

Abstract
The homeodomain family of transcription factors plays a fundamental role in a diverse set of functions that include body plan specification, pattern formation and cell fate determination during metazoan development. Members of this family are characterized by a helix–turn–helix DNA-binding motif known as the homeodomain. Homeodomain proteins regulate various cellular processes by specifically binding to the transcriptional control region of a target gene. These proteins have been conserved across a diverse range of species, from yeast to human. A number of inherited human disorders are caused by mutations in homeodomain-containing proteins. In this study, we present an evolutionary classification of 129 human homeodomain proteins. Phylogenetic analysis of these proteins, whose sequences were aligned based on the three-dimensional structure of the homeodomain, was performed using a distance matrix approach. The homeodomain proteins segregate into six distinct classes, and this classification is consistent with the known functional and structural characteristics of these proteins. An ancestral sequence signature that accurately describes the unique sequence characteristics of each of these classes has been derived. The phylogenetic analysis, coupled with the chromosomal localization of these genes, provides powerful clues as to how each of these classes arose from the ancestral homeodomain.
INTRODUCTION
The homeobox was first identified in 1984 as a conserved DNA sequence within the homeotic selector genes of Drosophila (1,2). The 180 bp homeobox region encodes a helix–turn–helix DNA-binding motif known as the homeodomain. Soon after the discovery of homeobox genes in Drosophila, homologous genes were also identified in yeast, mouse and human (for a review see 3). Homeodomain-containing proteins are transcription factors that regulate axial patterning, segment or cell identity and proliferation. A class of homeobox genes, the HOM-C complex in Drosophila, and their mammalian counterparts, the Hox genes, are organized in linked chromosomal clusters and show a striking colinearity in their 5′→3′ chromosomal position. They also show remarkably similar expression patterns along the anterior–posterior axis, determining the basic body plan during embryogenesis. In addition, homeobox genes appear to be either clustered or dispersed in the genome. The homeobox gene family is a large and diverse one, playing a fundamental role in metazoan development.
Homeodomain proteins regulate diverse developmental programs by modulating expression patterns of target genes in a temporal, spatial and tissue-specific manner (3). The DNA-binding mode of homeodomains has been extensively studied through both structural and site-directed mutagenesis experiments (4–12). X-ray crystallographic and NMR spectroscopic studies on several members of this family revealed that they contain three helical regions that are folded into a compact, globular structure having an N-terminal extension. Helices I and II lie parallel to each other and across from the third helix; this third helix is also known as the recognition helix. It has been shown that the third helix binds to the major groove of DNA, where it makes specific contacts with the DNA bases (13). The N-terminal arm also makes additional specific contacts to DNA bases in the adjacent minor groove. In addition, the conserved loop between helices I and II establishes specific contacts with the phosphate backbone. The third helix, in conjunction with the N-terminal arm, confers the DNA-binding specificity of individual homeodomain proteins. Amino acid conservation is highest within the recognition helix (14).
In previous analyses of homeodomain proteins, the family has been sub-classified into smaller groups (14). Within each sub-class, homeodomain proteins from a wide range of species show very high sequence conservation (80–100%) as well as conserved genetic function. Although the absolute sequence similarity varies among different groups, two positions in helix I (Leu 16 and Phe 20) and five positions in helix III (Trp 48, Phe 49, Asn 51, Arg 53 and Lys/Arg 55) are almost always conserved. A class of ‘atypical’ homeodomain proteins have also been identified; their primary sequence diverges considerably from the homeodomain consensus, having insertions or deletions within the 60 residue homeodomain motif. Despite this, X-ray crystallographic studies of the atypical MATα homeodomain revealed that it essentially forms the canonical homeodomain structural motif, even though it has three extra residues in the loop between the first and second α-helices (12). It has been shown that a wide range of sequence variations can be accommodated in formation of the homeodomain (15).
