δ)). On the basis of the cld1Δ spectra, we determined that both major substrates of the remodeling originated from CL biosynthesis. For LA-supplemented cells, we found that CL(18:2/18:2/18:2/18:2) was the major product (Figure 4C, wt-cld1Δ), while CL(16:0_18:2_16:0_18:2) and CL(16:0_18:2_18:2_18:2) were the major substrates of the remodeling process (Figure 4C, -(wt-cld1Δ)). Similar to OA supplementation, both major remodeling substrates represented the products of CL biosynthesis. Further, the differential analysis demonstrated that after OA or LA supplementation, Cld1 had preferentially hydrolyzed C16:0- and C16:1-fatty acid residues over C18:1-and C18:2-containing CL species. As a result, the remodeling process tended to convert heteroacylated CL to homoacylated CLs, provided C18:1 or C18:2 were available.
As all major phospholipids can serve as acyl sources for Taz1-catalyzed remodeling, we further looked at the content of several phospholipids as well as their monolyso-forms in wt and taz1Δ mutant cells. Included in our phospholipidomics analyses were several major classes of phospholipids: (i) a biosynthetic precursor of CL, phosphatidylglycerol (PG); (ii) two major phospholipids present in both mitochondria and extra-mitochondrial compartments, PE and PC, and (iii) an extra-mitochondrial phospholipid, phosphatidylserine (PS). We found that the effects of TAZ1 deletion were CL-specific. Other phospholipids such as PC, PG, PS were not sensitive to TAZ1 deletion.
Exogenous FAs Do Not Affect Molecular Speciation of CL in Remodeling of Mutant S. cerevisiae Cells
While Cld1 and Taz1 represent the two major components of the machinery involved in the CL remodeling process, the immediate motive force(s) triggering the process remain unknown. As exogenously added C18 FAs caused efficient remodeling of CL to generate mostly homoacylated tetra-18:1 and tetra-18:2 molecular species in wt cells, we further explored the CL composition in mutant cells. The dramatic changes in molecular speciation of CLs induced by exogenous C18-FAs in wt cells were essentially not found in any of the remodeling mutant cells, including cld1Δ, taz1Δ, and cld1Δtaz1Δ (Figure 1).
Because Cld1 is specific for the hydrolysis and remodeling of CLs but Taz1 can participate in remodeling of other phospholipids,12 we were interested in assessing the effects of deletion of CLD1 as well as TAZ1 on the phospholipidomes of the mutant cells grown in the presence of 18:1 and 18:2 FAs. Our findings clearly demonstrate that deletion of CLD1 did not cause any changes in PE and PC molecular species in either 18:1- or 18:2-supplemented cells. The absence of Taz1 also did not substantially affect the content or molecular speciation of PC and PE (except for the accumulation of 16:0/16:1 and 16:1/16:1 PC molecular species but in cells supplemented with C18:1).
Specificity of Cld1 for Different Molecular Species of CLs
Because of the apparent preference of CL remodeling triggered by exogenous fatty acids for C18 species, we further investigated the specificity of Cld1 in the hydrolysis of different molecular species of CLs. We used commercially available homoacylated CLs—(14:0)4-CL, (18:1)4-CL, (18:2)4-CL— and also three chemically synthesized CLs—(16:0)4-CL, (18:0)4-CL, and (16:1)(16:0)3-CL. The multistep preparations followed a modular approach that allowed the incorporation of each glycerol unit sequentially. Selectively protected glycerol building blocks were coupled to the desired fatty acid in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and N,N-dimethyl-4-aminopyridine (DMAP). Mild acidic deprotection using a resin-bound sulfonic acid gave the desired 1,2-diacylglycerols. Following a sequence of phosphoramidite coupling/oxidation, selective deprotection, and a second phosphoramidite coupling/oxidation, the fully protected CL precursor could be obtained. Successive removal of the phosphate and secondary alcohol protecting groups, followed by treatment with ammonium hydroxide, gave the CL diammonium salts that were used for biological analysis.
We found that (14:0)4-CL, (18:1)4-CL, and (18:2)4-CL were very poor substrates for CLD1 (Figure 6). Both (16:0)4-CL and (16:1)(16:0)3-CL and also (18:0)4-CL were readily hydrolyzed by Cld1 (Figure 6). To verify the selectivity of Cld1 toward CL containing palmitic acid, we performed experiments using several types of mixtures of different CLs. In particular, we were interested in testing Cld1 substrate specificity toward different CL species most commonly present in S. cerevisae cells, as well as long-chain polyunsaturated CL species “unusual” for these cells. To this end, we prepared two types of liposomes, dioleoyl-PC liposomes containing either CLs isolated from the cld1Δ yeast mutant (to obtain the most diversified set of yeast CLs in conditions of suppressed remodeling process) or long chain polyunsaturated CLs isolated from fish heart (northern red snapper, Lutjanus campechanus; Figure 7). These experiments provided interesting details regarding the specificity of Cld1 toward different molecular species of CLs as substrates. We found that hydrolysis of yeast CLs by Cld1 resulted in the release of predominantly palmitic acid (16:0) and the formation of four major MLCL species such as 12:0_14:0_18:1, 14:0_14:0__18:1, 16:0_16:1_18:1, and 18:1/18:1/18:1. We did not observe significant accumulation of MLCL and free fatty acids or decreased CL content when liposomes containing fish heart CLs (with highly diversified CLs containing C22:5 and C22:6 fatty acid residues) were treated with Cld1. Thus, Cld1 selectively hydrolyzed CL species containing palmitic acid.
Figure 6.
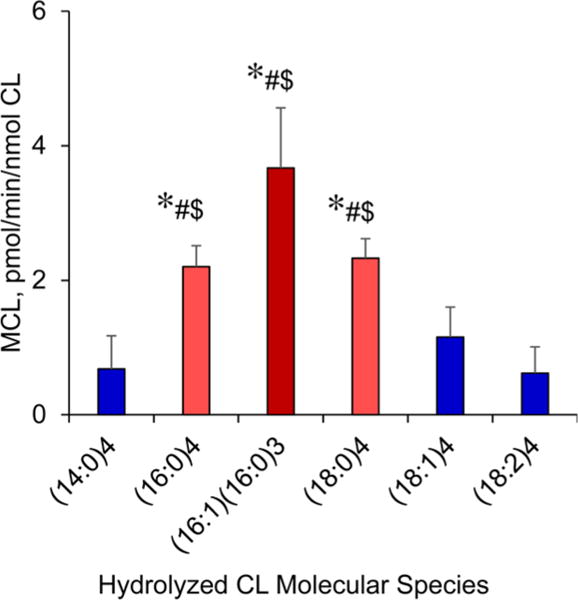
Effect of Cld1 isolated from S. cerevisae on accumulation of monolyso-CLs after hydrolysis of different CLs in a biochemical system in vitro. *p < 0.05 vs (14:0)3, #p < 0.002 vs (18:1)3, $p < 0.005 vs (18:2)3 (n = 4–10).
Figure 7.
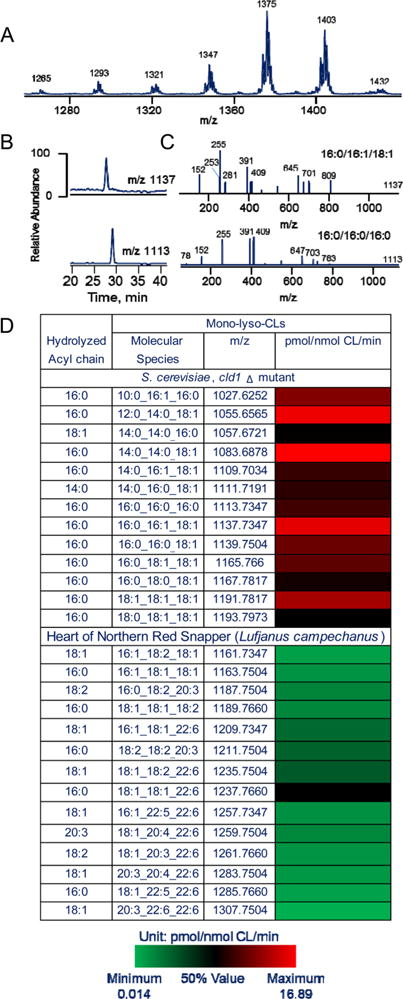
Typical LC-ESI-MS spectrum of CL isolated from cld1Δ mutants (A), LC-MS profiles (B), and MS/MS spectra (C) of MLCLs after CL hydrolysis by Cld1 and heat map of monolyso-CLs molecular species (D) after the release of fatty acids from CLs induced by Cld1 in the CLs isolated from cld1Δ mutant and from the heart of northern red snapper (Lutjanus campechanus).
Computational Studies of the Interactions of Homoacylated-CL with Cld1
To get insights into specifics of Cld1 hydrolysis of CL and its molecular species, a homology model of full-length structure of the protein was generated using Hsad, a steroid-degrading hydrolase, from Mycobacterium tuberculosis as a template (Figure 8A).13 This choice was based on the sequence analysis using the Protein BLAST tool14 and resulted in five templates with the highest levels of identities to Cld1. Accordingly, five homology models of Cld1 were built using the I-TASSER15 Web server. The model based on Hsad, a steroid-degrading hydrolase from Mycobacterium tuberculosis (PDB ID: 2VF213), demonstrated the greatest C-score (C-score was equal to −1.78). Therefore, it was selected for molecular docking modeling. The known lipase motif (228AHSLG232) and acyl transferase motif (424HHLYLD429) are shown in Figure 8B,C, respectively. According to Baile et al.,2 S230, H424, and D329 are the catalytic triad of Cld1 (Figure 8D). Intriguingly, the overlay of cavity and cartoon representation of the model structure revealed a channel toward the catalytic site. Subsequently, the molecular docking method was utilized to investigate the interactions of three homoacylated-CL species: (14:0)4-CL, (16:0)4-CL, and (18:2)4-CL. Notably, a palmitoyl residue (16:0) fit perfectly in this cavity (Figure 9). However, neither chains of tetra-myristoyl CL nor chains of tetra-linoleoyl CL penetrated into the active site channel. This indicates that the channel can introduce a CL acyl chain to the active site. Moreover, the length of the channel may function as a “ruler” defining the specificity of Cld1 toward particular molecular species of CLs—in reasonable agreement with the experimentally tested homo- and heteroacylated CLs.
Figure 8.
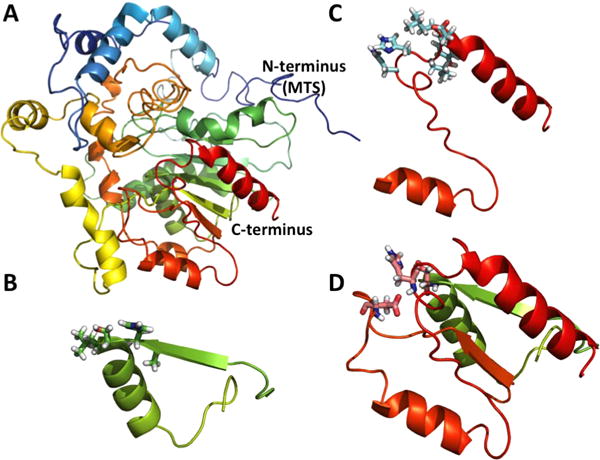
A homology model of Cld1, generated using iTassar server. (A) A full-length structure of Cld1 and its N-terminus region called mitochondrial target signal (MTS). (B) The known lipase motif (228AHSLG232). (C) Acyl transferase motif (424HHLYLD429). (D) The catalytic triad, including S230, D392, and H424, introduced by Baile et al.2
Figure 9.
