

Abstract
The synaptonemal complex (SC) is a proteinaceous macromolecular assembly that forms during meiotic prophase I and mediates adhesion of paired homologous chromosomes along their entire lengths. Although prompt disassembly of the SC during exit from prophase I is a landmark event of meiosis, the underlying mechanism regulating SC destruction has remained elusive. Here, we show that DDK (Dbf4‐dependent Cdc7 kinase) is central to SC destruction. Upon exit from prophase I, Dbf4, the regulatory subunit of DDK, directly associates with and is phosphorylated by the Polo‐like kinase Cdc5. In parallel, upregulated CDK1 activity also targets Dbf4. An enhanced Dbf4‐Cdc5 interaction pronounced phosphorylation of Dbf4 and accelerated SC destruction, while reduced/abolished Dbf4 phosphorylation hampered destruction of SC proteins. SC destruction relieved meiotic inhibition of the ubiquitous recombinase Rad51, suggesting that the mitotic recombination machinery is reactivated following prophase I exit to repair any persisting meiotic DNA double‐strand breaks. Taken together, we propose that the concerted action of DDK, Polo‐like kinase, and CDK1 promotes efficient SC destruction at the end of prophase I to ensure faithful inheritance of the genome.
Keywords: Dbf4‐dependent Cdc7 kinase, homologous recombination, meiosis, Polo‐like kinase, synaptonemal complex
Subject Categories: Cell Cycle; DNA Replication, Repair & Recombination
Introduction
Meiosis is central to the continuity of life in sexually reproducing organisms through the production of gametes. In meiosis, a single round of DNA replication is followed by two successive rounds of nuclear division, leading to the reduction of genetic material by exactly half. The unique aspect of meiosis lies in meiosis I, where homologous chromosomes (homologs) separate; this is in sharp contrast to mitosis or meiosis II, where sister chromatids separate (Petronczki et al, 2003).
Meiotic chromosomes undergo dynamic morphological changes as homologs align with one another (Cahoon & Hawley, 2016). Sister chromatids are organized around a proteinaceous axis, which is juxtaposed at close proximity along its entire length with the axis of the homolog. The incorporation of a proteinaceous transverse filament between axes leads to the formation of a meiosis‐specific chromosomal structure called the synaptonemal complex (SC). By adhering homologous axes in such a way, the SC provides a structural platform to promote efficient formation of crossovers between homologs, a process that is catalyzed by the homologous recombinases Rad51 and Dmc1.
The function and structure of the SC have been the subject of extensive research in budding yeast (Tsubouchi et al, 2016). SC components associated with chromosomal axes are highly relevant for repressing usage of Rad51, which is involved in homologous recombination (HR) during both mitosis and meiosis (Shinohara et al, 1992). Unlike Rad51, Dmc1 is only produced during meiosis, where it is thought to play a specialized role in promoting interhomolog interactions in meiotic HR (Bishop et al, 1992). Thus, preferential usage of Dmc1 serves to promote interhomolog HR (Schwacha & Kleckner, 1997; Lao et al, 2013). Red1 and Hop1, structural components of meiotic chromosome axes, and Mek1, a meiosis‐specific protein kinase functioning with Red1 and Hop1, are essential for repressing HR in the absence of Dmc1 (Schwacha & Kleckner, 1997; Wan et al, 2004). The phosphorylation of Hop1, which is under the control of the recombination checkpoint (see below), is also critical for repressing Rad51 (Carballo et al, 2008).
Given the central role of the SC in regulating meiotic HR, it is of particular importance to understand the regulation of SC dynamics. Prophase I is divided into substages based on SC morphology (Roeder, 1997). During early‐prophase I, newly replicated homologs start to condense (leptotene) and pairing of homologs initiates SC formation (zygotene). SC formation is considered complete when all paired homologs are incorporated along their entire lengths into the SC structure in mid‐prophase I (pachytene). The SC is then disassembled in late prophase I (diplotene), before entry into metaphase I. SC behavior during the passage from pachytene to diplotene (referred to as pachytene exit hereafter) warrants special attention as it encompasses the time when SC disassembly takes place. Timely SC disassembly is essential for proper segregation of homologs at anaphase I, as the SC would otherwise oppose the microtubule forces that are responsible for separating homologs (Cahoon & Hawley, 2016).
Pachytene exit also coincides with the maturation of recombination intermediates into interhomolog crossovers (Sourirajan & Lichten, 2008). Consistently, the timing of pachytene exit is closely coordinated with the progression of HR by the recombination checkpoint, also known as the pachytene checkpoint (Hochwagen & Amon, 2006). The recombination checkpoint is highly related to the DNA damage checkpoint operating in mitotic cells, except that a major downstream target of the signaling cascade is Ndt80, a meiosis‐specific transcriptional activator that governs the mid‐to‐late stages of meiosis and sporulation including pachytene exit (Xu et al, 1995; Chu et al, 1998). Budding yeast cells that progress past the recombination checkpoint make an irreversible commitment to meiosis and swiftly disassemble the SC as they enter metaphase I (Tsuchiya et al, 2014). Thus, pachytene exit represents a key event in the prophase I–metaphase I transition and commitment to the meiotic nuclear divisions.
The mechanisms governing SC disassembly have just begun to emerge. One major factor is Cdc5 (homolog of PLK1 and the only Polo‐like kinase in budding yeast), whose production is induced in an Ndt80‐dependent manner as cells exit pachytene (Chu et al, 1998; Sourirajan & Lichten, 2008; Acosta et al, 2011; Okaz et al, 2012). Cdc5 has also been shown to play a central role in regulating the resolution of recombination intermediates in both mitosis and meiosis (Sourirajan & Lichten, 2008; Matos et al, 2011, 2013; Szakal & Branzei, 2013). Production of Cdc5 before pachytene exit triggers untimely disassembly of the SC and resolution of recombination intermediates (Sourirajan & Lichten, 2008), arguing that Cdc5 is a major regulator of these events. During the prophase I–metaphase I transition, Cdc5 was shown to interact with another fundamental cell cycle kinase complex called Dbf4‐dependent Cdc7 kinase (DDK; Matos et al, 2008). DDK has drawn comparisons to cyclin‐dependent kinases (CDKs) as Cdc7 comprises the catalytic subunit, whereas Dbf4 fulfills a crucial regulatory role within the complex (Matthews & Guarné, 2013). Although its major role in vegetative cells is in controlling the initiation of DNA replication, DDK also has meiosis‐specific roles in DSB formation and chromosome segregation (Matos et al, 2008; Sasanuma et al, 2008; Wan et al, 2008; Murakami & Keeney, 2014). However, unlike Cdc5, which functions after pachytene exit (Okaz et al, 2012), DDK is believed to function primarily before pachytene exit.
Here, we show that DDK is central to the control of SC destruction in budding yeast. Dbf4 serves as the regulator of SC destruction by directly associating with and being phosphorylated by Cdc5. In parallel, Dbf4 is also regulated through phosphorylation by Cdc28 (homolog of CDK1 and the only CDK in budding yeast). We propose that the concerted action of DDK, CDK1, and Polo ensures SC destruction, with Dbf4 serving as the hub of the signaling pathway at the prophase I–metaphase I boundary, leading to the timely removal of a major physical obstacle to chromosome segregation. Interestingly, this coordinated mechanism leads to the reactivation of Rad51, which promotes the repair of any persisting DSBs before chromosomes are separated during anaphase I. By facilitating removal of the SC and triggering Rad51‐dependent DSB repair, we propose that fundamental cell cycle kinases collaborate at the prophase I–metaphase I transition to ensure faithful inheritance of the genome.
Results
DDK and Polo interact to regulate the meiotic cell cycle
Meiotic HR is initiated by the topoisomerase‐like protein Spo11, which continuously forms meiotic DNA double‐strand breaks (DSBs) before pachytene exit (Keeney et al, 1997). Unlike mitotic HR, meiotic HR is intricately regulated so that homologous chromosomes are connected through crossovers. Defects in meiotic HR lead to an accumulation of recombination intermediates, such as DSBs, which slowdown or arrest the meiotic cell cycle. In order to obtain further insight into the mechanism coordinating meiotic HR with cell cycle progression, we conducted a genetic screen to identify genes whose overexpression bypassed the cell cycle arrest caused by defects in meiosis‐specific recombination factors (see Materials and Methods for experimental details). This screen revealed that overexpression of DBF4 can suppress pachytene arrest in several recombination mutants (Appendix Fig S1A).
To understand how a high dose of Dbf4 suppresses the cell cycle arrest phenotype, we set out to isolate DBF4 point mutants that phenocopy this suppression effect. Randomly mutagenized versions of DBF4 were cloned into a single‐copy plasmid to produce a DBF4 mutant library. Clones that were able to suppress the cell cycle arrest phenotype were screened. A single clone, carrying a mutation that changes the Glu at the 86th position to Val, was isolated (dbf4‐E86V hereafter; Appendix Fig S1B).
This amino acid falls within residues 83–88 at Dbf4's N‐terminus, which were previously shown to mediate the direct interaction between Dbf4 and the Polo‐box domain (PBD) of Cdc5 (Fig 1A; Chen & Weinreich, 2010). Of the residues within this region, Arg83, Ile85, Gly87, and Ala88 are essential for the interaction, whereas Glu86 is not. Furthermore, the mutant polypeptide with Glu86 changed to Lys (dbf4‐E86K hereafter) interacts more strongly with Cdc5 than the wild‐type polypeptide (Chen & Weinreich, 2010). These observations raised the possibility that the mechanism responsible for suppression of meiotic arrest involves an interaction between Dbf4 and Cdc5. Thus, we employed two known DBF4 mutations: Arg83 to Glu (dbf4‐R83E hereafter) and dbf4‐E86K, which abolish and enhance the Dbf4‐Cdc5 interaction, respectively (Fig 1A; Chen & Weinreich, 2010). dbf4‐R83E did not suppress cell cycle arrest, whereas dbf4‐E86K showed a similar level of suppression to dbf4‐E86V (Appendix Fig S1C), leading to the robust upregulation of late‐stage proteins associated with cell cycle progression (Ndt80 and Cdc5) in dmc1Δ, a recombination deficient mutant that undergoes pachytene arrest (Fig 1B).
Figure 1. An enhanced interaction between DDK and Cdc5 suppresses pachytene arrest.
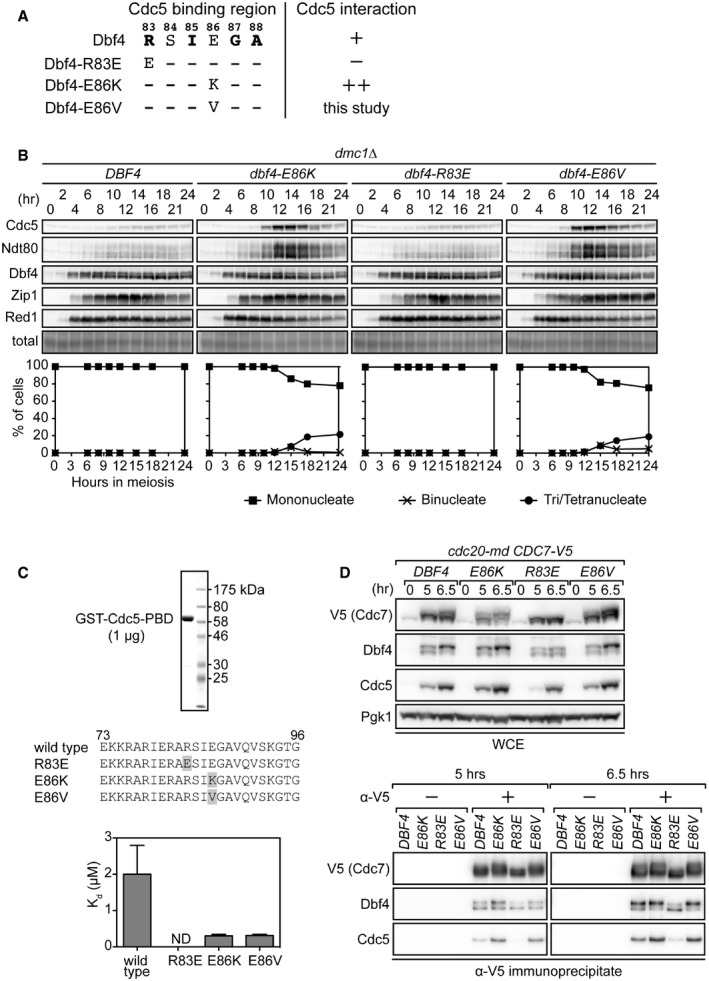
- Schematic depicting the Cdc5 binding region of Dbf4. Residues in bold are essential for the interaction. +, wild‐type interaction; −, no interaction detected; ++, enhanced interaction.
- Strains were induced to synchronously enter meiosis. At the indicated time points, cells were harvested for detection of proteins by immunoblotting (panels) and determination of cell cycle kinetics by DAPI staining of nuclei (graphs). Induction of Ndt80 and Cdc5 serves as a marker for pachytene exit. Total, total protein levels (Ponceau S staining). Mononucleate cells have not completed any nuclear divisions, binucleate cells have only completed the first nuclear division (anaphase I), and tri/tetranucleate cells have completed both nuclear divisions (anaphase I and II).
- A fragment of Cdc5 containing the PBD was N‐terminally GST‐tagged and purified to near homogeneity, as determined by Coomassie staining (upper panel). Various Dbf4 peptides corresponding to sequences spanning the Cdc5 binding region were synthesized with a fluorescein tag (middle panel). Mutations are highlighted in gray. Measurements obtained from fluorescence polarization assays depicted in Appendix Fig S2B were used to calculate the dissociation constants (K d) for each peptide‐PBD interaction (lower graphs). ND, not determined due to lack of detectable interaction (see Appendix Fig S2B).
- Strains were induced to synchronously enter meiosis. Cells harvested at the indicated time points were used to examine the interaction between DDK and Cdc5 by immunoprecipitating Cdc7‐V5 using anti‐V5 antibody. WCE, whole‐cell extract. “−” and “+” indicate the exclusion and inclusion of antibody for IP, respectively, with the no antibody condition serving as a negative control.
To test whether suppression of cell cycle arrest by dbf4‐E86K/V requires Cdc5, we wanted to deplete Cdc5 during meiosis. Since CDC5 is an essential gene, deletion mutants are not viable, so a conditional mutant was generated instead by transplacement of the native CDC5 promoter with the CLB2 promoter (cdc5‐md, meiotic depletion; Lee & Amon, 2003). CLB2 is expressed during vegetative growth but downregulated during meiosis. Under this condition, Cdc5 was barely detectable within prophase I and the dbf4‐E86K mutation was no longer able to suppress the pachytene arrest of the dmc1Δ mutant (Appendix Fig S2A), confirming the requirement for Cdc5 in Dbf4‐mediated suppression of cell cycle arrest.
