

Abstract
The survival rate for pancreatic ductal adenocarcinoma (PDAC) remains low. More therapeutic options to treat this disease are needed, for the current standard of care is ineffective. Using an animal model of aggressive PDAC (Kras/p48TGFβRIIKO), we discovered an effect of TGFβ signaling in regulation of G-CSF secretion in pancreatic epithelium. Elevated concentrations of G-CSF in PDAC promoted differentiation of Ly6G+ cells from progenitors, stimulated IL10 secretion from myeloid cells, and decreased T-cell proliferation via upregulation of Arg, iNOS, VEGF, IL6, IL1b from CD11b+ cells. Deletion of csf3 in PDAC cells or use of a G-CSF blocking antibody decreased tumor growth. Anti G-CSF treatment in combination with the DNA synthesis inhibitor gemcitabine reduced tumor size, increased the number of infiltrating T cells, and decreased the number of Ly6G+ cells more effectively than gemcitabine alone. Human Analysis of human datasets from The Cancer Genome Atlas and tissue microarrays (TMAs) correlated with observations from our mouse model experiments, especially in patients with grade I, stage II disease. We propose that in aggressive PDAC, elevated G-CSF contributes to tumor progression through promoting increases in infiltration of neutrophil-like cells with high immunosuppressive activity. Such a mechanism provides an avenue for a neoadjuvant therapeutic approach for this devastating disease.
Keywords: PDAC, G-CSF, immune response, p48/Kras/TGFβRII-KO mice
Introduction
Pancreatic ductal adenocarcinoma (PDAC), one of the most lethal types of cancers, is characterized by stromal expansion with marked fibrosis. We generated pancreatic epithelium–specific (p48 = Ptf1a) TGFβ receptor type II (TβRII) knockout mice in the context of activated Kras (Kras/p48TβRII-KO) and found that aggressive PDAC develops recapitulates the histological features of the human disease (1). These mice have shorter tumor latency and survival rates compared to Kras/p48 mice and other models such as Pdx/Smad4 or Pdx/p53 (KPC) (2,3). Abrogation of TGFβ signaling in Kras/p48 mice elevates epithelial STAT3 activity and correlates with decreased survival and advanced tumor stage in patients with PDAC. Treatment of mice with a STAT3 inhibitor and gemcitabine enhances drug delivery and therapeutic response through stroma remodeling without depletion of tumor stromal content (4,5). With STAT3 being a component of inflammatory signaling, the influence of the immune system on the development of these tumors could play a role in the progression of the disease. As such, these PDAC tissues show expression of CXCL 1, 2, and 5 chemokines, which are involved in the chemotaxis of myeloid cells (6). Treatment of these mice with a CXCR2 inhibitor decreases tumor progression, angiogenesis, and increases overall survival (6). In KPC mice, the combination of a CXCR2 inhibitor with a PD1-blocking antibody improved T-cell infiltration and extended survival (7). From this we can conclude that immune cell infiltration plays a role in tumor progression and also impedes antitumor immunity. Treatment based on stromal depletion in Kras/p48TβRII-KO mice resulted in invasive, undifferentiated tumors with enhanced hypoxia, EMT, and decreased animal survival. However, although these mice are not responsive to gemcitabine, anti-CTLA4 immunotherapy was effective at reducing tumor growth (8). Given the inefficacy of current immune modulating therapies, an in-depth understanding of immune interactions as well as the stromal context in these models could uncover therapeutic options.
The presence and function of myeloid cells in tumor tissue plays a role in regulation of an antitumorigenic immune response as well as drug resistance. Tumors developing in the Kras/p48TβRII-KO mice have an increased number of tumor-associated macrophages (TAMs) in the tumor tissue and have increased expression of CXCL1, 2, 5, and 16 chemokines, which affects the functions of immune cells (1,6). Accumulation of TAMs in pancreatic tumor tissue induces chemoresistance by reducing gemcitabine-induced apoptosis via upregulation of cytidine deaminase (9). Targeting TAMs and inflammatory monocytes by inhibiting either CSF1R or CCR2 decreases the number of tumor-initiating cells and improves chemotherapeutic response (10). Similarly, blocking myeloid growth factors such as M-CSF (orthotopic model) and GM-CSF (KPC mice) during pancreatic cancer development decreases tumor growth and improves responses to T-cell checkpoint immunotherapy (3,11). Such evidence supports the idea that tumor-associated macrophages represent an impediment towards effective antitumoral immune responses. Alternatively, targeted depletion of G-MDSCs by 1A8 Ab in KPC mice can increase intratumoral CD8+ T cells, apoptosis of tumor cells, and remodeling of the tumor stroma (12). In patients, pancreatic cancer converts CD14+ blood monocytes to monocyte-like myeloid derived suppressor cells (M-MDSC) by activation of the STAT3 pathway. These cells enhance stemness and mesenchymal properties of tumor tissue (13,14). Also, more granulocyte-like MDSCs (G-MDSCs), with high expression of arginase 1, but not M-MDSC were found in human pancreatic tumor tissue vs. circulating peripheral blood (15). As these cells affect T-cell activity, they could provide an additional layer of T-cell inhibition to be overcome for effective antitumorigenic immunity to occur. Given the ineffectiveness of checkpoint inhibitors in PDAC clinical trials, finding therapeutics to overcome these additional hurdles is imperative.
In the present study, we investigated the immune response during aggressive PDAC development. In Kras/p48 mice with abrogated TGFβ signaling (TβRII deletion), we determine that different subpopulations of immune cells influence tumor progression. We analyzed mechanisms involved in increasing the number of Ly6G+ cells and mechanisms regulating their immunosuppressive functions. By using csf3 shRNA and blocking antibody to G-CSF (anti–G-CSF), we demonstrate a positive therapeutic effect of blocking G-CSF in combination with gemcitabine. Analysis of human TCGA data sets and TMAs helped us to identify a group of patients with similar changes as we found in the mouse model.
Materials and Methods
Cell lines and mice
All studies were performed on Kras/p48 and Kras/p48TGFbRII-KO mice which were established and maintained as described (1,6). p48-Cre mice were used to delete TGFβ receptor II in the pancreatic epithelium and crossed with the KrasG12D mice to promote tumorigenesis. These mice have pure BALB/c backgrounds and have spontaneous tumor formation in the pancreas by 3-4–weeks-old. For additional experiments 8-10 week-old female C57BL/6 and BALB/c mice were purchased from Harlan Inc. (Indianapolis, IN).
Kras/p48 and Kras/p48TGFbRII-KO pancreatic tumor cell lines were derived from primary tumors of Kras/p48 and Kras/p48TGFbRII-KO mice, respectively, established and cultured in DMEM/10% FBS as previously described (1,6). These tumor cells were implanted to the pancreas (2.5×105/25 ul of collagen gel) or s.c. injected (5×105/100ul of PBS). For experiments using MDSCs (CD11b+Gr1+), 4T1 tumor cells (5×105/100ul of PBS) were s.c. injected to naïve BALB/c mice. The spleen of these mice was isolated on the 4th week after tumor injection for future MDSC isolation by magnetic microbeads (Miltenyi Biotec, San Diego, CA) (16). 4T1 breast cancer cell line (CRL-2539) was obtained from ATCC (Manassas, VA) and maintained following standard protocol. To examine the effect of neutralizing G-CSF on tumor growth, mice were i.p. injected with anti–G-CSF (R&D Systems, Inc., Minneapolis, MN) twice/week, 10-50 ug/mouse as previously described (17). Gemcitabine (EMD Millipore, Billerica, MA) was injected i.p. twice/week in 50 mg/kg (6). For CD4 and CD8 depletion experiment, CD4 Ab (clone GK1.5), CD8 Ab (clone 2.43), and a rat IgG2b isotype control (clone LTF-2), were purchased from BioXCell (West Lebanon, NH). Mice were injected i.p. once/week with 100 ug of Ab. The studies were approved by IACUC at Vanderbilt University Medical Center.
