

Abstract
Patients with malignant peripheral nerve sheath tumor (MPNST), a rare soft tissue cancer associated with loss of the tumor suppressor neurofibromin (NF1), have poor prognosis and typically respond poorly to adjuvant therapy. We evaluated the effect of 299 clinical and investigational compounds on seven MPNST cell lines, two primary cultures of human Schwann cells, and five normal bone marrow aspirates, to identify potent drugs for MPNST treatment with few side effects. Top hits included Polo‐like kinase 1 (PLK1) inhibitors (volasertib and BI2536) and the fluoronucleoside gemcitabine, which were validated in orthogonal assays measuring viability, cytotoxicity, and apoptosis. DNA copy number, gene expression, and protein expression were determined for the cell lines to assess pharmacogenomic relationships. MPNST cells were more sensitive to BI2536 and gemcitabine compared to a reference set of 94 cancer cell lines. PLK1,RRM1, and RRM2 mRNA levels were increased in MPNST compared to benign neurofibroma tissue, and the protein level of PLK1 was increased in the MPNST cell lines compared to normal Schwann cells, indicating an increased dependence on these drug targets in malignant cells. Furthermore, we observed an association between increased mRNA expression of PLK1,RRM1, and RRM2 in patient samples and worse disease outcome, suggesting a selective benefit from inhibition of these genes in the most aggressive tumors.
Keywords: drug screen, MPNST, pharmacology, Schwann cell
Abbreviations
- AKTAKT
serine/threonine kinase
- BRAF
B‐Raf proto‐oncogene, serine/threonine kinase
- CI
confidence interval
- CTG
CellTiter‐Glo®, viability assay
- CTX
CellTox‐Green®, cytotoxicity assay
- DMEM
Dulbecco's modified Eagle medium
- DMSO
dimethyl sulfoxide
- DSRT
drug sensitivity and resistance testing
- DSS
drug sensitivity score
- ERK
mitogen‐activated protein kinase
- HR
hazard ratio
- HSC
human Schwann cells (two batches, HSC1 and HSC2)
- HS‐PSS
MPNST cell line
- HS‐Sch‐2
MPNST cell line
- IC50
half maximal inhibitory concentration
- KRAS
V‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog
- MEK
mitogen‐activated protein kinase kinase
- MIM
Mendelian inheritance in Man
- MPNST
malignant peripheral nerve sheath tumor
- mTOR
mammalian target of rapamycin
- NAE
NEDD8‐activating enzyme
- NEDD8
neural precursor cell expressed, developmentally down‐regulated 8
- NF1
neurofibromatosis type 1 (disease)
- NF1
neurofibromin 1 (gene/protein)
- PI3K
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase
- PLK1
polo‐like kinase 1
- QC
quality control
- RAF
see BRAF
- RAS
see KRAS
- RPPA
reverse‐phase protein array
- RRM1
ribonucleotide reductase catalytic subunit M1
- RRM2
ribonucleotide reductase catalytic subunit M2
- S1507‐2
MPNST cell line
- S462
MPNST cell line
- SCFβTrCP
Skp, Cullin, F‐box‐containing complex, E3 ligase
- SCGF
Schwann cell growth supplement
- SCM
Schwann cell medium
- SSMD
strictly standardized mean difference
- ST8814
MPNST cell line
- STR
short tandem repeats
- STS26T
MPNST cell line
- TOP2A
topoisomerase (DNA) II alpha
- TP53
tumor suppressor protein p53
- YST‐1
MPNST cell line
Introduction
Malignant peripheral nerve sheath tumors (MPNST) are rare and aggressive soft tissue cancers that arise from cells of neuroectodermal origin in the peripheral nervous system. MPNST often strikes young adults and adolescents, and nearly half of all cases are associated with the genetic syndrome neurofibromatosis type 1 (NF1, MIM 162200). The median age for the NF1‐associated cases is around 25 years, while the sporadic cases have a median in the forties (Kolberg et al., 2013). The 5‐year overall survival rate for MPNST is less than 50%, and in recent years, the prognosis has been similar for NF1 and non‐NF1 patients (Kolberg et al., 2013). There are currently no consensus guidelines for adjuvant treatment with curative intent for MPNST (Bradford and Kim, 2015), and there is a critical need for new treatment options.
Management of MPNST is currently based on general soft tissue sarcoma guidelines and involves surgery and occasionally chemo‐ and radiotherapy (ESMO, 2014). Some relapse control has been reported following radiotherapy (Yang et al., 1998); however, radiation itself can be a significant risk factor, especially for patients with NF1 (Sharif et al., 2006). The rareness of MPNST and other soft tissue cancers precludes robust clinical trials, and the trials will often include several sarcoma entities with different genetic composition, tumor biology, and hence different drug response. There are 62 interventional trials listed in the US‐based National Institutes of Health (NIH) database that are eligible for patients with MPNST, and 16 of these are currently open or are recruiting patients (Table S1). However, most trials include several different sarcomas, often leaving the numbers of MPNSTs too low, lacking statistical power to conclusively document any benefit for these patients. Only a handful of trials have focused specifically on MPNST with focus on compounds that target the biological processes involved in MPNST development (Table 1). So far, however, none of these trials have compelled changes in the management of this malignancy. Notably, the SARC006 trial, which tested the effect of the TOP2A inhibitors doxorubicin and etoposide in combination with ifosfamide, reported an overall response rate of 33% in the sporadic and 17% in NF1‐associated MPNST, respectively, although both were below the set target of 40% (Widemann et al., 2013). The TOP2A gene has previously been identified as amplified and upregulated in a large subset of MPNST patient samples (Skotheim et al., 2003), which could explain the positive effect of TOP2A inhibition in these patients.
Table 1.
Clinical trials with main focus on MPNSTa
| Trial ID | Intervention | Drug class | Phase | Patient enrollment | Status and results |
|---|---|---|---|---|---|
| NCT01661283 (SARC016) | Bevacizumab/everolimus | Cell surface receptor antibody and mTOR inhibitor | II | 17 NF1‐associated MPNST, 8 sporadic MPNST |
Active, not recruiting Clinical benefit rate (CBR): 12% (3 of 25) (Widemann et al., 2016) |
| NCT00464620 (SARC009) | Dasatinib | Kinase inhibitor (KIT Src) | II | 14 MPNST |
Active, not recruiting All progressed within 4 months (Schuetze et al., 2016) |
| NCT00304083 (SARC006) | Doxorubicin/ifosfamide followed by etoposide/ifosfamide | Conventional chemotherapy | II | 33 NF1‐associated MPNST, 15 sporadic MPNST |
Completed Overall response rate 17% in NF1, 33% in sporadic (Widemann et al., 2013) |
| NCT00068367 | Erlotinib | Kinase inhibitor | II | 24 MPNST; 20 patients were evaluable for response. |
Completed One stable disease; 19 no response. Median progression‐free survival: 2 months. Median overall survival: 4 months (Albritton et al., 2006) |
| NCT02008877 (SARC023) | Ganetespib, sirolimus | HSP inhibitor and mTOR inhibitor | I/II | 38 MPNST | Active, not recruiting |
| NCT01418001 | Pazopanib in combination with gemcitabine and docetaxel | Kinase inhibitor and conventional chemotherapy | I/II | 5 sarcoma | Terminated |
| NCT00427583 | Imatinib mesylate | Kinase inhibitor | II/III | 7 MPNST |
Terminated All were taken off study; 5 due to progressive disease, one due to toxicity, and one withdrawal (Chugh et al., 2009) |
| NCT02691026 | Pembrolizumab | Cell surface receptor antibody | II | 18 MPNST | Recruiting |
| NCT02584647 | Pexidartinib (PLX3397), sirolimus | Kinase inhibitor, mTOR inhibitor | I/II | 49 MPNST | Recruiting |
| NCT00837148 | Sorafenib, dacarbazine | Kinase inhibitors (BRAF and VEGFR) and conventional chemotherapy | II | 12 MPNST evaluated |
Completed. All progressed within 12 months. Mean PFS: 1.7 months. Two patients with MPNST had regression or cystification of metastatic disease without a RECIST response (Maki et al., 2009) |
Data from clinicaltrials.gov.