The identical suppression phenotype of dbf4‐E86K and dbf4‐E86V suggested that, like the E86K mutation (Chen & Weinreich, 2010), the E86V mutation enhances the interaction between Dbf4 and Cdc5. To directly test this possibility, the fluorescence polarization assay was employed. The interaction strength between the C‐terminal half of Cdc5 containing the Polo‐box domain (PBD) and polypeptides corresponding to residues 73–96 of Dbf4 (wild type and mutants) was determined (Fig 1C, see Appendix Supplementary Methods for experimental details). Consistent with previous work (Chen & Weinreich, 2010), the wild‐type Dbf4 peptide interacted with Cdc5‐PBD with a K d of ~2 μM (Fig 1C and Appendix Fig S2B). The E86K and E86V peptides showed a stronger interaction than wild type, with K d values of ~0.3 μM, while the R83E peptide showed little/no interaction (Appendix Fig S2B), suggesting that Dbf4‐E86K/V proteins interact more strongly with Cdc5 than wild‐type Dbf4.
To validate these in vitro results and further correlate Dbf4‐Cdc5 interaction strength with suppression of pachytene arrest, we performed co‐immunoprecipitation (co‐IP) experiments to examine Dbf4‐Cdc5 complex formation in meiosis. Cdc7 was C‐terminally tagged with 9× copies of the V5 epitope and introduced into a genetic background in which cells arrest uniformly at the end of metaphase I due to meiosis‐specific depletion of the anaphase promoting complex/cyclosome (APC/C) activator Cdc20 (cdc20‐md; Matos et al, 2008). Cdc7 was immunoprecipitated from DBF4, dbf4‐R83E, and dbf4‐E86K/V strains 5 h and 6.5 h into meiosis, when Cdc5 levels were low and high, respectively. Cdc7, Dbf4, and Cdc5 were then detected by immunoblotting. Importantly, comparable amounts of Dbf4 were seen to co‐IP with Cdc7 in all four strains (Fig 1D), suggesting that the mutations examined here do not affect the interaction between Cdc7 and Dbf4. In sharp contrast, the amount of Cdc5 that co‐IP'd with Dbf4‐E86K/V was increased at both time points compared to wild‐type Dbf4. Furthermore, very low levels of Cdc5 were seen to co‐IP with Dbf4‐R83E, even at 6.5 h, when intracellular Cdc5 levels were high. These in vitro and in vivo data indicate that the dbf4‐E86K/V mutations enhance DDK‐Cdc5 complex formation, whereas the dbf4‐R83E mutation reduces DDK‐Cdc5 complex formation (Fig 1C and D, Appendix Fig 2B). We also noted that the migration of both Cdc7 and Dbf4 was affected by the dbf4 mutations. Posttranslational modification of Dbf4 was subjected to further investigation (see below).
We also created a condition where the Dbf4‐Cdc5 interaction is forced by fusing the two genes in‐frame and expressing this fusion construct within prophase I by employing the DBF4 promoter. The Cdc5‐Dbf4 fusion protein did not interfere with wild‐type meiosis, as judged by spore viability (99% without the transgene and 98% with the transgene, 80 spores examined per strain). Consistent with previous observations, production of Cdc5 alone within prophase I was able to suppress the pachytene arrest of dmc1Δ cells (Fig 2A and column 3 in Fig 2B; Acosta et al, 2011). Interestingly, this was dependent on the ability of Cdc5 to interact with Dbf4, as the suppression effect was lost in the dbf4‐R83E background (Fig 2A and column 4 in Fig 2B). Taking this into account, we wanted to eliminate the possibility that production of the Cdc5‐Dbf4 fusion protein within prophase I would simply mimic production of Cdc5, effectively rendering the fused Dbf4 fragment obsolete and leading to fusion‐independent suppression of dmc1Δ arrest. Thus, we employed the dbf4‐R83E background. Furthermore, we chose to express a Cdc5‐Dbf4‐R83E fusion protein. This would abolish interactions between the Dbf4 fragment of one fusion protein and the Cdc5 fragment of another fusion protein, which could also mimic the expression of Cdc5 alone and lead to fusion‐independent suppression of dmc1Δ arrest. Hence, expressing Cdc5‐Dbf4‐R83E in the dbf4‐R83E background allowed us to assess the sole impact of tethering Cdc5 to Dbf4. Notably, the fusion of Cdc5 to Dbf4‐R83E, which could not suppress the cell cycle arrest of the dmc1Δ mutant on its own (Fig 1B), was able to promote cell cycle progression (Fig 2A and column 6 in Fig 2B). This result further supports the idea that an enhanced interaction between Dbf4 and Cdc5 suppresses the pachytene arrest of dmc1Δ cells.
Figure 2. The Cdc5‐Dbf4 fusion protein can suppress pachytene arrest.

- Strains were induced to synchronously enter meiosis. Proteins were detected, and cell cycle kinetics was monitored as in Fig 1B.
- Cells were incubated for 48 h on sporulation plates, and sporulation percentage was determined by light microscopy. White labels depict the native DBF4 locus, and gray labels depict the ectopic locus.
Since Dbf4 interacts simultaneously with Cdc7 and Cdc5, and Cdc5 does not interact directly with Cdc7 (Matos et al, 2008), these data strongly suggest that Dbf4 mediates the interaction between Cdc5 and DDK to regulate progression of the cell cycle during meiosis.
Cell cycle progression is associated with unshackling of Rad51 activity
The cell cycle progression of dmc1Δ brought about by enhancing the Dbf4‐Cdc5 interaction could be mediated through different mechanisms. For example, it could be caused by a defect in the recombination checkpoint, which coordinates DSB repair with the cell cycle (Hochwagen & Amon, 2006). Alternatively, activation of a Dmc1‐independent pathway could repair DSBs, ultimately leading to cell cycle progression. To examine whether the cell cycle progression seen in the dmc1Δ mutant background is associated with DSB repair, the kinetics of meiotic DSBs was directly measured by pulsed‐field gel electrophoresis and Southern blotting with a chromosome II‐specific probe. This technique allows for the observation of intact and broken chromosome molecules. We found that adding extra copies of dbf4‐E86V dramatically improved cell cycle progression of the dmc1Δ dbf4‐E86V mutant, thus this strain was also included. This strain contains a dbf4‐E86V allele integrated homozygously at the URA3 locus and shows vastly improved spore formation compared to the dbf4‐E86V strain without additional copies of dbf4‐E86V (52% versus 18%, respectively. < 1% in the negative control strain (DBF4)). In both dbf4‐E86V strains, broken chromosomes accumulated to a level similar to the dmc1Δ single mutant but eventually decreased/disappeared (Fig 3A). The reappearance of intact parental chromosomes indicated that broken chromosomes were repaired. Consistent with the aforementioned sporulation data, repair was more efficient in the strain with extra copies of dbf4‐E86V. Moreover, broken chromosomes were no longer repaired if the RAD51 gene was deleted (Fig 3A), indicating that DSBs were repaired by a Rad51‐dependent mechanism.
Figure 3. DDK and Cdc5 interact to relieve Rad51 of its meiotic inhibition.
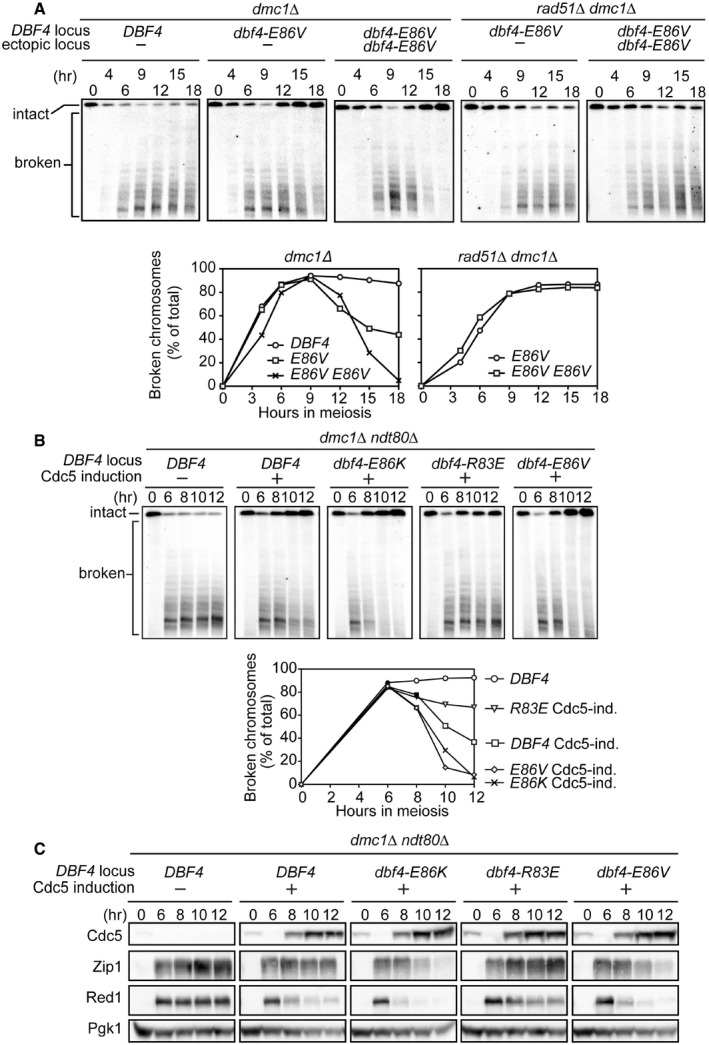
-
AStrains were induced to synchronously enter meiosis. At the indicated time points, cells were harvested for analysis of meiotic chromosomes by pulsed‐field gel electrophoresis followed by Southern blotting with a probe recognizing chromosome II. Southern blots (panels) were quantified to determine the percentage of signal corresponding to broken chromosomes (graphs; see Materials and Methods).
-
B, CStrains were induced to synchronously enter meiosis. At 6 h, β‐estradiol was added to induce Cdc5 production (Cdc5 induction or Cdc5‐ind.). Cells were harvested at the indicated time points to (B) monitor meiotic chromosomes as in (A), or (C) detect proteins as in Fig 1B. “−” and “+” denote the absence or presence of an inducible CDC5 allele at the URA3 locus, respectively.
These findings were supported by cytological observations in which DSB markers (Rad51 and RPA) that accumulate in a meiotic recombination mutant (hop2Δ) were no longer detected at metaphase I in the dbf4‐E86K/V mutants (Fig EV1A and B), suggesting that DSBs have been repaired in these strains before the onset of metaphase I. This contrasts with the results obtained in a checkpoint mutant (rad17Δ), where 100% of cells that progressed to metaphase I contained DSB markers (Fig EV1A and B; Lydall et al, 1996).
Figure EV1. Rad51‐dependent DSB repair occurs before metaphase I.
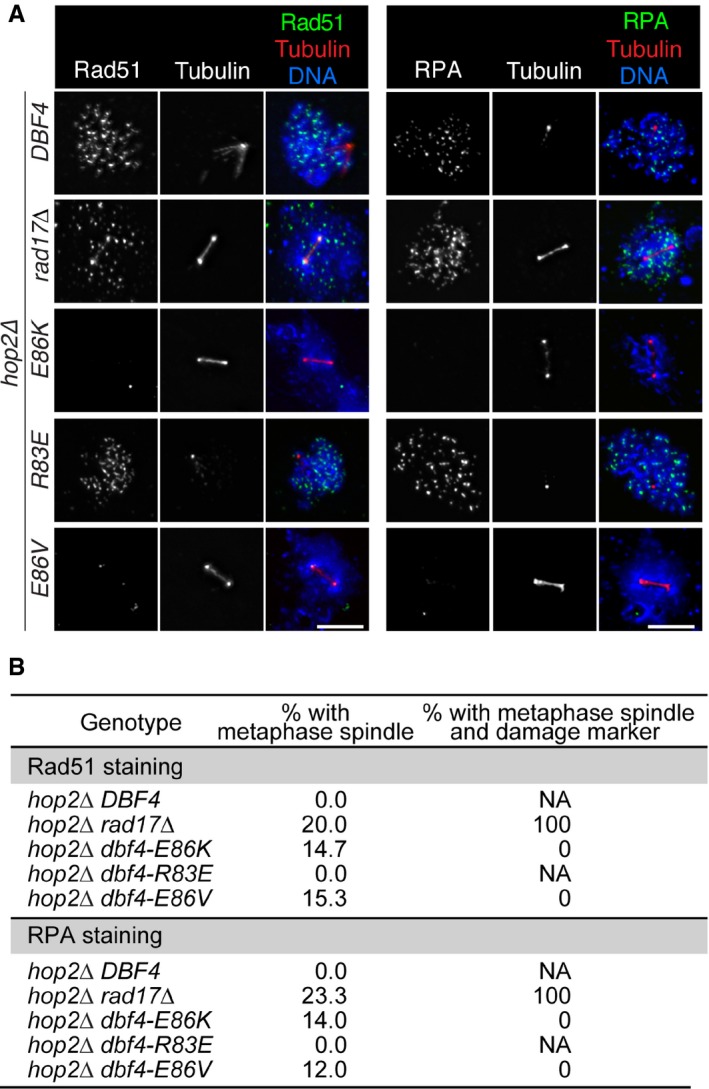
- hop2Δ strains in the BR1919 background were transferred to sporulation media. At 22 h (hop2Δ rad17Δ) or 30 h (all other strains), cells were harvested and meiotic chromosomes were spread for immunofluorescence microscopy as in Fig 4B. Scale bar, 5 μm.
- Quantification of the results in (A). The percentage of nuclei with metaphase spindle was determined for each strain; this represents cells that have exit pachytene and progressed into metaphase I. From nuclei with metaphase spindles, the percentage that are positive for DNA damage markers was determined; this represents cells that have progressed into metaphase I with unrepaired DSBs (e.g., the hop2Δ rad17Δ strain, in which the DNA damage checkpoint gene RAD17 has been deleted). NA, not applicable. At least 150 nuclei were scored for each strain.
Previous work has suggested that Rad51‐dependent DSB repair in meiosis does not lead to efficient crossover formation, resulting in reduced spore viability due to chromosome nondisjunction (Lao et al, 2013). Consistent with this notion, despite most/all DSBs being repaired by 18 h in both dmc1Δ dbf4‐E86V strains (Fig 3A), tetrads dissected after 48 h showed relatively low spore viability (< 20%; Fig EV2A). This low spore viability combined with the requirement for Rad51 (Fig 3A) suggested that DSB repair did not result in efficient crossover formation. To directly measure crossover formation in the two dmc1Δ dbf4‐E86V strains, we introduced the HIS4‐LEU2 recombination hotspot (Hunter & Kleckner, 2001). Due to restriction site polymorphisms in the parental chromosomes, it is possible to measure the efficiency of interhomolog crossing over within the population by Southern blotting (Fig EV2B). In the presence of Dmc1, efficient crossover formation was observed 12 h into meiosis, with ~17% of the total DNA corresponding to recombinant DNA molecules (Fig EV2C and D). In contrast, when both dmc1Δ dbf4‐E86V strains were examined at 26 h into meiosis, a time point by which most/all DSBs had been repaired (Fig 3A), a ~2.5‐fold reduction in recombinant DNA molecules was observed. These results indicate that, in the absence of Dmc1, enhancing the interaction between Dbf4 and Cdc5 leads to the majority of DSBs being repaired through a Rad51‐dependent HR pathway, which might be similar to the mitotic mode of HR, where noncrossover products are favored to reduce deleterious genomic rearrangements and loss of heterozygosity (Bzymek et al, 2010).