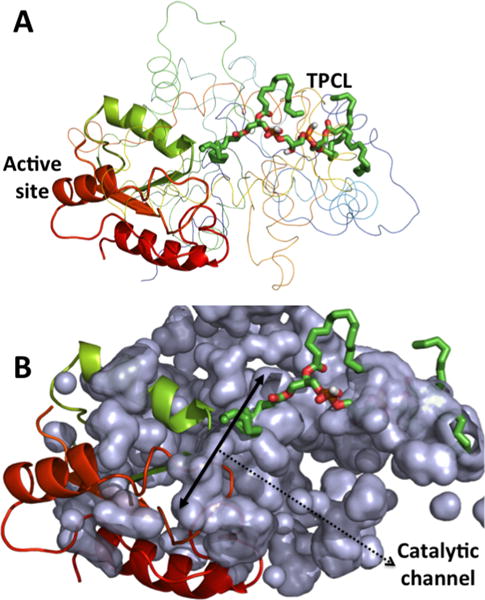
Interactions of (C16:0)4-CL with Cld1 structure, generated using homology modeling. (A) One of the acyl chains of TPCL aligned toward the active site. (B) A channel represents the CL acyl chain to the active site, which may define the specificity of Cld1 toward particular fatty acids based upon its size.
Effects of C18:1, C18:2, and C20:4 FAs Supplementation on Mitochondrial Bioenergetic Function in S. cerevisiae Cells
As CL is an essential structural component of the OxPhos machinery, we tested one possible effect of fatty acid supplementation on intact cell respiration of yeast cells. Interestingly, yeast cells analyzed in stationary phase in YPD media showed 45% and 21% increased respiration rates when the medium was supplemented with linoleic acid and arachidonic acid, respectively, but not with oleic acid, where respiration was decreased by −26% compared to control (Figure 10). These data suggest that CL composition plays an important role for mitochondrial function at the level of the oxidative phosphorylation system and that linoleic acid supplementation produces the highest respiration rates.
Figure 10.
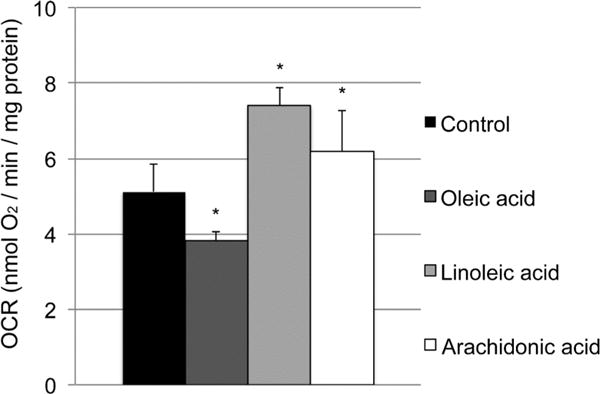
Yeast cells were grown in synthetic fermentable (YPD) media supplemented with tergitol alone (control) or tergitol plus oleic acid, linoleic acid, or arachidonic acid. Respiration of intact cells was analyzed via the polarographic method. *p < 0.05 versus control (n = 3).
DISCUSSION
Predominantly localized in mitochondria, CLs interact with numerous mitochondrial proteins,3 thus defining their role and significance in mitochondrial structural organization and metabolic/signaling functions. Similar to other phospholipids, CLs are subject to continuous turnover, resulting in changes in their molecular species that may be associated with their modified interactions with proteins. The molecular mechanisms and the significance of these changes—also called remodeling—in normal physiology and disease conditions are the subject of intensive investigation.16 We discovered that nutritional “pressure” by high levels of free FAs in the growth medium triggered massive synthesis of unusual homoacylated dioleoyl and dilinoleoyl molecular species in all major classes of phospholipids, including direct CL precursors as well as donors of acyl groups for the remodeling reactions. Not surprisingly, this resulted in the preponderant accumulation of the respective homoacylated CL species.
By employing detailed phospholipidomics analysis, this work established that the availability of specific FA precursors is the major factor defining the diversity and molecular speciation of CLs in S. cerevisiae. Supplementation with C18 FAs resulted in the predominant accumulation of homotetra-acylated 18:1 and 18:2 species of CL. On the basis of the engagement of only one biosynthetic and one remodeling pathway for CLs in S. cerevisiae, the specific contributions of these respective mechanisms to the overall molecular diversification/speciation of CLs in different growth conditions as well as in mutant forms can be established. For example, we established that the remodeling process is the major contributor to the pool of homoacylated (C18:1)4-CL and (C18:2)4-CL in cells supplemented with these FAs.
The CL hydrolysis is a major part of the remodeling pathway. To convert nascent CL, containing short saturated FA, to homoacylated CL with long chain polyunsaturated FAs, CL first has to undergo deacylation. In yeast, Cld1 fulfills this function and has been identified as a CL-specific hydrolase enzyme. Modeling data revealed the presence of a channel adjacent to the active site of Cld1, which may introduce a FA of CL to the active site to be hydrolyzed. In line with LC/MS data demonstrating preferable hydrolysis of 16:0 FA by Cld1, and modeling results revealing most likely interactions of 16:0 FA with the channel, it is tempting to speculate that the channel acts as a “molecular ruler” defining the specificity of Cld1 to particular FA residues in CLs.
By appropriately designing genetic manipulations (e.g., creating and producing appropriate deletion mutants such as cld1Δ and targeted transfection with mammalian remodeling genes such as iPLA2γ or iPLA2D), yeast cells supplemented with desired PUFA can be used as effective models in studies of mammalian CL metabolism. Similarly, the taz1Δ mutant transfected with acyltransferases alcat1 or mlcat1 may lead to the elucidation of their specific contributions to CL metabolic processes. In all these cases, the approach based on differential (subtractive) lipidomics analysis may be particularly powerful in examination of remodeling and biosynthesis.
Supplementation with exogenous C18 FAs stimulated CL remodeling. As FAs can be integrated into yeast CLs only via MLCL/Taz1-driven reactions, the acyl-CoA-dependent pathways are not utilized to remodel CL. However, it has been reported that acyl-CoAs are essential for reacylation of other phospholipids.17 Thus, detailed lipidomic analyses of other phospholipids in addition to CL are essential for comprehensive analysis of CL remodeling pathways.
It has been well established that taz1Δ cells are characterized by lower levels of CLs and markedly increased content of MLCL relative to wt cells.12 Our results are in full agreement with these earlier studies. In terms of molecular speciation of CLs, the same altered features of CL molecular speciation were also characteristic of taz1Δ mutant and double mutant cld1Δtaz1Δ cells, suggesting that the selectivity of Cld1 for C16-containing CLs is the major factor defining the overall molecular speciation and remodeling of CLs in S. cerevisiae. This is clearly illustrated by the analysis of MLCL molecular species that accumulate in taz1Δ cells, in which 16:1_16:1_16:0, 16:1_16:0_18:1, and 16:1_18:1_18:1 individual species dominated. Only minimal amounts of MLCLs were detected in cld1Δ and cld1Δtaz1Δ mutant cells, reconfirming that the Taz1-catalyzed reaction was the major contributor to the reacylation/remodeling of CLs in S. cerevisiae.
A recent study demonstrated that simultaneous deletion of cld1Δ and taz1Δ provided protection against mitochondrial injury triggered by single deletion of taz1Δ.12 However, molecular speciation of CLs in wt and double mutant cells cld1Δtaz1Δ was markedly different. This suggests that the accumulated MLCL—rather than the changed CL speciation—are the major contributors to mitochondrial dysfunction. Another recent study demonstrated that unremodeled CLs are functionally indistinguishable from remodeled CLs for yeast major bioenergetic functions.2 In contrast, we found that mitochondrial respiration rates were altered in the presence of different FA. Surprisingly, monounsaturated oleic acid had a small inhibitory effect on intact cell respiration, whereas the PUFAs—linoleic acid and arachidonic acid—significantly increased mitochondrial respiration. This may give yeast cells a survival advantage by shifting metabolism from glycolysis to the much more efficient OxPhos process under nonproliferating conditions. Interestingly, CL with four linoleic acid chains is the primary CL species in some mammalian tissues including cow heart,18 a tissue that fully depends on aerobic energy production, suggesting that linoleic acid is the evolutionarily preferred CL side chain to support optimal high levels of oxidative phosphorylation. It should be noted that the effect of the studied FAs on mitochondrial function can be the result of several independent mechanisms. One primary mechanism is likely through changes in CL FA composition (i.e., length and saturation versus unsaturation state), which will affect the structure of the OxPhos complexes. Another mechanism may be through cell signaling. For example, it is known that arachidonic acid, a well-studied signaling molecule in higher organisms, also affects cell signaling in yeast, where it leads to changes in the phosphorylation state of many proteins and in particular protein kinase C (PKC) signaling.19 PKC has been shown to regulate mitochondrial respiration by acting on OxPhos complexes both in mammals and yeast,20 suggesting that cell signaling may contribute to the observed changes in the presence of arachidonic acid.
Interestingly, these general considerations can be applied to specific pathogenic pathways in disease conditions such as, for example, the one likely operating in patients suffering from the X-linked disease Barth syndrome (BTHS), leading to a rare childhood cardiomyopathy with cyclic neutropenia.16 Recent detailed studies have identified seven functional classes of BTHS mutations with several distinct loss-of-function mechanisms leading to a partial or total loss of catalytic activity.21 It has also been demonstrated that accumulation of MLCL (decreased CL/MLCL ratio) rather than deficiency of “correct” unsaturated CL species is the major reason for mitochondrial dysfunction in yeast cells.12 On the basis of our data on the effective remodeling of CLs supplemented with PUFA, one may wonder whether the supplementation with C18:2 may be of therapeutic value, particularly in cases with a partial loss of TAZ catalytic activity.22
It has been long believed that remodeling of PUFA phospholipids is driven, at least in part, by their peroxidation with subsequent hydrolysis of oxidatively modified FA residues and reacylation.23 The results of this work clearly indicate that the remodeling process occurs even in the absence of oxidizable PUFA. This suggests that different mechanisms, independent of peroxidation, may drive the CL recycling process, yielding the molecular species of CL optimized in response to specific cell/tissue demands. Our results are compatible with the dominant role of Taz in reacylation of Cld1-hydrolyzed CLs and explain the high selectivity of the overall remodeling process toward homoacylated 18:2 species of CLs and the role of the tazΔ mutations in the pathogenesis of Barth syndrome. However, further detailed studies of the specificity and the role of PLAs in the reacylation of oxidized vs nonoxidized polyunsaturated CL species may be necessary to better understand these intriguing and important general mechanisms of biological adaptation.
METHODS
Yeast Strains, Media, and Growth Conditions
The Saccharomyces cerevisiae strains used in this work are listed in Table 1. Single deletion mutants were obtained from the yeast knockout deletion collection (Invitrogen). Double mutants were obtained by tetrad dissection. Complex media contained yeast extract (1%), peptone (2%), and glucose (2%; YPD) or glycerol (3%) and ethanol (1%; YPGE). The effect of fatty acids was determined in YPD or YPGE medium supplemented with 0.2% oleic, arachidonic, linoleic, or docosahexaenoic acid solubilized in the media with tergitol (1%). Unsupplemented controls contained only tergitol. Synthetic complete (SC) medium contained all the essential components of Difco yeast nitrogen base, 2% glucose, 0.2% ammonium sulfate, vitamins, adenine (20.25 mg/L), arginine (20 mg/L), histidine (20 mg/L), leucine (60 mg/L), lysine (200 mg/Liter), methionine (20 mg/L), threonine (300 mg/L), tryptophan (20 mg/L), uracil (20 mg/L), and inositol (75 μM). For solid medium, 2% agar was added. For selection of cells carrying plasmids, appropriate amino acids were omitted from the media. Yeast strains were grown at 30 °C. E. coli strain DH5 was used for plasmid maintenance and amplification. Bacteria were grown at 37 °C in LB medium (0.5% yeast extract, 1% tryptone, 1% NaCl), supplemented with ampicillin (100 μg/mL) for selection purposes. Solid medium contained 1.5% agar. Growth in liquid cultures was monitored spectrophotometrically. Cells were harvested and centrifuged at 4 °C (4300 rpm, 5 min). Supernatant was discarded, and glass beads (0.3 g) were added to the centrifuge tubes. Samples were vortexed (5 × 1 min intervals) and frozen at −80 °C for lipidomics analysis.