Malignant peripheral nerve sheath tumors are highly complex malignancies with multiple copy number alterations (Brekke et al., 2010; Lothe et al., 1996; Mertens et al., 2000) including alterations in several clinically relevant target genes at chromosome arm 17q (Kolberg et al., 2015; Skotheim et al., 2003; Storlazzi et al., 2006). Inactivating mutations in the NF1 tumor suppressor gene are found in both NF1‐associated and sporadic MPNST (Bottillo et al., 2009; Upadhyaya et al., 2008). Loss of NF1 activity leads to activation of RAS and consequently contributes to the PI3K/AKT/mTOR and RAF/MEK/ERK signaling in MPNST (Ågesen et al., 2005; Berner et al., 1999; Brems et al., 2009; Danielsen et al., 2015; Endo et al., 2013; Nielsen et al., 1999). Components of this network have been investigated as potential therapeutic targets (Fig. S1). In preclinical MPNST models, some drugs targeting these pathways have shown encouraging results, including the mTOR inhibitors everolimus and AZD8055 (De Raedt et al., 2011; Endo et al., 2013; Varin et al., 2016), and the multikinase Raf inhibitor sorafenib (Ambrosini et al., 2008; Castellsague et al., 2015), as well as a selection of other drugs that inhibit pathways associated with NF1, such as gemcitabine (Schoeler et al., 2007), erlotinib (Mahller et al., 2007), imatinib (Aoki et al., 2007; Patwardhan et al., 2014), pexidartinib (Patwardhan et al., 2014), and sunitinib (Zietsch et al., 2010). However, clinical trials have not confirmed any therapeutic benefit for the limited number of drug candidates identified by a knowledge‐based approach (Table 1). Of note, a recent study suggests the effect of MEK inhibitor selumetinib against inoperable plexiform neurofibromas in children with NF1 (Dombi et al., 2016).
As a complement to the knowledge‐based drug discovery approach, we here present a comprehensive high‐throughput approach to identify new therapeutic opportunities for MPNST among a large panel of clinical and investigational drugs. We identify and rank the compounds with the highest effect and specificity for MPNST cells by pharmacological analysis of seven MPNST cell lines using two normal Schwann cell cultures and bone marrow aspirates from healthy donors as controls. Candidate drugs showing the highest selectivity were subjected to validation in independent experiments.
Material and Methods
Cell lines, primary cultures, and patient material
The original drug testing assay included two primary cultures of human Schwann cells (HSC) termed HSC1 and HSC2 that were isolated from human spinal nerves (ScienCell, Carlsbad, CA, USA) and four MPNST cell lines, STS26T (Dahlberg et al., 1993) and ST8814 (Reynolds et al., 1992) (kindly provided by Nancy Ratner, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA), and S462 (Frahm et al., 2004) and S1507‐2 (Spyra et al., 2011) (kindly provided by Lan Kluwe, University Medical Center Hamburg‐Eppendorf, Germany). Later, the three MPNST cell lines HS‐PSS, HS‐Sch‐2 (Sonobe et al., 2000), and YST‐1 (Nagashima et al., 1990) (Riken BioResource Center, Ibaraki, Japan) were assayed with an extended and updated drug library (see below). STS26T and YST1 were derived from non‐NF1 patients and have been reported to express wild‐type NF1 (Miller et al., 2006; Nagashima et al., 1990), and the remaining cell lines are derived from NF1 patients and do not express NF1.
All cancer cell lines were maintained in DMEM‐F12 medium supplemented with 10% fetal bovine serum, 2 mm l‐glutamine, 100 units·mL−1 penicillin, and 100 μg·mL−1 streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). The HSC were maintained in Schwann cell medium (SCM, Cat. no. 1701, ScienCell) supplemented with Schwann cell growth supplement (SCGS, Cat. no. 1752, ScienCell) according to the suppliers’ recommendations.
The identity of the cell lines was validated by genotyping of the isolated DNA (Table S2, Appendix S1) according to the protocol of the AmpFLSTR Identifiler PCR Amplification Kit (Life Technologies by Thermo Fisher Scientific). For the cell lines YST‐1, HS‐PSS, HS‐Sch‐2, identical STR profiles were provided by Riken (also available at www.expasy.org/cellosaurus), and for STS26T and ST8814, identical STR profiles were obtained by N. Ratner (personal communication). All the cell lines were tested and found negative for mycoplasma contamination using the MycoAlert detection kit (Lonza Ltd, Basel, Switzerland).
Fresh‐frozen tumor material was available from 30 MPNSTs (17 NF1‐associated and 13 non‐NF1 cases) and eight benign neurofibromas (seven dermal and one plexiform) from Oslo University Hospital, Oslo, Norway, and Skåne University Hospital, Lund Sweden, as previously described (Kolberg et al., 2015). Briefly, DNA and RNA were extracted from tissue sections consisting of >90% representative tumor tissue as identified by a reference sarcoma pathologist. Informed consent was obtained from all living patients, and the study was approved by the South‐Eastern Norway Regional Health Authority and the Regional Ethics Committee at Lund University according to national legislation. Bone marrow aspirates were collected from five healthy donors after informed consent using approved study protocols (Helsinki Ethical Committee 239/13/03/00/2010 and 303/13/03/01/2011).
Drug sensitivity and resistance testing and data analysis
Drug sensitivity and resistance testing (DSRT) was performed as described earlier (Pemovska et al., 2013) on all seven MPNST cell lines and two normal HSC cultures. The initial drug library contained 309 compounds, while three MPNST cell lines were screened with 527 compounds (303 overlapping). Reference DSRT data were also available for 299 overlapping drugs for five primary cultures of adult human bone marrow cells derived from healthy donors, and from a reference collection of cell lines from different cancer types, including colorectal (n = 36), ovarian (n = 30), and acute myeloid leukemia (n = 28) (Mpindi et al., 2016). Briefly, the compounds were dissolved in 100% dimethyl sulfoxide (DMSO) or water (Table S3) and dispensed on tissue culture‐treated 384‐well plates (Cat. No. 3707, Corning, Tewksbury, MA, USA) using an acoustic liquid handling instrument, Echo 550 (Labcyte Inc., Sunnyvale, CA, USA). The compounds were plated in five concentrations using 10‐fold dilutions covering a 10 000‐fold concentration range (e.g., 1–10 000 nmol·L−1). The preprinted plates were kept in pressurized StoragePods (Roylan Developments Ltd., Fetcham, UK) under inert nitrogen gas until needed. Five microlitre of CellTox‐Green (CTX) Cytotoxicity Assay Reagent (Promega, Fitchburg, WI, USA), 1 : 200 dilution in growth media, was added to each 384‐well plate prior to seeding of cells to achieve a final concentration of 1 : 1000. The CTX assay is based on quantification of fluorescence‐labeled DNA released from disrupted cells. Plates were subsequently centrifuged briefly and put on an orbital shaker for 10 min. Twenty microliters of single‐cell suspension (750–1000 cells) was transferred to each well using a Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific). Proliferation rates and growth patterns of the cell lines were evaluated prior to the experiments in order to determine the optimal cell seeding density to assure logarithmic growth throughout the 72‐h incubation period. The plates were incubated in a humidified environment at 37 °C and 5% CO2, and after 72 h, cell cytotoxicity and cell viability (assessed using CellTiter‐Glo (CTG) Luminescent Cell Viability Assay, Promega) were measured according to the manufacturer's instructions with a PHERAstar FS microplate reader (BMG Labtech GmbH, Ortenberg, Germany). The CTG assay generates luminescence proportional to the amount of ATP that is extracted from living cells in the culture. The data were normalized to negative control (0.1% DMSO) and positive control wells (containing 100 μm benzethonium chloride, effectively killing all cells). Quality control (QC) metrics, Z′, and strictly standardized mean difference (SSMD, i.e., the effect size) for each plate were calculated as described (Mpindi et al., 2015; Yadav et al., 2014). Drug sensitivity scores (DSS) were calculated for both the CTG assay (DSSCTG) and the CTX assay (DSSCTX) by fitting of the dose–response curves on the basis of a four‐parameter logistic fit function defined by the top and bottom asymptote, the slope, and the inflection point (IC50). In the curve fitting, the bottom asymptote of the curve was fixed to 0% inhibition (=100% viability), whereas the top asymptote was allowed to float above 10% inhibition (i.e., drugs causing < 10% inhibition were considered inactive, DSS≡0), and the slope was allowed to float between 0 and 2.5 (Mpindi et al., 2015; Yadav et al., 2014). For validation, the orthogonal ApoToxGlo Triplex assay (Promega) was performed according to the manufacturer's instructions, in parallel with the CTG assay (using 15 dilutions from 1 000 nm to 0.0625 nm for each drug). The Triplex assay allows for luminescence measurements of caspase 3/7 activity as a measure of apoptosis levels in addition to viability and cytotoxicity in response to drug treatment. Cells treated with 0.1% DMSO were used as negative controls, 10 μm staurosporin was used as positive control for apoptosis, while 100 μm benzethonium chloride was used as positive control for viability and cytotoxicity.
The average quality control value (Z′) for all cell lines in the viability assay was 0.72 ± 0.04 and average SSMD was 14.1 ± 2.4 for each drug plate (Table S4A). The two cell lines S462 and S1507‐2 were re‐tested for technical validation demonstrating high reproducibility of the CTG data [Pearson's correlation r = 0.990 and 0.975, respectively (Fig. S2A and S2B)]. For the two independent normal HSC cultures, the Pearson's correlation was r = 0.982 (Fig. S2C). Overall, there was also a strong correlation in drug response patterns between the MPNST cell lines and the HSC primary cultures (Pearson's correlation r = 0.924) and to a lesser extent between MPNST and bone marrow (Pearson's correlation r = 0.752) (Fig. S2D). For the cytotoxicity assay, three cell lines (S462, YST‐1, and HS‐Sch‐2) failed the quality control. The remaining cell lines had an average Z′ of 0.58 ± 0.13 and an average SSMD of −10.5 ± 3.0 (Table S4B).
