

Abstract
Objective
Chronic HIV-1 infection leads to widespread inflammation and immune dysregulation. The gastrointestinal mucosa, a primary site for HIV-1 replication, is thought to play a significant role in this response. MicroRNAs (miRs) are small non-coding RNAs that regulate gene expression, including immune activation and inflammation. Here we investigate miR expression and function in the colonic mucosa during HIV-1 infection.
Design and Methods
Using miR profiling, we examined miR expression in the colonic mucosa of HIV-infected subjects. These miRs were further parsed to identify those that most likely function in HIV-related inflammation. Using bioinformatics tools, we identified potential target genes which were confirmed using in vitro functional testing.
Results
We identified twelve miRs that were differentially expressed in the colonic mucosa of HIV-infected subjects with high versus undetectable plasma viral concentrations. Of these, both miR-26a and miR-29a were down-regulated in untreated HIV-1 infection, yet not in the colonic mucosa from inflammatory bowel disease. This down-regulation occurs within the first hours after infection. These miRs were further shown to directly target IL-6 and STAT3, respectively, with similar changes confirmed in an ex vivo explant infection model.
Conclusions
miR-26a and miR-29a levels are decreased in the colonic mucosa during chronic HIV-1 infection, and this change may be initiated during acute infection. Both miRs de-repress the IL-6/STAT3 signaling pathway, which could contribute to increased inflammation during infection. These miRs may represent novel therapeutic targets for HIV-1 associated inflammation in the colonic mucosa.
Keywords: microRNAs, inflammation, HIV-1, gastrointestinal tract, intestinal mucosa
INTRODUCTION
The gastrointestinal (GI) tract is a primary site for both HIV-1 transmission and pathogenesis. As the largest secondary lymphoid organ in the body, the GI tract harbors the majority of the body’s T lymphocyte population, the primary target cell for HIV-1 infection[1]. The rectal mucosa is uniquely susceptible to HIV-1 infection[2], and rectal transmission accounts for over 60% of new infections in the United States[3]. During acute HIV-1 infection the GI mucosa is the site of rapid T cell depletion, regardless of transmission site[4–7]. The GI compartment is one of the largest latent reservoirs harboring persistent HIV-1 during chronic infection[8, 9].
The degree of mucosal inflammation plays a pivotal role in HIV-1 transmission and replication. Activated mucosal CD4+ T lymphocytes are among the major initial HIV-1 target cells[10, 11]. At baseline the rectal mucosa represents a physiologically inflamed environment, with an abundance of activated lamina propria and intraepithelial CD4+ T lymphocytes which could in part explain the increased susceptibility of this compartment[12, 13]. Indeed, our group has previously shown that rectal mucosal cytokine profiles correlate with tissue HIV-1 levels in vivo, supporting the importance of the immune milieu in fostering viral replication in this compartment[14]. During the course of untreated HIV-1 infection, the colonic mucosa exhibits chronically active inflammation comparable to that seen in the active phases of inflammatory bowel disease (IBD)[15]. This chronic inflammatory state, related at least in part to GI mucosal inflammation and dysfunction, leads to increased morbidity and mortality due to inflammation-related conditions in HIV-positive patients, such as cardiovascular risk, frailty, and non-AIDS related malignancies[16]. Therefore, therapies which target this inflammatory cascade could potentially reduce the incidence of these HIV comorbidities.
MicroRNAs (miRs) are small non-coding RNAs that exert post-transcriptional regulation of gene expression via direct binding in the 3′ untranslated regions (3′UTRs) of mRNA targets[17, 18]. Although miRs constitute a small fraction of the human genome, they play an ever increasing role in inflammation and disease[19, 20]. MiRs help regulate immune activation, including T cell activation and cytokine signaling[21, 22]. Increasing evidence supports a role of miRs in HIV-1 infection. While there are conflicting reports on the existence of HIV-encoded viral miRs (reviewed in[23]), the roles of cellular miRs are better understood. Suppression of miR processing machinery in HIV-infected cells increases HIV-1 replication, suggesting that host cell miRs may influence HIV-1 pathogenesis[24, 25]. Indeed, miRs can regulate HIV-1 infection both by direct interaction with viral mRNA[25, 26], as well as regulation of other cellular proteins[27–29]. Changes in miR expression have also been associated with elite controller and HIV-exposed seronegative phenotypes[30–33]. The majority of the data investigating miRs in HIV-1 infection has focused on early infection and peripheral blood cells. Less is known about the role of miRs in more chronic HIV-1 infection, and changes in expression at sites other than peripheral blood.
Given the central role of the GI tract in HIV-1 infection and inflammation, we sought to examine the expression of miRs at this site in patients with chronic HIV-1 infection. miRs have a known role in regulation of GI mucosal inflammation, and deregulation of specific miRs has been associated with other inflammatory disease states such as inflammatory bowel disease (IBD)[34]. Since HIV-1 infection similarly induces mucosal inflammation, we sought to identify unique miRs associated with HIV-1 related inflammation which could provide new therapeutic targets to reduce inflammation. Here we identify a specific miR signature present in the GI tract of HIV-1 infected subjects, and further identify and validate gene targets in the inflammatory pathway.
METHODS
Ethics Statement
This study was approved by the UCLA Office of the Human Research Protection Program Institutional Review Board (IRB) and all subjects signed written informed consent at the time of biopsy collection.
Study subjects and colon tissue samples
Sigmoid colon tissue samples from HIV-infected (n=30), active inflammatory bowel disease (n=19), and healthy control (n=7) subjects for use in microRNA-array and PCR experiments were obtained from the UCLA Mucosal Immunology Core Laboratory biorepository (UCLA IRB #11-001592). Sigmoid colon mucosal biopsies for explant experiments were obtained from healthy HIV seronegative subjects from the UCLA Mucosal Immunology Core Laboratory registry (UCLA IRB #10-000528). Sigmoid colon mucosal biopsies (approximately 8 mm × 2 mm × 1 mm) were collected via flexible sigmoidoscopy using large cup endoscopic biopsy forceps (Microvasive Radial Jaw #1589, outside diameter 3.3 mm) taken 25–30 cm from the anal verge. Biopsies were immediately placed in RPMI 1640 media then used for explant set-up. For HIV-infected participants, viral load, CD4 count, and antiretroviral medications were recorded at the time of collection.
