

Abstract
Background
In this review, we examine the continually expanding and increasingly compelling data linking radiation and various chemicals in our environment to the current high incidence of breast cancer.
Abstract
Singly and in combination, these toxicants may have contributed significantly to the increasing rates of breast cancer observed over the past several decades. Exposures early in development from gestation through adolescence and early adulthood are particularly of concern as they re-shape the program of genetic, epigenetic and physiological processes in the developing mammary system, leading to an increased risk for developing breast cancer. In the 8 years since we last published a comprehensive review of the relevant literature, hundreds of new papers have appeared supporting this link, and in this update, the evidence on this topic is more extensive and of better quality than that previously available.
Conclusion
Increasing evidence from epidemiological studies, as well as a better understanding of mechanisms linking toxicants with development of breast cancer, all reinforce the conclusion that exposures to these substances – many of which are found in common, everyday products and byproducts – may lead to increased risk of developing breast cancer. Moving forward, attention to methodological limitations, especially in relevant epidemiological and animal models, will need to be addressed to allow clearer and more direct connections to be evaluated.
Keywords: Breast cancer, Environmental toxicants, Endocrine disrupting compounds, Bisphenol a, Light-at-night, Radiation
Background
In this review, we examine the continually expanding and increasingly compelling data linking radiation and various chemicals in our environment to the current high incidence of breast cancer. We acknowledge the importance of many widely understood risk factors for breast cancer including: primary genetic mutations, reproductive history, and lifestyle factors such as weight gain, alcohol consumption and lack of physical exercise [1, 2]. Yet we begin with an understanding that in total, these factors do not address a considerable portion of the risk for the disease [2–5]. A substantial body of scientific evidence indicates that exposures to common chemicals and radiation, singly and in combination, also contribute to the increasingly high incidence of breast cancer observed over the past several decades. Although rates have leveled off overall in the past few years for some subsets of women, there was a significant and progressive rise in the incidence of breast cancer in the decades following World War II [6, 7], the same decades that saw exponential increases in the use of chemicals for production of pesticides, herbicides, plastics, cosmetics and other commonly used materials and products [8–10].
This report focuses on these environmental issues. In the 8 years since we last published a comprehensive review of the relevant literature [11], hundreds of new papers have been published supporting this link, and the evidence on this topic is more extensive and of better quality than that previously available. After describing our methodology for selecting scientific reports and reporting of statistical findings, we present introductory sections on breast cancer statistics and subtypes as well as critical concepts for framing the complex data we are exploring. We then examine the literature on exposures to environmental toxicants and risk for developing breast cancer, dividing the evidence discussion into seven major sections: (1) Hormones: Pharmaceutical agents & personal care products; (2) Endocrine disrupting compounds (EDCs); (3) Hormones in food: Natural and additives; (4) Non-EDC industrial chemicals; (5) Tobacco smoking: Active and passive; (6) Shift work, light-at-night and melatonin; and (7) Radiation. We conclude with a brief synopsis and reflection on the state of the evidence, including methodological limitations and promises, as well as directives for future research needs.
Methodology
Article selection process
The goal of this review is to offer a broad overview of the scientific literature examining the potential connections between exposure to environmental toxicants and changes in the risk for developing breast cancer, updating our last review of this topic published in 2009. To fully incorporate the relevant materials, we entered the following search terms into both PubMed and Scopus: ‘breast cancer’ and ‘mammary tumors’ in conjunction with ‘environment’, ‘endocrine disruptors/endocrine disrupting compounds’ and all of the individual toxicants covered in this report.
In selecting epidemiological studies, we emphasized work from the past 10 years. When studies were follow-up reports from large, longitudinal studies, we also reported the earlier data as the length of time between exposures and outcome assessments could lead to different conclusions, or recognition of different results as the participants in the study reached later ages and, especially, progressed from pre-menopausal to post-menopausal status.
Over the 8 years since our last report, there has been a substantial increase in the amount of information focused on mechanisms underlying the complex relationships between exposures and risk for developing breast cancer. This is especially true in the growing field examining exposures to endocrine disrupting compounds and disease risk. We therefore focused on articles from the past 8 years. While we did not report every gene whose expression might be affected by a particular exposure, we did try to give a full overview of the current understanding of physiological, developmental, genetic, epigenetic and endocrine processes that are affected by exposures relevant to a change in risk for developing breast cancer. Although the emphasis was on the most recent data, we included earlier results when they were needed as background or to provide a fuller picture of the evidence.
Exceptions to our primary reliance on very recent literature are found in the sections on non-endocrine disrupting industrial chemicals and some pesticides and herbicides. Much of the relevant data for these toxicants comes from studies from 25 to 30 years ago, when the National Toxicology Program (NTP) and International Agency for Research on Cancer (IARC) were determining possible carcinogenicity of these chemicals.
Finally, in selecting studies to report, we took care to include studies that had negative results, that is, those that reported no significant relationship between exposures and risk for developing breast cancer. Where possible, we then explored possible differences in study design or methods that might account for differences in results across studies.
Reporting of statistics for epidemiological studies
We report the statistics (e.g., RR, OR, HR, etc.), along with 95% confidence levels, as offered by the authors of the individual reports. Where explicit adjustments were made, we note the type of statistic used and the variable of adjustment. More often though, factors including age, menopausal status, breast cancer subtype (by receptor status, ductal vs. lobular, in situ vs. invasive, etc.), racial/ethnic identity, are reported as main factors to be analyzed, along with effects of particular exposures. Significant main effects and interactions between exposures and these other variables are reported in this review.
Introduction
In this introductory section, we provide basic statistics and a brief exploration of the several subtypes of breast cancer – recognizing that the term ‘breast cancer’ is often used as a proxy for several distinct genetic, histopathological, and hormonal profiles for the disease. We then introduce a series of key framing concepts necessary for appreciating the complex evidence supporting (or not) a growing understanding of the data implicating specific environmental toxicants in an increased risk for developing breast cancer. These framing concepts include: (a) low-dose and non-monotonic responses; (b) interactions between environmental toxicants; (c) gene-environment interactions and epigenetic changes; (d) cell-cell interactions and the Tissue Organization Field Theory; and (e) timing of exposures. We conclude with a schematic model of the complexity of factors influencing risk for developing breast cancer, with an emphasis on environmental factors.
Breast cancer statistics
The Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute (NCI) predicted that in 2015 in the U.S., 40,290 women and 440 men would die of breast cancer and 231,840 women and 2350 men would be diagnosed with invasive breast cancer; another 60,290 women would be diagnosed with breast cancer in situ. As of early 2016, the NCI estimated that approximately 3,560,570 U.S. women are living with a prior diagnosis of breast cancer [12].
The most recent year for which accurate data exist related to breast cancer incidence and mortality is 2012. In addition to total national incidence and mortality reports, SEER data are broken down by major census self-described categories of race/ethnicity. The average incidence rates (number of women diagnosed per 100,000 women, age-adjusted and normalized to the 2000 standardized U.S. population) across the 5 years from 2008 to 2012 differed across census categories, as did the trends across time. Five-year average incidence rates for whites were the highest (126.1), with rates for black women only slightly lower (124.1). However, in 2012 for the first time since SEER began collecting data in 1975, incidence for these two groups converged; historically black women had a significantly lower rate of the disease. Average 5-year incidence rates were lower for American Indian/Native American (91.9), Hispanic (91.9) and Asian-Pacific Island (88.3) women [12].
Across racial and ethnic groups in the U.S., mortality rates (deaths per 100,000 women, age-adjusted and normalized to the 2000 standardized U.S. population) from breast cancer have decreased since their peak in the mid-late 1990s. Despite this apparent good news, significant racial/ethnic disparities have remained consistent over the last several decades. In the U.S., black women have the highest breast cancer mortality rate (31.0) of any racial/ethnic group. Asian/Pacific Islander women have the lowest mortality rates (11.4), with white (21.9), Hispanic (14.5) and American Indian/Native American (15.0) women having intermediate rates. Despite the universal drop in mortality rates across the past two decades and the similarity in incidence rates, over the same time period the disparities between mortality rates for white and black women have grown significantly; the mortality rate for black women diagnosed with breast cancer is 42% higher than the comparable rate for white women [12, 13].
Breast cancer subtypes
Breast cancer is not a singular disease, and it will be important throughout this report to examine, where possible, the subtype(s) of the disease most affected by exposures to environmental toxicants. Several classification systems have been developed to distinguish different subtypes of the disease including age of patient (usually split by pre or post-menopausal, with age 50 often as the proxy for the shift between reproductive phases); in situ, localized, regional or metastatic presentation; morphological characteristics; histological grade and cellular proliferation rate; or gene expression profile [14–17].
Of particular relevance to the discussion of environmental exposures, especially to endocrine disrupting compounds (EDCs), is the classification based on expression of the estrogen receptor (ER), progesterone receptor (PR) or the HER2 oncogene. Two luminal subtypes (A and B) express ER but not HER2, with Luminal A co-expressing PR and having a low proliferation rate and Luminal B having either high proliferation rate or low PR expression. Luminal B-like (HER2 positive) expresses ER and high HER2 levels, with any proliferation and PR profile. The HER2 positive subtype has overexpression of HER2 but without ER or PR being present. Triple negative breast cancer has no expression of ER, PR or HER2 [18].
Breast cancer subtypes are not randomly distributed across the population and there are differences found when diagnoses are stratified by age, race/ethnicity, reproductive history, body mass index, socioeconomic status, or geographical location [17, 19–22]. For example, younger women in general, and younger black women in particular, are more likely to present with the triple negative (ER-, PR-, and HER2-) subtype of the disease, a diagnosis that is both more aggressive and less responsive to treatment than ER+/PR+ or HER2+ tumors [12, 23, 24]. Like young black women, Latinas are also disproportionately affected by aggressive triple-negative tumors [17, 24, 25].
Race and ethnicity
The existence of differences across self-identified race and ethnic categories do not necessarily imply genetic differences. Indeed, they reflect the complexity of geographic location; social and socioeconomic status; personal and community stress and security; lifestyle factors including diet, exercise, alcohol and pharmaceuticals use; physiological responses to life factors; gene-environment interactions; and epigenetic changes — all factors that may change over the lifetime of the individual and may vary considerably among people who self-identify in a particular race/ethnicity category [26, 27]. Because of the confounds of economic and social factors, people of different racial/ethnic identities may also experience different environmental and occupational exposures to disease-affecting toxicants [28–30].
Racial and ethnic minorities often are exposed to disproportionately high levels and varieties of environmental pollutants in the U.S. [31], as are people living in poverty [32]. There are racial/ethnic differences in the body burden of different environmental chemicals that have been associated with increased risk for breast cancer. Blacks have higher body burden levels than whites or Mexican Americans of many chemicals including many polychlorinated biphenyls (PCBs), mercury, polyaromatic hydrocarbons (PAHs), and phthalates. Mexican Americans have higher levels of the pesticide dichlorodiphenyltrichloroethane (DDT) [33]. Varying body burdens of some chemicals including bisphenol A (BPA), polyfluorinated chemicals (PFCs) and triclosan, all commonly found in household products, are associated with both race/ethnicity and socioeconomic status [27, 34, 35]. Yet as Nelson points out, socioeconomic status and race/ethnicity most probably serve independently as markers for other activities or circumstances that influence the level of exposures to potentially toxic chemicals [27].
Framing concepts
Building on and extending the ‘Hallmarks of Cancer’ framework proposed by Hanahan and Weinberg [36], an international team of 170 scientists participating in the Halifax project recently evaluated the contributions to carcinogenesis of low-dose exposures to individual compounds and mixtures of environmental chemicals on each of the proposed hallmark phenotypes [37]. Other recent reviews have focused on the importance of evaluating: non-monotonic dose-response relationships, especially between EDCs and health outcomes [38]; timing of exposures to environmental toxicants, with an emphasis on fetal to adolescent exposures to EDCs and later development of diseases [39–41]; environmental carcinogenesis from the perspective of disruptions of cell-cell (e.g., stromal-epithelia) interactions [42, 43]; gene-environment interactions [44, 45]; the importance of using the principles of basic endocrinology in establishing mechanistic models for examining health impacts of exposures to EDCs [46, 47] and the relevance for these mechanisms in understanding the growing appreciation of the links between environmental toxicants and increased risk for many diseases, including breast cancer [48–52].
In this current paper, we will not offer comprehensive overviews of these framing concepts, but refer the reader to the reviews cited above. Instead we will briefly introduce the main concepts with a couple of examples relevant to exploring the following evidence linking exposures to environmental chemicals toxicants with increased risk for development of breast cancer. While some of the chemicals of concern are traditionally defined carcinogens, many more fall into the class of endocrine disrupting compounds (EDCs), a group of exogenous compounds that exert at least part of their impacts on health outcomes by altering the activity of the endocrine system.
Low-dose and non-monotonic responses
EDCs disrupt the endocrine system. As such, their mechanisms of action and properties are different than most non-EDC carcinogens for which the toxicological model is that higher doses are more damaging than are lower doses; the relationship between dose and damage is functionally linear; and there may be safe levels below which no negative impact is observed (the No-Observed-Adverse-Effect Level or NOAEL) [53, 54]. Instead, in many ways, EDCs act much as natural hormones do: at very low doses, especially during critical periods of development, and often following non-monotonic response (NMRs) curves [38, 47]. Thus sub-cellular and physiological responses to low dose exposures may be greater than, or at least different from, exposures to higher doses.
For example, many animal studies have demonstrated that prenatal or neonatal exposures to bisphenol A (BPA) lead to changes in mammary tissue development that increase the likelihood of the later development of mammary tumors. Yet some of these effects are dependent on dose, but not in a linear fashion. In one study, prenatal exposures to low (and environmentally relevant) doses of BPA had significant effects on the mammary gland gene expression profile just prior to the onset of puberty, while higher exposure levels altered expression of different genes and at a much later age [55]. In another report, rat dams were exposed via gavage to no BPA, or doses ranging from 0.025 to 50 mg BPA/kg bw/d from day 7 of gestation through weaning of their pups. As adults, female offspring that had been exposed to the 0.25 mg dose had increased incidence of intraductal hyperplasia, although no similar effects were found for either higher or lower exposures [56].
Blei et al. examined the lifelong effect of dietary exposures to two different amounts of soy-derived isoflavones, choosing doses that yielded concentrations similar to the highest and lowest plasma levels of isoflavones in Asian women. Although both low and high exposure levels led to an earlier onset of puberty, only low levels of exposure led to increased expression of the proliferation marker Ki67 in mammary glands of 97-day old adults. On the other hand, only higher exposure levels led to significant decreases in the expression of the proliferation marker PCNA in mammary tissue from ovariectomized rats that had been treated with estradiol. In these animals, estradiol administration led to additive stimulation of PR induction in animals that were exposed to the low dose exposures, while the high dose exposure levels inhibited the estradiol-induced expression of PR in mammary gland [57].
Interactions between environmental toxicants
Numerous animal studies indicate that the kinds of mixtures to which an animal is exposed matter in determining ultimate risk [58]. Only a relatively few combinations and doses of chemicals have been tested. This is perhaps not surprising: One estimate predicts that it would require 166 million experiments to test all combinations of three out of the 1000 most common synthetic chemicals currently in use [59]. While only a small portion of those studies have actually been conducted, there are several reports demonstrating that mixtures of environmental chemicals or chemicals and radiation, may alter biological processes and possibly lead to increases in breast cancer risk.
For example, the E-screen assay uses ER+ human breast cancer tumor cells (MCF-7 cells) that are dependent on estrogens for cell growth and proliferation [60], and single studies can examine the effects of scores of chemicals at multiple doses, alone and in combination on breast cancer cell proliferation [61, 62]. An examination of the combined effects of 11 different environmental contaminants – all added at NOAEL concentrations – showed that the chemicals had additive effects with each other and also with naturally occurring estradiol [63]. At levels found in our environment, the ubiquitous plasticizer bisphenol A also significantly increased the effects of estradiol [64].
Payne et al. used the yeast estrogen screen (YES), an in vitro assay of estrogen receptor activation, to examine the combined effects of a pesticide residue (o,p’-DDT), a plant estrogen (genestien, found in soy) and two alkylphenol surfactants (sudsing agents and chemical dispersers; 4-n-octylphenol and 4-nonlyphenol). Clear additive effects of the four chemicals were found [65].
Rivero et al. examined the effects of two mixtures of organochlorine pesticides, the first composed to mimic the chemical profile found in healthy women and the second to mimic the pesticide profile found in breast cancer patients. Both mixtures down-regulated genes whose expression is involved in the binding of ATP in normal human mammary epithelial cells, but there were very different effects of the two mixture profiles on the expression of oncogenes and tumor suppressor genes [66, 67]. Similarly, combinations of different organochlorine pesticides, mixed to mimic combinations found in human samples, increased cytotoxic effects in a cell line derived from normal human breast epithelial cells [68].
In a study of mammary tissue development, mixtures of chemicals commonly found in the environment made rat mammary tissue more susceptible to exposures to dietary estrogens after birth, leading to tissue abnormalities that have been associated with mammary tumors [69]. And pre-treatment of young rats with a low dose of radiation resulted in earlier occurrence and increased frequency of mutated mammary tumors after subsequent exposure to a known chemical carcinogen [70].
Gene-environment interactions and epigenetic changes
Several studies have reported an increased risk for developing breast cancer in women with either BRCA1 or BRCA2 mutations following exposure to medical radiation, either through mammography or radiation therapy [71–74]. Another report found that a combination of multiple variants in genes associated with DNA repair mechanisms led to an increase in mammography-associated risk for developing breast cancer [75].
Other studies have reported an interaction between various gene variants associated with breast cancer risk and exposures to environmental exposures [76]. But overall, the relevant literature is mixed, with different single-nucleotide polymorphisms (SNPs) and different environmental toxicants being tested. A comprehensive overview of the field concluded that these studies were too few and underpowered for any clear demonstration of interactions between particular SNPs or clusters of SNPs and environmental factors in affecting breast cancer risk, given that most large epidemiological studies yield, at best, very small effects that are often nonreplicable [45]. Nevertheless, the authors concluded that, ‘Presently, we should consider hereditary variants and environmental factors as multiplicative/additive factors in the prediction of breast cancer risk’ [45].
In addition to genetic polymorphisms influencing the effects of environmental toxicants on inter- and intracellular responses, environmental chemicals, especially EDCs, can alter the regulation of genes involved in cell proliferation, apoptosis signaling pathways, etc. through epigenetic processes [77, 78]. Through mechanisms including altered DNA methylation, modifications of histones and expression of small regulatory RNAs (microRNAs), chemical and radiation exposures can have profound effects on the structure and function of the developing mammary gland [79–82].
For example, Kutanzi and Kovalchuk reported that concurrent treatment of adult ACI rats with exogenous sources of estradiol and radiation resulted in increased mammary gland methylation and acetylation of H3 and H4 histones, and significantly increased induction of MAPK and p38 pathways, known biomarkers for chromosome instability [83]. And in normal MCF-7 human ER+ cell line, addition of the growth promoter, zeranol, led to stimulatory effects on cell growth. These results were driven, at least in part, by down-regulation of the tumor suppressor gene p53, a process that was accompanied by up-regulation of DNA-methyltransferase 1 [84].
Hussain et al. explored the effects of BPA on the expression of HOXC6, a homeobox-containing gene that is associated with mammary cell growth and development and which is overexpressed in many breast cancers. Both in MCF-7 cell lines and in mammary tissue from adult Sprague-Dawley ovariectomized rats, BPA exposure increased histone methylation and acetylation and recruited RNA polymerase II at the HOXC6 promoter, resulting in HOXC6 overexpression [85]. Similarly, Doherty et al. demonstrated in both MCF-7 cells and in mammary glands from neonatally exposed mice that either BPA or diethylstilbestrol (DES) treatment led to a 2–3 fold increase in expression of the breast cancer associated histone methyltransferase, Enhancer of Zeste Homolog 2 (EZH2) mRNA expression and subsequent EZH2 synthesis. These changes were accompanied by increased trimethylation of histone H3, both in vivo and in vitro [86].
Cell-cell interactions and the tissue organization field theory
Rather than modeling cancer development as a result of accumulated DNA mutations, with consequent hallmark changes in cell physiology building on the initial genetic instability [36, 37], the Tissue Organization Field Theory (TOFT) of carcinogenesis [87, 88] is based on a more ecological view of cellular functioning and tissue organization. TOFT begins by recognizing that cell proliferation is the default state for cells, with processes and chemical signals critically regulating the rate of proliferation, and also that cells work in constant interaction with neighboring cells in the various tissues within an organ [87]. Perturbations of the reciprocal signals and disruption of cell-to-cell interactions, specifically between the mesenchyme/stroma and the parenchyma/epithelial compartments of the developing mammary gland, may underlie the development of breast cancer [39].
Much of the work exploring this model has been done examining the effects of prenatal or neonatal exposure to BPA and morphological changes in the stromal and epithelial compartments of the rodent mammary gland [40, 89–92]. For example, Wadia et al. explored the effects of low dose prenatal exposures to BPA on morphological changes in fetal mouse mammary glands using exposure levels that have previously been demonstrated to induce pre-neoplastic and cancerous tumors in adulthood. Neonatal BPA exposures led to changes in gene expression in both the epithelial and stromal compartments of developing mammary glands from gestational day 19 mice. Altered expression in the stromal fraction was found for genes involved in pathways mediating focal adhesion and adipogenesis, while in the epithelial fraction there were changes in expression of genes involved in apoptosis. Resulting morphological changes due to BPA exposure included advanced fat pad development and delayed epithelial lumen formation, effects that are eliminated in the absence of ERα. Together these data led the authors to propose that BPA (and estrogens, more generally) act directly on the stroma where prenatal estrogen receptors (ERα, ERβ, and GPR30) are expressed. In turn, signals from the stroma alter epithelial gene expression and, ultimately, the earliest morphological programming for the developing mammary gland [89].
Timing of exposures
A large body of research demonstrates that the timing of exposures across the lifespan can have an enormous influence on whether, how much, and how an environmental exposure might influence the risk for later development of breast cancer. Mammary cells are more susceptible to the carcinogenic effects of hormones, chemicals and radiation during early stages of development, from the prenatal period through puberty and adolescence, and on until the first full-term pregnancy. Particular concerns have been demonstrated for exposure during prenatal and early childhood periods. Much of this data comes from the use of animal models (reviewed in appropriate sections within this report), but there also are several sources of data that support this claim from the human clinical literature.
For example, daughters of mothers who suffered from preeclampsia during pregnancy, associated with lower levels of maternal estrogens, have decreased risk of developing breast cancer in adulthood [93, 94]. At birth, umbilical cord levels of estriol (E3) and estetrol (E4) – but not estradiol (E2) or estrone (E1) – have been shown to be lowered in neonates delivered from pregnancies associated with preeclampsia [95]. On the other hand, girls who are born with lower birth weight, associated with higher fetal estrogen exposures, have increased risk of later breast cancer diagnosis [96, 97].
And although it is rare to have exposure to exogenous chemicals only during fetal development, between 1938 and 1971 millions of fetuses were exposed to the synthetic estrogen, diethylstilbestrol (DES), when their pregnant mothers were prescribed the drug in order to prevent miscarriages and other complications of pregnancy. DES was banned when daughters of women who took the drug during pregnancy were found to have increased rates of an extremely rare clear-cell vaginal adenocarcinoma. DES exposure was also associated with an increased risk of breast cancer in the mothers [98–100].
In a follow-up study of daughters who were exposed prenatally to DES, a nearly twofold increase in breast cancer risk was observed in women older than age 40. An even greater effect was found for women over the age of 50, although relatively few of the daughters had yet reached that age at the time of the study [101, 102]. Women exposed in utero who had the most severe abnormalities of their vaginal epithelial cells (an indicator of exposures to higher doses of DES) also had a higher risk for developing breast cancer [99]. It now appears that granddaughters of women prescribed DES during pregnancy are also experiencing an elevated incidence of breast cancer [100].
In a case-control prospective study of 9300 women in a pregnancy cohort, stored postpartum maternal blood samples were analyzed for levels of dichlorodiphenyl-trichloroethane (DDT). Daughters were followed for 52 years and breast cancer diagnosis in this cohort was determined. Higher maternal DDT levels were associated with an almost 4-fold increase in occurrence of breast cancer in their daughters by age 52 [103].
A prospective, nested case-control study of 258 women explored their estimated historical DDT levels based on aggregate data from their year of birth as well as blood DDT levels at the time the women gave birth to their first child. Exposure to DDT during childhood and early adolescence (younger than 14 years) was associated with a 5-fold increase in the risk of developing breast cancer before age 50. The younger the women were when the heavy use of DDT was begun in 1945, the greater the risk [104].
Other studies have demonstrated that childhood and adolescence are particularly susceptible ages for exposure to medical radiation and later development of breast cancer. Decades of research have confirmed the link between radiation and breast cancer in women who were irradiated for many different medical conditions, including tuberculosis [105], benign breast disease [106, 107], acute postpartum mastitis [108], enlarged thymus [109, 110], skin hemangiomas [111], scoliosis [112], Hodgkin’s disease [113–116], non-Hodgkin’s lymphoma [117], acne [118], and prophylactic dental care [119]. Evidence from almost all conditions suggests that exposure to ionizing radiation during childhood and adolescence is particularly dangerous with respect to increased risk for breast cancer later in life [73, 120, 121].
Section summary: These framing concepts reveal the complexity of research examining relationships between environmental toxicants and risks for developing breast cancer. Breast cancer does not present with a single biomarker profile; incidence rates differ across ethnic/racial and resource-level groups; concentrations of exposures may make a difference, as do possible mixtures and interactions. And specific timing and duration of exposures, especially when they happen early in development, may cause more detrimental effects than later exposures.
As we move into examining the scientific literature addressing the relationship between various toxicants and breast cancer risk, we offer an interactive model to help situate these data (Fig. 1). While not meant to be fully comprehensive, this model challenges the reader to consider the effects of environmental exposures on disease risk within a complex web-like framework of often interconnected factors, each of which may exert direct, indirect, and interactive effects on cellular processes in mammary tissues [11].
Fig. 1.
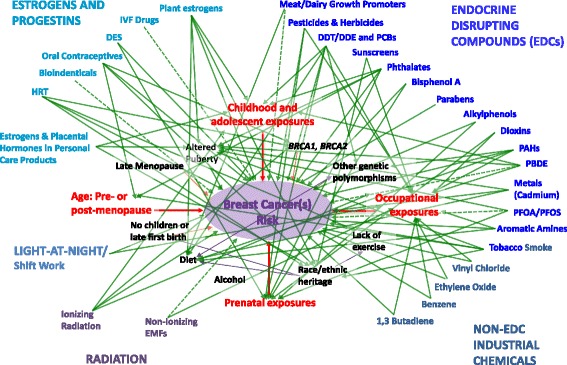
Complexity of factors affecting risk for developing breast cancer. This synopsis of much of the evidence described in this report demonstrates the complexities of the potential connections between exposures to environmental toxicants and development of breast cancer, all embedded in a web-like framework of interconnected factors. Solid arrows indicated connections that have been demonstrated directly between exposures and breast cancer risk, or, as appropriate, mediated through factors described in the framing section of this review. These relationships reflect results of the combined human epidemiological and/or animal studies discussed. Dashed arrows indicate connections between exposures and risk for breast cancer that are more ambiguous, with evidence coming from non-human or -animal studies, but without the in vivo data to support more directly the link. Arrows are not weighted to indicate relative strength of links. Rather the purpose of this model is to demonstrate the complexity of the relationships between environmental factors and breast cancer. (Updated and modified from Gray et al. 2009 [11])
Evidence linking environmental factors and breast cancer
We turn now to the evidence addressing possible connections between exposures to environmental toxicants and risk for developing breast cancer. In exploring the scientific literature, we draw from relevant human, animal, cell-culture, and high throughput studies. Where possible, we address explicitly the complicating themes raised in the framing section above. And where appropriate, we present conflicting data, especially from the epidemiological literatures, that make clear the nuances of methodology and results that complicate these relationships.
Hormones: pharmaceutical and personal care products
For decades, scientists have appreciated the positive relationship between lifetime exposures to estrogen and risk for developing breast cancer [122]. More recently it has become clear that long-term exposures to progesterone can also influence the possible development of breast cancer [123]. These exposures are often clumped under the category of ‘reproductive risk factors’ (e.g., age at menarche, menstruation, first full-term pregnancy, and whether or not children were breastfed) in the development of models and simple evaluative tests for determining breast cancer risk [124, 125].
In addition to variations in exposures to endogenous levels of both estrogens and progesterone, there are several other sources of natural and synthetic steroids, including those found in a number of pharmaceuticals and personal care products. Most of these hormonal agents have been designated as carcinogens by the IARC and the NTP (see Table 1). This section examines the relationships between use of these compounds and possible changes in risk for developing breast cancer.
Table 1.
Carcinogenicity classifications and sources of exposures for hormones in pharmaceuticals and personal care products
| Product | IARC | NTP | Source of exposure |
|---|---|---|---|
| Diethylstilbestrol | 1 | K | Formerly prescribed to pregnant women to sustain viable pregnancies |
| Hormone Replacement Therapy | 1 | Treatment of symptoms experienced in menopause | |
| Conjugated equine estrogens | 2A | ||
| Medroxyprogesterone acetate | |||
| Bioidentical hormones | 1 | ||
| Oral contraceptives | 1 | Contraception | |
| Infertility treatment drugs | Infertility treatment | ||
| Clomiphene citrate | 1 | ||
| Gonadotropins | |||
| Hormones in personal care products | 1 | Use of placental extracts in personal care products, especially products marketed to women of color |
International Agency for Research on Cancer (IARC) classifications: 1 = Carcinogenic to humans, 2A = Probably carcinogenic to humans, 2B = Possibly carcinogenic to humans, 3 = Not classifiable as to its carcinogenicity to humans; U.S. National Toxicology Program (NTP) classifications: K = Known to be a human carcinogen, RA = Reasonably anticipated to be a human carcinogen. Source of exposure list contains most common exposure sources
Diethylstilbestrol
The clearest evidence that a synthetic estrogen can increase risk for breast cancer decades later comes from the tragic experience with diethylstilbestrol (DES). From the 1940s until 1971, doctors prescribed DES for millions of pregnant women to prevent miscarriages and other complications of pregnancy. The drug was banned when daughters of women who took the drug were found to have higher rates of an extremely rare vaginal clear-cell adenosarcoma compared to those who were not exposed to DES in the womb. DES exposure was also associated with an increased risk of breast cancer in the mothers [98, 126, 127].
In a follow-up study of daughters who were exposed prenatally to DES, almost a twofold increase in breast cancer risk was observed in women older than age 40 years (HR = 1.82; 95% CI = 1.04–3.18) [99]. An even greater (three-fold) effect was found for women over the age of 50, although relatively few of the daughters had yet reached that age at the time of the study [101, 102]. Women exposed in utero who had the most severe abnormalities of their vaginal epithelial cells (an indicator of exposures to higher doses of DES) also had a higher risk for developing breast cancer [99].
Studies are just beginning on granddaughters of women prescribed DES during pregnancy, but since these women are only now reaching the ages when breast cancer incidence increases, data sets are too small to reach statistical significance [128]. Relevant rodent models, however, indicate that the F2 generation (granddaughters) of dams exposed to low doses of DES during pregnancy also developed several cancers, including mammary tumors, at rates significantly higher than expected [129].
Studies examining the mechanisms by which DES might be exerting its carcinogenic effects indicate that the compound activates the same subcellular pathways that estradiol does, both by altering cellular metabolism and interaction with DNA [130] and by increasing the rate of breast epithelial cell proliferation [131, 132]. In adult female rats, exposure to DES increased induction of HOTAIR transcription which produces an estrogen-responsive gene silencing protein implicated in the development of breast cancer [133]. DES further dysregulates the expression of estradiol regulated gene expression in adult females, again possibly contributing to an increased risk for breast cancer [134].
Prenatal exposures to DES lead to changes in the adult mammary gland epigenome through alterations in histone methylation, a process that leads to altered gene expression in puberty and adulthood [86, 133, 135]. These epigenetic changes could provide a mechanism for trans-generational effects of DES on breast cancer development [128, 136].
Hormone Replacement Therapy (HRT)
The Women’s Health Initiative (WHI) is a large (n = 16,608 women) randomized case control study designed to explore the benefits and risks of combined estrogen (conjugated equine estrogens) plus progestin (medroxyprogesterone acetate) HRT in post-menopausal women. In 2002, it was halted after a median follow-up of 5.5 years, three and half years before the intended end of the study period, because researchers observed a significant increase in the relative risk of breast cancer (HR = 1.26; 95% CI = 1.00–1.59) in addition to significant increases in the risk of heart disease, stroke and blood clots [137].
Analyses of a second arm of the WHI study clarified that the increased risk of breast cancer in the WHI study occurred in women taking the combined estrogen-progestin formula, but not for those women taking estrogen-only HRT supplements [138, 139] where a decreased risk for developing breast cancer was found (HR = 0.77; 95% CI = 0.62–0.95). It is critical to note that the estrogen-only option can only be offered to women who have previously undergone surgical hysterectomies because estrogen-only treatment leads to a highly significant increased risk for uterine cancer [140]. One difference between the estrogen-only contraceptives and the combined forms is in the type of estrogen in the formulation. Most often the estrogen in the mixed pill is the semisynthetic compound, ethinyl estradiol, while that in the estrogen only pill is a conjugated equine estrogen. The conjugated form is associated with lower rates of epithelial proliferation in post-menopausal breasts, providing one mechanism by which the two types of interventions might have different effects [141].
Longer-term (median of 13 years) follow-up of both arms of the WHI study indicate that for the women in the combined hormone arm, there was a time dependent and significant increase in risk for developing breast cancer (HR = 0.71; 95% CI = 0.47–1.08 at first year of intervention; 1.36 95% CI = 0.94–1.94 during third year of intervention; 1.65; 95% CI = 1.17–2.32 during the fifth year of intervention). Although there was a sharp decrease in risk after the first year of discontinued use of the mixed HRT formulation, for the full 8-year follow-up period after stopping the hormone treatment, HR values were above 1 (HR = 1.32; 95% CI = 1.08–1.61) [142]. The early, short-term finding is consistent with the rapid drop in post-menopausal breast cancer incidence in the US population since 2002, a decrease that has been attributed to the precipitous drop in HRT prescriptions in selected populations of women (white, middle/upper class, postmenopausal, ER+ tumors) following the release of the data from these large studies [143, 144].
For the estrogen-only arm, the decreased risk of breast cancer remained for the early post intervention phase (HR = 0.55; 95% CI = 73–1.87 for the first 3 years post-intervention) although the benefit disappears over the next 5 years (HR = 1.17; 95% CI = .73–1.87) [142].
Since the results of the original WHI were initially published, other large studies have supported its major conclusions. In 2003, Swedish researchers halted a study of HRT in women with a previous history of breast cancer. Originally planned as a 5-year study, the Swedish trial was stopped after 2 years because women taking combined estrogen-progestin HRT had a significantly increased rate of recurrence or new tumors compared to women who received other treatments for menopausal symptoms ((HR = 3.5; 95% CI = 1.5–8.1) [145].
Also in 2003, researchers in the Million Women Study (MWS) in the United Kingdom reported that the current use of all types of post-menopausal HRT significantly increased the risk of breast cancer (RR = 1.66; 95% CI = 1.58–1.75). Again, the risk was greatest among users of estrogen-progestin combination therapy (RR = 2.00; 95% CI = 1,88–2.12) [146].
Other research has confirmed the basic result that use of combined HRT increases risk of breast cancer in post-menopausal women, and that stopping use of the combination pill leads to decreased risk of developing breast cancer. One study in California found that county-wide decreased incidence in breast cancer was highest (22.6%) in counties with the greatest decline in using HRT, intermediate (13.9%) in counties with moderate decreases in HRT use, and smallest (8.8%) in counties with least decline in HRT use [147].
One study examined breast cancer incidence in BRCA1 mutation carriers who had undergone oophorectomy to prevent onset of ovarian cancer. Short-term use (median = 4.27 years) of HRT was not associated with any change in risk of developing breast cancer (OR = 0.80; 95% CI = 0.55–1.16), regardless of HRT formulation (estrogen alone or estrogen + progestin) [148].
Another study examining the possible interactions between use of HRT and race, weight, and breast density found that HRT use increased risk for breast cancer in white (OR = 1.21; 95% CI = 1.14–1.28), Asian (OR = 1.58; 95% CI = 1.18–2.11) and Hispanic (OR = 1.35; 95% CI = 1.09–1.67) women, but not Black women (OR = 0.91; 95% CI = 0.72–1.14). There was no interaction between HRT use and either BMI or breast density [149].
A meta-analysis that included 116,304 breast cancer cases demonstrated that women who engage in high levels of physical activity have a significantly reduced risk of developing breast cancer (SRR = 0.88; 95% CI = 0.85–0.90), with decreases being found in both ER+/PR+ and ER−/PR− cancers. However, women who used HRT had no decrease in breast cancer risk when they engaged in vigorous physical exercise [150].
Examination of cancer histology in women taking combined HRT at the time of diagnosis reveals an increased presentation of breast cancer of lobular origin [151–153], but also of cancers with low proliferation rates (mitotic indices) and favorable prognostic outcome [153, 154].
Bioidentical hormones
Following the results of the major studies implicating HRT as being causally related to postmenopausal breast cancer, many women turned to alternative sources of hormone therapy to treat their menopausal symptoms with hopes of finding safer options. For many women, this meant using ‘bioidentical hormones’ of some sort, hoping to mimic the effects of natural hormones without succumbing to the negative health outcomes associated with traditional HRT [155]. Unfortunately there have been very few studies examining the relationship between taking bioidentical hormones and later development of breast cancer. Perhaps more importantly, and confusing the conversation on this topic, the term ‘bioidentical hormones’ is used in many different ways with potentially different implications for associations with health outcomes [156]. The most conservative definition, adopted by the Endocrine Society, is for compounds that ‘have exactly the same chemical and molecular structure as hormones that are produced in the human body’ [156]. Bioidentical hormones may be synthesized or derived from plant sources.
A few types of bioidentical hormone composites or individual components have been tested and approved by the Food and Drug Administration (FDA). But the increasingly common use of individually compounded bioidentical hormone regimens has not been tested for safety or associated health outcomes and the consistency of prescribing and providing individualized compounded formulae varies enormously [156, 157].
The strongest evidence for a lack of association between use of bioidentical hormones and possible development of breast cancer comes from data examining the use of the natural hormone progesterone, instead of MPA or other synthetic progestins, as part of the HRT regime [158]. Research indicates that increased exposure to natural progesterone did not increase risk for breast cancer and, in some circumstances, might even be protective [159, 160]. In the single large-scale cohort study examining risks for breast cancer in women taking hormone replacement regimens with either natural progesterone or synthetic progestins compounded with estrogens, use of a progesterone-based replacement was associated with no added risk for breast cancer compared with controls (RR = 1.00; 95% CI = 0.83–1.22), while women who took combined HRTs that included synthetic progestins had significantly increased risk for developing the disease (RR = 1.69; 95% CI = 1.50–1.91) [161]. This difference was particularly prevalent in the incidence of ER+ tumors, especially ER+/PR− masses (RR = 2.6; 95% CI = 1.9–3.5) [162].
Less positive news comes from a study comparing the effects of conjugated equine estrogens, the major estrogenic component in traditional combined estrogen-progestin HRT, with natural estradiol in a primate model of postmenopausal breast cancer. In this study, natural estradiol induced greater proliferation of breast epithelial cells than did the conjugated form [141].
Oral contraceptives
Numerous studies have demonstrated an increased risk of breast cancer in women using oral contraceptives. The risk for breast cancer is greatest among current and recent users of oral contraceptives, particularly those who have used them for more than 5 years and initiated use at a young age [163–168]. For example, in a large prospective cohort-study, an increased incidence of breast cancer was found in women who were younger than age 50 at the time of diagnosis and had begun use of oral contraceptives before the age of 20 — as compared with those who started later (HR = 3.26; 95% CI = 1.06–10.01). Women who had begun use before the age of 20 and were older than age 50 at the time of diagnosis showed no increased risk compared to age similar cases who began use later (HR = 0.70; 95% CI = 0.33–1.46). Women in this study took contraceptives for an average of 6 years, although the duration of use varied from 2 ½ to 12 years [169].
Sweeney et al. examined possible effects of oral contraceptive use on later risk for breast cancer in Hispanic and non-Hispanic white women. Statistically, Hispanic women have somewhat lower rates of breast cancer than do white women and they are more likely to have ER− tumors. However, use of oral contraceptives during the previous 5 years led to significant increases in breast cancer incidence in both groups. The effect was magnified for women of both groups when oral contraceptive use continued for more than 20 years (OR = 2.23; 95% CI = 1.17–4.25 for ER− tumors). Mirroring other study evidence, and again for both Hispanic and non-Hispanic white women, significant increases in ER+ tumors were observed [170].
Researchers in the Black Women’s Health Study, a large (over 53,000 women) prospective study of women across the U.S., report that use of oral contraceptives by African American women was associated with a higher risk of receptor negative (ER−, PR−) cancer than women who did not use the pill (IRR = 1.65; 95% CI = 1.19–2.30). The risk for later diagnosis of ER−/PR− breast cancer increased as the duration of contraceptive use was prolonged among women who took the pill and were still using it within the past 5 years (trend p = 0.001). The only significant effect of oral contraceptive use on development of ER+/PR+ cancers in this cohort was for women who had taken the pill for more than 10 years (IRR = 1.45; 95% CI = 1.02–2.07) [171].
Women with BRCA1 or BRCA2 mutations, as well as women with family histories of breast or ovarian cancer, have an increased susceptibility to the risk-inducing effects of oral contraceptive usage [166, 172, 173]. Paternal contribution (as compared to maternal contribution) of the BRCA mutation confers greater risk for women with this genetic variation who also use oral contraceptives (HR = 1.84; 95% CI = 1.46–2.34) [174]. One mechanism by which the interaction between BRCA gene status and use of oral contraceptives may influence breast cancer risk, is by altering the sensitivity and activity of progesterone in breast cancer cells, both by increasing the synthesis of PR in the cells and by enhancing the responsiveness of progesterone-regulated genes [175].
Use of oral contraceptives is associated with an increase in later-stage (type II or greater) breast tumors [176], tumors originating in the lobular tissue [171], as well as with the ER− profile of the disease [171, 177]. Significant associations between use of oral contraceptives and development of the aggressive triple negative (ER−/PR−/Her-2R-) form of the disease was found in a primarily White cohort (OR = 2.5; 95% CI = 1.4–4.3) [178] as well as in a cohort of African American women (OR = 1.78; 95% CI = 1.25–2.53) [179]. Use of oral contraceptives for 10 or more years has also been associated with a diagnosis of comedo DCIS (OR = 1.31; 95% CI = 0.70–2.47) [180], the most aggressive form of DCIS which is sometimes confused with early forms of invasive breast cancer [181].
Post-menopausal women who used oral contraceptives for eight or more years, but who have discontinued use for at least a decade, show no significant increase in breast cancer rates [182, 183].
Two studies have examined the relationship between use of injectable progestin-only contraceptives and breast cancer incidence. Both studies found increases in breast cancer risk that were significant, but rates decreased to normal within a few years after stopping use of the drugs [184, 185].
Infertility treatment drugs
Despite the substantial evidence linking HRT and oral contraceptive use with increased incidence of breast cancer, neither the condition of subfertility nor the use of infertility-treatment (ovulation-stimulation) drugs appears to have a clear link to the disease [186–189]. This is true also when the study involves infertile women who are also BRCA carriers [190]. Where a link has been found, it has been for women who gave birth to more than one infant as a result of their IVF treatment (HR = 1.44; 95% CI = 1.06–1.97) [191] and those who have been treated with high doses of clomiphene citrate.
Two studies found increased risk of breast cancer for women who have been treated for ovarian infertility with drugs including gonadotropins or clomiphene citrate. However, the results were significant only when the incidence of breast cancer was compared with the general population of women, but not with the more appropriate control of women with ovarian infertility who have not been treated with fertility drugs [192, 193]. Two other studies, however, have found statistically significant increases in breast cancer rates in women taking clomiphene citrate compared with rates for infertile women taking no infertility treatment (HR = 1.42; 95% CI = 0.99–2.55) [194]; (OR = 2.7; 95% CI = 1.3–5.7] [195]. A smaller subgroup of women whose infertility was not ovarian in origin and who underwent multiple treatments with high doses of clomiphene citrate, had increased risk of later developing breast cancer compared with women in the general population (OR = 3.0; 95% CI = 1.35–6.67) [188].
Another study complicates the story, however. Within the cohort of women with fertility problems, there was no difference in the rate of breast cancer when general comparisons were made between women who had taken fertility drugs and those who had not. But when age of treatment was factored in, a significant increase in risk for breast cancer was found in women who had begun infertility drug treatments before the age of 24, as compared with infertile women of the same age who had not undergone drug IVF and associated drug treatments (HR = 1.59; 95% CI = .1.05–2.42). Increased risks for breast cancer were not associated with infertility treatment in older women (after aged 40 years) who underwent IVF protocols [196]. These data are consistent with a model in which younger adult breast cells are more sensitive to the perturbations and/or protections resulting from altered exposures to both endogenous and exogenous sources of hormones.
Hormones in personal care products
Placental extracts, probably with high concentrations of progesterone [197] and estrogenic chemicals [198] are sometimes used in cosmetics and hair care products, particularly products marketed to women of color. Addition of hormones and extracts is advertised to promote growth and thickness of hair. However, research indicates that use of these products in infants and children may also be linked to precocious puberty or early sexual maturation [191, 199, 200], a risk factor for later life breast cancer [201]. Scientists have proposed that use of these hormone-altered products might be contributing to the increased incidence of breast cancer, especially among young African American women who use these products more than their white counterparts [202, 203].
Seven of eight extracts from skin and hair products commonly used by African American women had effects on proliferation of MCF-7 cells in culture; four of the seven were estrogenic while three showed antiestrogenic activity [204].
Hormones, especially estrogens, are also regularly added to anti-aging creams [205], because of their effectiveness in raising collagen count, as well as skin hydration. Together, these two factors are thought to decrease wrinkling of the skin [206], but they can also increase women’s total lifetime exposure to estrogen.
Section summary: There is clear evidence that exposure to DES during gestation increases the risk for developing breast cancer in the women who were exposed in utero, and also for their mothers and possibly their daughters. Post-menopausal use of HRT compounded with synthetic estrogens and progestins also increases the likelihood of developing breast cancer although use of estrogen-only HRT has protective effects for those women who have undergone a hysterectomy. Compounding HRT drugs with the natural hormone, progesterone, does not appear to have detrimental effects on breast cancer risk, although use of the natural estrogen, estradiol, may increase breast cell proliferation and consequent risk for developing breast cancer. There is little consistent evidence that use of hormonal drugs in IVF procedures alters risk for breast cancer, although there are numerous methodological issues in these studies. Finally, several personal care products, especially those marketed primarily to communities of color, have estrogenic and progestin additives, increasing lifetime exposures to these hormones.
Endocrine disrupting compounds (EDCs)
Although intentional use of natural and synthetic hormones has been a practice for decades, if not centuries, it is only in the past two decades that scientists have come to recognize that many common products also contain chemicals that are disruptive to the exquisitely sensitive endocrine system [207]. These chemicals, found in products as different as plastics, pesticides, fire retardants, and sunscreen, were added to the manufactured products for reasons not intentionally related to their endocrine-related properties. Nevertheless, many compounds have been shown to fit the Endocrine Society’s definition of an endocrine disrupting compound (EDC), “an exogenous chemical, or mixture of chemicals, that interferes with any aspect of hormone action” [47].
By interfering with the actions of natural hormones, exposures to EDCs have been shown to contribute to the development of a wide variety of disease states [49, 51]. Often these effects are most profound when exposures are low-dose [38] and during early development [48]. This section addresses the growing literature on the connections between several important EDCs and the risk of developing breast cancer, mainly – but not exclusively – from non-human models. Although we mostly treat the chemicals independently, as is true of the research literature, we recognize the importance of exposures to mixtures of EDCs as these substances infuse the products we use, and also the air we breathe, the water we drink, and the surfaces on which we work and play. While most of these EDCs have not been formally evaluated for carcinogenicity, Table 2 demonstrates the almost ubiquitous presence of these chemicals in our environment.
Table 2.
Carcinogenicity classifications and sources of exposures for endocrine-disrupting compounds (EDCs)
| EDC | IARC | NTP | Sources of exposure |
|---|---|---|---|
| Bisphenol A | Polycarbonate plastic, epoxy resins linked food cans, dental sealants, thermal receipts | ||
| Phthalates | Fragrance ingredients in personal care and cleaning products, plastics. Also pharmaceuticals, building materials, insecticides and food packaging/food processing. | ||
| di(2-ethylhexyl)phthalate (DEHP) | 2B | RA | |
| di-n-butylphthalate (DNP/DBP) | |||
| monoethyl phthalate (MEHP) | |||
| diethyl phthalate (DEP) | |||
| butyl benzyl phthalate, (BBP) | 3 | ||
| di-n-octyl phthalate, (DOP) | |||
| di-i-butyl phthalate (DiBP) | |||
| monomethyl phthalate | |||
| Parabens | Antimicrobial preservatives in food, personal care products, soaps and detergents, and pharmaceuticals | ||
| methyl-paraben | |||
| propyl-parabens | |||
| butyl-parabens | |||
| Alkylpenols | Detergents and cleaning products, antioxidants in plastic and rubber products | ||
| 4-nonylphenol (4-NP) | |||
| 4-octylphenol (4-OP) | |||
| Triclosan & Triclocarban | Antimicrobials in liquid hand soap, other personal care products and household items | ||
| EDCs found in sunscreens | UV filters | ||
| 3-(4-methylbenzylidene)-camphor (4-MBC) | |||
| octyl-methoxycinnamate | |||
| octyl-dimethyl-PABA (OD-PABA) | |||
| benzophenone-3 (Bp-3) | |||
| homosalate (HMS) | |||
| Perfluorooctanoic Acid (PFOA) & Perfluorooctanoic Sulfate (PFOS) | 2B | Stain resistant coatings, non-stick coatings, commercial products including firefighting foams. | |
| Polycyclic Aromatic Hydrocarbons (PAHs) | RA | Byproducts of combustion resulting from fossil fuel production, diesel exhaust, grilled meats, cigarettes. | |
| Pyrene | |||
| benz[a]anthracene | 2B | RA | |
| benzo[a]pyrene | 1 | RA | |
| Triazine herbicides | Weed control for corn and sorghum crops. | ||
| Atrazine | |||
| Simazine | |||
| Cyanazine | |||
| Other Pesticides & Herbicides | |||
| Heptachlor | 2B | Insecticide, now banned | |
| Dieldrin and Aldrin | 2A | Insecticide for corn and cotton, now banned | |
| Chlordane | 2B | Home termites, general crop pesticide | |
| Malathion | 2A | Residential, recreational, crop pesticide | |
| 2,4-D | 2B | Broadleaf weed herbicide | |
| 2,4,5-trichlorophenoxypropionic acid (2,4,5-TP) | Woody plant and broadleaf weed herbicide, now banned | ||
| Persistent organochlorines | |||
| Dichloro-diphenyl-trichloroethane (DDT)/DDE | 2A | RA | Insecticide, now banned |
| PCBs | 1 | RA | Electrical insulation, fluid coolants, plasticizer in paints, dyes & inks |
| Dioxins: 2,3,7,8-tetra chlorodibenzo-para-dioxin (TCDD) | 1 | Byproduct of burning of chlorine-based chemicals | |
| Polybrominated Diphenyl Ether (PBDEs) | Flame retardants, previously used in furniture and electronics; most have been banned or voluntarily phased out | ||
| Aromatic amines | |||
| o-toluidine | 1 | K | Hair dyes |
| 4-aminobiphenyl (ABP) | 1 | K | Azo dyes in textiles |
| p-phenylenediamine | Hair dyes | ||
| 2-amino-phenylimidazo[4,5-b]pyridine (PhIP) | Cooked meats | ||
| heterocyclic aromatic amines | Hair dyes | ||
| Metals | Naturally occurring elements; contaminants in naturally derived colorants, clays, and other metals, found in cosmetics, toys, and other products. | ||
| Copper | |||
| Cobalt | PO | RA | |
| Nickel | PO | ||
| Lead | 2B | RA | |
| Mercury | |||
| Methylmercury | 2B | ||
| Tin | |||
| Cadmium | 1 | K | |
| Zinc | |||
| Iron | 1 | ||
International Agency for Research on Cancer (IARC) classifications: 1 = Carcinogenic to humans, 2A = Probably carcinogenic to humans, 2B = Possibly carcinogenic to humans, 3 = Not classifiable as to its carcinogenicity to humans; U.S. National Toxicology Program (NTP) classifications: K = Known to be a human carcinogen, RA = Reasonably anticipated to be a human carcinogen. Source of exposure list contains most common exposure sources
Bisphenol A (BPA)
The ubiquitous synthetic chemical bisphenol A (BPA) is the main component used in the manufacturing of polycarbonate plastic and is found in many common household products. It is also found in dental sealants, thermal receipts, food packaging, and epoxy resins lining food cans. Significant levels of BPA have been measured in ambient air [208], house dust [209], river and drinking water [210].