Reverse‐phase protein array analyses
Expression of 297 cancer‐related proteins and phosphoproteins was evaluated by reverse‐phase protein array analyses (RPPA) at the MD Anderson RPPA core facility (Houston, TX, USA) in the four MPNST cell lines, S1507‐2, S462, ST8814, and STS26T, and normal HSC1 according to the published protocol (Tibes et al., 2006). Later, the MPNST cell lines YST‐1, HS‐PSS, and HS‐Sch‐2, as well as a replicate of the HSC1, were submitted to the same analysis using an updated RPPA version including 306 antibodies, of which 271 were overlapping with the initial 297 antibodies.
Gene expression analysis
The genome‐wide gene expression levels of the seven MPNST cell lines, HS‐PSS, HS‐Sch‐2, S1507‐2, S462, ST8814, STS26T, and YST‐1, as well as the normal HSC1, were assessed by synthesis of cDNA from isolated RNA and subsequent hybridization to the GeneChip Human Transcriptome Array 2.0 according to the supplier's protocol (Affymetrix, Thermo Fisher Scientific Inc.) (see Appendix S1).
DNA copy number analyses
DNA from four MPNST cell lines, S1507‐2, S462, ST8814, and STS26T, were individually processed and hybridized on Genome‐Wide Human SNP Array 6.0 from Affymetrix (Thermo Fisher Scientific Inc.) as described in the Affymetrix Cytogenetics Copy Number Assay User Guide (P/N 702607 Rev. 2) (see Appendix S1).
Mutation analyses
The genes TP53 (exon 2–11) and BRAF (exon 15) were sequenced using DNA extracts of the four MPNST cell lines S1507‐2, S462, ST8814, and STS26T by Sanger sequencing using in‐laboratory‐established protocols (Ahlquist et al., 2008; Berg et al., 2010) (see Appendix S1).
Statistical analyses
Association between gene expression and disease‐specific survival was analyzed using Cox proportional hazards regression modeling with Wald test to provide univariate hazard ratios (HR) and confidence intervals (CI) and visualized by Kaplan–Meier plots. Comparison of gene expression differences between MPNST cell lines and other cell lines, and between MPNST patient samples and neurofibromas, was assessed by two‐tailed Student's t‐test for independent samples, and correlations between drug screen data from repeated or separate runs were assessed by Pearson's test. Spearman's correlation was used to compare IC 50 from different screening platforms to reduce influence of outliers. All statistical analyses were performed using the spss 21 software (IBM Corporation, Armonk, NY, USA).
Results
Identification of MPNST‐specific drugs
Four MPNST cell lines S1507‐2, S462, ST8814, and STS26T and two HSC primary cultures were subjected to high‐throughput DSRT with 309 emerging and clinical oncology compounds. Three additional MPNST cell lines HS‐PSS, HS‐Sch‐2, and YST‐1 were screened using an updated compound library of 527 compounds (with 303 compounds overlapping between both libraries). Data for 299 of these drugs were also available from normal bone marrow aspirates from healthy donors. As drug sensitivity readout, we used two chemically different assays measuring cell viability (CTG; Fig. 1A; Table S5) and cytotoxicity (CTX; Fig. S3, Table S6).
Figure 1.
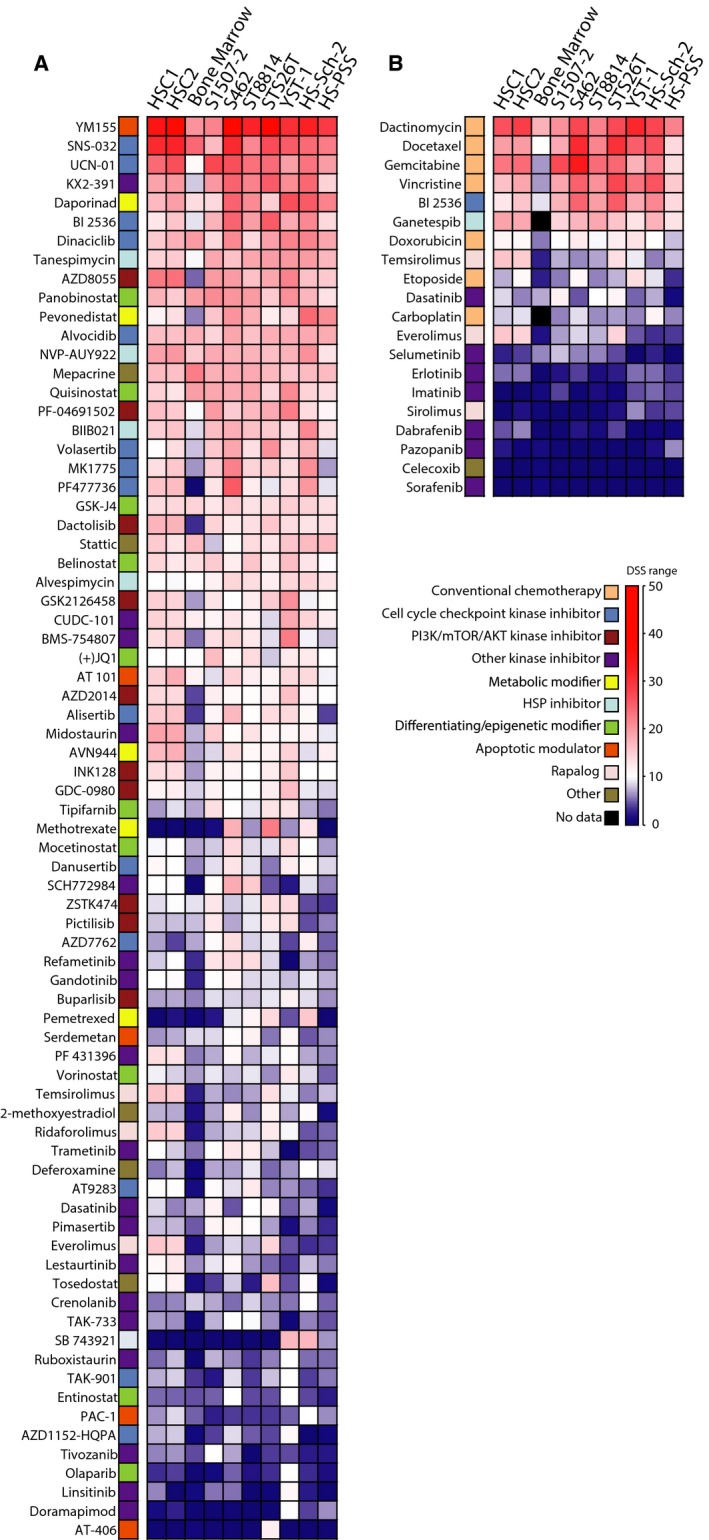
Drug response heatmaps from CellTiter‐Glo (CTG) viability assay for seven MPNST cell lines, two normal human Schwann cell (HSC) cultures, and bone marrow (mean result from five healthy individuals). Drug responses for targeted drugs (excluding chemotherapeutic drugs) with a drug sensitivity score (DSSCTG) of 10, or more, in at least one MPNST cell line (A), and chemotherapeutic and other targeted drugs that have been used in clinical treatment of patients with MPNST (B). The same color coding and DSS gradient is used for both heatmaps.
Twenty of the tested compounds are used in the clinic to treat MPNST (ESMO, 2014), or have been tested in recent clinical trials including patients with MPNST (MPNST trials: Table 1; sarcoma trials: Table S1). Twelve of these showed strong to moderate response in MPNST cells (DSSCTG > 5) in our assay, and they also showed differential response in MPNST cells as compared to bone marrow cells (missing data for ganetespib and carboplatin in bone marrow) (Fig. 1B). Only three, docetaxel, vincristine, and BI2536, showed selectively higher response in MPNST cells as compared to HSC (Fig. 1B, Table S5). Strikingly, both mTOR inhibitors temsirolimus and everolimus appear to be more effective in normal HSC than in MPNST cells, while sirolimus did not inhibit any of the cells in our assay at the concentrations used.