A number of earlier studies have addressed the evolutionary history of the homeodomain family (16–20). In most instances, the analysis was performed on a smaller number of sequences or on an individual class of homeodomains. Here, a near complete data set of all human homeodomain sequences has been analyzed. The purpose of this study was to rigorously derive a phylogenetic relationship among the human homeodomain proteins and to better understand the patterns of conservation and divergence among different classes. The sequences of 129 human homeodomain sequences have been compiled and aligned in accordance with thermodynamic results from homology model building experiments. Therefore, the alignment is based on the three-dimensional structure of the homeodomain, rather than using traditional sequence homology methods. This new structural alignment was then used to infer the clustering relationships between the human homeodomain proteins. The homeodomain proteins segregate into six distinct classes and this classification is consistent with the known functional and structural characteristics of these proteins. This new phylogenetic study provides a rational evolutionary framework with which to classify homeodomains and, more importantly, identify new members based on conserved sequence features, as more information becomes available from the human and other systematic sequencing projects.
MATERIALS AND METHODS
Homeodomain sequence extraction and alignment
The homeodomain sequences used in this study were compiled from various source databases using the PSI-BLAST algorithm (21), as well as from the literature. A PSI-BLAST search, using the sequence of HOXA1 (accession no. P49639) as the query, identified 427 entries from the non-redundant database at NCBI and 122 entries from the SWISS-PROT database. Duplicate sequences, sequence fragments and homeodomains formed at translocation break points were eliminated from the data set. A final data set of 129 human homeodomain proteins was selected for further analysis. A multiple sequence alignment of the homeodomain of these 129 proteins was made using CLUSTAL W (22). The resulting multiple sequence alignment was manually refined to reflect the thermodynamic information obtained from homology model building experiments (15). All accession numbers are given in Figure 1. The complete data set can be accessed through the World Wide Web at http://genome.nhgri.nih.gov/homeodomain/phylogeny.
Figure 1.

Multiple sequence alignment of the homeodomain classes. The abbreviations for each protein correspond to those in Table 1. The amino acid residues that are absolutely conserved across all homeodomain classes are shown in white with a blue background and the residues that are conservatively substituted are highlighted in yellow. For each of the homeodomain classes, the conserved residues are indicated by a downward arrow. The numbering scheme at the top of the figure refers to the amino acid positions within the 60 residue canonical homeodomain. The positions of the three α-helices found in the X-ray structure of engrailed (8) are shown above the figure. ALSCRIPT v.2.04 was used to format the alignment (85).
Phylogenetic analysis
Phylogenetic trees for the homeodomain family were constructed using algorithms contained within the PHYLIP Phylogeny Inference Package v.3.5c (23). PROTDIST was used on these 129 sequences to calculate a distance matrix according to the Dayhoff PAM probability model (24), as well as the categories model (23). The computed distances represent the expected fraction of amino acid substitutions between each pair of sequences. The distance matrix was then used to estimate phylogenies using the neighbor joining (NJ) method (25). Bootstrapping was carried out using SEQBOOT (1000 replicates for the PAM model of substitution and 100 replicates for the categories model of substitution). To independently confirm the results using alternative methodologies, a subset of 54 sequences was analyzed by both the NJ method applied as above on 1000 replicates as well as by the weighted least squares distance method of Fitch and Margoliash (23) on 100 replicates. All FITCH runs were performed with global rearrangements. CONSENSE was used to compute the consensus tree by the majority rule method. The final unrooted tree diagram was generated using TREETOOL (http://ftp.sunet.se/pub/Science/Molecular_Biology/unix/treetool/).