Figure EV2. The Rad51‐dependent DSB repair facilitated by Dbf4‐Cdc5 does not efficiently produce interhomolog crossovers.
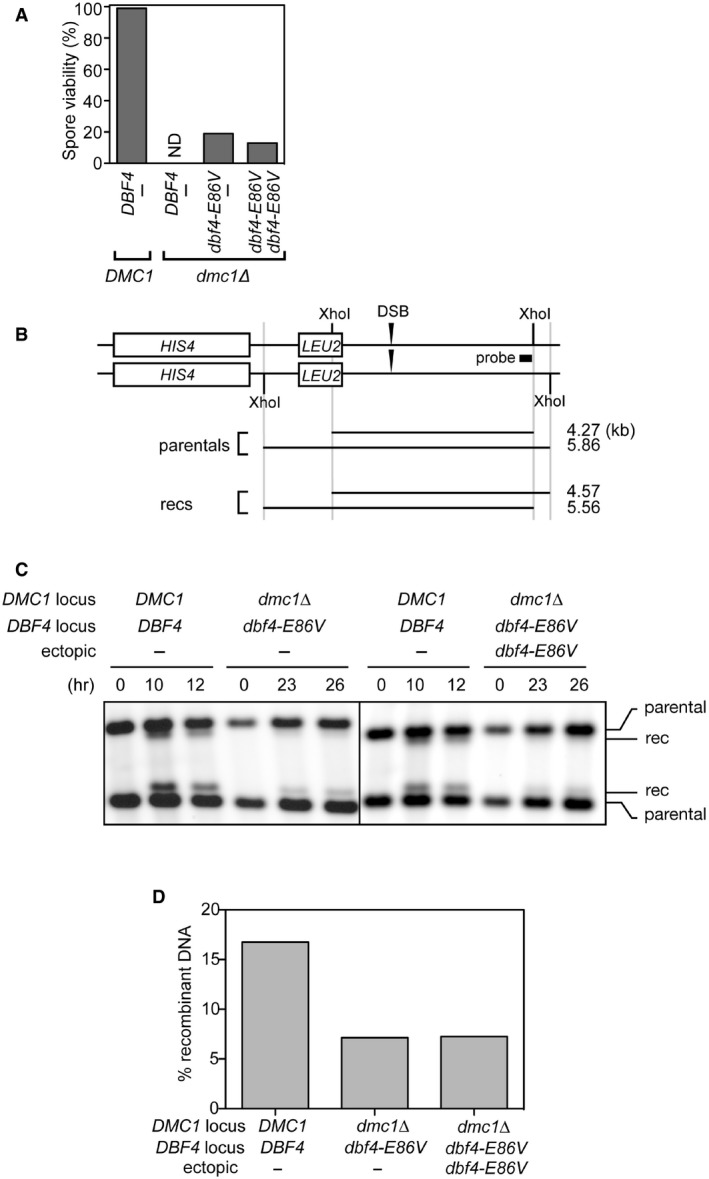
- Cells were incubated for 48 h on sporulation plates, and tetrads were dissected. After 3 days at 30°C, the number of colonies was counted and expressed as a percentage of spore viability. ND, not determined due to very low levels of sporulation; the spore viability of the dmc1Δ mutant was previously shown to be < 1% (Tsubouchi & Roeder, 2006).
- A schematic of the HIS4‐LEU2 recombination hotspot that was utilized to monitor crossover formation. The site where DSBs are formed and the probe for Southern blotting are shown. Note the XhoI polymorphisms between parental chromosomes. rec, recombinant.
- Strains were induced to synchronously enter meiosis. Cells were harvested at the indicated time points. Genomic DNA was extracted and digested with XhoI. Resultant DNA molecules were separated by gel electrophoresis and detected by Southern blotting using the probe shown in (B). rec, recombinant.
- Quantification of the results in (C). The percentage of total DNA corresponding to recombinant DNA molecules was plotted. Data are represented as means from two experiments.
Source data are available online for this figure.
Taken together, these data suggest that the mechanism to repress the mitotic recombination machinery during meiosis is alleviated if Dbf4 and Cdc5 interact with high affinity, leading to Rad51‐dependent repair of meiotic DSBs and progression from prophase I to metaphase I.
Destruction of SC components is mediated by DDK and Polo
SC components associated with chromosome axes are highly relevant to repressing Rad51 in prophase I (Schwacha & Kleckner, 1997; Wan et al, 2004). Furthermore, Cdc5 functions to induce SC destabilization (Sourirajan & Lichten, 2008), raising the possibility that alleviation of Rad51 inhibition is related to SC dynamics. Thus, we examined the relationship between the Dbf4‐Cdc5 interaction and SC proteins. Disassembly of the SC occurs as cells exit pachytene, when Ndt80, the master transcription factor that governs pachytene exit, upregulates ~300 late meiotic genes including Cdc5 (Xu et al, 1995; Chu et al, 1998). In order to focus solely on the effect of Cdc5, the ndt80Δ mutation was introduced into the dmc1Δ background so that the cell cycle permanently arrests at pachytene without the upregulation of late meiotic genes. DSB repair and SC destruction were monitored as Cdc5 was induced in the presence of Dbf4 proteins that display various interaction strengths with Cdc5. Transcription of CDC5 was controlled by the GAL promoter in a cell constitutively producing a fusion protein consisting of the Gal4 transcriptional activator and the estradiol receptor (GAL4‐ER; Benjamin et al, 2003). In this system, Cdc5 induction is triggered upon addition of β‐estradiol to the cell culture. The induction of Cdc5 alone was sufficient to trigger Dmc1‐independent DSB repair (Fig 3B), which is reminiscent of the previous observation that accumulation of DSBs was reduced in the dmc1Δ ama1Δ double mutant (Okaz et al, 2012). Ama1 is a meiosis‐specific activator of the APC/C and is indirectly responsible for suppressing Cdc5 production before pachytene exit. Thus, in the absence of Ama1, various M phase regulators such as Cdc5 are produced before pachytene exit, leading to a reduction in the number of DSBs.
Remarkably, the efficiency of DSB repair was positively correlated with Dbf4‐Cdc5 interaction strength (Fig 3B). Similarly, the kinetics of SC component destruction precisely mirrored the interaction strength (Fig 3C), especially that of Red1, a major component of meiotic chromosome axes responsible for repressing the mitotic recombination machinery (Schwacha & Kleckner, 1997). Similar results were obtained in the Dmc1‐positive background (Fig 4A). When Cdc5 was not induced, the levels of SC proteins did not decline even in the dbf4‐E86K/V strains, thus confirming that Cdc5 is an essential component of the Dbf4‐mediated SC destruction mechanism (Fig EV3A). These results in the dmc1Δ ndt80Δ background contrast with our findings in the dmc1Δ background, where we did not observe a clear reduction in the levels of Red1 and Zip1 despite dbf4‐E86K/V suppressing pachytene arrest (Fig 1B). This is because only a subset of the population exits pachytene in the dmc1Δ dbf4‐E86K/V strains; the decline in SC protein levels in this fraction of the population (~20%) is masked by the persistent SC proteins in the population of cells that remain arrested in pachytene (~80%).
Figure 4. Cdc5‐dependent phosphorylation of Dbf4 drives SC disassembly.
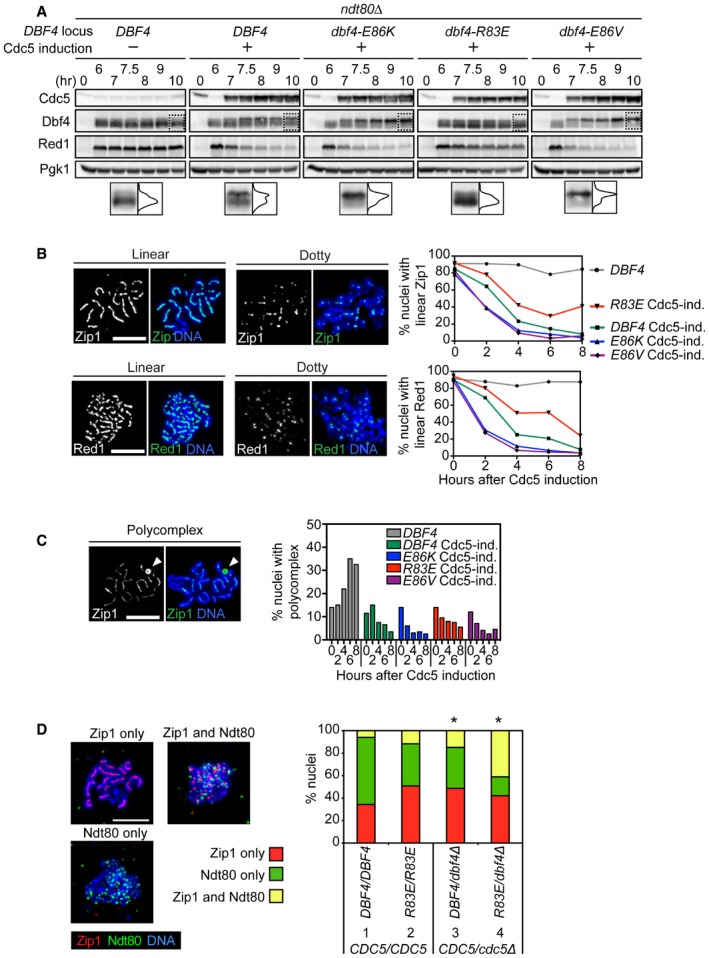
- Strains were induced to synchronously enter meiosis. At 6 h, β‐estradiol was added to induce Cdc5 production. Proteins were detected as in Fig 1B. Images within the dotted boxes are expanded, and the signal quantified to illustrate the distribution of Dbf4 for that lane. The horizontal axis represents the percentage of signal, and the vertical axis corresponds to the source of that signal. The total area under the curve is set to be equal between strains. “−” and “+” denote the absence or presence of an inducible CDC5 allele at the URA3 locus, respectively.
- ndt80Δ strains in the BR1919 background were transferred to sporulation media. At 20 h, β‐estradiol was added to induce Cdc5 production (confirmed in Fig EV3B). Cells were harvested, and meiotic chromosomes were spread for immunofluorescence microscopy at 2‐h intervals after addition of β‐estradiol. Representative images depicting the criteria for categorization of nuclei are shown (panels). Nuclei were categorized according to these criteria (graphs).
- Spread meiotic chromosomes were prepared as in (B). Cells are from the same cultures as (B). A nucleus is shown with the polycomplex depicted by a white arrowhead (panels). Nuclei were categorized according to this criterion (graphs).
- Strains in the BR1919 background containing NDT80‐6xHA were transferred to sporulation media. At 16 h, spread meiotic chromosomes were prepared as in (B). Representative images depicting the criteria for categorization of nuclei are shown (panels). Nuclei were categorized according to these criteria (graphs). *P < 0.05 (chi‐squared test), in comparison with the wild‐type ratio positive for both Zip1 and Ndt80.
Figure EV3. Production of Cdc5 leads to downregulation of DSB formation.
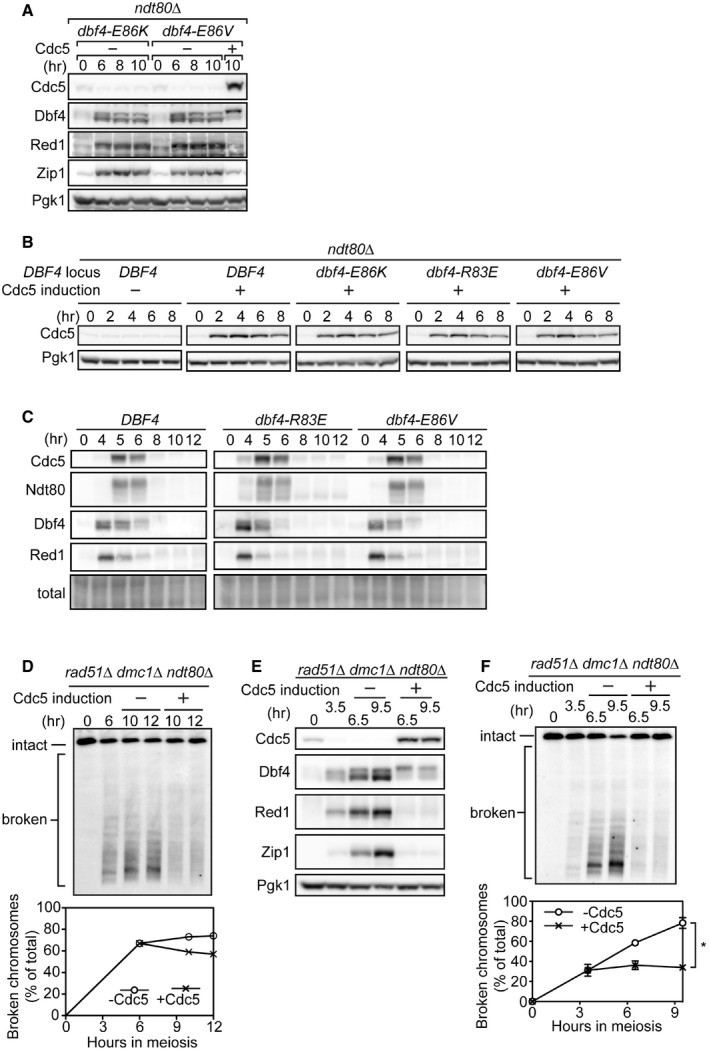
-
AStrains were induced to synchronously enter meiosis. At 6 h, the dbf4‐E86V culture was split and either carrier (− Cdc5) or β‐estradiol (+ Cdc5) was added. The dbf4‐E86K culture only received carrier. Cells were harvested at the indicated time points, and proteins were detected as in Fig 1B.
- B
-
CStrains were induced to synchronously enter meiosis. Cells were harvested at the indicated time points, and proteins were detected as in Fig 1B. Total, total protein levels (Ponceau S staining).
-
DCells were induced to synchronously enter meiosis. At 6 h, the culture was split and either carrier (− Cdc5 induction) or β‐estradiol (+ Cdc5 induction) was added. Cells were harvested at the indicated time points, and meiotic chromosomes were monitored as in Fig 3A. Data are represented as the results of an individual experiment (see Fig 5B for the duplicate experiment).
-
E, FCells were induced to synchronously enter meiosis. At 3.5 h, the culture was split and either carrier (− Cdc5 induction) or β‐estradiol (+ Cdc5 induction) was added. Cells were harvested at the indicated time points to detect proteins as in Fig 1B (E) or monitor meiotic chromosomes as in Fig 3A (F). Cells are from the same culture. Data are represented as mean ± SEM from three experiments (*P < 0.05, paired t‐test).
Source data are available online for this figure.