To systematically identify the most potent drugs, the DSSCTG values were filtered according to MPNST specificity and off‐target toxicity (Fig. 2). Of the 299 drugs tested in all cell types, including bone marrow, 111 had DSSCTG ≥ 10 in at least one MPNST cell line. Eighty‐one of these were well, or moderately, tolerated in bone marrow cells (DSSCTG < 10 in bone marrow), and of these, 49 drugs showed differential sensitivity in MPNST cells with five or more DSSCTG units higher in MPNST cells as compared to normal bone marrow. Nine of these drugs also showed higher selectivity for MPNST cells as compared to HSC (ΔDSSCTG(MPNST vs. HSC) ≥ 5). These included the polo‐like kinase 1 (PLK1) inhibitor BI2536, three tubulin/kinesin inhibitors (vinorelbine, vincristine, and SB 743921), two nucleoside analogs (floxuridine and thioguanine), two folate analogs (methotrexate and pemetrexed), as well as one proteasome‐ubiquitin inhibitor [NEDD8‐activating enzyme (NAE) inhibitor pevonedistat]. Four of these nine compounds were selected for validation: BI2536 was selected as a targeted kinase inhibitor, floxuridine was selected due to its association with thymidine kinase 1 (TK1), previously identified as a prognostic biomarker (Kolberg et al., 2015), while methotrexate and pemetrexed were selected for their differential response in MPNST cell lines. In addition, a second PLK1 inhibitor, volasertib, and a second fluoronucleoside, gemcitabine, were included from the list of 49 compounds with selectivity toward MPNST cells over bone marrow cells. Three of these six compounds, BI2536, volasertib, and gemcitabine, also showed a strong cytotoxic effect (DSSCTX > 10) in the MPNST cells, while methotrexate, pemetrexed, and floxuridine showed limited or no cytotoxicity in the CTX assay (Table S6).
Figure 2.
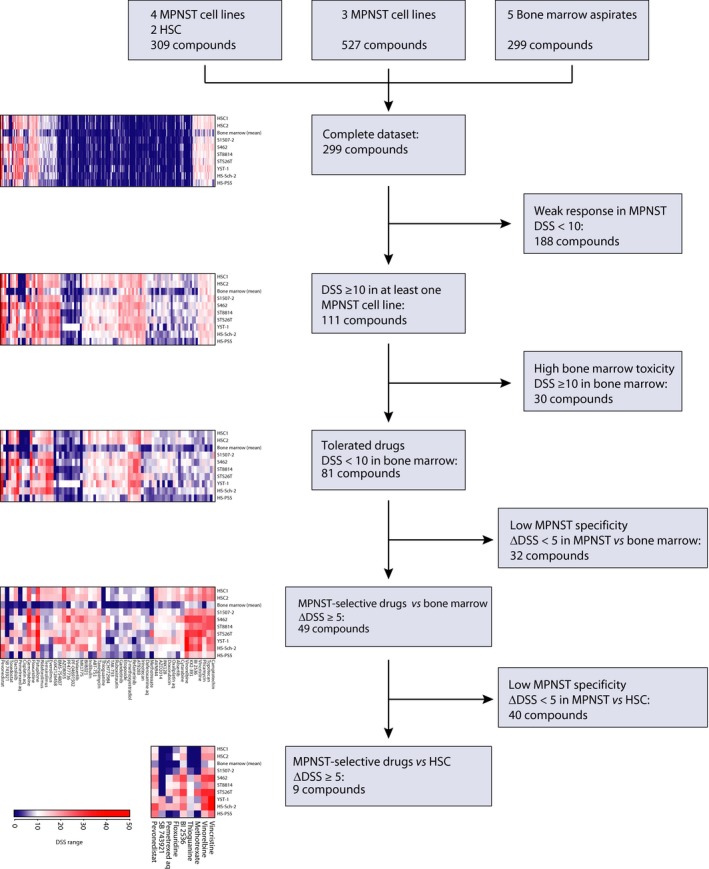
Identification of candidate drugs for MPNST treatment. Filtering steps used to identify drugs with high specificity and selectivity toward MPNST as compared to bone marrow and normal human Schwann cells (HSC) based on drug sensitivity scores from CellTiter‐Glo viability assay (DSSCTG).
The validation experiments for BI2536, volasertib, gemcitabine, methotrexate, pemetrexed, and floxuridine were performed using orthogonal assays in four MPNST cell lines (Fig. S4). A good correlation between the initial screen and the validations was observed for the two PLK1 inhibitors, BI2536 and volasertib, as well as for gemcitabine (Fig. 3A). However, the high drug responses for methotrexate, pemetrexed, and floxuridine observed in the initial DSRT were not confirmed (Fig. S4). Interestingly, the DSSCTG values of BI2536 and volasertib appear to be slightly higher for the seven MPNST cell lines as compared to the DSSCTG values of a panel of 94 cell lines from colon and ovarian cancer and leukemia (Fig. 3B), and significantly higher in MPNST cells for gemcitabine. Notably, in the extended drug panel consisting of 527 compounds tested on the three MPNST cell lines HS‐PSS, HS‐Sch‐2, and YST‐1 only, another PLK1 inhibitor, GSK‐461364, showed even higher DSSCTG values for all three cell lines than BI2536 and volasertib (Table S5), while the PLK1 inhibitor TAK‐960 did not inhibit these cells. High DSS values were also observed for the kinase inhibitor rigosertib, which is an inhibitor of both PLK1 and PI3K.
Figure 3.
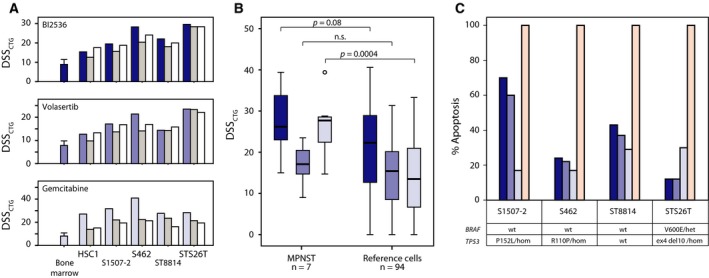
Independent validation and apoptosis assay of MPNST‐specific drugs. Comparison of drug sensitivity scores from initial (blue bars, including average data and standard deviation for the five bone marrow samples) CellTiter‐Glo viability assay (DSSCTG), and two subsequent validation rounds [manual (medium gray bars); custom plate (white bars)] (A). DSSCTG obtained for the three drugs BI2536 (dark blue), volasertib (medium blue), and gemcitabine (light blue) from MPNST cell lines in comparison with a reference set of 94 cancer cell lines (colon, ovarian, and leukemia); two‐tailed P‐values from independent samples t‐test, assuming unequal variance (B). The maximum level of apoptosis measured by a luminescence‐based caspase‐3/7 activation assay, induced by BI2536 (dark blue), volasertib (medium blue), and gemcitabine (light blue), in comparison with staurosporin (100% apoptosis, pink) and 0.1% DMSO (0% apoptosis) (C). The mutation status of TP53 and BRAF in each cell line is shown (het—heterozygous; hom—homozygous).
Cellular responses on specific compounds
The apoptotic response to specific compounds was measured using a photometric caspase 3/7 assay in the four cell lines S1507‐2, S462, ST8814, and STS26T after 72 h of drug exposure normalized to cells treated with staurosporin as positive control. We found that both PLK1 inhibitors, BI2536 and volasertib, induced apoptosis in the TP53‐mutant cell lines S1507‐2 and S462, as well as in the TP53 wild‐type cells ST8814, while the STS26T cell line, which harbors a homozygous 10‐bp deletion in exon 4 of TP53, had the lowest level of apoptosis induced by PLK1 inhibition (Fig. 3C). In the presence of gemcitabine, all the four cell lines showed a moderate apoptotic response at 15–30% under our assay conditions (Fig. 3C).
One of the MPNST cell lines, STS26T, had an oncogenic V600E mutation in BRAF (Fig. 3C), which is a known marker for benefit of BRAF inhibition in melanoma. We only detected a weak sensitivity against the five tested BRAF inhibitors, RAF265, vemurafenib, regorafenib, dabrafenib, and sorafenib in STS26T, with similar results found for the BRAF wild‐type cell lines. Actually, the normal HSCs were moderately more sensitive than all the MPNST cell lines.
Gene and protein expression of drug targets in MPNST
The expression of drug targets in the MPNST cell lines and HSC was examined by exon‐level gene expression arrays and protein expression arrays in vitro. On the gene expression level, there was little variation in the expression of PLK1 between MPNST cell lines and HSC (Fig. S5A). On the protein level, however, we found that the expression of PLK1 was higher in the MPNST cell lines as compared to normal HSC (Fig. 4A). Among all the 271 tested proteins on the RPPA array, PLK1 ranked among the top 10 with respect to difference between MPNST and normal cells (Table S7), suggesting that PLK1 is an accessible target in MPNST cells. The increased expression of PLK1 in MPNST as compared to HSC was not associated with gain of gene copy number, as assessed in four MPNST cell lines. Actually, two of the cell lines, S1507‐2 and ST8814, had genomic losses from a chromosomal region covering PLK1 (16p12.2), and for ST8814, this may partly explain the relatively low PLK1 protein level as compared to the other MPNST cell lines (Fig. 4B).
Figure 4.