RESULTS AND DISCUSSION
Sequence alignment
All available human sequences belonging to the homeodomain family were compiled as described above. After elimination of partial, incomplete or artificial sequences, a final data set of 129 proteins were selected for further analysis (Table 1). Multiple sequence alignments of the homeodomain proteins were performed based on the results of threading experiments using the structure of engrailed (pdb|1HDD) as the template for structure prediction (data not shown; cf. 15). The method of alignment used here is significantly different from traditional methods based on sequence similarity scores. This method produced an alignment that was able to accommodate the atypical homeodomain proteins, placing their ‘additional’ amino acids within the loop region between helices II and III. The full alignment of 129 sequences can be found at http://genome.nhgri.nih.gov/homeodomains/phylogeny/. An alignment of a subset of the proteins analyzed (74 of 129), representing all the resulting classes within the homeodomain family, is shown in Figure 1. In this figure, the sequences are arranged by class to show the conserved features characteristic for each class (see below). This alignment is based on the experimentally determined structure of Drosophila engrailed and the positions of the α-helices determined from the crystal structure are shown above the alignment. It is obvious from this alignment that, while there may not be absolute identity at many positions among the sequences examined, there are discrete regions of high similarity present across all the individual classes. The extended N-terminal arm of the homeodomain is rich in basic amino acids, with most of the proteins having a hydrophobic residue at position 8 prior to the beginning of helix I. Specific positions within helix I (positions 16 and 20) and helix II (positions 34 and 40) are usually occupied by a hydrophobic amino acid. A number of positions that are absolutely conserved within the homedomain include Trp 48, Phe 49, Asn 51 and Arg 53, all of which are in helix III. The high conservation of these residues in the third helix has major implications for DNA binding and overall stability of the tertiary structure of the homeodomain. In the X-ray structure of engrailed, the third helix fits directly into the major groove, with these invariant residues lying closest to the DNA. The invariant hydrophobic residues (Trp 48 and Phe 49) maintain the hydrophobic core by establishing favorable interactions with hydrophobic amino acids in helices I and II, thereby stabilizing the three-helical bundle structure of the homeodomain (15). Asn 51 and Arg 53, along with several neighboring residues in helix III, make critical contacts with the DNA and constitute the DNA recognition face of the homeodomain. The major groove contacts made by Asn 51 and Arg 53 have been observed in all DNA-bound homeodomain structures solved so far (13), as well as in the atypical class of homeodomains where sequence conservation is quite low compared to the other classes (12). The residue at position 50, along with two others (positions 47 and 54) is critical for DNA-binding specificity of the homeodomain and serves to define the sub-classes of the homeodomain family (26).
Table 1. The homeodomain family of proteins.
 |
Phylogenetic analysis
The evolutionary relationships between 129 structurally aligned human homeodomain proteins were determined in this study. This data set included the atypical homeodomain proteins containing insertions within the canonical 60 residue homeodomain. The NJ algorithm from the PHYLIP Phylogeny Inference Package was used to perform this analysis, using the Dayhoff PAM distance matrix as the basis for calculations (24). The resulting unrooted tree is shown in Figure 2 (see the World Wide Web supplement at http://genome.nhgri.nih.gov/homeodomain/phylogeny for bootstrap support as described in the Materials and Methods). A different distance matrix obtained for this data set using the categories model of amino acid substitution (23) generated the same tree topology (data not shown). Again, it is immediately apparent that the data set segregates into six classes. Although this phylogenetic reconstruction was performed only with the DNA-binding portion of the homeodomain sequences, the classifications do indeed correspond to the known functions of each of the full-length proteins. This classification of homeodomain proteins was further confirmed by analysis of a subset of 54 sequences representing all the classes of this family. A NJ analysis of this smaller data set with 1000 replicates generated essentially the same tree topology with better bootstrap support (data not shown). A more robust analysis of these 54 sequences was performed using FITCH, a least squares distance method employing global rearrangement that conducts an exhaustive search for the best tree. Under this method, the sum of the branch lengths between any two species is expected to equal the distance between the species found in the calculated distance matrix. The consensus FITCH tree obtained from 100 bootstrap replicates confirms the branching pattern observed in the larger NJ tree (see World Wide Web supplement).
Figure 2.

Phylogenetic relationships between the members of the homeodomain protein family. Unrooted neighbor joining trees were generated using the PHYLIP Phylogeny Inference Package v.3.5c (23). TREETOOL, a freestanding tree editor and formatter, was used to generate the tree diagram. The homeodomain classes are color coded and the abbreviations used here correspond to those in the alignment presented in Figure 1.