Table 1.
Strains and Plasmids Used in This Study
| strains/plasmid | characteristics or genotype | source or reference |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Invitrogen |
| cld1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 cld1Δ::KanMX6 | Invitrogen |
| taz1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 taz1Δ::KanMX6 | Invitrogen |
| cld1Δtaz1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 cld1Δ::KanMX6 taz1Δ::KanMX6 | Ye et al.12 |
| pYES2/CT | Multicopy shuttle vector containing URA3 marker | Invitrogen |
| pYES2/CT-CLD1 | Multicopy shuttle vector containing URA3 marker | this study |
Construction of Plasmids and Expression of His6-tagged CLD1 in S. cerevisiae
To construct a His6-tagged CLD1 overexpression plasmid, a 1373-bp sequence containing the entire open reading frame without a stop codon of CLD1 was amplified from yeast genomic DNA using an EcoRI-tagged forward primer CLD1_EcoR-I_F3 (5′-TGATTAATAAGAATTCAACACAATGTTCAA-GTCAACTTTAAACTC-3′) and an XbaI-tagged reverse primer CLD1_XbaI_R3 (5′-ATTTTGAGATTCTAGATATT-TTTTGCATTTCTTTCG-3′). The PCR products were purified using the Wizard SV gel and PCR cleanup system (Promega). The purified DNA fragments were ligated into pYES2/CT cut with EcoRI and XbaI downstream of the GAL1 promoter. All the plasmids were amplified and extracted using standard protocols. The plasmids were transformed into yeast strains using a one-step transformation protocol.
Purification of His6-tagged Cld1
S. cerevisiae strain BY4741 was used to express the recombinant His6-tagged Cld1. Cells were grown at 30 °C in synthetic complete raffinose medium lacking uracil. Galactose (2%) was added to induce expression of the recombinant protein. Cell extracts were prepared by disrupting cells with glass beads (0.5 mm diameter), vortexing for 5 min intermittently in disruption buffer containing 50 mM Tris-Cl (pH 8.0) and 300 mM NaCl. Protease inhibitor (cOmplete EDTA-free, Roche) was added to the disruption buffer before breaking the cells. His6-tagged Cld1 was purified with Millipore PureProteome nickel magnetic beads following the manufacturer’s instructions. Millipore UFC503024 Amicon Ultra-0.5 centrifugal filter concentrator (30 KD) was used to concentrate the protein and remove imidazole. Protein concentration was determined with a BIO-RAD DC protein assay kit using bovine serum albumin as the standard.
LC/MS Assessment of Phospholipids
Lipids were extracted from yeast using the Folch procedure,24 and lipid phosphorus was determined using a micromethod.25 LC/MS of phospholipids was performed in negative mode using a Dionex UltimateTM 3000 RSLCnano system coupled online Q-Exactive hybrid quadrupole-orbitrap mass spectrometer (ThermoFisher Scientific, San Jose, CA). Total lipids were separated on a normal phase column (Silica Luna 3 μm, 100A, 150 × 2 mm, (Phenomenex, Torrance CA)) with a flow rate of 0.2 mL/min using gradient solvents containing 5 mM CH3COONH4 (A, n-hexane/2-propanol/water, 43:57:1 (v/v/v) and B, n-hexane/2-propanol/water, 43:57:10 (v/v/v)). The gradient conditions (all linear) were as follows: 0–23 min (10% B to 32% B); 23–32 min (32% B to 65% B); 32–35 min (65% B to 100% B); 35–62 min (hold at 100% B); 62–64 min (100% B to 10% B); 64–80 min (10% B). Flow rate was maintained at 200 μL/min except for the 35–62 min time frame where the flow rate was increased to 225 μL/min. MS analysis was performed in negative ion mode (profile) at a resolution of 140 000 for the full MS scan and 17 500 for the MS2 scan in a data-dependent mode with appropriate inclusion and exclusion lists. The scan range for MS analysis was 400–1800 m/z with a maximum injection time of 128 ms using one microscan. A maximum injection time of 500 ms was used for MS2 (high energy collisional dissociation (HCD)) analysis with collision energy set to 24. An isolation window of 1.0 Da was set for the MS2 scans. Capillary spray voltage was set at 3.5 kV, and capillary temperature was 320 °C. The S-lens Rf level was set to 60 with a sheath gas flow of 8. To quantitatively assess phospholipids, PE-(17:0/17:0), PC-(17:0/17:0), CL-(14:0/14:0/14:0/14:0), LPE-(17:1/0:0), and LPC-(17:1/0:0) from Avanti Polar Lipids were used as internal standards. Reference standards PE-(18:1/18:1), PC-(18:1/18:1), CL-(18:2/18:2/18:2/18:2), LPE-(18:0/0:0), and LPC-(18:0/0:0) were used for calibrations and were also from AvantiPolar Lipids. Both internal and reference standards of MLCLs were prepared from CL-(14:0/14:0/14:0/14:0) and CL-(18:2/18:2/18:2/18:2), respectively, as described.26 Precursor ions [M–H] were identified with a mass accuracy less than 5 ppm. Therefore, phospholipids, including CL, were identified by accurate mass and MS2 fragmentation analysis. The fragmentation pattern provided information on fatty acids but not on their position. Assuming that the sn-1 position in phospholipids is commonly occupied by saturated and monounsaturated acids, we used this information to identify PE and PC molecular species. Clearly, this is a more difficult task for CL molecular species containing four acyl chains. Therefore, we presented CL molecular species based on the information provided in the paper by Liebisch et al.27 Spectra subtraction was done using the Background Subtraction Module in Xcalibur (Thermo Fisher Scientific). By defining one file as background (subtractor) and another file as foreground (minuend), the difference can be evaluated from the differential spectra. Since negative spectra cannot be calculated and exported from Xcalibur, we performed swapping of the subtractor and the minuend, which afforded the calculations and presentation of the negative spectra.
Respiration Measurements
The oxygen consumption rate (ORC) of intact yeast cells was analyzed via the polarographic method in a closed 500 μL chamber equipped with a micro Clark-type oxygen electrode (Oxygraph Plus System, Hansatech Instruments) at 30 °C. Yeast cells were grown in synthetic YPD media supplemented with oleic acid, linoleic acid, or arachidonic acid. Cells were harvested during the stationary phase. Cells were mixed in fresh media using a final protein concentration of 0.5 mg mL−1 following measurements of respiration. KCN (0.2 mM) was added at the end of the experiment to inhibit cytochrome c oxidase to correct for cytochrome c oxidase-independent oxygen utilization. Oxygen consumption was recorded on a computer and analyzed with the Oxygraph plus software. Oxygen consumption rate (ORC) is defined as consumed O2 (nmol)/min/total protein (mg).
Synthesis of Cardiolipins
General Experimental Conditions
All air- and moisture-sensitive reactions were carried out under a nitrogen or argon atmosphere. All reactions carried out above RT were performed using an oil bath set to the specified temperature and monitored with an external thermometer. THF and diethyl ether were distilled from sodium/benzophenone ketyl. Dichloromethane was distilled from CaH2. Diisopropylethylamine (DIPEA) and diisopropylamine were distilled from and stored over KOH. Pyridine was filtered through activated basic alumina and stored over freshly activated 4 Å molecular sieves for at least 24 h prior to use. Amberlite-IR120H resin was activated by stirring with 6 N HCl for 30 min, filtering, and rinsing once with equal volumes of water and methanol. All other reagents were used as received. Concentrating under reduced pressure refers to removing solvents by the use of a rotary evaporator connected to a PIAB Lab Vac H40. Reactions were monitored by thin layer chromatography analysis (precoated silica gel 60 F254 plates, 250 μm layer thickness) and visualized with a 254 nm UV light or by staining with a KMnO4 solution. Flash chromatography on SiO2 (Silicycle, 40–63 μm) was used for purification where indicated. All products were placed under a high vacuum to remove trace solvents. Melting points were determined using a Laboratory Devices Mel-Temp II and are uncorrected. Infrared spectra were obtained from neat solids or oils on a PerkinElmer ATR IR or Smiths Detection IdentifyIR FT-IR spectrometer. High-resolution mass spectra were obtained on a Thermo Scientific Exactive Orbitrap mass spectrometer using electrospray ionization. Low-resolution mass spectra were obtained on an Advion Expression L compact mass spectrometer using electrospray ionization. 1H NMR spectra were obtained on a Bruker Avance at 300, 400, or 500 MHz in CDCl3, CD2Cl2, or CD3OD. Chemical shifts (δ) were reported in parts per million with the residual solvent peak used as an internal standard δ 1H/13C (solvent): 7.26/77.00 (CDCl3) and 5.32/53.84 (CD2Cl2). 1H NMR spectra were obtained and are tabulated as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, m = multiplet, bs = broad singlet), number of protons, and coupling constant(s). 13C NMR spectra were recorded using a proton-decoupled pulse sequence run at 100 or 125 MHz and are tabulated by observed peaks. 31P NMR spectra were recorded using a complete decoupled pulse sequence run at 162 MHz.
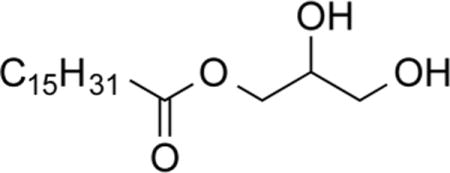
1-O-Palmitoylglycerol (1a)28
General Procedure A
A solution of palmitic acid (5.5 g, 21.4 mmol, 1.0 equiv), 50% glycidol in CH2Cl2 (3.0 mL, 22.8 mmol, 1.06 equiv), and tributylamine (0.1 mL, 0.4 mmol, 0.02 equiv) was heated to 85 °C and stirred for 5 h. The crude product was recrystallized from i-Pr2O:Et2O (1:1) to yield 1-O-palmitoylglycerol (1a, 4.88 g, 14.8 mmol, 69%) as a colorless solid. Mp: 71.9–72.5 °C. IR: 3293, 2913, 2848, 1728 cm−1. 1H NMR (CDCl3, 300 MHz): δ 4.22 (dd, 1 H, J = 4.8, 11.7 Hz), 4.15 (dd, 1 H, J = 6.0, 11.4 Hz), 3.97–3.90 (m, 1 H), 3.70 (dd, 1 H, J = 4.2, 11.7 Hz), 3.60 (dd, 1 H, J = 5.7, 11.4 Hz), 2.35 (t, 2 H, J = 7.7 Hz), 1.66–1.58 (m, 2 H), 1.28–1.25 (m, 25 H), 0.85 (t, 3 H, J = 6.8 Hz). 13C NMR (CDCl3, 100 MHz): δ 174.4, 70.3, 65.2, 63.3, 34.1, 31.9, 29.7–29.6 (multiple peaks), 29.4, 29.3, 29.1, 24.9, 22.7, 14.1.