We next examined the rate at which Zip1 and Red1 dissociate from meiotic chromosomes upon Cdc5 induction in recombination proficient ndt80Δ strains. Meiotic chromosomes were surface spread, and the behavior of chromosomally associated Zip1 and Red1 proteins was monitored by immunofluorescence. In this experiment, BR1919 strains were employed due to improved spreading of chromosomes. We found that dissociation of SC components from meiotic chromosomes also correlated closely with Dbf4‐Cdc5 interaction strength (Figs 4B and EV3B), arguing that removal of these SC components from meiotic chromosomes is related to protein destruction. We then monitored formation of the polycomplex (PC), which is an aggregate of SC proteins that can form in wild‐type cells but has a particular tendency to accumulate in pachytene‐arrested cells (Fig 4C). The percentage of cells with PC declined upon Cdc5 induction in all of the strains tested. The efficiency of decline was mildly correlated with Dbf4‐Cdc5 interaction strength, although the correlation was less pronounced than the dissociation kinetics from chromosomes. This is possibly because Zip1 requires rigid chromosomal axes to be associated with chromosomes, but the foundation of the axes is provided by Red1, which also undergoes prompt destabilization upon Cdc5 induction (Fig 4B).
Surprisingly, the dbf4‐R83E mutation had no noticeable effect on an otherwise unperturbed meiosis and spore viability remained high (98% in dbf4‐R83E compared to 99% in wild type, 160 spores examined per strain). The kinetics of Red1 and Ndt80 induction in dbf4‐R83E and dbf4‐E86V were comparable to wild type, suggesting that meiotic entry and progression are not impaired in these mutants (Fig EV3C). Moreover, the kinetics of Red1 destruction was indistinguishable from wild type. This prompted us to closely examine the events at the prophase I–metaphase I boundary. Around this period, SC disassembly swiftly follows the induction of Ndt80, which can be monitored by its accumulation on meiotic chromosomes (Fig 4D, left panels; Tung et al, 2000; Wang et al, 2011). Thus, in wild‐type cells, Ndt80 is rarely seen together with the SC (Fig 4D, column 1 in graph). In dbf4‐R83E, there was a marginal increase in the fraction of cells displaying Ndt80 and the major SC component Zip1, and a similar increase was seen in heterozygous diploids containing one copy each of DBF4 and CDC5 (Fig 4D, columns 2 and 3 in graph). However, when the only DBF4 gene in this heterozygous diploid was dbf4‐R83E, the majority of cells with Ndt80 signal still retained Zip1, indicating that Zip1 is more persistent in this genetic background (Fig 4D, column 4 in graph). These data suggest that, in an otherwise wild‐type meiosis, reducing the amount of intracellular DDK‐Cdc5 complexes results in inefficient SC disassembly. The relatively mild phenotypes of dbf4‐R83E, however, point toward the presence of another mechanism(s) involved in timely destruction of SC components at the prophase I–metaphase I transition.
Having established that Dbf4‐Cdc5 interaction strength plays a pivotal role in regulating removal of the SC, we wanted to better characterize the ensuing DSB repair. To examine whether the DSB repair seen in the dmc1Δ ndt80Δ mutant background (Fig 3B) is mechanistically equivalent to that seen in the dmc1Δ mutant background (Fig 3A), the rad51Δ dmc1Δ ndt80Δ triple mutant background was employed and Cdc5 production was induced. Importantly, despite causing degradation of SC components, Cdc5 was unable to induce efficient DSB repair; although a mild reduction in broken chromosome molecules was observed, there was no increase in intact chromosome molecules following Cdc5 induction at 6 h (Figs 5A and B, and EV3D). This uncoupling of SC destruction and DSB repair highlights the requirement for Rad51 in repairing DSBs that persist following destruction of the SC in the dmc1Δ mutant. The induction of Cdc5‐N209A, a catalytically inactive mutant (‐kd, kinase dead; Bartholomew et al, 2001), did not trigger Zip1 and Red1 destruction (Fig 5C). Nor did it lead to repair of DSBs (Fig 5D), arguing that the kinase activity of Cdc5 is essential for triggering SC component destruction and subsequent DSB repair at the end of pachytene.
Figure 5. Cdc5 kinase activity is required for destruction of SC components, unshackling of Rad51, and phosphorylation of Dbf4.

-
A, BStrains were induced to synchronously enter meiosis. At 6 h, the culture was split and either carrier (Cdc5 induction −) or β‐estradiol (Cdc5 induction +) was added. Cells were harvested at the indicated time points to detect proteins as in Fig 1B (A) or monitor meiotic chromosomes as in Fig 3A (B). Cells are from the same cultures.
-
C, DStrains were induced to synchronously enter meiosis. At 6 h, β‐estradiol was added to induce production of Cdc5 or Cdc5‐kd (kinase dead, Cdc5‐N209A). Cells were harvested at the indicated time points to detect proteins as in Fig 1B (C) or monitor meiotic chromosomes as in Fig 3A (D). Cells from the cdc5‐kd set of strains are from the same cultures. “−” denotes the absence of an inducible CDC5 or cdc5‐kd allele at the URA3 locus.
-
EStrains were induced to synchronously enter meiosis. At 6 h, β‐estradiol was added to induce production of Cdc5. Cells were harvested at 10 h and resolved by SDS–PAGE in gels containing the indicated amounts of Phos‐tag reagent. Cells are from the same cultures as Fig 4A. “−” denotes the absence of an inducible CDC5 allele at the URA3 locus.
While conducting the experiments in Fig 5B, we noticed that further accumulation of broken chromosomes was mildly reduced upon induction of Cdc5. Since both Dmc1 and Rad51 are absent in this strain, meaning that DSB repair is essentially nonexistent, we suspected that Cdc5 induction might affect DSB formation, as DSBs are continuously formed up until Ndt80 production triggers pachytene exit (Argunhan et al, 2013; Carballo et al, 2013; Gray et al, 2013; Rockmill et al, 2013; Thacker et al, 2014; Subramanian et al, 2016). When this result was compared with a duplicate experiment where Cdc5 was induced at the same time point (6 h), we noticed a correlation between the level of DSBs that had already formed and the magnitude of Cdc5's inhibitory effect on further DSB formation. When broken chromosomes comprised ~90% of total chromosomes at the time of Cdc5 induction, a reduction in broken chromosomes of ~10% was observed by 12 h (Fig 5B). However, when broken chromosomes comprised only ~70% of total chromosomes at the time of Cdc5 induction, a ~20% reduction in broken chromosomes was observed by 12 h (Fig EV3D). These observations prompted us to examine the effect of inducing Cdc5 at a much earlier time point, when even fewer DSBs have formed. Thus, Cdc5 was induced at 3.5 h and its impact on further DSB formation was examined in triplicate cultures. Interestingly, inducing Cdc5 at a time when ~30% of chromosomes were broken resulted in a ~45% reduction in further DSB formation (Fig EV3E and F).
These results suggest that Cdc5 acts during the prophase I–metaphase I transition to shut off meiotic DSB formation. It is formally possible that induction of Cdc5 at 3.5 h may have interfered with any ongoing DNA replication, which itself could result in inhibition of DSB formation, although previous reports have suggested that DNA has been mostly/completely replicated by this time point (Valentin et al, 2006; Murakami & Keeney, 2014). Moreover, such potential interference is unlikely when Cdc5 was induced at 6 h, a time point by which ~80% of chromosomes had already been broken (Figs 5B and EV3D).
Taken together, we conclude that the upregulation of Cdc5 upon pachytene exit is sufficient to drive SC destruction, which coincides with unshackling of the mitotic recombinase Rad51. This is accomplished through a direct interaction with DDK, with the interaction strength proving to be an important parameter in determining the efficiency of SC destruction. Concomitantly, Cdc5 is likely involved in repressing further DSB formation as cells exit from prophase I.
Polo‐dependent phosphorylation of Dbf4 is associated with SC destruction
Dbf4 is known to be phosphorylated in a Cdc5‐dependent manner during mitosis (Hardy & Pautz, 1996; Weinreich & Stillman, 1999). Furthermore, Dbf4 was shown to migrate as a doublet in immunoblotting experiments as a consequence of this phosphorylation (Ferreira et al, 2000). Consistently, Matos et al (2008) demonstrated that, in metaphase I‐arrested cells lacking Cdc5, Dbf4 migrated with increased electrophoretic mobility, suggesting a reduction in phosphorylation. To rule out the possibility that this phosphorylation involved other Ndt80‐dependent factors, Cdc5 was induced in pachytene‐arrested cells (ndt80Δ) and Dbf4 was detected by immunoblotting. We saw that, much like SC destruction efficiency, the electrophoretic mobility of Dbf4 varied with Dbf4‐Cdc5 interaction strength (Fig 4A), with enhanced interaction mutants displaying more slow‐migrating species. Furthermore, this band shift was not seen when kinase‐dead Cdc5 was induced (Fig 5C). To verify the notion that these species corresponded to phosphorylated Dbf4, we resolved them on gels containing the Phos‐tag reagent, which specifically retards the migration of phosphorylated proteins (Kinoshita‐Kikuta et al, 2014). Consistently, the migration of Dbf4 was significantly retarded only when Cdc5 was induced, with increasing concentrations of Phos‐tag exaggerating this effect (Fig 5E). Taken together, these data suggest that Cdc5 phosphorylates Dbf4 through direct binding via residues 83–88 in Dbf4.
Given that the dbf4‐R83E single mutant does not show obvious defects during an unperturbed meiosis, it is possible that Dbf4 is redundantly regulated through phosphorylation in a Cdc5‐independent manner. Consistent with this possibility, we noticed that Dbf4 occupies a range of electrophoretic mobilities within prophase I even in the absence of Cdc5 (Appendix Fig S2A) and when cells make the natural progression through meiosis (Fig EV3C). Moreover, the migration of Dbf4 in Phos‐tag gels was mildly retarded with increasing concentrations of Phos‐tag reagent even in the absence of Cdc5 induction (Fig 5E). In our co‐IP experiments, we saw a similar trend in the electrophoretic mobility of Cdc7 (Fig 1D). However, since Cdc5 is known to bind directly to Dbf4 but not Cdc7 (Matos et al, 2008), we focused our attention on Dbf4.
CDK1 is essential for efficient phosphorylation of Dbf4 and Polo‐driven SC component destruction
One potential candidate that could be responsible for Cdc5‐independent phosphorylation of Dbf4 is Cdc28, the budding yeast homolog of CDK1. In order to address this possibility, we took advantage of the cdc28‐as1 allele, which encodes a conditional mutant of Cdc28 that can be catalytically inactivated through the addition of an ATP analog, 1NM‐PP1 (‐as, analog sensitive; Bishop et al, 2000). The cdc28‐as1 allele was combined with the cdc20‐md allele to arrest the meiotic cell cycle at metaphase I and restrict the APC/C‐dependent degradation of Dbf4 that occurs at the onset of anaphase I.
Strikingly, upon Cdc28 inhibition in mid‐prophase I, phosphorylation of Dbf4 was greatly reduced and both Zip1 and Red1 continued to accumulate despite the production of Cdc5 (Fig 6A), although we note that induction of Cdc5 was itself mildly compromised by the inactivation of Cdc28. Given that relatively little Cdc5 is able to trigger efficient SC destruction (e.g., Figs 3C and EV3C), along with experiments in which Cdc5 was more uniformly induced in Cdc28‐inactivated pachytene‐arrested cells (see below), the above results support the notion that Cdc28 is important for efficient Dbf4 phosphorylation and Cdc5‐driven destruction of SC proteins.
Figure 6. CDK1 kinase activity and DDK are required for efficient SC destruction.
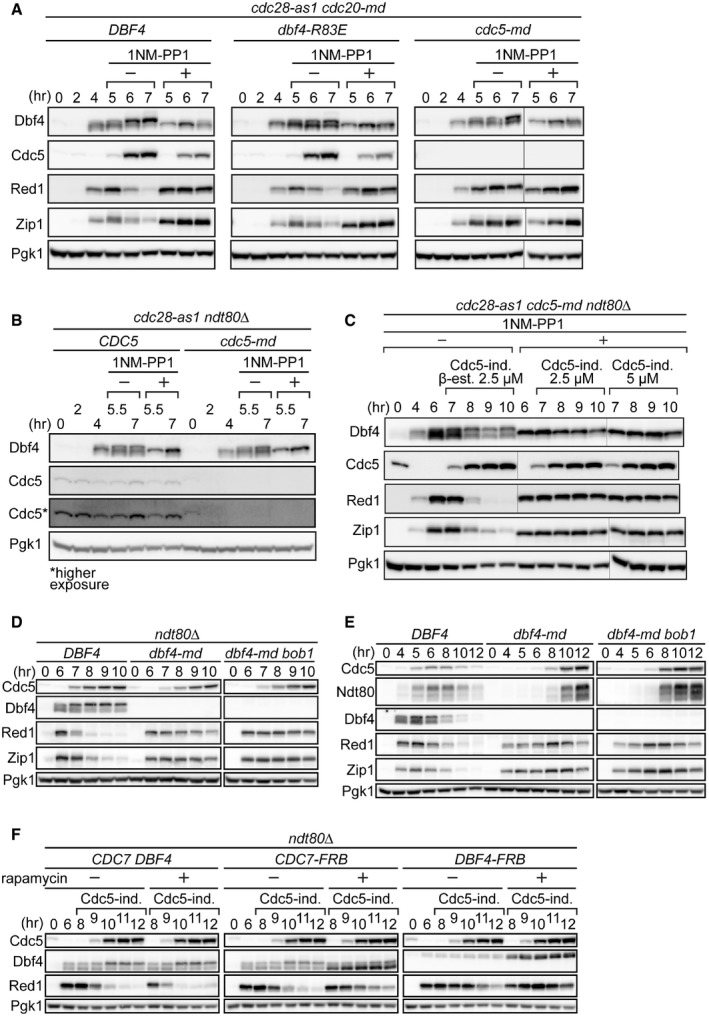
-
A, BStrains were induced to synchronously enter meiosis. At 4 h, cultures were split and either carrier or 1NM‐PP1 (an ATP analog that specifically inhibits Cdc28‐as1) was added (denoted “−” and “+” under 1NM‐PP1, respectively). Cells were harvested at the indicated time points, and proteins were detected as in Fig 1B.
-
CStrains were induced to synchronously enter meiosis. At 4 h, the culture was split and either carrier or 1NM‐PP1 was added. At 6 h, β‐estradiol was added to all cultures at the indicated concentrations to induce production of Cdc5. Cells were harvested at the indicated time points, and proteins were detected as in Fig 1B.
-
D, EStrains were induced to synchronously enter meiosis. In (D), β‐estradiol was added at 6 h to induce production of Cdc5. Cells were harvested at the indicated time points, and proteins were detected as in Fig 1B. The bob1 mutation was included as it bypasses the essential requirement for DDK in DNA replication.
-
FStrains were induced to synchronously enter meiosis. Cultures were split at 6 h, and either carrier (− rapamycin) or rapamycin (+ rapamycin) was added. β‐estradiol was added to all cultures at 8 h to induce production of Cdc5. Cells were harvested at the indicated time points, and proteins were detected as in Fig 1B. These strains were constructed in the anchor‐away background, where rapamycin triggers the nuclear deportation of FRB‐tagged proteins, which in this case corresponds to Cdc7 and Dbf4.