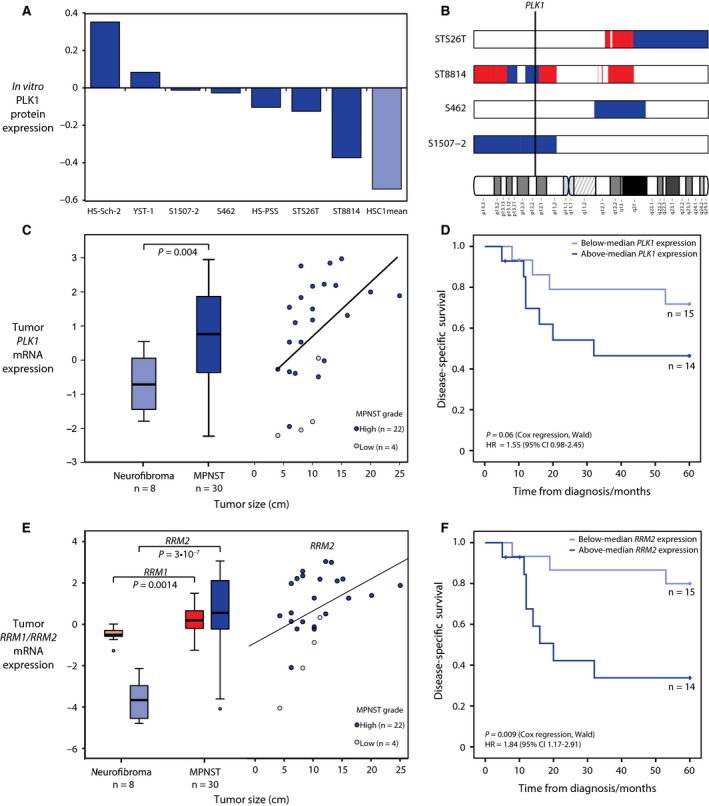
Expression of the drug targets PLK1, RRM1, and RRM2 in MPNST cell lines and prognostic relevance in patients with MPNST. Normalized RPPA protein expression of PLK1 in MPNST cell lines (median = 0 across 271 cancer‐relevant proteins) indicates an increased expression of PLK1 in seven MPNST cell lines as compared to the mean of duplicate runs of normal Schwann cell HSC1 (A). Regions of genomic gain (red) are not observed in the region on chromosome 16 harboring PLK1 (16p12.2) in the tested MPNST cell lines, while regions of loss (blue) are observed in the two cell lines S1507‐2 and ST8814 (B). Gene expression of PLK1 is significantly higher in MPNST tumor samples as compared to benign neurofibromas (C, left panel), and high expression in MPNST is associated with high tumor grade and large tumor size (C, right panel). MPNST patients with high expression of PLK1 in the tumor had worse outcome than patients with low expression, although not significantly at a 5% level (the P‐value and hazard ratio (HR) for PLK1 expression as a continuous variable in univariate Cox regression analysis for five‐year disease‐specific survival are shown) (D). The gene expression of gemcitabine target RRM1 and its activator RRM2 is significantly higher in MPNST tumor samples as compared to benign neurofibromas (E, left panel), and high expression of RRM2 in MPNST is associated with high tumor grade and large tumor size (E, right panel). MPNST patients with high expression of RRM2 in the tumor had significantly worse outcome than patients with low expression (P‐value and hazard ratio (HR) for gene expression as a continuous variable in univariate Cox regression analysis for 5‐year disease‐specific survival are shown) (F).
Gene expression data were also available for 30 MPNST patient samples and eight benign neurofibromas (Kolberg et al., 2015), and in these patient samples, the gene expression of PLK1 was significantly upregulated in malignant tumors as compared to benign tumors (P = 0.004, two‐sided independent samples t‐test with equal variance; Fig. 4C, left panel). Among the 30 MPNST samples, a high level of PLK1 expression was associated with large tumor size and high tumor grade (Fig. 4C, right panel). Patients with higher PLK1 expression also showed worse outcome in univariate analysis, although slightly above the 5% significance level (Fig. 4D). The mechanism of action is more complex for gemcitabine, but one of its direct targets is RRM1 where gemcitabine acts as a suicide substrate (Kolberg et al., 2004; Pereira et al., 2004). We did not observe any significant difference in RRM1 or its activator and binding partner RRM2 in the MPNST cell lines as compared to HSC1 (Fig. S5A); however, these genes were both significantly upregulated in MPNST patient tumor samples as compared to benign neurofibromas (Fig. 4E). The level of RRM2 was positively correlated with tumor grade and size, and strongly associated with poor patient outcome (Fig. 4E,F). For RRM1, there was also an association with poor outcome, although not statistically significant in our patient sample series (Fig. S5E). There was no significant difference in gene expression of the three genes in patient samples from non‐NF1‐ and NF1‐associated MPNSTs (Fig. S5B–D).
Discussion
There is a need for improved treatment options against MPNST, and to this end, we have systematically tested a comprehensive library of approved and investigational compounds to identify drug candidates that show differential inhibition of MPNST cell growth compared to normal Schwann cells and bone marrow cells. This approach was chosen to identify drugs with low neuro‐ and myelotoxicity, which are common dose‐limiting side effects in the clinical setting. Due to the young age at onset and long life expectancy after curative treatment, avoiding systemic side effects is of particular importance for patients with MPNST. The selection thresholds for the identification of potential new drug candidates were chosen to ensure robust selection of candidate drugs based on true biological differences between MPNST cell lines, normal nerve sheath cells, and bone marrow cells. Other dose‐limiting side effects associated with the drugs, such as gastrointestinal, dermatological, and liver toxicities, were not tractable in our preclinical models.
Recently, several landmark studies have demonstrated how cell lines recapitulate the major molecular phenotypes of cancer and have substantiated their value as preclinical model systems to assess a variety of pharmacogenomic relationships with potential therapeutic impact (Barretina et al., 2012; Garnett et al., 2012; Greshock et al., 2010; Haverty et al., 2016). Unfortunately, no MPNST cell lines were included in these studies, highlighting the need for a large systematic screen of available and emerging drugs in a panel of MPNST cell lines to select promising candidates for clinical testing. In our study, we present drug response as DSS values which are derived from the area under the dose–response curves for each drug, and this measure has recently been demonstrated to provide better agreement when comparing results from different laboratories than the inflection points of the dose–response curve (IC 50) (Mpindi et al., 2016).
The most promising drug candidates identified here include the PLK1 inhibitors volasertib and BI2536, and the fluoronucleoside gemcitabine. PLK1 plays an important role in progression of the cell cycle and is known to be overexpressed in many different cancer types, which makes this gene an interesting therapeutic target (Abbou et al., 2016; Gjertsen and Schoffski, 2015; Gutteridge et al., 2016). In patient biopsies of MPNST, we and others have shown expression changes in various cell cycle‐associated proteins (Ågesen et al., 2005; Berner et al., 1999; Endo et al., 2011; Kourea et al., 1999; Nielsen et al., 1999). Here, we report that PLK1 is overexpressed in MPNST compared to benign patient samples; in MPNST cell lines, PLK1 protein expression is higher than in normal HSC. However, the increased expression of PLK1 cannot be explained by DNA copy number aberrations, neither in patient samples (Brekke et al., 2010) nor in the cell lines reported here. Furthermore, there was no clear difference in mRNA levels between the mean of the MPNST cells and HSC1 cells. This suggests that the PLK1 is stabilized at the protein level, at least in the MPNST cell lines. A possible mechanism for PLK1 stabilization might be the deregulation of the SCFβTrCP/proteasome degradation pathway, which has recently been described as a degradation pathway for PLK1 (Giraldez et al., 2017).
Ongoing efforts aim to develop PLK1 inhibitors with improved pharmacokinetic and dynamic profiles, and volasertib is currently the most clinically advanced PLK1 inhibitor (Gjertsen and Schoffski, 2015). BI2536 was among the first PLK1 inhibitors to be tested in the clinic, but the efficacy was limited, partly due to the short terminal half‐life. Nevertheless, the drug seemed to be well tolerated (Schoffski et al., 2010). The effect of the PLK1 inhibitor TAK‐960 was recently assessed in a panel of sarcoma cell lines, including two MPNST cell lines (Nair and Schwartz, 2015). Nair and Schwartz found that all tested cell lines were sensitive to TAK‐960 at nanomolar concentrations. The authors also found that inhibition of PLK1 in the TP53‐mutant MPNST cell lines, both by small compound inhibition and by siRNA‐mediated gene knockdown, led to the induction of polyploidy, which was in contrast to TP53 wild‐type or TP53 −/− sarcoma cell lines where PLK1 inhibition led to G2 arrest and apoptosis (Nair and Schwartz, 2015). In our study, however, we also observed apoptosis in the two TP53‐mutant cell lines S1507‐2 and S462, which carry point mutations P152L and R110P, respectively (Fig. 3C). Apparently, the TP53 status is not sufficient to explain the relationship between PLK1 inhibition and induction of apoptosis. The available data from other cancer cell lines indicated that MPNST cells have a uniquely high sensitivity toward gemcitabine and PLK1 inhibitors (Fig. 3B), which suggests that the biological processes inhibited by these drugs cannot be easily compensated by other pathways in MPNST cells, at least not within the timeframe of the compound screen. The high sensitivity toward PLK1 inhibitors in MPNST as compared to other cancer cell lines may at least in part be linked to the increased RAS signaling due to the loss of tumor suppressor NF1. This is supported by studies of colon cancer models showing increased sensitivity toward PLK1 inhibition in KRAS‐mutated cells (Luo et al., 2009; Wang et al., 2016).