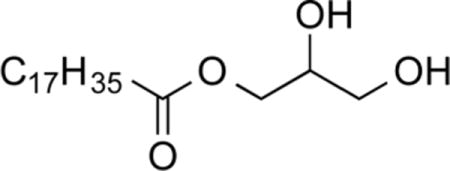
1-O-Stearoylglycerol (1b)28
Following General Procedure A but using stearic acid, 1-O-stearoylglycerol (1b, 4.36 g, 12.2 mmol, 71%) was obtained as a colorless solid. 1H NMR (CDCl3, 300 MHz): δ 4.21 (dd, 1 H, J = 4.8, 11.7 Hz), 4.14 (dd, 1 H, J = 5.7, 11.4 Hz), 3.96–3.90 (m, 1 H), 3.70 (dd, 1 H, J = 3.9, 11.4 Hz), 3.60 (dd, 1 H, J = 6.0, 11.4 Hz), 2.71 (bs, 2 H), 2.35 (t, 2 H, J = 7.7 Hz), 1.65–1.60 (m, 2 H), 1.28–1.25 (m, 29 H), 0.88 (t, 3 H, J = 6.6 Hz).
1,2-O-Dipalmitoyl-3-O-dimethoxytritylglycerol (2a)
General Procedure B
To a solution of 1a (4.57 g, 13.8 mmol, 1.0 equiv) in pyridine/CH2Cl2 (1:2, 35 mL) at 0 °C was added dimethoxytrityl chloride (DMTCl, 4.82 g, 14.1 mmol, 1.0 equiv) and dimethylaminopyridine (DMAP, 0.177 g, 1.43 mmol, 0.1 equiv). The orange solution was stirred at this temperature for 15 min, then warmed to RT and stirred for 13 h. The orange reaction mixture was quenched with water (30 mL) and extracted with CH2Cl2 (2 × 50 mL). The organic portion was rinsed with water (40 mL) and brine (40 mL), dried (MgSO4), and concentrated. The residue was then concentrated from toluene (2 × 20 mL) and the resulting orange oil used without further purification. A solution of 1-ethyl-3-(3-(dimethylamino)-propyl)carbodiimide (EDCI, 2.93 g, 15.3 mmol, 1.1 equiv), palmitic acid (3.91 g, 15.3 mmol, 1.1 equiv), and DMAP (0.851 g, 6.90 mmol, 0.5 equiv) in CH2Cl2 (60 mL) was allowed to stir at RT under nitrogen for 15 min. Then, a solution of the previously obtained oil from the dimethoxytrityl protection (8.77 g, 13.9 mmol 1.0 equiv) in CH2Cl2 (30 mL) was added, and stirring was continued for 4 h. The reaction mixture was diluted with water (50 mL), and the organic layer was rinsed with water (50 mL) and brine (50 mL), dried (MgSO4), and concentrated. Purification by filtration through SiO2 (9:1 CH2Cl2/hexanes) gave 1,2-O-dipalmitoyl-3-O-dimethoxytritylglycerol (2a, 7.94 g, 9.11 mmol, 66%) as a colorless solid. Pure 2a can also be obtained by repeated trituration with MeOH. Mp: 64.5–66.0 °C. IR: ν 2915, 2846, 1729 cm−1. 1H NMR (CDCl3, 300 MHz): δ 7.42 (d, 2.0 H, J = 2 Hz), 7.40–7.20 (m, 7 H), 6.82 (d, 4 H, J = 8.7 Hz), 5.28–5.22 (m, 1 H), 4.34 (dd, 1 H, J = 3.6, 11.7 Hz), 4.25 (dd, 1 H, J = 6.6, 11.7 Hz), 3.79 (s, 6 H), 3.22 (d, 2 H, J = 5.1 Hz), 2.33 (t, 2 H, J = 7.5 Hz), 2.23 (t, 2 H, J = 7.5 Hz), 1.65–1.53 (m, 4 H), 1.28–1.25 (m, 48 H), 0.91 (t, 3 H, J = 6.6 Hz). 13C NMR (CDCl3, 125 MHz): δ 173.4, 173.0, 158.5, 144.5, 135.7, 130.0, 128.1, 127.8, 126.8, 113.1, 86.0, 70.5, 62.9, 62.0, 55.1 34.4, 34.1, 31.9, 29.7–29.1 (multiple peaks), 25.0, 24.8, 22.7, 14.1.
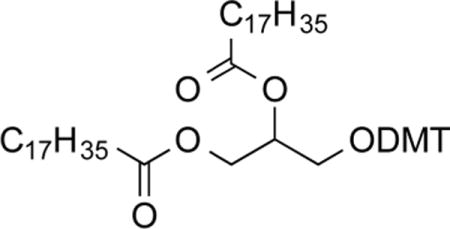
1,2-O-Distearoyl-3-O-dimethoxytritylglycerol (2b)
Following General Procedure B but using 1b and purification by trituration with MeOH gave 1,2-O-distearoyl-3-O-dimethoxytritylglycerol (2b, 6.68 g, 6.48 mmol, 53%) as a colorless solid. Mp: 67–68.4 °C. IR: ν 2915, 2848, 1730 cm−1. 1H NMR (CDCl3, 300 MHz): δ 7.45 (d, 2 H, J = 7.2 Hz), 7.35–7.24 (m, 7 H), 6.85 (d, 4 H, J = 9.0 Hz), 5.29–5.28 (m, 1 H), 4.35 (dd, 1 H, J = 8.1, 11.7 Hz), 4.25 (dd, 1 H, J = 6.6, 11.7 Hz), 3.82 (s, 6 H), 3.25 (d, 2 H, J = 3.3 Hz), 2.36 (t, 2 H, J = 7.5 Hz), 2.27 (t, 2 H, J = 7.5 Hz), 1.69–1.56 (m, 4 H), 1.29 (m, 58 H), 0.91 (t, 3 H, J = 6.6 Hz). 13C NMR (CDCl3, 125 MHz): δ 173.4, 173.0, 158.5, 144.6, 135.8, 130.1, 128.1, 127.8, 126.8, 113.1, 113.0, 86.1, 70.5, 62.9, 62.1, 55.2, 34.4, 34.1, 31.9, 29.7–29.1 (multiple peaks), 25.0, 24.8, 22.7, 14.1.
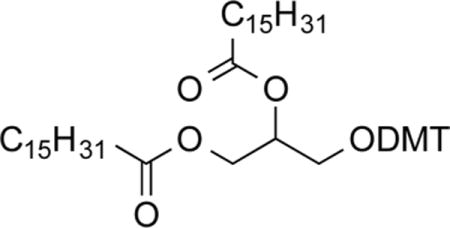
1-O-Palmitoyl-2-O-palmitoleoyl-3-O-dimethoxytritylglycerol (2c)
Following General Procedure B but using 1a and purification by chromatography on SiO2 (95:5, hexanes/ethyl acetate) gave 1-O-palmitoyl-2-O-palmitoleoyl-3-O-dimethoxytritylglycerol (2c, 0.619 g, 0.712 mmol, 65%) as a clear colorless oil. IR: ν 2923, 2853, 2093, 1741, 1250 cm−1. 1H NMR (CDCl3, 300 MHz): δ 7.43 (d, 2 H, J = 8.4 Hz), 7.31–7.25 (m, 7 H), 6.82 (d, 4 H, J = 8.7 Hz), 5.40–5.30 (m, 2 H), 5.29–5.22 (m, 1 H), 4.34 (dd, 1 H, J = 3.6, 11.7 Hz), 4.23 (dd, 1 H, J = 6.6, 12.0 Hz), 3.79 (s, 6 H), 3.23–3.21 (m, 2 H), 2.33 (t, 2 H, J = 7.5 Hz), 2.23 (t, 2 H, J = 7.7 Hz), 2.02–2.00 (m, 4 H), 1.67–1.53 (m, 4 H), 1.29 (m, 40 H), 0.88 (t, 6 H, J = 6.8 Hz). 13C NMR (CDCl3, 125 MHz): δ 173.4, 173.0, 158.5, 144.6, 135.8, 135.7, 130.0, 129.7 128.1, 127.8, 126.8, 113.1, 86.1, 70.5, 62.9, 62.0, 55.2, 34.4, 34.1, 31.9, 31.8, 29.8–29.0 (multiple peaks), 27.2, 25.0, 24.9, 22.7, 22.6, 14.1.
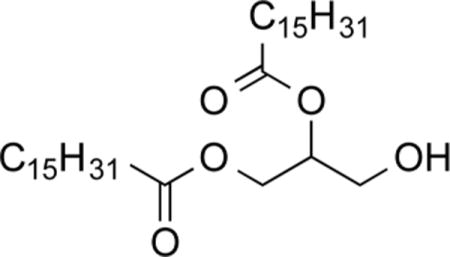
1,2-O-Dipalmitoylglycerol (3a)29
General Procedure C
To a solution of 2a (7.94 g, 9.11 mmol, 1.0 equiv) in CHCl3/MeOH (1:1, 90 mL) was added Amberlite IR-120H (4.0 g). The reaction mixture was stirred for 2.5 h, then filtered through basic alumina (EtOAc). The resulting yellow oil was precipitated from Et2O/hexanes (3:1) and filtered to give 1,2-O-dipalmitoylglycerol (3a, 3.75 g, 5.50 mmol, 60%, purity by 1H NMR: 90%) as a colorless solid. 1H NMR (CDCl3, 400 MHz): δ 5.10–5.06 (m, 1 H), 4.32 (dd, 1 H, J = 4.8, 12.0 Hz), 4.24 (dd, 1 H, J = 6.0, 12.0 Hz), 3.74–3.73 (m, 2 H), 2.33 (apparent q, 4 H, J = 8 Hz), 1.65–1.58 (m, 4 H), 1.28–1.25 (m, 48 H), 0.88 (t, 3 H, J = 6.6 Hz).
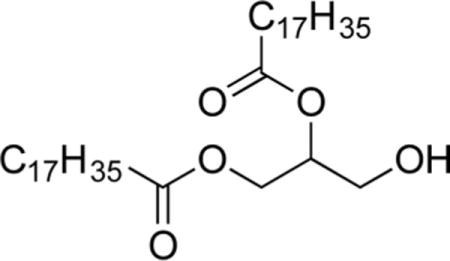
1,2-O-Distearoylglycerol (3b).30
Following General Procedure C but using 2b and purification by trituration with Et2O/hexanes (3:1) gave 1,2-O-distearoylglycerol (3b, 2.08 g, 3.39 mmol, 72%) as a colorless solid. 1H NMR (CDCl3, 500 MHz): δ 5.10–5.06 (m, 1 H), 4.32 (dd, 1 H, J1 = 4.5 Hz, J2 = 12 Hz), 4.24 (dd, 1 H, J1 = 5.5 Hz, J2 = 12 Hz), 3.79–3.72 (m, 2 H), 2.34 (m, 4 H), 1.99 (t, 1 H, J = 4.0 Hz), 1.64–1.54 (m, 4 H), 1.29–1.25 (m, 56 H), 0.88 (t, 3 H, J = 7.0 Hz).
