

Abstract
Following gene duplication, mRNA expression of the duplicated gene is reduced to maintain mRNA dosage. In mammals, this process is achieved with increased cytosine DNA methylation of the promoters of duplicated genes to suppress transcriptional initiation. However, not all animal species possess a full apparatus for cytosine DNA methylation. For such species, such as the roundworm (Caenorhabditis elegans, “worm” hereafter) or fruit fly (Drosophila melanogaster, “fly” hereafter), it is unclear how reduced expression of duplicated genes has been achieved evolutionarily. Here, we hypothesize that in the absence of a classical cytosine DNA methylation pathway, histone modifications play an increasing role in maintaining mRNA dosage following gene duplication. We initially verified that reduced gene expression of duplicated genes had occurred in the worm, fly, and mouse (Mus musculus). Next, several histone marks, with the capacity to control mRNA abundance in the models studied, were examined. In the worm and fly, but not in the mouse, multiple histone modifications were found to assist mRNA dosage maintenance following gene duplication events and the possible involvement of adenine DNA methylation in this process was excluded. Furthermore, the histone marks and acting regions that mediated the reduction in duplicated gene expression were found to be largely organism specific. Thus, it appears that many of the histone marks that maintain mRNA dosage were independently recruited during the evolution of worms and flies to compensate for the loss of cytosine DNA methylation machinery from their genomes.
Gene duplication frequently occurs during evolution (Lynch and Conery 2000), and it is an important mechanism that underlies the origin of organismal complexity (Freeling and Thomas 2006; Wagner et al. 2007). However, the resulting increase in total gene product from genes involved in duplication events can have immediate adverse effects on the fitness of an organism due to increased rates of promiscuous molecular interactions in cells (Vavouri et al. 2009; Yang et al. 2012) or stoichiometric imbalance (Papp et al. 2003; Birchler and Veitia 2007). To compensate for these deleterious effects, it has been found that the transcriptional activity per duplicated gene after a duplication event is reduced compared with the progenitor gene, and this reduction is more pronounced for genes that lead to reduced fitness when they are overexpressed, such as genes encoding subunits of protein complexes (Qian et al. 2010; Chang and Liao 2012). A reduction in expression levels could serve to prevent the loss and functional divergence of duplicated genes (Qian et al. 2010), thereby resulting in the long-term preservation of paralogous genes with genetic redundancy (Tischler et al. 2006; Vavouri et al. 2008). Given the central role that mRNA dosage maintenance has in determining the fate of duplicated genes and in shaping a genome, it is important to elucidate the molecular basis underlying this process.
A reduction in post-duplication mRNA expression has been discovered in both yeast and mammalian genes (Qian et al. 2010). This phenomenon implies that there is a common need for a variety of life forms to maintain gene product dosage following gene duplication events. Moreover, it is anticipated that the molecular mechanisms underlying this process differ among various lineages. DNA methylation is an epigenetic mechanism that involves the covalent addition of methyl groups to DNA. In particular, cytosine methylation is a predominant type of DNA methylation that occurs in plants and mammals to form the DNA base, 5-methylcytosine. This DNA base has been shown to mediate interactions between transcription factors and DNA, thereby regulating the transcriptional abundance of genes (Jones and Takai 2001; Suzuki and Bird 2008) and contributing to divergence in mRNA expression between duplicated genes (Keller and Yi 2014; Wang et al. 2014). In mammals, reduced mRNA expression of duplicated genes is achieved with increased cytosine methylation of the promoter region of the duplicated genes, presumably to inhibit transcriptional initiation (Chang and Liao 2012). However, in many animal lineages, the molecular mechanism of cytosine DNA methylation has degenerated. For example, the genes encoding key enzymes for cytosine methylation, including DNA (cytosine-5-)-methyltransferases 1 (DNMT1) and 3 (DNMT3) that maintain and establish the cytosine methylation landscape in various organisms, respectively, have been lost from the genomes of the fly (Drosophila melanogaster) and worm (Caenorhabditis elegans) (Gutierrez and Sommer 2004; Jurkowski and Jeltsch 2011). Correspondingly, in worms (Simpson et al. 1986), levels of 5-methylcytosine have been consistently undetectable. In the fruit fly genome (Lyko et al. 2000), although a very low level of cytosine DNA methylation is restricted to an early embryonic stage, it is unrelated to any known DNA methyltransferase, and its function is not obvious (Takayama et al. 2014). Therefore, for worms and flies, a mechanism other than cytosine methylation is potentially utilized to maintain mRNA dosage following gene duplication events.
Here, we hypothesized that epigenetic mechanisms are important for the process of post-duplicational mRNA expression reduction, and for organisms whose cytosine DNA methylation apparatus and pattern have degenerated over the course of their evolution, this process is achieved through increased engagement of histone modifications to provide additional levels of epigenetic control to inhibit the expression of duplicated genes, especially genes that are sensitive to changes in dosage. To test our hypothesis, we examined gene duplication events, as well as common and lineage-specific acting regions of several histone marks, that are capable of controlling the mRNA abundance of coding genes in worm, fly, or mouse genomes.
Results
Maintenance of mRNA dosage after gene duplication events in mammals, flies, and worms
The expression reduction model proposed that the deleterious effect of duplication-induced protein overexpression may be compensated evolutionarily by a reduction in gene expression, whereby the expression level of a duplicated gene is reduced until the sum of the total transcriptional output from both copies of duplicated genes approximates the original transcriptional level of the progenitor gene (Qian et al. 2010). In order to verify the general applicability of this model to animal evolution, we defined ortholog sets of two closely related species of mammals, flies, and worms (mammals: Homo sapiens–M. musculus; flies: D. melanogaster–Drosophila simulans; worms: C. elegans–Caenorhabditis briggsae) based on orthology information available at the Ensembl database (see below and Methods). These species pairs were chosen based on the availability of RNA sequencing (RNA-seq) data for each (see Methods), since RNA-seq data have been shown to be ideal for assessing the mRNA abundance of a gene in the presence of orthologs (Qian et al. 2010; Brawand et al. 2011; Liao and Chang 2014). An ortholog set is composed of a paralog group of one species and a paralog group of another species (with each paralog group containing a set of paralogs that arose from gene duplication events that occurred after the divergence of the two species examined). For each ortholog set, the difference in the mean mRNA expression level in a homologous tissue (H. sapiens–M. musculus: adult cerebellum), or in a whole organism at the same developmental stage (D. melanogaster–D. simulans: L3 larva; C. elegans–C. briggsae: L3 larva), was calculated as follows:
where NA is the number of genes in the paralog group of species A, NB is the number of genes in the paralog group of species B (and if NA or NB = 1, no duplication has occurred in the lineage), and ZA(i) is the normalized mRNA expression level of gene i in species A (or ZB(j) for gene j in species B; see Methods). The A species for the mammal, fly, and worm models examined were H. sapiens, D. melanogaster, and C. elegans, respectively; the corresponding B species were M. musculus, D. simulans, and C. briggsae, respectively. The ratio of paralog group size for species A to species B was calculated for each orthologous set as SA/B = NA/NB. Ortholog sets with gene duplication events that occurred after the divergence of the two species (i.e., NA ≠ 1 and/or NB ≠ 1) were examined. According to the expression reduction model, the extent of reduced expression in the paralogs should be proportionate to the number of duplication events in each ortholog set. Correspondingly, ΔEA-B and log(SA/B) were negatively correlated in the three pairs of model organisms that were examined (mammals: Spearman's correlation coefficient ρ = −0.332, P < 10−25; flies: ρ = −0.225, P < 10−5; worms: ρ = −0.360, P < 10−49).
To understand if the observed reduction in expression occurred exclusively in derived copies of the duplicated genes, ΔE′A-B was defined as the difference in mRNA expression abundance of the ortholog pair with the smallest nonsynonymous substitution rate (dN; see Methods) and the smallest promoter sequence divergence (dP; see Methods). This pair from each ortholog set was more likely to represent a pair of progenitor genes of the species compared because derived copies of duplicated genes often undergo rapid sequence evolution (or degeneration) and thus have a large dN value with the ortholog (Owens et al. 2013). The derived copies of duplicated genes also tend not to preserve the promoter sequences (e.g., retrogenes) and thus have a large dP with the ortholog. It was observed that ΔE′A-B and log(SA/B) remained negatively correlated in the three pairs of model organisms that were examined (mammals: ρ = −0.357, P < 10−6; flies: ρ = −0.302, P < 10−3; worms: ρ = −0.323, P < 10−4; ortholog sets whose ΔE′A-B could not be defined were excluded). These results suggest that the negative correlations between ΔEA-B and log(SA/B) are not fully explained by the fact that most duplicates were born with lower transcription level than their parent simply by chance, and that expansion of a gene family due to a duplication event induces reduced expression of the progenitor gene.