Flow Cytometry Analysis
To prepare single-cell suspensions, extracted tumors were chopped into small pieces, incubated in Dulbecco's modified Eagle's medium (DMEM) with 1 mg/ml collagenase P (Roche) for 30 min at 37°C, and then passed through a 70 μm cell strainer. After treatment with FcR Blocking Reagent (Miltenyi Biotec Inc., Auburn, CA), tumor single-cell suspensions (106 cells/ml) were labeled with specific antibody for 20 minutes on ice. All antibodies were obtained from Biolegend (San Diego, CA). The cells were analyzed on LSRII flow cytometry (Becton Dickinson) and the data were analyzed with FlowJo software. Nonviable cells were excluded by using DAPI. Cell sorting was performed on FACSAriaIII.
Magnetic Cell Separation
Tumor-infiltrating myeloid cells (CD11b+) and MDSCs (CD11b+Gr1+) were magnetically separated from tumor tissue of pancreas or spleen of tumor bearing mouse using CD11b or Gr1 magnetic microbeads following application protocols of the manufacturer (Miltenyi Biotec Inc., San Diego, CA).
Bone marrow isolation and differentiation
Bone marrow cells were harvested from the femurs of wild type mice. Hematopoietic progenitor cells (Lin−) were isolated using lineage cell depletion kit and LS columns from Miltenyi Biotec Inc. (San Diego, CA) according to the manufacturer's instructions. Hematopoietic progenitor cells were cultured on 24-well plates at 5 × 104 cells/mL concentration in RPMI medium containing 10% FBS, 20 mM HEPES, 50 μM 2-mercaptoethanol, 1× antibiotic-antimycotic solution (Sigma, St. Louis, MO) and supplemented with 25% conditioned media from pancreas carcinoma cells. For generation of dendritic cells (for MLR and IL10 studies), whole bone-marrow cells were cultured on 24-well plates at 5 × 105 cells/well concentration in RPMI medium containing 10% FBS, 20 mM Hepes, 50 μM 2-mercaptoethanol, 1× antibiotic-antimycotic solution (Sigma, St. Louis, MO) and supplemented with granulocyte-macrophage colony stimulating factor (GM-CSF; 20 ng/mL) and IL4 (10 ng/ml; both from R&D systems, Inc., Minneapolis, MN) for 5-6 days under humidified atmosphere of air/CO2 (19:1)at 37° C as previously described (18).
Functional T-cell assays
Dendritic cells were differentiated as described above. T cells from naïve BALB/c mice were isolated using R&D Systems Mouse T-cell Enrichment columns, according to the manufacturer's protocol. T cells were plated at 105 cells per well in 96 wells plate. Each well contained 100,000 T cells, 10,000 DCs, and various numbers of CD11b cells. Cells were incubated for 72 hours. 3[H]-thymidine was then added at 1μCi per 200uL of cells per well for an additional 18 hours followed by cell harvesting and radioactivity count using a liquid scintillation counter.
For non-specific T-cell stimulation, T cells from naïve BALB/c mice were isolated using R&D Systems Mouse T-cell Enrichment columns, according to the manufacturer's protocol, and mixed with CD3/CD28 Dynabeads (Invitrogen) according to the manufacturer's protocol and different number of CD11b+ cells. Cells were incubated for 72 hours. 3[H]-thymidine was then added at 1μCi per 200μL of cells per well for an additional 18 hours followed by cell harvesting and radioactivity count using a liquid scintillation counter.
Quantitative RT-PCR
Total RNA was extracted from sorted or cultured cells using an RNeasy Mini Kit (QIAGEN Inc., Valencia, CA). cDNA was synthesized using Invitrogen Superscript First-strand synthesis system for RT-PCR (Invitrogen Inc., Carlsbad, CA). Primers specific for IL4, IL10, IL6, IL2, IFNγ, TNFα, FoxP3, IL17a, Arg, iNOS, VEGF, MMP2, MMP9, G-CSF were used and relative gene expression was determined using ABI PRISM 7900HT Sequence Detection System (PE Applied Biosystems, Foster City, CA). The comparative threshold cycle method was used to calculate gene expression normalized to β-actin as a gene reference. Primer sequences are available upon request.
Tissue Microarray of Pancreatic Tissues
A tissue microarray (TMA) was purchased from US Biomax (Rockville, MD) and contained 10 normal pancreases, 11 chronic pancreatitis, 42 PDAC and 20 adjacent normal pancreas tissues. TMA slides were concurrently evaluated by Leica Biosystems software and by pathologist (C.S.) (4). Nuclear and cytoplasmic staining were scored as follows: the staining index was considered as the sum of the intensity score (0, no staining; 1+, weak; 2+, moderate; 3+, strong) and the distribution score (0, no staining; 1+, staining of <33% of cells; 2+, between 33% and 66% of cells; and 3+, staining of >66% of cells). Staining indices were classified as follows: 3+ or higher, strong staining; 1+ to 2+, weak staining; and 0, negative staining. CD15 and G-CSF were scored as positive if any detectable cytoplasmic staining was present.
ELISA and protein array
Cytokine levels in conditioned media of tumor cells, dendritic cells and shRNA transformed tumor cells were measured using the Mouse G-CSF and IL10 ELISA kits (R&D Systems, Minneapolis, MN) following the manufacturer's protocol. Proteome Profiler Mouse XL Cytokine Array with 111 different cytokine antibodies (R&D System) was used to measure proteins in conditioned media isolated CD11b cells from pancreas tumor tissue following the manufacturer's protocol.
Histology, IHC
Tissues were embedded directly in an optimal cutting temperature compound without fixation or placement in 10% formalin overnight, and then embedded in paraffin and sectioned at 5 μm. Sections were dewaxed in xylene and rehydrated in successive ethanol baths. For Immunohistochemistry (IHC), the MOM kit was used (Vector). Ki67, CD3, CD31, F4/80 and CD3 staining were performed in Translational Pathology Shared Resources (Vanderbilt University, Nashville). IHC sections were photographed using the AXIO Scope A1 ZEISS microscope and AxioVision 4.8 software. Human TMA slides were stained according manufacture protocol for CD15 (Abcam) and G-CSF (Abcam) and then were scanned by using Leica SCN400 Slide Scanner with 40× objective. Quantification of IHC was performed using NIH ImageJ (http://rsbweb.nih.gov/ij/docs/examples/stained-sections/index.html) as previously described (19).