Source data are available online for this figure.
Next, we examined pachytene‐arrested cells by introducing the ndt80Δ mutation. As with metaphase I‐arrested cells (cdc20‐md), phosphorylation of Dbf4 before pachytene exit was largely dependent on Cdc28 (Fig 6B). The electrophoretic mobility of Dbf4 in pachytene‐arrested cells was not affected by the absence of basal levels of Cdc5 (cdc5‐md strain, Fig 6B), further arguing that Cdc28 is the primary kinase responsible for Dbf4 phosphorylation before pachytene exit. We then combined the inducible Cdc5 expression system with the cdc28‐as1 allele to examine the interplay between Cdc5‐ and Cdc28‐dependent phosphorylation of Dbf4 in SC component destruction before pachytene exit. Unlike the experiments with the cdc20‐md strains described above, induction of Cdc5 when Cdc28 was inactivated was comparable to that when Cdc28 was active. Following Cdc28 inactivation, Cdc5 induction through addition of β‐estradiol at concentrations of 2.5 and 5 μM, with the latter causing slightly increased induction of Cdc5, led neither to Dbf4 phosphorylation nor SC component destruction (Fig 6C). These observations establish Cdc28 as an important component of the Cdc5‐driven SC destruction mechanism. However, we also noticed that, while Cdc5 is absolutely required for destruction of Red1 and Zip1, the Cdc28 dependency was conditional: Although both Red1 and Zip1 levels remained high for an extended period of time, Cdc28 inhibition did not block the decline of these proteins when Cdc20 was present (Fig EV4A and B), suggesting they can also be destroyed at or after the metaphase I‐anaphase I transition as long as Cdc5 is present.
Figure EV4. The requirement for CDK1 in SC destruction is conditional.

-
A, BStrains were induced to synchronously enter meiosis. At 4 h, cultures were split and either carrier or 1NM‐PP1 (an ATP analog that specifically inhibits Cdc28‐as1), was added (denoted “−” and “+” under 1NM‐PP1, respectively). Cells were harvested at the indicated time points, and proteins were detected as in Fig 1B.
Source data are available online for this figure.
DDK is part of the SC destruction mechanism triggered by Polo
Since our data strongly suggested the involvement of Dbf4 in the regulation of SC destruction, we wanted to determine whether DDK itself is required. We reasoned that employing the meiotic depletion allele of DBF4 (dbf4‐md) would abolish DDK activity specifically in meiosis, since Dbf4‐independent Cdc7 activity has not been reported (Matthews & Guarné, 2013). Under this condition, Dbf4 was not detectable by Western blotting (Fig 6D and E). Notably, destruction of SC components was severely compromised both within pachytene‐arrested cells, where Cdc5 was induced artificially, and during the natural transition through meiosis (Fig 6D and E, left panels). Dbf4 has an essential role in DNA replication, which can be bypassed by the bob1 mutation in the MCM5 gene (Hardy et al, 1997). Thus, we repeated these experiments in the presence of the bob1 mutation to exclude the possibility that these results were an artifact of defective DNA replication. This analysis verified our results, as the kinetics of SC protein destruction was still delayed in the absence of Dbf4 (Fig 6D and E, right panels).
DDK is also implicated in the initiation of meiotic recombination, which is essential for SC formation (Matos et al, 2008; Sasanuma et al, 2008; Wan et al, 2008). In order to examine the function of DDK once the SC has formed, we wanted to deplete DDK activity from meiotic nuclei after SC maturation. To achieve this, we employed the anchor‐away technique, which exploits tight ternary complex formation between the human FK506 binding protein (FKBP12) and the FKBP12‐rapamycin‐binding (FRB) domain of human mTOR (Haruki et al, 2008). In our assay, the ribosomal protein RPL13A was tagged with FKBP12 and proteins of interest were tagged with FRB. Since ribosomal proteins are actively deported from the nucleus upon assembly with rRNAs, proteins tagged with FRB rapidly bind to FKBP12‐tagged RPL13A upon addition of rapamycin, resulting in nuclear deportation. This technique has recently proven useful for stage‐specific depletion of proteins during budding yeast meiosis (Subramanian et al, 2016). We exploited the essential role of DDK in vegetative growth to confirm that both Dbf4 and Cdc7 could be conditionally inactivated using the anchor‐away technique (Fig EV5A).
Figure EV5. Conditional depletion of DDK before pachytene exit results in defective SC morphology.
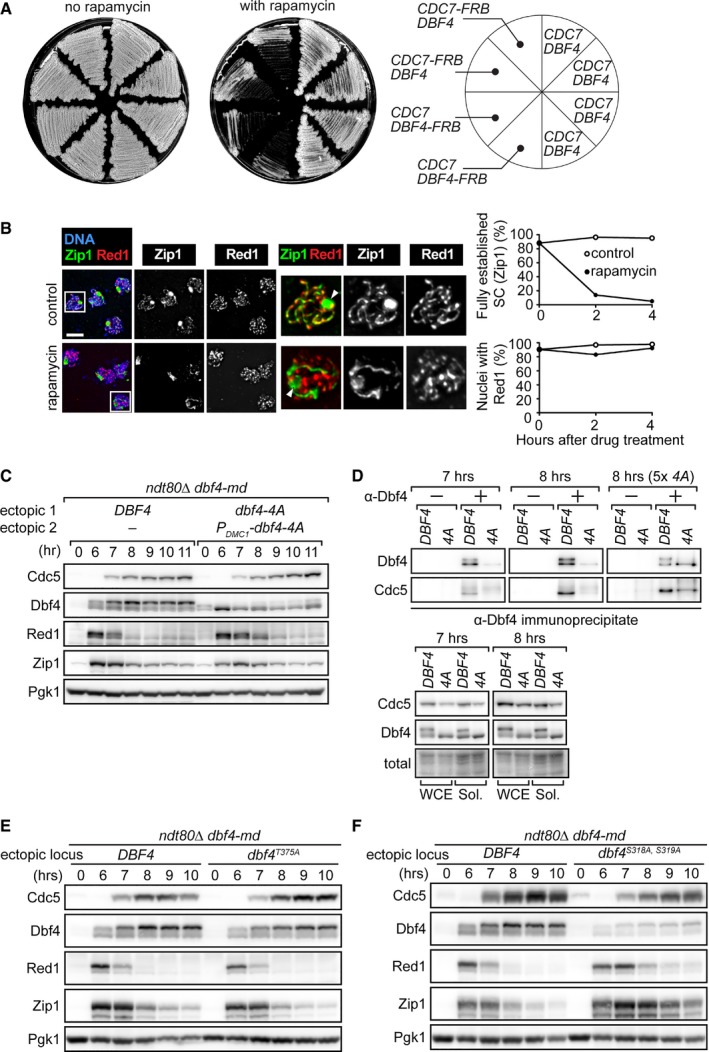
-
ASingle colonies from the indicated strains were streaked on rich media with or without rapamycin. Since DBF4 and CDC7 are both essential genes, the observed growth defect in the presence of rapamycin indicates conditional inactivation of DDK.
-
BThe ndt80Δ DBF4‐FRB strain was induced to synchronously enter meiosis. At 6 h, either carrier (control) or rapamycin (rapamycin) was added. Cells were harvested 2 and 4 h after drug treatment, and chromosomes were spread as in Fig 4B. To quantify these results, nuclei were scored for the presence of fully established SC, as judged by Zip staining, and the presence of Red1 staining (for representative images, see control nuclei). Nuclei in white boxes are expanded to the right. White arrowheads depict polycomplexes, which were larger than usual because of prolonged cell cycle arrest. At least 100 nuclei were scored in the presence and absence of rapamycin at each time point. Data are represented as means from two experiments. Scale bar, 5 μm.
-
C, DStrains were induced to synchronously enter meiosis. At 6 h, β‐estradiol was added to induce production of Cdc5. At the indicated time points, cells were harvested for detection of proteins as in Fig 1B (C). At the 7 and 8 h time points, cells were also harvested for immunoprecipitation with an anti‐Dbf4 antibody (D). 5× 4A denotes that fivefold more dbf4‐4A sample was loaded than DBF4. “−” and “+” indicate the exclusion and inclusion of antibody for IP, respectively, with the no antibody condition serving as a negative control. WCE, whole‐cell extract. Sol., soluble fraction.
-
E, FStrains were induced to synchronously enter meiosis. At 6 h, β‐estradiol was added to induce production of Cdc5. Cells were harvested at the indicated time points, and proteins were detected as in Fig 1B.
Source data are available online for this figure.
Cdc7‐FRB or Dbf4‐FRB was first depleted from pachytene nuclei 6 h into meiosis, by which time meiotic recombination had been induced and ~90% of nuclei showed fully established SC (Fig EV5B). β‐estradiol was then added to the media at 8 h to induce Cdc5 production. In both the CDC7‐FRB and DBF4‐FRB strains, the presence of rapamycin led to a delay in Red1 destruction compared to the untagged control strain (Fig 6F). This reduction in destruction efficiency was more prominent in the DBF4‐FRB strain, where Red1 destruction was delayed even in the absence of rapamycin, likely reflecting the fact that the FRB tag mildly affected Dbf4 functionality, as reported previously (Natsume et al, 2013). Nonetheless, the results presented here strongly suggest that DDK itself is required for efficient destruction of SC components. While conducting these experiments, we found that Dbf4 depletion within pachytene, without induction of Cdc5, compromised SC integrity: abnormally assembled Zip1 that was not associated with Red1 accumulated, while Red1 itself showed chromosomal association similar to cells without rapamycin treatment (Fig EV5B). It is possible that, before pachytene exit and Cdc5 induction, DDK has another role related to the maintenance of chromosomal Zip1 during early/mid‐prophase I. Consistent with this possibility, a recent study provided evidence that DDK is able to phosphorylate Zip1 in vitro, and in vivo experiments implicated this phosphorylation in regulating chromosome synapsis (Chen et al, 2015). Taken together, these findings suggest that DDK has dual roles in regulating the SC. Before pachytene exit, DDK is required for maintenance of SC integrity, but upon induction of Cdc5, DDK serves as an important component of the Cdc5‐driven SC destruction mechanism.
Dbf4 phosphorylation is important for efficient Polo‐driven SC destruction
The finding that efficient Dbf4 phosphorylation requires Cdc28 and Cdc5, combined with the observed correlation between Dbf4 phosphorylation and SC destruction efficiency, suggested that phosphorylation of Dbf4 is involved in SC destruction. However, CDKs and Polo kinases are known to have many targets (Barr et al, 2004; Enserink & Kolodner, 2010), raising the possibility that the defects in SC component destruction described above are not directly related to Dbf4 phosphorylation. To examine the role of Dbf4 phosphorylation per se, we set out to identify the Ser/Thr residues in Dbf4 required for its phosphorylation. Through multiple alignments of Dbf4 sequences from six species within the genus Saccharomyces, we identified conserved Ser/Thr resides among these species. These residues were then systematically mutated to Ala (see Appendix Supplementary Methods for details). The mutant genes were placed under the control of the DBF4 promoter and integrated at the URA3 locus in a strain that does not express DBF4 from its native locus during meiosis (i.e., dbf4‐md). Thus, these strains produce wild‐type Dbf4 during mitosis but mutated Dbf4 during meiosis. The inducible Cdc5 expression system and the ndt80Δ mutation were introduced.
We discovered that there was little/no observable band shift of Dbf4 before or after Cdc5 induction when S318, S319, S374, and T375 were mutated to Ala (dbf4‐4A; Fig 7A). Moreover, destruction of Red1 and Zip1 was severely delayed. However, Dbf4‐4A levels were reduced compared to wild‐type protein, suggesting that the Dbf4‐4A protein has reduced stability. We reasoned that if phosphorylation of Dbf4 is important for SC destruction, then circumventing the reduced stability of Dbf4‐4A by overproducing it should still affect SC destruction. To test this, we integrated an additional copy of the dbf4‐4A gene under the control of the strong meiosis‐specific DMC1 promoter at the HIS6 locus (P DMC1 ‐dbf4‐4A). Under this condition, the levels of Dbf4‐4A were similar to, if not more than, the strain expressing wild‐type Dbf4 (Fig EV5C). Nonetheless, we saw that destruction of Red1 was still delayed, supporting the notion that Dbf4 phosphorylation is important for the timely destruction of SC components, especially Red1. The relatively mild delay in Red1 destruction observed here, combined with the near normal kinetics of Zip1 destruction, could be due to excessive levels of Dbf4‐4A at the time of Cdc5 induction (compare 6 hrs in Fig EV5C).
Figure 7. Phosphorylation of Dbf4 is integral to the timely destruction of SC proteins.
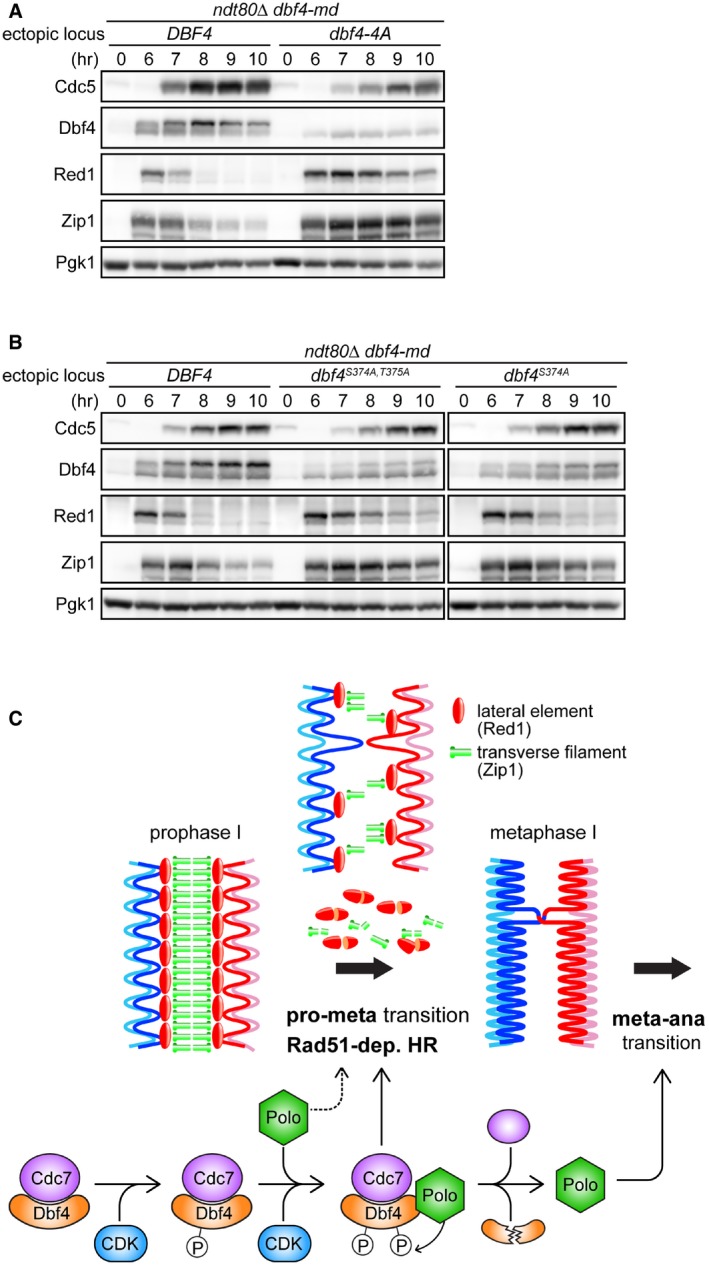
-
A, BStrains were induced to synchronously enter meiosis. At 6 h, β‐estradiol was added to induce production of Cdc5. Cells were harvested at the indicated time points, and proteins were detected as in Fig 1B. dbf4‐4A encodes four Ser/Thr to Ala mutations (S318A, S319A, S374A, T375A).