HOX class. The largest number of sequences examined in this analysis belong to the HOX class. Members of the HOX class are homologous to the clustered homeobox genes found in the antennapedia and bithorax complexes (HOM-C) of Drosophila. The homeotic selector genes in HOM-C are involved in the specification of developmental fate of body segments in Drosophila. Mutations in these genes often cause transformation of one body structure into another, structures that should normally be located elsewhere (27,28). In human and mouse, there are multiple orthologs of Drosophila HOM-C genes that are organized into four linked clusters at four different chromosomal locations in the genome. These genes co-localize to a single location in Drosophila. The similarity between the HOM-C genes in Drosophila and the mammalian Hox genes is carried through to the level of function, with genes towards the 5′-end of the clusters being transcribed at more posterior positions along the body axis and at later times during embryogenesis. It has been hypothesized that the HOX cluster probably originated from tandem duplications resulting in an array of genes linked on a single chromosome in the ancestral metazoan, followed by cluster duplications and rearrangement in early vertebrate evolution (29). Cluster duplication most likely occurred as multiple distinct events, with the four cluster state arising in three sequential steps (30). The present repertoire of HOX genes in each cluster is unique, probably due to gene loss during chromosomal rearrangement.
In human, there are 39 HOX genes organized in four paralogous clusters (HOXA, HOXB, HOXC and HOXD) in the genome. The chromosomal locations of the human HOX clusters are shown in Figure 3. Each cluster contains a subset of 13 paralogous genes (numbered 1–13 from the 3′-end) defined on the basis of sequence similarity. The HOX genes within each cluster show higher sequence similarity to paralogous genes in other HOX clusters, as compared to linked genes within the same cluster (31). This observation would be expected from duplication of a single ancient cluster. The paralogous genes in the HOX clusters also show conservation with respect to the functional domains found along the body axis. The genes at the 3′-end function in the anterior or the head region, the middle genes function in the trunk region and the 5′-genes function in the posterior or tail region of the embryo (32). This division into functional domains is also observed in this phylogenetic analysis. The HOX class members analyzed here further segregate into four sub-classes corresponding to their serial position within each cluster. Paralogous HOX genes 1 and 2 form the anterior sub-class, HOX 3 genes form sub-class 3, HOX genes 4–8 form the middle sub-class and HOX genes 9–13 form the posterior sub-class (Fig. 2), reflecting their expression domain along the body axis.
Figure 3.

Sequence signature for each of the homeodomain classes. Signatures are based on the multiple sequence alignment shown in Figure 1 and indicate positions within the homeodomain that are absolutely conserved within each class. The sequence signatures presented here unambiguously identify members within each homeodomain class. The numbering scheme at the top of the figure refers to the amino acid positions within the 60 residue canonical homeodomain.
A multiple sequence alignment of the human HOX class shows several conserved residues, including Arg5 in the N-terminal arm; Gln 12, Glu 15, Leu 16, Glu 17 and Glu 19, within the first helix, are also conserved in all members of this family. The recognition helix for the HOX class is highly conserved, with a Gln residue at position 50, a major determinant of the DNA recognition site. The HOX class members exhibit very similar DNA-binding specificities due to high conservation of amino acids in the recognition helix (33). Additional specificity in DNA recognition for the HOX members is achieved through cooperative DNA binding with other proteins (34).
A number of limb- and digit-related disorders are caused by mutations in a HOX gene. Targeted disruption of the mouse orthologs of HOXA10 and HOXA11 has established that these genes play a role in the skeletal development of the forearm (35). Mutation in HOXA11 is responsible for a malformation of the forearm (radio-ulnar synostosis) associated with amegakaryocytic thrombocytopenia in humans (35–37). The only two other HOX genes implicated in human inherited disorders are HOXD13 in synpolydactyly (38) and HOXA13 in hand/foot/genital syndrome (39,40). The phenotypic skeletal defects in the distal extremities for HOXA13 and HOXD13 and in the forearm for HOXA11 reveal the functional domains for each of these HOX class members. More severe HOX-related human disorders have not been observed, probably due to lethality of the mutation.