ShRNA Knockdown
Prepackaged lentiviral transduction particles containing verified MISSION shRNA constructs targeting the coding domain sequence of G-CSF and MISSION Non-Target shRNA Control Transduction Particles in pLKO.1-puro plasmid vectors were purchased from Sigma-Aldrich, UK. Cells were seeded at a density of 1 × 103 per well in a sterile 96-well plate and incubated for 24 hours before addition of 2 multiplicity of infection of lentiviral particles. Cells were maintained in 2 μg/mL puromycin media. Knockdown levels were confirmed by ELISA.
Statistical Analysis
Data were presented as mean ± SEM. Multiple comparisons between treatment groups and control untreated group were performed using one-way ANOVA followed by Dunnett's procedure for multiplicity adjustment. Two-group comparison was performed using two-sample t tests. Correlation analyses for the gene expression between the signatures of interest were performed using data representing 185 patients from TCGA data sets. The gene expression was normalized across all arrays. Gene symbols were assigned using the manufacturer-provided annotation, but the analysis was performed at probe level. The association between RFS and Smad4 was analyzed using the univariate Cox proportional hazard regression model. When an interaction was identified, a Kaplan-Meier survival curve was created for the “high versus low” of the Smad4 gene expression by dichotoming all the samples among the subtype (Fig. 4). All tests were two-tailed.
Figure 4. Increased number of CD15+ cells and expression of G-CSF correlates with Grade 1/Stage II in human pancreatic cancer.
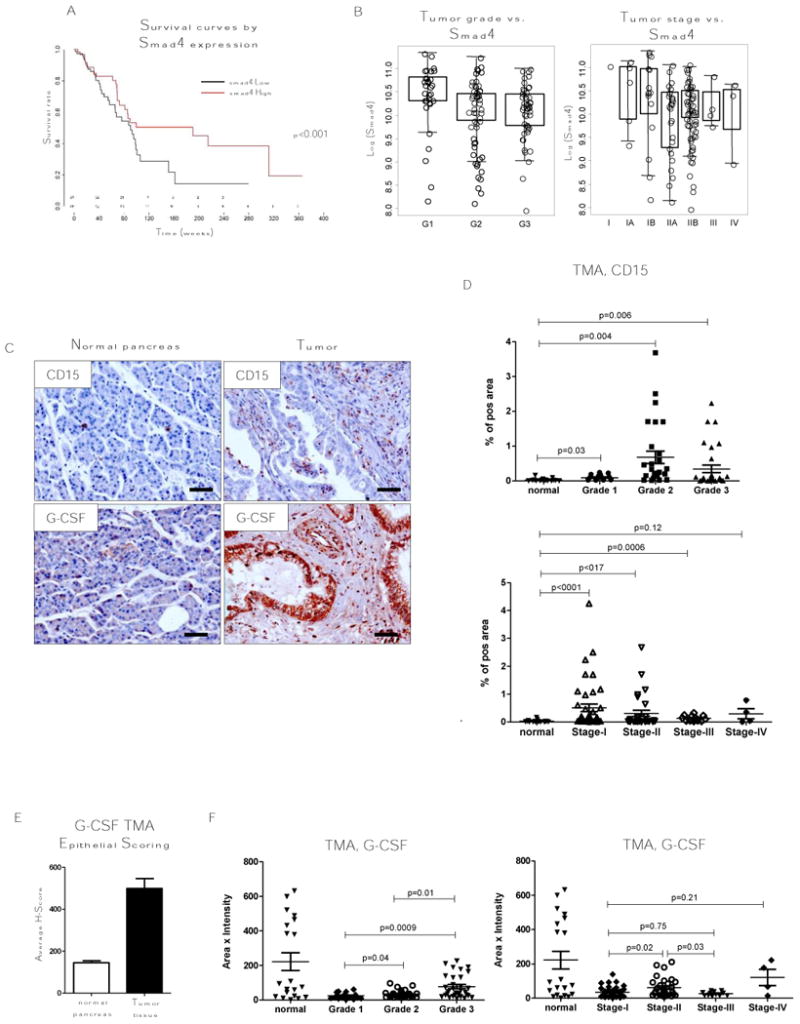
A, B, TCGA data set analysis of pancreatic cancer patient survival for Smad4 High and low expressing patients and analysis of Smad4 expression segregated by metastatic incidence, tumor grade, and tumor stage. C, Representative tissue section from TMA stained for CD15 and G-CSF by IHC. D, CD15 and F, G-CSF analysis of TMA staining grouped by grade and tumor stage. Normal, n = 17; tumor, n = 76; grade 1, n = 17; grade 2, n = 28; grade 3, n = 31; stage I, n = 43; stage II, n = 30; stage III, n = 12; stage IV, n = 4. E, Epithelial scoring of G-CSF in TMA used for C, D, F. Scale bars indicate 100 um.
Results
Tumor development correlates with excess G-CSF
Fibroblast function and mechanical properties influence Kras/p48TβRII-KO tumor progression (1,6,8). We sought to examine changes in immune infiltration and function during PDAC development, thus we performed flow cytometry analysis of pancreas tumor tissue from Kras/p48TβRII-KO animals in comparison with tissue from Kras/p48 mice. We observed an increase in the number of immune cells (CD45+) in pancreatic tissue of Kras/p48TβRII-KO vs. Kras/p48 and vs. normal tissue (Fig. 1A). After analyzing different subpopulations of immune cells, we found more CD11b+Gr1+ cells in pancreatic tissue, spleen, and bone marrow (Fig. 1B, C). There is a significant increase number of CD11b+Gr1+ cells in spleen of Kras/p48TβRII-KO mice vs. Kras/p48 and vs. control mice (P < 0.05) and also in Kras/p48 mice vs. control mice (P < 0.05). In the bone marrow, we found more of these cells only in Kras/p48TβRII-KO mice (P < 0.05). As Gr1 Ab recognizes both Ly6C and Ly6G proteins on the cell surface, we carried out additional flow cytometry analysis of the Gr1+ population of cells. We discovered an increased number of CD11b+Gr1high cells which represent Ly6G+ cells (neutrophils, G-MDSC). However, there was not an increase in Ly6C+ cells (Fig. 1D). Pancreatic carcinoma cell lines with deleted TβRII isolated from mice of mixed genetic background (C57BL/6, 129) have excess CXCL1/2/5/16, VCAM, and G-CSF (6). We validated these findings in our model using ELISA to measure levels of G-CSF in tissue and established carcinoma cell lines isolated from BALB/c animals. We observed increased G-CSF protein in tissue of Kras/p48TβRII-KO mice as well as in conditioned media from cultured pancreas carcinoma cell lines (Fig. 1E). To determine the functionality of this altered cytokine profile in promoting the differentiation of Ly6G+ cells, we supplemented progenitors (Lin- cells) derived from bone marrow with tumor-cell conditioned media during their differentiation. After 5 days of treatment, we found more myeloid (CD11b+) and Ly6G+ cells (Fig. 1F) if the differentiation was supplemented with Kras/p48TβRII-KO tumor-cell conditioned media than if the differentiation was supplemented with Kras/p48 cell conditioned media. Thus, TGFβ may regulate G-CSF in pancreas epithelium leading to accumulation of Ly6G+ cells during tumor progression.
Figure 1. Expansion of Ly6G+ cells in aggressive PDAC.