-
CSchematic model of how the CDK‐DDK‐Polo axis facilitates the transition from meiotic prophase I to metaphase I. dep., dependent; pro, prophase I; meta, metaphase I; ana, anaphase I; HR, homologous recombination.
Source data are available online for this figure.
The dbf4‐4A mutant provided a means to examine the effect of Cdc28‐dependent phosphorylation of Dbf4 on the Dbf4‐Cdc5 interaction. We employed the same strains as in Fig EV5C and utilized an anti‐Dbf4 antibody to directly immunoprecipitate Dbf4 and Dbf4‐4A and then monitored the amount of Cdc5 that was found to co‐IP. For unknown reasons, immunoprecipitation of Dbf4‐4A was inefficient compared to Dbf4 (Fig EV5D). This is not due to a difference in solubility between Dbf4 and Dbf4‐4A (Fig EV5D). Nevertheless, Cdc5 was found to co‐IP with Dbf4‐4A, arguing that Dbf4‐4A retains its ability to interact with Cdc5, although the interaction might be slightly compromised (Fig EV5D, 5× 4A). These observations suggest that the reduction in phosphorylation caused by the dbf4‐4A mutation does not dramatically affect the Dbf4‐Cdc5 interaction.
To further characterize the role of Dbf4 phosphorylation in SC destruction, we generated alleles of DBF4 with fewer mutations. Mutation of both S374 and T375 to Ala (dbf4 S374A,T375A) reduced phosphorylation to a level that is intermediate between wild‐type Dbf4 and Dbf4‐4A (Fig 7B, leftmost panels). Consistent with the notion that Dbf4 phosphorylation is important for efficient destruction of SC proteins, a milder delay in destruction of Red1 and Zip1 was seen in dbf4 S374A,T375A compared to the dbf4‐4A mutant. Furthermore, dbf4 S374A showed a level of phosphorylation between dbf4 S374A,T375A and wild type, and we observed only a modest delay in the destruction of Red1 and Zip1 (Fig 7B, rightmost panels). The dbf4 T375A mutation alone had no effect on Dbf4 phosphorylation, and consistent with a role for Dbf4 phosphorylation in SC destruction, we did not observe any delay in the destruction of Red1 and Zip1 (Fig EV5E). Similarly to dbf4‐4A, the dbf4 S318A,S319A mutant showed a reduction in the total levels of Dbf4, suggesting that mutation of these residues results in reduced protein stability (Fig EV5F). Thus, it is difficult to draw clear conclusions about the requirement for S318 and S319 in Dbf4 phosphorylation. Nonetheless, we note that phosphorylation was more evident in dbf4 S374A,T375A than in dbf4‐4A, suggesting that residues S318 and S319 do contribute to Dbf4 phosphorylation, perhaps indirectly. Taken together, our data suggest that Dbf4‐S374 and Dbf4‐T375 are synergistically involved in Cdc5/Cdc28‐dependent phosphorylation, and that this phosphorylation is required for efficient destruction of SC components.
Discussion
In this work, we demonstrated that three major cell cycle kinases, DDK, Polo, and CDK1, coordinate to dismantle the SC, a meiosis‐specific chromosomal structure, at the prophase I–metaphase I transition (Fig 7C). Before pachytene exit, the Dbf4 component of DDK undergoes CDK1‐dependent phosphorylation. As cells exit pachytene, Polo is upregulated and initiates SC destruction as it collaborates with CDK1 to hyperphosphorylate Dbf4. Hyperphosphorylated DDK greatly enhances the efficiency of SC destruction, which relieves Rad51 of its meiosis‐specific inhibition and allows rapid repair of any persisting DSBs. Upon completion of metaphase I, Dbf4 is targeted for degradation by the APC/C, and Polo promotes the metaphase I‐anaphase I transition independently of DDK by regulating key steps such as the destruction of cohesin. Our findings shed light on how SC destruction is temporally coordinated with the cell cycle and point toward the existence of a change in the mode of HR to promote faithful chromosome segregation and reinforce gamete viability. Below, we discuss the implications of these findings.
Fundamental cell cycle kinases collaborate during the prophase I–metaphase I transition
The formation of the SC in meiotic prophase I has been extensively studied (Tsubouchi et al, 2016). In comparison, little is known about how this macromolecular proteinaceous structure is removed from chromosomes following pachytene exit, despite the fact that persistence of the SC beyond prophase I would pose a major obstacle to homologous chromosome segregation (Cahoon & Hawley, 2016). A role for Cdc5 has been ascribed due to the finding that, even in the absence of Ndt80, which facilitates entry into the later stages of meiosis, the production of Cdc5 alone leads to efficient disassembly of the SC (Sourirajan & Lichten, 2008). Similarly, Polo‐like kinase was shown to be required for SC disassembly in the mouse and the worm, suggesting that the mechanism governing SC disassembly is evolutionarily conserved (Jordan et al, 2012; Machovina et al, 2016). However, the regulation of Cdc5‐driven SC disassembly and the involvement of other proteins had not been explored.
Here, we used budding yeast to demonstrate that two major cell cycle kinases, DDK (Dbf4‐dependent Cdc7 kinase) and CDK1 (Cdc28), constitute an important part of the SC destruction mechanism triggered by the induction of Polo (Cdc5) at the prophase I–metaphase I transition. We showed that this mechanism promotes disassembly of lateral element protein Red1 and transverse filament protein Zip1 (Fig 4B), which coincides with a reduction in the levels of these proteins (Figs 3C and 4A). Removal and/or destruction of major SC proteins provides an effective means for SC dismantlement from meiotic chromosomes. While both Red1 and Zip1 seem to be destroyed upon activation of the Cdc5‐triggered SC disassembly mechanism, Red1 could be a more important target in SC disassembly. Once Red1, a major component of the lateral elements of the SC, dissociates from chromosomes, Zip1, the transverse filament protein, loses the foundation for its chromosomal localization (Smith & Roeder, 1997). In this respect, it is interesting to note that destruction of Red1 is more responsive to Cdc5 induction than destruction of Zip1 (e.g., Figs 6D and EV5C), raising the possibility that the mechanism governing destruction of these two proteins is at least partially distinct. Zip1 destruction might require two steps: dissociation from chromosomes upon Red1 destruction, followed by destruction itself. With this in mind, it is relevant to note that incorporation of Zip1 into the SC is a dynamic feature, with Zip1 constantly dissociating from and reintegrating into the SC (Voelkel‐Meiman et al, 2012). It is possible that, following Cdc5 induction, this behavior of Zip1 might facilitate its destruction.
When the kinase activity of Cdc5 is ablated, SC destruction does not occur, regardless of the Dbf4‐Cdc5 interaction (Fig 5C). Similarly, although Cdc5‐independent phosphorylation of Dbf4 is important, it alone is not sufficient for triggering SC destruction (Fig 6A). Moreover, even if Cdc28 activity is inhibited, SC destruction can occur in a Cdc20‐ and Cdc5‐dependent manner (Fig EV4A and B). These findings build on previous reports by confirming the requirement for Cdc5's kinase activity (Sourirajan & Lichten, 2008). Notably, when either the regulatory subunit (Dbf4) or the catalytic subunit (Cdc7) of DDK was depleted, SC component destruction was severely compromised yet not completely repressed (Fig 6D–F), arguing that DDK activity is important but not an absolute requirement for this protein destruction mechanism. Since the influence of DDK is dependent on the kinase activity of Cdc5, one possibility is that DDK promotes SC destruction by enhancing the kinase activity of Cdc5. Our data suggest that Cdc28 contributes to SC destruction indirectly by phosphorylating Dbf4, although we cannot rule out the possibility that it has a more direct role in SC destruction. It is also possible that Cdc28 phosphorylates Cdc5, as in vitro experiments have indicated that Cdc28‐dependent phosphorylation of Cdc5 enhances its kinase activity and that this phosphorylation is essential for the role of Cdc5 in mitosis (Mortensen et al, 2005). What is the role played by Cdc28‐ and Cdc5‐dependent Dbf4 phosphorylation? One possibility is that, in the absence of Dbf4 phosphorylation, DDK is unable to enhance the kinase activity of Cdc5, thus mimicking the inefficient SC destruction seen in the absence of DDK activity. This would suggest that Cdc5 contributes to the establishment of a positive feedback loop by phosphorylating Dbf4.
The demonstration that the catalytic activities of Cdc5, DDK, and Cdc28 are required for SC destruction points toward a scenario in which SC components are phosphorylated directly by at least one of these kinases. Consistently, it was recently shown that partially purified DDK can phosphorylate Zip1 in vitro (Chen et al, 2015). Chen et al (2015) also provided evidence to suggest that DDK‐independent phosphorylation of Zip1 takes place in vivo, raising the possibility that Cdc5 and/or Cdc28 may phosphorylate Zip1. Consistently, it has been reported that mouse PLK1 phosphorylates SYCP1, the mouse homolog of Zip1, as well as another central element protein (Jordan et al, 2012). Additionally, Cdc28 has been shown to localize to meiotic chromosomes during prophase I, where it promotes the maturation of Zip1 into fully linear SC (Zhu et al, 2010). In contrast to Zip1, the phosphorylation of lateral element protein Red1 is well established (Bailis & Roeder, 1998), although the requirement for Red1 phosphorylation is debated (Lai et al, 2011). It is interesting to note that other members of the Ndt80 regulon are dispensable for SC destruction, strongly suggesting that the means to destroy the SC exists before pachytene exit but is unable to act in the absence of Cdc5 activity. Emphasis should be placed on identifying the targets of Cdc5, which likely include Red1 and/or Zip1, and possibly other central element proteins that are required for SC formation such as Ecm11 and Gmc2 (Humphryes et al, 2013; Voelkel‐Meiman et al, 2013; Leung et al, 2015).
Significance of the DDK‐Polo interaction in other contexts
The genetic and physical interaction between DDK and Cdc5 has been known for approximately two decades, although the molecular implications of this interaction have thus far remained elusive. Overproduction of Cdc5 can suppress the growth defect of several dbf4 temperature sensitive mutants, strongly suggesting that enhancing the physical interaction between Cdc5 and Dbf4 can suppress defects in the initiation of DNA replication (Kitada et al, 1993; Hardy & Pautz, 1996). More recently, it was shown that deletions/mutations in Dbf4 that abolish the Dbf4‐Cdc5 interaction are capable of suppressing the growth defect of the cdc5‐1 temperature sensitive mutant, which is unable to exit mitosis, suggesting that the Dbf4‐Cdc5 interaction may also regulate mitotic exit (Miller et al, 2009; Chen & Weinreich, 2010). We have now provided compelling evidence that the binding of Cdc5 through residues 83–88 on Dbf4 is important for phosphorylation of Dbf4, which in turn promotes efficient Cdc5‐driven SC destruction.
In addition to its role in destroying the SC and reactivating mitotic recombination mechanisms, our results suggest that Cdc5 downregulates DSB formation toward the end of prophase I (Figs 5A and B, and EV3D–F). Such an act would further facilitate the transition into metaphase I. Negative regulation of DSB formation by a Polo kinase has recently been reported in the worm (Nadarajan et al, 2017). We do not currently know whether other members of the Ndt80 regulon facilitate this role of Cdc5, although given the nature of the findings presented here, we speculate that Cdc5 does not act alone. The role of Cdc5 in regulating the resolution of joint molecules during meiosis and mitosis has been elucidated (Matos et al, 2011, 2013; Szakal & Branzei, 2013). Moreover, while we were in the process of preparing our manuscript, Princz et al (2017) provided compelling evidence that regulation of joint molecule resolution during mitosis also involves DDK. Taken together, it is becoming increasingly transparent that DDK and Cdc5 collaboratively regulate multiple events in both the mitotic and meiotic cell cycles in budding yeast. It remains a high priority to determine whether DDK and Polo show physical and/or functional interactions in other organisms, especially higher eukaryotes.
Switching of HR modes at the prophase I–metaphase I transition
A long‐standing question in the field of meiosis has concerned the requirement for two RecA orthologs. It has been proposed that Dmc1 preferentially catalyzes interhomolog recombination, whereas Rad51 primarily catalyzes intersister recombination during meiosis, although how the two recombinases serve their roles during meiosis is not clear. Recent studies have demonstrated that Dmc1 but not Rad51 can stabilize base pairing between the invading strand and the donor duplex despite the presence of mismatches (Lee et al, 2015; Qi et al, 2015), providing support for the argument that only Dmc1 can efficiently catalyze interhomolog recombination. It is also interesting to note that Dmc1 catalyzes branch migration in the opposite direction to Rad51 (Murayama et al, 2011). Rad51‐dependent HR does not abide by the regulatory processes that are characteristic of meiotic HR (Lao et al, 2013), leading to the proposal that it resembles mitotic HR. Conversely, several observations indicate that Rad51 is somewhat able to fulfill the same catalytic role as Dmc1, arguing that Rad51 can play a catalytic role in interhomolog recombination (Tsubouchi & Roeder, 2003, 2006; Busygina et al, 2008, 2012; Callender et al, 2016). Furthermore, in some organisms such as Caenorhabditis elegans and Drosophila melanogaster, Dmc1 does not exist and Rad51 is the sole recombinase in meiosis.
Cloud et al (2012) proposed that the catalytic activity of Rad51 during meiosis is dispensable, with its role being to support Dmc1. Although this may be the case for interhomolog crossover formation, our data point towards the possibility that Rad51 has an additional function following exit from pachytene. By this time, recombination intermediates that will give rise to interhomolog crossovers have already been established (Allers & Lichten, 2001), thus the meiotic bias to specifically engage homologs in HR, as opposed to sister chromatids, has served its purpose. Consequently, the SC, which is one of the key factors that promotes interhomolog recombination, can be dismantled. Rad51 is then able to efficiently repair any persisting DSBs before homologs are pulled by their kinetochores to opposite poles of the cells. Although it is formally possible that Dmc1 also participates in this DSB repair, its contribution is likely to be minimal because Dmc1 is dispensable for repairing DSBs in the absence of the SC (e.g., during the mitotic cell cycle). In our experimental system, where Dmc1 is absent, this DSB repair resembles mitotic HR as interhomolog crossovers represent a relatively small fraction of the possible repair outcomes (Fig EV2B–D). Consistently, despite the repair of most/all DSBs, > 80% of the resulting spores were inviable, likely due to mass aneuploidy (Figs 3A and EV2A). Taken together, these findings support the notion that a Rad51‐dependent mitotic mode of HR is activated at the prophase I–metaphase I boundary to repair any persisting DSBs.