RNA isolation from human colonic biopsies
RNA was extracted from snap frozen colonic biopsies after homogenization using Trizol (Invitrogen) according to the manufacturer’s instructions, as previously described[35]. The quality of the samples was evaluated by estimating the ratios of A260nm/A230nm and A260nm/A280nm for phenol and protein contamination, respectively, using microplate spectrophotometer (Synergy HT, BioTek); only samples with ratios ≥1.8 were used in further analysis. The integrity of the RNA was further evaluated using the Bioanalyzer 2100 (Agilent) and only samples with RNA integrity number (RIN) higher than 7.0 were included in the study.
MicroRNA-array analysis
MicroRNA-array analysis was performed using RNA extracted from colonic biopsies obtained from HIV-infected subjects with undetectable plasma viral load (<50 copies/ml, n=9) and high plasma viral load (>5000 copies/ml, n=8) using the miRCURY microRNA Array Profiling service (Exiqon Inc) with background subtraction and normalization performed. Differential expression of miRs between groups was determined using cutoff fold change >3 and p<0.05 (Student’s t test). Data was further parsed using DIANA microRNA pathway software (miRPath v3, http://snf-515788.vm.okeanos.grnet.gr/) to identify miRs with known roles in inflammatory pathways. The Targetscan algorithm (www.targetscan.org) was used to identify potential direct targets for candidate miRs.
Quantitative PCR analysis
MicroRNA levels were assessed by quantitative PCR (qPCR) on a CFX384 detection system (Bio-Rad) with SYBR Green using the Exiqon PCR primer sets according to manufacturer’s instructions (Exiqon Inc.). Data was normalized to reference gene expression U6 snRNA and 5S rRNA. All primers for microRNAs and the reference genes were purchased from Exiqon Inc. Quantitative PCR for cytokines and reference genes GAPDH or β-actin was performed similarly using CFX384 detection system (Bio-Rad) and SYBR Green. Primers were as noted (Supplementary Table 1) and data normalized to reference gene expression.
HIV-1 virus expansion
The following reagent was obtained from the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), NIH: HIV-1BaL from Dr. Suzanne Gartner, Dr. Mikulas Popovic and Dr. Robert Gallo. Stocks of HIV-1BaL were prepared by infection using PM1 cells. TCID50 was determined by titration using pooled PBMCs. The same viral stock was used throughout the study.
HIV-1 infection of explants and Jurkat cells
Gut mucosal explants were processed as previously described[36]. Briefly, sigmoid biopsies were collected and cultured using RPMI 1640 medium with 2.5 mg/ml amphotericin B and 0.1 mg/ml piperacillin-tazobactam in a 37 degree humidified incubator. Triplicate biopsies were infected with HIV-1BaL 1×104 TCID50 for 2 hours then thoroughly washed and replaced with fresh media on absorbable gelatin sponge in separate wells of standard tissue culture plates. Total RNA was then isolated at indicated time points for qPCR assays. Jurkat-CCR5 cells were kindly provided by Dr. Betsy Herold. 1×105 cells were infected with HIV-1BaL at MOI 0.1 for 2 hours then washed and replace with fresh media. Cells were harvested for RNA isolation at designated time points.
microRNA transfection
Jurkat-CCR5 cells were transfected using RNAiMAX reagent with 100nM of miR-26a or miR-29a mimics or the negative control #1 (miR-NC) (miRVana, Life Technologies) in OptiMEM medium. Medium was replaced with 10% FBS containing medium after overnight incubation.
3′-UTR luciferase assay
Luciferase reporter plasmids pLightSwitch carrying the 3′UTR of IL6 or STAT3 (Switchgear Genomics) were used for the 3′UTR luciferase experiments. Sequential transfection was performed; first, Jurkat-CCR5 cells were transfected with the reporter plasmid (4 μg plasmid per 1×106 cells) using FuGENE6 (Promega) and 24 hours later the same cells were transfected with a final concentration of 50nM of miR-26a or miR-29a or the respective negative controls. After 24 hours the cells were lysed and luciferase activity was measured using the Promega Luciferase Reporter Assay System.
STAT3 ELISA assay
The phosphorylation status of STAT3 in tyrosine 705 (p-STAT3) was assessed by sandwich ELISA assay (Bio-Rad) using cell lysates following transfection with mimics of miR-29a or appropriate negative controls as described above.
Statistical analysis
Data from clinical specimens were analyzed with nonparametric Mann-Whitney test (Figure 1). Experimental data were compared using unpaired t tests (Figures 2, 3 and 4). All statistical analyses were performed using GraphPad Prism (GraphPad Software).
Figure 1. microRNA profiling in HIV-infected colonic mucosa.