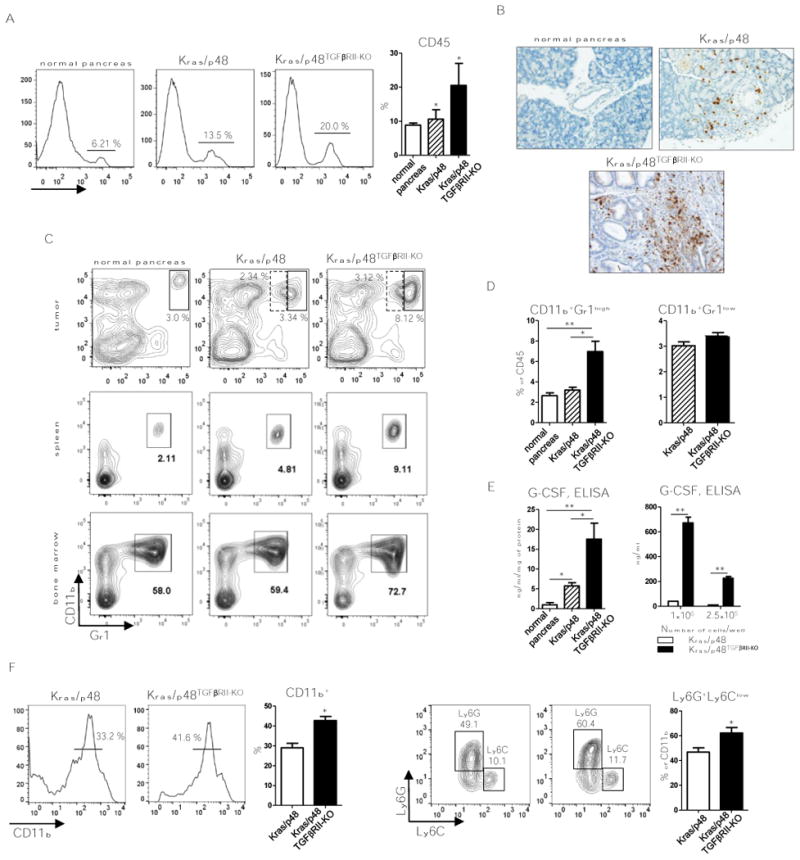
A, Flow cytometry analysis of CD45+ cells in normal, Kras/p48, and KRas/p48TGFβRII-KO tissue and percent to DAPI- live cells quantification. Plots are gated as DAPI-. 10 mice per group were analyzed.* P < 0.05. B, IHC for Gr1 (RB6-8C5) of normal pancreas, pancreas isolated from Kras/p48 mice and Kras/p48TGFβRII-KO mice at 5-6 weeks of age. C, representative flow cytometry plots of CD11b+Gr1+ cells in pancreatic tissue, spleen and bone marrow from normal, Kras/p48, and KRas/p48TGFβRII-KO mice. Plots are gated as CD45+DAPI-. 10 mice per group were analyzed. D, quantitative data of Gr1-high (solid line) and Gr1-low (dotted line) cells in tumor tissue showed on panel C, top. * P < 0.05, ** P < 0.01. 10 mice per group were analyzed. E, ELISA data of G-CSF levels in pancreatic tissue (left) and basal secretion in cultured cell lines (right). Data corresponds to the mean ± SEM of three individual mice from two experiments. Pancreatic cell lines from Kras/p48 mice and Kras/p48TGFβRII-KO mice were cultured in 6 wells plate in 3 ml of DMEM/10% FBS for 24 hr and level of G-CSF was measured in conditioned media. n = 3. * P < 0.05, ** P < 0.01. F, flow cytometry analysis of CD11b+ cells and Ly6G vs. Ly6C cells (gated as CD11b+) after differentiation of bone marrow progenitors (Lin- cells) for 5 days in presence of 25% conditioned media from Kras/p48 and Kras/p48TGFβRII-KO pancreatic carcinoma cell lines. Data correspond to the mean ± SEM of three individual mice from two experiments. * P < 0.05.
G-CSF upregulates immunosuppressive functions of myeloid cells
Ly6Ghigh cells isolated from tumors can be neutrophils or T-cell suppressive G-MDSCs (20). To analyze the functions of these infiltrating immune cells in PDAC tissue, we FACS-sorted various immune cell populations from tumor tissue to determine cellular function (Fig. 2A). qPCR analysis of sorted T cells (CD3+) showed shifts in gene expression toward a Th2 response through an increase in IL4, IL10, and IL6 expression (Fig. 2B). Markers of a Th1 response (IFNγ), Tregs (Foxp3) and Th17 (IL17) were not changed, but IL2 was downregulated (Fig. 2B). Analysis of sorted Ly6G+ cells showed increases in genes corresponding to M2-like/pro-tumorigenic responses – increased gene expression of iNOS, Arg, IL6, IL10, VEGF, and MMP9; however expression of MMP2 was decreased (Fig. 2C). Gene expression changes in Ly6C+ population of cells and Ly6C–Ly6G– (MF, DC) were similar to Ly6G+ cells.
Figure 2. Functions of myeloid cells.
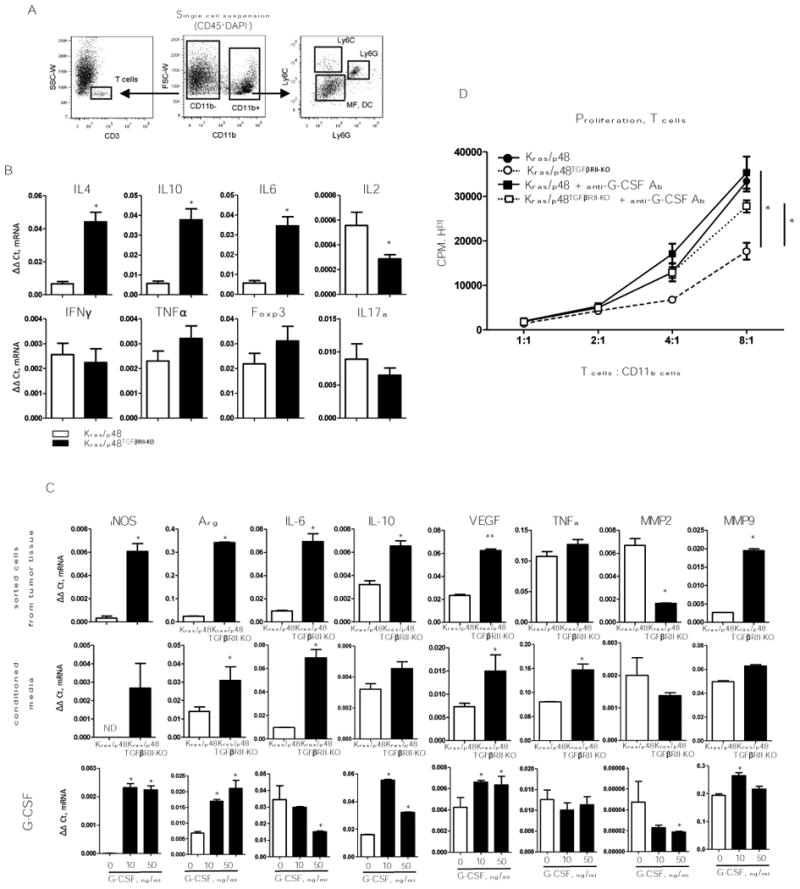
A, Cell sorting strategy. B, T-cell (CD3+) gene expression profile. Cells were sorted as shown in (A), top row: gene expression for Th2 cytokines presented as ΔΔCt normalized to Gapdh. Bottom row: gene expression for Th1 cytokines presented as ΔΔCt normalized to Gapdh. Data correspond to the mean ± SEM of three individual mice from two experiments. * P < 0.01. C, Gene expression analysis of pro-tumorigenic cytokines previously identified as cytokine mediators of MDSC immune suppression. top row: Gene expression measured by qPCR on sorted Ly6G; middle row: Gene expression analysis of Gr1+ cells, from the spleen of 4T1 tumor bearing mice, treated with conditioned media from Kras/p48 (open bars) or from Kras/p48TGFβRII-KO (closed bars) carcinoma cells. bottom row: Gene expression analysis of Gr1+ cells, from the spleen of 4T1 tumor-bearing mice, treated with G-CSF, 20 ng/ml for 6 hr. Data correspond to the mean ± SEM of three individual mice from two experiments. ** P < 0.05, * P < 0.01. D, T cells (CD3+) were isolated by negative selection from spleen of naïve BALB/c mice, mixed with CD11b cells isolated from pancreatic tissue by using CD11b magnetic microbeads (Miltenyi Biotec, San Diego, CA), anti–G-CSF was used in concentration 40 ng/ml. Data correspond to the mean ± SEM of three individual mice. * P < 0.05.