BPA is an unstable, lipophilic compound that can leach into food products, especially when heated [211], and a major source of exposure to BPA is thought to be through food products contaminated with the chemical [212, 213]. Two studies have explored the effects of increased ingestion of food and drink packaged in materials containing BPA. Both found rapid increases in BPA levels in urine and/or blood samples taken from subjects who intentionally increased their intake of common foods and drinks packaged in BPA-containing products [214, 215]. Another study took the opposite approach and demonstrated that just a 3-day period of limiting intake of packaged foods decreased the concentrations of BPA found in urine by an average 65% [216].
Samples taken from fasting people indicates that sources other than foods may also be responsible for the pervasive exposure to BPA, as levels of the chemical did not decrease as rapidly as would have been predicted were food the only source of contamination [217]. Of growing concern are the high levels of BPA that are transferred to our skin and then rapidly absorbed by holding BPA-containing thermal receipts [218].
Clearance of BPA from the body is quite rapid, with its urinary half-life on the order of hours to days [217]. Despite its rapid clearance rate, BPA was found in 93% of about 2500 urine samples from a broad national sample of adults through the NHANES study [219]. BPA has been found in the blood and urine of pregnant women [220–222], and in breast milk soon after women gave birth [223, 224]. BPA has also been found in blood samples from developing fetuses and the surrounding amniotic fluid [225]; in placental tissue and umbilical cord blood at birth [226, 227]; and in the urine of premature infants housed in neonatal ICUs [228].
Many studies using both rat and mouse models have demonstrated that even brief exposures to environmentally relevant doses of BPA during gestation or around the time of birth lead to changes in mammary tissue structure predictive of later development of tumors [90, 229, 230]. Early exposure to BPA led to abnormalities in mammary tissue development that were observable during gestation and were maintained into adulthood [92, 231, 232]. Many of these changes are similar to those observed after prenatal exposure to DES [132]. Prenatal exposure of rats to BPA resulted in increases in the number of pre-cancerous lesions and in situ carcinomas [233, 234], as well as an increased number of mammary tumors following adult exposures to sub-threshold doses of known carcinogens [235, 236] or without the addition of the additional carcinogen [234].
Prenatal exposure to BPA changes the gene transcription in both the epithelial and stromal compartments of the mouse fetal gland, through mechanisms that are mediated through both ER-dependent and ER-independent pathways [237, 238]. Both BPA and DES exposures alter the expression of several genes involved in extracellular matrix formation, as well as adipogenesis and lumen formation [237]. BPA acts on estrogen-independent pathways to alter the epithelial-mesenchymal transition (EMT) via down-regulation of FOXA1, a key regulator of hormone responses in breast cancer cells [238]. These data suggest that during gestation, BPA acts on stromal cells to alter the collagen fiber content and expression of several proteins including receptors mediating signaling pathways, which then alter epithelial gene expression and cell proliferation [237, 239].
Neonatal exposure of mice to BPA increased sensitivity to estradiol-mediated development of mammary gland structures at puberty [240] as well as increased synthesis of the progesterone receptor and activation of progesterone-regulated mammary-cell proliferation [132].
Changes in mammary development comparable to those observed in rodent models were also observed when female rhesus monkeys were exposed to environmentally relevant doses of BPA during gestation [241].
Some of the long-term effects of neonatal exposures to BPA may be dose dependent, with low- and high-dose exposures resulting in different timing and profiles of changes in gene expression in cells of the mammary gland. In one study, low-dose exposures had the most profound effect on rat mammary glands during the period just prior to the animals’ reaching reproductive maturity, while higher doses had more delayed effects, altering gene expression in mammary tissues from mature adults [55]. Prenatal exposure to low doses of BPA altered mammary gland development in adult rats, while higher doses did not [56]. In a study of chronic exposure of adult mice to different concentrations of BPA, only low doses decreased the latency of tumor appearance and increased the number of mammary tumors as well as their rate of metastasis. All doses enhanced the rate of mammary cell proliferation, but only relatively higher doses counteracted this increased proliferation with parallel increases in programmed cell death [242]. And in an evaluation of prenatal exposures to BPA in male rats, non-linear dose-response effects of BPA were found for development of mammary gland structures [243].
In addition to physical abnormalities in the developing mammary tissue of rodents treated perinatally with low levels of BPA, there are also functional deficits. Female rats exposed to BPA during gestation and suckling had physical abnormalities in their adult mammary tissue as well as decreases in yield and altered protein content of their own milk when, as new mothers, they were feeding their pups. Observed differences following BPA exposure were similar to those found in rats that had been similarly exposed to DES, a known breast carcinogen [244].
Studies using cultures of human breast cancer cells demonstrate that BPA acts, in part, through the same cellular response pathways as the natural estrogen estradiol [245, 246]. BPA binds weakly to the intracellular ER, and also affects cellular functions through interactions with the membrane ER (mER) [247, 248]. But BPA also exerts disruptive effects on cell processes, including changes in activation of signal transduction pathways in ER− cell lines [238, 249]. Beyond binding to ER, BPA binds to the orphan nuclear receptor estrogen-related receptor gamma (ERRγ), a protein to which estradiol does not bind [250–253]. The nuclear receptor family is involved in a wide scope of biological processes, from embryonic development and differentiation through normal maintenance of homeostatic systems to the dysregulation of these processes involved in the development of cancers [254]. BPA also binds to both the androgen receptor (AR) and the thyroid hormone receptor (TR) [255–257], altering activities of those hormone-regulated systems.
Prenatal exposure of mice to BPA also resulted in dysregulation of inflammatory cytokines in adult mammary tissue, a process that may lead to altered cell growth through inhibition of immune responses that commonly target developing cancer cells [258].
Exposure of normal and cancerous human breast cells to low levels of BPA led to altered expression of hundreds of genes including many involved in hormone-receptor-mediated processes, cell proliferation and apoptosis, and carcinogenesis [259–261]. In the presence of BPA, cells derived from the non-cancerous breast of women diagnosed with breast cancer had a gene-response profile associated with the development of highly aggressive tumors [262].
Effects of BPA on mammary tissue development may also be manifested via epigenetic mechanisms, leading to changes in gene regulation across the lifetime. Prenatal exposures of rats to low levels of BPA altered the epigenome in mammary tissue with different profiles being observed at weaning and post-puberty [135]. Exposures to either BPA or DES lead to similar changes in the adult mammary gland epigenome through alterations in histone methylation and gene silencing, processes that lead to altered gene expression in puberty and adulthood [86, 133–135].
BPA reduces the efficacy of common chemotherapy agents (cisplatin, doxirubicin and vinblastin) in their blocking the proliferation of human breast cancer cells when tested in vitro [263, 264].
Phthalates
Phthalates are a group of endocrine-disrupting chemicals commonly used to render plastics soft and flexible. They are found in a wide variety of common products including plastics (e.g., children’s toys), cosmetics, pharmaceuticals, baby care products, building materials, modeling clay, automobiles, cleaning materials and insecticides [265]. Phthalates are readily absorbed through the skin [266] and can also enter the body through inhalation or medical injection procedures [267]. Other major sources of at least one phthalate, di(2-ethylhexyl)phthalate (DEHP), are food packaging [268, 269] and fast food [270]. A dietary intervention study has demonstrated that just a 3-day period of limiting intake of packaged foods decreased by half the concentrations of DEHP found in urine [216]. Another dietary intervention in which study participants followed a 5-day monastic lifestyle, including a vegetarian diet, led to a significant decrease in urinary phthalate levels [271]. Significant levels of DEHP and another phthalate used in food packaging, di-n-butylphthalate (DNP), were found in cooked foods, both before and after packaging, that were served to children through school meal programs [272]. Many wines and liquors, as well as spices, are contaminated with phthalates resulting from leakage of the chemicals from storage containers [273, 274].
Phthalates have been found in indoor air and dust [275] and in human urine and blood samples from children, adolescents, and adults [216, 276–278], as well as in amniotic fluid from pregnant women [279]. Phthalates have also been detected in human breast milk and urine [280, 281]. Phthalates cross the human placenta, exposing fetuses to the hazards associated with exposure to an important class of EDCs during this critical period of development [282]. Young infants are also exposed to high levels of phthalates, with measurable levels of seven different phthalates being found in infants born between 2000 and 2005 [283].
A 2012 study examined whether or not there is a relationship between urinary levels of nine different phthalates and incidence of breast cancer. In this study, urinary phthalate metabolites were detected in 82% of the women, whether or not they had been diagnosed with breast cancer. Elevated levels of monoethyl phthalate (MEP), a urinary metabolite of the parent compound diethyl phthalate (DEP; often used in fragrance), was associated with increased risk of breast cancer (OR = 2.20; 95% CI = 1.33–3.63). This association was highest in premenopausal women (OR = 4.13; 95% CI = 1.60–10.70). Metabolites of two other common phthalates (butyl benzyl phthalate, BBP, and di-n-octyl phthalate, DOP) were negatively associated with breast cancer risk in this study (BBP: OR = 0.46; 95% CI = 0.27–0.79 and DOP: OR = 0.44; 95% CI = 0.24–0.80) [284]. Higher levels of urinary mono(2-ethylhexyl)phthalate (MEHP), a marker of DEP body burden, have also been associated with increased pregnancy loss in a study of Danish women [285].
Phthalates are considered to be endocrine disruptors because of their complex effects on several hormonal systems including the estrogen and androgen hormone systems. Some phthalates, including BBP and DBP, act as weak estrogens in cell culture systems. They can bind to estrogen receptors (ER), induce estrogen-appropriate cellular responses and act additively with estradiol in altering these systems [286, 287]. DBP, di-i-butyl phthalate (DiBP) and BBP also bind weakly to the androgen receptor (AR), disrupting the cellular actions ordinarily initiated by the androgens [288, 289]. In breast cancer cell lines, BBP promotes cancer stem cell growth through activation of the aryl hydrocarbon receptor (AhR) [290]. Phthalates can also induce proliferation, malignant invasion, and tumor formation in breast cancer cell lines that are receptor negative, indicating that at least some effects of these compounds are independent of their direct estrogenic or androgenic effects [291, 292].
The endocrine-disrupting properties of this class of chemicals have been well established in the offspring of mother rats who had been treated with phthalates while pregnant. Phthalates disrupt the development and functioning of male and female reproductive systems by interfering with the production of testosterone and estradiol, respectively [293, 294]. Abnormalities in male offspring exposed prenatally included nipple retention, shortened ano-genital distance and increased cryptorchidism [295, 296]. Exposure of human mothers to phthalates, as measured by analysis of their urine samples, has also been associated with shortened ano-genital distances in their newborn sons — a measure of feminization of external genitalia [297, 298].
A case-control study examined phthalate levels in apparently healthy girls who went through thelarche (breast development) before the age of 8, as compared with girls who underwent precocious puberty because of abnormalities in their neuroendocrine systems and with girls who were progressing through puberty at normal ages. Increased levels of monomethyl phthalate (MMP) were associated with early thelarche group, but not either of the comparison groups [299]. Early breast development in otherwise healthy girls is associated with an increased risk for breast cancer [300].
Exposure of very young rats to BBP resulted in increased cellular proliferation in the terminal end buds of mammary tissue. BBP-induced changes in mammary cell gene expression profile were consistent with abnormalities in cellular differentiation and cell-cell communication [301]. Similar structural irregularities were observed in post-natal development of mammary tissues when rats were exposed to the BBP only in utero when their mothers were fed low levels of the compound during the second half of their pregnancies [302].
DEHP has been shown to alter cellular mechanisms at a number of different levels, including inducing damage to DNA leading to altered rates of mitosis and apoptosis; increases in cell proliferation, tumor cell mobility and invasiveness; and decreased intercellular communication at gap junctions. DEHP also enhances the transition of epithelial cells to mesenchymal cells, thus gaining both migratory and invasive potentials [303]. Exposure of normal human breast epithelial cells to DBP resulted in changes in gene expression in pathways related to a number of systems, including immune responses, cell cycle regulation and antioxidant status of the cell [304].
BBP, DBP and DEHP all significantly increased cell proliferation in MCF-7 breast cancer cells. In addition, these three phthalates inhibited the anti-tumor action of tamoxifen in MCF-7 breast cancer cells [305]. BBP also decreased the efficacy of the chemotherapeutic agents, doxorubicin and cyclophosphamide [306].
Parabens
Parabens are a group of compounds widely used as antimicrobial preservatives in food, pharmaceutical and cosmetics products. Parabens are absorbed through intact skin and from the gastrointestinal tract and blood. Parabens have been found in almost all urine samples examined from a demographically diverse sample of U.S. adults through the National Health and Nutrition Examination Survey (NHANES) study. Adolescents and adult females had higher levels of urinary methyl paraben and propyl paraben than did similarly aged males [307]. Parabens are also found in amniotic samples during the second trimester of pregnancy [308].
Measurable concentrations of six different parabens have been identified in biopsy samples from breast tumors [309]. The particular parabens were found in relative concentrations that closely parallel their use in the synthesis of cosmetic products [310]. Higher levels of n-propylparaben were found in the axilla quadrant of the breast [311], the region in which the highest proportion of breast tumors are found, although concentrations were not related to the actual location of tumors in breasts of individual women. Several investigators have noted the importance of studying the effects of mixtures of parabens, in concentration profiles that are relevant to natural exposures to the compounds, to understand the complex effects of this class of chemicals on increased risk for developing breast cancer [312–314].
Parabens are weak estrogen mimickers, with the potency of the agonistic response being related to the alkyl side group structure [315, 316]. They can bind to both ERα and ERβ, with higher affinity to the ERβ site [316], and they increase the expression of several estrogen-responsive genes involved in cell growth and proliferation as well as inhibition of apoptosis [317–320]. Adding paraben mixtures at concentrations and combinations measured in breast biopsy tissue led to increases in MCF-7 growth and proliferation [321].
Methyl-, propyl- and butyl-parabens all stimulated proliferation in ER+ human breast cancer (MCF-7) cells, as well as in non-malignant human breast epithelial (MCF-10A) cells. The parabens increased estrogen secretion in the MCF-7 cells, but decreased it in the MCF-10A cells [321]. Follow-up work demonstrated that the proliferative effect of parabens on MCF-7 cells was independent of direct effects on either cell cycle or apoptosis gene expression. On the other hand, in the MCF-10A cells, the parabens mimicked estradiol in altering expression of genes involved in both cell cycle progression and apoptosis [322].
When added together to cultures of ERα- and HER2-positive human BT-474 breast cancer cells, butylparaben and heregulin, a natural HER ligand, led to a synergistic increase in the oncogene Myc mRNA expression and cell activity. These data indicate ligands for the two receptors can engage in crosstalk in breast cancer cells, increasing the effects of exposures to environmental parabens [323]. At concentrations lower than those found in breast cancer samples, parabens also exerted inverse antagonistic effects, thereby mimicking the effects of estrogenic stimulation, at the membrane ERRY [324].
Seventeen days treatment of nonmalignant human breast (MCF-10A) cells with methyl-, propyl-, or butyl-parabens led to induction of a transformed phenotype linked to the process of breast cell carcinogenesis [325]. Longer-term (>20 weeks) treatment of MCF-7 cells with the same parabens at the concentration that leads to maximal increase in cell proliferation enhanced migratory and invasive responses [326].
In breast epithelial cells derived from women at high risk for developing breast cancer, methylparaben countered the apoptotic effect of tamoxifen, a major adjuvant treatment of breast cancer [318].
Alkylphenols
Alkylphenols are industrial chemicals used in the production of detergents and other cleaning products, and as antioxidants in products made from plastics and rubber. They are also found in personal care products, especially hair products, and as an active component in many spermicides. In the Silent Spring Institute study of household contaminants, alkylphenols—especially 4-nonylphenol (4-NP) and its breakdown products—were found in all samples of house air and 80% of house dust samples [202]. Substantial concentrations of these chemicals have also been found in wastewater associated with domestic greywater and sewers, urban wastewater and municipal landfills [327–330].
NHANES data examining chemical levels in urine of American adults found 4-NP in 51% of samples evaluated [331] and 4-octylphenol (4-OP) in 57.4% of samples [219]. Similar results were found in serum samples of nursing Swedish women 3 weeks after they had given birth [332], and significant levels of both 4-NP and 4-OP were found in breast milk samples from Taiwanese women [333].
Alkylphenols, including 4-NP, have been shown to mimic the actions of estradiol, mediating their effects through the cellular estrogen receptor. They also bind to the cell membrane ER and mimic cellular signaling responses usually controlled by estradiol [334]. In a study examining the effects of 4-NP in human breast tumor cells (MCF-7) in vitro, changes in gene expression were observed in several genes involved in cell proliferation, DNA transcription and cell signaling—all systems that are disrupted in tumor formation [335–337].
Prenatal exposure of rats to 4-NP caused altered development of the mammary gland as well as changes in steroid-receptor populations in several reproductive tissues [338]. Treatment of mice with 4-NP led to an increased synthesis of estriol, a weak natural estrogen, by the livers of the treated animals. When compared with mice treated with equivalent amounts of estradiol, the mice exposed to 4-NP had an increased risk of mammary cancer [339].
Triclsoan and triclocarban
Triclosan and triclocarban are antimicrobial agents that have been used broadly in a wide range of personal care, household and industrial products over the past 40 years [340]. The chemical structure of triclosan has similarities to both thyroid hormone (T4) as well as several known endocrine disruptors including PCBs, DES and bisphenol A, while triclocarban has similar chemical properties to several pesticides and pharmaceuticals [341]. Both are found in freshwater samples, especially in lakes and downstream from wastewater treatment plants, in concentrations known to be harmful to wildlife [342–344].
In a study of adult American urine samples as part of the CDC NHANES study protocol, 75% of samples were found to have significant levels of triclosan and its metabolites. Higher levels were found in younger and more affluent adults [34]. A 10-year trend analysis of NHANES urinary triclosan levels found a small decrease in levels in the 6 years since they peaked in 2006. A parallel NHANES study examining chemical levels in pregnant women found measureable levels of triclosan in 87% of urine samples examined [345]. In a smaller study of American adult samples, triclocarban and its metabolites were detected in one third of urine and one half of serum samples that were tested [346]. Human autopsy analysis reveals that triclosan bioaccumulates in liver and adipose tissue, but not brain, the three tissues examined [347].
Although there has been very little work examining the direct effects of either triclosan or triclocarban on mammary system development or risk for developing breast cancer [348], considerable research demonstrates that these two compounds exert effects on hormonal systems in ways similar to established mechanisms for perturbing normal breast development and health. Depending on the concentration of chemical tested and the model system used, triclosan and triclocarban exert a complex combination of estrogenic and antiestrogenic, and androgenic and antiandrogenic effects, all mediated at least in part through interactions with the estrogen receptor (ER) and androgen receptor (AR) of target cells [340–342, 350, 351].
At even a very low dose, triclosan was estrogen-like in enhancing proliferation rates of cultured human breast cancer cells (MCF-7 BOS line), yet in combination with low doses of estradiol, triclosan was antiestrogenic in suppressing the full estradiol-induced increase in cell growth and proliferation. At higher, but still environmentally relevant concentrations, triclosan decreased the viability of the cells [351]. Also in MCF-7 cells, triclosan significantly enhanced both cyclin D1 and D2 activity and increased cell proliferation. These effects were blocked by concurrent treatment with ICI-182,780, a specific ER antagonist [352].
In addition to its effects exerted through the steroid receptor systems, triclosan has been shown to alter levels of thyroid hormone in pubertal rats [353, 354]. Treatment of mother rats with triclosan during the period of lactation led to a sustained 3-week decrease in thyroid hormone in the dams. However, pups, only had suppressed T4 levels for the first few days of suckling, with normal levels being recorded later in the period despite continued exposure to maternally ingested triclosan [355].
Hormonally active chemicals found in sunscreens (UV filters)
Concern about exposure to ultraviolet (UV) radiation from the sun and the risk of skin cancer has led to widespread use of sunscreens. Many sunscreens contain chemicals that are not only estrogenic but also lipophilic. Studies show these chemicals are accumulating in wildlife [356], and are found in human urine and breast milk samples [357, 358]. NHANES data indicate that over 96% of American adults have detectable levels of benzophenone-3 (Bp-3) in their urine [219], and that urinary levels have increased in the years between 2006 and 2012. Levels were higher in women and non-Hispanic whites than in other groups [351]. A recent occupational study of firefighters found that their Bp-3 levels were five times those reported in the NHANES studies [359].
Of six common sunscreen chemicals, five of them exerted significant estrogenic activity as measured by increased proliferation rates of human breast cancer cells (MCF-7 cells) grown in vitro. These chemicals were 3-(4-methylbenzylidene)-camphor (4-MBC), octyl-methoxycinnamate (OMC), octyl-dimethyl-PABA (OD-PABA), benzophenone-3 (Bp-3) and homosalate (HMS) [360, 361]. The results for 4-MBC have been replicated in another laboratory [362].
In a common yeast assay that measures the strength of a compound’s estrogenic response, mixtures of low concentrations (below the ‘no observed effect concentrations’ or NOEC) of chemicals similar to that found in sunscreens demonstrated additive synergistic effects. Other studies indicate that in addition to acting like estrogen in many cellular pathways, compounds found in sunscreens can also antagonize the effects of natural estradiol in other pathways [360].
Application of OMC to the skin of the rats enhanced the penetration of the endocrine-disrupting herbicide 2,4-D [363].
Perfluorooctanoic acid (PFOA) and Perfluorooctanoic sulfate (PFOS)
Perfluorooactanoic acid (PFOA) and perfluorooctanoic sulfate (PFOS) are used extensively in commercial applications for their chemical properties of being highly stable and having low surface tension. PFOA is found in compounds such as Teflon® and Gore-tex® as well as in other products including carpet and furniture protectants. PFOS is the main ingredient in Scotchguard® and other products aimed as treatments to provide resistance to soil or stains, especially in textiles.
PFOA and PFOS are ubiquitous, with measurable levels found in wildlife across the planet [364]. These chemicals are found in serum samples from over 95% of the U.S. adults tested in a NHANES study, although levels of PFOS have been decreasing over the past decade as the chemical has been phased out of use in the U.S. [365]. Another study found PFOA and PFOS in blood serum samples taken from adults from nine countries representing four continents [366]. In a study of umbilical cord blood samples from newborns in Baltimore, 100% of the samples had measurable levels of PFOA and PFOS [367]. Higher levels of the chemicals in cord blood were associated with both lower birth weight and smaller size, indicating an effect of PFOA on prenatal development [368]. There are strong correlations between maternal serum and amniotic fluid levels of PFOA. PFOS was also detected in the amniotic fluid, but not until maternal levels were relatively high [369].
Prenatal exposures to PFOA and PFOS have been associated with lower weights at birth, but higher weights at age 20 months in girls participating in the Avon Longitudinal Study of Parents and Children [370]. Follow-up of these same girls when they were 15 years old indicated that these prenatal exposures were associated with increases in serum testosterone levels in teens [371]. Testosterone and other androgens inhibit normal mammary development during adolescence [372].
In southeastern Ohio adolescents exposed to the perfluorinated chemicals, higher levels in blood serum were associated with delayed onset of menstruation in girls [373]. Other studies of Ohio girls demonstrated that exposures to higher levels of PFOA were associated with both later menarche [374] and later breast development [375]. While earlier breast development is a known risk factor for breast cancer, these data support a potential endocrine-disrupting effect of PFOA, which may lead to other health effects later in life. For example, higher levels of serum PFOA (or PFOS) are associated with longer delays in becoming pregnant in women trying to conceive [376].
Higher blood serum levels of PFOA and PFOS, as well as other perfluorinated compounds, were associated with an increased incidence of breast cancer in a study of Inuit women in Greenland. Levels of polychlorinated biphenyls (PCBs) were also elevated in the women who had been diagnosed with breast cancer [377].
In a series of studies examining the effects of gestational or neonatal exposures of mice to PFOA, abnormalities in the formation of mammary tissue were found in the dams during lactation as well as in the pups when they matured. Low-dose exposures to PFOA during pregnancy led to impaired differentiation during lactation, a process that is necessary for normal production and release of milk. In the female pups, mammary glands showed stunted development of epithelial branches before the animals had even been weaned [378]. In a follow-up study that used a cross-fostering design, pups exposed either in utero or in early postnatal life had enduring abnormalities in the development of their mammary tissues, and these abnormalities remained at least through the time of puberty, the latest time evaluated [379]. In a third study, gestational exposures to PFOA were shown to alter mammary development over three generations. In another group of mice, chronic exposures to PFOA in drinking water at levels similar to what is found in some contaminated human water supplies, led to similar negative developmental outcomes in the mammary tissues of the developing pups [378].
The complexity of PFOA’s effects is underscored by a study examining low-dose exposures of different mouse strains to the chemical in the period between weaning and puberty. In one strain (Balb/c), exposures to the chemical led to deficits in normal mammary development through puberty, while in the other strain (C57/BL6), low doses of PFOA exposure enhanced mammary development but higher doses were inhibitory [380]. It is not yet known what factors underlie the strain and dose differences.
On the other hand, when either CD-1 or C57/BL6 mice were exposed to low doses of PFOA prenatally, delays in mammary development were observed in both strains even though there were no observed differences in either ovarian hormone levels or time of onset of puberty. Importantly, these results indicate that the mammary gland is more sensitive to prenatal perturbations by PFOA than are other measures of pubertal status [381].
In hormone-dependent T47D human breast cancer cells, neither PFOA nor PFOS, by themselves, affected cell proliferation. However, in the presence of estradiol, both chemicals did enhance the effects of estradiol on cell growth as well as the expression of several estrogen-responsive genes and ERK1/2 activation [382].
Polycyclic aromatic hydrocarbons (PAHs)
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous byproducts of combustion, which enter the body from sources as varied as coal and coke burners, diesel-fueled engines, grilled meats and cigarettes. PAH residues are often associated with suspended particulate matter in the air, so inhalation is a major means of PAH exposure [383]. In a Silent Spring Institute study of environmental contaminants in house dust, three PAHs (pyrene, benz[a]anthracene and benz[a]pyrene) were found in more than three-quarters of the homes tested [209]. Although they are still found extensively in suspended particulate matter, federally imposed standards on vehicular emissions have led to a significant decrease in PAH release by vehicles compared to their highest levels in the 1970s [384].
Like many other environmental chemicals that are associated with breast cancer risk, PAHs are lipophilic and are stored in the fat tissue of the breast [385]. PAHs have been shown to increase risk for breast cancer through a variety of mechanisms. The most common PAHs are weakly estrogenic [386]. However, the major receptor-directed pathway is through interaction with a protein called the aryl hydrocarbon receptor (AhR), initiating a series of cell changes that lead to altered cell signaling and ultimately to increases in DNA mutations [387, 388]. Although it is currently unclear what the naturally occurring ligand for the AhR is, evidence suggests that the AhR system is important in regulating responses to cellular stress that can lead to disruption of normal cell functioning [389]. At least some of the effects of PAHs are mediated through complex interactions between the AhR-regulated and estrogen-receptor-regulated pathways [390]. PAHs can also be directly genotoxic, interacting directly with the genome and causing damage to DNA [391].
Several epidemiological studies have implicated PAH exposure in increased risk for breast cancer. For example, using traffic exposures estimates of one of the most potent PAHs (benzo[a]pyrene) as a proxy for all traffic related PAHs, researchers compared breast cancer incidence in women exposed to the top 5% of traffic exposure to those with below median exposure levels. Higher exposures to traffic-generated PAHs were associated with an increased incidence of breast cancer in women who ate low levels of fruits and vegetables (OR = 1.46; 95% CI = 0.89–2.40). A significant association was not found for women who ate high levels of these foods. The association with higher incidence of breast cancer was found only for ER−/PR− tumors [392].
In a case-control study, burning synthetic logs, but not wood-only logs, in woodstoves or fireplaces was also associated with an increased risk of breast cancer (OR = 1.42; 95% CI = 1.11–1.84). The association was stronger in women whose exposure was after age 20 and for at least 7 years duration of use. Women who burned synthetic logs and developed breast cancer were more likely to have at least two genetic variants in genes that regulate glutathione-S-transferases, enzymes which are important as cell detoxifiers (OR = 1.71; 95% CI = 1.09–2.69) [393].
One of the studies from the Long Island Breast Cancer Study Project found that women with the highest level of PAH-DNA adducts had a 50% increased risk of breast cancer [394]. More specifically, when PAHs interact with DNA and form an adduct, the result is the loss of one of the purine bases which, when not corrected, leads to gene mutations. These unstable PAH-adducts have been linked to the development of cancer [395].
In an earlier report, researchers explored the presence of PAH-DNA adducts in breast samples taken from women diagnosed with cancer as compared with those diagnosed with benign breast disease. Cancerous samples were twice as likely to have PAH-DNA adducts as were benign samples [396]. Follow-up work indicates that those women who had higher levels of PAH-DNA adducts may not necessarily have had higher exposures to PAHs, but instead were more sensitive to the exposures to PAHs because they had particular genetic profiles that encouraged the deficits in DNA repair [397]. Other studies support the existence of different genetic profiles in women who have increased numbers of PAH-DNA adducts, including polymorphisms in genes involved in cell metabolism, tumor-suppressor mechanisms and DNA repair [397, 398]. Differences were not found in the profiles of genes whose products are involved in the activation and deactivation of the PAHs themselves [399]. A population-based case-control study found that exposures to PAHs were associated with specific mutations of the p53 tumor suppressor gene in breast tumor samples [400].
Occupational exposure studies have looked at workers exposed regularly to gasoline fumes and vehicular exhaust, major sources of PAHs (as well as of benzene). These occupational exposures are associated with an increased risk of breast cancer for pre-menopausal women (low level exposure, OR = 1.56; 95% CI = 0.78–3.12; high dose exposure, OR = 2.40; 95% CI = 0.96–8.01) [401] and also for men. In the case of male breast cancer, PAHs may increase the risk of breast cancer specifically in men carrying a BRCA1 or BRCA2 mutation. In a small case-only study of 23 men with breast cancer, four of whom were carriers of a BRCA1/2 mutation, BRCA1/2 carrier status interacted with history of ever having driven trucks professionally to enhance the risk of developing breast cancer (COR = 25.5; 95% CI = 1.1–1415) [402].
A case-control study in western New York indicated that very early life exposure (around the time of birth) to high levels of total suspended particulates, a proxy measure for PAH levels, is associated with increased risk of breast cancer in post-menopausal women [375]. An extension of this study, examining PAH exposures at critical times in women’s reproductive histories, demonstrated a relationship between particulate exposures around the time of the first menstrual period and incidence of pre-menopausal breast cancer (OR = 2.05; 95% CI = 0.92–4.54), and a relationship between exposure level at the time a woman first gives birth and her risk of post-menopausal breast cancer (OR = 2.57; 95% CI = 1.16–5.69) [403].
The studies above all looked at breast cancer incidence. Two reports examined the relationship between PAH exposures and mortality. Using an ecological model exploring the association between suspended fine particulate matters in several municipalities in Taiwan, researchers found that women living in areas with high levels of particulate matter in the air had an increased probability of dying from breast cancer, as compared to those living in cleaner areas (RR = 1.19; 95% CI = 1.03–1.38) [404]. Another report examined PAH-DNA adduct levels and mortality among women who had been diagnosed with breast cancer. In an extension of the Long Island study described above, researchers found no overall relationship between survivorship and PAH-DNA adduct levels. Looking more closely at groups of women who had undergone different types of treatments, however, revealed a twofold increase in age-adjusted mortality rates from breast cancer among women with high PAH-DNA adduct levels who had received radiation treatment (HR = 2.47; 95% CI = 0.74–8.21). However, there was an increased survival rate for women with adducts who had received hormone therapy as part of the treatment for their breast cancers (HR = 0.52; 95% CI = 0.24–1.13) [405].
Pesticides and herbicides
A 2007 report from the Long Island Breast Cancer Study Project demonstrated that self-reported lifetime use of residential pesticides was associated with an increase in risk for breast cancer. The increase was found for women who had reported use of pesticides in the aggregate (ever having used any residential pesticides; OR = 1.39; 95% CI = 1.15–1.68), as well as specifically for use of lawn (OR = 1.48; 95% CI =1.20–1.82) and garden (OR = 1.58; 95% CI = 1.12–2.22) pesticides, although there were no relationships between perceived doses of exposures and risk for cancer [406]. These results are important because they address exposures to chemicals in the course of ordinary life, with all the complexities of mixtures and multiple sorts of uses. Many other studies focus on single chemicals or classes of chemicals, and the results are often contradictory depending on length and timing of exposures, types of chemical being studied and so forth. Despite that, many pesticides and herbicides have been labeled as human or animal carcinogens. Many are found in water supplies [406] as well as samples of air and dust from homes [209, 407].
Triazine herbicides: atrazine
Triazine herbicides (including atrazine, simazine, and cyanazine) are the most heavily used agricultural chemicals in the United States. Atrazine and simazine have been banned in the European Union, and cyanazine is labeled as a highly toxic mutagen, because of their high presence in drinking water, demonstrated harmful effects on wildlife, and potential health effects in humans. Cyanazine was phased out of use in the U.S beginning in 1996, although simazine and atrazine are still approved for use in the United States. More than 75 million pounds of atrazine, the most heavily used of the chemicals, were applied annually in the United States in 2008 (the most recent year for which the Environmental Protection Agency (EPA) has data), primarily to control broadleaf weeds in corn and sorghum crops in the Midwest [408].
Elevated levels of atrazine are found each spring and summer in both drinking water and groundwater in agricultural areas [409–411]. Atrazine is a known endocrine disruptor, causing dramatic damage to reproductive structures in frogs, fish and other wildlife [412, 413]. Although all three triazines have been shown to cause mammary cancer in laboratory rats [414] and increased proliferation of human breast cell lines in vitro [415], there is relatively little scientific data exploring the relationship between simazine or cyanazine and human breast cancer. The literature on atrazine is somewhat more substantial.
High levels of triazines, mainly atrazine, in contaminated waters were associated with an increased risk (OR = 1.20; 95% CI = 1.13–1.28) of breast cancer [416] although a further expansion of this study concluded that there was no relationship between atrazine exposures and risk for developing breast cancer [417]. Similarly contradictory results were found in another ecological study in which higher levels of mixed pesticides, including atrazine, were associated with increased breast cancer in one rural county in the UK, but not in the neighboring county [418]. Because these ecological studies tend to compare countywide average levels of atrazine contamination and incidence rates, rather than individual exposure histories and health outcomes, it is difficult to understand clearly the difference in results [419].
A weight-of-the evidence review of seven epidemiological studies, funded by Syngenta — the producer of atrazine, concluded that across study designs, there was no causal relationship between exposures to atrazine and development of breast cancer. However, availability of only relatively few studies, lack of attention to breast cancer subtypes and other methodological complications makes ruling out an association unsupportable [420].
Research in rodents has shown that atrazine exposure disrupts pituitary-ovarian function, resulting in decreases in circulating prolactin and luteinizing hormone levels, changes that contribute to the effects of this herbicide on increases in mammary tumors [414, 421]. Atrazine also exerts endocrine-disrupting effects by increasing the activity of the enzyme aromatase [422, 423], an enzyme that catalyzes the conversion of testosterone and other androgens to estrogens, including estradiol.
Exposure to atrazine or mixtures of atrazine metabolites during gestation delays development of the rat mammary gland in puberty, widening the window of sensitivity to breast carcinogens [424–426]. Similarly, exposure of rats late in pregnancy to a mixture of commonly formed metabolites of atrazine also leads to persistent changes in mammary gland development in pups exposed during gestation. These abnormalities persist into adulthood. Exposure of rats with existing mammary tumors to atrazine increases the rate of cell proliferation in those tumors [427]. The early changes in mammary gland development may reflect an indirect effect of atrazine on maternal health, especially atrazine-induced caloric restriction, during the time of pesticide exposure [428].
Although atrazine is an endocrine disruptor in estrogen-directed pathways, several studies have indicated that atrazine does not exert its effects through binding to ER [429, 430]. Other proposed mechanisms by which atrazine may alter estrogen pathways include through binding to and increasing expression of the membrane-bound GPR30 receptor [407, 423], activation of the steroidogenic factor-1 (SF-1) gene, activation of ERK phosphorylation, and direct or indirect amplification of cAMP [430–432].
Heptachlor
Heptachlor is an insecticide that was widely used in the United States throughout the 1980s, especially for termite control. In 1988, the U.S. EPA restricted use of heptachlor to certain applications for controlling fire ants, but agricultural use continued until 1993 because growers were allowed to use up existing stocks [433]. Heptachlor use was particularly high in Hawaii, where it was employed extensively on pineapple crops and consequently contaminated both local agricultural crops and dairy supplies. Breast cancer rates in Hawaii have increased dramatically for women of all ethnic groups over the past four decades [434]. In a relatively small (96 cases) case control study exploring possible relationships between serum levels of organochlorine pesticides, including heptachlor, and development of breast cancer, a trend (P = .078) between heptachlor concentrations and breast cancer risk was found.
Heptachlor still contaminates both soil and humans. Its breakdown product, heptachlor epoxide (HE), is known to accumulate in fat, including breast tissue. Levels are highest in women ages 20 and older, but HE is also found in the bodies of adolescents 12 to 19 years old [435] and in eight of ten samples of umbilical blood from newborn infants [436]. High levels of heptachlor in breast milk [437] and fat tissue from breast biopsies [438] have been shown to be associated with increased incidence of breast cancer.
Although HE does not act like estrogen, it affects the way the liver processes hormones, thereby allowing levels of circulating estrogens to rise and increasing breast cancer risk. HE also has been shown to disrupt cell-to-cell communication in human breast cells in culture [439] and to increase production of nitric oxide, a chemical that is found naturally in cells and known to cause damage to DNA [438].
Dieldrin and aldrin
From the 1950s until 1970, the pesticides dieldrin and aldrin (which breaks down to dieldrin, the active ingredient) were widely used for crops including corn and cotton. Because of concerns about damage to the environment and, potentially, to human health, in 1975 the EPA banned all uses of aldrin and dieldrin except in termite control; the EPA banned these pesticides altogether in 1987 [440]. Thus, most of the human body burden of this chemical comes either from past exposures or lingering environmental contamination.
Hoyer et al. showed a clear relationship between breast cancer incidence and dieldrin in their examination of a rare bank of blood samples taken from women before the development of breast cancer [441]. During the late 1970s and early 1980s, blood samples were taken from approximately 7500 Danish women age 30 to 75. Researchers detected organochlorine compounds in most of the 240 women who were diagnosed with breast cancer prior to the study’s publication in 2000. They found dieldrin in 78% of the women who were later diagnosed with breast cancer, with women who had the highest levels of dieldrin before diagnosis having more than double the chance of developing the disease than women with the lowest levels. Exposure to dieldrin correlated with the aggressiveness of breast cancer: highest levels of dieldrin were associated with higher breast cancer mortality (RR = 2.61; 95% CI + 0.97–7.01; Ptrend = <.01) [442].
Treatment of mice prenatally and neonatally to environmentally relevant doses of dieldrin increased the number and size of mammary tumors. These effects may have been mediated through changes in the cellular expression of the growth factor BDNF and cell-signal receptor Trks. Both of these were elevated in tumors from the dieldrin-treated animals [443].
Like many other pesticides found in the environment, dieldrin has been shown to be an endocrine disruptor, both by stimulating estrogen-regulated systems and by interfering with androgen-regulated pathways. Addition of dieldrin to human breast cancer (MCF-7) cells in vitro can stimulate their growth and proliferation [444].
Other pesticides
A case-control study of 128 Latina agricultural workers newly diagnosed with breast cancer in California and 640 cancer-free controls, identified three pesticides—chlordane, malathion and 2,4-D—associated with an increased risk of the disease. Scientists found that the risks associated with use of these chemicals were higher in young women and in those with early-onset breast cancer than in unexposed women [445].
Engel et al. studied the association between pesticide use and breast cancer risk in farmers’ wives in the NCI’s Agricultural Health Study. This large prospective cohort study enrolled more than 30,000 women in Iowa and North Carolina. Researchers found evidence of increased incidence of breast cancer in women using 2,4,5-trichlorophenoxypropionic acid (2,4,5-TP) (RR = 2.0; 95% CI = 1.2–3.2); a non-significant association was found for dieldrin and captan. Incidence was also modestly elevated in women whose homes were closest to areas of pesticide application (RR = 1.7; 95% CI = 1.0–1.9) [446].
Young children of farmers using 2,4,5-TP on their farms had high levels of the pesticide in their urine samples soon after the chemical had been applied to the fields [447]. This is of concern given the evidence of increased susceptibility of children and young adolescents to the carcinogenic effects of endocrine disrupting chemicals.
Treatment of female rats with malathion resulted in abnormal increases in mammary duct proliferation and induction of mammary tumors when animals were tested at 8 months of age [448].
Persistent organochlorines
DDT/DDE
Dichloro-diphenyl-trichloroethane (DDT) was the first widely used synthetic pesticide. It is credited on the one hand with the eradication of malaria in the United States and Europe and on the other with long-term devastating effects on reproductive success in wildlife and adverse health effects in humans [449]. Banned in most countries for agricultural use, DDT is still used for malaria control in many countries, especially in sub-Saharan Africa [450]. Because of its continued use and its persistence in the environment, DDT, and its main metabolite, DDE, are found worldwide. Most animals, including humans, ingest DDT and DDE-contaminated foods and retain the chemicals. Significant concentrations of DDT and DDE are found in the body fat of humans and animals as well as in human breast milk and placenta, even in regions where it has not been used for a long time [452–454].
Epidemiological data are mixed regarding the effects of DDT/DDE on breast cancer risk [454]. A case-control study from Tunisia found positive associations between serum DDE levels and breast cancer risk (OR = 9.65; 95% CI = 1.81–63.33; dose-response trend p = .02). On the other hand, a study from the Long Island Breast Cancer Study Project did not find an association between DDT/DDE (or polychlorinated biphenyls, PCBs) and breast cancer [456]. Both of these studies measured contaminant levels around the time of breast cancer diagnosis, without regard to possible exposures during critical early periods of breast development [457].
Two critical studies that examined early life (prenatal and childhood) exposures to DDT have demonstrated a clear association between exposures to DDT and increased risk for developing breast cancer [103, 104]. A prospective, nested case-control study of 129 women who had been diagnosed with breast cancer before age 50 and 129 age-matched controls, all participating in the Child Health and Development Studies (CHDS), explored the women’s estimated historical DDT levels based on aggregate data from their year of birth as well as blood DDT levels at the time the women gave birth to their first child. Researchers then monitored the women for the next 2 decades, noting when women either were diagnosed with breast cancer (invasive or noninvasive) before age 50 or died from breast cancer before age 50. Exposure to DDT during childhood and early adolescence (younger than 14 years) was associated with a 5-fold increase in the risk of developing breast cancer before age 50 [104].
In a case-control prospective study of 9300 women in the CHDS pregnancy cohort (daughters of the mothers in the larger CHDS cohort), stored postpartum maternal blood samples were analyzed for levels of DDT. Daughters were followed for 52 years and breast cancer diagnosis in this cohort was determined. DDT levels in perinatal blood of mothers from breast cancer cases were compared with levels in perinatal blood samples from mothers of age-matched controls. Higher maternal DDT levels were associated with a significant increase in occurrence of breast cancer in their daughters by age 52 (OR = 3.7; 95% CI = 1.5–9.0) [103].
A comparison of the association between disease risk and DDT use in developed countries (where DDT has been banned for several decades) and in developing countries (where DDT use is still prevalent) supports the premise that exposures to DDT are associated with increased risk of breast cancer. The association between DDT levels and breast cancer was much stronger in developing countries, where women of the age to be diagnosed with breast cancer also would have been exposed to DDT during critical periods of development [458].
A study from the Long Island Breast Cancer Study Project examined post-diagnosis mortality and serum DDT levels at time of diagnosis. Higher levels of DDT 5 years after diagnosis were associated with an increased mortality rate (HR = 2.72; 95% CI = 1.04–2.12) although the effect was not significant at 15 years post diagnosis [459].
Laboratory studies have found that in addition to being directly genotoxic or carcinogenic [460], the estrogen-like form of DDT enhances the growth of ER+ mammary tumors [461–463]. The percentage of breast tumors in the United States that are ER+ rose from 73% in 1973 to 78% in 1992 [464]. Although no direct relationship can be inferred, this change corresponds to the period when women exposed to DDT as young girls were expected to be exhibiting environmentally altered incidence in breast cancer related to DDT exposure. Another study, looking at chemical levels in breast fat tissue, did not find an association of DDT/DDE with ER+ tumors. However, data from this study indicated a significant association of higher concentrations of these compounds in breast tissue with tumors that were more aggressive and that had poorer prognoses (OR = 2.40; 95% CI = 1.0–5.4) [465].
PCBs
Although the EPA banned the use of PCBs in new products in 1976, substantial amounts of the insulation fluids, plastics, adhesives, paper, inks, paints, dyes and other products containing PCBs manufactured before the ban remained in use for decades [466]. About one-third was discarded, which means that these toxic compounds eventually made their way into landfills and waste dumps [467]. PCBs are found in the air and in aquifers and rivers, where they accumulate in the sediment but then are re-dissolved into the water where they contaminate and bioaccumulate across the food chain [468, 469].
Levels of PCBs were high before being banned in the United States, but generally their presence in the environment and in human tissues has decreased slowly over the past decades [470, 471]. Choi et al. found that PCB levels in neonatal cord serum were correlated with the distance of mothers’ residences from a Superfund site; levels were lower after site remediation [472]. However, exposure levels were high between childhood and young adulthood for many women who are now facing breast cancer diagnoses.
The more than 200 individual PCBs are classified in three types based on their cellular effects. Group I PCBs are estrogenic; Group II compounds are anti-estrogenic; and Group III PCBs appear not to be hormonally active, but can stimulate the enzyme systems of animals, including humans, in a manner similar to certain drugs (such as phenobarbital) and other toxic chemicals [473, 474]. Additionally, hydroxylated metabolites of PCBs alter the expression of genes involved in hormone synthesis, indicating that these compounds may act as endocrine disruptors through mechanisms not directly involving the estrogen receptor [475].
There are several epidemiological studies that have implicated exposures to PCBs as a risk factor for later development of breast cancer. Women who regularly ate PCB-contaminated pike or perch had higher risk for breast cancer than women who never ate these fish (perch: OR = 7.90; 95% CI = 1.01–61.9; pike: OR =9 .07; 95% CI = 1.10–74.4) [476]. Another study implicated PCBs in breast cancer recurrence among women with non-metastatic breast cancer. The study found that women with the highest levels of total PCBs (RR = 2.91; 95% CI = 1.0–8.2), as well as of PCB 118 (RR = 4.0; 95% CI = 1.3–4.9), in their fat tissues were almost three times as likely to have recurrent breast cancer as women with lower levels [477].
Most studies have looked at total PCB levels without identifying individual types. A few studies, however, have looked at relationships between cancer status and particular PCBs or groups of PCBs. For example, a recent meta-analysis demonstrated that there was no relationship between exposures to Group I PCBs, but there was a significantly increased risk of breast cancer with exposures to either Group II (OR = 1.23; 95% CI = 1.08–1.40) or Group III (OR = 1.25; 95% CI = 1.09–1.43) PCBs [474]. Another study examined PCB levels in breast tissue from disease-free women and women with metastatic breast cancer. Across all samples tested, higher levels of Group III PCBs were found, followed by Group II and then Group I compounds. These results were independent of disease status and were not associated with any pathological characteristics in the women who had been diagnosed with breast cancer [478]. A recent congener-specific meta-analysis examined association between representative congeners from three subgroups of PCBs and found Group III congeners PCB 99 and PCB 183 conferred a greater risk than Group I PCB 187 (respective ORs: 1.36; 95% CI = 1.02–1.80; 1.56; 95% CI = 1.25–1.95; 1.18; 95% CI = 1.01–1.39) [479].
In a case-control study, Aronson et al. measured several types of PCBs, along with DDE, in tissue samples from women scheduled for excision biopsy of the breast. Increased risk for breast cancer was associated with higher concentrations of Group II PCBs 105 and 118, with the ORs for these two PCBs increasing linearly with higher concentrations (p for trend <0.01) [480].
On the other hand, some studies have found no link between PCBs and breast cancer [481]. A 2009 review of the literature concluded that the overall picture was that PCBs, as a class (not considering the types of PCBs), were not associated with increased risk for breast cancer [482]. In a study examining occupational exposures to PCBs in electrical capacitor production workers and later breast cancer incidence, no overall relationship between exposure levels or duration and disease incidence was observed for female workers in general. But for non-white women, a significant relationship was found between incidence of breast cancer and earlier PCB exposure duration as well as cumulative exposure amounts (comparing highest vs. lowest categories of exposure, HR = 22.3; 95% CI = 2.38–209) [483]. More recently, Artacho-Cordón found no correlation between serum or adipose levels of PCBs and risk for being diagnosed with breast cancer [484].
Some of these compounds may have their greatest impact on women with particular susceptibilities and looking broadly at large samples will not tell the full story of cancer risk as influenced by PCB exposures. For example, researchers evaluating data from the Nurses’ Health Study revisited the issue of PCBs and breast cancer risk and revised their conclusion concerning the link between PCBs, DDE and breast cancer. In studies of PCBs and DDE in blood, they had previously concluded that exposure to these chemicals was unlikely to explain high breast cancer rates [485]. Newer evidence found that the complex interaction of high serum levels of PCBs and a particular variant (exon 7) of the CYP1A1 gene was associated with an increased risk for breast cancer (HR = 2.78; 95% CI = 0.99–7.82, compared to women with lower PCB levels and the wild-type genotype) [486].
As was true for the critiques of the DDT studies cited above, the methods used to test these relationships do not account for exposures to PCBs during earlier developmental times when mammary tissue is particularly sensitive to the toxic effects of many environmental chemicals [487]. The results from Cohn’s work on DDT and breast cancer make clear that this is a critical methodological issue [457].
Dioxins
Dioxins are formed by the incineration of products containing PVC, PCBs and other chlorinated compounds as well as from industrial processes that use chlorine and from the combustion of diesel and gasoline [488]. One of the dioxins (2,3,7,8-tetra chlorodibenzo-para-dioxin; TCDD) has been classified by IARC [489] and the U.S. EPA [490] as a carcinogen.
Dioxins accumulate in the body fat of wildlife and humans, and they break down very slowly, with a half-life of 7–11 years in body tissues [491]. People are exposed to dioxins primarily through consumption of animal products and human breast milk [488, 489]. Dioxin enters the food chain when vehicle exhaust or soot from incinerated chlorinated compounds falls on field crops later eaten by farm animals. It is then passed to humans through dairy and meat products. The body fat of every human being, including every newborn, is thought to contain dioxins [492].
There is a substantial decrease in the amount of dioxin remaining in a women’s breast fat tissue after she has breastfed because the chemicals have been passed on to her newborn via breast milk [493]. Although the presence of toxic chemicals in breast milk is potentially dangerous, the beneficial nutrients and immune system boosters that are transferred from mother to infant are thought to far outweigh the potential toxic transfers [494]. But in addition to potential transfer of dioxins to breast-feeding infants, the release of the chemicals from storage in breast fat cells, initiated by the process of milk synthesis, may actually trigger genotoxic effects in the mother’s breast tissue. Addition of breast milk extracts to MCF-7 cells led to a reprogramming of gene expression to a pattern typically found following estrogen stimulation, especially in the CYP1A1 and CPY1B1 genes [495].
A study of women exposed to dioxins during a chemical plant explosion in 1976 in Seveso, Italy demonstrated a time-dependent association between dioxin exposure and breast cancer [496, 497]. A tenfold increase in TCDD levels in blood samples taken at the time of the explosion was associated with more than twice the risk of breast cancer in the women who, in 1996, averaged 40 years old (HR = 2.1; 95% CI = 1.0–4.6), and whose breast cancer was diagnosed pre-menopausally. Follow-up of the cohort in 2008 revealed a non-significant increase in risk for developing breast cancer between the time the women were 40 and 12 years later, when they were 52, on average (HR = 1.44; 95% CI = 0.89–2.33). For all the breast cancer cases for which there were cancer profile data, more than 80% were ER+/PR+ cancers [497]. Continued follow-up of this cohort will examine risk of developing breast cancer as the women enter and continue into the post-menopausal years.