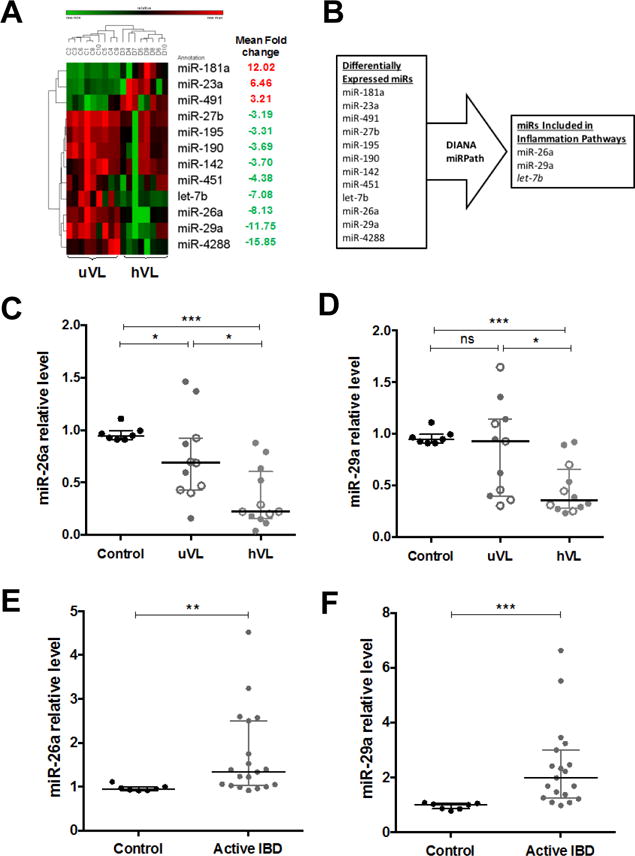
(A) Heatmap showing differentially expressed miRs in colonic mucosa between high viral load (hVL, n=8) and undetectable viral load (uVL, n=9) subjects. Mean fold-change of hVL vs uVL for each miR is indicated at right column. (B) Diagram of the selection criteria applied in order to select novel miRs for further analysis. (C) Confirmatory quantitative PCR (qPCR) for miR-26a and (D) miR-29a levels from colonic biopsies of a validation cohort of HIV-infected participants with hVL (n=11) and uVL (n=12) compared to healthy controls (n=7). Open circles indicate replicative samples from participants also included in the original array cohort for qPCR validation. (E) Relative specificity examined by qPCR for miR-26a and (F) miR-29a levels in colonic biopsies of active IBD participants (n=19). Relative levels are normalized to reference targets U6 snRNA and 5S rRNA. Line indicates the median with error bars showing interquartile range (IQR). *p < 0.05, **p < 0.01, ***p<0.001 and ns, not significant, using Mann-Whitney test.
Figure 2. HIV-1 infection rapidly decreases miRs-26a and 29a levels.
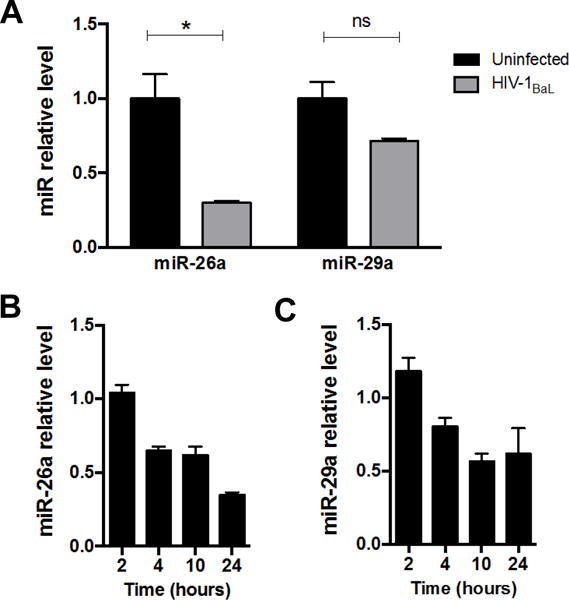
(A) Quantitative PCR for miR-26a and miR-29a levels in Jurkat-CCR5 cells infected with HIV-1 at 24 hours post-infection. Relative levels are normalized to reference targets U6 snRNA and 5S rRNA. (B) Quantitative PCR for miR-26a and (C) miR-29a levels in Jurkat-CCR5 cells infected HIV-1 at 2, 4, 10 and 24 hours post-infection. Relative levels normalized to non-infected cells. Data shown as mean ± SEM. *p < 0.05 and ns, not significant, using unpaired t tests.
Figure 3. miR-26a targets IL-6 and miR-29a targets STAT3.
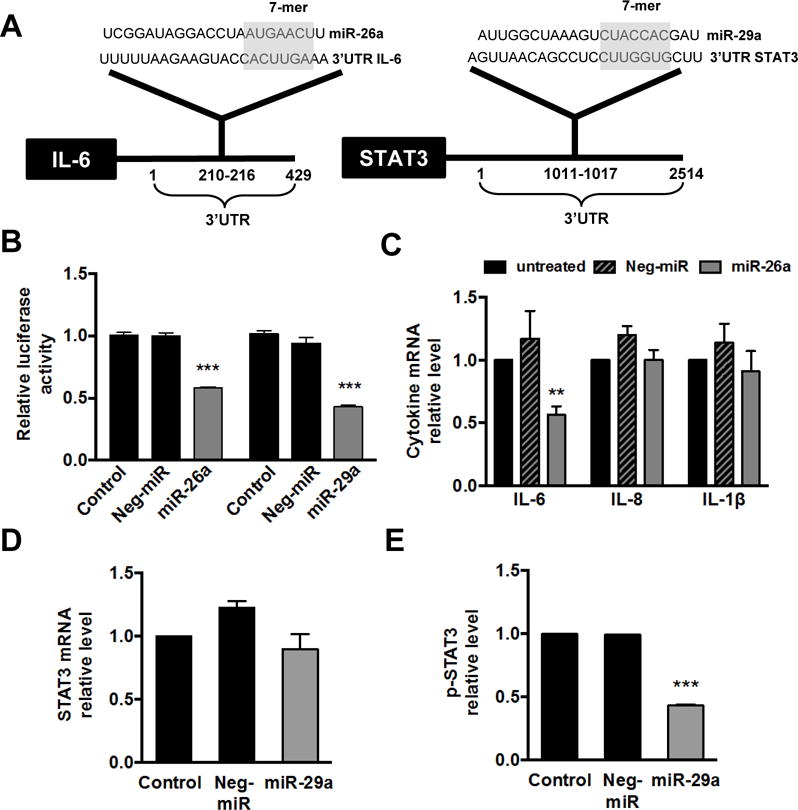
(A) Bioinformatics analysis identified complementarity of the “seed sequence” of miR-26a with the IL-6 mRNA 3′UTR nucleotides 210-216 and of miR-29a with the STAT3 mRNA 3′UTR nucleotides 1011-1017. (B) Luciferase reporter assay showing direct targeting of miR-26a and miR-29a with IL-6 and STAT3, respectively. 3′UTR from each gene was cloned downstream of a luciferase reporter, then luciferase activity measured following co-transfection with indicated miR or non-specific miR control (Neg-miR). Decreased luciferase activity indicates direct targeting of the miR with that 3′UTR. Data shown as mean ± SEM. (C) Quantitative PCR for cytokine mRNA levels in untreated or Jurkat-CCR5 cells transfected with negative control miR (Neg-miR) or miR-26a. (D) Quantitative PCR for STAT3 mRNA levels and (E) ELISA for phosphorylated STAT3 (p-STAT3) levels in untreated or Jurkat-CCR5 cells transfected with negative control miR (Neg-miR) or miR-29a. Data shown as mean ± SEM. **p < 0.01 and ***p < 0.001 using unpaired t tests.