G-CSF was increased in pancreas tumor tissue and cultured pancreas carcinoma cell lines (Fig. 1). Increased tissue stiffness in Kras/p48TβRII-KO mice can also elevate amounts of G-CSF (21). We sought to address the functional role for G-CSF in activity of Ly6G+ cells. First, we used a model for enrichment and isolation of MDSCs (CD11b+Gr1+) – we implanted the 4T1 tumor-cell line into BALB/c mice and isolated Gr1+ cells from spleen of these tumor-bearing mice. Isolated MDSCs were incubated with conditioned media collected from either Kras/p48 or Kras/p48TβRII-KO pancreas carcinoma cells. Secreted factors Arg, iNOS, IL6 and VEGF were upregulated from the tumor cells when incubated with the G-CSF rich carcinoma cell conditioned media (Fig. 2C). However, no differences in IL10, MMP2, and MMP9 expression were found. Second, using the same source of MDSCs we incubated them with various concentration of G-CSF and found similar changes in Arg, iNOS, IL10, MMP2, MMP9 and VEGF expression (Fig. 2C). These data highlighted the potential influence of IL10, as G-CSF regulates Th1/Th2 cells and influences the alternative polarization of macrophages through upregulation of IL10 (22). We performed analysis of myeloid cell secretion of IL10 in response to conditioned media from carcinoma cells. To do this, we differentiated dendritic cells from bone marrow progenitors and incubated them with conditioned media collected from pancreatic carcinoma cell lines. We found that carcinoma cells by themselves do not produce IL10 (Supplementary Fig. S1A). However, conditioned medium from Kras/p48TβRII-KO cell line stimulated secretion of IL10 from myeloid cells. This effect is primarily caused by G-CSF, as adding anti–G-CSF to the medium blocked upregulation of IL10 (Supplementary Fig. S1A). No effect on stimulation of IL10 secretion was observed using conditioned media from Kras/p48 carcinoma cell lines.
To analyze the ability of myeloid cells from PDAC tissue to modulate T-cell proliferation, we stimulated T-cell proliferation with CD3/CD28 beads and mixed them with CD11b+ cells isolated from Kras/p48TβRII-KO and Kras/p48 tumors with and without blocking G-CSF Ab. Additionally, we stimulated T-cell proliferation with dendritic cells from a different mouse background (allogeneic MLR) and mixed in CD11b+ cells from the same tumors (Supplementary Fig. S1B). We discovered that myeloid cells (CD11b+) from Kras/p48TβRII-KO PDAC tissue were better suppressors T-cells proliferation than CD11b+ cells isolated from Kras/p48 mice (Fig. 2D) that were dependent on G-CSF. We conclude that increased immunosuppressive function of myeloid cells in our PDAC in model was caused by G-CSF stimulation.
Inhibition of G-CSF suppress pancreatic tumor growth
Anti-G-CSF treatments decrease tumor growth of orthotopic lymphoma and lung cancer models (23). Anti-GM-CSF limits tumor growth in KPC mice (3), a model of spontaneous PDA in which expression of oncogenic KrasG12D and mutant p53R172H is targeted to the pancreas by Cre recombinase under the control of the pancreas-specific promoter Pdx-1. Moreover, inhibition of macrophage accumulation in tumor tissue by inhibition of CSF1R and CCR2 decreases tumor-initiating cells and improves chemotherapeutic responses in PAN02, KCM and Kras-INK pancreas tumor models (10). We subcutaneously (s.c.) implanted Kras/p48 and Kras/p48TβRII-KO pancreas carcinoma cell lines and treated mice with anti–G-CSF (Fig. 3A). After 4 weeks of anti–G-CSF treatment the average Kras/p48TβRII-KO tumor weight from mice receiving blocking Ab were decreased compared to mice receiving control IgG Ab injections (Fig. 3B). Tumor size of Kras/p48 vs. Kras/p48TβRII-KO was lower, which correlates with our data from Kras/p48TβRII-KO mice with spontaneous formation of pancreatic tumors (1,6). This model showed no effect of G-CSF blocking Ab on tumor weight of Kras/p48 tumors. Corresponding with the tumor weight data, the number of CD11b+Ly6G+ cells was decreased In Kras/p48TβRII-KO mice. No differences were observed in Kras/p48 animals (Fig. 3E, D). IHC analysis of tumor tissue showed no differences in CD3 staining but less proliferation (Ki-67) and angiogenesis (CD31) in Kras/p48TβRII-KO mice with anti–G-CSF treatment compared to control group (Fig. 3C). Because of the magnitude of changes in tumor growth observed in the Kras/p48TβRII-KO compared to growth in the Kras/p48 mice and no differences in anti–G-CSF Ab treatment, we decided to perform the next experiments using only the Kras/p48TβRII-KO cell line. We found no difference in blocking effect of anti–G-CSF in the s.c. model or the orthotopic transplantation model (Supplementary Fig. S2A, B). Due to the stress placed on the mice during the orthotopic transplants as well as the immune response observed from the tissue manipulation, our subsequent experiments used only the s.c. tumor model.
Figure 3. Anti-G-CSF treatment.