Extended HOX. A number of sequences analyzed in this study are closely related to the individual HOX sub-classes, which are not part of any of the four HOX clusters. The CDX members (CDX1, CDX2 and CDX3) segregate with the posterior HOX sub-class (Fig. 2). The CDX genes, the vertebrate orthologs of the Drosophila caudal gene, are involved in specification of axial skeletal identities (41) and intestinal development (42). Another non-linked HOX-related protein, IPF1, segregates with HOX sub-class 3 members (Fig. 2). Mutations in IPF1 are involved in several disorders, including agenesis of the pancreas, congenital pancreatic hypoplasia and diabetes mellitus. The dilemma of a close evolutionary relationship of these dispersed genes with the HOX sub-classes was resolved by finding linkage of the orthologous cephalochordate Amphioxus genes in a novel cluster (17). It has been hypothesized that these non-linked HOX-like genes are constituents of the ParaHox cluster, an evolutionary sister of the HOX cluster, that originated by duplication of the primordial ProtoHox cluster (17,43).
Several other homeodomain proteins also show close evolutionary relationships with the HOX class (Fig. 2). These include the EVX, MOX, EMX, HLXB9, GBX, EN and HESX1 proteins. Some of these proteins, in addition to being related phylogenetically to the HOX class, exhibit genetic linkage to the HOX clusters as well. It has been shown that human EVX1 and EVX2 are each linked to the 5′-end of the HOXA and HOXD clusters (Fig. 4), respectively, suggesting that the EVX and HOX genes were linked in an ancient cluster before duplication (44,45). The chromosomal location of the human MOX genes also imply a similar situation: MOX1 is tightly linked to the HOXB cluster on chromosome 17 and MOX2 is close to the HOXA cluster on chromosome 7 (46).
Figure 4.

Chromosomal distribution of the human homeobox gene family. The homeobox genes are color coded corresponding to the phylogenetic classes shown in Figure 2. The chromosomal map positions of the homeobox genes were obtained from LocusLink (http://www.ncbi.nlm.nih.gov/LocusLink/index.html). The centromere is shown in dark blue. The four HOX clusters, HOXA, HOXB, HOXC and HOXD, are distributed on chromosomes 7, 17, 12 and 2, respectively.
A triad of HOX-related genes (HLXB9, GBX1 and En-2) shows tight linkage at 7q36 (Fig. 4). The paralogous genes EN-1 (2q13) and GBX2 (2q36–q37) map close to the HOXD cluster, hinting at a common ancestry in the expanding HOX cluster (47). The other HOX-related genes, EMX1 and EMX2, are not linked to any of the HOX clusters; they may have been transposed away from the expanding HOX cluster during chromosomal rearrangements.
NK class. In this phylogenetic analysis, the NK class is most closely related to the HOX class. It has been hypothesized that the ancient NK gene cluster was chromosomally linked to an ancient HOX cluster prior to genome duplication and rearrangement (46). The sequence signature for the NK class includes conserved residues that are needed to stabilize the three-helical bundle structure of the homeodomain, as well as other conserved positions likely to be needed for protein–DNA interactions (Fig. 3). This signature is most closely related to the paralogous HOX sub-class 1, having the critical phenylalanine at position 8 prior to the beginning of the first helix. All other HOX proteins contain a tyrosine residue at position 8. A sub-class of the NK class, consisting of NKX2.2, NKX2.8, CSX, TTF1, BAPX1 and NKX3.1, contain a tyrosine residue at position 54 in the third helix, specifying the divergent DNA recognition sequence for this sub-class (48).
Many members of the NK class are involved in specification of muscle cell lineage. A well-studied member of this class, CSX, is expressed only in the heart and is critical for cardiogenensis (49). Targeted disruption of the mouse homolog of CSX causes early embryonic lethality, with cardiac development arrested at the linear heart tube stage, prior to looping (50). Identification of human mutations in CSX that cause congenital heart disease elucidates gene defects that perturb later stages of cardiac development (51,52). Also, the CSX ortholog in Drosophila, tinman, is expressed in the developing dorsal vessel and in the equivalent of the vertebrate heart. Mutations in this gene result in loss of heart formation in the embryo, suggesting that tinman is essential for Drosophila heart formation (53). The critical role played by CSX and its mouse and Drosophila orthologs during cardiogenesis indicates functional conservation during metazoan evolution.