Figure 4. Ex vivo infection of human rectal mucosal explants with HIV-1 leads to down-regulation of miR-26a and miR-29a and up-regulation of IL-6 mRNA levels.
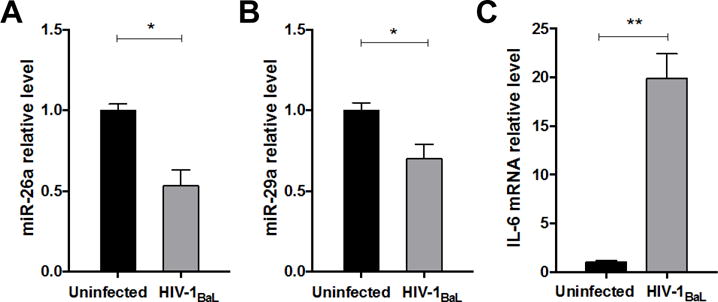
Quantitative PCR for (A) miR-26a levels, (B) miR-29a levels, and (C) IL-6 mRNA levels in human rectal explants from healthy (HIV-seronegative) individuals 24 hours post-infection with HIV-1BaL (1×104 TCID50) compared to uninfected explants. Data shown as mean ± SEM. *p < 0.05 and **p < 0.01 using unpaired t tests.
RESULTS
Study subjects
Our study aimed to identify unique miRs involved in colonic mucosal inflammation during chronic HIV-1 infection. To do this, we used sigmoid mucosal biopsies obtained from two cohorts of HIV-infected subjects: (1) undetectable viral load (uVL) (plasma viral concentration <50 copies/ml) on suppressive antiretroviral therapy (ART) and (2) high viral load (hVL) (plasma viral concentration >5000 copies/ml) either treatment-naïve or off ART for at least six months. As an inflammatory control, we used biopsies obtained from HIV-seronegative subjects with active IBD, both ulcerative colitis (UC) and Crohn’s disease colitis (CD). HIV-seronegative healthy control subjects were used as a baseline negative control. Demographics of these cohorts, including mean CD4 T cell counts and viral concentration for HIV-infected subjects, are shown in Table 1.
Table 1.
Study participant demographics
| Undetectable Viral Load N=15 |
High Viral Load N=15 |
Inflammatory Bowel Disease N=19 |
Healthy Control N=7 |
|
|---|---|---|---|---|
| Age (yrs) Mean ± SDa | 44 ± 9.2 | 43 ± 9.6 | 45.5 ± 13.1 | 39 ± 9.2 |
| Age Range (yrs) | 30-62 | 25-63 | 28-70 | 25-52 |
| Sex | ||||
| Male | 15 | 14 | 13 | 4 |
| Female | 0 | 1 | 6 | 3 |
| Race | ||||
| Asian | 1 (6.7%) | 0 | 1 (5.3%) | 0 |
| African American | 1 (6.7%) | 3 (20%) | 0 | 2 (28.5%) |
| Caucasian | 13 (86.6%) | 9 (60%) | 11 (57.9%) | 5 (71.5%) |
| Hispanic | 0 | 1 (6.7%) | 1 (5.3%) | 0 |
| Other/Unknown | 0 | 2 (13.3%) | 6 (31.6%) | 0 |
| CD4+ T cells (103 cells/ml) (range) | 603 (118-1020) |
357 (105-668) |
N/A | N/A |
| HIV RNA (copies/ml) (range) | <50 | 62,366 (7000-160000) |
N/A | N/A |
SD, standard deviation
Identification of 12-microRNA signature in colonic mucosa of HIV-infected patients
To examine the potential role of miRs in HIV-1 pathogenesis in the colonic mucosa, we performed miR profiling analysis in colonic tissues from HIV-infected patient with undetectable (n=9, uVL) and high (n=8, hVL) plasma viral concentrations. We chose to compare aviremic and viremic subjects in order to identify miRs expressed during the time of active viral replication, with the hypothesis that these miRs are more likely involved in direct pathogenesis of the virus and inflammation. Using a stringent 3-fold change and p<0.05 cutoff, this analysis revealed 12 miRs that were significantly differentially expressed between the uVL and hVL colonic tissue (Figure 1A). Specifically, three miRs (miR-181a, miR-23a, miR-491) were up-regulated, while nine miRs (miR-4288, miR-29a, miR-26a, let-7b, miR-451, miR-142, miR-190, miR-195, miR-27b) were down-regulated in high versus undetectable viremia tissues.
miR-26a and miR-29a are down-regulated in the colonic mucosa during active HIV-1 infection
From the miRs identified in the array analysis we were most interested in those that may be regulating the ongoing inflammation seen during chronic HIV-1 infection. To parse this list, we used DIANA miRPath to identify miRs with known regulatory roles in inflammatory pathways[37]. Three miRs were selected using these criteria (Figure 1B). As miR-26a and miR-29a have been previously shown to coordinately regulate pathways[38], including inflammatory[39], we chose to use these for further study. The role of let-7b is the subject of separate, ongoing investigation.