We have previously reported that enzymes in the nucleotide metabolism, in particular thymidine metabolism, are upregulated in MPNST (Kolberg et al., 2015). Several drugs interfering with this pathway were indicated as potential candidates in the drug screen performed here, including the fluoronucleosides gemcitabine and floxuridine, the thiopurines thioguanine and mercaptopurine, as well as the folate antagonists methotrexate and pemetrexed. Of these, we validated the effect of gemcitabine, an inhibitor of RRM1 and de novo DNA synthesis (Kolberg et al., 2004). Gemcitabine is already approved for many different cancer types, including sarcoma (Ducoulombier et al., 2016). Resistance against gemcitabine may partly be mediated by metabolic inactivation of gemcitabine catalyzed by cytidine deaminase (CDA) (Gilbert et al., 2006). However, we did not see any differences in CDA expression levels in the MPNST cells as compared to HSC.
The gene expression data from patients with MPNST and benign tumors suggest that the drug targets PLK1 and RRM1, as well as the RRM1 activator RRM2, are upregulated in malignant tumors and that the level of aggressiveness, as indicated by patient survival, is directly associated with the gene expression levels, especially for RRM2 (Fig. 4C–F). A continuous supply of deoxyribonucleotides provided by the RRM1/RRM2 complex is required in rapidly growing and dividing cells, and PLK1 is needed to promote cell cycle progression and to avoid apoptosis. Therefore, inhibition of these factors might be especially effective in the most aggressive tumors. In a clinical setting, PLK1 and RRM2 expression may have both prognostic and predictive values, as patients with high expression of these genes are most likely to experience disease progression, and at the same time, those with the highest levels are most likely to respond to PLK1 inhibitors and gemcitabine treatment. Interestingly, a recent study in pancreatic cancer cells showed that PLK1 inhibition enhances the effect of gemcitabine, also in gemcitabine‐resistant cells (Li et al., 2016). In view of the current results, a therapeutic combination strategy with gemcitabine and PLK1 inhibitors seems rational also for patients with MPNST.
Novel drug targets for MPNST have also been suggested by others based on preclinical findings (Semenova et al., 2017; Teicher et al., 2015; Yamashita et al., 2014). A recent drug screen of 63 sarcoma cell lines, including the two MPNST cell lines MPNST and ST8814, confirmed the heterogeneous responses among different soft tissue cancers (Teicher et al., 2015). None of the highlighted drugs in that study were found to be targeting MPNST cells, with the exception of a moderate inhibition by the PARP1 inhibitor talazoparib, and weak effect of selected aurora kinases (TAK‐901, SCH‐1473759, AS‐703569, and ABT‐348) (Teicher et al., 2015). However, in the available raw data, which were recorded after 96‐h drug exposure, both PLK1 inhibitors BI2536 and volasertib, as well as gemcitabine, were among the top‐ranked drugs with respect to low IC 50 values, which are in agreement with our own data (Fig. S6).
In conclusion, we have identified two PLK1 inhibitors, BI2536 and volasertib, and the DNA synthesis inhibitor gemcitabine as highly effective against MPNST cells, while being tolerable to normal HSC cells and bone marrow cells, and we propose these drugs as good candidates for future clinical testing, alone or in combination. The expression levels of target genes for these treatments also carry prognostic value, and we advocate for their potential as prognostic and predictive factors for future clinical trials to be further elucidated.
Author contributions
MK, JB, OK, and RAL involved in conception and design of the study. MK, JB, AM, CHB, MH, IAE, and SAD acquired data (cell culturing, cell assays, gene expression analysis, DNA copy number analysis, etc). All authors analyzed and interpreted the data. MK and JB drafted the manuscript. All authors reviewed and revised the manuscript. OK and RAL provided administrative, technical, or material support. RAL supervised the study.
Supporting information
Fig. S1. Overview of drugs tested in MPNST patients.
Fig. S2. Correlation between CellTiter‐Glo (CTG) viability experiments.
Fig. S3. Drug cytotoxicity response profiles of MPNST cell lines and normal HSCs.
Fig. S4. Dose–response curves.
Fig. S5. Gene expression of drug targets in cell lines and patient tumors, and association to patient survival.
Fig. S6. Correlation with public dataset.
Table S1. List of drugs in current or previous clinical testing against sarcoma, including MPNST (clinical trials.gov).
Table S2. STR profiles of MPNST cell lines*.
Table S3. List of tested compounds.
Table S4. (A) QC‐scores from viability assay (CTG). (B) QC‐scores from cytotoxicity assay (CTX).
Table S5. Cell viability assay (CTG) data.
Table S6. Cytotoxicity assay (CTX) data.
Table S7. Protein expression data from reverse phase protein lysate microarray (RPPA)a.
Table S8. Primer sequences.
Appendix S1. Supplementary methods.
Acknowledgements
The authors are grateful for the technical support from Jani Saarela and Laura Turunen at the High Throughput Biomedicine Unit (HTBU) at the Institute for Molecular Medicine Finland (FIMM) and to Disha Malani, Akira Hirasawa, Minna Suvela, and Alun Parsons at FIMM for generating data for the reference DSRT dataset. The authors also thank Mariliina Arjama at FIMM for excellent technical assistance with the drug testing, and Merete Hektoen, Mette Eknæs, and Sharmini Alagaratnam at the Department of Molecular Oncology (Oslo University Hospital) for excellent technical assistance with cell culturing and DNA analyses. This study was supported by grants to R.A.L. from the Research Council of Norway through its Centers of Excellence funding scheme (Project Number 179571, supporting M.K. as scientist) and through Norwegian Cancer Genomics Consortium (Project Number 218241, S.A.D scientist), from the Norwegian Cancer Society (Grant No. PR‐2006‐0442, J.B. post doc), from the South‐Eastern Norway Regional Health Authority (Grant No. 2016123, C.B. PhD student), from the Faculty of Medicine, University of Oslo (M.H. PhD student), and from Stiftelsen Kristian Gerhard Jebsen (I.A.E. PhD student); and to O.K. from the Academy of Finland (Center of Excellence for Translational Cancer Biology), Cancer Society of Finland, and Sigrid Jusélius Foundation.