A retrospective mortality study in Germany examined deaths from cancer among people who had worked in a chemical factory in which they were exposed to high levels of TCDD. As compared with national mortality rates in West Germany, 5 years after closure of the plant, there was no increase in overall mortality from cancer for female workers, although there was a significant increase in deaths from breast cancer among those who worked in high-exposure regions of the factory (SMR = 2.15) [498]. A later follow-up 23 years after the closing of the plant found a lower rate of mortality from all causes for women workers in the plant (SMR = 0.91; 95% CI =0.78–1.05), but a significant increase in mortality from breast cancer (SMR = 1.86; 95% CI = 1.12–2.91) in this cohort [499].
Several laboratory studies have demonstrated that the timing of exposures to dioxins matters. Although exposing animals to dioxins in adulthood may not affect cancer rates, earlier exposures may have profound effects. Administration of dioxins (especially TCDD) to pregnant rats leads to structural abnormalities in the development of their pups’ mammary tissues and higher incidence of tumors when the pups grow to adulthood [500–504]. TCDD may exert its cancer-causing effects both by decreasing the efficacy of tumor-suppressor mechanisms and by enhancing the estrogenic signaling within the mammary cells [505].
TCDD has been shown to inhibit estradiol-induced cell growth and proliferation as well as other pathways regulated by estrogens, including methylation of CYP1A1, in a variety of human breast cancer cell culture lines [506, 507]. Like the PAHs described above, dioxins like TCDD exert their effects by activating both the aryl hydrocarbon receptor (AhR), an important mediator of cell growth and proliferation [508] and anti-apoptosis pathways [509], as well other AhR-independent pathways including PR- mediated pathways [506, 510, 511]. Increasing evidence indicates that many of the effects of TCDD and other dioxins is the result of cross-talk between the AhR and ER, PR and even AR systems [512, 513].
Prenatal treatment of rats with low doses of TCDD led to increases in the number of terminal end buds (TEBs) counted in whole mount preparations of postnatal day 71 females. The treatment also resulted in a suppression of BRCA-1 expression, a result of increased BRCA-1 promoter hypermethylation [514].
Polybrominated Diphenyl Ether (PBDE) fire retardants
PBDEs are a complex group of chemicals that are structurally similar to the polychlorinated biphenyls (PCBs). Products containing PBDEs include polyurethane foam in furniture (penta-BDE) and electronic and plastic products (octa- and deca-BDEs) [515].
Although both penta- and octa-BDEs have been banned in the European Union and have not been produced in the United States since 2004, products containing them remain throughout the world. PBDEs are found ubiquitously in the environment and are detected in air, dust, soil and food as well as in many wildlife species. Although home exposures (as measured by dust levels) have decreased over the past decade, levels remain high enough to remain a serious health concern [516].
There is considerable geographic variability in exposures to the chemicals; people in California, with its historically stringent furniture flammability standards, have much higher levels of PBDE exposures than do people in Massachusetts. Within the California group, lower socioeconomic status was associated with higher PBDE levels [488, 517]. Mexican Americans living in California have significantly higher PBDE levels in blood serum than do Mexicans living in their homeland [518].
Data from young girls (ages 6 to 9) from California and Ohio support these findings. Although PBDEs were found in almost all samples tested, girls in California had significantly higher blood serum PBDE levels than did girls from Ohio, and young black African American girls had higher levels than either white or Hispanic girls [519]. In these cohorts, PBDE exposures are associated with delays in puberty in adolescent girls (RR = 1.05; 95% CI = 1.02–1.08), an effect that is not moderated by adjustment for BMI [520].
PBDEs have been found in human fat tissue, as well as in blood serum, breast tissue and milk [521–523]. PBDEs cross the placenta, resulting in exposures to developing fetuses [524]. PBDEs are endocrine-disrupting compounds, exerting effects on a number of hormonal systems, including the androgens, progestins and estrogens, although the major system affected by PBDEs is the thyroid hormone system [486]. Most studies of health outcomes after PBDE exposures have focused on neural development, given the prominent role of thyroid hormones (especially T4) in regulating brain development [525, 526].
Very few data directly address the possible effects of PBDEs on breast cancer risk. One case-control study found no relationship between PBDE levels in breast fat and breast cancer risk, but the sample was small and the chemical analysis was done around the time of diagnosis of breast cancer in the women who developed the disease [527]. However, at least some PBDEs have been shown to be as effective as many of the other endocrine-disrupting compounds in promoting estrogenic-like proliferation of human breast cancer cells in vitro [528]. Penta-BDE enhances tumor-cell proliferation in MCF-7 cells through estrogen-like effects on cell pathways that interrupt apoptosis [529]. The cell-proliferative and anti-apoptotic effects of PBDEs are additive with those of natural estradiol [530] and counteract the anti-cancer effects of tamoxifen in cultured breast cancer cells [531]. Given the extensive overlap and interaction of estrogen-mediated and thyroid-mediated responses in the regulation of breast cancer [532], PBDEs will be a class of chemicals of continued concern for scientists interested in understanding environmental links to breast cancer [533].
Even as PBDEs are being used less often as fire retardants in common consumer products, there is now evidence that the chemicals being used as substitutes — including Firemaster 550®, a common substitute with proprietary ingredients — are increasingly contaminating our environment [489, 534]. Although the physiological effects of exposures to Firemaster 550® have not yet been studied extensively, one study demonstrated that feeding rat dams low doses during pregnancy and lactation led to changes associated with exposures to other endocrine disrupting compounds. These effects included changes in thyroid hormone levels in the mothers, and changes in behavior, weight gain and earlier puberty in female pups [507].
Aromatic amines
Aromatic amines are a class of chemicals found in the plastic and chemical industries as byproducts of the manufacturing of compounds such as polyurethane foams, dyes, pesticides, pharmaceuticals and semiconductors [535]. They are also found in environmental pollution such as diesel exhaust, combustion of wood chips and rubber, tobacco smoke and grilled meats and fish [536, 537]. There are three types of aromatic amines: monocylic, polycyclic and heterocyclic.
Three monocyclic amines, including o-toluidine, have been identified in the breast milk of healthy lactating women [536], as well as in the urine of most people [535]. σ-Toluidine is known to cause mammary tumors in rodents [538, 539]. The carcinogenic aromatic amines, 2-amino-phenylimidazo[4,5-b]pyridine (PhIP) and 4-aminobiphenyl (ABP) are also found in human breast milk, as are DNA-adducts of these compounds [540].
Occupational exposures of female rubber-factory workers to another set of monocyclic aromatic amines derived from p-phenylenediamine are associated with an increased risk of breast cancer in the following several years. The amount of increased risk was correlated with total cumulative exposure levels to the aromatic amines, with lowest levels leading to a 3.7-fold increase in cancer and the highest levels of exposure increasing risk more than tenfold [541].
A case control study of women who used hair dyes, in comparison with those who have not, revealed an increased risk of breast cancer in the dye users (OR = 1.15; 95% CI = 1.06–1.24). In addition to intentional inclusion of p-phenylenediamine [542], major contaminants in many hair dyes include PhIP and ABP [543].
Heterocyclic aromatic amines (HAAs) are formed (along with PAHs) when meats or fish are grilled or otherwise cooked at high temperatures. In a case control study, Steck et al. found an association between higher lifetime consumption of grilled meats and fish and increased incidence of post-menopausal breast cancer (OR = 1.42; 95% CI = 1.12–1.92) [544]. Studies of both milk and cells from the ducts of women’s breast revealed the presence of DNA adducts in association with HAAs [545, 546].
Aromatic amines are metabolized by N-acetyltransferases. This metabolic process ultimately leads to other compounds that are thought to be the actual carcinogenic chemicals. There is an extensive literature examining whether or not genetic profiles that alter the efficacy or speed of N-acetyltransferase activation, especially through the N-acetyltransferase 2 (NAT2)-regulated pathway, might alter susceptibility to breast cancer. Studies have reached differing conclusions about the role of possible polymorphisms of the NAT2 gene on breast cancer susceptibility. A 2010 study tried to disentangle many of the possible confounding factors and found that eating grilled meat (and drinking coffee) resulted in greater risk for diagnosis of ER- breast tumors in women with the ‘slow-acetylator’ form of the NAT2 gene [547].
Laboratory studies of HAAs in systems using cultured breast cancer cells demonstrated that these chemicals can mimic estrogen, and they also can have direct effects on cell division processes in ways that might enhance the development of tumors [548].
Metals
Higher accumulations of iron, nickel, chromium, zinc, cadmium, mercury and lead have been found in cancerous breast biopsies as opposed to biopsies taken from the breasts of women without breast cancer. These metals also have been found in higher concentrations in serum and urine from women diagnosed with cancer as compared with those from healthy women [549–552].
Laboratory studies have shown that a number of metals including copper, cobalt, nickel, lead, mercury, methylmercury, tin, cadmium and chromium have estrogenic effects on breast cancer cells (MCF-7) cultured in vitro [553–555], with cadmium expressing the highest level of estrogenic activity [555]. The most extensive work on the relationship between breast cancer and metals has been done examining this metaloestrogen, cadmium.
Several epidemiological studies have demonstrated an association between higher cadmium levels in urine (OR = 2.29; 95% CI = 1.3–4.2) [556, 557] or blood [558] and increased breast cancer risk, although in a large prospective study using the WHI cohort, no association between urinary cadmium levels and risk for developing breast cancer was found [559]. Nevertheless, a recent meta-analysis reported a significant increase in risk of developing breast cancer as urinary cadmium levels increased (OR = 2.24; 95% CI = 1.49–3.35) [560].
Differences in the efficacy of establishing relationships between breast cancer and cadmium exposures as determined by dietary vs. urinary cadmium measures may reflect the observation that urinary cadmium levels are a stronger marker of lifetime exposures to the metal, given cadmium’s half-life of 12–30 years, while dietary exposure levels reflect a shorter term and potentially more variable marker [560].
Prospective studies of women’s dietary intake of cadmium and later diagnosis of cancers demonstrated a significant relationship between higher levels of dietary cadmium exposure and incidence of both endometrial cancers (RR = 1.39; 95% CI = 1.04–1.86) [561] and post-menopausal breast cancers (RR = 1.21; 95% CI = 1.07–1.36) [562]. With regard to breast cancer, the effect was significant for all sub-types combined, but more pronounced for ER+ tumors (OR = 1.94; 95% CI = 1.04–3.63 [563]; RR = 1.23; 95% CI = 1.02–1.49) [562]. On the other hand, studies examining dietary cadmium intake in Japanese [563, 564] and Danish [565] women and their risk of developing breast cancer found no relationship. A 2012 study in the United States that looked at dietary cadmium levels and breast cancer risk also did not find a significant relationship [566], nor did two recent meta-analyses that studied this relationship [567, 568].
In young rats, treatment with low doses of cadmium led to an increase in branching and bud formation in mammary tissue, and the induction of several estrogen-associated proteins. Prenatal exposure of rats to cadmium led to early onset of puberty and greater numbers of mammary terminal end buds, both known risk factors for breast cancer [569].
Estrogenic effects of cadmium have been studied in some detail, and the metal has been shown to interfere with a number of normal estrogen-sensitive pathways [570]: cadmium can bind to and activate mammary cell estrogen receptors; it also interacts with and regulates the transcription of estrogen-dependent genes affecting the synthesis of proteins and/or the activity of cell-signaling pathways in ways similar to the natural estrogen, estradiol [570, 571]. Cadmium exposure can also lead to malignant transformation of normal human breast epithelial cells (MCF-10A) through a mechanism that does not require the presence of ERα [572]. Other studies support the possibility that cadmium may also exert cellular effects through mechanisms beyond the conventional nuclear-ER directed pathways [573, 574], including possibly through binding to the membrane receptor, GPR30 [575].
In addition to hormone-mediated effects, cadmium may also promote the development of cancer through epigenetic processes including changes in DNA methylation patterns as well as possible modifications of gene-associated histones [576].
Like several other estrogen-mimicking endocrine disruptors, cadmium interferes with the efficacy of a common chemotherapeutic agent often prescribed for women who have been diagnosed with breast cancer. In a study examining the effects of cadmium, 5-fluorouracil (5-FU), and the two combined on subcellular structure and metabolic activity in cultured MCF-7 breast cancer cells, co-administration of cadmium negated the anti-cancer effects of 5-FU [577].
Section summary: The growing literature on exposures to EDCs, especially early in development, indicates an increased risk of developing breast cancer following exposure to many of these compounds, either alone or in combination. The most substantial human epidemiological data supporting this relationship come from prospective studies on women exposed to DDT during gestation or early childhood and increased development of premenopausal breast cancer [103, 104]. The largest non-human literature connects early, low-dose exposures to BPA to increased risk for developing mammary tumors. Both in situ and in vitro studies have added substantially to our understanding of the complex mechanisms underlying these relationships. Although not as robust as for the above chemicals, links between exposures, especially early in development, and many other EDCs have also been documented.
Hormones in foods: natural and additives
The prevailing evidence against synthetic estrogens must also be understood alongside evidence about the effects of plant estrogens (phytoestrogens) on risk for developing breast cancer. While most of the research in this area has focused on possible protective associations with soy-based isoflavones in a normal dietary regime, a growing literature is also examining the potential protective effects of the lignans, enterolactone and α-linolenic acid.
Mycoestrogens (estrogens found in fungal species) can contaminate agricultural and meat products, and this contamination may increase susceptibility to developing breast cancer. Also, exposures to growth enhancing compounds given to meat-producing and dairy animals have been linked to increased risk for developing breast cancer. This section explores the complicated profiles of exposures to food-based estrogens and risk for developing breast cancer. There has been no dermination on potential carcinogenicity of these substances by IARC or NTP.
Phytoestrogens (plant estrogens)
Foods such as whole grains, dried beans, peas, fruits, broccoli, cauliflower and especially soy products are rich in phytoestrogens. While most of the research in this area has focused on possible associations with soy-based isoflavones. Additionally, there is also a growing literature is also examining the possible role of the lignans, including enterolactone and α-linolenic acid, in affecting breast cancer risk.
Lignans
Lignans are polyphenolic compounds found widely in seeds and grains common to a Western diet. They are rapidly metabolized in the gut to the estrogenic compounds, enterolactone and enterodiol [578].
A matched case-control study of mainly premenopausal women in the Nurses’ Health Study II cohort found no overall relationship between plasma enterolactone levels and breast cancer risk. However, in those women whose follicular circulating estradiol levels were below the median, higher enterolactone was associate with a significant decrease in breast cancer risk (OR = 0.49; 95% CI = 0.27–0.91) [579]. A meta-analysis examining the possible relationship between serum enterolactone levels and either all-cause mortality or breast-cancer associated mortality found negative correlations between the circulating lignan levels and both outcome measures (HR = 0.57; 95% CI = 0.42–0.67 for all-cause mortality and HR = 0.54; 95% CI = 0.39–0.75 for breast cancer mortality [580].
A recent examination of possible mechanisms underlying the protective effect of enterolactone used the aggressive, highly invasive MDA-MB-321 human breast cancer cell line. Addition of the lignan to the cell culture led to decreased activity in mRNA levels of several genes associated with cell proliferation, as well as increased retention of mitotic cells in the S-phase, and decreased migration and invasion through interference with the cell cytoskeleton [580].
A systematic review of the literature on associations between α-linolenic acid and risk of breast cancer found significant negative relationships between higher intake of flax, a major source of α-linolenic acid, and both breast cancer incidence (OR = 0.82; 95% CI = 0.69–0.97) and mortality (HR = 0.69; 95% CI = 0.50–0.95) [581]. In women with recent diagnoses of breast cancer, higher intake of flax was associated with less-aggressive tumor profiles that had higher apoptotic indices, and lower HER2 expression and proliferative rates [581].
Soy and soy derivatives
Although scientific evidence suggests that soy-derived foods offer nutritional benefits and are associated with healthy diets [582, 583], the data are more conflicting as to whether the soy-based diets are beneficial, harmful or neutral when it comes to affecting breast cancer risk [584, 585].
Some of the disparity may be related to type of soy products consumed by individuals. Across Asian and Western diets, soy may be processed in scores of different ways, resulting in up to 100-fold differences in levels of particular phytoestrogens between products [583] and very different exposures levels for consumers. Diets high in products that contain higher levels of both the soy isoflavone, genistein, and its metabolite genistin, have been shown to affect breast tumor growth in a number of different models. In contrast, highly processed soy flour that does not contain isoflavones has no effect. Purified soy protein isolates are often processed to contain different concentrations of isoflavones, and their influence on mammary tumors is related to the amount of the isoflavone phytoestrogen, not the total amount of soy protein consumed [586].
Several epidemiological studies have shown that regular consumption of soy-based products, or other vegetables high in phytoestrogens, as part of a normal balanced diet can exert a protective influence with regards to later development of breast cancer. This effect has been studied extensively in China where soy intake is a regular part of the cultural diet. There, substantial evidence indicates that higher soy intake in adulthood or in adolescence is associated with a decreased risk of pre-menopausal breast cancer (OR = 0.41; 95% CI = 0.21–0.70 for soy intake; OR = 0.44; 95% CI = 0.26–0.73 for isoflavone intake [587]; OR = 0.68; 95% CI = 0.50–.93 for soy intake) [588]. Other studies have found protective effects of soy intake for both pre- and post-menopausal cancer, independent of receptor (ER and PR positive or negative) profile of the tumors (ORs ranging from 0.30 to 0.43) with a dose-dependent inverse relationship found across cancer subtypes (trend p < .0001) [589].
For Chinese women who were previously diagnosed with breast cancer, higher consumption of soy in its many forms found regularly in a woman’s diet, was correlated with decreased recurrence of cancer (OR = 0.68; 95% CI = 0.54–0.92) and longer survival (Or = 0.71; 95% CI = 0.54–0.87) [590]. Complicating the picture further is a study of Korean women who had previously been diagnosed and treated for breast cancer. Dietary soy intake was associated with a decreased rate of recurrence in women whose cancers were HER-2 negative (OR = 0.27; 95% CI = 0.13–0.57), and an increased rate of recurrence of cancer in women whose original tumors were HER-2 positive (trend p < .02) [591].
On the other hand, in a prospective study of women aged 43 to 55 years who had never been diagnosed with breast cancer but were considered to be at high risk, 6 months of dietary isoflavone (PTIG-2535, containing 150 mg genistein, 74 mg daidzein, and 11 mg glycitein) intake was associated with increased proliferation of breast cells. The effect was most pronounced in pre-menopausal women [592].
A series of reports from Japan looking at the relationship between soy intake and breast cancer risk have found an inverse relationship [593–595], with strong dose-response relationships being found for postmenopausal women (trend, p = .023 for soy intake and trend p = .046 for isoflavone intake) [596].
A study of Asian-American women living in California and Hawaii found significant decreases in breast cancer risk associated with greater soy intake during childhood (RR = 0.40 95% CI = 0.18–.83), adolescence (RR = 0.80 95% CI = 0.59–1.09), or adulthood (RR = 0.76 95% CI = 0.56–1.02) [597]. The protective effect of regular dietary soy intake during childhood was the strongest, and it was not mitigated when other variables like site of birth (Asian countries or U.S.), degree of continuing Asian lifestyle and cultural practices, reproductive factors or family history of breast cancer were factored into the analysis. In general, protective effects of dietary soy intake have been found to be strongest in association with childhood and early adolescent intake [598], especially in relationship to development of ER+/PR+ postmenopausal breast cancer (OR = 0.79; 95% CI = 0.65–0.96) [599]. One possible explanation for this association is that peri-pubertal exposures to genistein and other phytoestrogens may mimic the protective changes in breast development that are usually observed during the first pregnancy [600, 601].
Several studies have directly compared effects of consuming culturally appropriate soy diets in Asian and Western women. A 2012 meta-analysis that combined data from six studies found that regular dietary intake of soy during adolescence decreased the incidence of all later breast cancers (OR = 0.82 95% CI = 0.67–0.99), and was particularly effective in decreasing cancer incidence in pre-menopausal women (OR = 0.66 95% CI = 0.55–0.80). There was no reported difference in the effects of dietary intakes of soy during adolescence between Asian and American/European women [602]. On the other hand, a 2014 meta-analysis examining isoflavone intake in pre- and post-menopausal women from Asian and Western countries found protective effects of soy in both premenopausal (OR = 0.59; 95% CI = 0.48–0.69) and postmenopausal (OR = 0.59; 95% CI = 0.47–0.74) Asian women, with only very small and non-significant effects in both premenopausal and postmenopausal Western women [603]. A 2009 multiethnic study conducted in Hawaii demonstrated that the amount of soy in the diet might interact with associations between other phytoestrogens and protection against breast cancer. For Japanese Americans who had high soy content in their regular diets, there was a strong, significant but non-monotonic relationship for urinary genistein levels within the middle two quartiles (OR =0.88; 95% CI =0.78–0.99) with decreased risk of breast cancer. A similar strong relationship was not found for White women in the study who tended to eat diets lower in soy content [604]. Differences in responses between Asian and Western women may reflect both differences in the diet content as well as cultural differences in the ability to metabolize isoflavones, an effect that may result from both genetic differences as well as interactions with other dietary factors [584].
Data from studies on laboratory animals and cell culture models have indicated a more complicated story. In several studies, exposures to phytoestrogens have led to increases in mammary tumor proliferation and growth. In ACI rats, dietary exposure to total soy isoflavone content from conception through adulthood decreased incidence of mammary tumors in adult animals by 20% and multiplicity by 56%, while also decreasing the latency to tumor onset by 20% and almost tripling tumor volumes [605]. Exposure of Wistar rats to genistein from conception through weaning led to decreases in DMBA-induced tumor number, multiplicity and incidence at postnatal day 50 [606].
The soy phytoestrogens, genistein and daidzein, as well as their metabolites, cause oxidative DNA damage, a process that is thought to play a role in tumor initiation. Other data suggest that these two soy-based phytoestrogens may have opposing effects on the efficacy of the breast cancer drug, tamoxifen [607, 608].
The effects of the phytoestrogens may well be related to the particular components and doses in the diet [609], and cellular effects may vary depending on concentration and timing. In a study examining the effects of different types and concentrations of phytoestrogens on the expression of estrogen-dependent gene activity in human breast cancer cells grown in vitro (MCF-7 cells), low doses of genistein resulted in a pattern of expression that indicated increased cell proliferation, while somewhat higher concentrations led to increased apoptosis [610].
Isoflavones contained within natural soy flour have different effects than addition of isoflavones purified from the soy flour on gene expression patterns in induced tumors grown from MCF-7 cells in athymic nude mice. When mice were fed isolated isoflavones, the gene expression pattern was similar to that found in mice that had been treated with estradiol, while the pattern in animals treated with isoflavones included within a full soy flour were more like the negative control, suggesting an inhibitory effect of soy flour proteins on some of the proliferative effects of isolated isoflavones [611]. In addition to altering gene patterns associated with cell proliferation and carcinogenesis, genistein exposure also is a strong inhibitor of angiogenesis, an important process associated with tumor growth and metastasis. On the other hand, 30 days of treatment by gavage of peripubertal and adult ovariectomized rats with isoflavone supplements had no effect on cell proliferation or angiogenesis [612].
In cultured MCF-7 cells, the soy phytoestrogen daidzein slightly enhanced cell proliferation in the absence of natural estrogen (a possible model for post-menopausal breast cancer), while resveratrol (found in grapes and red wine) significantly decreased tumor cell proliferation [602]. These latter data are consistent with other studies finding anti-carcinogenic effects of resveratrol in several models [613, 614].
Concern has been raised about exposure of newborn babies to soy-based products, primarily through infant formulas. Although one study has shown that feeding only soy formula for the first 4 months of life was associated with a decrease in later development of breast cancer [615], animal studies have indicated deleterious effects of neonatal soy exposure on development of the female reproductive system and subsequent fertility [616].
Mycoestrogens (fungal estrogens)
Mycotoxins are compounds produced by several fungal species that contaminate agricultural and feed products, including corn silage and hay, both during before harvest and during later storage [617–619]. People are exposed to these compounds directly, by eating grains contaminated with the fungi, and indirectly, by eating meat from animals who have consumed contaminated feed [620].
Contamination of food by zearalenone (ZEA) and its natural metabolites has been associated with the development of precocious puberty, a known risk factor for breast cancer, in young girls [621, 622]. On the other hand, girls in the Jersey Girl Study who had higher urinary ZEA levels, resulting from recent intake of beef or popcorn, tended to be less likely to have reached the onset of breast development and to be of shorter stature. Almost 80% of girls in the study had detectable levels of mycoestrogens in their urine [623].
A case-control study of dogs revealed that higher dietary exposures to mycotoxins (aflatoxin G1 or G2) resulted in a significant increase in the number of mammary tumors (OR 2.74; 95% CI = 1.13–6.60 and OR 4.6; 95% CI = 2.2–7.8, respectively) [624].
In rat dams fed diets containing ZEA, both the compound and its metabolites crossed the placental barrier and also appeared in mother’s milk [625]. Exposure of female pups to environmentally relevant doses of ZEA during the last 2 (of 3) weeks of fetal development and the first few postnatal days resulted in long-term alterations in mammary gland development of the sort associated with increased risk for development of mammary tumors [626].
Rats treated on post-natal days 15–19 with ZEA and then with the carcinogenic substance, N-methyl-N-nitrosurea (MNU), at puberty developed fewer mammary tumors, with lower multiplicity, than matched controls treated only with the NMU carcinogen, although there was no difference in the latency to appearance of tumors in either group [627].
In cell culture models of human breast cancer, mycotoxins including Fusarin C and ZEA and their metabolites have been shown to be estrogenic [628]. For example, Fusarin C stimulates growth and proliferation of MCF-7 breast cancer cells via ER-mediated processes [629]. Similarly, ZEA also enhances proliferation of MCF-7 cells in vitro through estrogen-mediated pathways and activation of estradiol-induced gene expression [630, 631].
Natural, synthetic and genetically engineered hormones used in food production
Zeranol (Ralgro®)
The synthetic compound, zeranol (Ralgro®), is a potent non-steroidal growth promoter that mimics many of the effects of the natural hormone estradiol. Zeranol (ZER) is used extensively in the U.S. and Canada to promote rapid and more efficient growth rates in animals used as sources for meat [632].
As with the natural compound ZEA, ZER is a strongly estrogenic chemical as demonstrated by its ability to stimulate growth and proliferation of human breast tumor cells in vitro at potencies similar to the natural hormones, estradiol, and the known carcinogen, diethylstilbestrol (DES) [633]. A 2007 study demonstrated that adding ZER to cultured breast epithelial cells led to enhanced cell proliferation, accompanied by an upregulation or stimulation of the activity of protein disulfide isomerase, an enzyme whose activity is often increased in cancerous tissues [634].
Treatment of young adult female mice with ZER led to increased growth and branching of mammary glands, similar to what is found in mice treated with the natural hormone estradiol [635]. Increased ductile proliferation, in the absence of full maturation of the ducts through pregnancy and lactation, is associated with an increased risk for mammary (breast) tumors.
Brief (4-day) prepubertal exposure of mice or rats to either ZEA or ZER accelerated the onset of puberty, but did not affect development of the mammary gland structures through early adulthood [636, 637].
A series of studies examined estrogenic activity in normal breast epithelial cells and breast cancer cells treated with ZER. Abnormal cell growth was significant even at ZER levels almost 30 times lower than the FDA-established limit in beef [638]. Follow-up work demonstrated that ZER is comparable to natural estrogen (estradiol) and the synthetic estrogen diethylstilbestrol (DES) in its ability to transform MCF-10A human breast epithelial cells to a pre-cancerous profile in vitro [639]. Preliminary data indicate that serum from ZER-treated beef cattle can stimulate the proliferation of normal breast epithelial cells and the transformation of breast tumor cells in vitro [640, 641].
Bovine growth hormone (rBGH)/Recombinant Bovine Somatotropin (rBST)
Despite opposition from physicians, scientists and consumer advocacy groups, the Food and Drug Administration in 1993 approved Monsanto’s genetically engineered hormone product, recombinant bovine growth hormone (subsequently renamed recombinant bovine somatotrophin, rBST), for injection in dairy cows to increase milk production [642]. This hormone quickly found its way (without labeling) into the U.S. milk supply, and from there into ice cream, buttermilk, cheese, yogurt and other dairy products. Since its introduction, rBST has proven controversial because of its potential carcinogenic effects.
Drinking any type of cow’s milk noticeably raises body levels of insulin growth factor 1 (IGF-1), a naturally occurring hormone in both cows and humans. Injecting cows with rBST leads to an increase in IGF-1 levels in milk [643], although it is possible that the increased milk output by treated animals may dilute the excess production of hormone [644]. The content of IGF-1 in dairy milk is not altered by pasteurization [645].
Although the data are complex with some studies reaching different conclusions, several epidemiological studies have indicated a relationship between dairy consumption and breast cancer risk in pre-menopausal women [646]. Elevated levels of IGF-1, in particular, have been associated with increased risk of breast cancer [647–650]. A nested case-control study within a larger prospective study of American women found that pre-menopausal women with the highest levels of IGF-1 in their blood (drawn before cancer developed) were seven times as likely to develop breast cancer as women with the lowest levels when results were adjusted for plasma concentrations of the IGF binding protein [647]. No increased risk was noted in post-menopausal women. Three studies reported in 2005 by scientists in Sweden, the United Kingdom [651] and the United States [652] also showed an association between circulating levels of IGF-1 and the risk of breast cancer in pre-menopausal women.
One mechanism by which IGF-1 may raise risk in younger women is by increasing breast density in pre-menopausal women, a known risk factor for cancer [653]. In addition, laboratory studies have shown that IGF-1 can regulate the growth and increase the proliferation of breast cancer cells (MCF-7) grown in vitro [654] and decrease the death of mammary tumor cells in laboratory animals [655].
Proponents of rBST argue that IGF-1 is harmless because it occurs naturally in humans, is contained in human saliva and is broken down during digestion. However, animal evidence indicates that digestion does not break down IGF-1 in milk because casein, the principal protein in cow’s milk, protects IGF-1 from the action of digestive enzymes [656].
Section summary: When incorporated into regular nutritious diets, lignans and soy-based foods have been shown to be protective against breast cancer in numerous epidemiological studies. This protection is especially clear when dietary intake begins in childhood. On the other hand, both mycoestrogens and the stock animal growth enhancer zeranol are estrogenic in their interactions with human breast cells, including cells derived from cancers, in cell culture environments. The data are more ambiguous on possible effects of elevated IGF-1 levels, found after drinking cow’s milk.
Non-EDC industrial chemicals
Not all chemicals associated with increased risk for breast cancer exert their effects through endocrine disrupting mechanisms. This section examines the literatures linking a increased risk for developing breast cancer to a few industrial chemicals, all of which have been determined to be carcinogenic by IARC (see Table 3). These compounds and/or the DNA adducts formed following exposures to the compounds, are directly mutagenic.
Table 3.
Carcinogenicity classifications and sources of exposures of chemicals found in non-EDC industrial chemicals
| Chemical | IARC | NTP | Source of exposures |
|---|---|---|---|
| Non-EDC Industrial Chemicals | |||
| Benzene | 1 | K | Petrochemical solvent |
| Vinyl chloride | 1 | K | Monomer used in polyvinyl chloride (PVC) plastic |
| 1,3-Butadiene | 1 | K | Byproduct of combustion |
| Ethylene oxide | 1 | K | Sterilizer, byproduct contaminant in some cosmetics |
International Agency for Research on Cancer (IARC) classifications: 1 = Carcinogenic to humans, 2A = Probably carcinogenic to humans, 2B = Possibly carcinogenic to humans, 3 = Not classifiable as to its carcinogenicity to humans; U.S. National Toxicology Program (NTP) classifications: K = Known to be a human carcinogen, RA = Reasonably anticipated to be a human carcinogen. Source of exposure list contains most common exposure sources
Benzene
Benzene is one of the largest volume petrochemical solvents currently in production, and global production rates are expected to continue to grow over the next several years. Chemical industries estimate that more than 46 million metric tons (more than 115 billion pounds) of benzene will be consumed globally by the year 2020 [657]. Exposures to benzene come from inhaling gasoline fumes, automobile exhaust, or cigarette smoke (primary and secondary) and from industrial burning. Benzene presents a serious occupational hazard for people exposed through their work in chemical, rubber, shoe manufacturing, oil and gasoline refining industries. Both the NTP and IARC have designated benzene as a human carcinogen [658, 659].
Epidemiological studies of the effects of benzene on breast cancer risk are difficult to conduct, mainly because exposures to benzene occur in conjunction with exposures to other chemicals that are also released in combustion and manufacturing processes. Also, few of the occupational studies focusing on chemical and automotive industries have included women in substantial numbers to draw meaningful conclusions. One study that did look at relevant occupations among female Chinese workers, examined incidence by occupation as standardized by general breast cancer incidence rates in Shanghai and the number of women in each occupation according to the 1982 census. The occupations in which elevated risks for breast cancer were found included scientific research workers (SIR + 3.3); medical workers practicing Western style medicine (SIR =14.7, 95% CI = 5.9–30.3) or Chinese-Western style medicine (SIR = 7.2; 95% CI = 4.4–11.4); as well as workers with expected lower exposures such as teachers, librarians and accountants (SIRs 2.3–2.7). In the same study, looking across professions, benzene exposure was associated with an elevated risk of breast cancer [660]. A study of a fairly small sample of women for whom researchers have benzene exposure data from their work at a shoe factory in Florence, Italy, also supports a relationship between exposure to benzene and later development of breast cancer [661].
The largest study implicating benzene and associated chemicals comes from an occupational study looking at men who have been diagnosed with breast cancer. Men who had worked in professions that involved exposures to gasoline fumes and combustion had significantly increased rates of breast cancer. The effect was most pronounced among men who started at their jobs before the age of 40 [662].
Benzene administration to laboratory mice induces mammary tumors. Mice exposed to benzene have frequent mutations of genes that are responsible for suppressing the development of tumors [663, 664].
Vinyl chloride
Manufacturers use polyvinyl chloride (PVC) extensively to produce food packaging, medical products, appliances, cars, toys, credit cards and rainwear. When PVC is made, vinyl chloride may be released into the air or wastewater. Vinyl chloride has also been found in the air near hazardous waste sites and landfills and in tobacco smoke.
Vinyl chloride was one of the first chemicals designated as a human carcinogen by the NTP [665, 666]. Vinyl chloride has been linked to increased mortality from breast and liver cancer among workers involved in its manufacture [667, 668]. In the large prospective cohort California Teachers Study, exposure to vinyl chloride was associated with increased breast cancer risk. Analyses of subsets within the cohort reveal significant associations between vinyl chloride exposure (for highest quintile vinyl chloride exposure) and ER+/PR+ tumors (HR = 1.08; 95% CI = 0.98–1.19), and in women who had never, or were not currently, using HRT (HR = 1.27; 95% CI =1.04–1.54) [669]. In a case-control study of male breast cancer patients, all of whom had lived at Camp LeJeune during the decades when the drinking water was contaminated with several toxic solvents, exposures to vinyl chloride was associated with higher risk of developing breast cancer (OR = 1.20 (95% CI = .16–5.89) and earlier onset of the disease (OR = 2.14; 95% CI =0.31–14.81) [670].
Animals exposed long-term to low levels of airborne vinyl chloride show an increased risk of mammary tumors [671].
1,3-butadiene
1,3-butadiene is an air pollutant created by internal combustion engines and petroleum refineries. It is also a chemical used in the manufacture and processing of synthetic rubber products and some fungicides. In addition, 1,3-butadiene is found in tobacco smoke.
The EPA determined that 1,3-butadiene is carcinogenic to humans, with the main route of exposure being through inhalation. Women working in the synthetic rubber industry who had high exposures to 1,3-butadiene had increased risk of dying from breast cancer (RR = 2.6 m 95% CI = .9–7.3) [672]. The NTP classifies 1,3-butadiene as a known human carcinogen [673].
Data from research on animals indicate that females may be more vulnerable to the carcinogenic effects of 1,3-butadiene [674], which is known to cause mammary and ovary tumors in female mice and rats. This pollutant produces even greater toxic effects in younger rodent populations [675].
Ethylene oxide
Ethylene oxide is a fumigant used to sterilize surgical instruments and is also used in some cosmetic products [676]. Ethylene oxide is classified as a human carcinogen [677, 678] and one of 221 chemicals identified by researchers at the Silent Spring Institute as being associated with mammary tumors in animals [197].
Scientists from the National Institute for Occupational Safety and Health (NIOSH) studied breast cancer incidence in 7576 women exposed to ethylene oxide while working in commercial sterilization facilities. They found an increased incidence of breast cancer among these women in direct proportion to their cumulative exposure to ethylene oxide [679]. Although there are contradictory data in the recent literature [678], other occupational studies support the finding that exposure to ethylene oxide is associated with increased risk for breast cancer in women [680, 681].
Studies in which human breast cells grown in vitro were exposed to low doses of ethylene oxide demonstrated that the chemical exposure resulted in a significant increase in damage to the cells’ DNA [682]. These findings are supported by results of a study examining gene mutations in mammary tumors induced in mice by exposures to ethylene oxide. Common mutations included those in the tumor suppressor gene, p53, and the cell proliferation regulatory gene, H-ras [664].
Section summary: Epidemiological studies of both men and women exposed occupationally to benzene or vinyl chloride have higher risk for developing breast cancer. Limited human also indicate that exposures to 1,3-butadiene also have an increased risk for breast cancer, while the evidence supporting this relationship is more robust for ethylene oxide.
Tobacco smoking: active and passive
Increasing evidence indicat4es that exposure to the many chemicals included in tobacco smoke, both through active (first hand) and passive (second hand) means, can increase risk for developing breast cancer. We discuss this literature in this section. While exposures to smoke are often clustered with alcohol consumption and other lifestyle factors, in this category we only focus on tobacco smoke exposures as these are from chemicals polluting the environment, and the exposures are often involuntary.
Tobacco smoke contains polycyclic aromatic hydrocarbons (PAHs), as well as hundreds of other chemicals [683], including three known human carcinogens (polonium-210, a radioactive element; benzene; and vinyl chloride) as well as 1,3-butadiene and nicotine-derived nitrosamine ketone (NNK), all of which are known to cause mammary tumors in animals. NNK is a tobacco-specific carcinogen that has been shown to increase tumor cell proliferation and the transformation of healthy breast epithelial cells into cancer cells [684–686], at least in part via the nicotinic acetylcholine receptor [687] (see Table 4).
Table 4.
Carcinogenicity classifications of chemical exposures found in cigarette smoke
| Chemical | IARC | NTP |
|---|---|---|
| Tobacco smoking: Active and passive | K | |
| Polycyclic aromatic hydrocarbons (PAHs) | RA | |
| Polonium-210 | ||
| Benzene | 1 | K |
| Vinyl chloride | 1 | K |
| 1,3-butadiene | 1 | K |
| Nitrosamine ketone (NNK) |
International Agency for Research on Cancer (IARC) classifications: 1 = Carcinogenic to humans, 2A = Probably carcinogenic to humans, 2B = Possibly carcinogenic to humans, 3 = Not classifiable as to its carcinogenicity to humans; U.S. National Toxicology Program (NTP) classifications: K = Known to be a human carcinogen, RA = Reasonably anticipated to be a human carcinogen. Source of exposure list contains most common exposure sources
A large study of California teachers revealed an increased risk of breast cancer among smokers, particularly those who began smoking during adolescence (HR = 1.17; 95% CI = 1.05–1.30), at least 5 years before their first full-term pregnancy (HR = 1.13; 95% CI = 1.00–1.28), or who were longtime or heavy smokers (HR = 1.32; 95% CI = 1.10–1.57) [688]. Several earlier studies also suggest that women who begin smoking cigarettes as adolescents face increased risks of breast cancer [689–693].
Results from the Canadian National Breast Screening Study indicated that increased incidence of breast cancer was associated with longer duration of smoking (RR = 1.50; 95% CI = 1.19–1.89), number of cigarettes smoked per day (for 40 cigarettes/day: RR = 1.20; 95% CI = 1.00–1.44), and cumulative exposure to cigarette smoke (40 pack-years: RR = 1.17; 95% CI = 1.02–1.34) [694]. Similar results were recorded in reports from two large prospective studies: the Nurses Health Study [695] and the WHI study [696], which involved approximately 110,000 and 80,000 participants, respectively.
Although several more recent studies have reported that beginning smoking before a first full-term pregnancy (independent of age of onset of smoking) may make a woman increasingly susceptible to later diagnosis with breast cancer [691, 695, 697], a 2011 meta-analysis of 23 relevant research papers did not find a statistically significant relationship [698]. Complicating this picture is a report from the EPIC cohort reporting that the most important impact of active cigarette smoking was the number of pack-years (1 pack-year = 20 cigarettes/day for a full year) smoked from menarche to first full-term pregnancy (HR = 1.73, 95% CI = 1.29–2.32 for every increase of 20 pack-years). On the other hand, the number of pack-years smoked following menopause was significantly associated with a decreased risk for developing breast cancer (HR = 0.53; 95% CI = 0.34–0.82) [699]. Very different results were reported in a large study from the African American Breast Cancer Epidemiology and Risk (AMBER) study: as compared with women who never smoked, higher pack-years of active smoking in pre-menopausal women was associated with a decreased risk of breast cancer (OR = 0.80; 95% CI = 0.68–0.96), while higher pack years smoked in active postmenopausal smokers was associated with an increased risk of breast cancer (OR = 1.16; 95% CI = 1.01–1.33) [700]. The postmenopausal effect was strongest in women developing ER+ cancers. In post-menopausal women, an inverse relationship was found between active smoking and mammographic breast density, with the effect being magnified for women who started smoking before the age of 16 (OR = 0.79; 95% CI = 0.64–0.96) [701]. Lower breast density is associated with lower risk of developing breast cancer [702].
A population-based case control study examined effects of active smoking and risk of developing breast cancer based on whether tumors were luminal (ER+ and/or PR+) or basal (ER-, PR-, HER2-) types. Ever smoking led to an increased risk of developing luminal-type cancer (OR = 1.12; 95% CI = 0.92–1.33), but not basal-type, an effect that was most pronounced in Black women. Another study looking at inflammatory breast cancer incidence reported effects of active smoking on luminal (OR = 2.37; 95% CI = 1.24–4.52), but not other types of breast cancer [703]. However increased smoking duration was associated with an increased risk of developing basal type (OR = 1.51; 95% CI = 1.19–1.93), but not luminal, cancers [704].
Ethnic differences were reported in a study of Mexican and U.S. non-Hispanic white women. For Mexican women, a significant increase in breast cancer risk was found for former smokers (OR = 1.43; 95% CI = 1.04–1.96 vs. never smokers), and this effect was increased for former smokers with a history of alcohol consumption (OR = 2.30; 95% CI = 1.01–5.21). For U.S. non-Hispanic white women, current smoking of more than 20 cigarettes a day was associated with increased risk (OR = 1.61; 95% CI = 1.07–2.41). There were no significant effects found for U.S. Hispanic white women [705].
A prospective cohort study of 186,150 female AARP members, 7486 of whom developed breast cancer, found an increased risk of developing breast cancer in current active smokers (OR = 1.19; 95% CI = 1.10–1.28) as well as former active smokers (OR = 1.07; 95% CI = 1.01–1.13). For current smokers, the effect was significant in women with no family history of breast cancer (OR = 1.24; 95% CI = 1.15–1.35), but not in women with a family history. Later age of menarche was also associated with higher risk for developing breast cancer in active smokers (age of menarche x smoking status interaction, p < .03) [706].
Several recent studies have examined the effects of smoking at the time of breast cancer diagnosis and subsequent outcomes. Bérubé reported that smoking at the time of diagnosis led to an increase in all-cause mortality, as well as a significant increase in mortality from breast cancer (HR = 1.33; 95% CI = 1.12–1.58) [707]. Similar effects on breast-cancer mortality were reported (HR = 1.10; 95% CI = 0.73–1.68) in women diagnosed with localized breast cancer [708]. Continued active smoking after diagnosis was associated increase in breast cancer-related deaths (HR = 1.72; 95% CI = 1.13–2.60) [709].
In 309 female ER+ breast cancer patients being treated with aromatase inhibitors, smoking before surgical treatment for their disease was associated with increased numbers of breast cancer events (e.g., recurrence, new breast cancer diagnosis, metastasis; HR = 2.97; 95% CI = 1.44–6.13), distant metastases (HR = 4.19; 95% CI = 1.81–9.72), and mortality (HR = 3.52; 95% CI = 1.59–7.61). There was no relationship between smoking and breast cancer-related outcomes in women undergoing other forms of adjuvant therapy [710].
Two recent studies have examined smoking status and breast cancer outcomes in men. In a pooled case-study consortium with 2378 cases of male breast cancer in Florida, Cook et al. found no evidence of association between smoking status, pack-years, duration, or age at initiation of smoking and risk of developing breast cancer [711]. Another study of male breast cancer cases in Florida examined survival rates following diagnosis and stratified their analysis by race-ethnicity and socioeconomic status. Overall, as compared with never smokers, current smokers had higher mortality rates (HR = 1.63; 95% CI = 1.23–2.16), although there was no effect for past smokers who had given up the habit. There was a dose-response relationship between amount smoked and mortality risk (trend, p < .001). Similar effects were found for both White (but not Black) and non-Hispanic (but not Hispanic) men [712].
In addition to effects of active smoking on breast cancer incidence and mortality, a growing literature implicates exposures to second hand smoke (passive smoking) to increased risk for the disease. Until recently, more evidence linked secondhand smoke than active smoking to breast cancer risk. Current evidence suggests that both exposures increase breast cancer risk by about the same amount, even though women who are exposed to secondhand smoke receive a much lower dose of carcinogens than do active smokers [699, 713, 714]. Researchers at Japan’s National Cancer Center reported the results of a study involving 21,000 women ages 40 to 59. They found that the risk of breast cancer was elevated in pre-menopausal women who were either active smokers (RR = 3.9; 95% CI = 1.5–9.9) or exposed to second-hand environmental smoke (RR = 2.6; 95% CI = 1.3–5.2) [715]. Other major studies, including the WHI, support the finding of a link between extensive exposure to passive smoking lasting more than 10 years and increased risk for breast cancer (HR = 1.32; 95% CI = 1.04–1.67) [696]. Exposures to passive smoke at home, but not at work, increases risk of developing breast cancer (OR = 1.30; 95% CI = 1.05–1.61) and the amount of exposure at home is linked in a dose-response fashion (p = .009) [716].
A meta-analysis of eight studies of Chinese women exposed to passive smoking who were never active smokers themselves showed a significant increase in risk of developing breast cancer (OR = 1.67; 95% CI = 1.27–2.21) [717]. A more detailed analysis of Chinese women who had never smoked showed a significant effect of more than 4 pack-years of exposure to passive smoke (OR = 1.71; 95% CI = 1.17–2.50). The effect was found in women with ER+/PR+ tumors, but not for other tumor subtypes [718].
In trying to understand the mechanisms by which active and/or passive smoking might affect risk for developing breast cancer, several gene expression studies have been conducted. Studies exploring links between smoking and breast cancer incidence, recurrence and mortality have identified several polymorphisms associated with increased risk. The most consistent data implicate a ‘slow acetylator’ n-acetyltransferase 2 (NAT2) phenotype [719, 720], although specific polymorphisms of the BRCA1 [719, 721] and the CYP1A1 and COMT [719] genes have also been reported to be associated with increased incidence and/or mortality in active smokers.
Other physiological disruptions resulting from exposures to smoke include damaging the structure and function of the ovaries, thereby lowering estrogen levels in pre-menopausal women. While lower levels of estrogen would decrease breast cancer risk, at the same time carcinogens in cigarette smoke would increase risk of developing breast cancer [722].
A cross sectional study which is a part of a large, on-going prospective project (the EPIC cohort), examined the association between tobacco smoking and sex hormone levels in post-menopausal women, whose ovaries are no longer the major source of their of circulating hormones. Smoking was related to higher levels of testosterone, estradiol and other steroid hormones [723]. The increased levels of circulating estradiol were only statistically significant for women who were considerably overweight. By itself, obesity is a known risk factor for postmenopausal breast cancer. Adipose tissue is the main site of aromatization of testosterone to estradiol in men and postmenopausal women, and increased adipose tissue can thus contribute to increased circulating estrogens. Greater activation of breast fat cell metabolic pathways by tobacco-containing chemicals may enhance the development of breast cancer [724].
Section summary: There is now a substantial literature indicating that past and current active cigarette smoking is associated with a higher risk for developing breast cancer. For women who are smokers at the time of diagnosis, there is also an increased risk in mortality from breast cancer. These effects are complicated by interactions with race/ethnicity, history of alcohol consumption and subtype of breast cancer being evaluated.
Shift work, light-at-night and melatonin
In 2007, IARC concluded that shift work is ‘probably carcinogenic to humans’ based in large part on the growing association between shift working and increased incidence of breast cancer [725] (see Table 5). Several occupational studies have demonstrated that women who consistently work night shifts have increased breast cancer risk, although not all reports have found evidence for this relationship. Methodological differences between studies, including varied definitions of “shift work” and “night,” as well as lack of consistent attention to confounding factors may explain some of the differences in results between individual studies [726, 727].
Table 5.
Carcinogenicity classifiations and exposure sources of light-at night and radiation
| Exposure | IARC | NTP | Use |
|---|---|---|---|
| Shift Work, Light-at-Night | PR | Shift work or ambient light pollution | |
| Ionizing Radiation | K | K | Diagnostic medical tests; nuclear medicine procedures; nuclear power plants, research protocols |
| Non-ionizing radiation (electromagnetic fields) | Lighting, computers, cell phones and other electronic sources |
International Agency for Research on Cancer (IARC) classifications: 1 = Carcinogenic to humans, 2A = Probably carcinogenic to humans, 2B = Possibly carcinogenic to humans, 3 = Not classifiable as to its carcinogenicity to humans; U.S. National Toxicology Program (NTP) classifications: K = Known to be a human carcinogen, RA = Reasonably anticipated to be a human carcinogen. Source of exposure list contains most common exposure sources
Associations between long-term (> 20–30 years) night-shift work and breast cancer were reported in a 2008 comprehensive review of 13 studies [728]. Four other reviews that included meta-analyses reached similar conclusions although the strength of the associations varied considerably. Based on analysis of 13 studies, Megdal and colleagues reported an aggregated estimate of breast cancer risk (RR) for both airline attendants and others working on night shift work as 1.48 (95% CI = 1.36–1.61) [729]. Kamdar and colleagues included 15 case-control and cohort studies examining the possible relationship between night shift work and breast cancer risk, and reported a pooled RR of 1.21 (95% CI = 1.00–1.47) for individuals with any experience with night shift work as compared with those without such experience [730]. He et al. included a more heterogeneous group of 28 studies that examined circadian rhythm disruptions, but defined in various ways (shift work, short sleep duration, occupation as flight attendant, light-at-night exposure). They reported an aggregate RR of 1.14 (95% CI = 1.08–1.21), with similar RRs when analyses were limited to just studies examining shift work (RR = 1.19; 95% CI = 1.08–1.32) or light-at-night exposure (RR = 1.12; 95% CI = 1.12–1.12), but a considerably higher risk when studies of just flight attendants were analyzed (RR =1.56 (95% CI = 1.10–2.56). Shorter duration of sleep did not confer a change in risk for developing breast cancer in this analysis [731]. Lin, et al. analyzed 16 prospective cohort studies and reported an aggregate RR of 1.09 (95% CI = 1.02–1.17) for night shift workers compared with day workers. A linear trend (p = 0.010) was found for increased exposure length (< 5, 5–10, 10–20 and >20 years) and risk for developing breast cancer [732]. On the otherhand, a new meta-analysis of 10 prospective studies, including a total of 1.4 million women, found no effects on breast cancer risk for engaging in any shift work (RR = 0.99; 95% CI = 0.95–1.03), for 20 or more years of night shift work (RR = 1.01; 95% CI = 0.93–1.10), or for 30 or more years of night shift work (RR = 1.00; 95% CI = 0.87–1.14) [733]. A more recent evaluation of this meta-analysis called into question several methodological criteria and data interpretations within the report [734].
A record linkage study of occupation and cancer in Britain calculated that for 2012, the population attributable factor (PAF) of night shift work may account for 4.5% (95% CI = 3.2–5.9) of breast cancer diagnoses (1957 cases; 95% CI = 1395–2547) and deaths (552 cases; 95% CI = 393–724) [735]. A similar analysis in 2015 calculated a PAF for shift work of 5.7% (95% CI = 0.0—11.9) of U.S. women being diagnosed with breast cancer (attributable breast cancer cases = 11,777; 95% CI = 0–24,625) [736].