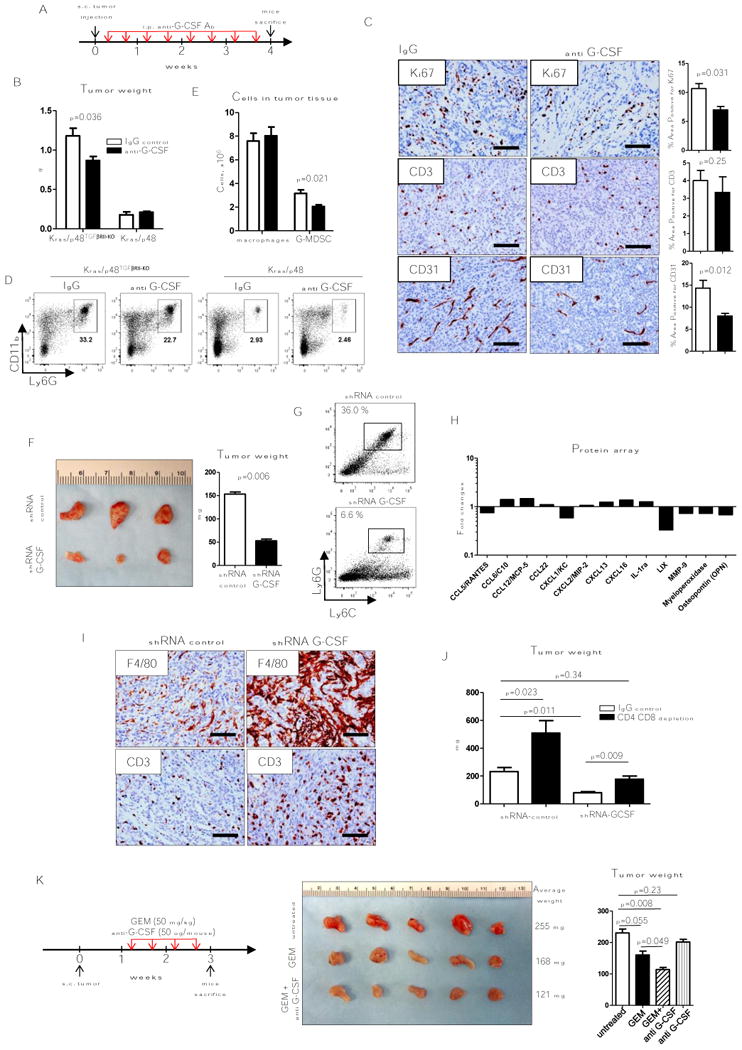
A, B, Schematic of treatment with either control (open bars) or anti G-CSF (10 ug/mouse, MAB414; closed bars) and KRas/p48TGFβRII-KO or Kras/p48, tumor weight 4 weeks after s.c. injection of cells (5×105), n = 5. C, IHC for Ki-67, CD3, and CD31 of tumor tissue isolated from mice treated IgG (control group, left column) and mice treated with anti G-CSF (right column) 4 weeks after tumor injection. D, Representative flow cytometry plots of tumor infiltrated immune cells. Cells are gated as CD45+. F, representative photograph of tumor tissue isolated from mice 3 weeks after s.c. injection of shRNA control (clone 1) and shRNA G-CSF (clone 1) carcinoma cells (left) and tumor weight (right), n = 5. G, Representative flow cytometry plots of Ly6G/Ly6C analysis of CD45+CD11b+ cells from tumor tissue shown in F. H, Cytokine array data analysis. CD11b+ cells were isolated from tumor tissue showed on F for collection of conditioned media. Data shows fold changes in shRNA G-CSF tumors vs. shRNA control. Graph shows only proteins with statistically significant changes from total 111 proteins (see details in Material and Methods section), n = 3. I, IHC for F4/80 and CD3 of tumors shown in F. J, Weight of tumor tissue isolated from mice 4 weeks after s.c. injection of shRNA control (clone 2) and shRNA G-CSF (clone 5) carcinoma cells treated in parallel with IgG control (open bars) or depletion due to use of CD4 and CD8 antibody (closed bars). K, Scheme of mice treatment with gemcitabine (GEM) and anti G-CSF. Tumor tissue isolated from mice 3 weeks after s.c. injection of Kras/p48TGFβRII-KO carcinoma cell line and treatment during last 2 weeks (left) and tumor weight (right), n = 5. Scale bars indicate 100 um.
G-CSF can be secreted by many types of cells, including myeloid cells, fibroblasts, and endothelial cells. To determine how important epithelial G-CSF secretion is, we used shRNA to target G-CSF in Kras/p48TGFβRIIKO cell lines (Supplementary Fig. S2C). Three weeks after tumor cells were s.c. injected, we found that the tumor weight was decreased in mice with G-CSF knockdown tumors (Fig. 3F). Using flow cytometry analysis of G-CSF–KO tumor tissue, we found Ly6G+Ly6Clow cells number were reduced by approximately 84% compared to control tumors (Fig. 3G). To determine if G-CSF knockdown also altered myeloid cell functions, we isolated CD11b+ cells from the implanted tumor tissue and measured concentrations of secreted proteins. We found that myeloid cells isolated from tumor tissue with knockdown of G-CSF produced less pro-tumorigenic proteins such as CXCL1 (KC), CXCL5 (LIX), MMP9, MPO, and CCL5 (RANTES) (Fig. 3H). IHC analysis showed more F4/80+ cells and more T cells (CD3+) in G-CSF knockdown tumor tissue (Fig. 3I). Based on in vitro immunosuppressive function of G-MDSC cells isolated from Kras/p48TβRII-KO mice indicated above (Fig. 2) we sought to establish the role of T cells during Kras/p48TβRII-KO tumor progression. We s.c. implanted Kras/p48TβRII-KO shRNA control and shRNA G-SCF pancreatic carcinoma cell lines into mice with depleted CD4 and CD8 cells (Fig. 3J, Supplementary Fig. S2D). We discovered that depletion of T cells reverted the growth inhibition effect in shRNA G-CSF cell lines demonstrating the immunosuppressive role of G-CSF in establishing a pro-tumorigenic microenvironment which promotes tumor progression.
Typical treatment of pancreatic cancer involves gemcitabine (24), even though this and other chemotherapies are not very effective and myeloid cells can mediate gemcitabine resistance (9). We sought to determine whether neoadjuvant myeloid depletion using anti–G-CSF would enhance the efficacy of gemcitabine. Thus, we subcutaneously implanted Kras/p48TGFβRIIKO cell lines and let the tumors grow for one week (Fig. 3K). A week after implantation, we began treatment with anti–G-CSF Ab and/or gemcitabine twice/week for the next 2 weeks. Mice were sacrificed 3 weeks after implantation. Tumor growth with gemcitabine alone or gemcitabine in combination with anti–G-CSF was decreased when compared to untreated mice. Tumor weight decreased more in mice receiving gemcitabine and anti–G-CSF than in mice receiving chemotherapy alone. Treatment with only anti–G-CSF did not affect tumor growth or tumor weight. From this we conclude that G-CSF inhibition during pancreatic tumor growth not only decreases the number of granulocytes and converts TAMs to an M1-like (antitumorigenic) type, but also increases effectiveness of chemotherapy.