To confirm our array findings, we quantified miR-26a and miR-29a levels using a second validation cohort of HIV-1-infected subjects (n=11, uVL and n=12, hVL) using quantitative PCR (qPCR). To establish the validity of qPCR as a confirmatory readout, this second cohort also included replicate RNA from the same individuals as used in the array when available (designated with open circles in Figure 1C and 1D). These findings were in agreement with our array data, showing both miR-26a and miR-29a had reduced levels in colonic biopsies from viremic subjects compared to aviremic subjects (Figure 1C and D). To assess relative specificity of these miRs in HIV-induced mucosal inflammation, we examined these miRs in active IBD. Using mucosal biopsies from patients with active colorectal IBD, either UC (n=13) or CD (n=6), we found that, in contrast to the HIV-infected colonic tissues, miR-26a and miR-29a were increased compared to healthy controls (n=7) (Figure 1E and F).
HIV-1 infection rapidly reduces miR-26a and miR-29a levels in T cells
While decreased miR-26a and miR-29a levels were seen in colonic tissues from chronic HIV-1 infection (Figure 1), the cell types involved are not clear. To examine the expression levels of these two miRs in T cells, the primary target for HIV-1 infection, we used the HIV-susceptible Jurkat-CCR5 T-cell line (Jurkat-CCR5) to examine miR expression following in vitro HIV-1 infection. In agreement with our observations using whole tissue biopsies, we found decreased miR-26a and miR-29a levels in HIV-infected Jurkat-CCR5 cells 24 hours post infection (Figure 2A). We next examined the time course of miR-26a and miR-29a expression at 2, 4, 10 and 24 hours post-infection with HIV-1. Interestingly, we found that HIV-1 infection leads to decreased miR-26a and miR-29a levels very early (4 hours post-infection) (Figure 2B and C), suggesting that miR-26a and miR-29a downregulation is an early event during HIV-1 infection.
miR-26a and miR-29a directly target the IL6-STAT3 signaling pathway
miRs regulate gene expression via direct binding on the 3′UTR of their target genes to induce mRNA degradation or inhibit protein translation[17], and potential targets can be predicted through bioinformatics methods. We were interested in identifying miR-26a and miR-29a direct gene targets that may be involved in inflammatory signaling pathways during HIV-1 infection. The TargetScan algorithm predicted 45 genes targeted by miR-26a and miR-29a, however only two genes (IL-6 and STAT3) were related to inflammatory-immune signaling pathways involved in HIV-1 infection. Both miR-26a and miR-29a have potential binding sites in the 3′UTR of IL6 and STAT3 genes, respectively (Figure 3A). To confirm these genes are direct targets of miR-26a and miR-29a, we used a 3′UTR luciferase reporter system. Specifically, the 3′UTR from each gene was cloned into the reporter plasmid downstream of the luciferase gene, then co-transfected with either miR-26a or miR-29a. In agreement with other studies[40–42], we found that IL-6 and STAT3 are indeed direct targets of miR-26a and miR-29a, as evidenced by decreased luciferase activity following transfection with the miR (Figure 3B).
To further examine these direct interactions in cells relevant to HIV-1 infection, we overexpressed miR-26a and miR-29a in Jurkat-CCR5 T cells then assessed mRNA levels or protein expression of the target genes. As shown, miR-26a overexpression specifically reduced IL-6 mRNA levels, yet did not significantly alter mRNA levels of other inflammatory cytokines IL-8 and IL-1β (Figure 3C). Similarly, miR-29a overexpression decreased STAT3 mRNA, though not significantly (Figure 3D), but did significantly decrease STAT3 phosphoprotein levels assessed by ELISA (Figure 3E). These data show that both miR-26a and miR-29a are indeed direct regulators of IL6/STAT3 pathway in Jurkat-CCR5 T cells, consistent with previous studies in other cell types[40–42]. Our data further suggests that miR-26a regulates IL-6 at the transcriptional level, while miR-29a regulates STAT3 post-transcriptionally, which has not been previously demonstrated. Taken together, our results indicate that HIV-1 infection leads to decreased miR-26a and miR-29a levels in the HIV-inflamed colonic mucosa, thereby enabling activation of the pro-inflammatory IL6-STAT3 signaling pathway.
HIV-1 reduces miR-26a and miR-29a expression levels in HIV-infected colonic explants
To confirm our findings in a human tissue model, we infected freshly-acquired human colonic explants with HIV-1BaL then assessed expression of miR-26a, miR-29a, and identified target genes. Using qPCR, we again found that HIV-1 infection significantly reduced both miR-26a and miR-29a levels (Figure 4A and B), with an expected increased IL-6 mRNA (Figure 4C). These results support our model of decreased negative miR regulation, resulting in increased IL-6 production in colonic tissue during HIV-1 infection.
DISCUSSION
While treatment of HIV-1 with combination antiretroviral therapy results in sustained viremic control and immune reconstitution, indices of systemic immune activation do not fully normalize[43–46]. Widespread inflammation of the GI tract is one suspected driver of ongoing immune activation via mucosal barrier dysfunction and alterations in intestinal microbiome[47–50]. Given the comorbidities associated with chronic inflammation, therapeutic efforts to further dampen this systemic response could have great benefit to chronic HIV-infected patients. Here we show marked changes in miR expression specific to HIV-1, and show direct action of these miRs on inflammatory mediators. This may represent an attractive target for adjunctive therapy in HIV-1 infected patients.
Using bioinformatics and molecular approaches we showed that miR-26a and miR-29a can directly regulate the IL-6/STAT3 inflammatory pathway in the context of HIV-1 infection, and do so using both transcriptional and translational mechanisms. Plasma IL-6 levels predict morbidity and mortality in treated HIV-1 infection[51–53], underscoring the importance of this pathway in HIV-1 pathogenesis. One potential mechanism linking this pathway to chronic mucosal HIV-1 infection is that IL-6/STAT3 axis favors Th17 T cell development, which are key cells regulating gut dysfunction and systemic inflammation. Th17 cells are also early targets for HIV-1 infection[54, 55], suggesting that promotion of Th17 development, via induction of IL-6/STAT3 resulting from miR-26a and miR-29a suppression shortly after infection, would be advantageous to the virus by increasing available target cells and inflammatory signals. However, from a clinical perspective, therapeutic manipulation of this IL-6/STAT3 pathway during chronic treated HIV-1 infection could further decrease Th17 cells, which may have deleterious effects on the barrier function of the gastrointestinal mucosa[56]. This potential consequence of impacting the IL-6/STAT3 pathway should be an important consideration for future studies and therapeutic development.