In a study of Danish nurses, effects of night shift work on breast cancer risk were greatest for women who worked rotating hours that include the overnight, as opposed to evening shift (OR = 1.8; 95% CI = 1.2–2.8) and for those who worked 12-h shifts that alternated day and night work, as compared to shorter work periods (OR = 2.9; 95% CI = 1.1–8.0) [737].
Risk of developing breast cancer increased for women who worked night shifts for more than 4.5 to 5 years (OR = 1.40; 95% CI = 1.01–1.92), and for those who regularly engaged in night work for at least 4 years prior to their first pregnancy (OR = 1.95; 95% CI = 1.13–3.35), therefore before the time when their mammary cells had fully differentiated [738].
The most thoroughly studied mechanism to explain these effects of night shift work is the light-at-night (LAN) hypothesis [739]. Increasing exposure to light, especially bright indoor light, at times outside of normal daylight hours, decreases secretion of melatonin by the pineal gland. Normal high levels of melatonin at nighttime are important for regulation of both pituitary and ovarian hormones, for suppressing the local production of estrogen resulting from aromatization of androgens in breast tumor cells, and for maintaining normal metabolic profiles and body weight [740–743].
Clinical studies have demonstrated that there is a decrease in the peak amount of melatonin secreted in women with metastatic cancer, as compared with healthy women, and larger tumors are associated with lower levels of melatonin [740]. Blind women who are completely unable to perceive the presence of environmental light, and concommitantly have no daily decreases in melatonin levels, have significantly lower risk of diagnosis of breast cancer than do blind women who do perceive light and have regular changes in melatonin secretion over the normal 24-h cycle (OR = 0.52; 95% CI = 0.27–1.01) [744].
One proposed pathway by which reduced melatonin might affect breast cancer risk is enhancement of the production or secretion of estradiol and other ovarian hormones. Nagata et al. reported that postmenopausal women who worked night shifts that went beyond midnight had significantly increased serum concentrations of estradiol both during their night shift phases and when they rotated to regular day awake periods, as compared with controls who did not engage in late night shift work [745]. Davis et al. tested urinary levels of 6-sulfatoxymelatonin (the major metabolite of melatonin), LH, FSH and estrone conjugate across sleep and work cycles for premenopausal nurses working both day and night shifts. As compared with nurses working day shifts, in night shift workers, LH and FSH levels were both significantly higher (35 and 38% higher, respectively) while 6-sulfatoxymelatonin levels were significantly decreased (69% lower); no significant differences were found in estrone conjugate [746]. However, one study that examined the possible link between changes in melatonin levels and changes in reproductive hormone levels did not find a relationship once other factors like age, menstrual status and body mass index were factored into the analysis [747].
In rodent models, higher levels of melatonin are associated with decreased incidence and size of mammary tumors, and when they do occur, the latency period of tumor development is lengthened [739]. In human mammary tumors that had been grafted into mice, perfusion with blood taken from women at night (when melatonin is high) decreased proliferation and growth of mammary tumors, as compared to the use of samples collected during the day when melatonin levels are naturally lower [748].
Mechanistically, night pulses of melatonin enhance the activity of endocrine, metabolic and immune-related pathways that can prevent the development of cancer [749]. These protective effects of melatonin are mediated by epigenetic changes in many of the genes involved in regulation of cell growth and proliferation, as well as in the synthesis and activation of the estrogen receptor [750]. Genes that are associated with the regulation of the daily melatonin cycle also regulate other pathways that may be involved in the development of breast cancer. Structural variation in one such gene, Per3, is associated with higher breast cancer rates in young women [751]. Per2, another gene associated with the control of daily rhythms, is also poorly regulated in many women with breast cancer, with normal structure and expression of this gene being associated with lower effectiveness of estradiol in altering cellular activity. In healthy cells, Per2 also may act directly as a tumor-suppressor gene, decreasing the activity of pathways associated with tumor formation [752]. A rare polymorphism of the CLOCK gene has been associated with an increased risk for developing breast cancer (OR = 3.53; 95% CI +1.09–11.42), and there was a positive interaction between the presence of this genotype and night shift work on risk for developing breast cancer (p = .02) [753]. However, a case-control study that evaluated 100 SNPs of 14 clock-related genes in interaction with shift work history found no associations for any of the SNPs [754].
Other recent studies have greatly complicated the light-at-night and breast cancer story. Relationships between night shift work and melatonin levels may be mediated by race/ethnic background. In a large population-based study of Chinese women, no association between shift work and breast cancer incidence was reported [755]. Asian and Asian-American women who work night shifts have less melatonin suppression than their white counterparts [756]. Effects of night shift work may also be limited to specific breast tumor types. In a population based case-control study of night shift workers, adjusting for chronotype (mid-sleep point on days when participants could choose when to sleep), resulted in increased risk of invasive (vs. in situ) tumors (OR = 1.23; 95% CI = 1.05–1.99) and increased risk of premenopausal ER+/PR+ tumors (OR = 1.44; 95% CI = 1.05–1.99) [757].
Several authors have proposed that factors associated with night shift work, beyond decreases in melatonin levels, need to be considered in understanding better the links with increased risk for breast cancer. Other possible consequences of shift work, including phase shift sleep disruption, lifestyle factors, changes in metabolism, desynchronization between central neural and peripheral systems, or decreased vitamin D production, may also be linked to increased cancer rates. These factors need to be studied as both single and possibly interacting factors in altered risk for developing breast cancer [758–762].
Finally, as with several endocrine disrupting chemicals, light-at-night decreases significantly the effectiveness of major chemotherapeutic agents used in the treatment of breast cancer. In rat models with MCF-7 breast cancer cells xenografts, addition of dim light exposures during the dark phase of the cycle led to decreased melatonin secretion during the dark phase, decreased latency to tumor progression, increased tumor growth, and complete resistance to both tamoxifen and doxorubicin [763, 764].
Section summary: Extensive experience with night shift work, and therefore higher exposure to light-at-night (LAN), has been shown to increase risk for breast cancer, although there may be ethnic differences in this response. The most studied underlying mechanism for the effect of LAN exposures is the accompanying change in patterns of melatonin secretion. Other lifestyle and physiological factors associated with shift work have also been proposed to alter risk for developing breast cancer.
Radiation
Exposure to ionizing radiation, from both military and medical sources, is the best known and longest established environmental cause of breast cancer in both women and men. Exposures early in life, during childhood through adolescence, are particularly important. Data for potential links between electromagnetic fields, or non-ionizing radiation, and breast cancer are mixed and inconclusive.
Ionizing radiation
Ionizing radiation is any form of radiation with enough energy to break off electrons from atoms (to ionize the atoms). This radiation can break the chemical bonds in molecules, including DNA molecules, thereby disturbing their normal functioning. X-rays and gamma rays are the only major forms of radiation with sufficient energy to penetrate and damage body tissue below the surface of the skin.
Among the many sources of ionizing radiation are traditional X-rays, computed tomography (CT) scans, fluoroscopy and other medical radiological procedures. Sources of gamma rays include emissions from nuclear power plants, scientific research involving radionuclides, military weapons testing and nuclear medicine procedures such as bone, thyroid and lung scans [765].
In 2005, the National Toxicology Program classified X-radiation and gamma radiation as known human carcinogens [766] (see Table 5). Although some scientists challenge this premise [767], most agree that no safe dose of radiation has been identified [768, 769]. Radiation damage to genes is cumulative over a lifetime [770]. Repeated low-dose exposures over time may have the same harmful effects as a single high-dose exposure.
Exposure to ionizing radiation is the best- and longest-established environmental cause of human breast cancer in both women and men. The link between radiation exposure and breast cancer has been demonstrated in atomic bomb survivors [771–774]. Rates of breast cancer were highest among women who were younger than age 20 when the United States dropped atomic bombs on Hiroshima and Nagasaki [773]. In addition, Ron et al. reported a significant association between ionizing radiation exposure and the incidence of male breast cancer in Japanese atomic bomb survivors [775].
Ionizing radiation can increase the risk for breast cancer through a number of different mechanisms, including direct mutagenesis, genomic instability [776, 777] and changes in breast cell micro-environments that can lead to damaged regulation of cell-cell interactions within the breast [778–780]. Ionizing radiation not only affects cells that are directly exposed, but it can also alter the DNA, cell growth and cell-cell interactions of neighboring cells, referred to as the ‘bystander effect.’ [767, 781]. A G2 micronucleus assay of blood samples from asymptomatic women carrying the BRCA1 mutation have deficits in many of these cell processes and a heightened sensitivity to the effects of radiation exposures, as compared with samples from healthy women without the mutation [782].
Interactions Between Radiation and Other Factors. There are a number of factors that may interact with radiation to increase the potency of its carcinogenic effects. Some of these factors include a woman’s age at exposure, genetic profile and possibly estrogen levels. Studies of women exposed to military, accidental or medical sources of radiation have demonstrated clearly that children and adolescents who are exposed are more seriously affected in their later risk for breast cancer than are older women [769]. In addition, recent genetic data indicate that women with some gene mutations (e.g., ATM, TP53 and BRCA1/2) are more likely to develop breast cancer and may be especially susceptible to the cancer-inducing effects of exposures to ionizing radiation [71, 783–785].
Studies using animal and in vitro human breast tumor cell culture models have demonstrated that the effects of radiation on mammary carcinogenesis may be additive with effects of estrogens [786–788]. This is of particular concern given the widespread exposure to estrogen-mimicking chemicals in our environment and the multiple sources of ionizing radiation.
Occupational exposures
Female radiology technologists who had sustained daily exposures to ionizing radiation demonstrated an increased risk of breast cancer for those women who began working during their teens or, independent of age, working in the field before the 1940s, when exposure levels were substantially higher than they have been in more recent decades [789, 790]. Follow-up of this cohort for another decade revealed an increased mortality rate for technologists who began work before 1950, with a significant trend (P = .01) for correlation with earlier year beginning work. Technologists who began working before 1950 and had worked for at least 5 years had an increased mortality rate from breast cancer (HR = 2.25; 95% CI = .95–6.68) [791]. In a subset of this cohort who had worked with fluoroscopically guided interventional procedures, there was an increased incidence of breast cancer compared to technologists who had not engaged in this work (HR = 1.166; 95% CI = 1.02–1.32), but no effect on mortality from breast cancer [792]. The susceptibility of radiologists to later diagnosis of breast cancer may be affected by common variants in genes that are involved in the metabolism of circulating estrogens [793].
A review and analysis of all existing related studies found that women who work as airline flight attendants had increased levels of breast cancer [794]. A meta-analysis of seven studies examining cancer incidence in flight attendants reported an elevated incidence of breast cancer (SIR = 1.40; 95% CI = 1.19–1.65) [795]. Factors that could explain this increase may include lifestyle and reproductive histories, light-at-night exposures, as well as increased exposures to cosmic (atmospheric) ionizing radiation.
Medical radiation: risks and benefits
Medical X-rays: Use of X-rays to examine the spine, heart, lungs, ribs, shoulders and esophagus also exposes parts of the breast to radiation. X-rays and fluoroscopy of infants irradiate the whole body [796]. Decades of research have confirmed the link between radiation and breast cancer in women who were irradiated for many different medical conditions, including tuberculosis [105], benign breast disease [106, 107], acute postpartum mastitis [108], enlarged thymus [109, 110], skin hemangiomas [111], scoliosis [112], Hodgkin’s disease [113–116], non-Hodgkin’s lymphoma [117], acne [118], and prophylactic dental care [119].
Anytime use of diagnostic chest X-rays before the age of 50 years in women carrying the BRCA1/2 gene mutations is associated with an increased risk of breast cancer (BRCA1 OR = 1.16; 95% CI = .64–2.11; BRCA2 OR = 1.22; 95% CI = .62–2.42) [797].
Evidence from almost all conditions suggests that exposure to ionizing radiation during childhood and adolescence is particularly dangerous with respect to increased risk for breast cancer later in life [75, 120, 121, 126] and that there is a significant dose-response relationship between the dosage of childhood radiation and the increased incidence of breast cancer (trend p < .001) [798]. Importantly, use of radiation in pediatric medicine leads to higher effective dose for children than for adults given the equivalent radiation exposure, a reflection of their smaller body sizes [799, 800].
Computed Tomography (CT) Scans: There is credible evidence that medical X-rays (including mammography, fluoroscopy and CT scans) are an important and controllable cause of breast cancer [119, 801]. Although there has been a substantial decrease in exposures to ionizing radiation from individual X-rays over the past several decades, there has been a six-fold increase in exposure to medical sources of radiation from the mid-1980s through 2007, with an annual increase of 16%, primarily from the increased use of CT scans and nuclear medicine [802, 803]. In 2007, approximately 72 million CT scans were conducted in the United States [804]. When a CT scan is directed to the chest, the individual receives the equivalent radiation of 30 to 442 chest X-rays [805]. Modeling estimates have indicated that use of chest CTs and CT angiography in 2007 alone will lead to an additional 5300 cases of lung and breast cancer within the next two to three decades [804]. Other modeling suggests that 1 in 150 women who are 20 years old when they undergo CT angiograms of the chest, and 1 in 270 women of all ages having the procedure, will subsequently develop cancers of the chest, including breast cancer [806].
CT angiography, a source of comparatively high radiation to the chest, has been associated with a significant increase in risk for developing breast cancer, especially in pre-menopausal women [807, 808].
Mammography: Many experts believe that the low-dose exposures to radiation received as a result of mammographic procedures are not sufficient to increase risk for breast cancer. However, damage from lower-energy sources of X-rays, including those used in mammography, cannot be predicted by estimating risk from models based on higher doses [75, 809]. Evidence indicates that the lower-energy X-rays provided by mammography resulted in substantially greater damage to DNA than would be predicted by those models. Evidence also suggests that risk of breast cancer caused by exposure to mammography radiation may be greatly underestimated [808].
As with other risk factors for breast cancer, both age at exposure and the individual’s genetic profile influence the degree of increased risk for disease in women exposed to multiple mammograms. For example, women who had multiple mammograms more than 5 years prior to diagnosis had an increased risk for breast cancer, but the effect was only statistically significant for women whose first mammograms began before age 35 [119].
This age effect is of particular concern, as it is often recommended that high-risk women, including carriers of either of the BRCA mutations, begin annual mammography screening at age 25 to 30. But, young women with these mutations are actually more vulnerable to the cancer-inducing effects of early and repeated mammograms. This increased vulnerability has been reported in women with BRCA1/2 mutations [71, 72] as well in women with other relatively uncommon variations in genes known to be involved in the process of DNA repair [75]. For women with BRCA2 mutations, meta-analysis of seven articles found low-dose exposures from either mammography or chest X-rays led to an increase risk for breast cancer (OR = 1.3; 95% CI = .9–1.8). Exposure before the age of 20 increased risk (OR = 2.0; 95% CI = 1.3–3.1), as did greater than 5 years of exposures (OR = 1.8; 95% CI = 1.1–3.0) [67]. Diagnostic radiation has been shown to increase risk for developing breast cancer in a dose-dependent manner [73].
The detrimental risks from mammography might also be heightened in older women, whose breast epithelial cells have gone through several decades of cell division. Cells derived from older women’s breast tissue were more sensitive to the DNA-damaging effects of low-energy radiation, increasing the likelihood of later conversion to cancerous cells [810].
In 2009, the U.S. Preventive Services Task Force (USPSTF) recommended against the use of routine mammography screening before the age of 50 (Nelson, [811]; USPSTF, 2009) but supported the use of biennial screening between the ages of 50 and 75 [811]. These recommendations were based on models using a number of factors, including positive and negative test results and the psychological consequences of those results on women; number of follow-up imaging procedures and biopsies; actual diagnoses; and, ultimately, mortality rates from breast cancer. Not considered in the analysis was the contribution of radiation from either single or repeated mammograms or other follow-up tests [812]. Several analyses suggest that for women over the age of 40 who are not at high risk, the trade-offs between diagnostic efficacy of mammography and radiation exposure lean more in the favor of regular mammography screening [813–815]. In 2016, the USPSTF updated their recommendations for women between 40 and 49 years, leaving the decision on whether or not to start mammographic screening up to the individual woman [816]. As women are now facing the need to make their own decisions about whether to undergo routine screening mammography, it is critical that both physicians and women are better educated about mammography’s potential harms, along with its potential benefits [72, 817].
Radiation therapy: Some studies suggest that doctors and patients should carefully evaluate the risks and benefits of radiation therapy for survivors of early-stage breast cancer, particularly older women. Women older than 55 derive less benefit from radiation therapy in terms of reduced rate of local recurrence [818] and may face increased risks of radiation-induced cardiovascular complications [819], as well as secondary cancers such as leukemias and cancers of the lung, esophagus, stomach and breast [820, 821]. Using NCI’s Surveillance, Epidemiology and End Results (SEER) data, researchers showed a 16-fold increased relative risk of angiosarcoma of the breast and chest wall following irradiation of a primary breast cancer [822]. Angiosarcomas of the breast are associated with relatively poor prognosis [823].
More recent data indicate that women younger than 45 who received the higher radiation exposure associated with post-lumpectomy radiotherapy (as compared to post-mastectomy radiation) had a 1.5–2.5-fold increase in later contralateral breast cancer diagnoses. This effect was especially prominent in younger women with a substantial family history of breast cancer [824–826]. Bernstein et al. studied a cohort of women, nested within the large WECARE study, who had developed contralateral breast cancer (as compared to breast cancer patients who did not develop contralateral breast cancer). They found main effects for both gene status and treatment, with significant elevations for BRCA1/2 carriers (RR = 4.5; 95% CI = 3.0–6.6), and ever treatment with radiotherapy (RR = 1.2; 95% CI = 1.0–6.6), but no significant interaction between the two factors [74].
Non-ionizing radiation (electromagnetic fields)
Electromagnetic waves are a type of low frequency, non-ionizing radiation without enough energy to break off electrons from their orbits around atoms and ionize the atoms. Microwaves, radio waves, radar and radiation produced by electrical transmission are examples of radiation sources that generate electromagnetic fields (EMF). Fluorescent lighting, computers and many other types of wired and wireless electronic equipment (e.g., cell phones) all create electromagnetic fields of varying strengths.
Both IARC and the National Institute of Environmental Health Sciences (NIEHS) EMF Working Group have classified EMF exposures as a possible human carcinogen based on the scientific literature related to EMF and childhood leukemias [827]. More recently, data have suggested a link between EMF exposure, especially from cell phone use, and development of brain cancer and acoustic neuromas, although the strength of these connections remain controversial [828]. Concensus has been even more been more difficult to reach about the relationship between EMF and breast cancer.
Although many epidemiological or occupational studies have not found significant relationships between exposures to EMF and risk for breast cancer, others have reported data supporting these effects [829, 830]. Methodological issues may account for some of the discrepancies, given the relatively small effects that are found and the ubiquitous nature of “background” EMF in our daily lives [831].
Kliukiene et al. reported an increased risk of breast cancer among Norwegian female radio and telegraph operators exposed to radiofrequency (one type of EMF) and extremely low frequency EMF. Pre-menopausal women showed an increased risk of estrogen-receptor-positive tumors (OR = 1.78; 95% CI = 0.59–5.41) and post-menopausal women had an increased risk of estrogen-receptor-negative tumors (OR = 2.37; 95% CI = 0.88–6.36) [832].
In an occupational study that looked at women in job setting with the potential for high, medium or low electromagnetic exposures, high exposures were associated with an increased risk for developing breast cancer (OR = 1.43; 95% CI = 0.99–2.09). Pre-menopausal women appear to be at higher risk (OR = 1.98; 95% CI = 1.04–3.78) than post-menopausal women (OR = 1.33; 95% CI = 0.82–2.17) [833].
Studies of residential and occupational EMF exposure found a significant increase (OR = 1.58; 95% CI = 1.30–1.92) in breast cancer risk among women of all ages living near high-voltage power lines. Similar effects were found for women with ER+ and ER– tumors. Occupational exposure also increased risk (OR = 1.13; 95% CI = 0.91–1.40) [834]. Women younger than age 50 who were exposed to EMF both at home and at work had a modest increase in risk of breast cancer [835].
Nevertheless, meta-analyses of 15 studies concluded that there is no clear relationship between EMF exposure and breast cancer in women (OR = 0.99; 95% CI = 0.90–1.09) [836]. Another meta-analysis that examined a subset of published studies that specified mode of exposure reported a small increase in breast cancer rates in premenopausal women associated with increased residential exposure to EMF (OR = 1.18; 95% CI = 1.02–1.37) [837].
Although breast cancer is rare in men, numerous occupational exposure studies point to a connection between EMF exposure and male breast cancer [838–842].
In the laboratory, EMF can cause increased mammary tumors in animals and proliferation in systems in which human breast cell tumors are grown in culture. Importantly, effects in rodents are found in some strains of animals but not others, indicating that subtle differences in genetic background might make some animals more susceptible to the carcinogenic effects of EMF [843]. In an in vitro cell system, EMF exposure of human breast tumor (MCF-7) cells led to an activation of genes that have been associated with the induction of metastasis in breast cancer cells [844].
Section summary: Exposure to ionizing radiation is a known cause of increased risk for breast cancer. Victims of military use of nuclear bombs have increased risk, as do women who had X-ray treatments for medical purposes, especially when they were young. Women carrying the BRCA1/2 mutations are particularly susceptible to the effects of X-rays, including those emitted by routine mammography.
More mixed results come from studies of women exposed to non-ionizing radiation, either because of occupational or residential exposures.
Discussion and conclusions
In the 8 years since we last published an extensive review of the relevant literature, hundreds of new papers have appeared addressing the link between exposures to environmental toxicants and an increased risk for developing breast cancer; the majority of the studies support the existence of this link for the agents discussed in this review. Not only has the corpus of the literature expanded in size over the past several years, but it has also been enhanced by greater depth, breadth, and complexity. The growing literature on developmental exposures to EDCs and later development of breast cancer is especially strong.
Epidemiological data strongly support the link between increased risk of developing breast cancer and early developmental exposures to DES, DDT and radiation, as well as adult exposures to oral contraceptives and HRT. A growing literature also implicates engaging in night shiftwork as an important factor leading to increased risk for breast cancer. On the other hand, a substantial literature examining the effects of consuming soy products and lignans as part of a regular diet, especially starting early in life, indicates they can have a protective effect against later development of breast cancer.
Animal and other in vitro models support the hypothesis that many other chemicals found in commonly used consumer products, as well as in our air, water and dust, all are associated with increased risk for predisposing mammary tissue to develop tumors. These data support the strong links described above for EDCs. Data from epidemiological studies suggest connections between exposures and later development of cancer, although methodological limitations often constrain the conclusions that can be drawn. Of particular concern for most epidemiological studies in this field is the lack of direct measurement of toxicant exposure levels in individuals, especially in the years (or decades) prior to diagnosis of the breast cancer [845]. As the literature has documented clearly, there is often a long latency between exposures and diagnosis, and earlier developmental exposures can be especially powerful in affecting development of breast cancer, even decades later [457]. To enlarge the body of relevant work, it will be important for large cohort studies to regularly collect exposure information across much of the lifespan, and to develop the technologies necessary to quantify exposure levels, biomarkers, and health outcomes at large-scale levels [845, 846].
Importantly, through animal, cell culture, high throughput and other non-epidemiological models, mechanisms are being elucidated by which exposures to various toxicants may lead to increased risk for developing cancer. This literature has been slow to develop, because regulatory toxicological studies examining reproductive and developmental consequences of exposures to various drugs or potential toxicants have not required examination of mammary tissue endpoints [847]. There are not standardized protocols for determining appropriate times of exposures, ranges of doses, or mammary gland endpoints to study and later potential carcinogenic, genotoxic, or endocrine disrupting effects of these exposures. In order to claim more definitively the connections between the many chemicals that have been implicated in increased risk for development of breast cancer and causal links to the disease, it will be important to develop a series of endpoints to be studied routinely. Critical endpoints to be evaluated include altered mammary gland development; activity of various biomarkers including PR, HER, other endocrine factors; and different subtypes of various hormone receptors, each of which can have different effects on cellular activity when activated [848].
Despite these critical methodological limitations and concerns, the breadth and strength of the evidence cited in this review, when taken as a whole, reinforce the conclusion that exposures to a wide variety of toxicants – many of which are found in common, everyday products and byproducts – can lead to increased risk for development of breast cancer. As concluded by the reports of the Presidential Cancer Panel [4] and the Interagency Breast Cancer and Environment Research Coordinating Committee [2], it is critical to recognize the growing literature demonstrating connections between exposures to environmental toxicants and later development of disease, including breast cancer, and to prioritize prevention both at the research and the public health levels.
Acknowledgements
The authors would like to acknowledge Nora Kaye and Monica Raiss for their help with citations.
Funding
Not applicable.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- 2,4,5-TP
2,4,5-trichlorophenoxypropionic acid
- 4-NP
4-nonylphenol
- ABP
4-aminophenyl
- AhR
Aryl hydrocarbon receptor
- AR
Androgen receptor
- BPA
Bisphenol A
- CHDS
Child Health and Development Study
- DCIS
Ductal carcinoma in situ
- DDE
Dichlorodiphenyldichloroethylene
- DDT
Dichlorodiphenyltrichloroethane
- DEHP
Di(2-ethylhexyl)phthalate
- DEP
Diethyl phthalate
- DES
Diethylstilbestrol
- DiBP
Di-i-butyl phthalate
- EDC
Endocrine disrupting compound
- EMT
Epithelial-mesenchymal transition
- EPA
Environmental Protection Agency
- ER
Estrogen receptor
- ERR
Estrogen-related receptor
- FDA
Food and Drug Administration
- HE
Heptachlor epoxide
- HHA
Heterocyclic aromatic amines
- HRT
Hormone Replacement Therapy
- IARC
International Agency for Research on Cancer
- IGF-1
Insuling growth factor-1
- IVF
In vitro fertilization
- LAN
Light-at-night
- MEHP
Mono(2-ethylhexyl)phthalate
- MMP
Monomethyl phthalate
- NAT2
N-acetyltransferase 2
- NCI
National Cancer Institute
- NHANES
National Health and Nutrition Examination Survey
- NMR
Non-monotonic response
- NNK
Nitrosamine ketone
- NOAEL
No-observed-adverse-effect-level
- NTP
National Toxicology Program
- OMC
Octyl-methoxycinnamate
- PAH
Polyaromatic hydrocarbon
- PBDE
Polybrominated Diphenyl Ether
- PCB
Polychlorinated biphenyl
- PFC
Polyfluorinated chemical
- PFOA
Perfluorooctanic acid
- PFOS
Perfluorooctanic sulfate
- PhIP
2-amino-phenylimidazo-[4.5.-b]pyridine
- PR
Progesterone receptor
- PVC
Polyvinyl chloride
- rBST
Recombinant Bovine Somatotropin
- SEER
Surveillance, Epidemiology and End Results
- SNP
Single-nucleotide polymorphism
- T4
Thyroid hormone
- TCDD
2,3,7,8-tetrachlorodibenzo-para-dioxin
- TEB
Terminal end buds
- TOFT
Tissue Organization Field Theory
- WHI
Women’s Health Initiative
- YES
Yeast estrogen screen
- ZEA
Zearalenone
- ZER
Zeranol
Authors’ contributions
JG is the lead author and wrote the full drafts of the manuscript and its revisions. SR, CE and JR edited and advised on the final manuscripts. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Janet M. Gray, Email: grayj@vassar.edu
Sharima Rasanayagam, Email: sharima@bcpp.org.
Connie Engel, Email: connie@bcpp.org.
Jeanne Rizzo, Email: jeanne@bcpp.org.
References
- 1.Colditz GA, Bohlke K. Priorities for the primary prevention of breast cancer. CA Cancer J Clin. 2014;64:186–194. doi: 10.3322/caac.21225. [DOI] [PubMed] [Google Scholar]
- 2.Interagency Breast Cancer and Environment Research Coordinating Committee. Breast Cancer and the Environment: Prioritizing Prevention. 2013. https://www.niehs.nih.gov/about/assets/docs/breast_cancer_and_the_environment_prioritizing_prevention_508.pdf. Accessed 25 Oct 2016.
- 3.Kruk J, Aboul-Enein HY. Environmental exposure, and other behavioral risk factors in breast cancer. Curr Cancer Ther Rev. 2006;2:3–21. doi: 10.2174/157339406775471795. [DOI] [Google Scholar]
- 4.President’s Cancer Panel; National Cancer Institute. Reducing Environmental Cancer Risk: What We Can Do Now. 2010. https://deainfo.nci.nih.gov/advisory/pcp/annualreports/pcp08-09rpt/pcp_report_08-09_508.pdf. Accessed 25 Oct 2016.
- 5.Fenton SE. A special issue dedicated to a complex tissue. Reprod Toxicol. 2015;54:1–5. doi: 10.1016/j.reprotox.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 6.American Cancer Society. Breast Cancer Facts & Figures 2015–2016, vol. 2015. Atlanta: American Cancer Society, Inc.
- 7.Sondik EJ. Breast cancer trends: Incidence, mortality, and survival. Cancer. 1994;74(Suppl 1):995–999. doi: 10.1002/1097-0142(19940801)74:3+<995::AID-CNCR2820741504>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 8.Timmons FL. A history of weed control in the United States and Canada. Weed Sci. 2005;53:748–761. doi: 10.1614/0043-1745(2005)053[0748:AHOWCI]2.0.CO;2. [DOI] [Google Scholar]
- 9.Murmann JP. Chemical industries after 1850. Oxford Encyclopedia of Economic History. 2002. http://www.professor-murmann.net/murmann_oeeh.pdf. Accessed 16 May 2017.
- 10.Frienkel S. A brief history of plastic’s conquest of the world. Sci Am. 2011. https://www.scientificamerican.com/article/a-brief-history-of-plastic-world-conquest/. Accessed 16 May 2017.
- 11.Gray J, Evans N, Taylor B, Rizzo J, Walker M. State of the evidence: The connection between breast cancer and the environment. Int J Occup Environ Health. 2009;15:43–78. doi: 10.1179/oeh.2009.15.1.43. [DOI] [PubMed] [Google Scholar]
- 12.DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women: Breast cancer statistics, 2015. CA Cancer J Clin. 2016;66:31–42. doi: 10.3322/caac.21320. [DOI] [PubMed] [Google Scholar]
- 13.DeSantis CE, Siegel RL, Sauer AG, Miller KD, Fedewa SA, Alcaraz KI, et al. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities: Cancer statistics for African Americans, 2016. CA Cancer J Clin. 2016;66:290–308. doi: 10.3322/caac.21340. [DOI] [PubMed] [Google Scholar]
- 14.Alizart M, Saunus J, Cummings M, Lakhani SR. Molecular classification of breast carcinoma. Diagn Histopathol. 2012;18:97–103. doi: 10.1016/j.mpdhp.2011.12.003. [DOI] [Google Scholar]
- 15.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast cancer: Epidemiology and etiology. Cell Biochem Biophys. 2015;72:333–338. doi: 10.1007/s12013-014-0459-6. [DOI] [PubMed] [Google Scholar]
- 17.Kohler BA, Sherman RL, Howlader N, Jemal A, Ryerson AB, Henry KA, et al. Annual report to the nation on the status of cancer, 1975–2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. J Natl Cancer Inste. 2015;107:djv048. [DOI] [PMC free article] [PubMed]
- 18.Inic Z, Zegarac M, Inic M, Markovic I, Kozomara Z, Djurisic I, et al. Difference between luminal a and luminal B subtypes according to Ki-67, tumor size, and progesterone receptor negativity providing prognostic information. Clin Med Insights Oncol. 2014;8:107–111. doi: 10.4137/CMO.S18006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang XR, Sherman ME, Rimm DL, Lissowska J, Brinton LA, Peplonska B, et al. Differences in risk factors for breast cancer molecular subtypes in a population-based study. Cancer Epidemiol Biomarkers Prev. 2007;16:439–443. doi: 10.1158/1055-9965.EPI-06-0806. [DOI] [PubMed] [Google Scholar]
- 20.Akinyemiju TF, Pisu M, Waterbor JW, Altekruse SF. Socioeconomic status and incidence of breast cancer by hormone receptor subtype. SpringerPlus. 2015;4:508. [DOI] [PMC free article] [PubMed]
- 21.Turkoz FP, Solak M, Petekkaya I, Keskin O, Kertmen N, Sarici F, et al. Association between common risk factors and molecular subtypes in breast cancer patients. Breast. 2013;22:344–350. doi: 10.1016/j.breast.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Anderson KN, Schwab RB, Martinez ME. Reproductive risk factors and breast cancer subtypes: A review of the literature. Breast Cancer Res Treat. 2014;144:1–10. doi: 10.1007/s10549-014-2852-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bowen R, Stebbing J, Jones L. A review of the ethnic differences in breast cancer. Pharmacogenomics. 2006;7:935–942. doi: 10.2217/14622416.7.6.935. [DOI] [PubMed] [Google Scholar]
- 24.Chen J-Q, Russo J. ERα-negative and triple negative breast cancer: Molecular features and potential therapeutic approaches. Biochim Biophys Acta. 2009;1796:162–175. doi: 10.1016/j.bbcan.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer registry. Cancer. 2007;109:1721–1728. doi: 10.1002/cncr.22618. [DOI] [PubMed] [Google Scholar]
- 26.Lorusso L, Bacchini F. A reconsideration of the role of self-identified races in epidemiology and biomedical research. Stud Hist Philos Biol Biomed Sci. 2015;52:56–64. doi: 10.1016/j.shpsc.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Nelson JW, Scammell MK, Hatch EE, Webster TF. Social disparities in exposures to bisphenol a and polyfluoroalkyl chemicals: A cross-sectional study within NHANES 2003–2006. Environ Health. 2012;11:1. doi: 10.1186/1476-069X-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans GW, Kantrowitz E. Socioeconomic status and health: The potential role of environmental risk exposure. Annu Rev Public Health. 2002;23:303–331. doi: 10.1146/annurev.publhealth.23.112001.112349. [DOI] [PubMed] [Google Scholar]
- 29.Forastiere F, Stafoggia M, Tasco C, Picciotto S, Agabiti N, Cesaroni G, et al. Socioeconomic status, particulate air pollution, and daily mortality: Differential exposure or differential susceptibility. Am J Ind Med. 2007;50:208–216. doi: 10.1002/ajim.20368. [DOI] [PubMed] [Google Scholar]
- 30.Quinn MM, Sembajwe G, Stoddard AM, Kriebel D, Krieger N, Sorensen G, et al. Social disparities in the burden of occupational exposures: Results of a cross-sectional study. Am J Ind Med. 2007;50:861–875. doi: 10.1002/ajim.20529. [DOI] [PubMed] [Google Scholar]
- 31.Brulle RJ, Pellow DN. Environmental justice: Human health and environmental inequalities. Annu Rev Public Health. 2006;27:103–124. doi: 10.1146/annurev.publhealth.27.021405.102124. [DOI] [PubMed] [Google Scholar]
- 32.Rauh VA, Landrigan PJ, Claudio L. Housing and health: Intersection of poverty and environmental exposures. Ann N Y Acad Sci. 2008;1136:276–288. doi: 10.1196/annals.1425.032. [DOI] [PubMed] [Google Scholar]
- 33.Center for Disease Control and Prevention: Fourth National Report on Human Exposure to Environmental Chemicals. 2015. http://www.cdc.gov/biomonitoring/pdf/FourthReport_UpdatedTables_Feb2015.pdf. Accessed 25 Oct 2016.
- 34.Calafat AM, Ye X, Wong L-Y, Reidy JA, Needham LL. Urinary concentrations of triclosan in the U.S. population: 2003-2004. Environ Health Perspect. 2008;116:303–307. doi: 10.1289/ehp.10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mortensen ME, Calafat AM, Ye X, et al. Urinary concentrations of environmental phenols in pregnant women in a pilot study of the National Children’s study. Environ Res. 2014;129:32–38. doi: 10.1016/j.envres.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 37.Goodson WH, III, Lowe L, Carpenter DO, Gilbertson M, Ali AM, de CS, et al. Assessing the carcinogenic potential of low-dose exposures to chemical mixtures in the environment: The challenge ahead. Carcinogenesis. 2015;36:S254–S296. doi: 10.1093/carcin/bgv039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Lee D-H, et al. Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocr Rev. 2012;33:378–455. doi: 10.1210/er.2011-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soto AM, Brisken C, Schaeberle C, Sonnenschein C. Does cancer start in the womb? Altered mammary gland development and predisposition to breast cancer due to in utero exposure to endocrine disruptors. J Mammary Gland Biol Neoplasia. 2013;18:199–208. doi: 10.1007/s10911-013-9293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paulose T, Speroni L, Sonnenschein C, Soto AM. Estrogens in the wrong place at the wrong time: Fetal BPA exposure and mammary cancer. Reprod Toxicol. 2015;54:58–65. doi: 10.1016/j.reprotox.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fenton SE, Birnbaum LS. Timing of environmental exposures as a critical element in breast cancer risk. J Clin Endocrinol Metab. 2015;100:3245–3250. doi: 10.1210/jc.2015-2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sonnenschein C, Soto AM. The aging of the 2000 and 2011 hallmarks of cancer reviews: A critique. J Biosci. 2013;38:651–663. doi: 10.1007/s12038-013-9335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soto AM, Sonnenschein C. The tissue organization field theory of cancer: A testable replacement for the somatic mutation theory. BioEssays. 2011;33:332–340. doi: 10.1002/bies.201100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mechanic LE, Hutter CM. Gene-environment interactions in human health. In: Su LJ, Chiang T, editors. Environmental Epigenetics. London: Springer London; 2015. pp. 229–249. [Google Scholar]
- 45.Rudolph A, Chang-Claude J, Schmidt MK. Gene–environment interaction and risk of breast cancer. Br J Cancer. 2016;114:125–133. doi: 10.1038/bjc.2015.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Lee D-H, et al. Regulatory decisions on endocrine disrupting chemicals should be based on the principles of endocrinology. Reprod Toxicol. 2013;38:1–15. doi: 10.1016/j.reprotox.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zoeller RT, Brown TR, Doan LL, Gore AC, Skakkebaek NE, Soto AM, et al. Endocrine-disrupting chemicals and public health protection: A statement of principles from the Endocrine Society. Endocrinology. 2012;153:4097–4110. doi: 10.1210/en.2012-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diamanti-Kandarakis E, Bourguignon J-P, Giudice LC, Hauser R, Prins GS, Soto AM, et al. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Coster S, van Larebeke N. Endocrine-disrupting chemicals: Associated disorders and mechanisms of action. J Environ Public Health. 2012;2012:1–52. doi: 10.1155/2012/713696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Renzo GC, Conry JA, Blake J, DeFrancesco MS, DeNicola N, Martin JN, et al. International Federation of Gynecology and Obstetrics Opinion on reproductive health impacts of exposure to toxic environmental chemicals. Int J Gynecol Obstet. 2015;13:218–225. doi: 10.1016/j.ijgo.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, et al. EDC-2: The Endocrine Society’s second scientific statement on endocrine-disrupting chemicals. Endocrinol Rev. 2015;36:1–150. doi: 10.1210/er.2015-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergman Å, Heindel JJ, Kasten T, Kidd KA, Jobling S, Neira M, et al. The impact of endocrine disruption: A consensus statement on the state of the science. Environ Health Perspect. 2013;121:A101–A106. doi: 10.1289/ehp.1205448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.National Research Council. Risk Assessment in the Federal Government: Managing the Process. 1983. https://www.nap.edu/catalog/366/risk-assessment-in-the-federal-government-managing-the-process. Accessed 25 Oct 2016.