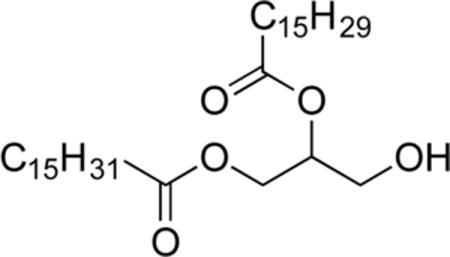
1-O-Palmitoyl-2-O-palmitoleoylglycerol (3c)
Following General Procedure C but using 2c and purification by chromatography on SiO2 (1:0 to 5:1, CH2Cl2/ethyl acetate) gave 1-O-palmitoyl-2-O-palmitoleoylglycerol (3c, 64 mg, 0.113 mmol, 98%) as a light yellow oil. IR: ν 3502, 2919, 2850, 2093, 1737, 1163 cm−1. 1H NMR (CDCl3, 300 MHz): δ 5.40–5.30 (m, 2 H), 5.10–5.05 (m, 1 H), 4.32 (dd, 1 H, J = 4.8, 12.3 Hz), 4.24 (dd, 1 H, J = 5.4, 11.7 Hz), 3.73 (m, 2 H), 2.33 (apparent q, 4 H, J = 7.3 Hz), 2.02–2.00, (m, 4 H), 1.64–1.54 (m, 4 H), 1.30–1.27 (m, 40 H), 0.88 (t, 6 H, J = 6.7 Hz). 13C NMR (CDCl3, 125 MHz): δ 173.8, 173.4, 130.0, 129.7 128.1, 72.1, 62.0, 61.6, 34.3, 34.1, 31.9, 29.7–29.0 (multiple peaks), 27.2, 25.0, 24.9, 22.7, 22.6, 14.1. HRMS (ESI+) m/z calculated for C35H67O5 [M + H]: 567.4983. Found: 567.4960.
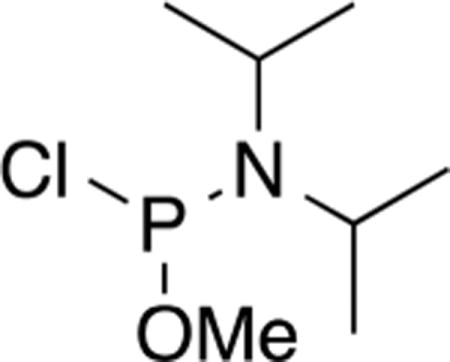
N,N-Diisopropylamidomethylchlorophosphite (4).31
To a solution of PCl3 (3.2 mL, 36 mmol, 1.04 equiv) and pyridine (2.9 mL, 36 mmol, 1.04 equiv) in Et2O (120 mL) at −78 °C under argon was added dry MeOH (1.4 mL, 35 mmol, 1.0 equiv) in Et2O (4 mL) over 30 min. The reaction mixture was stirred for 3 h at RT, diluted with Et2O (40 mL), and cooled to 0 °C. Diisopropylamine (9.6 mL, 67.9 mmol, 2.0 equiv) was added over 0.5 h, and stirring was continued for 22 h at RT. The salts were filtered off under argon and rinsed with Et2O (50 mL), and the solvent was distilled off. The resulting yellow oil was purified by vacuum distillation to give N,N-diisopropylamidomethylchlorophosphite (4, 3.73 g, 18.9 mmol, 56%) as a clear colorless oil. Bp: 88 °C (12 Torr). 1H NMR (CD2Cl2, 400 MHz): δ 3.74–3.64 (m, 2 H), 3.51 (d, 3 H, J = 14.4 Hz), 1.20–1.16 (m, 12 H). 31P NMR (CD2Cl2, 162 MHz): δ 183.8.
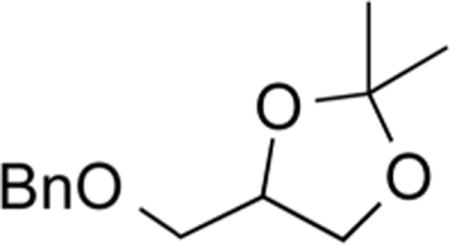
4-((Benzyloxy)methyl)-2,2-dimethyl-1,3-dioxolane (5).30
A slurry of 60% NaH in mineral oil (360 mg, 9.00 mmol, 1.2 equiv) in THF (10 mL) at 0 °C under nitrogen was added to a solution of 2,2-dimethyl-1,3-dioxolane-4-methanol (1.0 mL, 7.8 mmol, 1.0 equiv) in THF (5 mL). The reaction mixture was stirred for 30 min; then BnBr (0.98 mL, 1.4 mmol, 1.05 equiv) was added in a dropwise fashion. The reaction mixture was warmed to RT and stirred for 2.5 h. The reaction was quenched with water (8 mL) and diluted with Et2O (20 mL), and the layers were separated. The aqueous layer was extracted with Et2O (25 mL), and the combined organic layers were rinsed with brine (10 mL), dried (MgSO4), and concentrated. Purification by chromatography on SiO2 (95:5 to 9:1 to 85:15, hexanes/ethyl acetate) gave 4-((benzyloxy)methyl)-2,2-dimethyl-1,3-dioxolane (5, 1.15 g, 5.17 mmol, 67%) as a clear colorless oil. 1H NMR (CDCl3, 400 MHz): δ 7.35–7.29 (m, 5 H), 4.59, 4.56 (AB, 2 H, J = 12.0 Hz), 4.34–4.28 (m, 1 H), 4.06 (dd, 1 H, J = 6.4, 8.0 Hz), 3.75 (dd, 1 H, J = 6.4, 8.4 Hz), 3.56, 3.45 (ABd, 2 H, J = 5.6, 9.6 Hz), 3.46 (dd, 1 H, J = 5.6, 9.6 Hz), 1.42 (s, 3 H), 1.37 (s, 3 H).
1-O-Benzyl-2,3-O-bis(tert-butyldimethylsilyl)glycerol (6)
A solution of 5 (1.15 g, 5.17 mmol, 1.0 equiv) in THF/H2O (2:1, 10 mL) was treated with 12 M HCl (2.1, 25 mmol, 4.9 equiv), and the reaction mixture was stirred for 1 h at RT, poured into saturated NaHCO3 (30 mL), and extracted with Et2O (3× 25 mL). The combined organic layers were rinsed in brine (25 mL), dried (MgSO4), and concentrated. The resulting yellow oil was dissolved in CH2Cl2 (8.7 mL) at 0 °C under nitrogen and treated with Et3N (1.4 mL, 9.9 mmol, 2.3 equiv) and TBSOTf (2.2 mL, 9.5 mmol, 2.2 equiv). The cloudy white reaction mixture was then stirred for 3 h at RT, quenched with H2O (8 mL), and diluted with CH2Cl2 (10 mL). The organic layer was rinsed with brine (8 mL), dried (MgSO4), and concentrated. Purification by chromatography on SiO2 (99:1 to 95:5, hexanes:ethyl acetate) gave 1-O-benzyl-2,3-O-bis(tert-butyldimethylsilyl)glycerol (6, 1.72, 4.31 mmol, 83% over 2 steps) as a clear colorless oil. 1H NMR (CDCl3, 500 MHz): δ 7.35–7.33 (m, 4 H), 7.28–7.26 (m, 1 H), 4.54 (s, 2 H), 3.89–3.84 (m, 1 H), 3.61 (dd, 1 H, J = 6.0, 10.0 Hz), 3.57–3.52 (m, 2 H), 3.42 (dd, 1 H, J = 5.5, 10.0 Hz), 0.87 (s, 18 H), 0.070 (d, 6 H, J = 1.5 Hz), 0.042 (s, 6 H).
2,3-O-Bis(tert-butyldimethylsilyl)glycerol (7).32
To a solution of 6 (1.62 g, 4.06 mmol, 1.0 equiv) in ethyl acetate (20 mL) was added 10% Pd/C (0.161 g, 0.152 mmol, 0.037 equiv). The reaction mixture was submitted to hydrogenation on a Parr hydrogenator under H2 (5 atm) for 1 h, filtered through Celite (ethyl acetate), and concentrated to give 2,3-O-bis(tert-butyldimethylsilyl)glycerol (7, 1.15 g, 3.59 mmol, 88%) as a clear colorless oil that was used without further purification. An analytically pure sample was obtained by chromatography on SiO2 (95:5 to 9:1, hexanes/ethyl acetate). 1H NMR (CDCl3, 300 MHz): δ 3.79–3.74 (m, 1 H), 3.69–3.53 (m, 4 H), 2.10 (bs, 1 H), 0.90 (s, 18 H), 0.1–0.06 (m, 12 H).
3-(((2,3-Bis((tert-butyldimethylsilyl)oxy)propoxy)(methoxy)-phosphoryl)oxy)propane-1,2-diyl Dipalmitate (8a)7
General Procedure D
To a solution of 3a (1.73 g, 2.89 mmol, 1.0 equiv) and diisopropylethylamine (DIPEA, 0.53 mL, 3.2 mmol, 1.1 equiv) in CH2Cl2 (30 mL) under nitrogen was added 7 (6.0 mL, 3.0 mmol, 1.1 equiv). The reaction mixture was stirred for 1 h and treated dropwise with a solution of 6 (0.921 g, 2.87 mmol, 0.99 equiv) and 4,5-dicyanoimidazole (0.700 g, 5.80 mmol, 2 equiv) in THF (4.9 mL). The solution was stirred for 1 h, cooled to 0 °C, and treated with Bu4NIO4 (1.46 g, 3.36 mmol, 1.2 equiv) and stirring was continued for 30 min. The mixture was diluted with water (15 mL), and the combined organic layers were rinsed with saturated Na2S2O3 (15 mL), saturated NaHCO3 (15 mL), and brine (15 mL); dried (MgSO4); and concentrated. The crude residue was filtered through a plug of SiO2 (1:1, hexanes/ethyl acetate) and concentrated. Purification by chromatography on SiO2 (5:1 to 3:1, hexanes/ethyl acetate) gave 3-(((2,3-bis((tert-butyldimethylsilyl)oxy)propoxy)(methoxy)-phosphoryl)oxy)propane-1,2-diyl dipalmitate (8a, 1.42 g, 1.47 mmol, 51%) as a clear colorless oil. IR: ν 2922, 2854, 1741, 1252, 835 cm−1. 1H NMR (CDCl3, 300 MHz): δ 5.25–5.19 (m, 1 H), 4.36–4.30 (m, 1 H), 4.19–4.09 (m, 4 H), 3.97–3.83 (m, 2 H), 3.76 (d, 3 H, J = 11.1 Hz; diasteromers), 3.56–3.54 (m, 2 H), 2.34–2.27 (m, 4 H), 2.23 (t, 2 H, J = 7.5 Hz), 1.61–1.56 (m, 4 H), 1.28–1.25 (m, 48 H), 0.88–0.85 (m, 24 H), 0.08 (s, 3 H), 0.07 (s, 3 H), 0.05 (s, 6 H). 13C NMR (CDCl3, 125 MHz): δ 173.2, 172.7, 72.0 (d, J = 7.5 Hz), 69.4 (d, J = 7.5 Hz), 69.1 (d, J = 6.5 Hz), 65.4–65.3 (multiple peaks), 64.0, 61.6, 55.4 (d, J = 6.3 Hz) 34.1, 34.0, 31.9, 31.6, 29.7–29.1 (multiple peaks), 25.8, 25.7, 24.8, 22.6, 18.3, 18.1,14.1, −4.7, −4.8, −5.5. HRMS (ESI+) m/z calculated for C51H106O10PSi2 [M + H]: 965.7057. Found: 965.7086.