A more significant decrease in expression occurs for dosage-sensitive genes than for dosage-insensitive genes following gene duplications events
The expression reduction model (Qian et al. 2010; Chang and Liao 2012) also predicts that dosage-sensitive genes will exhibit a more significant decrease in gene expression after a gene duplication event than will dosage-insensitive genes. The dosage sensitivity of a gene can be accurately predicted by the number of binary interacting partners that the encoding protein exhibits in sensitive protein–protein interaction assays exclusively: Genes encoding proteins with a greater number of weakly interacting partners are more likely to be dosage sensitive (Vavouri et al. 2009). Accordingly, based on protein–protein interactions detected exclusively by sensitive yeast-two hybrid experiments (following the method of Vavouri et al. 2009), we defined genes exhibiting weak interactions with at least three other proteins encoded in the genome as “dosage-sensitive” genes and the genes encoding proteins exhibiting weak interactions with one to two other proteins as “dosage-insensitive” genes (see Methods). We could not define genes without an annotated interacting partner because it could not be determined whether the absence of interactions was because the potentiality of the encoding protein to form interaction with other proteins has not yet been examined or because there was genuinely no interaction mediated by the encoding protein. To examine if the mRNA levels of the dosage-sensitive genes are more strictly maintained evolutionarily compared with the mRNA levels of the dosage-insensitive genes after gene duplication events, two groups of ortholog sets were defined: (1) those containing at least one dosage-sensitive gene (referred to as “dosage-sensitive” ortholog sets) and (2) those containing only dosage-insensitive genes (referred to as “dosage-insensitive” ortholog sets). Consistent with our predictive model, a stronger negative correlation between ΔEA-B and SA/B was observed for the dosage-sensitive ortholog sets compared with the dosage-insensitive ortholog sets in all model organisms investigated (Table 1; Supplemental Fig. S1A).
Table 1.
Comparison of Spearman's correlation coefficient (ρ) between ΔEA-B and SA/B for the dosage-sensitive and dosage-insensitive ortholog sets
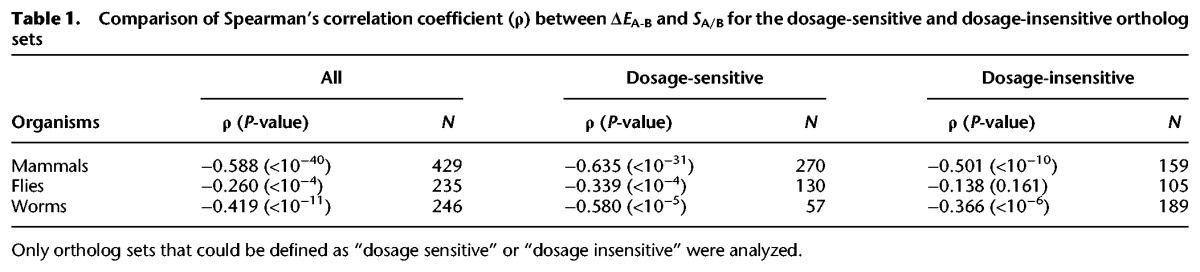
To understand if the observed ρ between the ΔEA-B and SA/B values for the dosage-sensitive ortholog sets is statistically stronger than that for the dosage-insensitive ortholog sets, a bootstrap test was performed. In this test, the dosage-sensitive and dosage-insensitive ortholog sets were each randomly resampled 10,000 times (the same number of dosage-sensitive ortholog sets and dosage-insensitive ortholog sets were sampled as indicated in Supplemental Fig. S1B), and ρ of ΔEA-B versus log(SA/B) was calculated for each resampled data set. The resulting two ρ distributions indicated that the correlations between ΔEA-B and SA/B for the dosage-sensitive ortholog sets were statistically more negative than those for the dosage-insensitive ortholog sets for the mammal, fly, and worm models (P < 10−300, Mann-Whitney U test) (Supplemental Fig. S1B). Genes that encode proteins that are subunits of protein complexes are also sensitive to dosage changes due to the potential for stoichiometric imbalances to occur when gene duplication events occur (Papp et al. 2003; Qian et al. 2010). Previous studies have utilized various approaches to identify genes that encode protein complex subunits. We obtained previously annotated lists of such genes for mammals and the fruit fly and predicted genes encoding complex subunits for the worm (see Methods). All of the ortholog sets were classified into two groups: (1) those containing at least one gene encoding a protein complex subunit (referred to as “complex” ortholog sets) and (2) those that did not contain any genes encoding known members of a protein complex (referred to as “noncomplex” ortholog sets). Based on the analysis that was performed for the dosage-sensitive and dosage-insensitive ortholog sets (Table 1; Supplemental Fig. S1), a similar analysis of the complex and noncomplex ortholog sets showed statistically more negative correlations between ΔEA-B and SA/B in the complex ortholog sets than in the noncomplex ortholog sets for all of the species examined (Supplemental Table S1; Supplemental Fig. S2).
The above-mentioned observations agree with the expression reduction model and also suggest that although expression reduction of duplicated genes could be a neural or adaptive process, the mRNA dosages of a proportion of the genes duplicated after the divergence of D. melanogaster–D. simulans and C. elegans–C. briggsae have been adaptively maintained as previously described for the mammals.
Maintenance of mRNA dosage after duplication events that occurred in flies and worms does not involve DNA methylation
To date, cytosine DNA methylation has not been detected in C. elegans, and it is extremely rare in D. melanogaster (Simpson et al. 1986; Jeltsch 2010). Regarding the latter, the level of genomic cytosine DNA methylation is very low, and it has only been detected during an early stage of embryonic development (e.g., in 0- to 4-h embryos) (Lyko et al. 2000). However, in the present study, reduced expression of duplicate genes was observed in the L3 larva stage of D. melanogaster (Table 1; Supplemental Fig. S1). Hence, the observed phenomena of reduced mRNA expression of duplicated genes in the worm and fly models examined (Table 1) are unrelated to the cytosine DNA methylation process.
Methylated adenines (N6-methyladenines) have been considered a feature of prokaryotic DNA, although they have also been discovered in the DNA of protists and plants (Ratel et al. 2006). Recently, N6-methyladenines have been detected in the genomes of C. elegans and D. melanogaster (Greer et al. 2015; Zhang et al. 2015). Despite these intriguing findings, however, the functional role of adenine DNA methylation in these two invertebrates remains to be confirmed. Adenine DNA methylation has been shown to mediate transcriptional regulation in Escherichia coli (Oshima et al. 2002) and plants (Rogers and Rogers 1995). Based on these findings, it is possible that methylation of adenine DNA could mediate the down-regulated expression of duplicated genes. To examine this possibility, we compared the levels of adenine methylation between duplicated genes with at least one recent paralog and unduplicated genes in the genomes of C. elegans and D. melanogaster. We found lower adenine methylation signals in the promoter regions of the duplicated genes (categorized as 1000, 500, or 250 nucleotides [nt] upstream of the transcriptional start site [TSS]) in the fly (Fig. 1A) and in the genic regions of both the fly and worm (Fig. 1B). However, when highly expressed genes were compared with weakly expressed genes within the paralog groups (RNA-seq data of fly ovary or worm of mixed stages were used to define mRNA expression level because adenine DNA methylation data of fly or worm, respectively, were obtained under such a condition; see Methods), the extent of adenine methylation did not differ in the fly promoter regions and genic regions (P = 0.14, Mann-Whitney U test) (Fig. 1B). Moreover, in the genic regions (Fig. 1A) of the worm genes, lower adenine methylation signals were found for the strongly expressed genes, thereby suggesting that the reduced levels of adenine methylation that characterize duplicated genes are unrelated to the observed transcriptional repression. Furthermore, if adenine methylation–mediated mRNA maintenance occurs, then dosage-sensitive genes would be targeted more often by this mechanism compared with dosage-insensitive genes after gene duplication events. However, the adenine methylation levels of the promoters and genic regions of the unduplicated genes versus the duplicated genes did not markedly differ between the dosage-sensitive and dosage-insensitive ortholog sets (Fig. 1). These results suggested that adenine DNA methylation does not contribute to the reduced mRNA expression observed for duplicated genes in C. elegans and D. melanogaster.
Figure 1.