G-CSF in human pancreatic cancers
To determine the relevance of these findings to human pancreatic cancer, we analyzed publicly available TCGA gene-expression profiles of pancreatic cancer tissues with clinical data related to metastasis, tumor stage and tumor grade over a 10-year period. As our model of PDAC showed that TGFβ signaling abrogation in pancreatic epithelial cells resulted in immune modulation, we sought to correlate expression of Smad4, a key gene of TGFβ signaling, with patient survival. We found that survival is decreased with low expression of Smad4 (Fig. 4A). Although no differences were found in patients with low vs. high metastasis, expression of Smad4 lowered as tumor stage and grade increased (Fig. 4B). Next, we used a TMA of human pancreatic tissue with 208 tissue cores (US Biomax, Inc) to determine differences in neutrophil infiltration. TMAs were stained for CD15, a marker of neutrophils, and for G-CSF (Fig. 4C). TMA IHC slides were validated by Leica software in parallel with a pathologist. We found that tumor tissue has a high number of CD15+ cells when compared to normal tissue, chronic pancreatitis (CP), or adjacent normal tissue (ANT). Segregating tumors by stage and grade, we found that tumors with grade 2 and 3 or stage II and III have the highest number of CD15+ cells (Fig. 4D). In the analysis of G-CSF, we found a lower level of this protein in tumor tissue compared to normal, CP, or ANT likely due to more fibrosis in tumor tissue and less epithelial cells which secrete G-CSF. However, epithelial scoring showed more G-CSF in tumor epithelium than in normal tissue (Fig. 4E). Analysis of different stages and grades showed us that tumors with grade 3, stage II have more G-CSF (Fig. 4F). Correlation analysis between CD15 and G-CSF was only in grade 1 tumors (r = 0.75, P = 0.0002, α = 0.05) and stage II tumors (r = 0.36, P = 0.049, α = 0.05) (Supplementary Fig. S3). These findings indicate that in human pancreatic tumors, as well as in mouse models, decreased TGFβ signaling was associated with increases in the number of neutrophils (CD15+ cells) and level of G-CSF in malignant pancreatic epithelium.
Our data suggest that downregulation of TGFβ signaling in human pancreatic cancer (usually through inactivating mutations in Smad4) is associated with decreased survival of patients and with increased G-CSF levels with accompanying increased number of CD15 cells which, based on our mouse model, have tumor-promoting properties. Collectively, our findings indicate a strategy for targeting pancreatic tumor growth by blocking G-CSF protein, especially in patients with grade 1/stage II tumors.
Discussion
Transforming growth factor-beta (TGFβ) signaling promotes PDAC progression, as indicated by the fact that Smad4, which encodes a signal mediator downstream from TGFβ, is deleted or mutated in 55% of patients (25) and low expression predicts poor patient survival (Fig. 4a). We generated an animal model of aggressive ductal carcinoma by deleting TGFβ receptor II (TβRII) in the pancreatic epithelium (p48) along with a KrasG12D mutation (1). These mice developed well-differentiated PDAC with 100% penetrance and a median survival of 59 d. The clinical and histopathological manifestations of the combined KrasG12D and deletion of TβRII in mice recapitulated human PDAC. Using the same mouse model in comparison with KPC mice, it was found that the loss of TGFβ signaling engages a positive feedback loop whereby epithelial STAT3 signaling modulates pancreatic cancer malignancy by increasing matricellular fibrosis and tissue tension (8). In PDAC patient biopsies, higher matricellular protein deposition and increased activated Stat3 is associated with SMAD4 mutation and shorter survival (5). Deletion of TβRII in pancreatic epithelium is associated with increased expression of CXCL1/5/16 chemokines, which in turn induce CTGF expression from CXCR2+ pancreatic stromal fibroblasts. Using a CXCR2 inhibitor reduces tumor progression in the Kras/p48TGFβRIIKO model (6). With such a robust change in inflammatory signaling and the efficacy of immune modulating CXCR2 treatments, using this model we could dissect the mechanisms by which tumors circumvent antitumoral immunity and develop strategies to combat them. Pancreatic cancer is associated with desmoplasia, which is a negative prognostic factor for patients (8). In Kras/p48TβRII-KO mice, depletion of fibroblasts in the context of pancreatic cancer led to invasive, undifferentiated tumors with diminished survival (6). Taken together, these data support the concept that alterations in TGFB signaling may be used as a determinant for differential therapeutic options. Given the stromal response observed in these models, targeting the altered cytokine profile resulting from attenuation of the signaling pathway could be efficacious.
STAT3 is the main transcription factor regulating the expansion of MDSCs (CD11b+Gr1+) in tumor tissue (26). Ablation of STAT3 expression in conditional STAT3 knockout mice or selective STAT3 inhibitors reduces the expansion of MDSCs and increases T-cell responses in tumor-bearing mice (26). Tumor-induced STAT3 activation in MDSCs enhances stemness and mesenchymal properties in human pancreatic cancer (13). MDSCs consist of two subsets of CD11b+Ly6G+Ly6Clow granulocytic and CD11b+Ly6G-Ly6Chigh monocytic cells. Almost all tumors demonstrate expansion of the granulocytic subset of MDSCs (27). In our study, we also found an increased number of granulocytic (Ly6G+) MDSCs in pancreatic tissue (Fig. 1) in parallel with increased secretion of G-CSF from the pancreatic epithelium with deleted TβRII when compared with cells with intact TGFβ signaling. In vitro studies demonstrated the ability of conditioned media from Kras/p48TGFβRIIKO carcinoma cells to increase differentiation of CD11b+ cells, especially the Ly6G+ subset. These cells in pancreatic tumors have increased expression of immune-suppressive genes and they suppress T-cell proliferation in vitro and elicit a polarized response toward pro-tumorigenic Th2 cells (Fig. 2). We found that secreted G-CSF from pancreatic epithelial cells upregulates secretion of IL10 from myeloid cells. This protein is an immunosuppressive cytokine with the ability to switch responsive myeloid cells from M1 to M2 and Th cells from Th1 to Th2 (28,29). Thus, the data presented here and by others suggests that inhibition of STAT3 signaling should be revisited, with patients identified as those who have abrogated TGFβ signaling and high levels of infiltrating myeloid cells. New treatments targeting MDSC differentiation through G-CSF could be utilized.
In the KPC mouse tumor model, (3) excess GM-CSF drives development of CD11b+Gr1+ cells with T-cell suppressive properties, but it is unknown what subset of MDSCs is responsible for these effects, how blocking of GM-CSF might affect standard chemotherapy, or how GM-CSF correlates with stage and grade of human tumors. Inhibition of GM-CSF decreases tumor growth and the number of CD11b+Gr1+ cells, and increases the number of cytotoxic CD8+ T cells (3,30). Targeted deletion of granulocytic MDSC in PDAC increased intratumoral accumulation of activated CD8+ T cells and apoptosis of tumor epithelial cells and also remodeling of the tumor stroma (12). However, suppression not only of Gr1+ cells but also monocytes and macrophages have positive antitumorigenic effects on pancreatic cancer. Targeting TAMs and monocytes by inhibiting CSF1R (M-CSF receptor) or CCR2 (CCL2/MCP-1 receptor) decreased a number of tumor-infiltrating cells in pancreatic tumors (14). These treatments also improve chemotherapeutic efficacy. CSF1R blockade also upregulates T-cells checkpoint proteins PDL1 and CTLA4 and treatment together with PDL1/CTLA4 antagonist reduces tumor growth (11). Blocking CSF1R also increases sensitivity of tumor cells to gemcitabine through elimination of cytidine deaminase that is secreted by TAMs, which causes resistance of tumor cells to chemotherapy (9). In our studies, we found that knockdown of G-CSF in pancreatic carcinoma cells reduced tumor growth in parallel with decreased number of Ly6G+ cells and increased number of T cells and antitumorigenic M1-like macrophages (F4/80+) based on protein array data. Combination treatment of gemcitabine with blocking G-CSF was more efficient in decreasing tumor growth compared to gemcitabine alone (Fig. 4). From this we conclude that anti–G-CSF provides an alternative means of neoadjuvantly increasing the efficacy of conventional chemotherapeutic interventions.