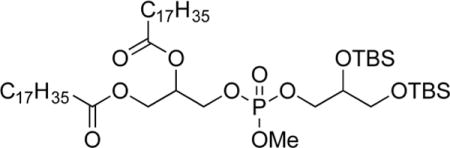
3-(((2,3-Bis((tert-butyldimethylsilyl)oxy)propoxy)(methoxy)-phosphoryl)oxy)propane-1,2-diyl Distearate (8b).32
Following General Procedure D but using 3b and purification by chromatography on SiO2 (5:1, hexanes/ethyl acetate) gave 3-(((2,3-bis((tert-butyldimethylsilyl)oxy)propoxy)(methoxy)phosphoryl)oxy)propane-1,2-diyl distearate (8b, 0.605 g, 0.592 mmol, 67%) as a waxy colorless solid. Mp: 31.0–31.9 °C. IR: ν 2923, 2852, 1743, 1252, 835 cm−1. 1H NMR (CDCl3, 300 MHz): δ 5.24–5.20 (m, 1 H), 4.37–4.31 (m, 1 H), 4.22–4.09 (m, 4 H), 3.97–3.84 (m, 2 H), 3.76 (d, 3 H, J = 11.1 Hz; diasteromers), 3.57–3.54 (m, 2 H), 2.31 (apparent q, 4 H, J = 7.0 Hz), 1.61–1.57 (m, 4 H), 1.28–1.25 (m, 56 H), 0.88–0.86 (m, 24 H), 0.09 (s, 3 H), 0.08 (s, 3 H), 0.05 (s, 6 H). 13C NMR (CDCl3, 125 MHz): δ 173.2, 172.2, 72.0 (d, J = 8.8 Hz), 69.4 (d, J = 7.5 Hz), 69.2–69.1 (m), 65.4–65.3 (m), 64.0, 61.6, 54.4 (d, J = 5.0 Hz), 34.2, 34.0, 31.9, 29.7–29.1 (multiple peaks), 25.9, 25.7, 24.8, 22.7, 22.6, 18.3, 18.1,14.1, −4.7, −4.8, −5.4, −5.5. HRMS (ESI+) m/z calculated for C55H114O10PSi2 [M + H]: 1021.7688. Found: 1021.7766.
3-(((2-((tert-Butyldimethylsilyl)oxy)-3-hydroxypropoxy)-(methoxy)phosphoryl)oxy) Propane-1,2-diyl Dipalmitate (9a)
General Procedure E
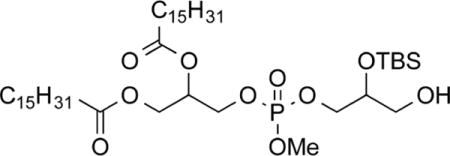
To a solution of 8a (800 mg, 0.829 mmol, 1.0 equiv) in CH2Cl2/MeOH (1:1, 8.3 mL) at 0 °C was added camphorsulfonic acid (195 mg, 0.823 mmol, 1.0 equiv). The reaction mixture was maintained between −5 and 5 °C and stirred for 5.5 h. The solution was filtered through a small plug of basic alumina (ethyl acetate) and concentrated. Purification by chromatography on SiO2 (3:1 to 1:1, hexanes/ethyl acetate) gave 3-(((2-((tert-butyldimethylsilyl)oxy)-3-hydroxypropoxy)(methoxy)phosphoryl)-oxy)propane-1,2-diyl dipalmitate (9a, 435 mg, 0.511 mmol, 62%) as a colorless solid. Mp: 27.8–28.0 °C. IR: ν 2922, 2854, 1741, 1252, 835 cm−1. 1H NMR (CDCl3, 400 MHz): δ 5.29–5.20 (m, 1 H), 4.33 (dd, 1 H, J = 4.4, 12.0 Hz), 4.21–4.14 (m, 3 H), 4.06–4.01 (m, 2 H), 3.93–3.89 (m, 1 H), 3.80–3.76 (m, 3 H), 3.67–3.50 (m, 2 H), 2.35–2.31 (m, 5 H), 1.62–1.60 (m, 4 H), 1.28–1.25 (m, 48 H), 0.90–0.86 (m, 15 H), 0.11 (s, 6 H). 13C NMR (CDCl3, 100 MHz): δ 173.2, 172.8, 71.1 (d, J = 7.0 Hz), 69.4–69.3 (multiple peaks), 67.7 (m), 65.6–65.5 (multiple peaks), 63.0, 61.6, 54.6 (multiple peaks), 34.1, 34.0, 31.9, 29.7–29.1 (multiple peaks), 25.7, 24.8, 22.7, 18.0, 14.1, −4.8, −4.9. HRMS (ESI+) m/z calculated for C45H92O10PSi [M + H]: 851.6192. Found: 851.6180.
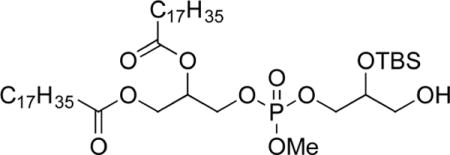
3-(((2-((Tert-Butyldimethylsilyl)oxy)-3-hydroxypropoxy)-(methoxy)phosphoryl)oxy) Propane-1,2-diyl distearate (9b)
Following General Procedure E but using 8b and purification by chromatography on SiO2 (4:1 to 1:1, hexanes/ethyl acetate) gave 3-(((2-((tert-butyldimethylsilyl)oxy)-3-hydroxypropoxy)(methoxy)-phosphoryl)oxy)propane-1,2-diyl distearate (9b, 470 mg, 0.518 mmol, 55%) as a waxy colorless solid. Mp: 41.2–41.7 °C. IR: ν 3535, 2921, 2850, 1739, 733 cm−1. 1H NMR (CDCl3, 300 MHz): δ 5.25–5.22 (m, 1 H), 4.34 (dd, 1 H, J = 4.2, 12.0 Hz), 4.19–4.11 (m, 3 H), 4.05–4.02 (m, 2 H), 3.95–3.90 (m, 1 H), 3.78 (d, 3 H, J = 11.4 Hz; diasteromers), 3.67–3.60 (m, 2 H), 2.36–2.29 (m, 5 H), 1.62–1.58 (m, 4 H), 1.28–1.25 (m, 56 H), 0.90–0.86 (m, 15 H), 0.11 (s, 6 H). 13C NMR (CDCl3, 125 MHz): δ 173.3, 172.8, 71.1 (d, J = 7.1 Hz), 69.4 (multiple peaks), 67.7 (multiple peaks), 65.6 (multiple peaks), 63.0, 61.6, 54.6 (multiple peaks), 34.2, 34.0, 31.9, 29.7–29.1 (multiple peaks), 25.7, 24.8, 22.7, 18.0, 14.1, −4.7, −4.9. HRMS (ESI+) m/z calculated for C49H100O10PSi [M + H]: 907.6818. Found: 907.6785.
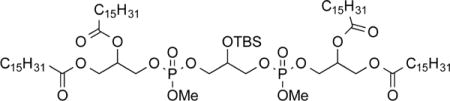
3-(((3-(((2,3-(Palmitoyloxy)propoxy)(methoxy)phosphoryl)oxy)-2-((tert-butyldimethylsilyl)oxy)propoxy)(methoxy)phosphoryl)oxy)-propane-1,2-diyl dipalmitate (10a).32
To a solution of 9a (322 mg, 0.378 mmol, 1.0 equiv) in CH2Cl2 (3.7 mL) at 0 °C under nitrogen was added DIPEA (0.069 mL, 0.42 mmol, 1.1 equiv) and a 0.5 M solution of 7 in CH2Cl2 (0.83 mL, 0.42 mmol, 1.1 equiv). The reaction mixture was warmed to RT, stirred for 1.5 h, and treated with a solution of 3a (364 mg, 0.416 mmol, 1.1 equiv) and 4,5-dicyanoimidazole (90 mg, 0.76 mmol, 2.0 equiv) in THF (1.5 mL) in a drowpwise fashion. The reaction mixture was stirred for 2 h, cooled to 0 °C, treated with Bu4NIO4 (186 mg, 0.429 mmol, 1.1 equiv), and stirred for 40 min. The mixture was diluted with water (3 mL) and CH2Cl2 (5 mL), and the organic layer was rinsed with saturated Na2S2O3 (3 mL) and brine (3 mL), dried (MgSO4), and concentrated. The crude product was filtered through a plug of SiO2 (1:1 hexanes/ethyl acetate) and concentrated. Purification by chromatography on SiO2 (3:1 to 2:1 to 1:1, hexanes/ethyl acetate) gave 3-(((3-(((2,3-(palmitoyloxy)propoxy)(methoxy)phosphoryl)-oxy)-2-((tert-butyldimethylsilyl)oxy)propoxy)(methoxy)phosphoryl)-oxy)propane-1,2-diyl dipalmitate (10a, 381 mg, 0.255 mmol, 67%) as a colorless solid. Mp: 36.5–37.5 °C. IR: ν 2913, 2848, 1737, 1465, 913, 744 cm−1. 1H NMR (CDCl3, 400 MHz): δ 5.25–5.22 (m, 2 H), 4.36–4.31 (m, 2 H), 4.22–4.12 (m, 6 H), 4.05–3.97 (m, 5 H), 3.78 (d, 6 H, J = 11.2 Hz; diasteromers), 2.32 (apparent q, 8 H, J = 7.9 Hz), 1.61–1.57 (m, 8 H), 1.28–1.25 (m, 96 H), 0.89–0.86 (m, 21 H), 0.11 (s, 6 H). 13C NMR (CDCl3, 125 MHz): δ 173.1, 172.7, 69.8 (multiple peaks), 69.4 (d, J = 6.3 Hz), 67.8 (2, J = 6.3 Hz), 65.7 (multiple peaks), 61.6, 54.6 (d, J = 6.3 Hz), 34.2, 34.0, 31.9, 29.7–29.1 (multiple peaks), 25.6, 24.9, 22.7, 18.0, 14.1, −4.9. HRMS (ESI+) m/z calculated for C81H161O17P2Si [M + H]: 1496.0973. Found: 1496.0903.
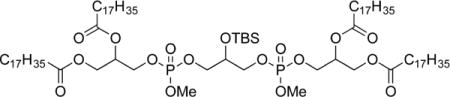
3-(((3-(((2,3-(Stearoyloxy)propoxy)(methoxy)phosphoryl)oxy)-2-((tert-butyldimethylsilyl)oxy)propoxy)(methoxy)phosphoryl)oxy)-propane-1,2-diyl distearate (10b)
General Procedure F.33
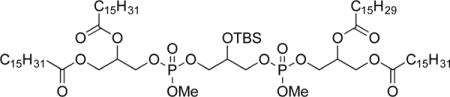
To a solution of 3b (356 mg, 0.570 mmol, 1.0 equiv) and DIPEA (0.10 mL, 0.63 mmol, 1.1 equiv) under nitrogen was added a 0.5 M solution of 7 (1.2 mL, 0.63 mmol, 1.1 equiv) in CH2Cl2. The reaction was stirred for 1 h and treated drowpwise with a solution of 9b (470 mg, 0.518 mmol, 1.0 equiv) and 4,5-dicyanoimidazole (125 mg, 1.04 mmol, 2.0 equiv) in THF (1.5 mL). The mixture was stirred for 3 h, cooled to 0 °C, treated with Bu4NIO4 (173 mg, 0.399 mmol, 1.2 equiv), stirred for 45 min, and diluted with water (2 mL) and CH2Cl2 (5 mL). The organic layer was rinsed with saturated Na2S2O3 (2 × 3 mL), saturated NaHCO3 (3 mL), and brine (3 mL); dried (MgSO4); and concentrated. The crude residue was filtered through a plug of SiO2 (1:1, hexanes/ethyl acetate), concentrated, dissolved in CH2Cl2 (4 mL), and treated with TBSCl (43 mg, 0.29 mmol 0.5 equiv), imidazole (21 mg, 0.31 0.6 equiv), and DMAP (3 mg, 0.024 mmol 0.04 equiv). The reaction mixture was stirred for 4.5 h, quenched with water (4 mL), and diluted with CH2Cl2 (4 mL). The organic layer was rinsed with brine (4 mL), dried (MgSO4), and concentrated. Purification by chromatography on SiO2 (5:1 to 3:1 to 1:1, hexanes/ethyl acetate) gave 3-(((3-(((2,3-(stearoyloxy)propoxy)(methoxy)phosphoryl)oxy)-2-((tert-butyldimethylsilyl)oxy)propoxy)(methoxy)phosphoryl)oxy)-propane-1,2-diyl distearate (10b, 335 mg, 0.208 mmol, 40%) as a waxy colorless solid. Mp: 48.1–48.2 °C. IR: ν 2918, 2851, 1741, 1043 cm−1. 1H NMR (CDCl3, 300 MHz): δ 5.25–5.22 (m, 2 H), 4.35–4.30 (m, 2 H), 4.29–4.04 (m, 11 H), 3.78 (d, 6 H, J = 11.4 Hz; diasteromers), 2.32 (apparent q, 8 H, J = 7.2 Hz), 1.61–1.57 (m, 8 H), 1.25 (m, 112 H), 0.89–0.86 (m, 21 H), 0.11 (s, 6 H). 13C NMR (CDCl3, 125 MHz): δ 173.2, 172.8, 69.7 (multiple peaks), 69.4 (multiple peaks), 67.9 (multiple peaks), 65.6 (multiple peaks), 61.6, 54.6 (d, J = 5.9 Hz), 34.1, 34.0, 31.9, 29.7–29.1 (multiple peaks), 25.6, 24.8, 22.7, 18.0, 14.1, −4.9. HRMS (ESI+) m/z calculated for C89H177O17P2Si [M + H] 1608.2225. Found: 1608.2190.