Violin plots of the adenine DNA methylation levels (m6A) detected in the following: (A) the promoter regions (1000, 500, and 250 nt upstream of the TSS) and (B) gene bodies (from the TSS to the TTS) of D. melanogaster (fly; top) and C. elegans (worm; bottom). P-values were determined with the Mann-Whitney U test and are associated with arched gray lines at the top of each panel that indicate the values that were compared. (Und) unduplicated; (Dup) duplicated; (Strong) strongly expressed; (Weak) weakly expressed; (DS) ortholog sets containing dosage-sensitive genes; (DI) ortholog sets containing only dosage-insensitive genes.
Defining common and lineage-specific acting regions of histone marks in mice, worms, and flies
To test our hypothesis that the maintenance of mRNA dosages of duplicated genes is accomplished in worms and flies by enhanced inhibition, or decreased activation, of duplicated gene expression according to the histone modifications present, we examined genome-wide patterns of histone modifications in our animal models. Histone modification data can be obtained with chromatin immunoprecipitation combined with DNA microarray analysis (ChIP-chip) (e.g., Bernstein et al. 2005) or with chromatin immunoprecipitation followed by sequencing (ChIP-seq) (e.g., Barski et al. 2007). ChIP-seq data are characterized to have higher resolution, greater coverage, and reduced noise (Park 2009). Therefore, we defined the roles of histone modifications in regulating mRNA expression based on the summarized result of Ho et al. (2014), who exploited ChIP-seq approaches to systematically and simultaneously investigate 11 histone marks of flies, worms, and humans (Homo sapiens). These 11 histone modifications include mono-, di-, and trimethylation of histone H3 on lysine 4 (H3K4me1, H3K4me2, and H3K4me3, respectively); monomethylation of histone H3 on lysine 9 (H3K9me1); monomethylation of histone H4 on lysine 20 (H4K20me1); dimethylation of histone H3 on lysine 79 (H3K79me2); trimethylation of histone H3 on lysine 9, lysine 27, and lysine 36 (H3K9me3, H3K27me3, and H3K36me3, respectively); and acetylation of histone H3 on lysine 9 and lysine 27 (H3K9ac andH3K27ac, respectively). To evaluate the histone marks detected for each gene, the chromosomal region of each gene was divided into five segments: (1) “TSS−1000 to TSS,” the region encompassing 1000 nt upstream of the TSS; (2) “TSS to TSS+500,” the region from the TSS to 500 nt downstream; (3) “TSS+500 to TTS−500,” the region encompassing 500 nt downstream from the TSS to 500 nt upstream of the transcriptional termination site (TTS); (4) “TTS−500 to TTS,” the region including 500 nt upstream of the TTS; and (5) “TTS to TTS+1000,” the region including 1000 nt downstream from the TTS. According to Ho et al. (2014), a histone mark was considered to play an active (or repressive) role in regulating transcription when it exhibited an increased (or decreased) scaled ChIP-fold enrichment in the focal regions of expressed genes. Conversely, decreased (or increased) scaled ChIP-fold enrichment was observed in the focal regions of silent genes. The former and the latter marks were termed “active histone marks” and “repressive histone marks,” respectively (Supplemental Fig. S3).
To understand the relationships between mRNA expression evolution and histone modifications in animals, we analyzed data from the whole body or tissues/organs rather than immortalized cell lines in order to avoid the inclusion of any phenomena that result from the process of establishing a cell line. For flies and worms, both histone and RNA-seq data obtained from L3 larva were analyzed. For mammals, instead of using human data from cell lines, histone and gene expression data from adult mouse tissues were used (Supplemental Tables S2, S3) by assuming that the human and mouse genomes use the same histone codes for regulating mRNA abundance. Of the 11 histone marks profiled by Ho et al. (2014), six were found to have equivalent data in the adult mouse tissues (i.e., H3K4me1, H3K4me3, H3K27ac, H3K9ac, H3K79me2, and H3K27me3) (Supplemental Table S2). Therefore, we used these six marks to define common and lineage-specific histone marks in mammals, worms, and flies (see Supplemental Fig. S3 and below). Despite equivalent data not being available for H3K9me3 in mouse tissues (Supplemental Table S2), the role of H3K9me3 in regulating mRNA abundance had been characterized and found to be specific for flies and worms (see below and Supplemental Fig. S3). Thus, H3K9me3 was included in our analysis, and a total of seven histone marks (Supplemental Table S2; Supplemental Fig. S3) were examined for their potential roles in reducing mRNA expression of duplicated genes in mammals, worms, and flies.
The regulatory roles of the histone marks simultaneously investigated in flies, worms, and mammals indicated that the region containing an active or repressive mark in one species could differ from that in another species (Supplemental Fig. S3). When a particular region containing histone modifications was found to consistently correlate with an effect on gene expression across three species, this region was referred to as a common acting region. In contrast, when a region contained a particular histone modification that had a unique effect on gene expression in only one or two species, this region was referred to as a lineage-specific acting region. Interestingly, we found that the histone marks that are utilized in the mouse only acted on common regions that were present in the other two invertebrates examined. Meanwhile, lineage-specific acting regions were exclusively found in flies and roundworms for all of the histone marks that were studied (Supplemental Fig. S3). We termed the lineage-specific acting regions that were shared between worms and flies as “invertebrate-specific” acting regions, while those used by worms alone were termed as “worm-specific” acting regions. Since the mouse has a typical cytosine DNA methylation apparatus and landscape, while flies and worms do not, this observation implies that histone marks have a greater role in regulating mRNA expression when cytosine methylation is degenerated within a genome.
Testing the hypothesis that mRNA dosage maintenance is mediated by histone modifications
The transcriptional activity of a gene is regulated and evolutionarily shaped by many factors, including the production of sufficient mRNA molecules to perform gene functions (Liao and Weng 2015), an avoidance of undesired gene products and interactions in a cell (Liao and Zhang 2008; Yang et al. 2012), and control of protein production noise/speed (Fraser et al. 2004). However, dosage maintenance is only associated with a few of the above-mentioned factors, and different regulatory mechanisms may underlie different biological processes. Therefore, a histone mark that is defined as active or repressive (as shown in Supplemental Fig. S3) may not necessarily play a role in post-duplication dosage maintenance. If a histone modification mark is utilized to reduce the transcription of duplicate genes and if this reduction partially results from an adaptive process, the mark should exhibit an intensity distribution that is consistent with “all” three predicted patterns as follows:
Predicted Pattern I—An active (or repressive) mark has a lower (or higher) signal intensity for genes that have undergone recent duplication events compared with genes that have not undergone duplication (according to the Mann-Whitney U test). Here, recently duplicated genes were defined as those that underwent duplication after the divergence of the two species that were used to define the orthologs sets mentioned above. The focal species examined for differences between duplicated genes and unduplicated genes in mammals, worms, and flies were M. musculus, C. elegans, and D. melanogaster, respectively.
Predicted Pattern II—Based on recently duplicated genes in the same paralog group, active (or repressive) marks should be more (or less) prevalent for strongly expressed versus weakly expressed paralogs (see Methods). Consequently, the weakly expressed genes from all of the paralog groups should have a collectively lower active mark intensity (or higher repressive mark intensity) compared with the highly expressed genes (according to the Mann-Whitney U test).
Predicted Pattern III—First, the difference in histone modifications between duplicated genes and unduplicated genes (DHMd-u) is calculated as (hd − hu)/(hd + hu), where hd and hu are the average intensities of the histone marks examined for duplicated and unduplicated genes, respectively. Then, for an active histone mark, the DHMd-u for the dosage-sensitive genes should be negative and have a lesser value than the DHMd-u for the dosage-insensitive genes. Conversely, for a repressive histone mark, the DHMd-u for the dosage-sensitive genes should be positive and have a greater value than the DHMd-u for the dosage-insensitive genes. To examine if the two compared groups are statistically significant, we randomly resampled the dosage-sensitive and dosage-insensitive ortholog sets 10,000 times without changing the sample size of each. Distributions of DHMd-u for the two compared groups were subsequently analyzed with the Mann-Whitney U test.
One of the major aims of this study was to understand the commonality and diversity of mechanisms that are exploited by different animal lineages to maintain mRNA dosages. To avoid uncertainties that are associated with the various differential analytical approaches that can be used to interpret the results obtained, we analyzed the differences in the changes of histone modifications between the dosage-sensitive genes and the dosage-insensitive genes according to Predicted Pattern III. We did not analyze this differences between the genes encoding protein complex subunits versus the genes encoding proteins that do not participate in protein complexes because the approaches used to generate the data used to define these two groups of genes varied among the three animals examined (see Methods). In contrast, the approaches used to generate the data used to define the dosage-sensitive and dosage-insensitive genes were the same for all three animal models investigated (see Methods).