Effects on the IL-6/STAT3 pathway is but one potential functional readout of these miRs on HIV-1 pathogenesis. In addition to regulating inflammatory response, miR-29a can directly interact with HIV-1 via downregulation of Nef protein and decrease in HIV-1 replication[26], though this targeting may be limited by secondary structure[57]. Suppression of miR-29a during HIV-1 infection, as seen in our study and also in peripheral blood[57, 58], is advantageous both by increasing inflammation and avoiding the direct inhibitory effects of this miR on the virus. The mechanism of the decreased miR-26a and miR-29a expression is not clear and may include both direct interactions mediated by the virus or rather a host response to the infection. While beyond the scope of the current study, further investigation into these mechanisms both in the context of HIV-1 or other gastrointestinal infections is warranted.
Studies of miRs have led to the development of novel therapeutics with great promise for clinical utility. The locked nucleic acid miR-122 inhibitor miravirsen has shown efficacy reducing hepatitis C viremia in chimpanzees[59], and more recently in a Phase IIa trial[60]. A recent study from our group showed that miR-214 has a critical role in UC and transformation to malignancy, and that direct inhibition of this miR not only improved colitis but also reduced the tumor size and burden[61], underscoring the potential impact of miR therapeutics across diseases.
Most miRs function as fine tuners of gene expression and therefore alterations of only one to two-fold can be highly significant[62]. It is rare to identify in human tissue samples miRs that are deregulated greater than five-fold; this includes landmark papers in cancer or autoimmune diseases[63–66]. Our study identified miRs that are deregulated three to sixteen-fold in HIV-1-infected colonic tissue (Figure 1A), suggesting these may have a profound impact on mucosal inflammatory mechanisms. Further, these miRs were decreased in HIV-1-infected participants, but not deregulated in healthy or inflammatory (IBD) controls. While this study is not an exhaustive comparison of all inflammatory conditions, including other enteric infections, it may suggest a unique mechanism in HIV-1 infection worthy of future investigation. If confirmed in wider studies, these miRs would be attractive targets for adjunctive therapy to potentially further reduce the inflammation associated with chronic HIV-1 infection.
Supplementary Material
Acknowledgments
We are grateful to all the participants for their time and willingness to participate in this study. We also thank Dr. Otto Yang for helpful comments and discussions, Justin Akin for administrative support, and Dr. Betsy Herold for reagents. Author contributions: J.A.F., G.K., M.K., J.E., A.D., performed experiments and analyzed the data. J.A.F., G.K., A.D., D.I., P.A.A., conceived and designed the study. J.A.F., G.K., M.K., D.I., P.A.A., wrote and edited the paper.
Source of Funding: This work was supported in part by UCLA CURE Digestive Diseases Research Center (National Institutes of Health (NIH) P30 DK041301). J.A.F. was supported in part by NIH KL2 TR001882 (UCLA CTSI). The UCLA Center for AIDS Research (CFAR) Mucosal Immunology Core provided developmental funds and technical support (NIH P30 AI028697). Additional support provided by the UCLA Immune Innovation Fund.
Footnotes
Conflicts of Interest: There are no declared conflicts of interest.
References
- 1.Mowat AM, Viney JL. The anatomical basis of intestinal immunity. Immunological reviews. 1997;156:145–166. doi: 10.1111/j.1600-065x.1997.tb00966.x. [DOI] [PubMed] [Google Scholar]
- 2.Patel P, Borkowf CB, Brooks JT, Lasry A, Lansky A, Mermin J. Estimating per-act HIV transmission risk: a systematic review. AIDS. 2014;28(10):1509–1519. doi: 10.1097/QAD.0000000000000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prevention CfDCa. Estimated HIV incidence among adults and adolescents in the United States, 2007–2010. HIV Surveillance Supplemental Report. 2012;17(4) [Google Scholar]
- 4.Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280(5362):427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- 5.Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434(7037):1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, et al. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005;434(7037):1148–1152. doi: 10.1038/nature03513. [DOI] [PubMed] [Google Scholar]
- 7.Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, et al. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. 2004;200(6):761–770. doi: 10.1084/jem.20041196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis. 2008;197(5):714–720. doi: 10.1086/527324. [DOI] [PubMed] [Google Scholar]
- 9.Poles MA, Boscardin WJ, Elliott J, Taing P, Fuerst MM, McGowan I, et al. Lack of decay of HIV-1 in gut-associated lymphoid tissue reservoirs in maximally suppressed individuals. J Acquir Immune Defic Syndr. 2006;43(1):65–68. doi: 10.1097/01.qai.0000230524.71717.14. [DOI] [PubMed] [Google Scholar]
- 10.Miller CJ, Li Q, Abel K, Kim EY, Ma ZM, Wietgrefe S, et al. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J Virol. 2005;79(14):9217–9227. doi: 10.1128/JVI.79.14.9217-9227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. 1999;286(5443):1353–1357. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
- 12.Anton PA, Elliott J, Poles MA, McGowan IM, Matud J, Hultin LE, et al. Enhanced levels of functional HIV-1 co-receptors on human mucosal T cells demonstrated using intestinal biopsy tissue. AIDS. 2000;14(12):1761–1765. doi: 10.1097/00002030-200008180-00011. [DOI] [PubMed] [Google Scholar]