- 54.DeSesso J, Watson R. The case for integrating low dose, beneficial responses into US EPA risk assessments. Hum Exp Toxicol. 2006;25:7–10. doi: 10.1191/0960327106ht578oa. [DOI] [PubMed] [Google Scholar]
- 55.Moral R, Wang R, Russo IH, Lamartiniere CA, Pereira J, Russo J. Effect of prenatal exposure to the endocrine disruptor bisphenol a on mammary gland morphology and gene expression signature. J Endocrinol. 2008;196:101–112. doi: 10.1677/JOE-07-0056. [DOI] [PubMed] [Google Scholar]
- 56.Mandrup K, Boberg J, Isling LK, Christiansen S, Hass U. Low-dose effects of bisphenol a on mammary gland development in rats. Andrology. 2016;4:673–683. doi: 10.1111/andr.12193. [DOI] [PubMed] [Google Scholar]
- 57.Blei T, Soukup ST, Schmalbach K, Pudenz M, Möller FJ, Egert B, et al. Dose-dependent effects of isoflavone exposure during early lifetime on the rat mammary gland: Studies on estrogen sensitivity, isoflavone metabolism, and DNA methylation. Mol Nutr Food Res. 2015;59:270–283. doi: 10.1002/mnfr.201400480. [DOI] [PubMed] [Google Scholar]
- 58.Kortenkamp A. Breast cancer, oestrogens and environmental pollutants: A re-evaluation from a mixture perspective. Int J Androl. 2006;29:193–198. doi: 10.1111/j.1365-2605.2005.00613.x. [DOI] [PubMed] [Google Scholar]
- 59.Koppe J, Bartonova A, Bolte G, Bistrup ML, Busby C, Butter M, et al. Exposure to multiple environmental agents and their effect. Acta Paediatr Suppl. 2006;95:106–113. doi: 10.1080/08035320600886646. [DOI] [PubMed] [Google Scholar]
- 60.Soto AM, Chung KL, Olea N. The E-SCREEN assay as a tool to identify estrogens: An update on estrogenic environmental pollutants. Environ Health Perspect. 1995;103:113–122. doi: 10.1289/ehp.95103s7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Silva E, Scholze M, Kortenkamp A. Activity of xenoestrogens at nanomolar concentrations in the E-screen assay. Environ Health Perspect. 2007;115:91–97. doi: 10.1289/ehp.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Meeuwen JA, ter Burg W, Piersma AH, van den Berg M, Sanderson JT. Mixture effects of estrogenic compounds on proliferation and pS2 expression of MCF-7 human breast cancer cells. Food Chem Toxicol. 2007;45:2319–2330. doi: 10.1016/j.fct.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 63.Rajapakse N, Silva E, Kortenkamp A. Combining xenoestrogens at levels below individual no-observed-effect concentrations dramatically enhances steroid hormone action. Environ Health Perspect. 2002;110:917–921. doi: 10.1289/ehp.02110917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajapakse N, Ong D, Kortenkamp A. Defining the impact of weakly estrogenic chemicals on the action of steroidal estrogens. Toxicol Sci. 2001;60:296–304. doi: 10.1093/toxsci/60.2.296. [DOI] [PubMed] [Google Scholar]
- 65.Payne J, Rajapakse N, Wilkins M, Kortenkamp A. Prediction and assessment of the effects of mixtures of four xenoestrogens. Environ Health Perspect. 2000;108:983. doi: 10.1289/ehp.00108983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boada LD, Zumbado M, Henríquez-Hernández LA, Almeida-González M, Álvarez-León EE, Serra-Majem L, et al. Complex organochlorine pesticide mixtures as determinant factor for breast cancer risk: A population-based case–control study in the Canary Islands (Spain) Environ Health. 2012;11:1. doi: 10.1186/1476-069X-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rivero J, Luzardo OP, Henríquez-Hernández LA, Machín RP, Pestano J, Zumbado M, et al. In vitro evaluation of oestrogenic/androgenic activity of the serum organochlorine pesticide mixtures previously described in a breast cancer case–control study. Sci Total Environ. 2015;537:197–202. doi: 10.1016/j.scitotenv.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 68.Valerón PF, Pestano JJ, Luzardo OP, Zumbado ML, Almeida M, Boada LD. Differential effects exerted on human mammary epithelial cells by environmentally relevant organochlorine pesticides either individually or in combination. Chem Biol Interact. 2009;180:485–491. doi: 10.1016/j.cbi.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 69.Foster WG. Mammary gland morphology in Sprague-Dawley rats following treatment with an organochlorine mixture in utero and neonatal genistein. Toxicol Sci. 2003;77:91–100. doi: 10.1093/toxsci/kfg247. [DOI] [PubMed] [Google Scholar]
- 70.Imaoka T, Nishimura M, Teramoto A, Nishimura Y, Ootawara M, Osada H, et al. Cooperative induction of rat mammary cancer by radiation and 1-methyl-1-nitrosourea via the oncogenic pathways involving c-Myc activation and H-ras mutation. Int J Cancer. 2005;115:187–193. doi: 10.1002/ijc.20904. [DOI] [PubMed] [Google Scholar]
- 71.Berrington de Gonzalez A, Berg CD, Visvanathan K, Robson M. Estimated risk of rradiation-induced breast cancer from mammographic screening for young BRCA mutation carriers. J Natl Cancer Inst. 2009;101:205–209. doi: 10.1093/jnci/djn440. [DOI] [PubMed] [Google Scholar]
- 72.Jansen-van der Weide MC, Greuter MJW, Jansen L, Oosterwijk JC, Pijnappel RM, de Block GH. Exposure to low-dose radiation and the risk of breast cancer among women with a familial or genetic predisposition: A meta-analysis. Eur Radiol. 2010;20:2547–2556. doi: 10.1007/s00330-010-1839-y. [DOI] [PubMed] [Google Scholar]
- 73.Pijpe A, Andrieu N, Easton DF, et al. Exposure to diagnostic radiation and risk of breast cancer among carriers of BRCA1/2 mutations: Retrospective cohort study (GENE-RAD-RISK). Brit Med J. 2012;345:e5660. [DOI] [PMC free article] [PubMed]
- 74.Bernstein JL, Thomas DC, Shore RE, Robson M, Boice JD, Stovall M, et al. Contralateral breast cancer after radiotherapy among BRCA1 and BRCA2 mutation carriers: A WECARE study report. Eur J Cancer. 2013;49:2979–2985. doi: 10.1016/j.ejca.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Millikan RC. Polymorphisms in DNA repair genes, medical exposure to ionizing radiation, and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2005;14:2326–2334. doi: 10.1158/1055-9965.EPI-05-0186. [DOI] [PubMed] [Google Scholar]
- 76.Fletcher O, Dudbridge F. Candidate gene-environment interactions in breast cancer. BMC Med. 2014;12:195. doi: 10.1186/s12916-014-0195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bernal AJ, Jirtle RL. Epigenomic disruption: The effects of early developmental exposures: Epigenomic reactions to early exposures. Birth Defects Res A Clin Mol Teratol. 2010;88:938–944. doi: 10.1002/bdra.20685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wild CP, Scalbert A, Herceg Z. Measuring the exposome: A powerful basis for evaluating environmental exposures and cancer risk. Environ Mol Mutagen. 2013;54:480–499. doi: 10.1002/em.21777. [DOI] [PubMed] [Google Scholar]
- 79.Treviño LS, Wang Q, Walker CL. Hypothesis: Activation of rapid signaling by environmental estrogens and epigenetic reprogramming in breast cancer. Reprod Toxicol. 2015;54:136–140. doi: 10.1016/j.reprotox.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luzhna L, Kutanzi K, Kovalchuk O. Gene expression and epigenetic profiles of mammary gland tissue: Insight into the differential predisposition of four rat strains to mammary gland cancer. Mutat Res. 2015;779:39–56. doi: 10.1016/j.mrgentox.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 81.Samantarrai D, Dash S, Chhetri B, Mallick B. Genomic and epigenomic cross-talks in the regulatory landscape of miRNAs in breast cancer. Mol Cancer Res. 2013;11:315–328. doi: 10.1158/1541-7786.MCR-12-0649. [DOI] [PubMed] [Google Scholar]
- 82.Khan SI, Aumsuwan P, Khan IA, Walker LA, Dasmahapatra AK. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem Res Toxicol. 2012;25:61–73. doi: 10.1021/tx200378c. [DOI] [PubMed] [Google Scholar]
- 83.Kutanzi K, Kovalchuk O. Exposure to estrogen and ionizing radiation causes epigenetic dysregulation, activation of mitogen-activated protein kinase pathways, and genome instability in the mammary gland of ACI rats. Cancer Biol Ther. 2013;14:564–573. doi: 10.4161/cbt.24599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ye W, Xu P, Jen R, Feng E, Zhong S, Li H, et al. Zeranol down-regulates p53 expression in primary cultured human breast cancer epithelial cells through epigenetic modification. Int J Mol Sci. 2011;12:1519–1532. doi: 10.3390/ijms12031519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hussain I, Bhan A, Ansari KI, Deb P, Bobzean SAM, Perrotti LI, et al. Bisphenol-a induces expression of HOXC6, an estrogen-regulated homeobox-containing gene associated with breast cancer. Biochim Biophys Acta. 2015;1849:697–708. doi: 10.1016/j.bbagrm.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Doherty LF, Bromer JG, Zhou Y, Aldad TS, Taylor HS. In utero exposure to diethylstilbestrol (DES) or bisphenol-a (BPA) increases EZH2 expression in the mammary gland: An epigenetic mechanism linking endocrine disruptors to breast cancer. Horm Cancer. 2010;1:146–155. doi: 10.1007/s12672-010-0015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Soto AM, Sonnenschein C. The somatic mutation theory of cancer: Growing problems with the paradigm? BioEssays. 2004;26:1097–1107. doi: 10.1002/bies.20087. [DOI] [PubMed] [Google Scholar]
- 88.Soto AM, Maffini MV, Sonnenschein C. Neoplasia as development gone awry: The role of endocrine disruptors. Int J Androl. 2008;31:288–293. doi: 10.1111/j.1365-2605.2007.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wadia PR, Cabaton NJ, Borrero MD, et al. Low-dose BPA exposure alters the mesenchymal and epithelial transcriptomes of the mouse fetal mammary gland. PLoS One. 2013;8:e63902. [DOI] [PMC free article] [PubMed]
- 90.Markey CM, Luque EH, Munoz de Toro M, Sonneschein C, Soto AM. In utero exposure to bisphenol a alters the development and tissue organization of the mouse mammary gland. Biol Reprod. 2001;65:1215–1223. doi: 10.1093/biolreprod/65.4.1215. [DOI] [PubMed] [Google Scholar]
- 91.Vandenberg LN, Wadia PR, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. The mammary gland response to estradiol: Monotonic at the cellular level, non-monotonic at the tissue-level of organization? J Steroid Biochem Mol Biol. 2006;101:263–274. doi: 10.1016/j.jsbmb.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 92.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-a alters development of the fetal mouse mammary gland. Endocrinology. 2007;148:116–127. doi: 10.1210/en.2006-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tamimi R, Lagiou P, Vatten LJ, Mucci L, Trichopoulos D, Hellerstein S, et al. Pregnancy hormones, pre-eclampsia, and implications for breast cancer risk in the offspring. Cancer Epidemiol Biomarkers Prev. 2003;12:647–650. [PubMed] [Google Scholar]
- 94.Vatten LJ, Forman MR, Nilsen TIL, Barrett JC, Romundstad PR. The negative association between pre-eclampsia and breast cancer risk may depend on the offspring’s gender. Br J Cancer. 2007;180:1436–1438. doi: 10.1038/sj.bjc.6603688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hickey M, Hart R, Keelan JA. The relationship between umbilical cord estrogens and perinatal characteristics. Cancer Epidemiol Biomarkers Prev. 2014;23:946–952. doi: 10.1158/1055-9965.EPI-13-1321. [DOI] [PubMed] [Google Scholar]
- 96.Ekbom A, Adami H-O, Lan S-J, Trichopoulos D, Hsieh C-C. Evidence of prenatal influences on breast cancer risk. Lancet. 1992;340:1015–1018. doi: 10.1016/0140-6736(92)93019-J. [DOI] [PubMed] [Google Scholar]
- 97.Trichopoulos D. Is breast cancer initiated in utero? Epidemiology. 1990;1:95–96. doi: 10.1097/00001648-199003000-00001. [DOI] [PubMed] [Google Scholar]
- 98.Titus-Ernstoff L, Hatch E, Hoover R, Palmer J, Greenberg E, Ricker W, et al. Long-term cancer risk in women given diethylstilbestrol (DES) during pregnancy. Br J Cancer. 2001;84:126–133. doi: 10.1054/bjoc.2000.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL, et al. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N Engl J Med. 2011;365:1304–1314. doi: 10.1056/NEJMoa1013961. [DOI] [PubMed] [Google Scholar]
- 100.Hilakivi-Clarke L. Maternal exposure to diethylstilbestrol during pregnancy and increased breast cancer risk in daughters. Breast Cancer Res. 2014;16:208. doi: 10.1186/bcr3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Palmer JR. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:1509–1514. doi: 10.1158/1055-9965.EPI-06-0109. [DOI] [PubMed] [Google Scholar]
- 102.Troisi R, Hatch EE, Titus-Ernstoff L, Hyer M, Palmer JR, Robboy SJ, et al. Cancer risk in women prenatally exposed to diethylstilbestrol. Int J Cancer. 2007;121:356–360. doi: 10.1002/ijc.22631. [DOI] [PubMed] [Google Scholar]
- 103.Cohn BA, La M, Krigbaum NY, Yeh G, Park J-S, Zimmermann L, et al. DDT exposure in utero and breast cancer. J Clin Endocrinol Metab. 2015;100:2865–2872. doi: 10.1210/jc.2015-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cohn BA, Wolff MS, Cirillo PM, Sholtz RI. DDT and breast cancer in young women: New data on the significance of age at exposure. Environ Health Perspect. 2007;115:1406–1414. doi: 10.1289/ehp.10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mackenzie I. Breast cancer following multiple fluoroscopies. Br J Cancer. 1965;19:1–8. doi: 10.1038/bjc.1965.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Golubicic I, Borojevic N, Pavlovic T. Risk factors for breast cancer: Is ionizing radiation among them? J BUON. 2008;13:487–494. [PubMed] [Google Scholar]
- 107.Mattson A, Ruden BI, Palmgren J, Rutgvist LE. Dose- and time-response for breast cancer risk after radiation therapy for benign breast disease. Br J Cancer. 1995;72:1054–1061. doi: 10.1038/bjc.1995.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shore RE, Hildreth N, Woodard E, Dvoretsky P, Hempelmann L, Pasternack B. Breast cancer among women given X-ray therapy for acute postpartum mastitis. J Natl Cancer Inst. 1986;77:689–696. doi: 10.1093/jnci/77.3.689. [DOI] [PubMed] [Google Scholar]
- 109.Adams MJ, Dozier A, Shore RE, Lipshultz SE, Schwartz RG, Constine LS, et al. Breast cancer risk 55+ years after irradiation for an enlarged thymus and its implications for early childhood medical irradiation today. Cancer Epidemiol Biomarkers Prev. 2010;19:48–58. doi: 10.1158/1055-9965.EPI-09-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hildreth NG, Shore RE, Dvoretsky PM. The risk of breast cancer after irradiation of the thymus in infancy. N Engl J Med. 1989;321:1281–1284. doi: 10.1056/NEJM198911093211901. [DOI] [PubMed] [Google Scholar]
- 111.Lundell M, Mattsson A, Karlsson P, Holmberg E, Gustafsson A, Holm L-E. Breast ancer risk after radiotherapy in infancy: A pooled analysis of two Swedish cohorts of 17,202 infants. Radiat Res. 1999;151:626. doi: 10.2307/3580039. [DOI] [PubMed] [Google Scholar]
- 112.Morin Doody M, Lonstein JE, Stovall M, Hacker DG, Luckyanov N, Land CE. Breast cancer mortality after diagnostic radiography: Findings from the U.S. scoliosis cohort study. Spine. 2000;25:2052–2063. doi: 10.1097/00007632-200008150-00009. [DOI] [PubMed] [Google Scholar]
- 113.Bhatia S. High risk of subsequent neoplasms continues with extended follow-up of childhood Hodgkin’s disease: Report from the late effects study group. J Clin Oncol. 2003;21:4386–4394. doi: 10.1200/JCO.2003.11.059. [DOI] [PubMed] [Google Scholar]
- 114.Guibout C, Adjadj E, Rubino C, Shamsaldin A, Grimaud E, Hawkins M, et al. Malignant breast tumors after radiotherapy for a first cancer during childhood. J Clin Oncol. 2005;23:197–204. doi: 10.1200/JCO.2005.06.225. [DOI] [PubMed] [Google Scholar]
- 115.Horwich A, Swerdlow AJ. Second primary breast cancer after Hodgkin’s disease. Br J Cancer. 2004;90:294–298. doi: 10.1038/sj.bjc.6601499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wahner-Roedler DL, Petersen IA. Risk of breast cancer and breast cancer characteristics in women after treatment for Hodgkin lymphoma. Drugs Today (Barc) 2004;40:865–879. doi: 10.1358/dot.2004.40.10.863746. [DOI] [PubMed] [Google Scholar]
- 117.Tward JD, Wendland MMM, Shrieve DC, Szabo A, Gaffney DK. The risk of secondary malignancies over 30 years after the treatment of non-Hodgkin lymphoma. Cancer. 2006;107:108–115. doi: 10.1002/cncr.21971. [DOI] [PubMed] [Google Scholar]
- 118.El-Gamal H, Bennett RG. Increased breast cancer risk after radiotherapy for acne among women with skin cancer. J Am Acad Dermatol. 2006;55:981–989. doi: 10.1016/j.jaad.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 119.Ma H, Hill CK, Bernstein L, Ursin G. Low-dose medical radiation exposure and breast cancer risk in women under age 50 years overall and by estrogen and progesterone receptor status: Results from a case-control and a case-case comparison. Breast Cancer Res Treat. 2008;109:77–90. doi: 10.1007/s10549-007-9625-5. [DOI] [PubMed] [Google Scholar]
- 120.John EM, Phipps AI, Knight JA, Milne RL, Dite GS, Hopper JL, et al. Medical radiation exposure and breast cancer risk: Findings from the breast cancer family registry. Int J Cancer. 2007;121:386–394. doi: 10.1002/ijc.22668. [DOI] [PubMed] [Google Scholar]
- 121.Koo E, Henderson MA, Dwyer M, Skandarajah AR. Management and prevention of breast cancer after radiation to the chest for childhood, adolescent, and young adulthood malignancy. Ann Surg Oncol. 2015;22:545–551. doi: 10.1245/s10434-015-4596-y. [DOI] [PubMed] [Google Scholar]
- 122.Russo J, Russo IH. Molecular basis of breast cancer. Berlin: Springe-Verlag; 2004. The breast as a developing organ. [Google Scholar]
- 123.Briskin C. Progesterone signaling in breast cancer: A neglected hormone coming into the limelight. Nat Rev Cancer. 2013;13:385–396. doi: 10.1038/nrc3518. [DOI] [PubMed] [Google Scholar]
- 124.Gail MH, Brinton LA, Byar DP, Corle DK, Green SB, Schairer C, et al. Projecting individualized probabilities of developing breasat cancer for white females who are being examined annually. J Natl Cancer Inst. 1989;81:1879–1886. doi: 10.1093/jnci/81.24.1879. [DOI] [PubMed] [Google Scholar]
- 125.Gail MH. Twenty-five years of breast cancer risk models and their applications. J Natl Cancer Inst. 2015;107:djv042. doi: 10.1093/jnci/djv042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hoover RN, Troisi RJ. Understanding mechanisms of breast cancer prevention. J Natl Cancer Inst. 2001;93:1119–1120. doi: 10.1093/jnci/93.15.1119. [DOI] [PubMed] [Google Scholar]
- 127.Hilakivi-Clarke L, de Assis S, Warri A. Exposures to synthetic estrogens at different times during the life, and their effect on breast cancer risk. J Mammary Gland Biol Neoplasia. 2013;18:25–42. doi: 10.1007/s10911-013-9274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Reed CE, Fenton SE. Exposure to diethylstilbestrol during sensitive life stages: A legacy of heritable health effects. Birth Defects Res C Embryo Today. 2013;99:134–146. doi: 10.1002/bdrc.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Walker BE, Haven MI. Intensity of multigenerational carcinogenesis from diethylstilbestrol in mice. Carcinogenesis. 1997;18:791–793. doi: 10.1093/carcin/18.4.791. [DOI] [PubMed] [Google Scholar]
- 130.Saeed M, Rogan E, Cavalieri E. Mechanism of metabolic activation and DNA adduct formation by the human carcinogen diethylstilbestrol: The defining link to natural estrogens. Int J Cancer. 2009;124:1276–1284. doi: 10.1002/ijc.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Larson PS, Ungarelli RA, de Las MA, Cupples LA, Rowlings K, Palmer JR, et al. In utero exposure to diethylstilbestrol (DES) does not increase genomic instability in normal or neoplastic breast epithelium. Cancer. 2006;107:2122–2126. doi: 10.1002/cncr.22223. [DOI] [PubMed] [Google Scholar]
- 132.Ayyanan A, Laribi O, Schuepbach-Mallepell S, Schrick C, Gutierrez M, Tanos T, et al. Perinatal exposure to bisphenol a increases adult mammary gland progesterone response and cell number. Mol Endocrinol. 2011;25:1915–1923. doi: 10.1210/me.2011-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bhan A, Hussain I, Ansari KI, Bobzean SAM, Perrotti LI, Mandal SS. Bisphenol-a and diethylstilbestrol exposure induces the expression of breast cancer associated long noncoding RNA HOTAIR in vitro and in vivo. J Steroid Biochem Mol Biol. 2014;141:160–170. doi: 10.1016/j.jsbmb.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bhan A, Hussain I, Ansari KI, Bobzean SAM, Perrotti LI, Mandal SS. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-a and diethylstilbestrol. J Mol Biol. 2014;426:3426–3441. doi: 10.1016/j.jmb.2014.07.025. [DOI] [PubMed] [Google Scholar]
- 135.Dhimolea E, Wadia PR, Murray TJ, et al. Prenatal exposure to BPA alters the epigenome of the rat mammary gland and increases the propensity to neoplastic development. PLoS One. 2014;9:e99800. doi: 10.1371/journal.pone.0099800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Knower KC, To SQ, Leung Y-K, Ho S-M, Clyne CD. Endocrine disruption of the epigenome: A breast cancer link. Endocr Relat Cancer. 2014;21:T33–T55. doi: 10.1530/ERC-13-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the women’s health initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 138.Anderson GL, Chlebowski RT, Aragaki AK, Kuller LH, Manson JE, Gass M, et al. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal women with hysterectomy: Extended follow-up of the Women’s health initiative randomised placebo-controlled trial. Lancet Oncol. 2012;13:476–486. doi: 10.1016/S1470-2045(12)70075-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SAA, Black H, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s health initiative randomized controlled trial. J Am Med Assoc. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 140.Montgomery BE, Daum GS, Dunton CJ. Endometrial hyperplasia: A review. Obstet Gynecol Surv. 2004;59:368–378. doi: 10.1097/00006254-200405000-00025. [DOI] [PubMed] [Google Scholar]
- 141.Wood CE, Clarkson TB, Chen H, Veenstra TD, Xu X, Scott L, et al. Comparative effects of oral conjugated equine estrogens and micronized 17beta-estradiol on breast proliferation: A retrospective analysis. Menopause. 2008;15:890–898. doi: 10.1097/gme.0b013e318168f0ad. [DOI] [PubMed] [Google Scholar]
- 142.Chlebowski RT, Rohan TE, Manson JE, Aragaki AK, Kaunitz A, Stefanick ML, et al. Breast cancer after use of estrogen plus progestin and estrogen alone: Analyses of data from 2 Women’s health initiative randomized clinical trials. JAMA Oncol. 2015;1:296–305. doi: 10.1001/jamaoncol.2015.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Krieger N, Chen JT, Waterman PD. Decline in US breast cancer rates after the Women’s health initiative: Socioeconomic and racial/ethnic differentials. Am J Public Health. 2010;100(Suppl 1):S132–S139. doi: 10.2105/AJPH.2009.181628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Verkooijen HM, Bouchardy C, Vinh-Hung V, Rapiti E, Hartman M. The incidence of breast cancer and changes in the use of hormone replacement therapy: A review of the evidence. Maturitas. 2009;64:80–85. doi: 10.1016/j.maturitas.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 145.Holmberg L, Anderson H, HABITS steering and data monitoring committees HABITS (hormonal replacement therapy after breast cancer—is it safe?), a randomized comparison: Trial stopped. Lancet. 2004;363:453–455. doi: 10.1016/S0140-6736(04)15493-7. [DOI] [PubMed] [Google Scholar]
- 146.Million Women Study Collaborators Breast cancer and hormone replacement therapy in the million women study. Lancet. 2003;362:419–427. doi: 10.1016/S0140-6736(03)14065-2. [DOI] [PubMed] [Google Scholar]
- 147.Robbins AS, Clarke CA. Re: Declines in invasive breast cancer and use of postmenopausal hormone therapy in a screening mammography population. J Natl Cancer Inst. 2007;99:1815. doi: 10.1093/jnci/djm209. [DOI] [PubMed] [Google Scholar]
- 148.Kotsopoulos J, Huzarski T, Gronwald J, Moller P, Lynch HT, Neuhausen SL, et al. Hormone replacement therapy after menopause and risk of breast cancer in BRCA1 mutation carriers: A case–control study. Breast Cancer Res Treat. 2016;155:365–373. doi: 10.1007/s10549-016-3685-3. [DOI] [PubMed] [Google Scholar]
- 149.Hou N, Hong S, Wang W, Olopade OI, Dignam JJ, Huo D. Hormone replacement therapy and breast cancer: Heterogeneous risks by race, weight, and breast density. J Natl Cancer Inst. 2013;105:1365–1372. doi: 10.1093/jnci/djt207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pizot C, Boniol M, Mullie P, Koechlin A, Boniol M, Boyle P, et al. Physical activity, hormone replacement therapy and breast cancer risk: A meta-analysis of prospective studies. Eur J Cancer. 2016;52:138–154. doi: 10.1016/j.ejca.2015.10.063. [DOI] [PubMed] [Google Scholar]
- 151.Biglia N, Sgro L, Defabiani E, De Rosa G, Ponzone R, Marenco D, et al. The influence of hormone replacement therapy on the pathology of breast cancer. Eur J Surg Oncol. 2005;31:467–472. doi: 10.1016/j.ejso.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 152.Borgquist S, Anagnostaki L, Jirström K, Landberg G, Manjer J. Breast tumours following combined hormone replacement therapy express favourable prognostic factors. Int J Cancer. 2007;120:2202–2207. doi: 10.1002/ijc.22542. [DOI] [PubMed] [Google Scholar]
- 153.Reeves GK, Beral V, Green J, Gathani T, Bull D, Million Women Study Collaborators Hormonal therapy for menopause and breast-cancer risk by histological type: A cohort study and meta-analysis. Lancet Oncol. 2006;7:910–918. doi: 10.1016/S1470-2045(06)70911-1. [DOI] [PubMed] [Google Scholar]
- 154.Schuetz F, Diel IJ, Pueschel M, von Holst T, Solomayer EF, Lange S, et al. Reduced incidence of distant metastases and lower mortality in 1072 patients with breast cancer with a history of hormone replacement therapy. Am J Obstet Gynecol. 2007;196:342.e1–342.e9. doi: 10.1016/j.ajog.2006.10.901. [DOI] [PubMed] [Google Scholar]
- 155.Davis R, Batur P, Thacker HL. Risks and effectiveness of compounded bioidentical hormone therapy: A case series. J Women’s Health (Larchmt) 2014;23:642–648. doi: 10.1089/jwh.2014.4770. [DOI] [PubMed] [Google Scholar]
- 156.Files JA, Ko MG, Pruthi S. Bioidentical hormone therapy. Mayo Clin Proc. 2011;86:673–680. doi: 10.4065/mcp.2010.0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rosenthal MS. The Wiley protocol: An analysis of ethical issues. Menopause. 2008;15:1014–1022. doi: 10.1097/gme.0b013e318178862e. [DOI] [PubMed] [Google Scholar]
- 158.L’hermite M, Simoncini T, Fuller S, Genazzani AR. Could transdermal estradiol + progesterone be a safer postmenopausal HRT? A review. Maturitas. 2008;60:185–201. doi: 10.1016/j.maturitas.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 159.Campagnoli C, Clavel-Chapelon F, Kaaks R, Peris C, Berrino F. Progestins and progesterone in hormone replacement therapy and the risk of breast cancer. J Steroid Biochem Mol Biol. 2005;96:95–108. doi: 10.1016/j.jsbmb.2005.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Holtorf K. The bioidentical hormone debate: Are bioidentical hormones (estradiol, estriol, and progesterone) safer or more efficacious than commonly used synthetic versions in hormone replacement therapy? Postgrad Med. 2009;121:73–85. doi: 10.3810/pgm.2009.01.1949. [DOI] [PubMed] [Google Scholar]
- 161.Fournier A, Berrino F, Clavel-Chapelon F. Unequal risks for breast cancer associated with different hormone replacement therapies: Results from the E3N cohort study. Breast Cancer Res Treat. 2008;107:103–111. doi: 10.1007/s10549-007-9523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fournier A, Fabre A, Mesrine S, Boutron-Ruault M-C, Berrino F, Clavel-Chapelon F. Use of different postmenopausal hormone therapies and risk of histology- and hormone receptor-defined invasive breast cancer. J Clin Oncol. 2008;26:1260–1268. doi: 10.1200/JCO.2007.13.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Althuis MD, Brogan DR, Coates RJ, Daling JR, Gammon MD, Malone KE, et al. Hormonal content and potency of oral contraceptives and breast cancer risk among young women. Br J Cancer. 2003;88:50–57. doi: 10.1038/sj.bjc.6600691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Delort L, Kwiatkowski F, Chalabi N, Satih S, Bignon Y-J, Bernard-Gallon DJ. Risk factors for early age at breast cancer onset—the “COSA program” population-based study. Anticancer Res. 2007;27:1087–1094. [PubMed] [Google Scholar]
- 165.Dai Q, Liu B, Du Y. Meta-analysis of the risk factors of breast cancer concerning reproductive factors and oral contraceptive use. Front Med China. 2009;3:452–458. doi: 10.1007/s11684-009-0080-z. [DOI] [Google Scholar]
- 166.Pasanisi P, Hédelin G, Berrino J, Chang-Claude J, Hermann S, Steel M, et al. Oral contraceptive use and BRCA penetrance: A case-only study. Cancer Epidemiol Biomark Prev. 2009;18:2107–2113. doi: 10.1158/1055-9965.EPI-09-0024. [DOI] [PubMed] [Google Scholar]
- 167.Kumle M, Weiderpass E, Braaten T, Persson I, Adami H-O, Lund E. Use of oral contraceptives and breast cancer risk: The Norwegian-Swedish Women’s lifestyle and health cohort study. Cancer Epidemiol Biomark Prev. 2002;11:1375–1381. [PubMed] [Google Scholar]
- 168.Rosenberg L, Boggs DA, Wise LA, Adams-Campbell LL, Palmer JR. Oral contraceptive use and estrogen/progesterone receptor-negative breast cancer among African American women. Cancer Epidemiol Biomark Prev. 2010;19:2073–2079. doi: 10.1158/1055-9965.EPI-10-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Huzell L, Persson M, Simonsson M, Markkula A, Ingvar C, Rose C, et al. History of oral contraceptive use in breast cancer patients: Impact on prognosis and endocrine treatment response. Breast Cancer Res Treat. 2015;149:505–515. doi: 10.1007/s10549-014-3252-8. [DOI] [PubMed] [Google Scholar]
- 170.Sweeney C, Giuliano AR, Baumgartner KB, Byers T, Herrick JS, Edwards SL, et al. Oral, injected and implanted contraceptives and breast cancer risk among U.S. Hispanic and non-Hispanic white women. Int J Cancer. 2007;121:2517–2523. doi: 10.1002/ijc.22970. [DOI] [PubMed] [Google Scholar]
- 171.Rosenberg L, Zhang Y, Coogan PF, Strom BL, Palmer JR. A case-control study of oral contraceptive use and incident breast cancer. Am J Epidemiol. 2009;169:473–479. doi: 10.1093/aje/kwn360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Haile RW, Thomas DC, McGuire V, Felberg A, John EM, Milne RL, et al. BRCA1 and BRCA2 mutation carriers, oral contraceptive use, and breast cancer before age 50. Cancer Epidemiol Biomark Prev. 2006;15:1863–1870. doi: 10.1158/1055-9965.EPI-06-0258. [DOI] [PubMed] [Google Scholar]
- 173.Narod SA, Dubé M-P, Klijn J, et al. Oral contraceptives and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst. 2002;94:1773–1779. doi: 10.1093/jnci/94.23.1773. [DOI] [PubMed] [Google Scholar]
- 174.Bernholtz S, Laitman Y, Kaufman B, Paluch Shimon S, Friedman E. Cancer risk in Jewish BRCA1 and BRCA2 mutation carriers: Effects of oral contraceptive use and parental origin of mutation. Breast Cancer Res Treat. 2011;129:557–563. doi: 10.1007/s10549-011-1509-z. [DOI] [PubMed] [Google Scholar]
- 175.Calvo V, Beato M. BRCA1 counteracts progesterone action by ubiquitination leading to progesterone receptor degradation and epigenetic silencing of target promoters. Cancer Res. 2011;71:3422–3431. doi: 10.1158/0008-5472.CAN-10-3670. [DOI] [PubMed] [Google Scholar]
- 176.Jia X, Liu G, Mo M, Cheng J, Shen Z, Shao Z. Reproductive factors and hormone receptor status among very young (<35 years) breast cancer patients. Oncotarget. 2015;6:24571–24580. doi: 10.18632/oncotarget.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Newcomer LM, Newcomb PA, Trentham-Dietz A, Longnecker MP, Greenberg ER. Oral contraceptive use and risk of breast cancer by histologic type. Int J Cancer. 2003;106:961–4. [DOI] [PubMed]
- 178.Dolle JM, Daling JR, White E, Brinton LA, Doody DR, Porter PL, et al. Risk factors for triple-negative breast cancer in women under the age of 45 years. Cancer Epidemiol Biomark Prev. 2009;18:1157–1166. doi: 10.1158/1055-9965.EPI-08-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bethea TN, Rosenberg L, Hong C-C, Troester MA, Lunetta KL, Bandera EV, et al. A case-control analysis of oral contraceptive use and breast cancer subtypes in the African American breast cancer epidemiology and risk consortium. Breast Cancer Res. 2015;17:22. doi: 10.1186/s13058-015-0535-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Phillips LS, Millikan RC, Schroeder JC, Barnholtz-Sloan JS, Levine BJ. Reproductive and hormonal risk factors for ductal carcinoma in situ of the breast. Cancer Epidemiol Biomark Prev. 2009;18:1507–1515. doi: 10.1158/1055-9965.EPI-08-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pervez S, Khan H. Infiltrating ductal carcinoma breast with central necrosis closely mimicking ductal carcinoma in situ (comedo type): A case series. J Med Case Rep. 2007;1:83. doi: 10.1186/1752-1947-1-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Calle EE, Heath J, Miracle-McMahill HL, Coates RJ, Liff JM, Franceschi S, et al. Breast cancer and hormonal contraceptives: Collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet. 1996;347:1713–1727. doi: 10.1016/S0140-6736(96)90806-5. [DOI] [PubMed] [Google Scholar]
- 183.Vessey M, Painter R. Oral contraceptive use and cancer. Findings in a large cohort study, 1968-2004. Br J Cancer. 2006;95:385–389. doi: 10.1038/sj.bjc.6603260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Beaber EF, Buist DSM, Barlow WE, Malone KE, Reed SD, Li CI. Recent oral contraceptive use by formulation and breast cancer risk among women 20 to 49 years of age. Cancer Res. 2014;74:4078–4089. doi: 10.1158/0008-5472.CAN-13-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Urban M, Banks E, Egger S, Canfell K, O’Connell D, Beral V, et al. Injectable and oral contraceptive use and cancers of the breast, cervix, ovary, and endometrium in black south African women: Case-control study. PLoS Med. 2012;9:e1001182. doi: 10.1371/journal.pmed.1001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gauthier E, Paoletti X, Clavel-Chapelon F, E3N group Breast cancer risk associated with being treated for infertility: Results from the French E3N cohort study. Hum Reprod. 2004;19:2216–2221. doi: 10.1093/humrep/deh422. [DOI] [PubMed] [Google Scholar]
- 187.Klip H, Burger CW, Kenemans P, van Leeuwen FE. Cancer risk associated with subfertility and ovulation induction: A review. Cancer Causes Control. 2000;11:319–344. doi: 10.1023/A:1008921211309. [DOI] [PubMed] [Google Scholar]
- 188.Orgéas CC, Sanner K, Hall P, Conner P, Holte J, Nilsson SJ, et al. Breast cancer incidence after hormonal infertility treatment in Sweden: A cohort study. Am J Obstet Gynecol. 2009;200:72.e1–72.e7. doi: 10.1016/j.ajog.2008.08.066. [DOI] [PubMed] [Google Scholar]
- 189.Kessous R, Davidson E, Meirovitz M, Sergienko R, Sheiner E. The risk of female malignancies after fertility treatments: A cohort study with 25-year follow-up. J Cancer Res Clin Oncol. 2016;142:287–293. doi: 10.1007/s00432-015-2035-x. [DOI] [PubMed] [Google Scholar]
- 190.Kotsopoulos J, Librach CL, Lubinski J, Gronwald J, Kim-Sing C, Ghadirian P, et al. Infertility, treatment of infertility, and the risk of breast cancer among women with BRCA1 and BRCA2 mutations: A case-control study. Cancer Causes Control. 2008;19:1111–1119. doi: 10.1007/s10552-008-9175-0. [DOI] [PubMed] [Google Scholar]
- 191.Krul IM, Groeneveld E, Spaan M, van den Belt-Dusebout AW, Mooij TM, Hauptmann M, et al. Increased breast cancer risk in in vitro fertilisation treated women with a multiple pregnancy: A new hypothesis based on historical in vitro fertilisation treatment data. Eur J Cancer. 2015;51:112–120. doi: 10.1016/j.ejca.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 192.Brinton LA, Scoccia B, Moghissi KS, Westhoff CL, Althuis MD, Mabie JE, et al. Breast cancer risk associated with ovulation-stimulating drugs. Hum Reprod. 2004;19:2005–2013. doi: 10.1093/humrep/deh371. [DOI] [PubMed] [Google Scholar]
- 193.Silva I dos S, Wark PA, McCormack VA, Mayer D, Overton C, Little V, et al. Ovulation-stimulation drugs and cancer risks: A long-term follow-up of a British cohort. Br J Cancer. 2009;100:1824–1831. doi: 10.1038/sj.bjc.6605086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Calderon-Margalit R, Friedlander Y, Yanetz R, Kleinhaus K, Perrin MC, Manor O, et al. Cancer risk after exposure to treatments for ovulation induction. Am J Epidemiol. 2009;169:365–375. doi: 10.1093/aje/kwn318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lerner-Geva L, Keinan-Boker L, Blumstein T, Boyko V, Olmar L, Mashiach S, et al. Infertility, ovulation induction treatments and the incidence of breast cancer—a historical prospective cohort of Israeli women. Breast Cancer Res Treat. 2006;100:201–212. doi: 10.1007/s10549-006-9238-4. [DOI] [PubMed] [Google Scholar]
- 196.Stewart LM, Holman CDJ, Hart R, Bulsara MK, Preen DB, Finn JC. In vitro fertilization and breast cancer: Is there cause for concern? Fertil Steril. 2012;98:334–340. doi: 10.1016/j.fertnstert.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 197.Rudel RA, Attfield KR, Schifano JN, Brody JG. Chemicals causing mammary gland tumors in animals signal new directions for epidemiology, chemicals testing, and risk assessment for breast cancer prevention. Cancer. 2007;109:2635–2666. doi: 10.1002/cncr.22653. [DOI] [PubMed] [Google Scholar]
- 198.Tiwary CM. Premature sexual development in children following the use of estrogen- or placenta-containing hair products. Clin Pediatr (Phila) 1998;37:733–739. doi: 10.1177/000992289803701204. [DOI] [PubMed] [Google Scholar]
- 199.Li S-TT, Lozano P, Grossman DC, Graham E. Hormone-containing hair product use in prepubertal children. Arch Pediatr Adolesc Med. 2002;156:85–86. doi: 10.1001/archpedi.156.1.85. [DOI] [PubMed] [Google Scholar]
- 200.Tiwary CM, Ward JA. Use of hair products containing hormone or placenta by US military personnel. J Pediatr Endocrinol Metab. 2003;16:1025–1032. doi: 10.1515/JPEM.2003.16.7.1025. [DOI] [PubMed] [Google Scholar]
- 201.Hsieh CC, Trichopoulos D, Katsouyanni K, Yuasa S. Age at menarche, age at menopause, height and obesity as risk factors for breast cancer: Associations and interactions in an international case-control study. Int J Cancer. 1990;46:796–800. doi: 10.1002/ijc.2910460508. [DOI] [PubMed] [Google Scholar]
- 202.Donovan M, Tiwary CM, Axelrod D, Sasco AJ, Jones L, Hajek R, et al. Personal care products that contain estrogens or xenoestrogens may increase breast cancer risk. Med Hypotheses. 2007;68:756–766. doi: 10.1016/j.mehy.2006.09.039. [DOI] [PubMed] [Google Scholar]
- 203.James-Todd T, Senie R, Terry MB. Racial/ethnic differences in hormonally-active hair product use: A plausible risk factor for health disparities. J Immigr Minor Health. 2012;14:506–511. doi: 10.1007/s10903-011-9482-5. [DOI] [PubMed] [Google Scholar]
- 204.Myers SL, Yang CZ, Bittner GD, Witt KL, Tice RR, Baird DD. Estrogenic and anti-estrogenic activity of off-the-shelf hair and skin care products. J Expo Sci Environ Epidemiol. 2015;25:271–277. doi: 10.1038/jes.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Olson AC, Link JS, Waisman JR, Kupiec TC. Breast cancer patients unknowingly dosing themselves with estrogen by using topical moisturizers. J Clin Oncol. 2009;27:e103–e104. doi: 10.1200/JCO.2009.23.1225. [DOI] [PubMed] [Google Scholar]
- 206.Draelos ZD. Topical and oral estrogens revisited for antiaging purposes. Fertil Steril. 2005;84:291–292. doi: 10.1016/j.fertnstert.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 207.Marty MS, Carney EW, Rowlands JC. Endocrine disruption: Historical perspectives and its impact on the future of toxicology testing. Toxicol Sci. 2010;120:S93–108. doi: 10.1093/toxsci/kfq329. [DOI] [PubMed] [Google Scholar]
- 208.Matsumoto H, Adachi S, Suzuki Y. Bisphenol a in ambient air particulates responsible for the proliferation of MCF-7 human breast cancer cells and its concentration changes over 6 months. Arch Environ Contam Toxicol. 2005;48:459–466. doi: 10.1007/s00244-003-0243-x. [DOI] [PubMed] [Google Scholar]
- 209.Rudel RA, Camann DE, Spengler JD, Korn LR, Brody JG. Phthalates, alkylphenols, pesticides, polybrominated diphenyl ethers, and other endocrine-disrupting compounds in indoor air and dust. Environ Sci Technol. 2003;37:4543–4553. doi: 10.1021/es0264596. [DOI] [PubMed] [Google Scholar]
- 210.Rodriguez-Mozaz S, de Alda ML, Barceló D. Analysis of bisphenol a in natural waters by means of an optical immunosensor. Water Res. 2005;39:5071–5079. doi: 10.1016/j.watres.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 211.Brotons JA, Olea-Serrano MF, Villalobos M, Pedraza V, Olea N. Xenoestrogens released from lacquer coatings in food cans. Environ Health Perspect. 1995;103:608–612. doi: 10.1289/ehp.95103608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lakind JS, Naiman DQ. Daily intake of bisphenol a and potential sources of exposure: 2005-2006 National Health and nutrition examination survey. J Expo Sci Environ Epidemiol. 2011;21:272–279. doi: 10.1038/jes.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol a (BPA) Reprod Toxicol. 2007;24:139–177. doi: 10.1016/j.reprotox.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 214.Carwile JL, Luu HT, Bassett LS, Driscoll D, Yuan C, Chang J, et al. Polycarbonate bottle use and urinary bisphenol a concentrations. Environ Health Perspect. 2009;117:1368–1372. doi: 10.1289/ehp.0900604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Smith R, Lourie . Slow death by rubber duck: The secret danger of everyday things. Berkeley: Counterpoint; 2009. [Google Scholar]
- 216.Rudel RA, Gray JM, Engel CL, Rawsthorne TW, Dodson RE, Ackerman JM, et al. Food packaging and bisphenol a and bis(2-ethyhexyl) phthalate exposure: Findings from a dietary intervention. Environ Health Perspect. 2011;119:914–920. doi: 10.1289/ehp.1003170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Stahlhut RW, Welshons WV, Swan SH. Bisphenol a data in NHANES suggest longer than expected half-life, substantial nonfood exposure, or both. Environ Health Perspect. 2009;117:784–789. doi: 10.1289/ehp.0800376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Vom Saal FS, Welshons WV. Evidence that bisphenol a (BPA) can be accurately measured without contamination in human serum and urine and that BPA causes numerous hazards from multiple routes of exposure. Mol Cell Endocrinol. 2014;398:101–113. doi: 10.1016/j.mce.2014.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Calafat AM, Ye X, Wong L-Y, Reidy JA, Needham LL. Exposure of the U.S. population to bisphenol a and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect. 2008;116:39–44. doi: 10.1289/ehp.10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ye X, Pierik FH, Angerer J, Meltzer HM, Jaddoe VWV, Tiemeier H, et al. Levels of metabolites of organophosphate pesticides, phthalates, and bisphenol a in pooled urine specimens from pregnant women participating in the Norwegian mother and child cohort study (MoBa) Int J Hyg Environ Health. 2009;212:481–491. doi: 10.1016/j.ijheh.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Padmanabhan V, Siefert K, Ransom S, Johnson T, Pinkerton J, Anderson L, et al. Maternal bisphenol-a levels at delivery: A looming problem? J Perinatol. 2008;28:258–263. doi: 10.1038/sj.jp.7211913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Teeguarden JG, Twaddle NC, Churchwell MI, Doerge DR. Urine and serum biomonitoring of exposure to environmental estrogens I: Bisphenol a in pregnant women. Food Chem Toxicol. 2016;92:129–142. doi: 10.1016/j.fct.2016.03.023. [DOI] [PubMed] [Google Scholar]
- 223.Kuruto-Niwa R, Ito T, Goto H, Nakamura H, Nozawa R, Terao Y. Estrogenic activity of the chlorinated derivatives of estrogens and flavonoids using a GFP expression system. Environ Toxicol Pharmacol. 2007;23:121–128. doi: 10.1016/j.etap.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 224.Sun Y, Irie M, Kishikawa N, Wada M, Kuroda N, Nakashima K. Determination of bisphenol a in human breast milk by HPLC with column-switching and fluorescence detection. Biomed Chromatogr. 2004;18:501–507. doi: 10.1002/bmc.345. [DOI] [PubMed] [Google Scholar]
- 225.Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol a concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod. 2002;17:2839–2841. doi: 10.1093/humrep/17.11.2839. [DOI] [PubMed] [Google Scholar]
- 226.Environmental Working Group . Guide to BPA. 2009. [Google Scholar]
- 227.Schönfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol a accumulation in the human maternal-fetal-placental unit. Environ Health Perspect. 2002;110:A703–A707. doi: 10.1289/ehp.021100703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Calafat AM, Weuve J, Ye X, Jia LT, Hu H, Ringer S, et al. Exposure to bisphenol a and other phenols in neonatal intensive care unit premature infants. Environ Health Perspect. 2009;117:639–644. doi: 10.1289/ehp.0800265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Maffini MV, Rubin BS, Sonnenschein C, Soto AM. Endocrine disruptors and reproductive health: The case of bisphenol-a. Mol Cell Endocrinol. 2006;254–255:179–186. doi: 10.1016/j.mce.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 230.Muñoz-de-Toro M, Markey CM, Wadia PR, Luque EH, Rubin BS, Sonnenschein C, et al. Perinatal exposure to bisphenol-a alters peripubertal mammary gland development in mice. Endocrinology. 2005;146:4138–4147. doi: 10.1210/en.2005-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Vandenberg LN, Maffini MV, Schaeberle CM, Ucci AA, Sonnenschein C, Rubin BS, et al. Perinatal exposure to the xenoestrogen bisphenol-a induces mammary intraductal hyperplasias in adult CD-1 mice. Reprod Toxicol. 2008;26:210–219. doi: 10.1016/j.reprotox.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Betancourt AM, Mobley JA, Russo J, Lamartiniere CA. Proteomic analysis in mammary glands of rat offspring exposed in utero to bisphenol a. J Proteome. 2010;73:1241–1253. doi: 10.1016/j.jprot.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 233.Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal bisphenol a exposure. Reprod Toxicol. 2007;23:383–390. doi: 10.1016/j.reprotox.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Acevedo N, Davis B, Schaeberle CM, Sonnenschein C, Soto AM. Perinatally administered bisphenol a as a potential mammary gland carcinogen in rats. Environ Health Perspect. 2013;121:1040–1046. doi: 10.1289/ehp.1306734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Durando M, Kass L, Piva J, Sonnenschein C, Soto AM, Luque EH, et al. Prenatal bisphenol a exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect. 2007;115:80–86. doi: 10.1289/ehp.9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Jenkins S, Raghuraman N, Eltoum I, Carpenter M, Russo J, Lamartiniere CA. Oral exposure to bisphenol a increases dimethylbenzanthracene-induced mammary cancer in rats. Environ Health Perspect. 2009;117:910–915. doi: 10.1289/ehp.11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wadia PR, Cabaton NJ, Borrero MD, Rubin BS, Sonnenschein C, Shioda T, et al. Low-dose BPA exposure alters the mesenchymal and epithelial transcriptomes of the mouse fetal mammary gland. PLoS One. 2013;8:e63902. doi: 10.1371/journal.pone.0063902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhang X-L, Wang H-S, Liu N, Ge L-C. Bisphenol a stimulates the epithelial mesenchymal transition of estrogen negative breast cancer cells via FOXA1 signals. Arch Biochem Biophys. 2015;585:10–16. doi: 10.1016/j.abb.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 239.Ibrahim MAA, Elbakry RH, Bayomy NA. Effect of bisphenol a on morphology, apoptosis and proliferation in the resting mammary gland of the adult albino rat. Int J Exp Pathol. 2016;97:27–36. doi: 10.1111/iep.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wadia PR, Vandenberg LN, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. Perinatal bisphenol a exposure increases estrogen sensitivity of the mammary gland in diverse mouse strains. Environ Health Perspect. 2007;115:592–598. doi: 10.1289/ehp.9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Tharp AP, Maffini MV, Hunt PA, VandeVoort CA, Sonnenschein C, Soto AM. Bisphenol a alters the development of the rhesus monkey mammary gland. Proc Natl Acad Sci U S A. 2012;109:8190–8195. doi: 10.1073/pnas.1120488109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Jenkins S, Wang J, Eltoum I, Desmond R, Lamartiniere CA. Chronic oral exposure to bisphenol a results in a nonmonotonic dose response in mammary carcinogenesis and metastasis in MMTV-erbB2 mice. Environ Health Perspect. 2011;119:1604–1609. doi: 10.1289/ehp.1103850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Vandenberg LN, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. The male mammary gland: A target for the xenoestrogen bisphenol a. Reprod Toxicol. 2013;37:15–23. doi: 10.1016/j.reprotox.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Kass L, Altamirano GA, Bosquiazzo VL, Luque EH, Muñoz-de-Toro M. Perinatal exposure to xenoestrogens impairs mammary gland differentiation and modifies milk composition in Wistar rats. Reprod Toxicol. 2012;33:390–400. doi: 10.1016/j.reprotox.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 245.Rivas A, Lacroix M, Olea-Serrano F, Laíos I, Leclercq G, Olea N. Estrogenic effect of a series of bisphenol analogues on gene and protein expression in MCF-7 breast cancer cells. J Steroid Biochem Mol Biol. 2002;82:45–53. doi: 10.1016/S0960-0760(02)00146-2. [DOI] [PubMed] [Google Scholar]
- 246.Welshons WV, Nagel SC, vom Saal FS. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol a at levels of human exposure. Endocrinology. 2006;147:S56–S69. doi: 10.1210/en.2005-1159. [DOI] [PubMed] [Google Scholar]
- 247.Watson CS, Bulayeva NN, Wozniak AL, Finnerty CC. Signaling from the membrane via membrane estrogen receptor-alpha: Estrogens, xenoestrogens, and phytoestrogens. Steroids. 2005;70:364–371. doi: 10.1016/j.steroids.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 248.Wozniak AL, Bulayeva NN, Watson CS. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-alpha-mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ Health Perspect. 2005;113:431–439. doi: 10.1289/ehp.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Castillo Sanchez R, Gomez R, Perez Salaezar E. Bisphenol a induces migration through a GPER-, FAK-, Src-, and ERK2-dependent pathway in MDA-MB-231 breast cancer cells. Chem Res Toxicol. 2016;29:285–295. doi: 10.1021/acs.chemrestox.5b00457. [DOI] [PubMed] [Google Scholar]
- 250.Okada H, Tokunaga T, Liu X, Takayanagi S, Matsushima A, Shimohigashi Y. Direct evidence revealing structural elements essential for the high binding ability of bisphenol a to human estrogen-related receptor-gamma. Environ Health Perspect. 2008;116:32–38. doi: 10.1289/ehp.10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Matsushima A, Teramoto T, Okada H, Liu X, Tokunaga T, Kakuta Y, et al. ERRgamma tethers strongly bisphenol a and 4-alpha-cumylphenol in an induced-fit manner. Biochem Biophys Res Commun. 2008;373:408–413. doi: 10.1016/j.bbrc.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 252.Takayanagi S, Tokunaga T, Liu X, Okada H, Matsushima A, Shimohigashi Y. Endocrine disruptor bisphenol a strongly binds to human estrogen-related receptor gamma (ERRgamma) with high constitutive activity. Toxicol Lett. 2006;167:95–105. doi: 10.1016/j.toxlet.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 253.Song H, Zhang T, Yang P, Li M, Yang Y, Wang Y, et al. Low doses of bisphenol a stimulate the proliferation of breast cancer cells via ERK1/2/ERRγ signals. Toxicol in Vitro. 2015;30:521–528. doi: 10.1016/j.tiv.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 254.Riggins RB, Mazzotta MM, Maniya OZ, Clarke R. Orphan nuclear receptors in breast cancer pathogenesis and therapeutic response. Endocr Relat Cancer. 2010;17:R213–R231. doi: 10.1677/ERC-10-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Hofmann PJ, Schomburg L, Köhrle J. Interference of endocrine disrupters with thyroid hormone receptor-dependent transactivation. Toxicol Sci. 2009;110:125–137. doi: 10.1093/toxsci/kfp086. [DOI] [PubMed] [Google Scholar]
- 256.Roy P, Salminen H, Koskimies P, Simola J, Smeds A, Saukko P, et al. Screening of some anti-androgenic endocrine disruptors using a recombinant cell-based in vitro bioassay. J Steroid Biochem Mol Biol. 2004;88:157–166. doi: 10.1016/j.jsbmb.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 257.Sun H, Xu L-C, Chen J-F, Song L, Wang X-R. Effect of bisphenol a, tetrachlorobisphenol a and pentachlorophenol on the transcriptional activities of androgen receptor-mediated reporter gene. Food Chem Toxicol. 2006;44:1916–1921. doi: 10.1016/j.fct.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 258.Fischer C, Mamillapalli R, Goetz LG, Jorgenson E, Ilagan Y, Taylor HS. Bisphenol a (BPA) exposure in utero leads to Immunoregulatory cytokine dysregulation in the mouse mammary gland: A potential mechanism programming breast cancer risk. Horm Cancer. 2016;7:241–251. doi: 10.1007/s12672-016-0254-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Goodson WH, Luciani MG, Sayeed SA, Jaffee IM, Moore DH, Dairkee SH. Activation of the mTOR pathway by low levels of xenoestrogens in breast epithelial cells from high-risk women. Carcinogenesis. 2011;32:1724–1733. doi: 10.1093/carcin/bgr196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Tilghman SL, Bratton MR, Segar HC, Martin EC, Rhodes LV, Li M, et al. Endocrine disruptor regulation of microRNA expression in breast carcinoma cells. PLoS One. 2012;7:e32754. doi: 10.1371/journal.pone.0032754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Weng Y-I, Hsu P-Y, Liyanarachchi S, Liu J, Deatherage DE, Huang Y-W, et al. Epigenetic influences of low-dose bisphenol a in primary human breast epithelial cells. Toxicol Appl Pharmacol. 2010;248:111–121. doi: 10.1016/j.taap.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Dairkee SH, Seok J, Champion S, Sayeed A, Mindrinos M, Xiao W, et al. Bisphenol a induces a profile of tumor aggressiveness in high-risk cells from breast cancer patients. Cancer Res. 2008;68:2076–2080. doi: 10.1158/0008-5472.CAN-07-6526. [DOI] [PubMed] [Google Scholar]
- 263.Lapensee EW, Tuttle TR, Fox SR, Ben-Jonathan N. Bisphenol a at low nanomolar doses confers chemoresistance in estrogen receptor-alpha-positive and -negative breast cancer cells. Environ Health Perspect. 2009;117:175–180. doi: 10.1289/ehp.11788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.LaPensee EW, LaPensee CR, Fox S, Schwemberger S, Afton S, Ben-Jonathan N. Bisphenol a and estradiol are equipotent in antagonizing cisplatin-induced cytotoxicity in breast cancer cells. Cancer Lett. 2010;290:167–173. doi: 10.1016/j.canlet.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Center for Disease Control and Prevention, National: Biomonitoring Program: Phthalates factsheet. 2015. http://www.cdc.gov/biomonitoring/phthalates_factsheet.html. Accessed 25 Oct 2016.