A group of homeodomain proteins closely related to the NK class constitutes the DL sub-class (Fig. 2). The DL sub-class consists of six DLX and two MSX members. The DLX and MSX genes are the vertebrate orthologs of the Drosophila Distal-less (Dll) and msh (muscle segment homeobox) genes, respectively. During mouse embryonic development, the dlx and msx genes are expressed in distinct and overlapping regions of the first and second branchial arches (54,55) that generate many structures, including bone, teeth and hair. Targeted deletion of mouse dlx and msx genes have revealed their importance in craniofacial and dental development (56,57). In humans, mutations in MSX1 cause selective tooth agenesis with or without orofacial clefting (58), while mutations in MSX2 are responsible for craniosynostosis manifested as premature closure of the cranial sutures (59) and skull ossification defects (60). Also, mutations in DLX3 are associated with tricho-dento-osseous syndrome, which is characterized by abnormal teeth, bone and hair development (61). The members of the DL sub-class play critical roles in the complex pathways involved in human craniofacial growth and development.
The DLX genes are organized in pairs and show an interesting association with the HOX clusters (Fig. 4). DLX1 and DLX2 are tightly linked to the HOXD cluster on chromosome 2 (62), while DLX3 and DLX4 are linked to the HOXB cluster on chromosome 17 (63,64). DLX5 and DLX6 and the HOXA cluster are all on chromosome 7, albeit not tightly linked (62). In this analysis, the DL sub-class is more closely related to the NK class than to the HOX class (Fig. 2). Furthermore, MSX1 is closely linked to two NK class members (HMX1 and BAPX1) on chromosome 4 and MSX2 is close to another NK class member (CSX) on chromosome 5. The chromosomal locations of the DL class members, together with its overall phylogenetic position (Fig. 2), lends support to the hypothesis that the NK class members were originally adjacent to the HOX cluster, from which the ancestral DL class arose (46).
Paired class. After the NK class, the Paired class appears most closely related to the HOX class based on this clustering analysis. Members of the Paired class show a high degree of sequence conservation, with 22 residues of the 60 residue homeodomain being absolutely conserved. The positions that are necessary to maintain the folded structure of the homeodomain (Phe8, Leu16, Phe20, Trp48 and Phe49) are all conserved in all members of this class. A conserved triad (Tyr–Phe–Asp) in the loop region between the first and the second helices is characteristic of this class. In addition, ∼50% of the residues in the third helix are identical in all members of this class. The Paired class sequence signature (Fig. 3) deduced from the sequence alignment recognizes proteins belonging to this class across diverse phyla.
Most members of this class are involved at different steps in eye morphogenesis; consequently, a number of human eye-related disorders are caused by mutations in a Paired class homeodomain gene. A list of human disorders caused by mutations in Paired class proteins is shown in Table 2. A subset of the Paired class, the PAX proteins, contains an additional DNA-binding domain, called the paired domain. It has been postulated that, during evolution, there was a homedomain-capturing event by an ancestral paired protein that generated the PAX class of transcription factors (16). The most extensively characterized member of the Paired class is PAX6; mutations in this protein cause aniridia, a severe eye disorder in humans (65). The finding that human aniridia, small eye of the mouse, and ey of Drosophila are encoded by orthologs of PAX6 suggests that eye organogenesis is under similar genetic control in both vertebrates and insects, in spite of the large differences in eye morphology and mode of development (66).
Table 2. Genomic disorders caused by mutations in a homeodomain protein.
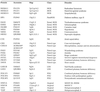 |
LIM class. Proteins belonging to the LIM class are characterized by an additional conserved region located N-terminal to the homeodomain (the lim domain). The lim domain contains a zinc-binding motif composed of 50–60 amino acid residues that form a pair of zinc fingers, separated by a linker region of two amino acids. This domain is recognized by a number of cofactors that mediate the function of the LIM class of transcription factors. The presence of this additional protein-binding domain further enhances the ability of LIM class factors to interact with other transcriptional modulators.