Only H3K79me2 is associated with mRNA dosage maintenance in the mouse
In the mouse, we analyzed six mammalian histone marks: H3K4me1, H3K4me3, H3K27ac, H3K9ac, H3K79me2, and H3K27me3. As mentioned above, all of the acting regions of these six histone marks in mice are targeted by the same marks in worms and flies, thus indicating they represent common acting regions. Specifically, “TSS−1000 to TSS” is associated with H3K4me1; “TSS to TSS+500” is associated with H3K4me3, H3K27ac, H3K9ac, and H3K79me2; and “TSS−1000 to TSS+500” is associated with H3K27me3. The observation that these acting regions have been conserved through evolution between vertebrates and invertebrates further suggests that our approach of investigating mouse tissues based on histone codes obtained from human cell lines should be acceptable. As shown in Figure 2A, all six marks that were examined exhibited distributions that were consistent with Predicted Patterns I and II, except for the repressive mark, H3K27me3. Only the distribution of H3K79me2 was consistent with Predicted Pattern III (Fig. 2A). It is known that H3K79me2 enhances transcriptional elongation by weakening nucleosome–DNA interactions and facilitating nucleosome removal and restoration in the wake of elongating RNA polymerase II (Pol II) (Mueller et al. 2007; Jonkers and Lis 2015). As a result, the movement of Pol II along DNA is more rapid, and this is consistent with the association of H3K79me2 with dosage-sensitive genes to control the transcriptional elongation of duplicated genes in mammals. Hence, it appears that cytosine DNA methylation and the histone modification, H3K79me2, cooperate to maintain mRNA dosage by inhibiting the processes of transcriptional initiation (Chang and Liao 2012) and elongation (H3K79me2 in Fig. 2A), respectively, when mammalian genes undergo duplication.
Figure 2.

Intensities of the various active or repressive histone marks indicated on common acting regions of mouse (A), fly (B), and worm (C) genes. Both active and repressive histone marks were compared according to Predicted Patterns I–III. At the top of each panel, the solid horizontal line is separated into five segments by four lines to represent the regions of “TSS−1000 to TSS,” “TSS to TSS+500,” “TSS+500 to TTS−500,” “TTS−500 to TTS,” and “TTS to TTS+1000,” from left to right, respectively. The gray shaded area represents the acting regions examined, and the solid versus hatched vertical lines represent boundaries of a gene and boundaries within a gene, respectively. The values of the histone modification intensities are presented as violin plots. If the three boxes at the top of each compared distribution are checked, they indicate that the observed patterns of histone marks are consistent with Predicted Patterns I, II, and III, respectively. P-values were determined with the Mann-Whitney U test and are associated with the horizontal square brackets at the top of each panel that indicate the values that were compared. (Und) unduplicated; (Dup) duplicated; (Strong) strongly expressed; (Weak) weakly expressed; (DS) ortholog sets containing dosage-sensitive genes; (DI) ortholog sets containing only dosage-insensitive genes; (DHMd-u) difference in histone modification (duplicated vs. unduplicated genes), distribution based on 10,000 resampled experiments.
H3K79me2, H3K4me1, H3K4me3, and H3K9ac maintain mRNA dosage after gene duplication events in fruit flies
In addition to the common acting regions that contain H3K4me1, H3K4me3, H3K27ac, H3K9ac, H3K79me2, and H3K27me3 (Fig. 2B), fruit flies also employ H3K4me1, H3K79me2, H3K27me3, and H3K9me3 in genic or downstream regions to regulate mRNA abundance (Fig. 3A). Worms, yet not mice, also employ the latter group of histone marks to regulate mRNA abundance (see H3K9me3 in Fig. 3A). The invertebrate-specific regions for the latter marks include “TSS+500 to TTS−500,” “TSS+500 to TTS−500,” “TSS+500 to TTS,” and “TTS to TTS+1000,” respectively, with H3K4me1 and H3K79me2 having active roles and with H3K27me3 and H3K9me3 having repressive roles in controlling mRNA abundance. Furthermore, none of the seven histone marks examined was found to regulate fly genes specifically (Fig. 3; Supplemental Fig. S3).
Figure 3.

Intensities of the various active or repressive histone marks that are indicated in the invertebrate-specific (A) and worm-specific (B) acting regions of fly (A) and worm (A,B) genes. Both active and repressive histone marks were compared according to Predicted Patterns I–III. For a detailed description of the figure, see the legend of Figure 2.
According to the histone mark distribution described by Predicted Patterns I–III, histone modifications with a potential role in maintaining the mRNA dosage of duplicated genes include the active marks, H3K79me2, H3K4me3, and H3K9ac in the common acting region “TSS to TSS+500” and include H3K4me1 in the common acting region “TSS−1000 to TSS” (Fig. 2B). While H3K79me2 also acts on the “TSS+500 to TTS−500” region in Drosophila, Predicted Pattern II was not observed in this region (Fig. 3A). Therefore, in the fly, dosage maintenance of duplicated genes may be partially achieved by suppressing the rate of early elongation via a decrease in H3K79me2 in the 5′ genic region, similar to that observed in the mouse (Fig. 2A). While both H3K4me1 and H3K4me3 represent methylations that occur at H3K4, these two methylations may not be coupled in the fly because they are catalyzed by different enzymes and they influence transcriptional processes via distinct mechanisms (Ardehali et al. 2011; Kusch 2012). In invertebrates, H3K4me1 marks promoters of actively transcribed genes (Liu et al. 2011), while H3K4me3 and H3K9ac control transcription after the initiation stage (Yin et al. 2011). Thus, our observations are consistent with the mechanisms previously characterized for these four marks. Interestingly, however, none of the histone marks that were present in the invertebrate-specific acting regions were found to play a role in evolutionary mRNA dosage maintenance in the fly (Fig. 3A).
It should be noted that among the histone marks analyzed here, H3K27me3 is the only histone mark that has been reported to date to underlie the diverse regulatory diversification of paralogous genes in Drosophila (Arthur et al. 2014). H3K27me3 has a distribution that is consistent with Predicted Pattern I, and not Predicted Pattern II or III, in the common acting region from “TSS−1000 to TSS+500” (Fig. 2B) and in the invertebrate-specific acting region from “TSS+500 to TTS” (Fig. 3A). Thus, H3K27me3 is unrelated to the process of gene dosage control. Our results based on H3K27me3 indicate that differential mechanisms have been employed over the evolution of Drosophila to create regulatory/functional divergence and to cope with increases in mRNA and protein that occur after gene duplication events.
Various histone marks are used to maintain mRNA dosage after gene duplication events in worms
To regulate transcription, several histone marks are utilized by C. elegans in the common acting regions shown in Figure 2C, in the invertebrate-specific acting regions shown in Figure 3A (“TSS+500 to TTS−500” by H3K4me1 and H3K79me2, “TSS+500 to TTS” by H3K27me3, and “TTS to TTS+1000” by H3K9me3), and in the worm-specific acting regions shown in Figure 3B (“TSS−1000 to TSS” by H3K27ac, H3K9ac, and H3K79me2; “TTS−500 to TTS” by H3K79me2; and “TSS−1000 to TTS” by H3K9me3). Analyses of the histone marks in worms have identified striking patterns that indicate that lineage-specific acting regions include both active marks and repressive marks that are intensively engaged to maintain mRNA dosage following gene duplication events (see below).
Among the six common acting regions (Fig. 2C), the distributions of H3K4me1 and H3K79me2 over the “TSS−1000 to TSS” and “TSS to TSS+500” regions, respectively, are consistent with Predicted Patterns I–III (Fig. 2C). The presence of H3K4me1 over the “TSS−1000 to TSS” region to maintain mRNA dosage was only observed in the fly and worm, while the role of H3K79me2 in the “TSS to TSS+500” region was observed in the same regions in the mouse, fly, and worm (Fig. 2). In contrast, the majority of lineage-specific acting regions (including three out of four invertebrate-specific acting regions [Fig. 3A] and all five worm-specific acting regions [Fig. 3B]) exhibited histone mark distribution patterns that are consistent with Predicted Patterns I–III. As a result, the acting regions, including the common and lineage-specific regions, of H3K4me1 that are involved in maintaining mRNA dosage in the worm include “TSS−1000 to TSS” (Fig. 2C) and “TSS+500 to TTS−500” (Fig. 3A) as discontinuous regions. In addition, the region associated with dosage maintenance for H3K79me2 was found to extend over the “TSS−1000 to TTS” region, which represents the concatenated regions of “TSS−1000 to TSS,” “TSS to TSS+500,” “TSS+500 to TTS−500,” and “TTS−500 to TTS” (Figs. 3B, 2C, 3A,B, respectively).