References
- Abbou S, Lanvers‐Kaminsky C, Daudigeos‐Dubus E, LE Dret L, Laplace‐Builhe C, Molenaar J, Vassal G, Geoerger B, within the IB Preclinical Evaluation C (2016) Polo‐like kinase inhibitor volasertib exhibits antitumor activity and synergy with vincristine in pediatric malignancies. Anticancer Res 36, 599–609. [PubMed] [Google Scholar]
- Ågesen TH, Flørenes VA, Molenaar WM, Lind GE, Berner JM, Plaat BE, Komdeur R, Myklebost O, van den Berg E and Lothe RA (2005) Expression patterns of cell cycle components in sporadic and neurofibromatosis type 1‐related malignant peripheral nerve sheath tumors. J Neuropathol Exp Neurol 64, 74–81. [DOI] [PubMed] [Google Scholar]
- Ahlquist T, Bottillo I, Danielsen SA, Meling GI, Rognum TO, Lind GE, Dallapiccola B and Lothe RA (2008) RAS signaling in colorectal carcinomas through alteration of RAS, RAF, NF1, and/or RASSF1A. Neoplasia 10(680–686), 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albritton KH, Rankin C, Coffin CM, Ratner N, Budd GT, Schuetze SM, Randall RL, Declue JE and Borden EC (2006) Phase II study of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumors (MPNST). J Clin Oncol 24, S9518. [Google Scholar]
- Ambrosini G, Cheema HS, Seelman S, Teed A, Sambol EB, Singer S and Schwartz GK (2008) Sorafenib inhibits growth and mitogen‐activated protein kinase signaling in malignant peripheral nerve sheath cells. Mol Cancer Ther 7, 890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki M, Nabeshima K, Koga K, Hamasaki M, Suzumiya J, Tamura K and Iwasaki H (2007) Imatinib mesylate inhibits cell invasion of malignant peripheral nerve sheath tumor induced by platelet‐derived growth factor‐BB. Lab Invest 87, 767–779. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D et al (2012) The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg M, Danielsen SA, Ahlquist T, Merok MA, Agesen TH, Vatn MH, Mala T, Sjo OH, Bakka A, Moberg I et al (2010) DNA sequence profiles of the colorectal cancer critical gene set KRAS‐BRAF‐PIK3CA‐PTEN‐TP53 related to age at disease onset. PLoS One 5, e13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berner JM, Sørlie T, Mertens F, Henriksen J, Sæter G, Mandahl N, Brogger A, Myklebost O and Lothe RA (1999) Chromosome band 9p21 is frequently altered in malignant peripheral nerve sheath tumors: studies of CDKN2A and other genes of the pRB pathway. Genes Chromosom Cancer 26, 151–160. [PubMed] [Google Scholar]
- Bottillo I, Ahlquist T, Brekke H, Danielsen SA, van den Berg E, Mertens F, Lothe RA and Dallapiccola B (2009) Germline and somatic NF1 mutations in sporadic and NF1‐associated malignant peripheral nerve sheath tumours. J Pathol 217, 693–701. [DOI] [PubMed] [Google Scholar]
- Bradford D and Kim A (2015) Current treatment options for malignant peripheral nerve sheath tumors. Curr Treat Options Oncol 16, 328. [DOI] [PubMed] [Google Scholar]
- Brekke HR, Ribeiro FR, Kolberg M, Ågesen TH, Lind GE, Eknaes M, Hall KS, Bjerkehagen B, van den Berg E, Teixeira MR et al (2010) Genomic changes in chromosomes 10, 16, and X in malignant peripheral nerve sheath tumors identify a high‐risk patient group. J Clin Oncol 28, 1573–1582. [DOI] [PubMed] [Google Scholar]
- Brems H, Beert E, de Ravel T and Legius E (2009) Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol 10, 508–515. [DOI] [PubMed] [Google Scholar]
- Castellsague J, Gel B, Fernandez‐Rodriguez J, Llatjos R, Blanco I, Benavente Y, Perez‐Sidelnikova D, Garcia‐Del Muro J, Vinals JM, Vidal A et al (2015) Comprehensive establishment and characterization of orthoxenograft mouse models of malignant peripheral nerve sheath tumors for personalized medicine. EMBO Mol Med 7, 608–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh R, Wathen JK, Maki RG, Benjamin RS, Patel SR, Meyers PA, Priebat DA, Reinke DK, Thomas DG, Keohan ML et al (2009) Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J Clin Oncol 27, 3148–3153. [DOI] [PubMed] [Google Scholar]
- Dahlberg WK, Little JB, Fletcher JA, Suit HD and Okunieff P (1993) Radiosensitivity in vitro of human soft tissue sarcoma cell lines and skin fibroblasts derived from the same patients. Int J Radiat Biol 63, 191–198. [DOI] [PubMed] [Google Scholar]
- Danielsen SA, Lind GE, Kolberg M, Holand M, Bjerkehagen B, Sundby Hall K, van den Berg E, Mertens F, Smeland S, Picci P et al (2015) Methylated RASSF1A in malignant peripheral nerve sheath tumors identifies neurofibromatosis type 1 patients with inferior prognosis. Neuro Oncol 17, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, Maertens O, Jeong SM, Bronson RT, Lebleu V et al (2011) Exploiting cancer cell vulnerabilities to develop a combination therapy for ras‐driven tumors. Cancer Cell 20, 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, Whitcomb P, Martin S, Aschbacher‐Smith LE, Rizvi TA et al (2016) Activity of selumetinib in neurofibromatosis type 1‐related plexiform neurofibromas. N Engl J Med 375, 2550–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducoulombier A, Cousin S, Kotecki N and Penel N (2016) Gemcitabine‐based chemotherapy in sarcomas: a systematic review of published trials. Crit Rev Oncol Hematol 98, 73–80. [DOI] [PubMed] [Google Scholar]
- Endo M, Kobayashi C, Setsu N, Takahashi Y, Kohashi K, Yamamoto H, Tamiya S, Matsuda S, Iwamoto Y, Tsuneyoshi M et al (2011) Prognostic significance of p14ARF, p15INK4b and p16INK4a inactivation in malignant peripheral nerve sheath tumors. Clin Cancer Res 17, 3771–3782. [DOI] [PubMed] [Google Scholar]
- Endo M, Yamamoto H, Setsu N, Kohashi K, Takahashi Y, Ishii T, Iida K, Matsumoto Y, Hakozaki M, Aoki M et al (2013) Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1‐related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res 19, 450–461. [DOI] [PubMed] [Google Scholar]
- ESMO (2014) Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 25(Suppl. 3), S102–S112. [DOI] [PubMed] [Google Scholar]
- Frahm S, Mautner VF, Brems H, Legius E, Debiec‐Rychter M, Friedrich RE, Knofel WT, Peiper M and Kluwe L (2004) Genetic and phenotypic characterization of tumor cells derived from malignant peripheral nerve sheath tumors of neurofibromatosis type 1 patients. Neurobiol Dis 16, 85–91. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J et al (2012) Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JA, Salavaggione OE, Ji Y, Pelleymounter LL, Eckloff BW, Wieben ED, Ames MM and Weinshilboum RM (2006) Gemcitabine pharmacogenomics: cytidine deaminase and deoxycytidylate deaminase gene resequencing and functional genomics. Clin Cancer Res 12, 1794–1803. [DOI] [PubMed] [Google Scholar]
- Giraldez S, Galindo‐Moreno M, Limon‐Mortes MC, Rivas AC, Herrero‐Ruiz J, Mora‐Santos M, Saez C, Japon MA, Tortolero M, Romero F (2017) G1/S phase progression is regulated by PLK1 degradation through the CDK1/betaTrCP axis. FASEB J, in press. https://doi.org/10.1096/fj.201601108R [DOI] [PubMed] [Google Scholar]
- Gjertsen BT and Schoffski P (2015) Discovery and development of the Polo‐like kinase inhibitor volasertib in cancer therapy. Leukemia 29, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greshock J, Bachman KE, Degenhardt YY, Jing J, Wen YH, Eastman S, McNeil E, Moy C, Wegrzyn R, Auger K et al (2010) Molecular target class is predictive of in vitro response profile. Cancer Res 70, 3677–3686. [DOI] [PubMed] [Google Scholar]
- Gutteridge RE, Ndiaye MA, Liu X and Ahmad N (2016) Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol Cancer Ther 15, 1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverty PM, Lin E, Tan J, Yu Y, Lam B, Lianoglou S, Neve RM, Martin S, Settleman J, Yauch RL et al (2016) Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 533, 333–337. [DOI] [PubMed] [Google Scholar]
- Kolberg M, Høland M, Ågesen TH, Brekke HR, Liestøl K, Hall KS, Mertens F, Picci P, Smeland S and Lothe RA (2013) Survival meta‐analyses for > 1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro‐Oncology 15, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolberg M, Høland M, Lind GE, Ågesen TH, Skotheim RI, Hall KS, Mandahl N, Smeland S, Mertens F, Davidson B et al (2015) Protein expression of BIRC5, TK1, and TOP2A in malignant peripheral nerve sheath tumours–A prognostic test after surgical resection. Mol Oncol 9, 1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolberg M, Strand KR, Graff P and Andersson KK (2004) Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta 1699, 1–34. [DOI] [PubMed] [Google Scholar]
- Kourea HP, Orlow I, Scheithauer BW, Cordon‐Cardo C and Woodruff JM (1999) Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am J Pathol 155, 1855–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang R, Schweickert PG, Karki A, Yang Y, Kong Y, Ahmad N, Konieczny SF and Liu X (2016) Plk1 inhibition enhances the efficacy of gemcitabine in human pancreatic cancer. Cell Cycle 15, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothe RA, Karhu R, Mandahl N, Mertens F, Sæter G, Heim S, Børresen‐Dale AL and Kallioniemi OP (1996) Gain of 17q24‐qter detected by comparative genomic hybridization in malignant tumors from patients with von Recklinghausen's neurofibromatosis. Cancer Res 56, 4778–4781. [PubMed] [Google Scholar]