The LIM class shows overall lower sequence conservation in both the N-terminal extension and within the first α-helix. Although positions 8 (Phe, Leu or Ile), 16 (Met, Leu or Phe) and 20 (Phe or Tyr) are all occupied by hydrophobic amino acids, when considered as a group they are much more heterogeneous in nature compared to other homeodomain proteins. A triad of residues at positions 38–40 (Thr–Gly–Leu) defines this class; this particular triplet has not been observed in any other class within the homeodomain family. A sequence signature deduced for the human LIM class, based on this triad and other conserved residues in the third helix, uniquely identifies family members from diverse organisms, including nematodes, flies and vertebrates.
Comparison of the expression patterns and functions of LIM class homeodomain proteins reveals several interesting features that are conserved across most species. Several members of this class are involved in neuronal patterning and proliferation. Knockout studies in mice have shown that the mouse ortholog of human LHX3 is responsible for a critical step in pituitary cell fate commitment and regulates proliferation and differentiation of pituitary-specific cell lineages (67). Not surprisingly, mutations in LHX3 lead to combined pituitary hormone deficiency in humans (68). Another member of this class, LMX1B, is essential for dorsoventral patterning during limb development. Mutations in LMX1B cause the human disorder nail patella syndrome characterized by limb patterning defects and, in some cases, kidney malfunctions (69).
POU class. Among the typical homeodomain proteins, the POU class members are evolutionarily furthest from the HOX class proteins. Members of the POU class are characterized by a bipartite DNA-binding domain, consisting of an ∼75 residue long POU-specific domain that is tethered by a flexible linker to the homeodomain. High affinity, site-specific DNA binding by POU class transcription factors requires both the POU-specific domain and the POU homeodomain. The presence of two structurally independent DNA-binding sub-domains enables POU class members to adopt various monomer configurations on DNA, thereby conferring additional variability in target recognition. POU class members can also homodimerize or heterodimerize to further enhance their functional repertoire.
Although the POU class homeodomain sequences show ∼25% sequence identity with the HOX class members, several features are uniquely conserved within the POU class. In the N-terminal arm of the POU homeodomain, the first five residues are either lysine or arginine, followed by a conserved threonine residue. This region of the homeodomain fits in the minor grove of the target DNA and makes contact near the 5′-end of the binding site (70). The recognition helix of the POU class homeodomains is highly conserved, with a distinguishing cysteine residue at position 50 in all the members of this class. In the X-ray structure of the POU class member Oct-1, the sulfhydryl group of Cys 50 makes a hydrogen bond with the carbonyl oxygen of Arg 46 (70). Also, Cys 50 makes van der Waals contact with the target DNA sequence. A sequence signature based on the conserved residues of the human POU class proteins, including Cys 50, accurately identifies family members across diverse phyla.
Transcription factors belonging to the POU class have been shown to be important regulators of tissue-specific gene expression in a broad array of developmental systems, including lymphoid, endocrine and early mammalian neurogenesis. Mutations in the POU1F1 gene in human and in its mouse ortholog, Pit1, are responsible for combined pituitary hormone deficiency, manifested by pleiotropic deficiencies of the growth hormone prolactin and thyroid-stimulating hormone. Two other members of this class, POU4F3 and POU3F4, are involved in inner ear development. Mutations in POU4F3 are associated with inherited progressive hearing loss in human (71), whereas POU3F4 mutations cause human X-linked mixed deafness (72).
Atypical class. As its name suggests, this class contains the divergent members of the homeodomain family. Members of the atypical class show very low sequence identity with other homeodomain classes, except for the invariant residues Trp48, Phe49, Asn51 and Arg53 in the third helix. A proline residue is also conserved in the loop region between the first and second helices. A sub-class of the atypical homeodomains clustered in this analysis that contains three additional residues in the first loop region has been characterized as the TALE class in a previous analysis (73). X-ray crystallographic studies on a TALE sub-class member, the MATα protein from yeast, revealed that it essentially forms the canonical homeodomain structure. Another highly divergent member of the atypical class with 21 extra amino acids (HNF1) also forms the three-helical bundle homeodomain structure, with the additional residues accommodated in the loop region between helices II and III (4). It therefore appears that a variety of divergent sequences are still capable of forming the evolutionarily conserved homeodomain structure.