The present data suggest that there is a common role for H3K4me1 in gene promoters for the maintenance of mRNA dosage in the fly and worm (Fig. 2A,B). However, a regulatory role for H3K4me1 in gene body regions has not been reported. The mechanism involving the presence of H3K4me1 in nematode gene bodies to regulate transcription and control mRNA dosage in the worm awaits further investigation. It does appear that the regulation provided by H3K79me2 in an early stage of transcriptional elongation is particularly important for the maintenance of mRNA dosage in higher eukaryotes due to the evolutionary conservation of this regulatory role in the mouse, fly, and worm (see H3K79me2 in Fig. 2A–C). In addition to the region of “TSS to TSS+500” (Fig. 2C) that is utilized by H3K79me2 in the mouse and fly, H3K79me2 in nematodes also appears to maintain mRNA dosage by decreasing its density along the “TSS−1000 to TSS” (Fig. 3B), “TSS+500 to TTS−500” (Fig. 3A), and “TTS−500 to TTS” (Fig. 3B) regions of duplicated, weakly expressed, and dosage-sensitive genes. Taken together, these observations imply that H3K79me2 may enhance dosage maintenance by additionally mediating the initiation of transcription and an intermediate stage of elongation of duplicated genes in worms. We also found that the distribution pattern of H3K27me3 on “TSS+500 to TTS” (Fig. 3A), yet not on “TSS to TSS+500” (a subregion of “TSS−1000 to TSS+500” marked in Fig. 2C), is consistent with Predicted Patterns I–III. This indicates that H3K27me3 maintains mRNA dosage in C. elegans by mediating both intermediate and late stages of the transcriptional elongation process in an organism-specific manner.
The other histone marks that are distributed among lineage-specific acting regions and involved in mRNA dosage maintenance following gene duplication events include the active marks H3K27ac (at “TSS−1000 to TSS”) and H3K9ac (at “TSS−1000 to TSS”) and the repressive mark H3K9me3 (at “TSS−1000 to TTS”) that exhibit Predicted Patterns I–III (Fig. 3). H3K4me3 has been found to colocalize with H3K27ac in the promoter regions of highly expressed genes, and it has been hypothesized that these marks have similar roles in regulating the expression of C. elegans genes (Liu et al. 2011). In the present study, a role for H3K27ac at promoters in maintaining gene dosage was evident, yet there was no evidence to support a similar role for H3K4me3. Thus, H3K4me3 and H3K27ac appear to regulate gene expression for different purposes. H3K9me3, a well-known epigenetic repressor whose functions may differ between species and whose association with H3K27me3 is organism specific in C. elegans (Ho et al. 2014), was also found to be distributed over the worm-specific acting region “TSS−1000 to TTS,” consistent with Predicted Patterns I–III. However, based on the observation that the acting region of H3K9me3 in gene promoters (e.g., “TSS−1000 to TSS”) does not overlap with the acting region of H3K27me3 that is associated with maintenance of mRNA dosage (i.e., H3K27me3 at “TSS+1000 to TTS”) (Fig. 3A) in C. elegans, it is possible that H3K9me3 may have an independent role in down-regulating gene expression following gene duplication events that involves transcriptional initiation. Additional studies are needed to confirm this possibility and to more fully characterize the regulatory mechanism of H3K9me3 in nematodes.
We found that the dosage-sensitive genes tended to exhibit higher (or lower) intensities for all of the active (or repressive) marks in the acting regions identified as underlying post-duplicational mRNA dosage maintenance in the mouse, fly, and worm (Supplemental Fig. S4). To examine the potential influence of this bias in hypothesis testing based on comparisons of the dosage-sensitive versus dosage-insensitive genes (i.e., testing Predicted Pattern III), we separated the ortholog sets into five equal-sized bins according to histone mark intensity (the highest mark intensity value among the genes of the paralog group of species A was used as the representative value), and a set of ortholog sets from the dosage-sensitive and dosage-insensitive groups were randomly selected while keeping the numbers of sampled ortholog sets from each bin of the two groups the same in order to ensure that the default histone mark intensities of the two compared ortholog sets were similar (Supplemental Fig. S5). This process was repeated 10,000 times, and DHMd-u was calculated each time. All of the marks reported to be related to dosage maintenance, except for worm H3K9ac at “TSS−1000 to TSS” showed a distribution of DHMd-u that was consistent with Predicted Pattern III (Supplemental Fig. S6). Based on this result, the distribution of worm H3K9ac that follows Predicted Pattern III may represent an artifact, and thus, H3K9ac may not underlie mRNA dosage maintenance in worms.
Discussion
The results of the present study demonstrate that flies and worms need to maintain mRNA dosages by reducing mRNA expression after gene duplication events, similar to the strategy used by mammals. Evidence supporting the expression reduction model (Table 1; Supplemental Table S1) are not only from observations of mammalian cerebellum and fly and worm L3 larva tissues. The same patterns were also observed in the breast, colon, heart, kidney, liver, lung, and ovary tissues from mammals, in ovary tissues from flies, and in mixed-stage worms (Supplemental Tables S4, S5). In addition to D. simulans, RNA-seq data of L3 larva of Drosophila pseudoobscura were also available (see Methods). The results obtained did not change when fly ortholog sets were defined with D. pseudoobscura rather than D. simulans, with the former being a sister species that is more divergent from D. melanogaster than D. simulans (Supplemental Table S6; Clark et al. 2007). These results suggest the broad applicability of the expression reduction model to duplicate gene evolution in animals, and they also suggest that maintenance of mRNA levels in cells is very important during the evolution of animal genomes and transcriptomes.
In addition to expression reduction model, previously proposed models of duplicate gene retention include neofunctionalization and subfunctionalization (Force et al. 1999). To determine which model is more prevalent in explaining duplicate gene retention, we compared mammalian ortholog sets characterized by NA = NB = 1 with ortholog sets characterized by NA = 1 and NB > 1. For each ortholog set, ΔEA-B.max = ZA − ZB.max was calculated, where ZA is the expression level of the unduplicated gene in species A that represents the progenitor gene, and ZB.max is the expression level of the duplicate gene copy that showed the highest expression in species B in the tissue concerned. Assuming that regulatory subfunctions are independently mutable, the redundancy of regulatory elements between paralogs will eventually be eliminated after subfunctionalization or neofunctionalization (Force et al. 1999). Consequently, only one copy of duplicated genes is expressed (with an expression level that is the same as the progenitor gene) under any of the conditions where the progenitor gene was originally expressed after sub- or neofunctionalization. Thus, when subfunctionalization or neofunctionalization occurs, a difference in ΔEA-B.max between two ortholog sets is not expected. However, we found that the ortholog sets characterized by NA = 1 and NB > 1 consistently exhibited higher ΔEA-B.max values than do the ortholog sets with NA = NB = 1 in the various mammalian tissues examined. In addition, in most of the tissues that exhibited this trend, the trend was exclusively associated with the subset of dosage-sensitive ortholog sets (Supplemental Fig. S7). These results are consistent with an expression reduction model, although they may also be due to the fact that the assumption of independently mutable regulatory subfunctions is often violated in nature. To distinguish among the possibilities, more sophisticated analyses need to be performed in the future. Moreover, in addition to expression reduction, neofunctionalization, and subfunctionalization mechanisms, duplicate genes may also be preserved by positive selection that favors an increased gene dosage (Daborn et al. 2002; Cardoso-Moreira et al. 2016). However, Cardoso-Moreira et al. (2016) reported that duplicate genes that were positively selected for increased dosage only constituted a very small proportion of the complete gene duplication mutations that segregated in fly populations, and more than half of the completely duplicated fly genes did not result in increased mRNA dosage. In another study, the increased fitness of duplicated genes in yeasts was found to be unrelated to increases in gene dosage (Qian and Zhang 2014). Based on these observations, expression reduction appears to be the more prevalent model for explaining duplicate gene retention, particularly for genes that are sensitive to changes in dosage.