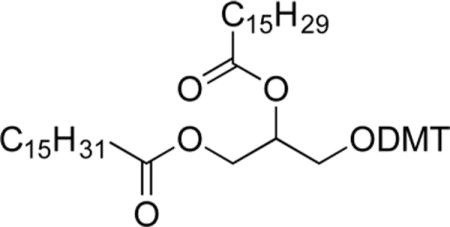
3-(((3-(((2-Palmitoleoyloxy-3-(palmitoyloxy)propoxy)(methoxy)-phosphoryl)oxy)-2-((tert-butyldimethylsilyl)oxy)propoxy)(methoxy)-phosphoryl)oxy)propane-1,2-diyl dipalmitate (10c)
Following General Procedure F using 3c and 9a and a purification by chromatography on SiO2 (5:1 to 3:1 to 1:1, hexanes/ethyl acetate) gave 3-(((3-(((2-palmitoleoyloxy-3-(palmitoyloxy)propoxy)-(methoxy)phosphoryl)oxy)-2-((tert-butyldimethylsilyl)oxy)propoxy)-(methoxy)phosphoryl)oxy)propane-1,2-diyl dipalmitate (10c, 145 mg, 0.0970 mmol, 39%) as a clear colorless oil. IR: ν 2919, 2850, 1741, 1465, 1034 cm−1. 1H NMR (CDCl3, 300 MHz): δ 5.35–5.33 (m, 2 H), 5.25–5.22 (m, 2 H), 4.36–4.32 (m, 2 H), 4.23–4.12 (m, 6 H), 4.05–4.00 (m, 5 H), 3.78 (d, 6 H, J = 11.4 Hz; diasteromers visible), 2.32 (apparent q, 8 H, J = 7.2 Hz), 2.02–2.00 (m, 4 H), 1.61–1.54 (m, 8 H), 1.30–1.26 (m, 88 H), 0.89–0.86 (m, 21 H), 0.11 (s, 6 H). 13C NMR (CDCl3, 125 MHz): δ 173.2, 172.8, 130.0, 129.7, 69.7 (m), 69.4 (d, J = 6.5 Hz), 67.8 (d, J = 5.5 Hz), 65.6 (m), 61.6, 54.6 (d, J = 6.0 Hz), 34.1, 34.0, 31.8, 29.7–29.0 (multiple peaks), 25.6, 24.8, 22.7, 22.6 18.0, 14.1, −4.9. HRMS (ESI+) m/z calculated for C81H159O17P2Si [M + H]: 1494.0816. Found: 1494.0825.
Tetrapalmitoylcardiolipin Diammonium Salt (11a)
General Procedure G
A solution of 10a and NaI (24 mg, 0.16 mmol, 4 equiv) in 2-butanone (0.8 mL) was heated at reflux for 13.5 h and concentrated. The crude waxy yellow solid was dissolved in 1 M HCl/THF/H2O (0.1:2:1, 1 mL), and the resulting yellow solution was stirred for 12 h and quenched with 30% NH4OH (2 mL). The resulting white slurry was extracted with CHCl3 (3 × 5 mL), concentrated, and triturated with Et2O to give tetrapalmitoylcardiolipin diammonium salt (11a, 40 mg, 0.029 mmol, 73%) as a colorless solid containing 5–7% (ESI-MS) monolyso (tripalmitoyl) cardiolipin as an impurity that was removed by thin layer chromatography. 1H NMR (CDCl3, 400 MHz): δ 7.46 (bs, 8 H), 5.21 (bs, 2 H), 4.37 (m, 2 H), 4.17–4.13 (m, 2 H), 3.91 (bs, 9 H), 2.29 (apparent q, 8 H, J = 8.5 Hz), 1.58 (m, 8 H), 1.28–1.25 (m, 98 H), 0.87 (t, 12 H, J = 6.8 Hz). HRMS (ESI−) m/z calculated for C73H141O17P2 [M−H]: 1351.9639. Found: 1351.9648.
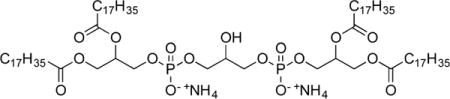
Tetrastearoylcardiolipin Diammonium Salt (11b).33
Following General Procedure G using 10b and purification by precipitation from hot THF gave tetrastearoylcardiolipin diammonium salt (11b, 53 mg, 0.035 mmol, 38%) as a colorless powder containing 5–7% (ESI-MS) monolyso (tristearoylcardiolipin) cardiolipin as an impurity that was removed by thin layer chromatography. 1H NMR (CDCl3, 400 MHz): δ 7.44 (bs, 8 H), 5.21 (bs, 2 H), 4.37–4.35 (m, 2 H), 4.17–4.13 (m, 2 H), 3.92 (bs, 9 H), 2.30 (apparent q, 8 H, J = 8.3 Hz), 1.59–1.58 (m, 8 H), 1.28–1.25 (m, 112 H), 0.88 (t, 12 H, J = 6.8 Hz). MS (ESI−) m/z calculated for C81H156O17P2 [M−2H]2−: 731.5. Found: 731.8.
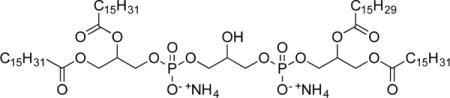
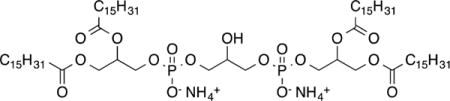
Tripalmitoyl-monopalmitoleoylcardiolipin Diammonium Salt (11c)
Following General Procedure G using 10c and purification by trituration with Et2O gave tripalmitoyl-monopalmitoleoylcardiolipin diammonium salt (11c, 83 mg, 0.060 mmol, 70%) as a colorless powder containing 5–7% (ESI-MS) monolyso-11c as an impurity that was removed by thin layer chromatography. 1H NMR (CDCl3, 400 MHz): δ 7.44 (bs, 8 H), 5.35–5.32 (m, 2 H), 5.21 (bs, 2 H), 4.39–4.35 (m, 2 H), 4.17–4.13 (m, 2 H), 3.84 (bs, 9 H), 2.30 (apparent q, 8 H, J = 8.1 Hz), 2.01–1.99 (m, 4 H), 1.66–1.58 (m, 8 H), 1.28–1.25 (m, 106 H), 0.88 (t, 12 H, J = 6.4 Hz). MS (ESI−) m/z calculated for C73H138O17P2 [M−2H]2−: 674.5. Found: 674.7.
Assessment of Substrate Specificity of Purified Cld1
Activity of Cld1 was detected in liposomes by accumulation of MLCL after CL hydrolysis. Liposomes containing DOPC and CL (10 μM, 1:1) were prepared by sonication (Ultrasonic Homogenizer 4710 series, Cole-Parmer Instrument Co., Chicago, IL) and treated with Cld1 (0.1–0.5 μg of protein per 250 μL sample) in 50 mM HEPES at pH 7.45, containing 100 μM DTPA for 20 min at 37 °C. After incubation with Cld1, lipids were extracted by Folch procedure and analysis of MLCL was performed using LC-ESI-MS. Internal standards of MLCLs were prepared from respective CLs in a PLA2 (porcine pancreatic, Sigma) driven reaction as described by Kim and Hoppel26 with light modifications.
Molecular Modeling
The homology model of Cld1 was built using the I-TASSER34 Web server. Hsad, a steroid-degrading hydrolase from Mycobacterium tuberculosis (PDB ID: 2VF2), was utilized as a template. C-score for the constructed model was equal to 1.78. The C-score is a confidence score to estimate the quality of predicted models by I-TASSER, in the range of [−5,2], where a C-score of higher value defines a model with a high confidence.34 The model showed a TM score equal to 0.5. TM score is a scale for measuring the structural similarity of two protein models, with lower sensitivity to the local error compared to RMSD (a TM score > 0.5 indicates a model of correct topology independent from protein length).35 The overlay of the cavities and cartoon representations was employed to explore the active site and a possible channel facilitating access of CL acyl chain to the hydrolase domain of Cld1. Three homoacylated-CL species, including (C14:0)4-CL, (C16:0)4-CL, and (C18:2)4-CL, were docked to the predicted structure of Cld1 using default settings by AutoDock Vina (http://vina.scripps.edu).36 The pdb into pdbqt format conversion of lipids and protein structures was performed using MGL Tools. A grid box was centered at coordinates 39.861, 40.057, and 74.975 with 100, 80, and 90 Å units in x, y, and z directions, respectively, to cover the entire protein structure. AutoDock Vina reports the 9 lowest energy conformations, which were inspected using PyMOL software (www.pymol.org).
Statistical Analysis
The results are presented as mean ± SD values from at least three experiments, and statistical analyses were performed using paired/unpaired Student’s t test. The statistical significance of differences was set at p < 0.05.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (HL114453, ES020693, U19AIO68021, NS076511, NS061817, NS052315, CA165065, HL117880), Human Frontier Science Program (HFSP-RGP0013/2014), and the Barth Syndrome Foundation, Inc. and the Barth Syndrome Foundation of Canada.
ABBREVIATIONS
- CL
cardiolipin
- MLCL
monolyso-cardiolipin
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- PS
phosphatidylserine
- PA
phosphatidic acid
- FA
fatty acid
- PUFA
polyunsaturated fatty acids
- C14:0
myristic acid
- 16:0
palmitic acid
- 16:1
palmitoleic acid
- C18:1
oleic acid
- C18:2
linoleic acid
- C20:4
arachidonic acid
- C22:6
docosahexaenoic acid
- OxPhos
oxidative phosphorylation
- ORC
oxygen consumption rate
- PLA2
phospholipase A2
- TAZ
tafazzin
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.6b00995.
ORCID
Valerian E. Kagan: 0000-0002-7245-1885
Notes
The authors declare no competing financial interest.