- 13.Poles MA, Elliott J, Taing P, Anton PA, Chen IS. A preponderance of CCR5(+) CXCR4(+) mononuclear cells enhances gastrointestinal mucosal susceptibility to human immunodeficiency virus type 1 infection. J Virol. 2001;75(18):8390–8399. doi: 10.1128/JVI.75.18.8390-8399.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGowan I, Elliott J, Fuerst M, Taing P, Boscardin J, Poles M, et al. Increased HIV-1 mucosal replication is associated with generalized mucosal cytokine activation. J Acquir Immune Defic Syndr. 2004;37(2):1228–1236. doi: 10.1097/01.qai.0000131846.12453.29. [DOI] [PubMed] [Google Scholar]
- 15.Olsson J, Poles M, Spetz AL, Elliott J, Hultin L, Giorgi J, et al. Human immunodeficiency virus type 1 infection is associated with significant mucosal inflammation characterized by increased expression of CCR5, CXCR4, and beta-chemokines. J Infect Dis. 2000;182(6):1625–1635. doi: 10.1086/317625. [DOI] [PubMed] [Google Scholar]
- 16.Deeks SG, Tracy R, Douek DC. Systemic effects of inflammation on health during chronic HIV infection. Immunity. 2013;39(4):633–645. doi: 10.1016/j.immuni.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boldin MP, Baltimore D. MicroRNAs, new effectors and regulators of NF-kappaB. Immunol Rev. 2012;246(1):205–220. doi: 10.1111/j.1600-065X.2011.01089.x. [DOI] [PubMed] [Google Scholar]
- 19.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10(10):704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129(1):147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nature reviews Immunology. 2007;7(6):454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 23.Swaminathan G, Navas-Martín S, Martín-García J. MicroRNAs and HIV-1 Infection: Antiviral Activities and Beyond. Journal of Molecular Biology. 2014;426(6):1178–1197. doi: 10.1016/j.jmb.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 24.Triboulet R, Mari B, Lin YL, Chable-Bessia C, Bennasser Y, Lebrigand K, et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. 2007;315(5818):1579–1582. doi: 10.1126/science.1136319. [DOI] [PubMed] [Google Scholar]
- 25.Nathans R, Chu CY, Serquina AK, Lu CC, Cao H, Rana TM. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol Cell. 2009;34(6):696–709. doi: 10.1016/j.molcel.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahluwalia JK, Khan SZ, Soni K, Rawat P, Gupta A, Hariharan M, et al. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology. 2008;5:117. doi: 10.1186/1742-4690-5-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiang K, Sung TL, Rice AP. Regulation of cyclin T1 and HIV-1 Replication by microRNAs in resting CD4+ T lymphocytes. J Virol. 2012;86(6):3244–3252. doi: 10.1128/JVI.05065-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sung TL, Rice AP. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009;5(1):e1000263. doi: 10.1371/journal.ppat.1000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen CJ, Jia YH, Tian RR, Ding M, Zhang C, Wang JH. Translation of Pur-alpha is targeted by cellular miRNAs to modulate the differentiation-dependent susceptibility of monocytes to HIV-1 infection. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26(11):4755–4764. doi: 10.1096/fj.12-209023. [DOI] [PubMed] [Google Scholar]
- 30.Bignami F, Pilotti E, Bertoncelli L, Ronzi P, Gulli M, Marmiroli N, et al. Stable changes in CD4+ T lymphocyte miRNA expression after exposure to HIV-1. Blood. 2012;119(26):6259–6267. doi: 10.1182/blood-2011-09-379503. [DOI] [PubMed] [Google Scholar]
- 31.Yahyaei S, Biasin M, Saulle I, Gnudi F, De Luca M, Tasca KI, et al. Identification of a Specific miRNA Profile in HIV-Exposed Seronegative Individuals. J Acquir Immune Defic Syndr. 2016;73(1):11–19. doi: 10.1097/QAI.0000000000001070. [DOI] [PubMed] [Google Scholar]
- 32.Reynoso R, Laufer N, Hackl M, Skalicky S, Monteforte R, Turk G, et al. MicroRNAs differentially present in the plasma of HIV elite controllers reduce HIV infection in vitro. Scientific reports. 2014;4:5915. doi: 10.1038/srep05915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egana-Gorrono L, Escriba T, Boulanger N, Guardo AC, Leon A, Bargallo ME, et al. Differential microRNA expression profile between stimulated PBMCs from HIV-1 infected elite controllers and viremic progressors. PLoS One. 2014;9(9):e106360. doi: 10.1371/journal.pone.0106360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dalal SR, Kwon JH. The Role of MicroRNA in Inflammatory Bowel Disease. Gastroenterol Hepatol (N Y) 2010;6(11):714–722. [PMC free article] [PubMed] [Google Scholar]
- 35.Koukos G, Polytarchou C, Kaplan JL, Morley-Fletcher A, Gras-Miralles B, Kokkotou E, et al. MicroRNA-124 regulates STAT3 expression and is down-regulated in colon tissues of pediatric patients with ulcerative colitis. Gastroenterology. 2013;145(4):842–852 e842. doi: 10.1053/j.gastro.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fletcher PS, Elliott J, Grivel JC, Margolis L, Anton P, McGowan I, et al. Ex vivo culture of human colorectal tissue for the evaluation of candidate microbicides. AIDS. 2006;20(9):1237–1245. doi: 10.1097/01.aids.0000232230.96134.80. [DOI] [PubMed] [Google Scholar]
- 37.Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, et al. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic acids research. 2015;43(W1):W460–466. doi: 10.1093/nar/gkv403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang H, Jiang X, Wang J, Li Y, Chen P, Arnovitz S, et al. Identification of MLL-Fusion/Myc⊣miR-26a/Mir-29a⊣Tet1 Signaling Circuit in <em>MLL</em>-Rearranged Leukemia. Blood. 2014;124(21):1011. [Google Scholar]
- 39.Kleinsteuber K, Heesch K, Schattling S, Kohns M, Sander-Jülch C, Walzl G, et al. Decreased Expression of miR-21, miR-26a, miR-29a, and miR-142-3p in CD4+ T Cells and Peripheral Blood from Tuberculosis Patients. PLoS One. 2013;8(4):e61609. doi: 10.1371/journal.pone.0061609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang X, Liang L, Zhang XF, Jia HL, Qin Y, Zhu XC, et al. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology (Baltimore, Md) 2013;58(1):158–170. doi: 10.1002/hep.26305. [DOI] [PubMed] [Google Scholar]