While cytosine DNA methylation helps to regain proper mRNA dosage following gene duplication events by mediating transcription initiation in mammals, histone modifications at promoter (i.e. “TSS−1000 to TSS”) regions are utilized to suppress transcriptional initiation of duplicate genes in flies and worms (Fig. 4). In flies, this modification is H3K4me1, and in worms these modifications include H3K4me1, H3K27ac, H3K79me2, and H3K9me3 (Fig. 4). Considering that some histone modifications can be predicted based on transcription factor binding sites (Benveniste et al. 2014) or DNA motifs (Whitaker et al. 2015), as well as the observation that epigenome evolution is coupled with promoter sequence evolution (Lowdon et al. 2016), it is possible that the intensity of changes in histone marks are related to the evolution of DNA sequences. At the present stage, it remains unclear whether changes in histone modifications or changes in transcription factor binding sites are the primary driver of mRNA dosage maintenance that involves transcriptional initiation. In this study, we also showed that maintenance of mRNA dosage is partially regulated by histone marks that control transcriptional elongation by acting on gene body regions (e.g., H3K79me2 in mouse; H3K4me3, H3K9ac, and H3K79me2 in flies; and H3K4me1, H3K79me2, H3K27me3, and H3K9me3 in worms) (Fig. 4). H3K79me2, the only mark currently shown to be involved in dosage maintenance in mammals, was profiled in the mouse liver and human hepatocytes differentiated from ES cells (see Methods). We assumed that the H3K79me2 distribution profile for human hepatocytes would be similar to that of human liver tissue. ΔHisH-M = HisH − HisM was calculated for each pair of human–mouse orthologous genes, where HisH and HisM represented the log histone intensity values in human hepatocytes and mouse liver, respectively, in the “TSS to TSS+500” region of the human gene and mouse ortholog, respectively. We focused on genes that are present as single copies in humans to compare the distributions of ΔHisH-M for human–mouse orthologs that remained as single copies in the mouse lineage (as unduplicated genes) and orthologs that are present in multiple copies in the mouse lineage (as duplicated genes). Our data indicate that ΔHisH-M of the duplicated orthologs was statistically greater than that of the unduplicated orthologs at “TSS to TSS+500” (Fig. 5). This result directly suggests that a reduction in H3K79me2 of 5′ gene bodies evolved after the duplication events. Confirmation of evolutionary changes in other marks in response to gene duplication requires that histone data are measured under the same conditions in multiple species, and these data may become available in the near future.
Figure 4.
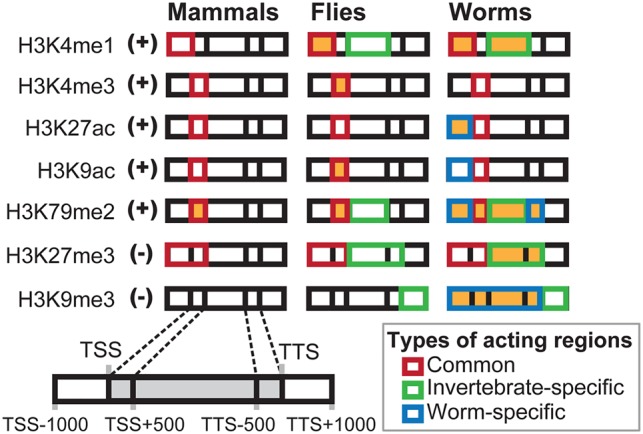
An overview of the histone marks (+: active; −: repressive) and the corresponding acting regions (shown in orange) that are involved in dosage maintenance in mammals, flies, and worms after gene duplication events. The boxes for each mark in each animal model are intended to represent five consecutive genic regions as diagrammed at the bottom left of the figure.
Figure 5.
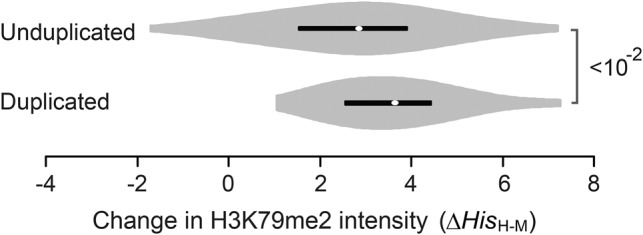
Violin plots of ΔHisH-M of H3K79me2 at “TSS to TSS+500” that were measured in mammalian liver tissues. Orthologs that underwent duplication events (duplicated) and those that did not (unduplicated) were compared in the mouse lineage. Only orthologs composed of a single copy of human genes with a H3K79me2 signal greater than zero were analyzed. P-values were determined with the Mann-Whitney U test.
The histone marks and corresponding acting regions involved in post-duplication mRNA dosage maintenance in the fly and worm models that were examined were found to be largely organism-specific (Fig. 4). It is possible that the species pairs used to define ortholog sets in mammals, flies, and worms have different divergent times (human vs. mouse: 90 Mya [million years ago]; D. melanogaster vs. D simulans: 5.9 Mya; C. elegans vs. C. briggsae: 60.2 Mya; see Methods), thereby making the duplicate genes that are included in the tree model systems representative of different stages of duplicate gene evolution and, therefore, not directly comparable. To address this issue, we calculated the rate of synonymous substitute (dS) for all the possible pairs of paralogs and calculated , the average value of the obtained dS, for each ortholog set. The worm ortholog set had substantially greater values than those calculated for the mammals and flies (Supplemental Fig. S8). When ortholog sets with a were excluded so that only ortholog sets constituting young duplicates were kept in the analysis, the observed trends that supported an expression reduction model for all of the species remained unchanged (Supplemental Tables S7, S8). The greatest number of histone marks involved in dosage maintenance remained observed in worms, followed by flies, and then a further marked decrease in histone mark number was observed in mammals. Moreover, most of the histone marks examined in the worm and fly were found to be lineage-specific (Supplemental Figs. S9, S10), indicating that the major observations of the present study are independent of the differential ages of the duplicated genes examined among the animal lineages examined. These results strongly suggest that many of the histone marks that are involved in the adaptive process of mRNA dosage maintenance have been independently and lineage specifically recruited over the evolution of flies and worms. However, due to the highly dynamic nature of the histone marks that were identified as contributing to the maintenance of mRNA dosage, it cannot be excluded that the histone marks and regions commonly used by multiple organisms are also recruited in a lineage-specific manner. Thus, additional studies are warranted to examine this possibility.
Worms are potentially an ideal model for understanding how different levels of epigenetic controls are integrated to maintain mRNA dosages during evolution. In C. elegans, there are many histone marks that are involved in dosage maintenance. These include both active marks and repressive marks, and most act on lineage-specific regions. It is hypothesized that the marks that are involved in maintaining dosage have been progressively recruited over the evolution of worms. The presence of methylated cytosines in the genome of Trichinella spiralis, as well as DNMT1 and DNMT3, suggest that the last common ancestor of the phylum Nematoda should be equipped with a complete set of DNA cytosine methylation machinery (Gao et al. 2012). However, this machinery is absent in the lineage of C. elegans. When sequencing-based transcriptomic and epigenomic data become available for multiple nematode species, including DNA methylation and histone modification data, it will be of great interest to investigate how DNA methylation was replaced with histone modifications for the maintenance of mRNA dosages following gene duplication events during the evolution of C. elegans.
Methods
Ortholog sets of mammal, fly, and worm genes
Reference genomes, gene coordinates, transcript annotations, and orthology information were obtained from Ensembl (v79, for mammalian genes; http://www.ensembl.org, last accessed March 2015) and Ensembl Metazoa (v25, for fly and worm genes; http://metazoa.ensembl.org, last accessed March 2015) through BioMart (http://www.biomart.org, last accessed November 2015) (Zhang et al. 2011). Our analyses were based on compiled human (GRCh38.p2)–mouse (GRCm38.p3), D. melanogaster (BDGP6)–D. simulans (GCA_000259055.1), and C. elegans (WBcel235)–C. briggsae (CB4) orthologous genes. The phylogenetic relationship and divergence time of the orthologs annotated by Ensembl enabled us to focus on genes that duplicated after the divergence of the two species being compared. By selecting paralogs arising from their latest common ancestral state (e.g., primates/Rodentia; D. melanogaster/D. simulans; C. elegans/C. briggsae) (Chang and Liao 2012), many of the analyses included paralogs that had undergone duplication after the evolutionary divergence of the two species pairs for mammalian, fly, and worm genes, respectively. When multiple isoforms were encoded by a gene, the longest form was considered the representative protein. The values of dN and dP were calculated for all ortholog pairs of one-to-many orthologs in order to determine the pair of progenitor genes in each ortholog set. The value of dS between paralogs was used to determine the age of the duplication event. Both the dN and dS values were estimated by codeml of PAML (Yang 2007) with the following settings: runmode = −2, seqtype = 1, CodonFreq = 0, model = 0, and NSsites = 0. The dP value was defined as −3/4 × loge[1-4/3(1-I)], where I is the nucleotide sequence identity calculated from a ClustalW pairwise alignment (with default parameters) of 5000 nt upstream of the TSS of the gene that was annotated by Ensembl. The TSS and TTS of a gene were defined as the positions of the first and last nucleotide, respectively, of the longest transcript of the focal gene annotated by Ensembl. The absolute divergent time of the two compared species pairs were obtained by using TimeTree (http://www.timetree.org/) (Kumar et al. 2017).