References
- 1.(a) Kagan VE, Tyurina YY, Tyurin VA, Mohammadyani D, Angeli JP, Baranov SV, Klein-Seetharaman J, Friedlander RM, Mallampalli RK, Conrad M, Bayir H. Cardiolipin signaling mechanisms: collapse of asymmetry and oxidation. Antioxid Redox Signaling. 2015;22(18):1667–80. doi: 10.1089/ars.2014.6219. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res. 2008;49:1607–1620. doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baile MG, Sathappa M, Lu YW, Pryce E, Whited K, McCaffery JM, Han X, Alder NN, Claypool SM. Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J Biol Chem. 2014;289:1768–1778. doi: 10.1074/jbc.M113.525733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Sharpley MS, Shannon RJ, Draghi F, Hirst J. Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry. 2006;45:241–248. doi: 10.1021/bi051809x. [DOI] [PubMed] [Google Scholar]; (b) Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007;26:1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Poyry S, Cramariuc O, Postila PA, Kaszuba K, Sarewicz M, Osyczka A, Vattulainen I, Rog T. Atomistic simulations indicate cardiolipin to have an integral role in the structure of the cytochrome bc1 complex. Biochim Biophys Acta, Bioenerg. 2013;1827:769–778. doi: 10.1016/j.bbabio.2013.03.005. [DOI] [PubMed] [Google Scholar]; (d) Eble KS, Coleman WB, Hantgan RR, Cunningham CC. Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J Biol Chem. 1990;265:19434–19440. [PubMed] [Google Scholar]; (e) Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873–52880. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]; (f) Mileykovskaya E, Dowhan W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem Phys Lipids. 2014;179:42–48. doi: 10.1016/j.chemphyslip.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang Y, Powers C, Madala SK, Greis KD, Haffey WD, Towbin JA, Purevjav E, Javadov S, Strauss AW, Khuchua Z. Cardiac metabolic pathways affected in the mouse model of barth syndrome. PLoS One. 2015;10:e0128561. doi: 10.1371/journal.pone.0128561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Beranek A, Rechberger G, Knauer H, Wolinski H, Kohlwein SD, Leber R. Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J Biol Chem. 2009;284:11572–11578. doi: 10.1074/jbc.M805511200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gu Z, Valianpour F, Chen S, Vaz FM, Hakkaart GA, Wanders RJ, Greenberg ML. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol Microbiol. 2004;51:149–158. doi: 10.1046/j.1365-2958.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- 6.Jiang F, Gu ZM, Granger JM, Greenberg ML. Cardiolipin synthase expression is essential for growth at elevated temperature and is regulated by factors affecting mitochondrial development. Mol Microbiol. 1999;31:373–379. doi: 10.1046/j.1365-2958.1999.01181.x. [DOI] [PubMed] [Google Scholar]
- 7.(a) Zhong Q, Li G, Gvozdenovic-Jeremic J, Greenberg ML. Up-regulation of the cell integrity pathway in saccharomyces cerevisiae suppresses temperature sensitivity of the pgs1Delta mutant. J Biol Chem. 2007;282:15946–15953. doi: 10.1074/jbc.M701055200. [DOI] [PubMed] [Google Scholar]; (b) Zhou J, Zhong Q, Li G, Greenberg ML. Loss of cardiolipin leads to longevity defects that are alleviated by alterations in stress response signaling. J Biol Chem. 2009;284:18106–18114. doi: 10.1074/jbc.M109.003236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S, Tarsio M, Kane PM, Greenberg ML. Cardiolipin mediates cross-talk between mitochondria and the vacuole. Mol Biol Cell. 2008;19:5047–5058. doi: 10.1091/mbc.E08-05-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Patil VA, Greenberg ML. Cardiolipin-mediated cellular signaling. Adv Exp Med Biol. 2013;991:195–213. doi: 10.1007/978-94-007-6331-9_11. [DOI] [PubMed] [Google Scholar]; (b) Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS, Kapralova VI, Vikulina AS, Jung MY, Epperly MW, Mohammadyani D, Klein-Seetharaman J, Jackson TC, Kochanek PM, Pitt BR, Greenberger JS, Vladimirov YA, Bayir H, Kagan VE. A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem. 2014;6:542–552. doi: 10.1038/nchem.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Schlame M. Cardiolipin remodeling and the function of tafazzin. Biochim Biophys Acta, Mol Cell Biol Lipids. 2013;1831(3):582–8. doi: 10.1016/j.bbalip.2012.11.007. [DOI] [PubMed] [Google Scholar]; (b) Lu YW, Claypool SM. Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front Genet. 2015;6:3. doi: 10.3389/fgene.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren M, Phoon CK, Schlame M. Metabolism and function of mitochondrial cardiolipin. Prog Lipid Res. 2014;55:1–16. doi: 10.1016/j.plipres.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Ye C, Lou W, Li Y, Chatzispyrou IA, Huttemann M, Lee I, Houtkooper RH, Vaz FM, Chen S, Greenberg ML. Deletion of the cardiolipin-specific phospholipase Cld1 rescues growth and life span defects in the tafazzin mutant: implications for Barth syndrome. J Biol Chem. 2014;289:3114–3125. doi: 10.1074/jbc.M113.529487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lack N, Lowe ED, Liu J, Eltis LD, Noble ME, Sim E, Westwood IM. Structure of HsaD, a steroid-degrading hydrolase, from Mycobacterium tuberculosis. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2008;64:2–7. doi: 10.1107/S1744309107065931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 15.Yang JY, Yan RX, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barth PG, Valianpour F, Bowen VM, Lam J, Duran M, Vaz FM, Wanders RJ. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am J Med Genet A. 2004;126A:349–354. doi: 10.1002/ajmg.a.20660. [DOI] [PubMed] [Google Scholar]
- 17.Rijken PJ, Houtkooper RH, Akbari H, Brouwers JF, Koorengevel MC, de Kruijff B, Frentzen M, Vaz FM, de Kroon AI. Cardiolipin molecular species with shorter acyl chains accumulate in Saccharomyces cerevisiae mutants lacking the acyl coenzyme A-binding protein Acb1p: new insights into acyl chain remodeling of cardiolipin. J Biol Chem. 2009;284:27609–27619. doi: 10.1074/jbc.M109.016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlame M, Brody S, Hostetler KY. Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur J Biochem. 1993;212:727–735. doi: 10.1111/j.1432-1033.1993.tb17711.x. [DOI] [PubMed] [Google Scholar]
- 19.Kuo WN, Walbey DL, Davis DL, McCall LK. Dual modulation of the phosphorylation of endogenous yeast proteins by arachidonic acid and phosphatidylinositol. Cytobios. 1994;78:241–247. [PubMed] [Google Scholar]
- 20.(a) Ogbi M, Johnson JA. Protein kinase Cepsilon interacts with cytochrome c oxidase subunit IV and enhances cytochrome c oxidase activity in neonatal cardiac myocyte preconditioning. Biochem J. 2006;393:191–199. doi: 10.1042/BJ20050757. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Machida K, Tanaka T, Fujita K, Taniguchi M. Farnesol-induced generation of reactive oxygen species via indirect inhibition of the mitochondrial electron transport chain in the yeast Saccharomyces cerevisiae. J Bacteriol. 1998;180:4460–4465. doi: 10.1128/jb.180.17.4460-4465.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Claypool SM, Whited K, Srijumnong S, Han X, Koehler CM. Barth syndrome mutations that cause tafazzin complex lability. J Cell Biol. 2011;192:447–462. doi: 10.1083/jcb.201008177. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Whited K, Baile MG, Currier P, Claypool SM. Seven functional classes of Barth syndrome mutation. Hum Mol Genet. 2013;22:483–492. doi: 10.1093/hmg/dds447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valianpour F, Wanders RJ, Overmars H, Vaz FM, Barth PG, van Gennip AH. Linoleic acid supplementation of Barth syndrome fibroblasts restores cardiolipin levels: implications for treatment. J Lipid Res. 2003;44:560–566. doi: 10.1194/jlr.M200217-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.(a) Kagan VE, Shvedova AA, Novikov KN. Participation of phospholipases in the “repair” of photoreceptor membranes subjected to peroxidation. Biofizika. 1978;23:279–284. [PubMed] [Google Scholar]; (b) Salgo MG, Corongiu FP, Sevanian A. Enhanced interfacial catalysis and hydrolytic specificity of phospholipase A2 toward peroxidized phosphatidylcholine vesicles. Arch Biochem Biophys. 1993;304:123–132. doi: 10.1006/abbi.1993.1330. [DOI] [PubMed] [Google Scholar]; (c) Vankuijk FJGM, Sevanian A, Handelman GJ, Dratz EA. A New Role for Phospholipase-A2 - Protection of Membranes from Lipid-Peroxidation Damage. Trends Biochem Sci. 1987;12:31–34. [Google Scholar]
- 24.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 25.Boettcher C, Pries C, Vangent CM. A Rapid and Sensitive Sub-Micro Phosphorus Determination. Anal Chim Acta. 1961;24:203. [Google Scholar]
- 26.Kim J, Hoppel CL. Monolysocardiolipin: improved preparation with high yield. J Lipid Res. 2011;52(2):389–392. doi: 10.1194/jlr.D010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liebisch G, Vizcaino JA, Kofeler H, Trotzmuller M, Griffiths WJ, Schmitz G, Spener F, Wakelam MJ. Shorthand notation for lipid structures derived from mass spectrometry. J Lipid Res. 2013;54:1523–1530. doi: 10.1194/jlr.M033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Lok CM, Mank APJ, Ward JP. Synthesis of Glycidol Esters and Mono-Di-Acylglycerols from Glycidol. Chem Phys Lipids. 1985;36:329–334. [Google Scholar]; (b) Mori K. Pheromone synthesis. Part 253: Synthesis of the racemates and enantiomers of triglycerides of male Drosophila fruit flies with special emphasis on the preparation of enantiomerically pure 1-monoglycerides. Tetrahedron. 2012;68:8441–8449. [Google Scholar]
- 29.Roodsari FS, Wu DP, Pum GS, Hajdu J. A new approach to the stereospecific synthesis of phospholipids. The use of L-glyceric acid for the preparation of diacylglycerols, phosphatidylcholines, and related derivatives. J Org Chem. 1999;64:7727–7737. [Google Scholar]
- 30.Lee JD, Ueno M, Miyajima Y, Nakamura H. Synthesis of boron cluster lipids: closo-dodecaborate as an alternative hydrophilic function of boronated liposomes for neutron capture therapy. Org Lett. 2007;9:323–326. doi: 10.1021/ol062840+. [DOI] [PubMed] [Google Scholar]
- 31.Stadlbauer S, Frank R, Maulana I, Lonnecke P, Kirchner B, Hey-Hawkins E. Synthesis and Reactivity of ortho-Carbaborane-Containing Chiral Aminohalophosphines. Inorg Chem. 2009;48:6072–6082. doi: 10.1021/ic900443c. [DOI] [PubMed] [Google Scholar]
- 32.Harried SS, Croghan MD, Kaller MR, Lopez P, Zhong W, Hungate R, Reider PJ. Stereoselective synthesis of anti-N-protected 3-amino-1,2-epoxides by nucleophilic addition to N-tert-butanesulfinyl imine of a glyceraldehyde synthon. J Org Chem. 2009;74:5975–5982. doi: 10.1021/jo900643b. [DOI] [PubMed] [Google Scholar]
- 33.Abe M, Niibayashi R, Koubori S, Moriyama I, Miyoshi H. Molecular mechanisms for the induction of peroxidase activity of the cytochrome c-cardiolipin complex. Biochemistry. 2011;50:8383–8391. doi: 10.1021/bi2010202. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins: Struct, Funct, Genet. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 36.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.