Patients with PDAC exhibit elevated level of MDSCs (CD33+HLA-DR-) upon progression of disease compared to healthy donors or patients with stable disease (31). MDSCs can be a marker of chemotherapy failure in pancreatic cancer patients (32). Circulating and tumor-infiltrating granulocytic (Lin-HLA-DR-CD33+CD15+), but not monocytic (Lin-HLA-DR-CD33+CD14+) MDSCs were detected in pancreatic cancer patients when compared to healthy donors and patients with chronic pancreatitis (15), which correlates with our mouse tumor model. Chemotherapy-derived inflammation accelerates the accumulation of MDSCs in human pancreatic cancer and blocking GM-CSF may benefit PDAC patients by downregulation of the immunosuppressive properties of MDSCs (20). PDAC is the fourth most common cause of cancer in North America and activating KRAS mutation and Smad4 loss occur in approximately 90% and 55% of PDAC, respectively. The combination of KRAS oncogene and TGFβ signaling alteration (via loss of Smad4) promotes malignant transformation of human pancreatic epithelial cells and metastasis (33). Our analysis of TCGA datasets showed no differences in Smad4 expression in patients with or without metastasis, but Smad4 is downregulated during tumor progression Smad4 mutation or loss associates with an increased number of CD15+ cells and G-CSF levels through attenuating TGFβ signaling as we discovered in our mouse model, especially in patients with Grade 1/Stage II tumor based on TMA analysis. Our data identify a cohort of patients most likely to benefit from anti–G-CSF treatment.
In summary, our results demonstrated that aggressive pancreatic adenocarcinoma tumors with deletion of TβRII in pancreatic epithelium had elevated expression of G-CSF. This resulted in an increase in the number of infiltrating CD11b+Ly6ClowLy6Ghigh cells into the tumor tissue. Also, G-CSF upregulated immunosuppressive genes such as iNOS, Arg, IL6, IL10, MMP9, and VEGF. These myeloid cells had an increased ability to suppress T-cell proliferation and as a result decreased the antitumor immune response (Fig. 4). Our studies provide insights into the development of aggressive ductal adenocarcinoma. Mutations in genes encoding TGFβ signaling molecules during PDAC development are very common, but manipulation of the TGFβ pathway during tumor progression remains controversial due to the dual roles of TGFβ on tumor progression. As G-CSF has protumorigenic effects on pancreatic cancer, the targeting of this cytokine could be considered when it is associated with mutations in TGFβ signaling components in patients with Grade 1/Stage II pancreatic cancer.
Supplementary Material
Figure 5. Schematic summarizing data and working hypotheses.

The following numbers refer to the number in the schematic. 1, Tgfbr2KO pancreatic epithelial cells secrete abundant CXCL1/CXCL5 due to lack of TGFβ suppression of chemokine expression (6). 2, The CXCL chemokines recruit to the tumor microenvironment myeloid cells (CD11b+). 3, In parallel with chemokines carcinoma cells secrete increased level of G-CSF. 4, G-CSF promotes differentiation of myeloid cells to Ly6G+ cells, upregulates immunosuppressive genes such as Arg, iNOS, VEGF and upregulate secretion of IL10 making (5) myeloid cells that are immunosuppressive with pro-tumorigenic properties. 6, The net result is enhanced tumor growth and decrease survival.
Acknowledgments
This work is supported by NIH grants CA200681 (to S.V. Novitskiy), CA151925 (to H. Moses) and the Robert J. and Helen C. Kleberg Foundation and the T.J. Martell Foundation to Hal Moses and CA068485 for core laboratory support.
Footnotes
The authors whose names are listed above have NO conflict of interest to report
References
- 1.Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20(22):3147–60. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herreros-Villanueva M, Hijona E, Cosme A, Bujanda L. Mouse models of pancreatic cancer. World J Gastroenterol. 2012;18(12):1286–94. doi: 10.3748/wjg.v18.i12.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology. 2015;149(7):1932–43 e9. doi: 10.1053/j.gastro.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016 doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B, Tada M, et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest. 2011;121(10):4106–17. doi: 10.1172/JCI42754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steele Colin W, K SA, Leach Joshua DG, Bailey Peter, Upstill-Goddard Rosanna, Rishi Loveena, Foth Mona, Bryson Sheila, McDaid Karen, Wilson Zena, Eberlein Catherine, Candido Juliana B, Clarke Mairi, Nixon Colin, Connelly John, Jamieson Nigel, Carter C Ross, Balkwill Frances, Chang David K, Evans TR Jeffry, Strathdee Douglas, Biankin Andrew V, Nibbs Robert JB, Barry Simon T, Sansom Owen J, Morton Jennifer P. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016 doi: 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–34. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene. 2014;33(29):3812–9. doi: 10.1038/onc.2013.357. [DOI] [PubMed] [Google Scholar]
- 10.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–41. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769–81. doi: 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panni RZ, Sanford DE, Belt BA, Mitchem JB, Worley LA, Goetz BD, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. 2014;63(5):513–28. doi: 10.1007/s00262-014-1527-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer research. 2013;73(10):3007–18. doi: 10.1158/0008-5472.CAN-12-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khaled YS, Ammori BJ, Elkord E. Increased levels of granulocytic myeloid-derived suppressor cells in peripheral blood and tumour tissue of pancreatic cancer patients. J Immunol Res. 2014;2014:879897. doi: 10.1155/2014/879897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106(16):6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16(8):880–6. doi: 10.1038/nm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pickup MW, Hover LD, Guo Y, Gorska AE, Chytil A, Novitskiy SV, et al. Deletion of the BMP receptor BMPR1a impairs mammary tumor formation and metastasis. Oncotarget. 2015;6(26):22890–904. doi: 10.18632/oncotarget.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol. 2012;91(1):167–81. doi: 10.1189/jlb.0311177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22(5):497–505. doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rutella S, Zavala F, Danese S, Kared H, Leone G. Granulocyte colony-stimulating factor: a novel mediator of T cell tolerance. J Immunol. 2005;175(11):7085–91. doi: 10.4049/jimmunol.175.11.7085. [DOI] [PubMed] [Google Scholar]
- 23.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106(16):6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.http://www.cancer.net/cancer-types/pancreatic-cancer/treatment-options
- 25.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosmann TR, Moore KW. The role of IL-10 in crossregulation of TH1 and TH2 responses. Immunol Today. 1991;12(3):A49–53. doi: 10.1016/S0167-5699(05)80015-5. [DOI] [PubMed] [Google Scholar]
- 30.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–47. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wachsmann MB, Pop LM, Vitetta ES. Pancreatic ductal adenocarcinoma: a review of immunologic aspects. J Investig Med. 2012;60(4):643–63. doi: 10.231/JIM.0b013e31824a4d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markowitz J, Brooks TR, Duggan MC, Paul BK, Pan X, Wei L, et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol Immunother. 2015;64(2):149–59. doi: 10.1007/s00262-014-1618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leung L, Radulovich N, Zhu CQ, Wang D, To C, Ibrahimov E, et al. Loss of canonical Smad4 signaling promotes KRAS driven malignant transformation of human pancreatic duct epithelial cells and metastasis. PLoS One. 2013;8(12):e84366. doi: 10.1371/journal.pone.0084366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.