RNA-seq gene expression
Gene expression data were obtained from RNA-seq data deposited in the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) or Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra/), and they included data from L3 larva of C. elegans (GSE53359), C. briggsae (GSE53359), D. melanogaster (GSE18068), D. simulans (GSE49945), D. pseudoobscura (GSE49945); ovary of D. melanogaster (GSE46100) and D. simulans (GSE31302); mixed-stage C. elegans (GSE22410) and C. briggsae (SRA050228); and adult cerebellum of human (GSE13652) and mouse (GSE49847). The sources of RNA-seq data of human and mouse breast, colon, heart, kidney, liver, lung, and ovary are listed in Supplemental Table S9. Sequencing reads were mapped to each genome using TopHat (v2.0.12) (Trapnell et al. 2009) with default parameters set to the reference genomes. The reference genomes of human (GRCh38.p2), mouse (GRCm38.p3), D. melanogaster (BDGP6), D. simulans (GCA_000259055.1), C. elegans (WBcel235), and C. briggsae (CB4) were downloaded from Ensembl (v79) and Ensembl Metazoa (v25) as described above. Normalized gene expression levels, measured in fragments per kilobase of exon per million fragments mapped (FPKM), were calculated using Cufflinks (v2.2.1) (Trapnell et al. 2010) with default parameters. When the “-N 0” option was used to map reference transcript annotations with zero mismatches or the “-g 0” option was used to exclude reads that mapped to multiple regions, consistent results were obtained (Supplemental Tables S9, S10). A pseudocount value of one (to preserve a 0 FPKM count after log transformation) was added to the expression values measured in FPKM that subsequently underwent a log2 transformation. Z-score normalization for each sample was performed as follows: Z = (log2R − TM)/TSD, where R is FPKM + 1, TM is the mean of all the log2R values within an RNA-seq sample of a species, and TSD is the standard deviation of all the log2R values within the sample. To define weakly or strongly expressed genes in a paralog group, RNA-seq expression data for each individual gene were compared with the average RNA-seq expression data in the paralog group. If the expression of a gene was higher than the mean Z-score for the transformed expression signals of its paralog group, the gene was classified as strongly expressed. Otherwise, the gene was classified as weakly expressed.
Ortholog sets of dosage-sensitive genes or genes that encode members of protein complexes
Annotations of protein interaction data detected by various approaches for the mouse, fly, and worm were obtained from BioGRID (version 3.4.139; https://thebiogrid.org/) (Chatr-Aryamontri et al. 2015). As described in a previous study (Vavouri et al. 2009), binary protein interactions for defining “dosage sensitivity” of a protein were compiled from physical interactions that were identified through a sensitive “two-hybrid” method, and the interactions overlapping physical interactions identified by a “Affinity Capture-MS” method were removed. This procedure tends to keep “promiscuous” interactions only. Accordingly, genes encoding proteins that promiscuously interact with at least three other proteins encoded in a genome were defined as dosage-sensitive genes, whereas genes encoding proteins that interact with at least one but fewer than three other proteins were defined as dosage-insensitive genes. Protein complex data for mouse were defined based on human ortholog proteins, and the subunits of human protein complexes were obtained from H-Invitational protein–protein interactions integrative data set (http://www.h-invitational.jp) (Kikugawa et al. 2012). Protein complex data for the fly were obtained from the Drosophila Protein Interaction Map (https://interfly.med.harvard.edu) (Guruharsha et al. 2011). Protein complex data for the worm were predicted by WCOACH algorithm (http://bioinformatics.aut.ac.ir/wcoach) (Kouhsar et al. 2015) with default parameters (weighted, minimum size of clusters = 3, NA threshold = 0.85) based on the weighted protein interaction data compiled from seven molecular interaction databases (Huang et al. 2016). For each ortholog set, its member genes were searched with Ensembl IDs against a list of genes encoding members of protein complexes. If at least one member of an ortholog set encoded a component of a protein complex, the ortholog set was defined as a “complex” ortholog set. Otherwise, an ortholog set was classified as “noncomplex.”
Adenine DNA methylation level
The data of N6-Methyladenine DNA modifications in C. elegans (GSE66504) and D. melanogaster (SRP055483) were obtained from NCBI GEO (http://www.ncbi.nlm.nih.gov/geo) and the DNA Data Bank of Japan (DDBJ, http://trace.ddbj.nig.ac.jp/), respectively. Raw reads were mapped to respective reference genomes with Bowtie (v1.1.2) (Langmead et al. 2009) with default options. Reads mapped to more than one position were removed by SAMtools (http://www.htslib.org/) (Li et al. 2009) in order to generate a “SAM-format” alignment that could be analyzed by the peak calling software MACS2 (https://github.com/taoliu/MACS) (Zhang et al. 2008). Levels of DNA adenine methylation modification enrichment were obtained by normalizing against the background input by “macs2 bdgcmp” on the bedGraph files generated by “macs2 callpeak” of MACS2 with the following option: -B --nomodel --SPMR, according to the method of Greer et al. (2015).
Histone modification data
Epigenomic ChIP-seq histone modification data sets for the mouse, fly, and worm were obtained from the ENCODE (https://www.encodeproject.org, last accessed November 2015) (Yue et al. 2014) and modENCODE (http://www.modencode.org, last accessed January 2016) databases (Ho et al. 2014) (see Supplemental Table S1). After mapping the ChIP-seq reads to an appropriate genome by TopHat, histone modification enrichments were identified by a peak calling software used by ENCODE and modENCODE to provide interpreted signal data files in broadPeak/GFF3/wiggle file formats (Landt et al. 2012). Mapping the ChIP-seq reads using the parameters “-N 0 -g 0” that do not allow mismatches and multiple hits did not significantly affect the results of hypothesis testing (Supplemental Figs. S11, S12). The magnitudes of the ChIP-seq histone modification enrichments were averaged over the genic regions analyzed, and the average intensities were compared among the groups of genes (Figs. 2, 3). Based on the patterns of histone modification that were observed for the expressed genes versus the silent genes in the human, fly, and worm genomes (shown in the extended data fig. 1 of Ho et al. 2014), we defined acting regions of the mouse, fly, and worm histone marks as active or repressive (Supplemental Fig. S3). Genic regions in which a histone mark consistently correlated with an effect on gene expression across different species were designated common acting regions, while regions in which a histone mark exhibited a lineage-specific effect on gene expression that differed across the compared species were designated lineage-specific acting regions (Supplemental Fig. S3). Both H3K79me2 ChIP-seq data of mouse liver (GSM1000152) and human hepatocytes (GSE96318) were obtained from GEO of NCBI.
Supplementary Material
Acknowledgments
This study was supported by intramural funding from the National Health Research Institutes and research grants from the Ministry of Science and Technology, Taiwan (MOST 101-2311-B-400-001-MY3, 104-2311-B-400-002-MY3, and 105-2314-B400-021-MY4) to B.Y.L.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.221739.117.