- 266.Janjua NR, Frederiksen H, Skakkebaek NE, Wulf HC, Andersson A-M. Urinary excretion of phthalates and paraben after repeated whole-body topical application in humans. Int J Androl. 2008;31:118–130. doi: 10.1111/j.1365-2605.2007.00841.x. [DOI] [PubMed] [Google Scholar]
- 267.Schettler T. Human exposure to phthalates via consumer products. Int J Androl. 2005;29:134–139. doi: 10.1111/j.1365-2605.2005.00567.x. [DOI] [PubMed] [Google Scholar]
- 268.Fromme H, Gruber L, Schlummer M, Wolz G, Böhmer S, Angerer J, et al. Intake of phthalates and di(2-ethylhexyl)adipate: Results of the integrated exposure assessment survey based on duplicate diet samples and biomonitoring data. Environ Int. 2007;33:1012–1020. doi: 10.1016/j.envint.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 269.Birnbaum LS, Schug TT. Phthalates in our food. Endocr Disrupt. 2013;1(1):e25078. doi: 10.4161/endo.25078. [DOI] [Google Scholar]
- 270.Zota AR, Phillips CA, Mitro SD. Recent fast food consumption and bisphenol a and phthalates exposures among the U.S. population in NHANES, 2003-2010. Environ Health Perspect. 2016; doi:10.1289/ehp.1510803. [DOI] [PMC free article] [PubMed]
- 271.Ji K, Lim Kho Y, Park Y, Choi K. Influence of a five-day vegetarian diet on urinary levels of antibiotics and phthalate metabolites: A pilot study with “temple stay” participants. Environ Res. 2010;110:375–382. doi: 10.1016/j.envres.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 272.Cirillo T, Fasano E, Castaldi E, Montuori P, Amodio Cocchieri R. Children’s exposure to di(2-ethylhexyl)phthalate and dibutylphthalate plasticizers from school meals. J Agric Food Chem. 2011;59:10532–10538. doi: 10.1021/jf2020446. [DOI] [PubMed] [Google Scholar]
- 273.Chatonnet P, Boutou S, Plana A. Contamination of wines and spirits by phthalates: Types of contaminants present, contamination sources and means of prevention. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2014;31:1605–1615. doi: 10.1080/19440049.2014.941947. [DOI] [PubMed] [Google Scholar]
- 274.Sathyanarayana S, Alcedo G, Saelens BE, Zhou C, Dills RL, Yu J, et al. Unexpected results in a randomized dietary trial to reduce phthalate and bisphenol a exposures. J Expo Sci Environ Epidemiol. 2013;23:378–384. doi: 10.1038/jes.2013.9. [DOI] [PubMed] [Google Scholar]
- 275.Rudel RA, Brody JG, Spengler JD, Vallarino J, Geno PW, Sun G, et al. Identification of selected hormonally active agents and animal mammary carcinogens in commercial and residential air and dust samples. J Air Waste Manag Assoc. 2001;51:499–513. doi: 10.1080/10473289.2001.10464292. [DOI] [PubMed] [Google Scholar]
- 276.Calafat AM, Wong L-Y, Silva MJ, Samandar E, Preau JL, Jia LT, et al. Selecting adequate exposure biomarkers of diisononyl and diisodecyl phthalates: Data from the 2005-2006 National Health and nutrition examination survey. Environ Health Perspect. 2011;119:50–55. doi: 10.1289/ehp.1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Frederiksen H, Aksglaede L, Sorensen K, Skakkebaek NE, Juul A, Andersson A-M. Urinary excretion of phthalate metabolites in 129 healthy Danish children and adolescents: Estimation of daily phthalate intake. Environ Res. 2011;111:656–663. doi: 10.1016/j.envres.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 278.Zota AR, Calafat AM, Woodruff TJ. Temporal trends in phthalate exposures: Findings from the National Health and nutrition examination survey, 2001–2010. Environ Health Perspect. 2014;122:235–241. doi: 10.1289/ehp.1408629R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Jensen MS, Nørgaard-Pedersen B, Toft G, Hougaard DM, Bonde JP, Cohen A, et al. Phthalates and perfluorooctanesulfonic acid in human amniotic fluid: Temporal trends and timing of amniocentesis in pregnancy. Environ Health Perspect. 2012;120:897–903. doi: 10.1289/ehp.1104522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Hines EP, Calafat AM, Silva MJ, Mendola P, Fenton SE. Concentrations of phthalate metabolites in milk, urine, saliva, and serum of lactating North Carolina women. Environ Health Perspect. 2009;117:86–92. doi: 10.1289/ehp.11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Meeker JD, Sathyanarayana S, Swan SH. Phthalates and other additives in plastics: Human exposure and associated health outcomes. Philos Trans R Soc Lond Ser B Biol Sci. 2009;364:2097–2113. doi: 10.1098/rstb.2008.0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Wittassek M, Angerer J, Kolossa-Gehring M, Schäfer SD, Klockenbusch W, Dobler L, et al. Fetal exposure to phthalates—a pilot study. Int J Hyg Environ Health. 2009;212:492–498. doi: 10.1016/j.ijheh.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 283.Sathyanarayana S, Karr CJ, Lozano P, Brown E, Calafat AM, Liu F, et al. Baby care products: Possible sources of infant phthalate exposure. Pediatrics. 2008;121:e260–e268. doi: 10.1542/peds.2006-3766. [DOI] [PubMed] [Google Scholar]
- 284.López-Carrillo L, Hernández-Ramírez RU, Calafat AM, Torres-Sánchez L, Galván-Portillo M, Needham LL, et al. Exposure to phthalates and breast cancer risk in northern Mexico. Environ Health Perspect. 2010;118:539–544. doi: 10.1289/ehp.0901091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Toft G, Jönsson BAG, Lindh CH, Jensen TK, Hjollund NH, Vested A, et al. Association between pregnancy loss and urinary phthalate levels around the time of conception. Environ Health Perspect. 2012;120:458–463. doi: 10.1289/ehp.1103552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Jobling S, Reynolds T, White R, Parker MG, Sumpter JP. A variety of environmentally persistent chemicals, including some phthalate plasticizers, are weakly estrogenic. Environ Health Perspect. 1995;103:582–587. doi: 10.1289/ehp.95103582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Kang SC, Lee BM. DNA methylation of estrogen receptor alpha gene by phthalates. J Toxicol Environ Health A. 2005;68:1995–2003. doi: 10.1080/15287390491008913. [DOI] [PubMed] [Google Scholar]
- 288.Borch J, Metzdorff SB, Vinggaard AM, Brokken L, Dalgaard M. Mechanisms underlying the anti-androgenic effects of diethylhexyl phthalate in fetal rat testis. Toxicology. 2006;223:144–155. doi: 10.1016/j.tox.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 289.Fang H, Tong W, Branham WS, Moland CL, Dial SL, Hong H, et al. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem Res Toxicol. 2003;16:1338–1358. doi: 10.1021/tx030011g. [DOI] [PubMed] [Google Scholar]
- 290.Wang Y-C, Tsai C-F, Chuang H-L, Chang Y-C, Chen H-S, Lee J-N, et al. Benzyl butyl phthalate promotes breast cancer stem cell expansion via SPHK1/S1P/S1PR3 signaling. Oncotarget. 2016;7:29563–29576. doi: 10.18632/oncotarget.9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Das MT, Singh MK, Thakur IS. Differential toxicological endpoints of di(2-ethylhexyl) phthalate (DEHP) exposure in MCF-7 and MDA-MB-231 cell lines: Possible estrogen receptor α (ERα) independent modulations. Indian J Exp Biol. 2014;52:1052–1061. [PubMed] [Google Scholar]
- 292.Hsieh T-H, Tsai C-F, Hsu C-Y, Kuo P-L, Lee J-N, Chai C-Y, et al. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J. 2012;26:778–787. doi: 10.1096/fj.11-191742. [DOI] [PubMed] [Google Scholar]
- 293.Jiang J, Ma L, Yuan L, Wang X, Zhang W. Study on developmental abnormalities in hypospadiac male rats induced by maternal exposure to di-n-butyl phthalate (DBP) Toxicology. 2007;232:286–293. doi: 10.1016/j.tox.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 294.Lovekamp-Swan T, Davis BJ. Mechanisms of phthalate ester toxicity in the female reproductive system. Environ Health Perspect. 2003;111:139–145. doi: 10.1289/ehp.5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Foster PMD. Disruption of reproductive development in male rat offspring following in utero exposure to phthalate esters. Int J Androl. 2006;29:140–147. doi: 10.1111/j.1365-2605.2005.00563.x. [DOI] [PubMed] [Google Scholar]
- 296.Latini G, Del Vecchio A, Massaro M, Verrotti A, De Felice C. Phthalate exposure and male infertility. Toxicology. 2006;226:90–98. doi: 10.1016/j.tox.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 297.Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM, et al. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect. 2005;113:1056–1061. doi: 10.1289/ehp.8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Bornehag C-G, Carlstedt F, Jönsson BAG, Lindh CH, Jensen TK, Bodin A, et al. Prenatal phthalate exposures and anogenital distance in Swedish boys. Environ Health Perspect. 2015;123:101–107. doi: 10.1289/ehp.1408163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Chou Y-Y, Huang P-C, Lee C-C, Wu M-H, Lin S-J. Phthalate exposure in girls during early puberty. J Pediatr Endocrinol Metab. 2009;22:69–77. doi: 10.1515/JPEM.2009.22.1.69. [DOI] [PubMed] [Google Scholar]
- 300.Steingraber S. The falling age of puberty in U.S. girls: What we know, what we need to know. San Francisco: Breast Cancer Fund; 2007. [Google Scholar]
- 301.Moral R, Wang R, Russo IH, Mailo DA, Lamartiniere CA, Russo J. The plasticizer butyl benzyl phthalate induces genomic changes in rat mammary gland after neonatal/prepubertal exposure. BMC Genomics. 2007;8:453. doi: 10.1186/1471-2164-8-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Moral R, Santucci-Pereira J, Wang R, Russo IH, Lamartiniere CA, Russo J. In utero exposure to butyl benzyl phthalate induces modifications in the morphology and the gene expression profile of the mammary gland: An experimental study in rats. Environ Health. 2011;10:5. doi: 10.1186/1476-069X-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Oral D, Erkekoglu P, Kocer-Gumusel B, Chao M-W. Epithelial-mesenchymal transition: A special focus on phthalates and bisphenol a. J Environ Pathol Toxicol Oncol. 2016;35:43–58. doi: 10.1615/JEnvironPatholToxicolOncol.2016014200. [DOI] [PubMed] [Google Scholar]
- 304.Gwinn MR, Whipkey DL, Tennant LB, Weston A. Gene expression profiling of di-n-butyl phthalate in normal human mammary epithelial cells. J Environ Pathol Toxicol Oncol. 2007;26:51–61. doi: 10.1615/JEnvironPatholToxicolOncol.v26.i1.60. [DOI] [PubMed] [Google Scholar]
- 305.Kim IY, Han SY, Moon A. Phthalates inhibit tamoxifen-induced apoptosis in MCF-7 human breast cancer cells. J Toxicol Environ Health A. 2004;6:2025–2035. doi: 10.1080/15287390490514750. [DOI] [PubMed] [Google Scholar]
- 306.Hsu Y-L, Hung J-Y, Tsai E-M, Wu C-Y, Ho Y-W, Jian S-F, et al. Benzyl butyl phthalate increases the chemoresistance to doxorubicin/cyclophosphamide by increasing breast cancer-associated dendritic cell-derived CXCL1/GROα and S100A8/A9. Oncol Rep. 2015;34:2889–2900. doi: 10.3892/or.2015.4307. [DOI] [PubMed] [Google Scholar]
- 307.Calafat AM, Ye X, Wong L-Y, Bishop AM, Needham LL. Urinary concentrations of four parabens in the U.S. population: NHANES 2005–2006. Environ Health Perspect. 2010;118:679–685. doi: 10.1289/ehp.0901560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Philippat C, Wolff MS, Calafat AM, Ye X, Bausell R, Meadows M, et al. Prenatal exposure to environmental phenols: Concentrations in amniotic fluid and variability in urinary concentrations during pregnancy. Environ Health Perspect. 2013;121:1225–1231. doi: 10.1289/ehp.1206335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Darbre P, Aljarrah A, Miller W. Concentrations of parabens in human breast tumours. J Appl Toxicol. 2004;24:5–13. doi: 10.1002/jat.958. [DOI] [PubMed] [Google Scholar]
- 310.Rastogi S, Schouten A, De Kruijf N, Weijland J. Contents of methylparaben, ethylparaben, propylparaben, butylparaben and benzylparaben in cosmetic products. Contact Dermatitis. 1995;32:28–30. doi: 10.1111/j.1600-0536.1995.tb00836.x. [DOI] [PubMed] [Google Scholar]
- 311.Barr L, Metaxas G, Harbach CAJ, Savoy LA, Darbre PD. Measurement of paraben concentrations in human breast tissue at serial locations across the breast from axilla to sternum. J Appl Toxicol. 2012;32:219–232. doi: 10.1002/jat.1786. [DOI] [PubMed] [Google Scholar]
- 312.Darbre PD, Harvey PW. Parabens can enable hallmarks and characteristics of cancer in human breast epithelial cells: A review of the literature with reference to new exposure data and regulatory status. J Appl Toxicol. 2014;34:925–938. doi: 10.1002/jat.3027. [DOI] [PubMed] [Google Scholar]
- 313.Karpuzoglu E, Holladay SD, Gogal RM. Parabens: Potential impact of low-affinity estrogen receptor binding chemicals on human health. J Toxicol Environ Health B Crit Rev. 2013;16:321–335. doi: 10.1080/10937404.2013.809252. [DOI] [PubMed] [Google Scholar]
- 314.Klopčič I, Kolšek K, Dolenc MS. Glucocorticoid-like activity of propylparaben, butylparaben, diethylhexyl phthalate and tetramethrin mixtures studied in the MDA-kb2 cell line. Toxicol Lett. 2015;232:376–383. doi: 10.1016/j.toxlet.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 315.Darbre PD, Harvey PW. Paraben esters: Review of recent studies of endocrine toxicity, absorption, esterase, and discussion of possible health risks. J Appl Toxicol. 2008;28:561–578. doi: 10.1002/jat.1358. [DOI] [PubMed] [Google Scholar]
- 316.Watanabe Y, Kojima H, Takeuchi S, Uramaru N, Ohta S, Kitamura S. Comparative study on transcriptional activity of 17 parabens mediated by estrogen receptor α and β and androgen receptor. Food Chem Toxicol. 2013;57:227–234. doi: 10.1016/j.fct.2013.03.036. [DOI] [PubMed] [Google Scholar]
- 317.Byford J, Shaw L, Drew M, Pope G, Sauer M, Darbre P. Oestrogenic activity of parabens in MCF7 human breast cancer cells. J Steroid Biochem Mol Biol. 2002;80:49–60. doi: 10.1016/S0960-0760(01)00174-1. [DOI] [PubMed] [Google Scholar]
- 318.Gopalakrishan K, Teitelbaum SL, Lambertini L, Wetmur J, Manservisi F, Luciani MG, Falcioni L, et al. Changes in mammary histology and transcriptome profils by low-dose exposure to environmental phenols at critical periods of development. Environ Res. 2017;152:232–43. [DOI] [PMC free article] [PubMed]
- 319.Pugazhendhi D, Sadler A, Darbre P. Comparison of the global gene expression profiles produced by methylparaben, n-butylparaben and 17B-oestradiol in mcf7 human breast cancer cells. J Appl Toxicol. 2007;27:67–77. doi: 10.1002/jat.1200. [DOI] [PubMed] [Google Scholar]
- 320.Wróbel A, Gregoraszczuk EL. Effects of single and repeated in vitro exposure of three forms of parabens, methyl-, butyl-and propylparabens on the proliferation and estradiol secretion in MCF-7 and MCF-10A cells. Pharmacol Rep. 2013;65:484–493. doi: 10.1016/S1734-1140(13)71024-7. [DOI] [PubMed] [Google Scholar]
- 321.Charles AK, Darbre PD. Combinations of parabens at concentrations measured in human breast tissue can increase proliferation of MCF-7 human breast cancer cells. J Appl Toxicol. 2013;33:390–398. doi: 10.1002/jat.2850. [DOI] [PubMed] [Google Scholar]
- 322.Wróbel AM, Gregoraszczuk EL. Differential effect of methyl-, butyl- and propylparaben and 17β-estradiol on selected cell cycle and apoptosis gene and protein expression in MCF-7 breast cancer cells and MCF-10A non-malignant cells. J App Toxicol. 2014;34:1041–1050. doi: 10.1002/jat.2978. [DOI] [PubMed] [Google Scholar]
- 323.Pan S, Yuan C, Tagmount A, Rudel RA, Ackerman JM, Yaswen P, et al. Parabens and human epidermal growth factor receptor ligand cross-talk in breast cancer cells. Environ Health Perspect. 2016;124:563–569. doi: 10.1289/ehp.1409200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Zhang Z, Sun L, Hu Y, Jiao J, Hu J. Inverse antagonist activities of parabens on human oestrogen-related receptor γ (ERRγ): In vitro and in silico studies. Toxicol Appl Pharmacol. 2013;270:16–22. doi: 10.1016/j.taap.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 325.Khanna S, Darbre PD. Parabens enable suspension growth of MCF-10A immortalized, non-transformed human breast epithelial cells. J Appl Toxicol. 2013;33:378–382. doi: 10.1002/jat.2753. [DOI] [PubMed] [Google Scholar]
- 326.Khanna S, Dash PR, Darbre PD. Exposure to parabens at the concentration of maximal proliferative response increases migratory and invasive activity of human breast cancer cells in vitro. J Appl Toxicol. 2014;34:1051–1059. doi: 10.1002/jat.3003. [DOI] [PubMed] [Google Scholar]
- 327.Deshayes S, Eudes V, Droguet C, Bigourie M, Gasperi J, Moilleron R. Alkylphenols and phthalates in greywater from showers and washing machines. Water Air Soil Pollut. 2015;226 doi:10.1007/s11270-015-2652-7.
- 328.Slack RJ, Gronow JR, Voulvoulis N. Household hazardous waste in municipal landfills: Contaminants in leachate. Sci Total Environ. 2005;337:119–137. doi: 10.1016/j.scitotenv.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 329.Pasquini L, Munoz J-F, Pons M-N, Yvon J, Dauchy X, France X, et al. Occurrence of eight household micropollutants in urban wastewater and their fate in a wastewater treatment plant. Statistical evaluation. Sci Total Environ. 2014;481:459–468. doi: 10.1016/j.scitotenv.2014.02.075. [DOI] [PubMed] [Google Scholar]
- 330.Swartz CH, Reddy S, Benotti MJ, Yin H, Barber LB, Brownawell BJ, et al. Steroid estrogens, nonylphenol ethoxylate metabolites, and other wastewater contaminants in groundwater affected by a residential septic system on cape cod, MA. Environ Sci Technol. 2006;40:4894–4902. doi: 10.1021/es052595+. [DOI] [PubMed] [Google Scholar]
- 331.Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL. Urinary concentrations of bisphenol a and 4-nonylphenol in a human reference population. Environ Health Perspect. 2005;113:391–395. doi: 10.1289/ehp.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Gyllenhammar I, Glynn A, Darnerud PO, Lignell S, van Delft R, Aune M. 4-Nonylphenol and bisphenol a in Swedish food and exposure in Swedish nursing women. Environ Int. 2012;43:21–28. doi: 10.1016/j.envint.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 333.Chen G-W, Ding W-H, Ku H-Y, Chao H-R, Chen H-Y, Huang M-C, et al. Alkylphenols in human milk and their relations to dietary habits in central Taiwan. Food Chem Toxicol. 2010;48:1939–1944. doi: 10.1016/j.fct.2010.04.038. [DOI] [PubMed] [Google Scholar]
- 334.Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. 2006;102:175–179. doi: 10.1016/j.jsbmb.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 335.Oh M. Comparative analysis of gene expression pattern after exposure to nonylphenol in human cell lines. Biochip J. 2009:2:261–8.
- 336.Amaro AA, Esposito AI, Mirisola V, Mehilli A, Rosano C, Noonan DM, et al. Endocrine disruptor agent nonyl phenol exerts an estrogen-like transcriptional activity on estrogen receptor positive breast cancer cells. Curr Med Chem. 2014;2:630–640. doi: 10.2174/09298673113209990169. [DOI] [PubMed] [Google Scholar]
- 337.Lee H-R, Hwang K-A, Nam K-H, Kim H-C, Choi K-C. Progression of breast cancer cells was enhanced by endocrine-disrupting chemicals, triclosan and octylphenol, via an estrogen receptor-dependent signaling pathway in cellular and mouse xenograft models. Chem Res Toxicol. 2014;27:834–842. doi: 10.1021/tx5000156. [DOI] [PubMed] [Google Scholar]
- 338.Moon HJ, Han SY, Shin J-H, Kang IH, Kim TS, Hong JH, et al. Gestational exposure to nonylphenol causes precocious mammary gland development in female rat offspring. J Reprod Dev. 2007;53:333–344. doi: 10.1262/jrd.18055. [DOI] [PubMed] [Google Scholar]
- 339.Acevedo R, Parnell PG, Villanueva H, Chapman LM, Gimenez T, Gray SL, et al. The contribution of hepatic steroid metabolism to serum estradiol and estriol concentrations in nonylphenol treated MMTVneu mice and its potential effects on breast cancer incidence and latency. J Appl Toxicol. 2005;25:339–353. doi: 10.1002/jat.1078. [DOI] [PubMed] [Google Scholar]
- 340.Dann AB, Hontela A. Triclosan: Environmental exposure, toxicity and mechanisms of action. J Appl Toxicol. 2011;31:285–311. doi: 10.1002/jat.1660. [DOI] [PubMed] [Google Scholar]
- 341.Ahn KC, Zhao B, Chen J, Cherednichenko G, Sanmarti E, Denison MS, et al. In vitro biologic activities of the antimicrobials triclocarban, its analogs, and triclosan in bioassay screens: Receptor-based bioassay screens. Environ Health Perspect. 2008;116:1203–1210. doi: 10.1289/ehp.11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Brausch JM, Rand GM. A review of personal care products in the aquatic environment: Environmental concentrations and toxicity. Chemosphere. 2011;82:1518–1532. doi: 10.1016/j.chemosphere.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 343.Venkatesan AK, Pycke BFG, Barber LB, Lee KE, Halden RU. Occurrence of triclosan, triclocarban, and its lesser chlorinated congeners in Minnesota freshwater sediments collected near wastewater treatment plants. J Hazard Mater. 2012;229–230:29–35. doi: 10.1016/j.jhazmat.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Yueh M-F, Tukey RH. Triclosan: A widespread environmental toxicant with many biological effects. Annu Rev Pharmacol Toxicol. 2016;56:251–272. doi: 10.1146/annurev-pharmtox-010715-103417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Woodruff TJ, Zota AR, Schwartz JM. Environmental chemicals in pregnant women in the united states: NHANES 2003–2004. Environ Health Perspect. 2011;119:878–885. doi: 10.1289/ehp.1002727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Ye X, Zhou X, Furr J, Ahn KC, Hammock BD, Gray EL, et al. Biomarkers of exposure to triclocarban in urine and serum. Toxicology. 2011;286:69–74. doi: 10.1016/j.tox.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Geens T, Neels H, Covaci A. Distribution of bisphenol-a, triclosan and n-nonylphenol in human adipose tissue, liver and brain. Chemosphere. 2012;87:796–802. doi: 10.1016/j.chemosphere.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 348.Dinwiddie MT, Terry PD, Chen J. Recent evidence regarding triclosan and cancer risk. Int J Environ Res Public Health. 2014;11:2209–2217. doi: 10.3390/ijerph110202209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Christen V, Crettaz P, Oberli-Schrämmli A, Fent K. Some flame retardants and the antimicrobials triclosan and triclocarban enhance the androgenic activity in vitro. Chemosphere. 2010;81:1245–1252. doi: 10.1016/j.chemosphere.2010.09.031. [DOI] [PubMed] [Google Scholar]
- 350.Gee RH, Charles A, Taylor N, Darbre PD. Oestrogenic and androgenic activity of triclosan in breast cancer cells. J Appl Toxicol. 2008;28:78–91. doi: 10.1002/jat.1316. [DOI] [PubMed] [Google Scholar]
- 351.Henry ND, Fair PA. Comparison of in vitro cytotoxicity, estrogenicity and anti-estrogenicity of triclosan, perfluorooctane sulfonate and perfluorooctanoic acid. J Appl Toxicol. 2013;33:265–272. doi: 10.1002/jat.1736. [DOI] [PubMed] [Google Scholar]
- 352.Lee H-R, Hwang K-A, Nam K-H, Kim H-C, Choi K-C. Progression of breast cancer cells was enhanced by endocrine-disrupting chemicals, triclosan and octylphenol, via an estrogen receptor-dependent signaling pathway in cellular and mouse xenograft models. Chem Res Toxicol. 2014;27:834–842. doi: 10.1021/tx5000156. [DOI] [PubMed] [Google Scholar]
- 353.Kim J-Y, Yi B-R, Go R-E, Hwang K-A, Nam K-H, Choi K-H. Methoxychlor and triclosan stimulates ovarian cancer growth by regulating cell cycle- and apoptosis-related genes via an estrogen-dependent pathway. Environ Toxicol Pharmacol. 2014;37:1264–74. [DOI] [PubMed]
- 354.Zorrilla LM, Gibson EK, Jeffay SC, Crofton KM, Setzer WR, Cooper RL, et al. The effects of triclosan on puberty and thyroid hormonezorillas in male wistar rats. Toxicol Sci. 2009;107:56–64. doi: 10.1093/toxsci/kfn225. [DOI] [PubMed] [Google Scholar]
- 355.Paul KB, Hedge JM, DeVito MJ, Crofton KM. Developmental triclosan exposure decreases maternal and neonatal thyroxine in rats. Environ Toxicol Chem. 2010;29:2840–2844. doi: 10.1002/etc.339. [DOI] [PubMed] [Google Scholar]
- 356.Hayden C, Roberts M, Benson H. Systemic absorption of sunscreen after topical application. Lancet. 1997;350:853–864. doi: 10.1016/S0140-6736(05)62032-6. [DOI] [PubMed] [Google Scholar]
- 357.Felix T, Hall BJ, Brodbelt JS. Determination of benzophenone-3 and metabolites in water and human urine by solid-phase microextraction and quadrupole ion trap GC-MS. Anal Chim Acta. 1998;371:195–203. doi: 10.1016/S0003-2670(98)00293-1. [DOI] [Google Scholar]
- 358.Kunz PY, Fent K. Estrogenic activity of UV filter mixtures. Toxicol Appl Pharmacol. 2006;217:86–99. doi: 10.1016/j.taap.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 359.Waldman JM, Gavin Q, Anderson M, Hoover S, Alvaran J, Ip HSS, et al. Exposures to environmental phenols in Southern California firefighters and findings of elevated urinary benzophenone-3 levels. Environ Int. 2016;88:281–287. doi: 10.1016/j.envint.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 360.Krause M, Klit A, Blomberg Jensen M, Søeborg T, Frederiksen H, Schlumpf M, et al. Sunscreens: Are they beneficial for health? An overview of endocrine disrupting properties of UV-filters. Int J Androl. 2012;35:424–436. doi: 10.1111/j.1365-2605.2012.01280.x. [DOI] [PubMed] [Google Scholar]
- 361.Schlumpf M, Cotton B, Conscience M, Haller V, Steinmann B, Lichtensteiger W. In vitro and in vivo estrogenicity of UV screens. Environ Health Perspect. 2001;109:239–244. doi: 10.1289/ehp.01109239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Klann A, Levy G, Lutz I, Muller C, Kloas W, Hildebrandt J. Estrogen-like effects of ultraviolet screen 3-(4methylbenzylidene)-camphor (Eusolex 6300) on cell proliferation and gene induction in mammalian and amphibian cells. Environ Res. 2005;97:274–281. doi: 10.1016/j.envres.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 363.Brand R, McMahon L, Jendrzejewski J, Charron A. Transdermal absorption of the herbicide 2,4-dichlorophenoxyacetic acid is enhanced by both ethanol consumption and sunscreen application. Food Chem Toxicol. 2007;45:93–97. doi: 10.1016/j.fct.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 364.Jensen AA, Leffers H. Emerging endocrine disrupters: Perfluoroalkylated substances. Int J Androl. 2008;31:161–169. doi: 10.1111/j.1365-2605.2008.00870.x. [DOI] [PubMed] [Google Scholar]
- 365.Kato K, Wong L-Y, Jia LT, Kuklenyik Z, Calafat AM. Trends in exposure to polyfluoroalkyl chemicals in the U.S. p[o]pulation:1999–2008. Environ Sci Technol. 2001;45:8037–8045. doi: 10.1021/es1043613. [DOI] [PubMed] [Google Scholar]
- 366.Kannan K, Corsolini S, Falandysz J, Fillmann G, Kumar KS, Loganathan BG, et al. Perfluorooctanesulfonate and related fluorochemicals in human blood from several countries. Environ Sci Technol. 2004;38:4489–4495. doi: 10.1021/es0493446. [DOI] [PubMed] [Google Scholar]
- 367.Apelberg BJ, Goldman LR, Calafat AM, Herbstman JB, Kuklenyik Z, Heidler J, et al. Determinants of fetal exposure to polyfluoroalkyl compounds in Baltimore, Maryland. Environ Sci Technol. 2007;41:3891–3897. doi: 10.1021/es0700911. [DOI] [PubMed] [Google Scholar]
- 368.Apelberg BJ, Witter FR, Herbstman JB, Calafat AM, Halden RU, Needham LL, et al. Cord serum concentrations of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in relation to weight and size at birth. Environ Health Perspect. 2007;115:1670–1676. doi: 10.1289/ehp.10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Stein CR, Wolff MS, Calafat AM, Kato K, Engel SM. Comparison of polyfluoroalkyl compound concentrations in maternal serum and amniotic fluid: A pilot study. Reprod Toxicol. 2012;34:312–316. doi: 10.1016/j.reprotox.2012.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Maisonet M, Terrell ML, McGeehin MA, Christensen KY, Holmes A, Calafat AM, et al. Maternal concentrations of polyfluoroalkyl compounds during pregnancy and fetal and postnatal growth in British girls. Environ Health Perspect. 2012;120:1432–1437. doi: 10.1289/ehp.1003096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Maisonet M, Calafat AM, Marcus M, Jaakkola JJK, Lashen H. Prenatal exposure to Perfluoroalkyl acids and serum testosterone concentrations at 15 years of age in female ALSPAC study participants. Environ Health Perspect. 2015;123:1325–1330. doi: 10.1289/ehp.1408847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Dimitrakakis C, Bondy C. Androgens and the breast. Breast Cancer Res. 2009;11:212. doi: 10.1186/bcr2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Lopez-Espinosa M-J, Fletcher T, Armstrong B, Genser B, Dhatariya K, Mondal D, et al. Association of Perfluorooctanoic Acid (PFOA) and Perfluorooctane sulfonate (PFOS) with age of puberty among children living near a chemical plant. Environ Sci Technol. 2011;45:8160–8166. doi: 10.1021/es1038694. [DOI] [PubMed] [Google Scholar]
- 374.Kristensen SL, Ramlau-Hansen CH, Ernst E, Olsen SF, Bonde JP, Vested A, et al. Long-term effects of prenatal exposure to perfluoroalkyl substances on female reproduction. Hum Reprod. 2013;28:3337–3348. doi: 10.1093/humrep/det382. [DOI] [PubMed] [Google Scholar]
- 375.Pinney SM, Windham GC, Biro FM, Kushi LH, Yaghjyan L, Calafat A, et al. Perfluorooctanoic acid (PFOA) and pubertal maturation in young girls. Epidemiology. 2009;20:S80. doi: 10.1097/01.ede.0000362949.30847.cb. [DOI] [Google Scholar]
- 376.Fei C, McLaughlin JK, Lipworth L, Olsen J. Maternal levels of perfluorinated chemicals and subfecundity. Hum Reprod. 2009;24:1200–1205. doi: 10.1093/humrep/den490. [DOI] [PubMed] [Google Scholar]
- 377.Bonefeld-Jorgensen EC, Long M, Bossi R, Ayotte P, Asmund G, Krüger T, et al. Perfluorinated compounds are related to breast cancer risk in Greenlandic Inuit: A case control study. Environ Health. 2011;10:88. doi: 10.1186/1476-069X-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.White SS, Calafat AM, Kuklenyik Z, Villanueva L, Zehr RD, Helfant L, et al. Gestational PFOA exposure of mice is associated with altered mammary gland development in dams and female offspring. Toxicol Sci. 2007;96:133–144. doi: 10.1093/toxsci/kfl177. [DOI] [PubMed] [Google Scholar]
- 379.White SS, Kato K, Jia LT, Basden BJ, Calafat AM, Hines EP, et al. Effects of perfluorooctanoic acid on mouse mammary gland development and differentiation resulting from cross-foster and restricted gestational exposures. Reprod Toxicol. 2009;27:289–298. doi: 10.1016/j.reprotox.2008.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Yang C, Tan YS, Harkema JR, Haslam SZ. Differential effects of peripubertal exposure to perfluorooctanoic acid on mammary gland development in C57Bl/6 and Balb/c mouse strains. Reprod Toxicol. 2009;27:299–306. doi: 10.1016/j.reprotox.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Tucker DK, Macon MB, Strynar MJ, Dagnino S, Andersen E, Fenton SE. The mammary gland is a sensitive pubertal target in CD-1 and C57Bl/6 mice following perinatal perfluorooctanoic acid (PFOA) exposure. Reprod Toxicol. 2015;54:26–36. doi: 10.1016/j.reprotox.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Sonthithai P, Suriyo T, Thiantanawat A, Watcharasit P, Ruchirawat M, Satayavivad J. Perfluorinated chemicals, PFOS and PFOA, enhance the estrogenic effects of 17β-estradiol in T47D human breast cancer cells. J Appl Toxicol. 2016;36:790–801. doi: 10.1002/jat.3210. [DOI] [PubMed] [Google Scholar]
- 383.Bonner M, Han D, Nie J, Rogerson P, Vena J, Muti P, et al. Breast cancer risk and exposure in early life to polycyclic aromatic hydrocarbons using total suspended particulates as a proxy measure. Cancer Epidemiol Biomark Prev. 2005;14:53–60. [PubMed] [Google Scholar]
- 384.Beyea J, Stellman S, Hatch M, Gammon M. Airborne emissions from 1961 to 2004 of benzo(a)pyrene from U.S. vehicles per km of travel based on tunnel studies. Environ Sci Technol. 2008;47:7315–7320. doi: 10.1021/es8000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Morris JJ, Seifter E. The role of aromatic hydrocarbons in the genesis of breast cancer. Med Hypoth. 1992;38:177–184. doi: 10.1016/0306-9877(92)90090-Y. [DOI] [PubMed] [Google Scholar]
- 386.Pliskova M, Vondracek J, Vojtesek B, Kozubik A, Machala M. Deregulation of cell proliferation by polycyclic aromatic hydrocarbons in human breast carcinoma MCF-7 cells reflects both genotoxic and nongenotoxic events. Toxicol Sci. 2005;83:246. doi: 10.1093/toxsci/kfi040. [DOI] [PubMed] [Google Scholar]
- 387.Kemp M, Liu W, Thorne P, Kane M, Selmin O, Romagnolo D. Induction of the transferring receptor gene by benzo[a]pyrene in breast cancer MCF-7 cells: Potential as a biomarker of PAH exposure. Environ Mol Mutagen. 2006;47:518–526. doi: 10.1002/em.20221. [DOI] [PubMed] [Google Scholar]
- 388.Santodonato J. Review of the estrogenic and antiestrogenic activity of polycyclic aromatic hydrocarbons: Relationship to carcinogenicity. Chemosphere. 1997;34:835–848. doi: 10.1016/S0045-6535(97)00012-X. [DOI] [PubMed] [Google Scholar]
- 389.Kung T, Murphy KA, White LA. The aryl hydrocarbon receptor (AhR) pathway as a regulatory pathway for cell adhesion and matrix metabolism. Biochem Pharmacol. 2009;77:536–546. doi: 10.1016/j.bcp.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Ohura T, Morita M, Kuruto-Niwa R, Amagai T, Sakakibara H, Shimoi K. Differential action of chlorinated polycyclic aromatic hydrocarbons on aryl hydrocarbon receptor-mediated signaling in breast cancer cells. Environ Toxicol. 2010;25:180–187. doi: 10.1002/tox.20488. [DOI] [PubMed] [Google Scholar]
- 391.Ralston S, Coffing S, Seidel A, Luch A, Platt K, Baird W. Stereoselective activation of dibenzo[a,l]pyrene and its trans-11,12-dihydrodiol to fjord region 11,12-diol 13,14-epoxides in a human mammary carcinoma MCF-7 cell-mediated V79 cell mutation assay. Chem Res Toxciol. 1997;10:687–693. doi: 10.1021/tx9700275. [DOI] [PubMed] [Google Scholar]
- 392.Mordukhovich I, Beyea J, Herring AH, Hatch M, Stellman SD, Teitelbaum SL, et al. Vehicular traffic–related polycyclic aromatic hydrocarbon exposure and breast cancer incidence: The long island breast cancer study project (LIBCSP) Environ Health Perspect. 2016;124:30–38. doi: 10.1289/ehp.1307736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.White AJ, Teitelbaum SL, Stellman SD, Beyea J, Steck SE, Mordukhovich I, et al. Indoor air pollution exposure from use of indoor stoves and fireplaces in association with breast cancer: A case-control study. Environ Health. 2014;13:108. doi: 10.1186/1476-069X-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Gammon MD, Santella RM, Neugut AI. Environmental toxins and breast cancer on Long Island. I. Polycyclic aromatic hydrocarbon DNA adducts. Cancer Epidemiol Biomark Prev. 2002;11:677–685. [PubMed] [Google Scholar]
- 395.Cavalieri E, Rogan E. The molecular etiology and prevention of estrogen-initiated cancers: Ockham’s razor: Pluralitas non est ponenda sine necessitate. Plurality should not be posited without necessity. Mol Asp Med. 2014;36:1–55. doi: 10.1016/j.mam.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Rundle A, Tang D, Hibshoosh H, Estabrook A, Schnabel F, Cao C, et al. The relationship between genetic damage from polycyclic hydrocarbons in breast tissue and breast cancer. Carcinogenesis. 2000;21:1281–1289. doi: 10.1093/carcin/21.7.1281. [DOI] [PubMed] [Google Scholar]
- 397.Gammon MD, Santella RM. PAH, genetic susceptibility and breast cancer risk: An update from the Long Island breast cancer study project. Eur J Cancer. 2008;44:636–640. doi: 10.1016/j.ejca.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 398.Mahadevan B, Keshava C, Musafia-Jeknic T, Pecaj A, Weston A, Baird W. Altered gene expression patterns in MCF-7 cells induced by the urban dust particulate complex mixture standard reference material 1649a. Cancer Res. 2005;65:1251–1258. doi: 10.1158/0008-5472.CAN-04-2357. [DOI] [PubMed] [Google Scholar]
- 399.McCarty K, Santella R, Steck S, Cleveland R, Ahn J, Ambrosone C, et al. PAH-DNA adducts, cigarette smoking, GST polymorphisms and breast cancer risk. Environ Health Perspect. 2009;117:552–555. doi: 10.1289/ehp.0800119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Mordukhovich I, Pavel R, Jr, Terry MB, Santella R, Zhang Y-J, Hibshoosh H, et al. Associations between polycyclic aromatic hydrocarbon-related exposures and p53 mutations in breast tumors. Environ Health Perspect. 2010;118:511–518. doi: 10.1289/ehp.0901233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Petralia SA, Vena JE, Freudenheim JL, Dosemeci M, Michalek A, Goldberg MS, et al. Risk of premenopausal breast cancer in association with occupational exposure to polycyclic aromatic hydrocarbons and benzene. Scand J Work Environ Health. 1999;25:215–221. doi: 10.5271/sjweh.426. [DOI] [PubMed] [Google Scholar]
- 402.Palli D, Masala G, Mariani-Costantini R, Zanna I, Saieva C, Sera F, et al. A gene-environment interaction between occupation and BRCA1/BRCA2 mutations in male breast cancer. Eur J Cancer. 2004;40:2474–2479. doi: 10.1016/j.ejca.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 403.Nie J, Beyea J, Bonner M, Han D, Vena J, Rogerson P, et al. Exposure to traffic emissions throughout life and risk of breast cancer: The western New York exposures and breast cancer (web) study. Cancer Causes Control. 2007;18:947–955. doi: 10.1007/s10552-007-9036-2. [DOI] [PubMed] [Google Scholar]
- 404.Hung L-J, Tsai S-S, Chen P-S, Yang Y-H, Liou S-H, Wu T-N, et al. Traffic air pollution and risk of death from breast cancer in Taiwan: Fine particulate matter (PM 2.5) as a proxy marker. Aerosol Air Qual Res. 2012;12:275–282. [Google Scholar]
- 405.Sagiv SK, Gaudet MM, Eng SM, Abrahamson PE, Shantakumar S, Teitelbaum SL, et al. Polycyclic aromatic hydrocarbon-DNA adducts and survival among women with breast cancer. Environ Res. 2009;109:287–291. doi: 10.1016/j.envres.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Teitelbaum SL, Gammon MD, Britton JA, Neugut AI, Levin B, Stellman SD. Reported residential pesticide use and breast cancer risk on Long Island, New York. Am J Epidemiol. 2007;165:643–651. doi: 10.1093/aje/kwk046. [DOI] [PubMed] [Google Scholar]
- 407.Dodson RE, Camann DE, Morello-Frosch R, Brody JG, Rudel RA. Semivolatile organic compounds in homes: Strategies for efficient and systematic exposure measurement based on empirical and theoretical factors. Environ Sci Technol. 2015;49:113–122. doi: 10.1021/es502988r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.U.S. Environmental Protection Agency: Interim reregistration eligibility decision for atrazine. 2008. https://archive.epa.gov/pesticides/reregistration/web/pdf/atrazine_ired.pdf. Accessed 24 Sept 2016.
- 409.Hua W, Bennett E, Maio X, Metcalfe C, Letcher R. Seasonality effects on pharmaceuticals and S-triazine herbicides in wastewater effluent and surface water from the Canadian side of the upper Detroit River. Environ Toxicol Chem. 2006;25:2356–2365. doi: 10.1897/05-571R.1. [DOI] [PubMed] [Google Scholar]
- 410.Miller S, Sweet C, Depinto J, Hornbuckle K. Atrazine and nutrients in precipitation from the Lake Michigan mass balance study. Environ Sci Technol. 2000;34:55–61. doi: 10.1021/es990486n. [DOI] [Google Scholar]
- 411.Villanueva C, Durand G, Coutté M, Chevrier C, Cordier S. Atrazine in municipal drinking water and risk of low birth weight, preterm delivery, and small-for-gestational-age status. Occup Environ Med. 2005;62:400–405. doi: 10.1136/oem.2004.016469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Hayes TB, Anderson LL, Beasley VR, et al. Demasculinization and feminization of male gonads by atrazine: Consistent effects across vertebrate classes. J Steroid Biochem Mol Biol. 2011;127:64–73. doi: 10.1016/j.jsbmb.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Rohr JR, McCoy KA. A qualitative meta-analysis reveals consistent effects of atrazine on freshwater fish and amphibians. Environ Health Perspect. 2010;118:20–32. doi: 10.1289/ehp.0901164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.O’Connor J, Plowchalk D, Van-Pelt C, Davis L, Cook J. Role of prolactin in chloro-S-triazine rat mammary tumorigenesis. Drug Chem Toxicol. 2000;23:575–601. doi: 10.1081/DCT-100101972. [DOI] [PubMed] [Google Scholar]
- 415.Florian CP, Mansfield SR, Schroeder JR. Differences in GPR30 regulation by Chlorotriazine herbicides in human breast cells. Biochem Res Int. 2016;2016:2984081. [DOI] [PMC free article] [PubMed]
- 416.Kettles MA, Browning SR, Prince TS, Horstman SW. Triazine herbicide exposure and breast cancer incidence: An ecologic study of Kentucky counties. Environ Health Perspect. 1997;105:1222–1227. doi: 10.1289/ehp.971051222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Hopenhayn-Rich C, Stump ML, Browning SR. Regional assessment of atrazine exposure and incidence of breast and ovarian cancers in Kentucky. Arch Environ Contam Toxicol. 2002;42:127–136. doi: 10.1007/s002440010300. [DOI] [PubMed] [Google Scholar]
- 418.Muir K, Rattanamongkolgul S, Smallman-Raynor M, Thomas M, Downer S, Jenkinson C. Breast cancer incidence and its possible spatial association with pesticide application in two counties of England. Public Health. 2004;118:513–520. doi: 10.1016/j.puhe.2003.12.019. [DOI] [PubMed] [Google Scholar]
- 419.Hunter L, Gadbury G, Huang Y. Atrazine exposure and breast cancer incidence: An ecologic study of Missouri counties. Toxicol Environ Chem. 2008;90:367–376. doi: 10.1080/02772240701529012. [DOI] [Google Scholar]
- 420.Boffetta P, Adami H-O, Berry SC, Mandel JS. Atrazine and cancer: A review of the epidemiologic evidence. Eur J Cancer Prev. 2013;22:169–180. doi: 10.1097/CEJ.0b013e32835849ca. [DOI] [PubMed] [Google Scholar]
- 421.Cooper R, Stoker T, Tyrey L, Goldman J, McElroy W. Atrazine disrupts the hypothalamic control of pituitary-ovarian function. Toxicol Sci. 2000;53:297–307. doi: 10.1093/toxsci/53.2.297. [DOI] [PubMed] [Google Scholar]
- 422.Fan WQ, Yanase T, Morinaga H, Gondo S, Okabe T, Nomura M, et al. Atrazine-induced aromatase expression is SF-1 dependent: Implications for endocrine disruption in wildlife and reproductive cancers in humans. Environ Health Perspect. 2007;115:720–727. doi: 10.1289/ehp.9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Sanderson J, Letcher R, Heneweer M, Giesy J, van den Berg M. Effects of chloro-S-triazine herbicides and metabolites on aromatase activity in various human cell lines and on vitellogenin production in male carp hepatocytes. Environ Health Perspect. 2001;109:1027–1031. doi: 10.1289/ehp.011091027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Enoch R, Stanko J, Greiner S. Mammary gland development as a sensitive end point after acute prenatal exposure to an atrazine metabolite mixture in female long-Evans rats. Environ Health Perspect. 2007;115:541–7. [DOI] [PMC free article] [PubMed]
- 425.Raynor J, Enoch R, Fenton S. Adverse effects of prenatal exposure to atrazine during a critical period of mammary gland growth. Toxicol Sci. 2005;87:255–66. [DOI] [PubMed]
- 426.Fenton SE, Reed C, Newbold RR. Perinatal environmental exposures affect mammary development, function, and cancer risk in adulthood. Ann Rev Pharmacol Toxicol. 2012;52:455–79. [DOI] [PMC free article] [PubMed]
- 427.Ueda M, Imai T, Takizawa T, Onodera H, Mitsumori K, Matsui T, et al. Possible enhancing effects of atrazine on growth of 7, 12-dimethylbenz(a) anthracene-induced mammary tumors in ovariectomized Sprague-Dawley rats. Cancer Sci. 2005;96:19–25. doi: 10.1111/j.1349-7006.2005.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Hovey RC, Coder PS, Wolf JC, Sielken RL, Jr, Tisde MO, Breckenridge CB. Quantitative assessment of mammary gland development in female long Evans rats following in utero exposure to atrazine. Toxicol Sci. 2011;119:380–390. doi: 10.1093/toxsci/kfq337. [DOI] [PubMed] [Google Scholar]
- 429.Eldridge JC, Stevens JT, Breckenridge CB. Atrazine interaction with estrogen expression systems. Journal or something? 2008;196:147–156. [DOI] [PubMed]
- 430.Albanito L, Lappano R, Madeo A, et al. Effects of atrazine on estrogen receptor α– And G protein–coupled receptor 30–mediated signaling and proliferation in cancer cells and cancer-associated fibroblasts. Environ Health Perspect. 2015;123:493–499. doi: 10.1289/ehp.1408586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Kucka M, Pogrmic-Majkic K, Fa S, Stojilkovic SS, Kovacevic R. Atrazine acts as an endocrine disrupter by inhibiting cAMP-specific phosphodiesterase-4. Toxicol Appl Pharmacol. 2012;265:19–26. doi: 10.1016/j.taap.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Suzawa M, Ingraham HA. The herbicide atrazine activates endocrine gene networks via non-steroidal NR5A nuclear receptors in fish and mammalian cells. PLoS One. 2008;3:e2117. [DOI] [PMC free article] [PubMed]
- 433.Siegel BZ. Pesticide Hazard Assessment 1981–1984. Honolulu: Pacific Biomedical Research Center, University of Hawaii; 1995. [Google Scholar]
- 434.Maskarinec G, Zhang Y, Takata Y, Pagano I, Shumay D, Goodman M, et al. Trends of breast cancer incidence and risk factor prevalence over 25 years. Breast Cancer Res Treat. 2006;98:45–54. doi: 10.1007/s10549-005-9129-0. [DOI] [PubMed] [Google Scholar]
- 435.Arrebola JP, Belhassen H, Artacho-Cordón F, Ghali R, Ghorbel H, Boussen H, et al. Risk of female breast cancer and serum concentrations of organochlorine pesticides and polychlorinated biphenyls: A case-control study in Tunisia. Sci Total Environ. 2015;520:106–13. [DOI] [PubMed]
- 436.Center for Disease Control and Prevention (CDC): Third National Report on Human Exposure to Environmental Chemicals. 2005. http://www.jhsph.edu/research/centers-and-institutes/center-for-excellence-in-environmental-health-tracking/Third_Report.pdf. Accessed 25 Oct 2016.
- 437.Samra NM, Selim AA. Organochlorine pesticides concentrations in maternal serum and their effects on umbilical cord serum pesticides concentrations, neonatal birth weight and gestational age. Aust J Basic Appl Sci. 2009;3:1972–83.
- 438.Khanjani N, English DR, Sim MR. An ecological study of organochlorine pesticides and breast cancer in rural Victoria, Astralie. Arch Environ Contam Toxicol. 2006;50:452–61. [DOI] [PubMed]
- 439.Cassidy RA, Natarajan S, Vaughan GM. The link between the insecticide heptachlor epoxide, estradiol, and breast cancer. Breast Cancer Res Treat. 2005;90:55–64. doi: 10.1007/s10549-004-2755-0. [DOI] [PubMed] [Google Scholar]
- 440.Dich J, Zahm S, Hanberg A, Adami H. Pesticides (heptachlor) and cancer. Cancer Causes Control. 1997;8:420–443. doi: 10.1023/A:1018413522959. [DOI] [PubMed] [Google Scholar]
- 441.Agency for Toxic Substances and Disease Registry. Fact Sheet: ToxFAQs for aldrin/dieldrin. 2002. https://www.atsdr.cdc.gov/toxfaqs/tf.asp?id=316&tid=56. Access date 10 Oct 2016.
- 442.Hoyer A, Grandjean P, Jorgensen T, Brock J, Hartvig H. Organochlorine exposure and risk of breast cancer. Lancet. 1998;352:1816–1820. doi: 10.1016/S0140-6736(98)04504-8. [DOI] [PubMed] [Google Scholar]
- 443.Hoyer A, Jorgensen T, Brock J, Grandjean P. Organochlorine exposure and breast cancer survival. J Clin Epidemiol. 2000;53:323–330. doi: 10.1016/S0895-4356(99)00165-1. [DOI] [PubMed] [Google Scholar]
- 444.Cameron H, Foster W. Developmental and lactational exposure to dieldrin alters mammary tumorigenesis in her2/neu transgenic mice. PLoS One. 2009;4:4303. doi: 10.1371/journal.pone.0004303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Andersen H, Vinggaard A, Rasmussen T, Gjermandsen I, Bonefeld-Jorgensen E. Effects of currently used pesticides in assays for estrogenicity, androgenicity, and aromatase activity in vitro. Toxicol Appl Pharmacol. 2002;179:1–12. doi: 10.1006/taap.2001.9347. [DOI] [PubMed] [Google Scholar]
- 446.Mills P, Yang R. Breast cancer risk in Hispanic agricultural workers in California. Int J Occup Environ Health. 2005;11:123–131. doi: 10.1179/oeh.2005.11.2.123. [DOI] [PubMed] [Google Scholar]
- 447.Engel LS, Hill DA, Hoppin JA, Lubin JH, Lynch CF, Pierce J, et al. Pesticide use and breast cancer risk among farmers’ wives in the agricultural health study. Am J Epidemiol. 2005;161:121–135. doi: 10.1093/aje/kwi022. [DOI] [PubMed] [Google Scholar]
- 448.Alexander B, Mandel J, Baker B, Burns C, Bartels M, Acquavella J, et al. Biomonitoring of 2,4-dichlorophenoxyacetic acid exposure and dose in farm families. Environ Health Perspect. 2007;115:370–376. doi: 10.1289/ehp.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Calaf GM, Garrido F. Catechol estrogens as biomarkers for mammary gland cancer. Int J Oncol. 2011;39:177–183. doi: 10.3892/ijo.2011.1008. [DOI] [PubMed] [Google Scholar]
- 450.Beard J. DDT and human health. Sci Total Environ. 2006;355:78–89. doi: 10.1016/j.scitotenv.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 451.McGinn AP. Malaria, Mosquitoes, and DDT. World Watch Magazine. 2002. http://www.worldwatch.org/node/517. Accessed 25 Oct 2016.
- 452.Rogan WJ, Ragan NB. Some evidence of effects of environmental chemicals on the endocrine system in children. Int J Hyg Environ Health. 2007;21:659–667. doi: 10.1016/j.ijheh.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Shen H, Main K, Virtanen H, Damggard I, Haavisto A, Kaleva M, et al. From mother to child: Investigation of prenatal and postnatal exposure to persistent bioaccumulating toxicants using breast milk and placenta biomonitoring. Chemosphere. 2007;67:236–262. doi: 10.1016/j.chemosphere.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 454.Zheng T, Holford T, Mayne S, Ward B, Carter D, Owens P, et al. DDE and DDT in breast adipose tissue and risk of female breast cancer. Am J Epidemiol. 1999;150:453–458. doi: 10.1093/oxfordjournals.aje.a010033. [DOI] [PubMed] [Google Scholar]
- 455.Brody JG, Moysich KB, Humblet O, Attfield KR, Beehler GP, Rudel RA. Environmental pollutants and breast cancer: Epidemiologic studies. Cancer. 2007;109:2667–2711. doi: 10.1002/cncr.22655. [DOI] [PubMed] [Google Scholar]
- 456.Gammon MD, Santella RM, Neugut AI. Environmental toxins and breast cancer on Long Island. II. Organochlorine compound levels in Blood. Cancer Epidemiol Biomark Prev. 2002;11:686–697. [PubMed] [Google Scholar]
- 457.Cohn BA. Developmental and environmental origins of breast cancer: DDT as a case study. Reprod Toxicol. 2011;31:302–311. doi: 10.1016/j.reprotox.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Shakeel MK, George PS, Jose J, Jose J, Mathew A. Pesticides and breast cancer risk: A comparison between developed and developing countries. Asian Pac J Cancer Prev. 2010;11:173–180. [PubMed] [Google Scholar]
- 459.Parada H, Wolff MS, Engel LS, White AJ, Eng SM, Cleveland RJ, et al. Organochlorine insecticides DDT and chlordane in relation to survival following breast cancer. Int J Cancer. 2016;138:565–575. doi: 10.1002/ijc.29806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Canales-Aguirre A, Padilla-Camberos E, Gómez-Pinedo U, Salado-Ponce H, Feria-Velasco A, De Celis R. Genotoxic effect of chronic exposure to DDT on lymphocytes, oral mucosa and breast cells of female parrats. Int J Environ Res Public Health. 2011;8:540–553. doi: 10.3390/ijerph8020540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Robison AK, Sirbasku DA, Stancel GM. DDT supports the growth of an estrogen-responsive tumor. Toxicol Lett. 1985;27:109–113. doi: 10.1016/0378-4274(85)90127-4. [DOI] [PubMed] [Google Scholar]
- 462.Scribner J, Mottet N. DDT acceleration of mammary gland tumors induced in the male Sprague-Dawley rat by 2-acetamidophenanthrene. Carcinogenesis. 1981;2:1235–1239. doi: 10.1093/carcin/2.12.1235. [DOI] [PubMed] [Google Scholar]
- 463.Uppala PT, Roy SK, Tousson A, Barnes S, Uppala GR, Eastmond DA. Induction of cell proliferation, micronuclei and hyperdiploidy/polyploidy in the mammary cells of DDT- and DMBA-treated pubertal rats. Environ Mol Mutagen. 2005;46:43–52. doi: 10.1002/em.20131. [DOI] [PubMed] [Google Scholar]
- 464.Pujol P, Hilsenbeck S, Chamness G, Elledge R. Rising levels of estrogen receptor in breast cancer over 2 decades. Cancer. 1994;74:1601–1606. doi: 10.1002/1097-0142(19940901)74:5<1601::AID-CNCR2820740517>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 465.Woolcott C, Aronson K, Hanna W, SenGupta S, McCready D, Sterns E, et al. Organochlorines and breast cancer risk by receptor status, tumor size and grade. Canada. Cancer Causes Control. 2001;12:395–404. doi: 10.1023/A:1011289905751. [DOI] [PubMed] [Google Scholar]
- 466.Davies H, Delistraty D. Evaluation of PCB sources and releases for identifying priorities to reduce PCBs in Washington state (USA) Environ Sci Pollut Res Int. 2016;23:2033–2041. doi: 10.1007/s11356-015-4828-5. [DOI] [PubMed] [Google Scholar]
- 467.Weber R, Watson A, Forter M, Oliaei F. Review article: Persistent organic pollutants and landfills - a review of past experiences and future challenges. Waste Manag Res. 2011;29:107–121. doi: 10.1177/0734242X10390730. [DOI] [PubMed] [Google Scholar]
- 468.U.S. Environental Protection Agency: Learn about polychlorinated biphenyls (PCBs). 2012. https://www.epa.gov/pcbs/learn-about-polychlorinated-biphenyls-pcbs. Accessed 24 Oct 2016.