- 41.Chen C-YA, Chang JT, Ho Y-F, Shyu A-B. MiR-26 down-regulates TNF-α/NF-κB signalling and IL-6 expression by silencing HMGA1 and MALT1. Nucleic acids research. 2016;44(8):3772–3787. doi: 10.1093/nar/gkw205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song X, Wang CT, Geng XH. MicroRNA-29a promotes apoptosis of monocytes by targeting STAT3 during sepsis. Genetics and molecular research: GMR. 2015;14(4):13746–13753. doi: 10.4238/2015.October.28.37. [DOI] [PubMed] [Google Scholar]
- 43.French MA, King MS, Tschampa JM, da Silva BA, Landay AL. Serum immune activation markers are persistently increased in patients with HIV infection after 6 years of antiretroviral therapy despite suppression of viral replication and reconstitution of CD4+ T cells. J Infect Dis. 2009;200(8):1212–1215. doi: 10.1086/605890. [DOI] [PubMed] [Google Scholar]
- 44.Lederman MM, Calabrese L, Funderburg NT, Clagett B, Medvik K, Bonilla H, et al. Immunologic failure despite suppressive antiretroviral therapy is related to activation and turnover of memory CD4 cells. J Infect Dis. 2011;204(8):1217–1226. doi: 10.1093/infdis/jir507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis. 2003;187(10):1534–1543. doi: 10.1086/374786. [DOI] [PubMed] [Google Scholar]
- 46.Neuhaus J, Jacobs DR, Jr, Baker JV, Calmy A, Duprez D, La Rosa A, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis. 2010;201(12):1788–1795. doi: 10.1086/652749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nature medicine. 2006;12(12):1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 48.Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199(8):1177–1185. doi: 10.1086/597476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dillon SM, Lee EJ, Kotter CV, Austin GL, Gianella S, Siewe B, et al. Gut dendritic cell activation links an altered colonic microbiome to mucosal and systemic T-cell activation in untreated HIV-1 infection. Mucosal Immunol. 2016;9(1):24–37. doi: 10.1038/mi.2015.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunt PW, Sinclair E, Rodriguez B, Shive C, Clagett B, Funderburg N, et al. Gut Epithelial Barrier Dysfunction and Innate Immune Activation Predict Mortality in Treated HIV Infection. J Infect Dis. 2014;210(8):1228–1238. doi: 10.1093/infdis/jiu238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald B, Moyo S, Gabaitiri L, Gaseitsiwe S, Bussmann H, Koethe JR, et al. Persistently elevated serum interleukin-6 predicts mortality among adults receiving combination antiretroviral therapy in Botswana: results from a clinical trial. AIDS Res Hum Retroviruses. 2013;29(7):993–999. doi: 10.1089/aid.2012.0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boulware DR, Hullsiek KH, Puronen CE, Rupert A, Baker JV, French MA, et al. Higher levels of CRP, D-dimer, IL-6, and hyaluronic acid before initiation of antiretroviral therapy (ART) are associated with increased risk of AIDS or death. J Infect Dis. 2011;203(11):1637–1646. doi: 10.1093/infdis/jir134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tenorio AR, Zheng Y, Bosch RJ, Krishnan S, Rodriguez B, Hunt PW, et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J Infect Dis. 2014;210(8):1248–1259. doi: 10.1093/infdis/jiu254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stieh DJ, Matias E, Xu H, Fought AJ, Blanchard JL, Marx PA, et al. Th17 Cells Are Preferentially Infected Very Early after Vaginal Transmission of SIV in Macaques. Cell Host Microbe. 2016;19(4):529–540. doi: 10.1016/j.chom.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alvarez Y, Tuen M, Shen G, Nawaz F, Arthos J, Wolff MJ, et al. Preferential HIV infection of CCR6+ Th17 cells is associated with higher levels of virus receptor expression and lack of CCR5 ligands. J Virol. 2013;87(19):10843–10854. doi: 10.1128/JVI.01838-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dandekar S, George MD, Baumler AJ. Th17 cells, HIV and the gut mucosal barrier. Current opinion in HIV and AIDS. 2010;5(2):173–178. doi: 10.1097/COH.0b013e328335eda3. [DOI] [PubMed] [Google Scholar]
- 57.Sun G, Li H, Wu X, Covarrubias M, Scherer L, Meinking K, et al. Interplay between HIV-1 infection and host microRNAs. Nucleic acids research. 2012;40(5):2181–2196. doi: 10.1093/nar/gkr961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel P, Ansari MY, Bapat S, Thakar M, Gangakhedkar R, Jameel S. The microRNA miR-29a is associated with human immunodeficiency virus latency. Retrovirology. 2014;11:108. doi: 10.1186/s12977-014-0108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327(5962):198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting microRNA. The New England journal of medicine. 2013;368(18):1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 61.Polytarchou C, Hommes DW, Palumbo T, Hatziapostolou M, Koutsioumpa M, Koukos G, et al. MicroRNA214 Is Associated With Progression of Ulcerative Colitis, and Inhibition Reduces Development of Colitis and Colitis-Associated Cancer in Mice. Gastroenterology. 2015;149(4):981–992.e911. doi: 10.1053/j.gastro.2015.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stagakis E, Bertsias G, Verginis P, Nakou M, Hatziapostolou M, Kritikos H, et al. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis. 2011;70(8):1496–1506. doi: 10.1136/ard.2010.139857. [DOI] [PubMed] [Google Scholar]
- 64.Hirsch HA, Iliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, et al. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer cell. 2010;17(4):348–361. doi: 10.1016/j.ccr.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353(17):1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 66.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.