References
- Ardehali MB, Mei A, Zobeck KL, Caron M, Lis JT, Kusch T. 2011. Drosophila Set1 is the major histone H3 lysine 4 trimethyltransferase with role in transcription. EMBO J 30: 2817–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur RK, Ma L, Slattery M, Spokony RF, Ostapenko A, Negre N, White KP. 2014. Evolution of H3K27me3-marked chromatin is linked to gene expression evolution and to patterns of gene duplication and diversification. Genome Res 24: 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837. [DOI] [PubMed] [Google Scholar]
- Benveniste D, Sonntag HJ, Sanguinetti G, Sproul D. 2014. Transcription factor binding predicts histone modifications in human cell lines. Proc Natl Acad Sci 111: 13367–13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ III, Gingeras TR, et al. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120: 169–181. [DOI] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA. 2007. The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell 19: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csardi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478: 343–348. [DOI] [PubMed] [Google Scholar]
- Cardoso-Moreira M, Arguello JR, Gottipati S, Harshman LG, Grenier JK, Clark AG. 2016. Evidence for the fixation of gene duplications by positive selection in Drosophila. Genome Res 26: 787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AY, Liao B-Y. 2012. DNA methylation rebalances gene dosage after mammalian gene duplications. Mol Biol Evol 29: 133–144. [DOI] [PubMed] [Google Scholar]
- Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, Chen D, Stark C, Breitkreutz A, Kolas N, O'Donnell L, et al. 2015. The BioGRID interaction database: 2015 update. Nucleic Acids Res 43: D470–D478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, et al. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218. [DOI] [PubMed] [Google Scholar]
- Daborn PJ, Yen JL, Bogwitz MR, Le Goff G, Feil E, Jeffers S, Tijet N, Perry T, Heckel D, Batterham P, et al. 2002. A single p450 allele associated with insecticide resistance in Drosophila. Science 297: 2253–2256. [DOI] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151: 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Hirsh AE, Wall DP, Eisen MB. 2004. Coevolution of gene expression among interacting proteins. Proc Natl Acad Sci 101: 9033–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M, Thomas BC. 2006. Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res 16: 805–814. [DOI] [PubMed] [Google Scholar]
- Gao F, Liu X, Wu XP, Wang XL, Gong D, Lu H, Xia Y, Song Y, Wang J, Du J, et al. 2012. Differential DNA methylation in discrete developmental stages of the parasitic nematode Trichinella spiralis. Genome Biol 13: R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizabal-Corrales D, Hsu CH, Aravind L, He C, Shi Y. 2015. DNA methylation on N6-adenine in C. elegans. Cell 161: 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha KG, Rual JF, Zhai B, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee DY, Cenaj O, et al. 2011. A protein complex network of Drosophila melanogaster. Cell 147: 690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A, Sommer RJ. 2004. Evolution of dnmt-2 and mbd-2-like genes in the free-living nematodes Pristionchus pacificus, Caenorhabditis elegans and Caenorhabditis briggsae. Nucleic Acids Res 32: 6388–6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn KA, Minoda A, Tolstorukov MY, Appert A, et al. 2014. Comparative analysis of metazoan chromatin organization. Nature 512: 449–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XT, Zhu Y, Chan LL, Zhao Z, Yan H. 2016. An integrative C. elegans protein–protein interaction network with reliability assessment based on a probabilistic graphical model. Mol Biosyst 12: 85–92. [DOI] [PubMed] [Google Scholar]
- Jeltsch A. 2010. Molecular biology: phylogeny of methylomes. Science 328: 837–838. [DOI] [PubMed] [Google Scholar]
- Jones PA, Takai D. 2001. The role of DNA methylation in mammalian epigenetics. Science 293: 1068–1070. [DOI] [PubMed] [Google Scholar]
- Jonkers I, Lis JT. 2015. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 16: 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowski TP, Jeltsch A. 2011. On the evolutionary origin of eukaryotic DNA methyltransferases and Dnmt2. PLoS One 6: e28104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller TE, Yi SV. 2014. DNA methylation and evolution of duplicate genes. Proc Natl Acad Sci 111: 5932–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikugawa S, Nishikata K, Murakami K, Sato Y, Suzuki M, Altaf-Ul-Amin M, Kanaya S, Imanishi T. 2012. PCDq: human protein complex database with quality index which summarizes different levels of evidences of protein complexes predicted from h-invitational protein–protein interactions integrative dataset. BMC Syst Biol 6(Suppl 2): S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouhsar M, Zare-Mirakabad F, Jamali Y. 2015. WCOACH: Protein complex prediction in weighted PPI networks. Genes Genet Syst 90: 317–324. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Suleski M, Blair Hedges S. 2017. TimeTree: A resource for timelines, timetrees, and divergence times. Mol Biol Evol 34: 1812–1819. [DOI] [PubMed] [Google Scholar]
- Kusch T. 2012. Histone H3 lysine 4 methylation revisited. Transcription 3: 310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22: 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao BY, Chang AYF. 2014. Accumulation of CTCF-binding sites drives expression divergence between tandemly duplicated genes in humans. BMC Genomics 15: S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B-Y, Weng MP. 2015. Unraveling the association between mRNA expressions and mutant phenotypes in a genome-wide assessment of mice. Proc Natl Acad Sci 112: 4707–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B-Y, Zhang J. 2008. Coexpression of linked genes in mammalian genomes is generally disadvantageous. Mol Biol Evol 25: 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung MS, Ercan S, Ikegami K, Jensen M, Kolasinska-Zwierz P, et al. 2011. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res 21: 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowdon RF, Jang HS, Wang T. 2016. Evolution of epigenetic regulation in vertebrate genomes. Trends Genet 32: 269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F, Ramsahoye BH, Jaenisch R. 2000. DNA methylation in Drosophila melanogaster. Nature 408: 538–540. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155. [DOI] [PubMed] [Google Scholar]
- Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, et al. 2007. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 110: 4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima T, Wada C, Kawagoe Y, Ara T, Maeda M, Masuda Y, Hiraga S, Mori H. 2002. Genome-wide analysis of deoxyadenosine methyltransferase-mediated control of gene expression in Escherichia coli. Mol Microbiol 45: 673–695. [DOI] [PubMed] [Google Scholar]
- Owens SM, Harberson NA, Moore RC. 2013. Asymmetric functional divergence of young, dispersed gene duplicates in Arabidopsis thaliana. J Mol Evol 76: 13–27. [DOI] [PubMed] [Google Scholar]
- Papp B, Pal C, Hurst LD. 2003. Dosage sensitivity and the evolution of gene families in yeast. Nature 424: 194–197. [DOI] [PubMed] [Google Scholar]
- Park PJ. 2009. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 10: 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Zhang J. 2014. Genomic evidence for adaptation by gene duplication. Genome Res 24: 1356–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Liao B-Y, Chang AY, Zhang J. 2010. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet 26: 425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratel D, Ravanat JL, Berger F, Wion D. 2006. N6-Methyladenine: the other methylated base of DNA. Bioessays 28: 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JC, Rogers SW. 1995. Comparison of the effects of N6-methyldeoxyadenosine and N5-methyldeoxycytosine on transcription from nuclear gene promoters in barley. Plant J 7: 221–233. [DOI] [PubMed] [Google Scholar]
- Simpson VJ, Johnson TE, Hammen RF. 1986. Caenorhabditis elegans DNA does not contain 5-methylcytosine at any time during development or aging. Nucleic Acids Res 14: 6711–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476. [DOI] [PubMed] [Google Scholar]
- Takayama S, Dhahbi J, Roberts A, Mao GX, Heo SJ, Pachter L, Martin DIK, Boffelli D. 2014. Genome methylation in D. melanogaster is found at specific short motifs and is independent of DNMT2 activity. Genome Res 24: 821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler J, Lehner B, Chen N, Fraser AG. 2006. Combinatorial RNA interference in Caenorhabditis elegans reveals that redundancy between gene duplicates can be maintained for more than 80 million years of evolution. Genome Biol 7: R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavouri T, Semple JI, Lehner B. 2008. Widespread conservation of genetic redundancy during a billion years of eukaryotic evolution. Trends Genet 24: 485–488. [DOI] [PubMed] [Google Scholar]
- Vavouri T, Semple JI, Garcia-Verdugo R, Lehner B. 2009. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 138: 198–208. [DOI] [PubMed] [Google Scholar]
- Wagner GP, Pavlicev M, Cheverud JM. 2007. The road to modularity. Nat Rev Genet 8: 921–931. [DOI] [PubMed] [Google Scholar]
- Wang J, Marowsky NC, Fan C. 2014. Divergence of gene body DNA methylation and evolution of plant duplicate genes. PLoS One 9: e110357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker JW, Chen Z, Wang W. 2015. Predicting the human epigenome from DNA motifs. Nat Methods 12: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang JR, Liao BY, Zhuang SM, Zhang J. 2012. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proc Natl Acad Sci 109: E831–E840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Sweeney S, Raha D, Snyder M, Lin H. 2011. A high-resolution whole-genome map of key chromatin modifications in the adult Drosophila melanogaster. PLoS Genet 7: e1002380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Haider S, Baran J, Cros A, Guberman JM, Hsu J, Liang Y, Yao L, Kasprzyk A. 2011. BioMart: a data federation framework for large collaborative projects. Database (Oxford) 2011: bar038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, et al. 2015. N6-Methyladenine DNA modification in Drosophila. Cell 161: 893–906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.