- 469.Martinez A, Schnoebelen DJ, Hornbuckle KC. Polychlorinated biphenyl congeners in sediment cores from the upper Mississippi River. Chemosphere. 2016;144:1943–1949. doi: 10.1016/j.chemosphere.2015.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Shunthirasingham C, Gawor A, Hung H, Brice KA, Su K, Alexandrou N, et al. Atmospheric concentrations and loadings of organochlorine pesticides and polychlorinated biphenyls in the Canadian Great Lakes Basin (GLB): Spatial and temporal analysis (1992–2012) Environ Pollut. 2016;217:124–133. doi: 10.1016/j.envpol.2016.01.039. [DOI] [PubMed] [Google Scholar]
- 471.Hagmar L, Wallin E, Vessby B, Jönsson BA, Bergman A, Rylander L. Intra-individual variations and time trends 1991–2001 in human serum levels of PCB, DDE and hexachlorobenzene. Chemosphere. 2006;64:1507–1513. doi: 10.1016/j.chemosphere.2005.12.054. [DOI] [PubMed] [Google Scholar]
- 472.Choi AL, Levy JI, Dockery DW, Ryan LM, Tolbert PE, Altshul LM, et al. Does living near a superfund site contribute to higher polychlorinated biphenyl (PCB) exposure? Environ Health Perspect. 2006;114:1092–1098. doi: 10.1289/ehp.8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Wolff MS, Camann D, Gammon M, Stellman SD. Proposed PCB congener groupings for epidemiological studies. Environ Health Perspect. 1997;105:13–14. doi: 10.1289/ehp.9710513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Zhang J, Huang Y, Wang X, Lin K, Wu K. Environmental polychlorinated biphenyl exposure and breast cancer risk: A meta-analysis of observational studies. PLoS One. 2015;10:e0142513. doi: 10.1371/journal.pone.0142513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Braathen M, Mortensen AS, Sandvik M, Skåre JU, Arukwe A. Estrogenic effects of selected hydroxy polychlorinated biphenyl congeners in primary culture of Atlantic Salmon (Salmo Salar) hepatocytes. Arch Environ Contam Toxicol. 2009;56:111–122. doi: 10.1007/s00244-008-9163-0. [DOI] [PubMed] [Google Scholar]
- 476.Helmfrid I, Berglund M, Löfman O, Wingren G. Health effects and exposure to polychlorinated biphenyls (PCBs) and metals in a contaminated community. Environ Int. 2012;44:53–58. doi: 10.1016/j.envint.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 477.Muscat JE, Britton JA, Djordjevic MV, Citron ML, Kemeny M, Busch-Devereaux E, et al. Adipose concentrations of organochlorine compounds and breast cancer recurrence in Long Island, New York. Cancer Epidemiol Biomark Prev. 2003;12:1474–1478. [PubMed] [Google Scholar]
- 478.Ellsworth RE, Mamula KA, Costantino NS, Deyarmin B, Kostyniak PJ, Chi L-H, et al. Abundance and distribution of polychlorinated biphenyls (PCBs) in breast tissue. Environ Res. 2015;138:291–297. doi: 10.1016/j.envres.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 479.Leng L, Li J, Luo X-M, Kim J-Y, Li Y-M, Guo X-M, et al. Polychlorinated biphenyls and breast cancer: A congener-specific meta-analysis. Environ Int. 2016;88:133–141. doi: 10.1016/j.envint.2015.12.022. [DOI] [PubMed] [Google Scholar]
- 480.Aronson KJ, Miller AB, Woolcott CG, Sterns EE, McCready DR, Lickley LA, et al. Breast adipose tissue concentrations of polychlorinated biphenyls and other organochlorines and breast cancer risk. Cancer Epidemiol Biomark Prev. 2000;9:55–63. [PubMed] [Google Scholar]
- 481.Salehi F, Turner MC, Phillips KP, Wigle DT, Krewski D, Aronson KJ. Review of the etiology of breast cancer with special attention to organochlorines as potential endocrine disruptors. J Toxicol Environ Health B Crit Rev. 2008;11:276–300. doi: 10.1080/10937400701875923. [DOI] [PubMed] [Google Scholar]
- 482.Golden R, Kimbrough R. Weight of evidence evaluation of potential human cancer risks from exposure to polychlorinated biphenyls: An update based on studies published since 2003. Crit Rev Toxicol. 2009;39:299–331. doi: 10.1080/10408440802291521. [DOI] [PubMed] [Google Scholar]
- 483.Silver SR, Whelan EA, Deddens JA, Steenland NK, Hopf NB, Waters MA, et al. Occupational exposure to polychlorinated biphenyls and risk of breast cancer. Environ Health Perspect. 2009;117:276–282. doi: 10.1289/ehp.11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Artacho-Cordón F, Fernández-Rodríguez M, Garde C, Salamanca E, Iribarne-Durán LM, Torné P, et al. Serum and adipose tissue as matrices for assessment of exposure to persistent organic pollutants in breast cancer patients. Environ Res. 2015;142:633–643. doi: 10.1016/j.envres.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 485.Laden F, Collman G, Iwamoto K, Alberg AJ, Berkowitz GS, Freudenheim JL, et al. 1,1-Dichloro-2,2-bis(p-chlorophenyl)ethylene and polychlorinated biphenyls and breast cancer: Combined analysis of five U.S. studies. J Natl Cancer Inst. 2001;93:768–776. doi: 10.1093/jnci/93.10.768. [DOI] [PubMed] [Google Scholar]
- 486.Laden F, Ishibe N, Hankinson SE, Wolff MS, Gertig DM, Hunter DJ, et al. Polychlorinated biphenyls, cytochrome P450 1A1, and breast cancer risk in the nurses’ health study. Cancer Epidemiol Biomark Prev. 2002;11:1560–1565. [PubMed] [Google Scholar]
- 487.Verner M-A, Bachelet D, McDougall R, Charbonneau M, Guénel P, Haddad S. A case study addressing the reliability of polychlorinated biphenyl levels measured at the time of breast cancer diagnosis in representing early-life exposure. Cancer Epidemiol Biomark Prev. 2011;20:281–286. doi: 10.1158/1055-9965.EPI-10-0992. [DOI] [PubMed] [Google Scholar]
- 488.Kulkarni PS, Crespo JG, Afonso CAM. Dioxins sources and current remediation technologies—a review. Environ Int. 2008;34:139–153. doi: 10.1016/j.envint.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 489.International Agency on Research on Cancer (IARC). Polychlorinated Dibenzo-dioxins and polychlorinated dibenzofurans. IARC Monogr Eval Carcin Effects Chem. 1997;69. http://monographs.iarc.fr/ENG/Monographs/vol69/index.php. Accessed 25 Oct 2016.
- 490.Agency for Toxic Substances and Disease Registry (ATSDR): Toxicological profile for chlorinated dibenzo-p-dioxins. 1998. https://www.atsdr.cdc.gov/toxprofiles/TP.asp?id=366&tid=63. Accessed 25 Oct 2016. [PubMed]
- 491.World Health Organization (WHO): Dioxins and their effects on human health. 2016. http://www.who.int/mediacentre/factsheets/fs225/en/. Accessed 24 Oct 2016.
- 492.Schecter A, Birnbaum L, Ryan JJ, Constable JD. Dioxins: An overview. Environ Res. 2006;101:419–428. doi: 10.1016/j.envres.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 493.Massart F, Harrell JC, Federico G, Saggese G. Human breast milk and xenoestrogen exposure: A possible impact on human health. J Perinatol. 2005;25:282–288. doi: 10.1038/sj.jp.7211251. [DOI] [PubMed] [Google Scholar]
- 494.Nickerson K. Environmental contaminants in breast milk. J Midwifery Womens Health. 2006;51:26–34. doi: 10.1016/j.jmwh.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 495.Dip R, Buterin T, Wenger D, Schmid P, Naegeli H. Genetic reprogramming of human mammary cells by environmental carcinogens released into breast milk. CHIMIA Int J Chem. 2008;62:410–416. doi: 10.2533/chimia.2008.410. [DOI] [Google Scholar]
- 496.Warner M, Eskenazi B, Mocarelli P, Gerthoux PM, Samuels S, Needham L, et al. Serum dioxin concentrations and breast cancer risk in the Seveso Women’s health study. Environ Health Perspect. 2002;110:625–628. doi: 10.1289/ehp.02110625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Warner M, Mocarelli P, Samuels S, Needham L, Brambilla P, Eskenazi B. Dioxin exposure and cancer risk in the Seveso Women’s health study. Environ Health Perspect. 2011;119:1700–1705. doi: 10.1289/ehp.1103720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Manz A, Berger J, Dwyer JH, Flesch-Janys D, Nagel S, Waltsgott H. Cancer mortality among workers in chemical plant contaminated with dioxin. Lancet. 1991;338:959–964. doi: 10.1016/0140-6736(91)91835-I. [DOI] [PubMed] [Google Scholar]
- 499.Manuwald U, Velasco Garrido M, Berger J, Manz A, Baur X. Mortality studies of chemical workers exposed to dioxins: Follow-up 23 years after chemical plant closure. Br Med J. 2012;69:636–642. doi: 10.1136/oemed-2012-100682. [DOI] [PubMed] [Google Scholar]
- 500.Brown NM, Manzolillo PA, Zhang JX, Wang J, Lamartiniere CA. Prenatal TCDD and predisposition to mammary cancer in the rat. Carcinogenesis. 1998;19:1623–1629. doi: 10.1093/carcin/19.9.1623. [DOI] [PubMed] [Google Scholar]
- 501.Fenton SE, Hamm JT, Birnbaum LS, Youngblood GL. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Toxicol Sci. 2002;67:63–74. doi: 10.1093/toxsci/67.1.63. [DOI] [PubMed] [Google Scholar]
- 502.Lewis BC, Hudgins S, Lewis A, Schorr K, Sommer R, Peterson RE, et al. In utero and lactational treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs mammary gland differentiation but does not block the response to exogenous estrogen in the postpubertal female rat. Toxicol Sci. 2001;62:46–53. doi: 10.1093/toxsci/62.1.46. [DOI] [PubMed] [Google Scholar]
- 503.Jenkins S, Rowell C, Wang J, Lamartiniere C. Prenatal TCDD exposure predisposes for mammary cancer in rats. Reprod Toxicol. 2007;23:391–396. doi: 10.1016/j.reprotox.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.La Merrill M, Kuruvilla BS, Pomp D, Birnbaum LS, Threadgill DW. Dietary fat alters body composition, mammary development, and cytochrome p450 induction after maternal TCDD exposure in DBA/2J mice with low-responsive aryl hydrocarbon receptors. Environ Health Perspect. 2009;117:1414–1419. doi: 10.1289/ehp.0800530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Seifert A, Taubert H, Hombach-Klonisch S, Fischer B, Navarrete Santos A. TCDD mediates inhibition of p53 and activation of ERalpha signaling in MCF-7 cells at moderate hypoxic conditions. Int J Oncol. 2009;35:417–424. [PubMed] [Google Scholar]
- 506.Yoshioka H, Hiromori Y, Aoki A, Kimura T, Fujii-Kuriyama Y, Nagase H, et al. Possible aryl hydrocarbon receptor-independent pathway of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced antiproliferative response in human breast cancer cells. Toxicol Lett. 2012;211:257–265. doi: 10.1016/j.toxlet.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 507.Marques M, Laflamme L, Gaudreau L. Estrogen receptor α can selectively repress dioxin receptor-mediated gene expression by targeting DNA methylation. Nucleic Acids Res. 2013;41:8094–8106. doi: 10.1093/nar/gkt595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Marlowe JL, Puga A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J Cell Biochem. 2005;96:1174–1184. doi: 10.1002/jcb.20656. [DOI] [PubMed] [Google Scholar]
- 509.Bekki K, Vogel H, Li W, Ito T, Sweeney C, Haarmann-Stemmann T, et al. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic Biochem Physiol. 2015;120:5–13. doi: 10.1016/j.pestbp.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Chen Y-J, Hung C-M, Kay N, Chen C-C, Kao Y-H, Yuan S-S. Progesterone receptor is involved in 2,3,7,8-tetrachlorodibenzo-p-dioxin-stimulated breast cancer cells proliferation. Cancer Lett. 2012;319:223–231. doi: 10.1016/j.canlet.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 511.Labrecque MP, Takhar MK, Hollingshead BD, Prefontaine GG, Perdew GH, Beischlag TV. Distinct roles for aryl hydrocarbon receptor nuclear translocator and ah receptor in estrogen-mediated signaling in human cancer cell lines. PLoS One. 2012;7:e29545. doi: 10.1371/journal.pone.0029545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Lo R, Matthews J. The aryl hydrocarbon receptor and estrogen receptor alpha differentially modulate nuclear factor erythroid-2-related factor 2 transactivation in MCF-7 breast cancer cells. Toxicol Appl Pharmacol. 2013;270:139–148. doi: 10.1016/j.taap.2013.03.029. [DOI] [PubMed] [Google Scholar]
- 513.Ohtake F, Fujii-Kuriyama Y, Kawajiri K, Kato S. Cross-talk of dioxin and estrogen receptor signals through the ubiquitin system. J Steroid Biochem Mol Biol. 2011;127:1027. doi: 10.1016/j.jsbmb.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 514.Papoutis AJ, Selmin OI, Borg JL, Romagnolo DF. Gestational exposure to AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: Preventive effects of resveratrol. Mol Carcinogen. 2015;54:261–269. doi: 10.1002/mc.22095. [DOI] [PubMed] [Google Scholar]
- 515.Zota A, Rudel R, Morello-Frosch R, Brody J. Elevated house dust and serum concentrations of PBDEs in California: Unintended consequences of furniture flammability standards? Environ Sci Technol. 2008;42:8158–8164. doi: 10.1021/es801792z. [DOI] [PubMed] [Google Scholar]
- 516.Dodson RE, Perovich LJ, Covaci A, Van Den Eede N, Ionas AC, Dirtu AC, et al. After the PBDE phase-out: A broad suite of flame retardants in repeat house dust samples from California. Environ Sci Technol. 2012;46:13056–13066. doi: 10.1021/es303879n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Petreas M, Nelson D, Brown FR, Goldberg D, Hurley S, Reynolds P. High concentrations of polybrominated diphenylethers (PBDEs) in breast adipose tissue of California women. Environ Int. 2011;37:190–197. doi: 10.1016/j.envint.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Eskenazi B, Fenster L, Castorina R, Marks AR, Sjödin A, Rosas LG, et al. A comparison of PBDE serum concentrations in Mexican and Mexican-American children living in California. Environ Health Perspect. 2011;119:1442–1448. doi: 10.1289/ehp.1002874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Windham G, Pinney S, Sjodin A, Lum R, Jones R, Needham L, et al. Body burdens of brominated flame retardants and other persistent organo-halogenated compounds and their descriptors in US girls. Environ Res. 2010;110:251–257. doi: 10.1016/j.envres.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Windham GC, Pinney SM, Voss RW, Sjödin A, Biro FM, Greenspan LC, et al. Brominated flame retardants and other persistent organohalogenated compounds in relation to timing of puberty in a longitudinal study of girls. Environ Health Perspect. 2015;123:1046–1052. doi: 10.1289/ehp.1408778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Costa L, Giordano G, Tagliaferri S, Caglieri A, Mutti A. Polybrominated diphenyl ether (PBDE) flame retardants: Environmental contamination, human body burden and potential adverse health effects. Acta Biomed. 2008;79:172–183. [PubMed] [Google Scholar]
- 522.Darnerud PO, Eriksen GS, Jóhannesson T, Larsen PB, Viluksela M. Polybrominated diphenyl ethers: Occurrence, dietary exposure, and toxicology. Environ Health Perspect. 2001;109(Suppl 1):49–68. doi: 10.1289/ehp.01109s149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.De Wit C. An overview of brominated flame retardants in the environment. Chemosphere. 2002;46:583–624. doi: 10.1016/S0045-6535(01)00225-9. [DOI] [PubMed] [Google Scholar]
- 524.Frederiksen M, Thomsen C, Froshaug M, Vorkamp K, Thomsen M, Becher G, et al. Polybrominated diphenyl ethers in paired samples of maternal and umbilical cord blood plasma and assocaitions with house dust in a Danish cohort. Int J Hyg Environ Health. 2010;13:233–242. doi: 10.1016/j.ijheh.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 525.Costa LG, Giordano G. Developmental neurotoxicity of polybrominated diphenyl ether (PBDE) flame retardants. Neurotoxicology. 2007;28:1047–1067. doi: 10.1016/j.neuro.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Talsness CE. Overview of toxicological aspects of polybrominated diphenyl ethers: A flame-retardant additive in several consumer products. Environ Res. 2008;108:158–167. doi: 10.1016/j.envres.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 527.Hurley S, Reynolds P, Goldberg D, Nelson DO, Jeffrey SS, Petreas M. Adipose levels of polybrominated diphenyl ethers and risk of breast cancer. Breast Cancer Res Treat. 2011;129:505–511. doi: 10.1007/s10549-011-1481-7. [DOI] [PubMed] [Google Scholar]
- 528.Meerts IA, Letcher RJ, Hoving S, Marsh G, Bergman A, Lemmen JG, et al. In vitro estrogenicity of polybrominated diphenyl ethers, hydroxylated PDBEs, and polybrominated bisphenol a compounds. Environ Health Perspect. 2001;109:399–407. doi: 10.1289/ehp.01109399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Yu L, Zhan P. Molecular mechanisms underlying proliferation and apoptosis in breast cancer MCF-7 cells induced by pentabrominated diphenyl ethers. Toxicol Environ Chem. 2009;91:665–670. doi: 10.1080/02772240802343370. [DOI] [Google Scholar]
- 530.Kwiecińska P, Wróbel A, Gregoraszczuk EŁ. Combinatory effects of PBDEs and 17β-estradiol on MCF-7 cell proliferation and apoptosis. Pharmacol Rep. 2011;63:189–194. doi: 10.1016/S1734-1140(11)70415-7. [DOI] [PubMed] [Google Scholar]
- 531.Li Z-H, Liu X-Y, Wang N, Chen J-S, Chen Y-H, Huang J-T, et al. Effects of decabrominated diphenyl ether (PBDE-209) in regulation of growth and apoptosis of breast, ovarian, and cervical cancer cells. Environ Health Perspect. 2012;120:541–546. doi: 10.1289/ehp.1104051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Davis F, Lin H-Y, Luidens M, Zhou M, Mousa S. Similar and shared nongenomic mechanisms of action of estrogen and thyroid hormone. Immunol Endocr Metabol Agents Med Chem. 2009;9:84–89. doi: 10.2174/187152209789000696. [DOI] [Google Scholar]
- 533.Birnbaum L. Halogenated flame retardants: Does the benefit justify the risk? Environ Health Perspect. 2009;117:A478. doi: 10.1289/ehp.0901417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Patisaul HB, Roberts SC, Mabrey N, Mccaffrey KA, Gear RB, Braun J, et al. Accumulation and endocrine disrupting effects of the flame retardant mixture Firemaster® 550 in rats: An exploratory assessment. J Biochem Mol Toxicol. 2013;27:124–136. doi: 10.1002/jbt.21439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Baan R, Straif K, Grosse Y, Secretan B, El G, Bouvard V, et al. Carcinogenicity of some aromatic amines, organic dyes, and related exposures. Lancet Oncol. 2008;9:322–323. doi: 10.1016/S1470-2045(08)70089-5. [DOI] [PubMed] [Google Scholar]
- 536.DeBruin LS, Pawliszyn JB, Josephy PD. Detection of monocyclic aromatic amines, possible mammary carcinogens, in human milk. Chem Res Toxicol. 1999;12:78–82. doi: 10.1021/tx980168m. [DOI] [PubMed] [Google Scholar]
- 537.DeBruin LS, Josephy PD. Perspectives on the chemical etiology of breast cancer. Environ Health Perspect. 2002;110(Suppl 1):119–128. doi: 10.1289/ehp.02110s1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.National Toxicology Program (NTP) Report on carcinogens. Thirteenth. Research Triangle Park: U.S. Department of Health and Human Services, Public Health Service; 2014. O-Toluidine. [Google Scholar]
- 539.Layton D, Bogen K, Knize M, Hatch F, Johnson V, Felton J. Cancer risk of heterocyclic amines in cooked foods: An analysis and implications for research. Carcinogenesis. 1995;16:39–52. doi: 10.1093/carcin/16.1.39. [DOI] [PubMed] [Google Scholar]
- 540.Ambrosone CB, Abrams SM, Gorlewska-Roberts K, Kadlubar FF. Hair dye use, meat intake, and tobacco exposure and presence of carcinogen-DNA adducts in exfoliated breast ductal epithelial cells. Arch Biochem Biophys. 2007;464:169–175. doi: 10.1016/j.abb.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 541.de Vocht F, Sobala W, Wilczynska U, Kromhout H, Szeszenia-Dabrowska N, Peplonska B. Cancer mortality and occupational exposure to aromatic amines and inhalable aerosols in rubber tire manufacturing in Poland. Cancer Epidemiol. 2009;33:94–102. doi: 10.1016/j.canep.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 542.Meyer A, Fischer K. Oxidative transformation processes and products of para-phenylenediamine (PPD) and para-tolyenediamine (PTD) – A review. Environ Sci Eur. 2015;27:11. doi: 10.1186/s12302-015-0044-7. [DOI] [Google Scholar]
- 543.Heikkinen S, Pitkäniemi J, Sarkeala T, Malila N, Koskenvuo M. Does hair dye use increase the risk of breast cancer? A population-based case-control study of Finnish women. PLoS One. 2015;10:e0135190. doi: 10.1371/journal.pone.0135190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Steck SE, Gaudet MM, Eng SM, Britton JA, Teitelbaum SL, Neugut AI, et al. Cooked meat and risk of breast cancer—lifetime versus recent dietary intake. Epidemiology. 2007;18:373–382. doi: 10.1097/01.ede.0000259968.11151.06. [DOI] [PubMed] [Google Scholar]
- 545.Thompson PA, DeMarini DM, Kadlubar FF, McClure GY, Brooks LR, Green BL, et al. Evidence for the presence of mutagenic arylamines in human breast milk and DNA adducts in exfoliated breast ductal epithelial cells. Environ Mol Mutagen. 2002;39:134–142. doi: 10.1002/em.10067. [DOI] [PubMed] [Google Scholar]
- 546.Turesky RJ. Formation and biochemistry of carcinogenic heterocyclic aromatic amines in cooked meats. Toxicol Lett. 2007;168:219–227. doi: 10.1016/j.toxlet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 547.Rabstein S, Brüning T, Harth V, Fischer H-P, Haas S, Weiss T, et al. N-acetyltransferase 2, exposure to aromatic and heterocyclic amines, and receptor-defined breast cancer. Eur J Cancer Prev. 2010;19:100–109. doi: 10.1097/CEJ.0b013e328333fbb7. [DOI] [PubMed] [Google Scholar]
- 548.Gooderham NJ, Creton S, Lauber SN, Zhu H. Mechanisms of action of the carcinogenic heterocyclic amine PhIP. Toxicol Lett. 2007;168:269–277. doi: 10.1016/j.toxlet.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 549.Ionescu JG, Novotny J, Stejskal V, Lätsch A, Blaurock-Busch E, Eisenmann-Klein M. Increased levels of transition metals in breast cancer tissue. Neuroendocrinol Lett. 2006;27:36–39. [PubMed] [Google Scholar]
- 550.Peng L, Huang Y, Zhang J, Peng Y, Lin X, Wu K, et al. Cadmium exposure and the risk of breast cancer in Chaoshan population of southeast China. Environ Sci Pollut Res. 2015;22:19870–19878. doi: 10.1007/s11356-015-5212-1. [DOI] [PubMed] [Google Scholar]
- 551.Strumylaite L, Bogusevicius A, Abdrachmanovas O, Baranauskiene D, Kregzdyte R, Pranys D, et al. Cadmium concentration in biological media of breast cancer patients. Breast Cancer Res Treat. 2011;125:511–7. doi: 10.1007/s10549-010-1007-8. [DOI] [PubMed] [Google Scholar]
- 552.Wu H-DI, Chou S-Y, Chen D-R, Kuo H-W. Differentiation of serum levels of trace elements in normal and malignant breast patients. Biol Trace Elem Res. 2006;113:9–18. [DOI] [PubMed]
- 553.Brama M, Gnessi L, Basciani S, Cerulli N, Politi L, Spera G, et al. Cadmium induces mitogenic signaling in breast cancer cell by an ERα-dependent mechanism. Mol Cell Endocrinol. 2007;264:102–8. [DOI] [PubMed]
- 554.Martin MB, Reiter R, Pham T, Avellanet YR, Camara J, Lahm M, et al. Estrogen-like activity of metals in MCF-7 breast cancer cells. Endocrinology. 2003;144:2425–2436. doi: 10.1210/en.2002-221054. [DOI] [PubMed] [Google Scholar]
- 555.Sukocheva OA, Yang Y, Gierthy JF, Seegal RF. Methyl mercury influences growth-related signaling in MCF-7 breast cancer cells. Environ Toxicol. 2005;20:32–44. doi: 10.1002/tox.20075. [DOI] [PubMed] [Google Scholar]
- 556.Choe S-Y, Kim S-J, Kim H-G, Lee JH, Choi Y, Lee H, et al. Evaluation of estrogenicity of major heavy metals. Sci Total Environ. 2003;312:15–21. doi: 10.1016/S0048-9697(03)00190-6. [DOI] [PubMed] [Google Scholar]
- 557.McElroy JA, Shafer MM, Trentham-Dietz A, Hampton JM, Newcomb PA. Cadmium exposure and breast cancer risk. J Natl Cancer Inst. 2006;98:869–873. doi: 10.1093/jnci/djj233. [DOI] [PubMed] [Google Scholar]
- 558.Gallagher C, Chen JJ, Kovach JS. Environmental cadmium and breast cancer. Aging. 2010;1:804–814. doi: 10.18632/aging.100226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Saleh F, Behbehani A, Asfar S, Khan I, Ibrahim G. Abnormal blood levels of trace elements and metals, DNA damage, and breast cancer in the state of Kuwait. Biol Trace Elem Res. 2011;141:96–109. doi: 10.1007/s12011-010-8724-z. [DOI] [PubMed] [Google Scholar]
- 560.Adams SV, Shafer MM, Bonner MR, LaCroix AZ, Manson JE, Meliker JR, et al. Urinary cadmium and risk of invasive breast cancer in the Women’s health initiative. Am J Epidemiol. 2016;183:815–823. doi: 10.1093/aje/kwv285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Lin J, Zhang F, Lei Y. Dietary intake and urinary level of cadmium and breast cancer risk: A meta-analysis. Cancer Epidemiol. 2016;42:101–107. doi: 10.1016/j.canep.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 562.Åkesson A, Julin B, Wolk A. Long-term dietary cadmium intake and postmenopausal endometrial cancer incidence: A population-based prospective cohort study. Cancer Res. 2008;68:6435–6441. doi: 10.1158/0008-5472.CAN-08-0329. [DOI] [PubMed] [Google Scholar]
- 563.Julin B, Wolk A, Bergkvist L, Bottai M, Åkesson A. Dietary cadmium exposure and risk of postmenopausal breast cancer: A population-based prospective cohort study. Cancer Res. 2012;72:1459–1466. doi: 10.1158/0008-5472.CAN-11-0735. [DOI] [PubMed] [Google Scholar]
- 564.Itoh H, Iwasaki M, Sawada N, Takachi R, Kasuga Y, Yokoyama S, et al. Dietary cadmium intake and breast cancer risk in Japanese women: A case-control study. Int J Hyg Environ Health. 2014;217:70–77. doi: 10.1016/j.ijheh.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 565.Sawada N, Iwasaki M, Inoue M, Takachi R, Sasazuki S, Yamaji T, et al. Long-term dietary cadmium intake and cancer incidence. Epidemiology. 2012;23:368–376. doi: 10.1097/EDE.0b013e31824d063c. [DOI] [PubMed] [Google Scholar]
- 566.Eriksen KT, Halkjær J, Sørensen M, Meliker JR, McElroy JA, Tjønneland A, et al. Dietary cadmium intake and risk of breast, endometrial and ovarian cancer in danish postmenopausal women: A prospective cohort study. PLoS One. 2014;9:e100815. [DOI] [PMC free article] [PubMed]
- 567.Adams SV, Newcomb PA, White E. Dietary cadmium and risk of invasive postmenopausal breast cancer in the VITAL cohort. Cancer Causes Control. 2012;23:845–854. doi: 10.1007/s10552-012-9953-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Van Maele-Fabry G, Lombaert N, Lison D. Dietary exposure to cadmium and risk of breast cancer in postmenopausal women: A systematic review and meta-analysis. Environ Int. 2016;86:1–13. doi: 10.1016/j.envint.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 569.Johnson MD, Kenney N, Stoica A, et al. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 2003;9:1081–1084. doi: 10.1038/nm902. [DOI] [PubMed] [Google Scholar]
- 570.Byrne C, Divekar SD, Storchan GB, Parodi DA, Martin MB. Metals and breast cancer. J Mammary Gland Biol Neoplasia. 2013;18:63–73. doi: 10.1007/s10911-013-9273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Silva N, Peiris-John R, Wickremasinghe R, Senanayake H, Sathiakumar N. Cadmium a metalloestrogen: Are we convinced? J Appl Toxicol. 2012;32:318–332. doi: 10.1002/jat.1771. [DOI] [PubMed] [Google Scholar]
- 572.Benbrahim-Tallaa L, Tokar EJ, Diwan BA, Dill AL, Coppin J-F, Waalkes MP. Cadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotype. Environ Health Perspect. 2009;117:1847–1852. doi: 10.1289/ehp.0900999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Ali I, Penttinen-Damdimopoulou PE, Mäkelä SI, Berglund M, Stenius U, Åkesson A, et al. Estrogen-like effects of cadmium in vivo do not appear to be mediated via the classical estrogen receptor transcriptional pathway. Environ Health Perspect. 2010;118:1389–1394. doi: 10.1289/ehp.1001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Ali I, Damdimopoulou P, Stenius U, Adamsson A, Mäkelä SI, Åkesson A, et al. Cadmium-induced effects on cellular signaling pathways in the liver of transgenic estrogen reporter mice. Toxicol Sci. 2012;127(1):66–75. doi: 10.1093/toxsci/kfs077. [DOI] [PubMed] [Google Scholar]
- 575.Yu X, Filardo EJ, Shaikh ZA. The membrane estrogen receptor GPR30 mediates cadmium-induced proliferation of breast cancer cells. Toxicol Appl Pharmacol. 2010;245:83–90. doi: 10.1016/j.taap.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 576.Martinez-Zamudio R, Ha HC. Environmental epigenetics in metal exposure. Epigenetics. 2011;6:820–827. doi: 10.4161/epi.6.7.16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Asara Y, Marchal JA, Bandiera P, Mazzarello V, Delogu LG, Sotgiu MA, et al. Cadmium influences the 5-fluorouracil cytotoxic effects on breast cancer cells. Eur J Histocyhem. 2012;56:1–6. doi: 10.4081/ejh.2012.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Seibold P, Vrieling A, Heinz J, Obi N, Sinn H-P, Flesch-Janys D, et al. Pre-diagnostic smoking behaviour and poorer prognosis in a german breast cancer patient cohort - differential effects by tumour subtype, NAT2 status, BMI and alcohol intake. Cancer Epidemiol. 2014;38:419–426. doi: 10.1016/j.canep.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 579.Xie J, Tworoger SS, Franke AA, Terry KL, Rice MS, Rosner BA, et al. Plasma enterolactone and breast cancer risk in the nurses’ health study II. Breast Cancer Res Treat. 2013;139:801–809. doi: 10.1007/s10549-013-2586-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Xiong X-Y, Hu X-J, Li Y, Liu C-M. Inhibitory effects of Enterolactone on growth and metastasis in human breast cancer. Nutr Cancer. 2015;67:1324–1332. doi: 10.1080/01635581.2015.1082113. [DOI] [PubMed] [Google Scholar]
- 581.Flower G, Fritz H, Balneaves LG, Verma S, Skidmore B, Fernandes R, et al. Flax and breast cancer: A systematic review. Integr Cancer Ther. 2014;13:181–192. doi: 10.1177/1534735413502076. [DOI] [PubMed] [Google Scholar]
- 582.Cederroth C, Nef S. Soy, phytoestrogens and metabolism: A review. Mol Cell Endocrinol. 2009;304:30–42. doi: 10.1016/j.mce.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 583.He F-J, Chen J-Q. Consumption of soybean, soy foods, soy isoflavones and breast cancer incidence: Differences between Chinese women and women in western countries and possible mechanisms. Food Sci Human Wellness. 2013;2:146–161. doi: 10.1016/j.fshw.2013.08.002. [DOI] [Google Scholar]
- 584.Andres S, Abraham K, Appel KE, Lampen A. Risks and benefits of dietary isoflavones for cancer. Crit Rev Toxicol. 2011;41:463–506. doi: 10.3109/10408444.2010.541900. [DOI] [PubMed] [Google Scholar]
- 585.Rice S, Whitehead SA. Phytoestrogens and breast cancer - promoters or protectors? Endocr Relat Cancer. 2006;13:995–1015. doi: 10.1677/erc.1.01159. [DOI] [PubMed] [Google Scholar]
- 586.Helferich WG, Andrade JE, Hoagland MS. Phytoestrogens and breast cancer: A complex story. Inflammopharmacology. 2008;16:219–226. doi: 10.1007/s10787-008-8020-0. [DOI] [PubMed] [Google Scholar]
- 587.Lee S-A, Shu X-O, Li H, Yang G, Cai H, Wen W, et al. Adolescent and adult soy food intake and breast cancer risk: Results from the shanghai Women’s health study. Am J Clin Nutr. 2009;89:1920–1926. doi: 10.3945/ajcn.2008.27361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Wu Y-C, Zheng D, Sun J-J, Zou Z-K, Ma Z-L. Meta-analysis of studies on breast cancer risk and diet in Chinese women. Int J Clin Exp Med. 2015;8:73–85. [PMC free article] [PubMed] [Google Scholar]
- 589.Zhang M, Yang H, Holman CDJ. Dietary intake of isoflavones and breast cancer risk by estrogen and progesterone receptor status. Breast Cancer Res Treat. 2009;118:553–563. doi: 10.1007/s10549-009-0354-9. [DOI] [PubMed] [Google Scholar]
- 590.Shu XO, Zheng Y, Cai H, Gu K, Chen Z, Zheng W, et al. Soy food intake and breast cancer survival. JAMA. 2009;302:2437–2443. doi: 10.1001/jama.2009.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Woo HD, Park KS, Ro J, Kim J. Differential influence of dietary soy intake on the risk of breast cancer recurrence related to HER2 status. Nutr Cancer. 2012;64:198–205. doi: 10.1080/01635581.2012.635261. [DOI] [PubMed] [Google Scholar]
- 592.Khan SA, Chatterton RT, Michel N, Bryk M, Lee O, Ivancic D, et al. Soy isoflavone supplementation for breast cancer risk reduction: A randomized phase ii trial. Cancer Prev Res. 2012;5:309–319. doi: 10.1158/1940-6207.CAPR-11-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Nagata C, Mizoue T, Tanaka K, Tsuji I, Tamakoshi A, Matsuo K, et al. Soy intake and breast cancer risk: An evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jap J Clin Oncol. 2014;44:282–295. doi: 10.1093/jjco/hyt203. [DOI] [PubMed] [Google Scholar]
- 594.Takagi A, Kano M, Kaga C. Possibility of breast cancer prevention: Use of soy isoflavones and fermented soy beverage produced using probiotics. Int J Mol Sci. 2015;16:10907–10920. doi: 10.3390/ijms160510907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Toi M, Hirota S, Tomotaki A, Sato N, Hozumi Y, Anan K, et al. Probiotic beverage with soy isoflavone consumption for breast cancer prevention: A case-control study. Curr Nutr Food Sci. 2013;9:194–200. doi: 10.2174/15734013113099990001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Wada K, Nakamura K, Tamai Y, Tsuji M, Kawachi T, Hori A, et al. Soy isoflavone intake and breast cancer risk in Japan: From the Takayama study. Int J Cancer. 2013;133:952–960. doi: 10.1002/ijc.28088. [DOI] [PubMed] [Google Scholar]
- 597.Korde LA, Wu AH, Fears T, Nomura AMY, West DW, Kolonel LN, et al. Childhood soy intake and breast cancer risk in Asian American women. Cancer Epidemiol Biomarkers Prev. 2009;18:1050–1059. doi: 10.1158/1055-9965.EPI-08-0405. [DOI] [PubMed] [Google Scholar]
- 598.Adlercreutz H. Phytoestrogens and breast cancer. J Ster Biochem Mol Biol. 2002;83:113–118. doi: 10.1016/S0960-0760(02)00273-X. [DOI] [PubMed] [Google Scholar]
- 599.Anderson LN, Cotterchio M, Boucher BA, Kreiger N. Phytoestrogen intake from foods, during adolescence and adulthood, and risk of breast cancer by estrogen and progesterone receptor tumor subgroup among Ontario women. Int J Cancer. 2013;132:1683–1692. doi: 10.1002/ijc.27788. [DOI] [PubMed] [Google Scholar]
- 600.Messina M, Wu A. Perspectives on the soy-breast cancer relation. Am Soc Nutr. 2009;89:1673–1679. doi: 10.3945/ajcn.2009.26736V. [DOI] [PubMed] [Google Scholar]
- 601.Warri A, Saarinen N, Makela S, Hilakivi-Clarke L. The role of early life genistein exposures in modifying breast cancer risk. Br J Cancer. 2008;98:1485–1493. doi: 10.1038/sj.bjc.6604321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Zhao Y, Pan J-H, Zhang L-L. Soy foods intake in adolescence and the risk of breast cancer: A systematic review. Chin J Evid-Based Med. 2012;12:550–556. [Google Scholar]
- 603.Chen M, Rao Y, Zheng Y, Wei S, Li Y, Guo T, et al. Association between soy isoflavone intake and breast cancer risk for pre- and post-menopausal women: A meta-analysis of epidemiological studies. PLoS One. 2014;9:e89288. doi: 10.1371/journal.pone.0089288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Goodman M, Shvetsov Y, Wilkens L, Franke A, Le Marchand L, Kakazu K, et al. Urinary phytoestrogen excretion and postmenopausal breast cancer risk: The multiethnic cohort study. Cancer Prev Res. 2009;2:887–894. doi: 10.1158/1940-6207.CAPR-09-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Möller FJ, Pemp D, Soukup ST, Wende K, Zhang X, Zierau O, et al. Soy isoflavone exposure through all life stages accelerates 17β-estradiol-induced mammary tumor onset and growth, yet reduces tumor burden, in ACI rats. Arch Toxicol. 2016;90:1907–1916. doi: 10.1007/s00204-016-1674-2. [DOI] [PubMed] [Google Scholar]
- 606.Phrakonkham P, Brouland JP, Saad HES, Bergès R, Pimpie C, Pocard M, et al. Dietary exposure in utero and during lactation to a mixture of genistein and an anti-androgen fungicide in a rat mammary carcinogenesis model. Reprod Toxicol. 2015;54:101–109. doi: 10.1016/j.reprotox.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 607.Liu B, Edgerton S, Yang X, Kim A, Ordonez-Ercan D, Mason T, et al. Low-dose dietary phytoestrogen abrogates tamoxifen-associated mammary tumor prevention. Cancer Res. 2005;65:879–886. [PubMed] [Google Scholar]
- 608.Constantinou A, White B, Tonetti D, Yang Y, Liang W, Li W, et al. The soy isoflavone daidzein improves the capacity of tamoxifen to prevent mammary tumors. Eur J Cancer. 2005;41:647–654. doi: 10.1016/j.ejca.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 609.Dip R, Lenz S, Gmuender H, Naegeli H. Pleiotropic combinatorial transcriptomes of human breast cancer cells exposed to mixtures of dietary phytoestrogens. Food Chem Toxicol. 2009;47:787–795. doi: 10.1016/j.fct.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 610.Sakamoto T, Horiguchi H, Oguma E, Kayama F. Effects of diverse dietary phytoestrogens on cell growth, cell cycle and apoptosis in estrogen-receptor-positive breast cancer cells. J Nutr Biochem. 2010;21:856–864. doi: 10.1016/j.jnutbio.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 611.Liu Y, Hilakivi-Clarke L, Zhang Y, Wang X, Pan Y-X, Xuan J, et al. Isoflavones in soy flour diet have different effects on whole-genome expression patterns than purified isoflavone mix in human MCF-7 breast tumors in ovariectomized athymic nude mice. Mol Nutr Food Res. 2015;59:1419–1430. doi: 10.1002/mnfr.201500028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Aparecida Santos M, Florencio-Silva R, Teixeira CP, Rodrigues Da Silva Sasso G, Souza Marinho D, Simões RS, et al. Effects of early and late treatment with soy isoflavones in the mammary gland of ovariectomized rats. Climacteric. 2016;19:77–84. doi: 10.3109/13697137.2015.1094783. [DOI] [PubMed] [Google Scholar]
- 613.Garvin S, Ollinger K, Dabrosin C. Resveratrol induces apoptosis and inhibits angiogenesis in human breast cancer xenografts in vivo. Cancer Lett. 2006;231:113–122. doi: 10.1016/j.canlet.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 614.Athar M, Back J, Kopelovich L, Bickers D, Kim A. Multiple molecular targets of resveratrol: Anti-carcinogenic mechanisms. Arch Biochem Biophys. 2009;486:95–102. doi: 10.1016/j.abb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Boucher B, Cotterchio M, Krieger N, Thompson L. Soy formula and breast cancer risk. Epidemiology. 2008;19:165–168. doi: 10.1097/EDE.0b013e31815c40ab. [DOI] [PubMed] [Google Scholar]
- 616.Jefferson W, Doerge D, Padilla-Banks E, Woodling K, Kissling G, Newbold R. Oral exposure to genistin, the glycosylated form of genistein, during neonatal life adversely affects the female reproductive system. Environ Health Perspect. 2009;117:1883–1889. doi: 10.1289/ehp.0900923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Benzoni E, Minervini F, Giannoccaro A, Fornelli F, Vigo D, Visconti A. Influence of in vitro exposure to mycotoxin zearalenone and its derivatives on swine sperm quality. Reprod Toxicol. 2008;25:461–467. doi: 10.1016/j.reprotox.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 618.Mirocha C, Schauerhamer B, Christensen C, Niku-Paavola M, Nummi M. Incidence of zearalenol (fusarium mycotoxin) in animal feed. Appl Environ Microbiol. 1979;38:749–750. doi: 10.1128/aem.38.4.749-750.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Aiko V, Mehta A. Occurrence, detection and detoxification of mycotoxins. J Biosci. 2015;40:943–954. doi: 10.1007/s12038-015-9569-6. [DOI] [PubMed] [Google Scholar]
- 620.Mukherjee D, Royce SG, Alexander JA, Buckley B, Isukapalli SS, Bandera EV, et al. Physiologically-based toxicokinetic modeling of zearalenone and its metabolites: Application to the Jersey girl study. PLoS One. 2014;9:e113632. doi: 10.1371/journal.pone.0113632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Massart F, Meucci V, Saggese G, Soldani G. High growth rate of girls with precocious puberty exposed to estrogenic mycotoxins. J Pediatr. 2008;152:690–5.e1. doi: 10.1016/j.jpeds.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 622.Massart F, Saggese G. Oestrogenic mycotoxin exposures and precocious pubertal development. Int J Androl. 2010;33:369–376. doi: 10.1111/j.1365-2605.2009.01009.x. [DOI] [PubMed] [Google Scholar]
- 623.Bandera EV, Chandran U, Buckley B, Lin Y, Isukapalli S, Marshall I, et al. Urinary mycoestrogens, body size and breast development in New Jersey girls. Sci Total Environ. 2011;409:5221–5227. doi: 10.1016/j.scitotenv.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Frehse MS, Martins MIM, Ono EYS, Bracarense APFRL, Bissoqui LY, Teixeira EMK, et al. Aflatoxins ingestion and canine mammary tumors: There is an association? Food Chem Toxicol. 2015;84:74–78. doi: 10.1016/j.fct.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 625.Videmann B, Koraichi F, Mazallon M, Lecoeur S. Effect of gender, pregnancy and exposure conditions on metabolism and distribution of zearalenone in rats. World Mycotox J. 2012;5:57–69. doi: 10.3920/WMJ2011.1319. [DOI] [Google Scholar]
- 626.Belli P, Bellaton C, Durand J, Balleydier S, Milhau N, Mure M, et al. Fetal and neonatal exposure to the mycotoxin zearalenone induces phenotypic alterations in adult rat mammary gland. Food Chem Toxicol. 2010;48:2818–2826. doi: 10.1016/j.fct.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 627.Nikaido Y, Yoshizawa K, Pei R-J, Yuri T, Danbara N, Hatano T, et al. Prepubertal zearalenone exposure suppresses N-methyl-N-nitrosourea-induced mammary tumorigenesis but causes severe endocrine disruption in female Sprague-Dawley rats. Nutr Cancer. 2003;47:164–170. doi: 10.1207/s15327914nc4702_9. [DOI] [PubMed] [Google Scholar]
- 628.Belhassen H, Jiménez-Díaz I, Arrebola JP, Ghali R, Ghorbel H, Olea N, et al. Zearalenone and its metabolites in urine and breast cancer risk: A case-control study in Tunisia. Chemosphere. 2015;128:1–6. doi: 10.1016/j.chemosphere.2014.12.055. [DOI] [PubMed] [Google Scholar]
- 629.Sondergaard TE, Hansen FT, Purup S, Nielsen AK, Bonefeld-Jørgensen EC, Giese H, et al. Fusarin C acts like an estrogenic agonist and stimulates breast cancer cells in vitro. Toxicol Lett. 2011;205:116–121. doi: 10.1016/j.toxlet.2011.05.1029. [DOI] [PubMed] [Google Scholar]
- 630.Parveen M, Zhu Y, Kiyama R. Expression profiling of the genes responding to zearalenone and its analogues using estrogen-responsive genes. FEBS Lett. 2009;583:2377–2384. doi: 10.1016/j.febslet.2009.06.035. [DOI] [PubMed] [Google Scholar]
- 631.Khosrokhavar R, Rahimifard N, Shoeibi S, Hamedani MP, Hosseini M-J. Effects of zearalenone and α-Zearalenol in comparison with Raloxifene on T47D cells. Toxicol Mech Methods. 2009;19:246–250. doi: 10.1080/15376510802455347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Al-Dobaib S, Mousa H. Benefits and risks of growth promoters in animal production. J Food Agric Environ. 2009;7:202–208. [Google Scholar]
- 633.Leffers H, Næsby M, Vendelbo B, Skakkebæk NE, Jørgensen M, Grandjean P, et al. Oestrogenic potencies of Zeranol, oestradiol, diethylstilboestrol, bisphenol-a and genistein: Implications for exposure assessment of potential endocrine disrupters. APMIS Suppl. 2001;109:S463–S472. doi: 10.1111/j.1600-0463.2001.tb05799.x. [DOI] [PubMed] [Google Scholar]
- 634.Updike MS, Sawdy JC, Wang L-S, Liu S, Huang Y-W, Ye W, et al. Primary cultured human breast epithelial cells up-regulate protein disulfide isomerase in response to zeranol. Anticancer Res. 2007;27:407–410. [PubMed] [Google Scholar]
- 635.Sheffield LG, Welsch CW. Zeranol (β-resorcylic acid lactone), a common residous component of natural foodstuffs, stimulates developmental growth of the mouse mammary Gland. Cancer Lett. 1985;28:77–83. doi: 10.1016/0304-3835(85)90095-3. [DOI] [PubMed] [Google Scholar]
- 636.Nikaido Y, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, Tsubura A. Effects of prepubertal exposure to xenoestrogen on development of estrogen target organs in female CD-1 mice. In Vivo. 2005;19:487–494. [PubMed] [Google Scholar]
- 637.Yuri T, Nikaido Y, Shimano N, Uehara N, Shikata N, Tsubura A. Effects of prepubertal zeranol exposure on estrogen target organs and N-methyl-N-nitrosourea-induced mammary tumorigenesis in female Sprague-Dawley rats. In Vivo. 2004;18:755–762. [PubMed] [Google Scholar]
- 638.Liu S, Kulp SK, Sugimoto Y, Jiang J, Chang H-L, Lin YC. Involvement of breast epithelial-stromal interactions in the regulation of protein tyrosine phosphatase-γ (PTPγ) mRNA expression by estrogenically active agents. Breast Cancer Res Treat. 2002;71:21–35. doi: 10.1023/A:1013343718942. [DOI] [PubMed] [Google Scholar]
- 639.Liu S, Lin Y. Transformation of MCF-10A human breast epithelial cells by zeranol and estradiol-17B. Breast J. 2004;10:514–521. doi: 10.1111/j.1075-122X.2004.21410.x. [DOI] [PubMed] [Google Scholar]
- 640.Xu P, Ye W, Jen R, Lin S, Kuo C, Lin Y. Mitogenic activity of zeranol in human breast cancer cells is enhanced by leptin and suppressed by gossypol. Anticancer Res. 2009;29:4621–4628. [PubMed] [Google Scholar]
- 641.Ye W, Xu P, Zhong S, Threlfall WR, Frasure C, Feng E, et al. Serum harvested from heifers one month post-zeranol implantation stimulates MCF-7 breast cancer cell growth. Exp Ther Med. 2010;1:963–968. doi: 10.3892/etm.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Eaton ML. Ethics and business of bioscience. Palo Alto: Stanford University Press; 2004. Ethics analysis applied to Monsanto and the labeling of rBST; pp. 70–105. [Google Scholar]
- 643.Daxenberger A, Breier BH, Sauerwein H. Increased milk levels of insulin-like growth factor 1 (IGF-1) for the identification of bovine somatotropin (bST) treated cows. Analyst. 1998;123:2429–2435. doi: 10.1039/a804923h. [DOI] [PubMed] [Google Scholar]
- 644.Collier R, Miller M, McLaughlin C, Johnson H, Baile C. Effects of recombinant bovine somatotropin (rbST) and season on plasma and milk insulin-like growth factors I (IGF-I) and II (IGF-II) in lactating dairy cows. Domest Anim Endocrinol. 2008;35:16–23. doi: 10.1016/j.domaniend.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 645.Collier R, Miller M, Hildebrandt J, Torkelson A, White T, Madsen K, et al. Factors affecting insulin-like growth factor-I concentration in bovine milk. J Dairy Sci. 1991;74:2095–2911. doi: 10.3168/jds.S0022-0302(91)78473-7. [DOI] [PubMed] [Google Scholar]
- 646.Outwater JL, Nicholson A, Barnard N. Dairy products and breast cancer: The IGF-I, estrogen, and bGH hypothesis. Med Hypotheses. 1997;48:453–461. doi: 10.1016/S0306-9877(97)90110-9. [DOI] [PubMed] [Google Scholar]
- 647.Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351:1393–1396. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- 648.Ciftci K, Su J, Trovitch PB. Growth factors and chemotherapeutic modulation of breast cancer cells. J Pharm Pharmacol. 2003;55:1135–1141. doi: 10.1211/002235703322277177. [DOI] [PubMed] [Google Scholar]
- 649.Fürstenberger G, Morant R, Senn HJ. Insulin-like growth factors and breast cancer. Onkologie. 2003;26:290–294. doi: 10.1159/000071627. [DOI] [PubMed] [Google Scholar]
- 650.Holly J. Insulin-like growth factor-I and new opportunities for cancer prevention. Lancet. 1998;351:1373–1375. doi: 10.1016/S0140-6736(05)79438-1. [DOI] [PubMed] [Google Scholar]
- 651.Allen N, Roddam A, Allen D, Fentiman I, Peto J, Holly J, et al. A prospective study of serum insulin-like growth factor-I (IGF-1), IGF-II, IGF-binding protein-3 and breast cancer risk. Br J Cancer. 2005;92:1283–1287. doi: 10.1038/sj.bjc.6602471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652.Schernhammer ES, Holly JM, Pollak MN, Hankinson SE. Circulating levels of insulin-like growth factors, their binding proteins, and breast cancer risk. Cancer Epidemiol Biomark Prev. 2005;14:699–704. doi: 10.1158/1055-9965.EPI-04-0561. [DOI] [PubMed] [Google Scholar]
- 653.Frydenberg H, Flote VG, Iversen A, Finstad SE, Furberg A-S, Torjesen PA, et al. Insulin-like growth factor-1, growth hormone, and daily cycling estrogen are associated with mammographic density in premenopausal women. Cancer Causes Control. 2014;25:891–903. doi: 10.1007/s10552-014-0389-z. [DOI] [PubMed] [Google Scholar]
- 654.Macaulay VM. Insulin-lie growth factors and cancer. Br J Cancer. 1992;65:311–320. doi: 10.1038/bjc.1992.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Resnicoff M, Abraham D, Yutanawiboonchai W, Rotman HL, Kajstura J, Rubin R, et al. The insulin-like growth factor I receptor protects tumor cells from apoptosis in vivo. Cancer Res. 1995;55:2463–2469. [PubMed] [Google Scholar]
- 656.Xian C. Degradation of IGF-1 in the adult rat gastrointestinal tract is limited by a specific antiserum or the dietary protein casein. J Endocrinol. 1995;146:215–225. doi: 10.1677/joe.0.1460215. [DOI] [PubMed] [Google Scholar]
- 657.TechSci Research: Global benzenemarket and opportunities, 2020. 2015. http://www.slideshare.net/TechSci_Research/global-benzene-market-forecast-and-opportunities-2020. Accessed 25 Oct 2016.
- 658.National Toxicology Program (NTP): Benzene. 2014. https://ntp.niehs.nih.gov/ntp/roc/content/profiles/benzene.pdf. Accessed 25 Oct 2016.