Distinct roles for the catalytic activities of Rad51 and Dmc1 during meiosis are further supported by observations in the fission yeast Schizosaccharomyces pombe. The absence of Dmc1 does not lead to a reduction in spore viability (Fukushima et al, 2000), despite a substantial reduction in crossovers (Grishchuk & Kohli, 2003), indicating that the number of crossovers formed is sufficient to support high levels of spore viability (~85%). The absence of either Rad55 or Rad57, which serve as activators for Rad51, results in a relatively small reduction in crossover formation (Grishchuk & Kohli, 2003). Nonetheless, despite forming more crossovers than the dmc1Δ mutant, both the rad55Δ and rad57Δ mutants show ~55% spore viability (Khasanov et al, 1999; Tsutsui et al, 2000), suggesting that defects in the Rad51 HR pathway can lead to a reduction in spore viability that is independent of the requirement for crossover formation. These observations point toward the existence of a role for HR in meiosis that is dependent on the mitotic HR machinery but independent of crossover formation. Consistent with this possibility, the disruption of Rad51 catalytic activity in budding yeast led to a ~12% reduction in spore viability despite DNA joint molecules forming at wild‐type levels (Cloud et al, 2012).
Based on the above discussion, we propose that two modes of HR exist during meiosis. In the first mode, Rad51 promotes interhomolog recombination, likely by assisting Dmc1 (Cloud et al, 2012). It is also possible that Rad51 makes a catalytic contribution to interhomolog recombination, although this is likely to be inefficient (Lao et al, 2013). In the second mode, Rad51 is responsible for the rapid repair of any persisting DSBs before paired bivalents align on the metaphase plate. This repair likely involves the sister chromatid or preferentially produces noncrossovers as it does not lead to efficient interhomolog crossover formation or rescue the spore inviability of the dmc1Δ mutant (Fig EV2). The recombination checkpoint, which controls exit from pachytene and commitment to the meiotic divisions, is likely to be the switch that triggers activation of the second mode of HR.
A similar change in the mode of HR has been reported in C. elegans, where the sole recombinase in meiosis is RAD‐51. The meiotic mode of HR is characterized by the competence to form crossovers and a dependence on RAD‐50 for efficient chromosomal loading of RAD‐51 (Hayashi et al, 2007). At the mid‐ to late‐pachytene transition, this RAD‐50 dependency is abruptly lost, along with the ability to form crossovers, pointing toward a change from the meiotic mode to a mitotic mode of HR. Thus, a change in the mode of HR at the prophase I–metaphase I transition might be a conserved mechanism operating among eukaryotes.
The necessity for the second mode of HR may arise from how pachytene exit is controlled by the recombination checkpoint. Before pachytene exit, interhomolog joint molecules accumulate as DSBs are intensively formed in chromosomes that have not yet been incorporated into the SC, thus constantly stimulating the recombination checkpoint (Hochwagen & Amon, 2006). Furthermore, even after SC maturation, DSBs continue forming, although at a much reduced rate (Argunhan et al, 2013; Carballo et al, 2013; Gray et al, 2013; Rockmill et al, 2013; Thacker et al, 2014; Subramanian et al, 2016). Nonetheless, the recombination checkpoint allows cells to exit pachytene most likely because of its “leaky” nature; low numbers of DSBs within pachytene are not detected by the recombination checkpoint and cells can still progress to metaphase I. This concept is supported by experiments involving the homing endonuclease VDE (Nogami et al, 2002). In response to four unrepairable DSBs, the recombination checkpoint was unable to enforce meiotic arrest, with viability among the resultant spores being reduced from ~90% (zero DSBs) to ~30%. Moreover, some recombination mutants such as rad51, rad55, and rad57 are able to complete meiosis with unrepaired DSBs, producing inviable spores (Game & Mortimer, 1974; Kupiec & Steinlauf, 1997). If cell cycle progression beyond pachytene with unrepaired DSBs is an intrinsic feature of the meiotic cell cycle, it is advantageous for cells to be equipped with a mechanism specifically targeted to repair this DNA damage. The utilization of Rad51 as the recombinase and the sister chromatid as the template would provide a convenient solution, considering the prevalence of this type of DSB repair in mitotic cells and the relatively little time between pachytene exit and metaphase I.
SC disassembly, which occurs at the prophase I–metaphase I boundary, precedes chromosome segregation triggered by removal of sister chromatid cohesion at the metaphase I‐anaphase I transition (Fig 7C). Why meiosis employs this two‐step dismantlement of chromosomal structures/proteins was not known. In light of the discussion above and the findings presented here, there is now substantial molecular evidence to suggest that a second mode of HR exists in the intervening period between SC disassembly and loss of sister chromatid cohesion. The characterization of this HR mechanism, and its exact contribution to ensuring the viability of the products of meiosis, should be the focal topic of future research.
Materials and Methods
Further details of the experimental procedures can be found in Appendix Supplementary Methods.
Yeast strains
All strains are derivatives of SK1 or BR1919. Further details can be found in Appendix Table S1. The dmc1Δ mutant was not examined in the BR1919 background as it does not cause efficient cell cycle arrest in that background.
Sporulation and sample preparation
Cells were sporulated in 2% potassium acetate. Protein extracts were prepared by the trichloroacetic acid method as described (Humphryes et al, 2013). Agarose plugs containing meiotic chromosomes were prepared as described (Farmer et al, 2011). Cdc5 induction was achieved through the addition of β‐estradiol at a final concentration of 5 μM (Okaz et al, 2012), except where indicated otherwise, using the system described by Benjamin et al (2003). Nuclear depletion of FRB‐tagged proteins was achieved through the addition of rapamycin (final concentration 1 μg/ml; Haruki et al, 2008). To inhibit Cdc28‐as1, 1NM‐PP1 was added to cultures at a final concentration of 10 μM (Bishop et al, 2000).
Isolation of dbf4‐E86V as a suppressor of the zip1‐4LA mutant
A diploid strain carrying the zip1‐4LA mutation and the can1/CAN1 cyh2/CYH2 heteroalleles was employed for multicopy suppressor screening (Tsubouchi & Roeder, 2002). zip1‐4LA is a nonnull mutant of ZIP1 that shows a severe cell cycle arrest phenotype at meiotic prophase I despite exhibiting apparently normal SC assembly (Mitra & Roeder, 2007). This cell cycle arrest is suppressed by eliminating DSB formation (spo11Δ), suggesting that it is caused by recombination events that occur following DSB formation. The zip1‐4LA diploid strain was transformed with a YEp24 yeast genomic library (Carlson & Botstein, 1982). These transformants were sporulated and replica plated onto medium containing canavanine and cycloheximide to screen for clones that displayed increased spore viability. Plasmids were recovered from such clones, and the identity of the genes in the genomic DNA fragment was determined. Approximately 50,000 colonies were screened, leading to the recovery of three isolates carrying DBF4 and 10 isolates carrying ZIP1.
In order to understand how overproduction of Dbf4 suppresses the cell cycle arrest of meiotic recombination mutants, we sought to isolate DBF4 point mutants that can phenocopy the overproduction effect. DBF4 was randomly mutagenized by PCR using a low fidelity Taq polymerase (DreamTaq DNA polymerase, Thermo Scientific). Amplified fragments were cloned into a single‐copy plasmid (YCplac33) to form a mutagenized DBF4 library. The zip1‐4LA diploid strain was screened with this DBF4 library as above. A single clone, encoding the dbf4‐E86V mutation, was isolated.
Protein analysis
A fragment of Cdc5 containing the PBD was N‐terminally tagged with GST (GST‐Cdc5‐PBD) and overexpressed in Escherichia coli strain Rosetta 2 (DE3). Following sonication, the lysate was clarified and GST‐Cdc5‐PBD was bound to glutathione resin and further purified by gel filtration. Fluorescein‐labeled peptides were purchased from Peptide Protein Research (Fareham, UK).
Proteins from meiotic cultures were separated by SDS–PAGE and transferred to PVDF membranes. Immunoprecipitations were performed essentially as described (Matos et al, 2008; see Appendix Supplementary Methods for more details). Immunodetection was performed with the following antibodies: Cdc5 (goat, 500‐fold dilution, Santa Cruz yC‐19), Dbf4 (goat, 200‐fold dilution, Santa Cruz yN‐15), Ndt80 (rabbit, 5,000‐fold dilution, Kirsten Benjamin; Benjamin et al, 2003), Zip1 (rabbit, 5,000‐fold dilution, Shirleen Roeder; Sym & Roeder, 1994), Red1 (rabbit, 5,000‐fold dilution, Shirleen Roeder; Smith & Roeder, 1997), Pgk1 (mouse, 5,000‐fold dilution, Invitrogen 459250), and V5 (mouse, 5,000‐fold dilution, Bio‐Rad MCA1360).
DNA analysis
Meiotic chromosomes were separated by pulsed‐field gel electrophoresis, and Southern blotting was performed with a probe recognizing chromosome II as described (Farmer et al, 2012). At each time point, the lane signal was background‐subtracted and the signal for the intact chromosome band and the smear corresponding to the broken chromosomes were combined to obtain the total lane signal. The amount of signal for the broken chromosomes was divided by the total lane signal to obtain a ratio for broken chromosomes, which was represented as a percentage.
Author contributions
BA, TT, and HT conceived and performed experiments, analyzed data, wrote the manuscript, and supervised the study. W‐KL, NA, YT, and YM provided reagents, performed experiments, and validated results. VVS, AH, and HI provided reagents.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We would like to thank Antony Oliver, Mohan Rajasekaran, Matthew Day, and Raquel Arribas for help with protein purification and the fluorescence polarization assay. We thank Shirleen Roeder, Kirsten Benjamin, Michael Lichten, Angelika Amon, and Bruce Stillman for strains/antibodies, and Katsuki Johzuka for sharing resources for Southern blotting. We would also like to extend our gratitude to Antony Carr for encouragement and support. This work was supported by grants from the Biotechnology and Biological Sciences Research Council (BB/I009159/1) and Japan Society for the Promotion of Science (JSPS; 16H07422) to H.T.; a Medical Research Council doctoral studentship to B.A.; a Grant‐in‐Aid for Scientific Research on Innovative Areas from JSPS (15H059749) to H.I.; a Grant‐in‐Aid for Young Scientists (A) from JSPS (16H06160) to Y.M.; and NIH grant GM111715 to A.H.
The EMBO Journal (2017) 36: 2488–2509
Contributor Information
Tomomi Tsubouchi, Email: ttsubo@nibb.ac.jp.
Hideo Tsubouchi, Email: htsubo@nibb.ac.jp.