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK and Elledge SJ (2009) A genome‐wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137, 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahller YY, Vaikunth SS, Currier MA, Miller SJ, Ripberger MC, Hsu YH, Mehrian‐Shai R, Collins MH, Crombleholme TM, Ratner N et al (2007) Oncolytic HSV and erlotinib inhibit tumor growth and angiogenesis in a novel malignant peripheral nerve sheath tumor xenograft model. Mol Ther 15, 279–286. [DOI] [PubMed] [Google Scholar]
- Maki RG, D'Adamo DR, Keohan ML, Saulle M, Schuetze SM, Undevia SD, Livingston MB, Cooney MM, Hensley ML, Mita MM et al (2009) Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol 27, 3133–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens F, Dal Cin P, De Wever I, Fletcher CD, Mandahl N, Mitelman F, Rosai J, Rydholm A, Sciot R, Tallini G et al (2000) Cytogenetic characterization of peripheral nerve sheath tumours: a report of the CHAMP study group. J Pathol 190, 31–38. [DOI] [PubMed] [Google Scholar]
- Miller SJ, Rangwala F, Williams J, Ackerman P, Kong S, Jegga AG, Kaiser S, Aronow BJ, Frahm S, Kluwe L et al (2006) Large‐scale molecular comparison of human schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res 66, 2584–2591. [DOI] [PubMed] [Google Scholar]
- Mpindi JP, Swapnil P, Dmitrii B, Jani S, Saeed K, Wennerberg K, Aittokallio T, Ostling P and Kallioniemi O (2015) Impact of normalization methods on high‐throughput screening data with high hit rates and drug testing with dose‐response data. Bioinformatics 31, 3815–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpindi JP, Yadav B, Östling P, Gautam P, Malani D, Murumägi A, Hirasawa A, Kangaspeska S, Wennerberg K, Kallioniemi O et al (2016) Consistency in drug response profiling. Nature 540, E5–E6. [DOI] [PubMed] [Google Scholar]
- Nagashima Y, Ohaki Y, Tanaka Y, Sumino K, Funabiki T, Okuyama T, Watanabe S, Umeda M and Misugi K (1990) Establishment of an epithelioid malignant schwannoma cell line (YST‐1). Virchows Arch B Cell Pathol Incl Mol Pathol 59, 321–327. [DOI] [PubMed] [Google Scholar]
- Nair JS and Schwartz GK (2015) Inhibition of polo like kinase 1 in sarcomas induces apoptosis that is dependent on Mcl‐1 suppression. Cell Cycle 14, 3101–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen GP, Stemmer‐Rachamimov AO, Ino Y, Moller MB, Rosenberg AE and Louis DN (1999) Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol 155, 1879–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan PP, Surriga O, Beckman MJ, de Stanchina E, Dematteo RP, Tap WD and Schwartz GK (2014) Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin Cancer Res 20, 3146–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemovska T, Kontro M, Yadav B, Edgren H, Eldfors S, Szwajda A, Almusa H, Bespalov MM, Ellonen P, Elonen E et al (2013) Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov 3, 1416–1429. [DOI] [PubMed] [Google Scholar]
- Pereira S, Fernandes PA and Ramos MJ (2004) Mechanism for ribonucleotide reductase inactivation by the anticancer drug gemcitabine. J Comput Chem 25, 1286–1294. [DOI] [PubMed] [Google Scholar]
- Reynolds JE, Fletcher JA, Lytle CH, Nie L, Morton CC and Diehl SR (1992) Molecular characterization of a 17q11.2 translocation in a malignant schwannoma cell line. Hum Genet 90, 450–456. [DOI] [PubMed] [Google Scholar]
- Schoeler D, Kunitz A and Reichard P (2007) Gemcitabine in heavily pretreated adult soft tissue sarcoma patients. J Clin Oncol 25, S20524. [Google Scholar]
- Schoffski P, Blay JY, De Greve J, Brain E, Machiels JP, Soria JC, Sleijfer S, Wolter P, Ray‐Coquard I, Fontaine C et al (2010) Multicentric parallel phase II trial of the polo‐like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI). Eur J Cancer 46, 2206–2215. [DOI] [PubMed] [Google Scholar]
- Schuetze SM, Wathen JK, Lucas DR, Choy E, Samuels BL, Staddon AP, Ganjoo KN, von Mehren M, Chow WA, Loeb DM et al (2016) SARC009: Phase 2 study of dasatinib in patients with previously treated, high‐grade, advanced sarcoma. Cancer 122, 868–874. [DOI] [PubMed] [Google Scholar]
- Semenova G, Stepanova D, Deyev SM, Chernoff J (2017) Medium throughput biochemical compound screening identifies novel agents for pharmacotherapy of neurofibromatosis type I. Biochimie 135, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME and Evans DG (2006) Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol 24, 2570–2575. [DOI] [PubMed] [Google Scholar]
- Skotheim RI, Kallioniemi A, Bjerkhagen B, Mertens F, Brekke HR, Monni O, Mousses S, Mandahl N, Soeter G, Nesland JM et al (2003) Topoisomerase‐II alpha is upregulated in malignant peripheral nerve sheath tumors and associated with clinical outcome. J Clin Oncol 21, 4586–4591. [DOI] [PubMed] [Google Scholar]
- Sonobe H, Takeuchi T, Furihata M, Taguchi T, Kawai A, Ohjimi Y, Iwasaki H, Kaneko Y and Ohtsuki Y (2000) A new human malignant peripheral nerve sheath tumour‐cell line, HS‐sch‐2, harbouring p53 point mutation. Int J Oncol 17, 347–352. [DOI] [PubMed] [Google Scholar]
- Spyra M, Kluwe L, Hagel C, Nguyen R, Panse J, Kurtz A, Mautner VF, Rabkin SD and Demestre M (2011) Cancer stem cell‐like cells derived from malignant peripheral nerve sheath tumors. PLoS One 6, e21099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storlazzi CT, Brekke HR, Mandahl N, Brosjo O, Smeland S, Lothe RA and Mertens F (2006) Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheath tumours. J Pathol 209, 492–500. [DOI] [PubMed] [Google Scholar]
- Teicher BA, Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, Ogle C, Reinhart R, Selby M et al (2015) Sarcoma cell line screen of oncology drugs and investigational agents identifies patterns associated with gene and microRNA expression. Mol Cancer Ther 14, 2452–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB and Kornblau SM (2006) Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 5, 2512–2521. [DOI] [PubMed] [Google Scholar]
- Upadhyaya M, Kluwe L, Spurlock G, Monem B, Majounie E, Mantripragada K, Ruggieri M, Chuzhanova N, Evans DG, Ferner R et al (2008) Germline and somatic NF1 gene mutation spectrum in NF1‐associated malignant peripheral nerve sheath tumors (MPNSTs). Hum Mutat 29, 74–82. [DOI] [PubMed] [Google Scholar]
- Varin J, Poulain L, Hivelin M, Nusbaum P, Hubas A, Laurendeau I, Lantieri L, Wolkenstein P, Vidaud M, Pasmant E et al (2016) Dual mTORC1/2 inhibition induces anti‐proliferative effect in NF1‐associated plexiform neurofibroma and malignant peripheral nerve sheath tumor cells. Oncotarget 7, 35753–35767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hu K, Guo J, Cheng F, Lv J, Jiang W, Lu W, Liu J, Pang X and Liu M (2016) Suppression of KRas‐mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat Commun 7, 11363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widemann BCR, Reinke DK, Helman LJ, Ludwig JA, Schuetze S, Staddon AP, Milhem MM, Rushing DA, Moertel CL, Goldman S et al (2013) SARC006: Phase II trial of chemotherapy in sporadic and neurofibromatosis type 1 (NF1)‐associated high‐grade malignant peripheral nerve sheath tumors (MPNSTs). J Clin Oncol 31, S10522. [Google Scholar]
- Widemann BC, Meyer CF, Cote GM, Chugh R, Milhem MM, van Tine BM, Kim A, Turpin B, Dombi E, Jayaprakash N et al (2016) SARC016: Phase II study of everolimus in combination with bevacizumab in sporadic and neurofibromatosis type 1 (NF1) related refractory malignant peripheral nerve sheath tumors (MPNST). J Clin Oncol 34, S11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav B, Pemovska T, Szwajda A, Kulesskiy E, Kontro M, Karjalainen R, Majumder MM, Malani D, Murumagi A, Knowles J et al (2014) Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep 4, 5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita AS, Baia GS, Ho JS, Velarde E, Wong J, Gallia GL, Belzberg AJ, Kimura ET and Riggins GJ (2014) Preclinical evaluation of the combination of mTOR and proteasome inhibitors with radiotherapy in malignant peripheral nerve sheath tumors. J Neurooncol 118, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Chang AE, Baker AR, Sindelar WF, Danforth DN, Topalian SL, DeLaney T, Glatstein E, Steinberg SM, Merino MJ et al (1998) Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16, 197–203. [DOI] [PubMed] [Google Scholar]
- Zietsch J, Ziegenhagen N, Heppner FL, Reuss D and von Deimling DA, Holtkamp N (2010) The 4q12 amplicon in malignant peripheral nerve sheath tumors: consequences on gene expression and implications for sunitinib treatment. PLoS One 5, e11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Overview of drugs tested in MPNST patients.
Fig. S2. Correlation between CellTiter‐Glo (CTG) viability experiments.
Fig. S3. Drug cytotoxicity response profiles of MPNST cell lines and normal HSCs.
Fig. S4. Dose–response curves.
Fig. S5. Gene expression of drug targets in cell lines and patient tumors, and association to patient survival.
Fig. S6. Correlation with public dataset.
Table S1. List of drugs in current or previous clinical testing against sarcoma, including MPNST (clinical trials.gov).
Table S2. STR profiles of MPNST cell lines*.
Table S3. List of tested compounds.
Table S4. (A) QC‐scores from viability assay (CTG). (B) QC‐scores from cytotoxicity assay (CTX).
Table S5. Cell viability assay (CTG) data.
Table S6. Cytotoxicity assay (CTX) data.
Table S7. Protein expression data from reverse phase protein lysate microarray (RPPA)a.
Table S8. Primer sequences.
Appendix S1. Supplementary methods.