- 659.IARC Benzene. IARC Monog Eval Carcinog Risk Chem Hum. 1987;Supl 7:120–122. [Google Scholar]
- 660.Petralia SA, Chow WH, McLaughlin J, et al. Occupational risk factors for breast cancer among women in shanghai. Am J Ind Med. 1998;34:477–483. doi: 10.1002/(SICI)1097-0274(199811)34:5<477::AID-AJIM8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 661.Costantini A, Gorini G, Consonni D, Miligi L, Giovannetti L, Quinn M. Exposure to benzene and risk of breast cancer among shoe factory workers in Italy. Tumori. 2009;95:8–12. doi: 10.1177/030089160909500102. [DOI] [PubMed] [Google Scholar]
- 662.Hansen J. Elevated risk for male breast cancer after occupational exposure to gasoline and vehicular combustion products. Am J Ind Med. 2000;37:349–352. doi: 10.1002/(SICI)1097-0274(200004)37:4<349::AID-AJIM4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 663.Huff JE, Haseman JK, DeMarini DM, Eustis S, Maronpot RR, Peters AC, et al. Multiple-site carcinogenicity of benzene in fisher 344 rats and B6C3F1 mice. Environ Health Perspect. 1989;82:125–163. doi: 10.1289/ehp.8982125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.Houle CD, Ton T-VT, Clayton N, Huff J, Hong H-HL, Sills RC. Frequent p53 and H-ras mutations in benzene- and ethylene oxide-induced mammary gland carcinomas from B6C3F1 mice. Toxicol Pathol. 2006;34:752–762. doi: 10.1080/01926230600935912. [DOI] [PubMed] [Google Scholar]
- 665.National Toxicology Program (NTP). Vinyl chloride. Report on carcinogens. Thirteenth ed. Research Triangle Park: U.S. Department of Health and Human Services, Public Health Service; 2014. Available at: https://ntp.niehs.nih.gov/ntp/roc/content/profiles/vinylhalides.pdf.
- 666.IARC Vinyl chloride (group 1) IARC Monog Eval Carcinog Risk Chem Hum. 1987;Suppl 7:383. [Google Scholar]
- 667.Chiazze L, Ference LD. Mortality among PVC-fabricating employees. Environ Health Perspect. 1981;41:137–143. doi: 10.1289/ehp.8141137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Infante PF, Pesák J. A historical perspective of some occupationally related diseases of women. J Occup Med. 1994;36:826–831. [PubMed] [Google Scholar]
- 669.Garcia E, Hurley S, Nelson DO, Hertz A, Reynolds P. Hazardous air pollutants and breast cancer risk in California teachers: A cohort study. Environ Health. 2015;14:14. doi: 10.1186/1476-069X-14-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Ruckart PZ, Bove FJ, Shanley E, Maslia M. Evaluation of contaminated drinking water and male breast cancer at marine Corps Base camp Lejeune, North Carolina: A case control study. Environ Health. 2015;14:74. doi: 10.1186/s12940-015-0061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Drew RT, Boorman GA, Haseman JK, McConnell EE, Busey WM, Moore JA. The effect of age and exposure duration on cancer induction by a known carcinogen in rats, mice, and hamsters. Toxicol Appl Pharmacol. 1983;68:120–130. doi: 10.1016/0041-008X(83)90361-7. [DOI] [PubMed] [Google Scholar]
- 672.Sathiakumar N, Delzell E. A follow-up of women in the synthetic rubber industry. Chem Biol Interact. 2007;166:25–28. doi: 10.1016/j.cbi.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 673.National Toxicology Program (NTP): 1,3-Butadiene. Services, Public Health Service. 2014. http://ntp.niehs.nih.gov/ntp/roc/content/profiles/butadiene.pdf. Accessed 25 Oct 2016.
- 674.Environmental Protection Agency (EPA): 1,3-Butadiene. 1987. https://cfpub.epa.gov/ncea/iris/iris_documents/documents/subst/0139_summary.pdf. Accessed 24 Sept 2016.
- 675.Melnick RL, Sills RC, Portier CJ, Roycroft JH, Chou BJ, Grumbein SL, et al. Multiple organ carcinogenicity of inhaled chloroprene (2-chloro-1,3-butadiene) in F344/N rats and B6C3F1 mice and comparison of dose-response with 1,3-butadiene in mice. Carcinogenesis. 1999;20:867–878. doi: 10.1093/carcin/20.5.867. [DOI] [PubMed] [Google Scholar]
- 676.Agency for Toxic Substances and Disease Registry (ATSDR): Toxicological profile for ethylene oxide. 1990. https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=734&tid=133. Accessed 25 Oct 2016. [PubMed]
- 677.National Toxicology Program (NTP): Ethylene oxide. 2014. https://ntp.niehs.nih.gov/ntp/roc/content/profiles/ethyleneoxide.pdf. Accessed 25 Oct 2016.
- 678.International Agency on tResearch on Cancer (IARC): 1,3-Butadiene, Ethylene Oxide and Vinyl Halides (Vinyl Fluoride, Vinyl Chloride and Vinyl Bromide). IARC Monogr Eval Carcinog Risks Hum. 2008. https://monographs.iarc.fr/ENG/Monographs/vol100F/mono100F-28.pdf. Accessed 25 Oct 2016. [PMC free article] [PubMed]
- 679.Steenland K, Whelan E, Deddens J, Stayner L, Ward E. Ethylene oxide and breast cancer incidence in a cohort study of 7576 women (United States) Cancer Causes Control. 2003;14:531–539. doi: 10.1023/A:1024891529592. [DOI] [PubMed] [Google Scholar]
- 680.Norman SA, Berlin JA, Soper KA, Middendorf BF, Stolley PD. Cancer incidence in a group of workers potentially exposed to ethylene oxide. Int J Epidemiol. 1995;24:276–284. doi: 10.1093/ije/24.2.276. [DOI] [PubMed] [Google Scholar]
- 681.Mikoczy Z, Tinnerberg H, Björk J, Albin M. Cancer incidence and mortality in Swedish Sterilant workers exposed to ethylene oxide: Updated cohort study findings 1972–2006. Inl J Environ Res Public Health. 2011;8:2009–2019. doi: 10.3390/ijerph8062009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Adám B, Bárdos H, Adány R. Increased genotoxic susceptibility of breast epithelial cells to ethylene oxide. Mutat Res. 2005;585:120–126. doi: 10.1016/j.mrgentox.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 683.California Environmental Protection Agency. Air Resources Board: Proposed identification of environmental tobacco smole as a toxic air contaminant. 2005. www.oehha.ca.gov/media/downloads/crnr/app3partb2005.pdf. Accessed 26 Oct 2016.
- 684.Chen Z, An Y, Wang Z, Zhang B, Liu L. Tobacco-specific carcinogen 4-(Methylnitrosoamino)-1-(3-pyridyl)-1-butanone(NNK) activating ERK1/2 MAP kinases and stimulating proliferation of human mammary epithelial cells. Chem Res Chin Univ. 2007;23:76–80. doi: 10.1016/S1005-9040(07)60015-4. [DOI] [Google Scholar]
- 685.Mei J, Hu H, McEntee M, Plummer H, Song P, Wang H-CR. Transformation of non-cancerous human breast epithelial cell line MCF10A by the tobacco-specific carcinogen NNK. Breast Cancer Res Treat. 2003;79:95–105. doi: 10.1023/A:1023326121951. [DOI] [PubMed] [Google Scholar]
- 686.Siriwardhana N, Choudhary S, Wang H-CR. Precancerous model of human breast epithelial cells induced by NNK for prevention. Breast Cancer Res Treat. 2008;109:427–441. doi: 10.1007/s10549-007-9666-9. [DOI] [PubMed] [Google Scholar]
- 687.Kalantari-Dehaghi M, Parnell EA, Armand T, Bernard H-U, Grando SA. The nicotinic acetylcholine receptor-mediated reciprocal effects of the tobacco nitrosamine NNK and SLURP-1 on human mammary epithelial cells. Int Immunopharmacol. 2015;29:99–104. doi: 10.1016/j.intimp.2015.04.041. [DOI] [PubMed] [Google Scholar]
- 688.Reynolds P, Hurley S, Goldberg DE, Anton-Culver H, Bernstein L, Deapen D, et al. Active smoking, household passive smoking, and breast cancer: Evidence from the California teachers study. J Natl Cancer Inst. 2004;96:29–37. doi: 10.1093/jnci/djh002. [DOI] [PubMed] [Google Scholar]
- 689.Band PR, Le ND, Fang R, Deschamps M. Carcinogenic and endocrine disrupting effects of cigarette smoke and risk of breast cancer. Lancet. 2002;360:1044–1049. doi: 10.1016/S0140-6736(02)11140-8. [DOI] [PubMed] [Google Scholar]
- 690.Calle EE, Miracle-McMahill HL, Thun MJ, Heath CW. Cigarette smoking and risk of fatal breast cancer. Am J Epidemiol. 1994;139:1001–1007. doi: 10.1093/oxfordjournals.aje.a116939. [DOI] [PubMed] [Google Scholar]
- 691.Gram IT, Park S-Y, Kolonel LN, Maskarinec G, Wilkens LR, Henderson BE, et al. Smoking and risk of breast cancer in a racially/ethnically diverse population of mainly women who do not drink alcohol: The MEC study. Am J Epidemiol. 2015;182:917–925. doi: 10.1093/aje/kwv092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Johnson KC, Hu J, Mao Y, Canadian Cancer Registries Epidemiology Research Group Passive and active smoking and breast cancer risk in Canada, 1994-97. Cancer Causes Control. 2000;11:211–221. doi: 10.1023/A:1008906105790. [DOI] [PubMed] [Google Scholar]
- 693.Marcus PM, Newman B, Millikan RC, Moorman PG, Baird DD, Qaqish B. The associations of adolescent cigarette smoking, alcoholic beverage consumption, environmental tobacco smoke, and ionizing radiation with subsequent breast cancer risk (United States) Cancer Causes Control. 2000;11:271–278. doi: 10.1023/A:1008911902994. [DOI] [PubMed] [Google Scholar]
- 694.Cui Y, Miller AB, Rohan TE. Cigarette smoking and breast cancer risk: Update of a prospective cohort study. Breast Cancer Res Treat. 2006;100:293–299. doi: 10.1007/s10549-006-9255-3. [DOI] [PubMed] [Google Scholar]
- 695.Xue F, Willett WC, Rosner BA, Hankinson SE, Michels KB. Cigarette smoking and the incidence of breast cancer. Arch Intern Med. 2011;171:125–133. doi: 10.1001/archinternmed.2010.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Luo J, Margolis KL, Wactawski-Wende J, Horn K, Messina C, Stefanick ML, et al. Association of active and passive smoking with risk of breast cancer among postmenopausal women: A prospective cohort study. Br Med J. 2011;342:536. doi: 10.1136/bmj.d1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 697.Bjerkaas E, Parajuli R, Engeland A, Maskarinec G, Weiderpass E, Gram IT. Social inequalities and smoking-associated breast cancer — Results from a prospective cohort study. Prev Med. 2015;73:125–129. doi: 10.1016/j.ypmed.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 698.DeRoo LA, Cummings P, Mueller BA. Smoking before the first pregnancy and the risk of breast cancer: A meta-analysis. Am J Epidemiol. 2011;174:390–402. doi: 10.1093/aje/kwr090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699.Dossus L, Boutron-Ruault M-C, Kaaks R, Gram IT, Vilier A, Fervers B, et al. Active and passive cigarette smoking and breast cancer risk: Results from the EPIC cohort. Int J Cancer. 2014;134:1871–1888. doi: 10.1002/ijc.28508. [DOI] [PubMed] [Google Scholar]
- 700.Park S-Y, Palmer JR, Rosenberg L, Haiman CA, Bandera EV, Bethea TN, et al. A case-control analysis of smoking and breast cancer in African American women: Findings from the AMBER consortium. Carcinogenesis. 2016;37:607–615. doi: 10.1093/carcin/bgw040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 701.Jacobsen KK, Lynge E, Vejborg I, Tjønneland A, von E-C, Andersen ZJ. Cigarette smoking and mammographic density in the Danish diet, cancer and health cohort. Cancer Causes Control. 2016;27:271–280. doi: 10.1007/s10552-015-0704-3. [DOI] [PubMed] [Google Scholar]
- 702.McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: A meta-analysis. Cancer Epidemiol Biomark Prev. 2006;15:1159–1169. doi: 10.1158/1055-9965.EPI-06-0034. [DOI] [PubMed] [Google Scholar]
- 703.Atkinson RL, El-Zein R, Valero V, Lucci A, Bevers TB, Fouad T, et al. Epidemiological risk factors associated with inflammatory breast cancer subtypes. Cancer Causes Control. 2016;27:359–366. doi: 10.1007/s10552-015-0712-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 704.Butler EN, Tse C-K, Bell ME, Conway K, Olshan AF, Troester MA. Active smoking and risk of luminal and basal-like breast cancer subtypes in the Carolina breast cancer study. Cancer Causes Control. 2016;27:775–786. doi: 10.1007/s10552-016-0754-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705.Connor AE, Baumgartner KB, Baumgartner RN, Pinkston CM, Boone SD, John EM, et al. Cigarette smoking and breast cancer risk in Hispanic and non-Hispanic white women: The breast cancer health disparities study. J Women’s Health. 2016;25:299–310. doi: 10.1089/jwh.2015.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Nyante SJ, Gierach GL, Dallal CM, Freedman ND, Park Y, Danforth KN, et al. Cigarette smoking and postmenopausal breast cancer risk in a prospective cohort. Br J Cancer. 2014;110:2339–2347. doi: 10.1038/bjc.2014.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707.Bérubé S, Lemieux J, Moore L, Maunsell E, Brisson J. Smoking at time of diagnosis and breast cancer-specific survival: New findings and systematic review with meta-analysis. Breast Cancer Res. 2014;16:R42. doi: 10.1186/bcr3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.Izano M, Satariano WA, Hiatt RA, Braithwaite D. Smoking and mortality after breast cancer diagnosis: The health and functioning in women study. Cancer Med. 2015;4:315–324. doi: 10.1002/cam4.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.Passarelli MN, Newcomb PA, Hampton JM, Trentham-Dietz A, Titus LJ, Egan KM, et al. Cigarette smoking before and after breast cancer diagnosis: Mortality from breast cancer and smoking-related diseases. J Clin Oncol. 2016;34:1315–1322. doi: 10.1200/JCO.2015.63.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.Persson M, Simonsson M, Markkula A, Rose C, Ingvar C, Jernström H. Impacts of smoking on endocrine treatment response in a prospective breast cancer cohort. Br J Cancer. 2016;115:382–390. doi: 10.1038/bjc.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.Cook MB, Guénel P, Gapstur SM, van den Brandt PA, Michels KB, Casagrande JT, et al. Tobacco and alcohol in relation to male breast cancer: An analysis of the male breast cancer pooling project consortium. Cancer Epidemiol Biomark Prev. 2015;24:520–531. doi: 10.1158/1055-9965.EPI-14-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Padron-Monedero A, Koru-Sengul T, Tannenbaum SL, Miao F, Hansra D, Lee DJ, et al. Smoking and survival in male breast cancer patients. Breast Cancer Res Treat. 2015;153:679–687. doi: 10.1007/s10549-015-3582-1. [DOI] [PubMed] [Google Scholar]
- 713.Ambrosone CB, Freudenheim JL, Graham S, Marshall JR, Vena JE, Brasure JR, et al. Cigarette smoking, N-acetyltransferase 2 genetic polymorphisms, and breast cancer risk. JAMA. 1996;276:1494–1501. doi: 10.1001/jama.1996.03540180050032. [DOI] [PubMed] [Google Scholar]
- 714.Macacu A, Autier P, Boniol M, Boyle P. Active and passive smoking and risk of breast cancer: A meta-analysis. Breast Cancer Res Treat. 2015;154:213–224. doi: 10.1007/s10549-015-3628-4. [DOI] [PubMed] [Google Scholar]
- 715.Hanaoka T, Yamamoto S, Sobue T, Sasaki S, Tsugane S, Japan Public Health Center-Based Prospective Study on Cancer and Cardiovascular Disease Study Group Active and passive smoking and breast cancer risk in middle-aged Japanese women. Int J Cancer. 2005;114:317–322. doi: 10.1002/ijc.20709. [DOI] [PubMed] [Google Scholar]
- 716.Li B, Wang L, Lu M-S, Mo X-F, Lin F-Y, Ho SC, et al. Passive smoking and breast cancer risk among non-smoking women: A case-control study in China. PLoS One. 2015;10:e0125894. doi: 10.1371/journal.pone.0125894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Chen Z, Shao J, Gao X, Li X. Effect of passive smoking on female breast cancer in China: A meta-analysis. Asia Pac J Public Health. 2015;27:NP58–NP64. doi: 10.1177/1010539513481493. [DOI] [PubMed] [Google Scholar]
- 718.Tong J, Li Z, Shi J, Li H, Wang Y, Fu L, et al. Passive smoking exposure from partners as a risk factor for ER+/PR+ double positive breast cancer in never-smoking Chinese urban women: A hospital-based matched case control study. PLoS One. 2014;9:e97498. doi: 10.1371/journal.pone.0097498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 719.Andres SA, Bickett KE, Alatoum MA, Kalbfleisch TS, Brock GN, Wittliff JL. Interaction between smoking history and gene expression levels impacts survival of breast cancer patients. Breast Cancer Res Treat. 2015;152:545–556. doi: 10.1007/s10549-015-3507-z. [DOI] [PubMed] [Google Scholar]
- 720.Kasajova P, Holubekova V, Mendelova A, Lasabova Z, Zubor P, Kudela E, et al. Active cigarette smoking and the risk of breast cancer at the level of N-acetyltransferase 2 (NAT2) gene polymorphisms. Tumour Biol. 2016;37:7929–7937. doi: 10.1007/s13277-015-4685-3. [DOI] [PubMed] [Google Scholar]
- 721.Ricks-Santi LJ, Nie J, Marian C, Ochs-Balcom HM, Trevisan M, Edge SB, et al. BRCA1 polymorphisms and breast cancer epidemiology in the western New York exposures and breast cancer (WEB) study. Genet Epidemiol. 2013;37:504–511. doi: 10.1002/gepi.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722.Dechanet C, Anahory T, Mathieu Daude JC, Quantin X, Reyftmann L, Hamamah S, et al. Effects of cigarette smoking on reproduction. Hum Reprod Update. 2011;17:76–95. doi: 10.1093/humupd/dmq033. [DOI] [PubMed] [Google Scholar]
- 723.Brand JS, Chan M-F, Dowsett M, Folkerd E, Wareham NJ, Luben RN, et al. Cigarette smoking and endogenous sex hormones in postmenopausal women. J Clin Endocrinol Metab. 2011;96:3184–3192. doi: 10.1210/jc.2011-1165. [DOI] [PubMed] [Google Scholar]
- 724.Wang Y-Y, Lehuédé C, Laurent V, Dirat B, Dauvillier S, Bochet L, et al. Adipose tissue and breast epithelial cells: A dangerous dynamic duo in breast cancer. Cancer Lett. 2012;324:142–151. doi: 10.1016/j.canlet.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 725.IARC. Shiftwork. IARC Monogr Eval Carcinog Risks Hum. https://monographs.iarc.fr/ENG/Monographs/vol98/mono98-8.pdf. 2007. Accessed 25 July 2017.
- 726.Bonde JP, Hansen J, Kolstad HA, Mikkelsen S, Olsen JH, Blask DE, et al. Work at night and breast cancer—report on evidence-based options for preventive actions. Scand J Work Environ Health. 2012;38:380–390. doi: 10.5271/sjweh.3282. [DOI] [PubMed] [Google Scholar]
- 727.Costa G, Haus E, Stevens R. Shift work and cancer–considerations on rationale, mechanisms, and epidemiology. Scand J Work Environ Health. 2010;36:163–179. doi: 10.5271/sjweh.2899. [DOI] [PubMed] [Google Scholar]
- 728.Kolstad HA. Nightshift work and risk of breast cancer and other cancers—a critical review of the epidemiologic evidence. Scand J Work Environ Health. 2008;34:5–22. doi: 10.5271/sjweh.1194. [DOI] [PubMed] [Google Scholar]
- 729.Megdal SP, Kroenke CH, Laden F, Pukkala E, Schernhammer ES. Night work and breast cancer risk: A systematic review and meta-analysis. Eur J Cancer. 2005;41:2023–2032. doi: 10.1016/j.ejca.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 730.Kamdar BB, Tergas AI, Mateen FJ, Bhayani NH, Oh J. Night-shift work and risk of breast cancer: A systematic review and meta-analysis. Breast Cancer Res Treat. 2013;138:291–301. doi: 10.1007/s10549-013-2433-1. [DOI] [PubMed] [Google Scholar]
- 731.He C, Anand ST, Ebell MH, Vena JE, Robb SW. Circadian disrupting exposures and breast cancer risk: A meta-analysis. Int Arch Occup Environ Health. 2015;88:533–547. doi: 10.1007/s00420-014-0986-x. [DOI] [PubMed] [Google Scholar]
- 732.Lin X, Chen W, Wei F, Ying M, Wei W, Xie X. Night-shift work increases morbidity of breast cancer and all-cause mortality: A meta-analysis of 16 prospective cohort studies. Sleep Med. 2015;16:1381–1387. doi: 10.1016/j.sleep.2015.02.543. [DOI] [PubMed] [Google Scholar]
- 733.Travis RC, Balkwill A, Fensom GK, Appleby PN, Reeves GK, Wang X-S, et al. Night shift work and breast cancer incidence: Three prespective studies and meta-analysis of the published studies. J Natl Cancer Inst. 2016;108:djw241. doi: 10.1093/jnci/djw169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.O’Neill R. Cancer all-clear for night work based on ‘bad science’, warn scientists. 2016. http://www.hazards.org/cancer/graveyardshift.htm. Hazards Magazine. Accessed 9 Feb 2017.
- 735.Slack R, Young C, Rushton L, British Occupational Cancer Burden Study Group Occupational cancer in Britain. Female cancers: Breast, cervix and ovary. Br J Cancer. 2012;107(Suppl 1):S27–S32. doi: 10.1038/bjc.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Purdue MP, Hutchings SJ, Rushton L, Silverman DT. The proportion of cancer attributable to occupational exposures. Ann Epidemiol. 2015;25:188–192. doi: 10.1016/j.annepidem.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 737.Hansen J, Stevens RG. Case-control study of shift-work and breast cancer risk in Danish nurses: Impact of shift systems. Eur J Cancer. 2012;48:1722–1729. doi: 10.1016/j.ejca.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 738.Menegaux F, Truong T, Anger A, Cordina-Duverger E, Lamkarkach F, Arveux P, et al. Night work and breast cancer: A population-based case-control study in France (the CECILE study) Int J Cancer. 2013;132:924–931. doi: 10.1002/ijc.27669. [DOI] [PubMed] [Google Scholar]
- 739.Stevens RG. Working against our endogenous circadian clock: Breast cancer and electric lighting in the modern world. Mutat Res. 2009;680:106–108. doi: 10.1016/j.mrgentox.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 740.Cos S, González A, Güezmes A, Mediavilla MD, Martínez-Campa C, Alonso-González C, et al. Melatonin inhibits the growth of DMBA-induced mammary tumors by decreasing the local biosynthesis of estrogens through the modulation of aromatase activity. Int J Cancer. 2006;118:274–278. doi: 10.1002/ijc.21401. [DOI] [PubMed] [Google Scholar]
- 741.Cos S, Sánchez-Barceló EJ. Melatonin and mammary pathological growth. Front Neuroendocrinol. 2000;21:133–170. doi: 10.1006/frne.1999.0194. [DOI] [PubMed] [Google Scholar]
- 742.Knower KC, To SQ, Takagi K, Miki Y, Sasano H, Simpson ER, et al. Melatonin suppresses aromatase expression and activity in breast cancer associated fibroblasts. Breast Cancer Res Treat. 2012;132:765–771. doi: 10.1007/s10549-012-1953-4. [DOI] [PubMed] [Google Scholar]
- 743.Fonken LK, Nelson RJ. The effects of light at night on circadian clocks and metabolism. Endocr Rev. 2014;35:648–670. doi: 10.1210/er.2013-1051. [DOI] [PubMed] [Google Scholar]
- 744.Flynn-Evans EE, Stevens RG, Tabandeh H, Schernhammer ES, Lockley SW. Total visual blindness is protective against breast cancer. Cancer Causes Control. 2009;20:1753–1756. doi: 10.1007/s10552-009-9405-0. [DOI] [PubMed] [Google Scholar]
- 745.Nagata C, Nagao Y, Yamamoto S, Shibuya C, Kashiki Y, Shimizu H. Light exposure at night, urinary 6-sulfatoxymelatonin, and serum estrogens and androgens in postmenopausal Japanese women. Cancer Epidemiol Biomark Prev. 2008;17:1418–1423. doi: 10.1158/1055-9965.EPI-07-0656. [DOI] [PubMed] [Google Scholar]
- 746.Davis S, Mirick DK, Chen C, Stanczyk FZ. Night shift work and hormone levels in women. Cancer Epidemiol Biomark Prev. 2012;21:609–618. doi: 10.1158/1055-9965.EPI-11-1128. [DOI] [PubMed] [Google Scholar]
- 747.Langley AR, Graham CH, Grundy AL, Tranmer JE, Richardson H, Aronson KJ. A cross-sectional study of breast cancer biomarkers among shift working nurses. BMJ Open. 2012;2:e000532. doi: 10.1136/bmjopen-2011-000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 748.Blask DE, Hill SM, Dauchy RT, Xiang S, Yuan L, Duplessis T, et al. Circadian regulation of molecular, dietary, and metabolic signaling mechanisms of human breast cancer growth by the nocturnal melatonin signal and the consequences of its disruption by light at night. J Pineal Res. 2011;51:259–269. doi: 10.1111/j.1600-079X.2011.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749.Blask DE, Brainard GC, Dauchy RT, Hanifin JP, Davidson LK, Krause JA, et al. Melatonin-depleted blood from premenopausal women exposed to light at night stimulates growth of human breast cancer xenografts in nude rats. Cancer Res. 2005;65:11174–11184. doi: 10.1158/0008-5472.CAN-05-1945. [DOI] [PubMed] [Google Scholar]
- 750.Zhu Y, Stevens RG, Hoffman AE, Tjonneland A, Vogel UB, Zheng T, et al. Epigenetic impact of long-term shiftwork: Pilot evidence from circadian genes and whole-genome methylation analysis. Chronobiol Int. 2011;28:852–861. doi: 10.3109/07420528.2011.618896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 751.Zhu Y, Stevens RG, Leaderer D, Hoffman A, Holford T, Zhang Y, et al. Non-synonymous polymorphisms in the circadian gene NPAS2 and breast cancer risk. Breast Cancer Res Treat. 2008;107:421–425. doi: 10.1007/s10549-007-9565-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Gery S, Virk RK, Chumakov K, Yu A, Koeffler HP. The clock gene Per2 links the circadian system to the estrogen receptor. Oncogene. 2007;26:7916–7920. doi: 10.1038/sj.onc.1210585. [DOI] [PubMed] [Google Scholar]
- 753.Rabstein S, Harth V, Justenhoven C, Pesch B, Plöttner S, Heinze E, et al. Polymorphisms in circadian genes, night work and breast cancer: Results from the GENICA study. Chronobiol Int. 2014;31:1115–1122. doi: 10.3109/07420528.2014.957301. [DOI] [PubMed] [Google Scholar]
- 754.Grundy A, Schuetz JM, Lai AS, Janoo-Gilani R, Leach S, Burstyn I, et al. Shift work, circadian gene variants and risk of breast cancer. Cancer Epidemiol. 2013;37:606–612. doi: 10.1016/j.canep.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 755.Pronk A, Ji B-T, Shu X-O, Xue S, Yang G, Li H-L, et al. Night-shift work and breast cancer risk in a cohort of Chinese women. Am J Epidemiol. 2010;171:953–959. doi: 10.1093/aje/kwq029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Bhatti P, Mirick DK, Davis S. Racial differences in the association between night shift work and melatonin levels among women. Am J Epidemiol. 2013;177:388–393. doi: 10.1093/aje/kws278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757.Papantoniou K, Castaño-Vinyals G, Espinosa A, Aragonés N, Pérez-Gómez B, Ardanaz E, et al. Breast cancer risk and night shift work in a case-control study in a Spanish population. Eur J Epidemiol. 2015;31:867–878. doi: 10.1007/s10654-015-0073-y. [DOI] [PubMed] [Google Scholar]
- 758.Anjum B. Associations of circadian disruption of sleep and nutritional factors with risk of cancer. Open Nutraceuticals J. 2012;5:124–135. doi: 10.2174/1876396001205010124. [DOI] [Google Scholar]
- 759.Fritschi L, Glass DC, Heyworth JS, Aronson K, Girschik J, Boyle T, et al. Hypotheses for mechanisms linking shiftwork and cancer. Med Hypotheses. 2011;77:430–436. doi: 10.1016/j.mehy.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 760.Sharat A, Tamrin ShBM, Daneshjoo A, Sadeghi H. The adverse health effects of shift work in relation to health/disease: A review. Acta Medica Bulg. 2015;42:63–72.
- 761.Aronson K, Grundy A, Korsiak J, Spinelli JJ. Causes of breast cancer: Could work at night really be a cause? Breast Cancer Manag. 2015;4:125–127. doi: 10.2217/bmt.15.4. [DOI] [Google Scholar]
- 762.Van Dycke KCG, Rodenburg W, van Oostrom CTM, van Kerkhof LWM, Pennings JLA, Roenneberg T, et al. Chronically alternating light cycles increase breast cancer risk in mice. Curr Biol. 2015;25:1932–1937. doi: 10.1016/j.cub.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 763.Dauchy RT, Xiang S, Mao L, Brimer S, Wren MA, Yuan L, et al. Circadian and melatonin disruption by exposure to light at night drives intrinsic resistance to tamoxifen therapy in breast cancer. Cancer Res. 2014;74:4099–4110. doi: 10.1158/0008-5472.CAN-13-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764.Xiang S, Dauchy RT, Hauch A, Mao L, Yuan L, Wren MA, et al. Doxorubicin resistance in breast cancer is driven by light at night-induced disruption of the circadian melatonin signal. J Pineal Res. 2015;59:60–69. doi: 10.1111/jpi.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 765.Environmental Protection Agency (EPA): Radiation basics. 2016. https://www.epa.gov/radiation/radiation-basics#tab-2. Accessed 25 Oct 2016.
- 766.National Toxicology Program. Ionizing radiation. 2014. https://ntp.niehs.nih.gov/ntp/roc/content/profiles/ionizingradiation.pdf. Accessed 25 Oct 2016.
- 767.Harbron RW. Cancer risks from low dose exposure to ionising radiation – Is the linear no-threshold model still relevant? Radiography. 2012;18:28–33. doi: 10.1016/j.radi.2011.07.003. [DOI] [Google Scholar]
- 768.Brenner DJ, Doll R, Goodhead DT, Hall EJ, Land CE, Little JB, et al. Cancer risks attributable to low doses of ionizing radiation: Assessing what we really know. Proc Natl Acad Sci U S A. 2003;100:13761–13766. doi: 10.1073/pnas.2235592100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 769.National Radiological Protection Board (NRPB) (Britain) Risk of radiation-induced cancer at low doses and low dose rates for radiation protection purposes. 1995. p. 25. [Google Scholar]
- 770.Boice JD. Radiation and breast carcinogenesis. Med Pediatr Oncol. 2001;36:508–513. doi: 10.1002/mpo.1122. [DOI] [PubMed] [Google Scholar]
- 771.Goto H, Watanabe T, Miyao M, Fukuda H, Sato Y, Oshida Y. Cancer mortality among atomic bomb survivors exposed as children. Environ Health Prev Med. 2012;17:228–234. doi: 10.1007/s12199-011-0246-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Pierce DA, Shimizu Y, Preston DL, Vaeth M, Mabuchi K. Studies of the mortality of atomic bomb survivors. Report 12, part I. Cancer: 1950–1990. Radiat Res. 1996;146:1–27. doi: 10.2307/3579391. [DOI] [PubMed] [Google Scholar]
- 773.Tokunaga M, Land CE, Tokuoka S, Nishimori I, Soda M, Akiba S. Incidence of female breast cancer among atomic bomb survivors, 1950–1985. Radiat Res. 1994;138:209–223. doi: 10.2307/3578591. [DOI] [PubMed] [Google Scholar]
- 774.Land CE. Studies of cancer and radiation dose among atomic bomb survivors. The example of breast cancer. JAMA. 1995;274:402–407. doi: 10.1001/jama.1995.03530050050031. [DOI] [PubMed] [Google Scholar]
- 775.Ron E, Ikeda T, Preston DL, Tokuoka S. Male breast cancer incidence among atomic bomb survivors. J Natl Cancer Inst. 2005;97:603–605. doi: 10.1093/jnci/dji097. [DOI] [PubMed] [Google Scholar]
- 776.Goldberg Z, Lehnert BE. Radiation-induced effects in unirradiated cells: A review and implications in cancer. Int J Oncol. 2002;21:337–349. [PubMed] [Google Scholar]
- 777.Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat Res. 2003;159:581–596. doi: 10.1667/0033-7587(2003)159[0581:NADEOE]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 778.Tsai KKC, Chuang EY-Y, Little JB, Yuan Z-M. Cellular mechanisms for low-dose ionizing radiation-induced perturbation of the breast tissue microenvironment. Cancer Res. 2005;65:6734–6744. doi: 10.1158/0008-5472.CAN-05-0703. [DOI] [PubMed] [Google Scholar]
- 779.Borrego-Soto G, Ortiz-López R, Rojas-Martínez A. Ionizing radiation-induced DNA injury and damage detection in patients with breast cancer. Genet Mol Biol. 2015;38:420–432. doi: 10.1590/S1415-475738420150019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780.Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci. 2016;17:102. doi: 10.3390/ijms17010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 781.Little JB. Genomic instability and radiation. J Radiol Prot. 2003;23:173–181. doi: 10.1088/0952-4746/23/2/304. [DOI] [PubMed] [Google Scholar]
- 782.Baert A, Depuydt J, Van Maerken T, Poppe B, Malfait F, Storm K, et al. Increased chromosomal radiosensitivity in asymptomatic carriers of a heterozygous BRCA1 mutation. Breast Cancer Res. 2016;18:52. doi: 10.1186/s13058-016-0709-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 783.Andrieu N, Easton DF, Chang-Claude J, Rookus MA, Brohet R, Cardis E, et al. Effect of chest X-rays on the risk of breast cancer among BRCA1/2 mutation carriers in the international BRCA1/2 carrier cohort study: A report from the EMBRACE, GENEPSO, GEO-HEBON, and IBCCS collaborators’ group. J Clin Oncol. 2006;24:3361–3366. doi: 10.1200/JCO.2005.03.3126. [DOI] [PubMed] [Google Scholar]
- 784.Pepe S, Pensabene M, Condello C. Modifiers of risk in BRCA1/2 mutation carriers. Curr Women’s Health Rev. 2012;8:23–29. doi: 10.2174/157340412799079246. [DOI] [Google Scholar]
- 785.Turnbull C, Mirugaesu N, Eeles R. Radiotherapy and genetic predisposition to breast cancer. Clin Oncol (R Coll Radiol) 2006;1:257. doi: 10.1016/j.clon.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 786.Calaf GM, Hei TK. Establishment of a radiation- and estrogen-induced breast cancer model. Carcinogenesis. 2000;21:769–776. doi: 10.1093/carcin/21.4.769. [DOI] [PubMed] [Google Scholar]
- 787.Imaoka T, Nishimura M, Iizuka D, Daino K, Takabatake T, Okamoto M, et al. Radiation-induced mammary carcinogenesis in rodent models: What’s different from chemical carcinogenesis? J Radiat Res. 2009;50:281–293. doi: 10.1269/jrr.09027. [DOI] [PubMed] [Google Scholar]
- 788.Segaloff A, Maxfield WS. The synergism between radiation and estrogen in the production of mammary cancer in the rat. Cancer Res. 1971;31:166–168. [PubMed] [Google Scholar]
- 789.Doody MM, Freedman DM, Alexander BH, Hauptmann M, Miller JS, Rao RS, et al. Breast cancer incidence in U.S. radiologic technologists. Cancer. 2006;106:2707–2715. doi: 10.1002/cncr.21876. [DOI] [PubMed] [Google Scholar]
- 790.Simon SL, Weinstock RM, Doody MM, Neton J, Wenzl T, Stewart P, et al. Estimating historical radiation doses to a cohort of U.S. radiologic technologists. Radiat Res. 2006;166:174–192. doi: 10.1667/RR3433.1. [DOI] [PubMed] [Google Scholar]
- 791.Liu JJ, Freedman DM, Little MP, Doody MM, Alexander BH, Kitahara CM, et al. Work history and mortality risks in 90,268 US radiological technologists. Occup Environ Med. 2014;71:819–835. doi: 10.1136/oemed-2013-101859. [DOI] [PubMed] [Google Scholar]
- 792.Rajaraman P, Doody MM, Yu CL, Preston DL, Miller JS, Sigurdson AJ, et al. JOURNAL CLUB: Cancer risks in U.S. radiologic technologists working with fluoroscopically guided interventional procedures, 1994. Am J Roentgenol. 2016;206:1101–1109. doi: 10.2214/AJR.15.15265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 793.Sigurdson AJ, Bhatti P, Chang S, Rajaraman P, Doody MM, Bowen L, et al. Polymorphisms in estrogen biosynthesis and metabolism-related genes, ionizing radiation exposure, and risk of breast cancer among US radiologic technologists. Breast Cancer Res Treat. 2009;118:177–184. doi: 10.1007/s10549-009-0307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 794.Ballard T, Lagorio S, De Angelis G, Verdecchia A. Cancer incidence and mortality among flight personnel: A meta-analysis. Aviat Space Environ Med. 2000;71:216–224. [PubMed] [Google Scholar]
- 795.Buja A, Mastrangelo G, Perissinotto E, Grigoletto F, Frigo AC, Rausa G, et al. Cancer incidence among female flight attendants: A meta-analysis of published data. J Women’s Health (Larchmt) 2006;15:98–105. doi: 10.1089/jwh.2006.15.98. [DOI] [PubMed] [Google Scholar]
- 796.Gofman J. Preventing breast cancer: The story of a major proven, preventable cause of this disease. 2. San Francisco: CNR Division, Committee for Nuclear Responsibility; 1996. [Google Scholar]
- 797.John EM, McGuire V, Thomas D, Haile R, Ozcelik H, Milne RL, et al. Diagnostic chest X-rays and breast cancer risk before age 50 years for BRCA1 and BRCA2 mutation carriers. Cancer Epidemiol Biomarkers Prev. 2013;22:1547–1556. doi: 10.1158/1055-9965.EPI-13-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 798.Inskip PD, Sigurdson AJ, Veiga L, Bhatti P, Ronckers C, Rajaraman P, et al. Radiation-related new primary solid cancers in the childhood cancer survivor study: Comparative radiation dose response and modification of treatment effects. Int J Radiat Oncol Biol Phys. 2016;94:800–807. doi: 10.1016/j.ijrobp.2015.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 799.Miglioretti DL, Johnson E, Williams A, Greenlee RT, Weinmann S, Solberg LI, et al. The use of computed tomography and the associated radiation exposure and estimated cancer risk. JAMA Pediatr. 2013;167:700–707. doi: 10.1001/jamapediatrics.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 800.Islam SMS, Abru AF, Al Obaidani S, Al-Shabibi S, Al FS. Trends in CT request and related outcomes in a pediatric emergency department. Oman Med J. 2016;31:365–369. doi: 10.5001/omj.2016.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 801.Gofman JW. Radiation from medical procedures in the pathogenesis of cancer and ischemic heart disease: Dose-response studies with physicians per 100,000 population. San Francisco: CNR Division, Committee for Nuclear Responsibility; 1999. [Google Scholar]
- 802.Larson DB, Johnson LW, Schnell BM, Salisbury SR, Forman HP. National trends in CT use in the emergency department: 1995-2007. Radiology. 2011;258:164–173. doi: 10.1148/radiol.10100640. [DOI] [PubMed] [Google Scholar]
- 803.Linet MS, Slovis TL, Miller DL, Kleinerman R, Lee C, Rajaraman P, et al. Cancer risks associated with external radiation from diagnostic imaging procedures. CA Cancer J Clin. 2012;62:75–100. doi: 10.3322/caac.21132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 804.Berrington de González A, Mahesh M, Kim K-P, Bhargavan M, Lewis R, Mettler F, et al. Projected cancer risks from computed tomographic scans performed in the United States in 2007. Arch Intern Med. 2009;169:2071–2077. doi: 10.1001/archinternmed.2009.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 805.Redberg RF. Cancer risks and radiation exposure from computed tomographic scans: How can we be sure that the benefits outweigh the risks? Arch Intern Med. 2009;169:2049. doi: 10.1001/archinternmed.2009.453. [DOI] [PubMed] [Google Scholar]
- 806.Smith-Bindman R. Radiation dose associated with common computed tomography examinations and the associated lifetime attributable risk of cancer. Arch Intern Med. 2009;169:2078. doi: 10.1001/archinternmed.2009.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 807.Huda W, Schoepf UJ, Abro JA, Mah E, Costello P. Radiation-related cancer risks in a clinical patient population undergoing cardiac CT. Am J Roetenol. 2011;196:W159–W165. doi: 10.2214/AJR.10.4981. [DOI] [PubMed] [Google Scholar]
- 808.Einstein AJ, Henzlova MJ, Rajagopalan S. Estimating risk of cancer associated with radiation exposure from 64-slice computed tomography coronary angiography. J Am Med Assoc. 2007;298:317–323. doi: 10.1001/jama.298.3.317. [DOI] [PubMed] [Google Scholar]
- 809.Heyes GJ, Mill AJ, Charles MW. Mammography—Oncogenecity at low doses. J Radiol Protect. 2009;29:A123–A132. doi: 10.1088/0952-4746/29/2A/S08. [DOI] [PubMed] [Google Scholar]
- 810.Soler D, Pampalona J, Tusell L, Genescà A. Radiation sensitivity increases with proliferation-associated telomere dysfunction in nontransformed human epithelial cells. Aging Cell. 2009;8:414–425. doi: 10.1111/j.1474-9726.2009.00488.x. [DOI] [PubMed] [Google Scholar]
- 811.Nelson HD, Tyne K, Naik A, Bougatsos C, Chan BK, Humphrey L. Screening for breast cancer: An update for the U.S. preventive services task force. Ann Intern Med. 2009;151:727–737. doi: 10.7326/0003-4819-151-10-200911170-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 812.Nelson HD, Pappas M, Cantor A, Griffin J, Daeges M, Humphrey L. Harms of breast cancer screening: Systematic review to update the 2009 U.S. preventive services task force recommendation. Ann Intern Med. 2016;164:256. doi: 10.7326/M15-0970. [DOI] [PubMed] [Google Scholar]
- 813.de Gelder R, Draisma G, Heijnsdijk EA, de Koning HJ. Population-based mammography screening below age 50: Balancing radiation-induced vs prevented breast cancer deaths. Br J Cancer. 2011;104:1214–1220. doi: 10.1038/bjc.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814.Yaffe MJ, Mainprize JG. Risk of radiation-induced breast cancer from mammographic screening. Radiology. 2011;258:98–105. doi: 10.1148/radiol.10100655. [DOI] [PubMed] [Google Scholar]
- 815.Miglioretti DL, Lange J, van den Broek JJ, Lee CI, van Ravesteyn NT, Ritley D, et al. Radiation-induced breast cancer incidence and mortality from digital mammography screening: A modeling study. Ann Intern Med. 2016;164:205. doi: 10.7326/M15-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 816.Siu AL. Screening for breast cancer: U.S preventive task forces recommendation statement. Ann Intern Med. 2016;164:279–296. doi: 10.7326/M15-2886. [DOI] [PubMed] [Google Scholar]
- 817.Gotzsche PC, Hartling OJ, Nielsen M, Brodersen J, Jorgensen KJ. Breast screening: The facts—or maybe not. Br Med J. 2009;338:b86. doi: 10.1136/bmj.b86. [DOI] [PubMed] [Google Scholar]
- 818.Veronesi U, Luini A, Del Vecchio M. Radiotherapy after breast-preserving surgery in women with localized cancer of the breast. N Engl J Med. 1993;328:1587–1591. doi: 10.1056/NEJM199306033282202. [DOI] [PubMed] [Google Scholar]
- 819.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) Favourable and unfavourable effects on long-term survival of radiotherapy for early breast cancer: An overview of the randomised trials. Early breast cancer Trialists’ collaborative group. Lancet. 2000;355:1757–1770. doi: 10.1016/S0140-6736(00)02263-7. [DOI] [PubMed] [Google Scholar]
- 820.Mellemkjær L, Friis S, Olsen JH, Scélo G, Hemminki K, Tracey E, et al. Risk of second cancer among women with breast cancer. Int J Cancer. 2006;118:2285–2292. doi: 10.1002/ijc.21651. [DOI] [PubMed] [Google Scholar]
- 821.Roychoudhuri R, Evans H, Robinson D, Møller H. Radiation-induced malignancies following radiotherapy for breast cancer. Brit J Cancer. 2004;91:868–872. doi: 10.1038/sj.bjc.6602084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 822.Huang J, Mackillop WJ. Increased risk of soft tissue sarcoma after radiotherapy in women with breast carcinoma. Cancer. 2001;92:172–180. doi: 10.1002/1097-0142(20010701)92:1<172::AID-CNCR1306>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 823.Shah S, Rosa M. Radiation-associated angiosarcoma of the breast: Clinical and pathologic features. Arch Pathol Lab Med. 2016;140:477–481. doi: 10.5858/arpa.2014-0581-RS. [DOI] [PubMed] [Google Scholar]
- 824.Hooning MJ, Aleman BMP, Hauptmann M, Baaijens MHA, Klijn JGM, Noyon R, et al. Roles of radiotherapy and chemotherapy in the development of contralateral breast cancer. J Clin Oncol. 2008;26:5561–5568. doi: 10.1200/JCO.2007.16.0192. [DOI] [PubMed] [Google Scholar]
- 825.Ng AK, Travis LB. Radiation therapy and breast cancer risk. J Natl Compr Cancer Netw. 2009;7:1121–1128. doi: 10.6004/jnccn.2009.0073. [DOI] [PubMed] [Google Scholar]
- 826.Stovall M, Smith SA, Langholz BM, Boice JD, Shore RE, Andersson M, et al. Dose to the contralateral breast from radiotherapy and risk of second primary breast cancer in the WECARE study. Int J Radiat Oncol Biol Phys. 2008;72:1021–1030. doi: 10.1016/j.ijrobp.2008.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 827.National Institute of Environmental Heal Sciences (NIEHS) Working group report: Assessment of health effects from exposure to power-line frequency electric and magnetic fields. Brooklyn Park: National Institute of Environmental Health; 1998. [Google Scholar]
- 828.Carpenter DO. Electromagnetic fields and cancer: The cost of doing nothing. Rev Environ Health. 2010;25:75–80. doi: 10.1515/REVEH.2010.25.1.75. [DOI] [PubMed] [Google Scholar]
- 829.McElroy JA, Egan KM, Titus-Ernstoff L, Anderson HA, Trentham-Dietz A, Hampton JM, et al. Occupational exposure to electromagnetic field and breast cancer risk in a large, population-based, case-control study in the United States. J Occup Environ Med. 2007;49:266–264. doi: 10.1097/JOM.0b013e318032259b. [DOI] [PubMed] [Google Scholar]
- 830.Peplonska B, Stewart P, Szeszenia-Dabrowska N, Rusiecki J, Garcia-Closas M, Lissowska J, et al. Occupation and breast cancer risk in polish women: A population-based case-control study. Am J Ind Med. 2007;50:97–111. doi: 10.1002/ajim.20420. [DOI] [PubMed] [Google Scholar]
- 831.Ahlbom IC, Cardis E, Green A, Linet M, Savitz D, Swerdlow A, et al. Review of the epidemiologic literature on EMF and health. Environ Health Perspect. 2001;109:911–933. doi: 10.2307/3454653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 832.Kliukiene J, Tynes T, Andersen A. Follow-up of radio and telegraph operators with exposure to electromagnetic fields and risk of breast cancer. Eur J Cancer Prev. 2003;12:301–307. doi: 10.1097/00008469-200308000-00010. [DOI] [PubMed] [Google Scholar]
- 833.Coogan PF, Clapp RW, Newcomb PA, Wenzl TB, Bogdan G, Mittendorf R, et al. Occupational exposure to 60-hertz magnetic fields and risk of breast cancer in women. Epidemiology. 1996;7:459–464. doi: 10.1097/00001648-199609000-00002. [DOI] [PubMed] [Google Scholar]
- 834.Kliukiene J, Tynes T, Andersen A. Residential and occupational exposures to 50-Hz magnetic fields and breast cancer in women: A population-based study. Am J Epidemiol. 2004;159:852–861. doi: 10.1093/aje/kwh116. [DOI] [PubMed] [Google Scholar]
- 835.Feychting M, Forssén U, Rutqvist LE, Ahlbom A. Magnetic fields and breast cancer in Swedish adults residing near high-voltage power lines. Epidemiology. 1998;9:392–397. doi: 10.1097/00001648-199807000-00008. [DOI] [PubMed] [Google Scholar]
- 836.Chen C, Ma X, Zhong M, Yu Z. Extremely low-frequency electromagnetic fields exposure and female breast cancer risk: A meta-analysis based on 24,338 cases and 60,628 controls. Breast Cancer Res Treat. 2010;123:569–576. doi: 10.1007/s10549-010-0782-6. [DOI] [PubMed] [Google Scholar]
- 837.Zhang Y, Lai J, Ruan G, Chen C, Wang DW. Meta-analysis of extremely low frequency electromagnetic fields and cancer risk: A pooled analysis of epidemiologic studies. Environ Int. 2016;88:36–43. doi: 10.1016/j.envint.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 838.Loomis D. Cancer of breast among men in electrical occupations. Lancet. 1992;339:1482–1483. doi: 10.1016/0140-6736(92)92078-T. [DOI] [PubMed] [Google Scholar]
- 839.Matanoski GM, Breysse PN, Elliott EA. Electromagnetic field exposure and male breast cancer. Lancet. 1991;337:737. doi: 10.1016/0140-6736(91)90325-J. [DOI] [PubMed] [Google Scholar]
- 840.Milham S. Most cancer in firefighters is due to radio-frequency radiation exposure not inhaled carcinogens. Med Hypotheses. 2009;73:788–789. doi: 10.1016/j.mehy.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 841.Milham S. A cluster of male breast cancer in office workers. Am J Ind Med. 2004;46:86–87. doi: 10.1002/ajim.20027. [DOI] [PubMed] [Google Scholar]
- 842.Tynes T, Andersen A, Langmark F. Incidence of cancer in Norwegian workers potentially exposed to electromagnetic fields. Am J Epidemiol. 1992;136:81–88. doi: 10.1093/oxfordjournals.aje.a116423. [DOI] [PubMed] [Google Scholar]
- 843.Fedrowitz M, Kamino K, Löscher W. Significant differences in the effects of magnetic field exposure on 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis in two substrains of Sprague-Dawley rats. Cancer Res. 2004;64:243–251. doi: 10.1158/0008-5472.CAN-03-2808. [DOI] [PubMed] [Google Scholar]
- 844.Girgert R, Emons G, Hanf V, Gründker C. Exposure of mcf-7 breast cancer cells to electromagnetic fields up-regulates the plasminogen activator system. Int J Gynecol Cancer. 2009;19:334–338. doi: 10.1111/IGC.0b013e31819f53ec. [DOI] [PubMed] [Google Scholar]
- 845.Rudel RA, Ackerman JM, Attfield KR, Brody JG. New exposure biomarkers as tools for breast cancer epidemiology, biomonitoring, and prevention: A systematic approach based on animal evidence. Environ Health Perspect. 2014;122:881–895. doi: 10.1289/ehp.1307455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 846.Khoury MJ, Lam TK, Ionnidis JPA, Hartge P, Spitz MR, Buring JE, et al. Transforming epidemiology for 21st century medicine and public health. Cancer Epidemiol Biomark Prev. 2013;22:508–516. doi: 10.1158/1055-9965.EPI-13-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847.Osborne G, Rudel R, Schwarzmann Evaluating chemical effects on mammary gland development: A critical need in disease prevention. Reprod Toxicol. 2014;54:148–155. doi: 10.1016/j.reprotox.2014.07.077. [DOI] [PubMed] [Google Scholar]
- 848.Schwarzman MR, Ackerman JM, Dairkee SH, Fenton SE, Johnson D, et al. Screening for chemical contributions to breast cancer risk: A case study for chemical safety evaluation. Environ Health Perspect. 2015;123:1255–1264. doi: 10.1289/ehp.1408337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.