References
- Acosta I, Ontoso D, San‐Segundo PA (2011) The budding yeast polo‐like kinase Cdc5 regulates the Ndt80 branch of the meiotic recombination checkpoint pathway. Mol Biol Cell 22: 3478–3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allers T, Lichten M (2001) Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106: 47–57 [DOI] [PubMed] [Google Scholar]
- Argunhan B, Farmer S, Leung WK, Terentyev Y, Humphryes N, Tsubouchi T, Toyoizumi H, Tsubouchi H (2013) Direct and indirect control of the initiation of meiotic recombination by DNA damage checkpoint mechanisms in budding yeast. PLoS ONE 8: e65875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis JM, Roeder GS (1998) Synaptonemal complex morphogenesis and sister‐chromatid cohesion require Mek1‐dependent phosphorylation of a meiotic chromosomal protein. Genes Dev 12: 3551–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FA, Silljé HH, Nigg EA (2004) Polo‐like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 5: 429–440 [DOI] [PubMed] [Google Scholar]
- Bartholomew CR, Woo SH, Chung YS, Jones C, Hardy CF (2001) Cdc5 interacts with the Wee1 kinase in budding yeast. Mol Cell Biol 21: 4949–4959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin KR, Zhang C, Shokat KM, Herskowitz I (2003) Control of landmark events in meiosis by the CDK Cdc28 and the meiosis‐specific kinase Ime2. Genes Dev 17: 1524–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DK, Park D, Xu L, Kleckner N (1992) DMC1: a meiosis‐specific yeast homolog of Escherichia coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69: 439–456 [DOI] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JS, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM (2000) A chemical switch for inhibitor‐sensitive alleles of any protein kinase. Nature 407: 395–401 [DOI] [PubMed] [Google Scholar]
- Busygina V, Sehorn MG, Shi IY, Tsubouchi H, Roeder GS, Sung P (2008) Hed1 regulates Rad51‐mediated recombination via a novel mechanism. Genes Dev 22: 786–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busygina V, Saro D, Williams G, Leung WK, Say AF, Sehorn MG, Sung P, Tsubouchi H (2012) Novel attributes of Hed1 affect dynamics and activity of the Rad51 presynaptic filament during meiotic recombination. J Biol Chem 287: 1566–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzymek M, Thayer NH, Oh SD, Kleckner N, Hunter N (2010) Double Holliday junctions are intermediates of DNA break repair. Nature 464: 937–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon CK, Hawley RS (2016) Regulating the construction and demolition of the synaptonemal complex. Nat Struct Mol Biol 23: 369–377 [DOI] [PubMed] [Google Scholar]
- Callender TL, Laureau R, Wan L, Chen X, Sandhu R, Laljee S, Zhou S, Suhandynata RT, Prugar E, Gaines WA, Kwon Y, Börner GV, Nicolas A, Neiman AM, Hollingsworth NM (2016) Mek1 down regulates Rad51 activity during yeast meiosis by phosphorylation of Hed1. PLoS Genet 12: e1006226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo JA, Johnson AL, Sedgwick SG, Cha RS (2008) Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell 132: 758–770 [DOI] [PubMed] [Google Scholar]
- Carballo JA, Panizza S, Serrentino ME, Johnson AL, Geymonat M, Borde V, Klein F, Cha RS (2013) Budding yeast ATM/ATR control meiotic double‐strand break (DSB) levels by down‐regulating Rec114, an essential component of the DSB‐machinery. PLoS Genet 9: e1003545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M, Botstein D (1982) Two differentially regulated mRNAs with different 5′ ends encode secreted with intracellular forms of yeast invertase. Cell 28: 145–154 [DOI] [PubMed] [Google Scholar]
- Chen YC, Weinreich M (2010) Dbf4 regulates the Cdc5 Polo‐like kinase through a distinct non‐canonical binding interaction. J Biol Chem 285: 41244–41254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Suhandynata RT, Sandhu R, Rockmill B, Mohibullah N, Niu H, Liang J, Lo HC, Miller DE, Zhou H, Börner GV, Hollingsworth NM (2015) Phosphorylation of the synaptonemal complex protein Zip1 regulates the crossover/noncrossover decision during yeast meiosis. PLoS Biol 13: e1002329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I (1998) The transcriptional program of sporulation in budding yeast. Science 282: 699–705 [DOI] [PubMed] [Google Scholar]
- Cloud V, Chan YL, Grubb J, Budke B, Bishop DK (2012) Rad51 is an accessory factor for Dmc1‐mediated joint molecule formation during meiosis. Science 337: 1222–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enserink JM, Kolodner RD (2010) An overview of Cdk1‐controlled targets and processes. Cell Div 5: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer S, Leung WK, Tsubouchi H (2011) Characterization of meiotic recombination initiation sites using pulsed‐field gel electrophoresis. Methods Mol Biol 745: 33–45 [DOI] [PubMed] [Google Scholar]
- Farmer S, Hong EJ, Leung WK, Argunhan B, Terentyev Y, Humphryes N, Toyoizumi H, Tsubouchi H (2012) Budding yeast Pch2, a widely conserved meiotic protein, is involved in the initiation of meiotic recombination. PLoS ONE 7: e39724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MF, Santocanale C, Drury LS, Diffley JF (2000) Dbf4p, an essential S phase‐promoting factor, is targeted for degradation by the anaphase‐promoting complex. Mol Cell Biol 20: 242–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima K, Tanaka Y, Nabeshima K, Yoneki T, Tougan T, Tanaka S, Nojima H (2000) Dmc1 of Schizosaccharomyces pombe plays a role in meiotic recombination. Nucleic Acids Res 28: 2709–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Game JC, Mortimer RK (1974) A genetic study of x‐ray sensitive mutants in yeast. Mutat Res 24: 281–292 [DOI] [PubMed] [Google Scholar]
- Gray S, Allison RM, Garcia V, Goldman AS, Neale MJ (2013) Positive regulation of meiotic DNA double‐strand break formation by activation of the DNA damage checkpoint kinase Mec1(ATR). Open Biol 3: 130019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk AL, Kohli J (2003) Five RecA‐like proteins of Schizosaccharomyces pombe are involved in meiotic recombination. Genetics 165: 1031–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy CF, Pautz A (1996) A novel role for Cdc5p in DNA replication. Mol Cell Biol 16: 6775–6782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy CF, Dryga O, Seematter S, Pahl PM, Sclafani RA (1997) mcm5/cdc46‐bob1 bypasses the requirement for the S phase activator Cdc7p. Proc Natl Acad Sci USA 94: 3151–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK (2008) The anchor‐away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31: 925–932 [DOI] [PubMed] [Google Scholar]
- Hayashi M, Chin GM, Villeneuve AM (2007) C. elegans germ cells switch between distinct modes of double‐strand break repair during meiotic prophase progression. PLoS Genet 3: e191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochwagen A, Amon A (2006) Checking your breaks: surveillance mechanisms of meiotic recombination. Curr Biol 16: R217–R228 [DOI] [PubMed] [Google Scholar]
- Humphryes N, Leung WK, Argunhan B, Terentyev Y, Dvorackova M, Tsubouchi H (2013) The Ecm11‐Gmc2 complex promotes synaptonemal complex formation through assembly of transverse filaments in budding yeast. PLoS Genet 9: e1003194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N, Kleckner N (2001) The single‐end invasion: an asymmetric intermediate at the double‐strand break to double‐Holliday junction transition of meiotic recombination. Cell 106: 59–70 [DOI] [PubMed] [Google Scholar]
- Jordan PW, Karppinen J, Handel MA (2012) Polo‐like kinase is required for synaptonemal complex disassembly and phosphorylation in mouse spermatocytes. J Cell Sci 125: 5061–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, Kleckner N (1997) Meiosis‐specific DNA double‐strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88: 375–384 [DOI] [PubMed] [Google Scholar]
- Khasanov FK, Savchenko GV, Bashkirova EV, Korolev VG, Heyer WD, Bashkirov VI (1999) A new recombinational DNA repair gene from Schizosaccharomyces pombe with homology to Escherichia coli RecA. Genetics 152: 1557–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita‐Kikuta E, Kinoshita E, Matsuda A, Koike T (2014) Tips on improving the efficiency of electrotransfer of target proteins from Phos‐tag SDS‐PAGE gel. Proteomics 14: 2437–2442 [DOI] [PubMed] [Google Scholar]
- Kitada K, Johnson AL, Johnston LH, Sugino A (1993) A multicopy suppressor gene of the Saccharomyces cerevisiae G1 cell cycle mutant gene dbf4 encodes a protein kinase and is identified as CDC5 . Mol Cell Biol 13: 4445–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupiec M, Steinlauf R (1997) Damage‐induced ectopic recombination in the yeast Saccharomyces cerevisiae . Mutat Res 384: 33–44 [DOI] [PubMed] [Google Scholar]
- Lai YJ, Lin FM, Chuang MJ, Shen HJ, Wang TF (2011) Genetic requirements and meiotic function of phosphorylation of the yeast axial element protein Red1. Mol Cell Biol 31: 912–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao JP, Cloud V, Huang CC, Grubb J, Thacker D, Lee CY, Dresser ME, Hunter N, Bishop DK (2013) Meiotic crossover control by concerted action of Rad51‐Dmc1 in homolog template bias and robust homeostatic regulation. PLoS Genet 9: e1003978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Amon A (2003) Role of Polo‐like kinase CDC5 in programming meiosis I chromosome segregation. Science 300: 482–486 [DOI] [PubMed] [Google Scholar]
- Lee JY, Terakawa T, Qi Z, Steinfeld JB, Redding S, Kwon Y, Gaines WA, Zhao W, Sung P, Greene EC (2015) Base triplet stepping by the Rad51/RecA family of recombinases. Science 349: 977–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung WK, Humphryes N, Afshar N, Argunhan B, Terentyev Y, Tsubouchi T, Tsubouchi H (2015) The synaptonemal complex is assembled by a polySUMOylation‐driven feedback mechanism in yeast. J Cell Biol 211: 785–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydall D, Nikolsky Y, Bishop DK, Weinert T (1996) A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature 383: 840–843 [DOI] [PubMed] [Google Scholar]
- Machovina T, Mainpal R, Daryabeigi A, McGovern O, Paouneskou D, Labella S, Zetka M, Jantsch V, Yanowitz J (2016) A surveillance system ensures crossover formation in C. elegans . Curr Biol 26: 2873–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos J, Lipp JJ, Bogdanova A, Guillot S, Okaz E, Junqueira M, Shevchenko A, Zachariae W (2008) Dbf4‐dependent Cdc7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell 135: 662–678 [DOI] [PubMed] [Google Scholar]
- Matos J, Blanco MG, Maslen S, Skehel JM, West SC (2011) Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147: 158–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos J, Blanco MG, West SC (2013) Cell‐cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Rep 4: 76–86 [DOI] [PubMed] [Google Scholar]
- Matthews LA, Guarné A (2013) Dbf4: the whole is greater than the sum of its parts. Cell Cycle 12: 1180–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CT, Gabrielse C, Chen YC, Weinreich M (2009) Cdc7p‐Dbf4p regulates mitotic exit by inhibiting Polo kinase. PLoS Genet 5: e1000498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra N, Roeder GS (2007) A novel nonnull ZIP1 allele triggers meiotic arrest with synapsed chromosomes in Saccharomyces cerevisiae . Genetics 176: 773–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen EM, Haas W, Gygi M, Gygi SP, Kellogg DR (2005) Cdc28‐dependent regulation of the Cdc5/Polo kinase. Curr Biol 15: 2033–2037 [DOI] [PubMed] [Google Scholar]
- Murakami H, Keeney S (2014) Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell 158: 861–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Tsutsui Y, Iwasaki H (2011) The fission yeast meiosis‐specific Dmc1 recombinase mediates formation and branch migration of Holliday junctions by preferentially promoting strand exchange in a direction opposite to that of Rad51. Genes Dev 25: 516–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadarajan S, Lambert TJ, Altendorfer E, Gao J, Blower MD, Waters JC, Colaiácovo MP (2017) Polo‐like kinase‐dependent phosphorylation of the synaptonemal complex protein SYP‐4 regulates double‐strand break formation through a negative feedback loop. Elife 6: e23437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Müller CA, Katou Y, Retkute R, Gierliński M, Araki H, Blow JJ, Shirahige K, Nieduszynski CA, Tanaka T (2013) Kinetochores coordinate pericentromeric cohesion and early DNA replication by Cdc7‐Dbf4 kinase recruitment. Mol Cell 50: 661–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogami S, Fukuda T, Nagai Y, Yabe S, Sugiura M, Mizutani R, Satow Y, Anraku Y, Ohya Y (2002) Homing at an extragenic locus mediated by VDE (PI‐SceI) in Saccharomyces cerevisiae . Yeast 19: 773–782 [DOI] [PubMed] [Google Scholar]
- Okaz E, Argüello‐Miranda O, Bogdanova A, Vinod PK, Lipp JJ, Markova Z, Zagoriy I, Novak B, Zachariae W (2012) Meiotic prophase requires proteolysis of M phase regulators mediated by the meiosis‐specific APC/CAma1. Cell 151: 603–618 [DOI] [PubMed] [Google Scholar]
- Petronczki M, Siomos MF, Nasmyth K (2003) Un ménage à quatre: the molecular biology of chromosome segregation in meiosis. Cell 112: 423–440 [DOI] [PubMed] [Google Scholar]
- Princz LN, Wild P, Bittmann J, Aguado FJ, Blanco MG, Matos J, Pfander B (2017) Dbf4‐dependent kinase and the Rtt107 scaffold promote Mus81‐Mms4 resolvase activation during mitosis. EMBO J 36: 664–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Redding S, Lee JY, Gibb B, Kwon Y, Niu H, Gaines WA, Sung P, Greene EC (2015) DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160: 856–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockmill B, Lefrançois P, Voelkel‐Meiman K, Oke A, Roeder GS, Fung JC (2013) High throughput sequencing reveals alterations in the recombination signatures with diminishing Spo11 activity. PLoS Genet 9: e1003932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder GS (1997) Meiotic chromosomes: it takes two to tango. Genes Dev 11: 2600–2621 [DOI] [PubMed] [Google Scholar]
- Sasanuma H, Hirota K, Fukuda T, Kakusho N, Kugou K, Kawasaki Y, Shibata T, Masai H, Ohta K (2008) Cdc7‐dependent phosphorylation of Mer2 facilitates initiation of yeast meiotic recombination. Genes Dev 22: 398–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwacha A, Kleckner N (1997) Interhomolog bias during meiotic recombination: meiotic functions promote a highly differentiated interhomolog‐only pathway. Cell 90: 1123–1135 [DOI] [PubMed] [Google Scholar]
- Shinohara A, Ogawa H, Ogawa T (1992) Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA‐like protein. Cell 69: 457–470 [DOI] [PubMed] [Google Scholar]
- Smith AV, Roeder GS (1997) The yeast Red1 protein localizes to the cores of meiotic chromosomes. J Cell Biol 136: 957–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourirajan A, Lichten M (2008) Polo‐like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev 22: 2627–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian VV, MacQueen AJ, Vader G, Shinohara M, Sanchez A, Borde V, Shinohara A, Hochwagen A (2016) Chromosome synapsis alleviates Mek1‐dependent suppression of meiotic DNA repair. PLoS Biol 14: e1002369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sym M, Roeder GS (1994) Crossover interference is abolished in the absence of a synaptonemal complex protein. Cell 79: 283–292 [DOI] [PubMed] [Google Scholar]
- Szakal B, Branzei D (2013) Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J 32: 1155–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker D, Mohibullah N, Zhu X, Keeney S (2014) Homologue engagement controls meiotic DNA break number and distribution. Nature 510: 241–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi H, Roeder GS (2002) The Mnd1 protein forms a complex with Hop2 to promote homologous chromosome pairing and meiotic double‐strand break repair. Mol Cell Biol 22: 3078–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi H, Roeder GS (2003) The importance of genetic recombination for fidelity of chromosome pairing in meiosis. Dev Cell 5: 915–925 [DOI] [PubMed] [Google Scholar]
- Tsubouchi H, Roeder GS (2006) Budding yeast Hed1 down‐regulates the mitotic recombination machinery when meiotic recombination is impaired. Genes Dev 20: 1766–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi H, Argunhan B, Tsubouchi T (2016) Shaping meiotic chromosomes with SUMO: a feedback loop controls the assembly of the synaptonemal complex in budding yeast. Microbial Cell 3: 126–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya D, Yang Y, Lacefield S (2014) Positive feedback of NDT80 expression ensures irreversible meiotic commitment in budding yeast. PLoS Genet 10: e1004398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui Y, Morishita T, Iwasaki H, Toh H, Shinagawa H (2000) A recombination repair gene of Schizosaccharomyces pombe, rhp57, is a functional homolog of the Saccharomyces cerevisiae RAD57 gene and is phylogenetically related to the human XRCC3 gene. Genetics 154: 1451–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung KS, Hong EJ, Roeder GS (2000) The pachytene checkpoint prevents accumulation and phosphorylation of the meiosis‐specific transcription factor Ndt80. Proc Natl Acad Sci USA 97: 12187–12192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin G, Schwob E, Della Seta F (2006) Dual role of the Cdc7‐regulatory protein Dbf4 during yeast meiosis. J Biol Chem 281: 2828–2834 [DOI] [PubMed] [Google Scholar]
- Voelkel‐Meiman K, Moustafa SS, Lefrancois P, Villeneuve AM, MacQueen AJ (2012) Full‐length synaptonemal complex grows continuously during meiotic prophase in budding yeast. PLoS Genet 8: e1002993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel‐Meiman K, Taylor LF, Mukherjee P, Humphryes N, Tsubouchi H, Macqueen AJ (2013) SUMO localizes to the central element of synaptonemal complex and Is required for the full synapsis of meiotic chromosomes in budding yeast. PLoS Genet 9: e1003837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, de los Santos T, Zhang C, Shokat K, Hollingsworth NM (2004) Mek1 kinase activity functions downstream of RED1 in the regulation of meiotic double strand break repair in budding yeast. Mol Biol Cell 15: 11–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Niu H, Futcher B, Zhang C, Shokat KM, Boulton SJ, Hollingsworth NM (2008) Cdc28‐Clb5 (CDK‐S) and Cdc7‐Dbf4 (DDK) collaborate to initiate meiotic recombination in yeast. Genes Dev 22: 386–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chang C‐Y, Wu J‐F, Tung K‐S (2011) Nuclear localization of the meiosis‐specific transcription factor Ndt80 is regulated by the pachytene checkpoint. Mol Biol Cell 22: 1878–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich M, Stillman B (1999) Cdc7p‐Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J 18: 5334–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Ajimura M, Padmore R, Klein C, Kleckner N (1995) NDT80, a meiosis‐specific gene required for exit from pachytene in Saccharomyces cerevisiae . Mol Cell Biol 15: 6572–6581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Mori S, Oshiumi H, Matsuzaki K, Shinohara M, Shinohara A (2010) Cyclin‐dependent kinase promotes formation of the synaptonemal complex in yeast meiosis. Genes Cells 15: 1036–1050 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7