A number of inherited developmental disorders are caused by mutations in atypical class members. HNF1A and HNF1B are responsible for maturity onset diabetes of the young types III and V, respectively. The atypical class member SIX3 is essential for development of the anterior neural plate and eyes in humans and four different mutations in the homeodomain of SIX3 are associated with holoprosencephaly (74). The human SIX proteins are the orthologs of the Drosophila sine oculis (so) gene, which encodes a nuclear homeoprotein required for eye development. Another atypical class member TGIF is expressed during early brain development in mice (75) and four missense mutations in TGIF have also been identified in patients with holoprosencephaly (76).
Genomic perspective
The recent publication of the initial analysis of the human genome sequence (77) provides some interesting insights as to the evolutionary origin of the human genome in general, and to the origin of portions of the homeodomain family in particular. One interesting observation is the fact that the loci containing the four major homeodomain gene clusters on chromosomes 2, 7, 12 and 17 contain few to no repeat sequences. In fact, these regions of the human genome have the absolutely lowest density of repeats, with the interspersed repeats accounting for <2% of the sequence. It is speculated that the lack of repeats indicates that the region contains critical large-scale cis-regulatory elements that cannot be interrupted by insertions such as repeat sequences. This is consistent with the nature of the cellular roles played by the homeodomain proteins, particularly with respect to development.
Another interesting observation coming from the human genome sequence (77) regards the possible evolutionary origin of the four major clusters listed above, having to do with the concept of segmental duplication (78–80). This mode of duplication, where 10–300 kb of sequence can be moved from location to location within the genome, may account for interchromosomal duplications amongst non-homologous chromosomes. These events, which allow for copies of genes to undergo evolutionary drift and possibly acquire new functions, may explain the pattern of distribution of some of the homeobox genes as well as the array of functions that can be performed by homeodomain proteins.
One final speculation that has arisen from having the human genome sequence in hand concerns whole genome duplication, where part or all of an organism goes from being diploid to being tetraploid (77). This process is often cited as the underlying cause for large-scale evolutionary shifts (81–84). While there is no direct evidence in this study or elsewhere regarding the HOX genes specifically, it is interesting to speculate as to whether this process did, in fact, give rise to the four major HOX gene clusters seen on chromosomes 2, 7, 12 and 17 or whether the fact that there are four HOX gene clusters (mimicking the tetraploid state) is purely coincidental.
What is sure is that availability of the full, finished human genome sequence, along with that of closely related organisms, will allow a much more in-depth analysis of questions such as these, analyses which may shed some new light on the evolutionary development of the human species.
References
- 1.Scott M.P. and Weiner,A.J. (1984) Structural relationships among genes that control development: sequence homology between the Antennapedia, Ultrabithorax and fushi tarazu loci of Drosophila. Proc. Natl Acad. Sci. USA, 81, 4115–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGinnis W., Levine,M.S., Hafen,E., Kuroiwa,A. and Gehring,W.J. (1984) A conserved DNA sequence in homoeotic genes of the Drosophila Antennapedia and bithorax complexes. Nature, 308, 428–433. [DOI] [PubMed] [Google Scholar]
- 3.Gehring W.J., Affolter,M. and Burglin,T. (1994) Homeodomain proteins. Annu. Rev. Biochem., 63, 487–526. [DOI] [PubMed] [Google Scholar]
- 4.Ceska T., Lamers,M., Monaci,P., Nicosia,A., Cortese,R. and Suck,D. (1993) The X-ray structure of an atypical homeodomain present in the rat liver transcription factor LFB1/HNF1 and implications for DNA binding. EMBO J., 12, 1805–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dekker N., Cox,M., Boelens,R., Verrijzer,C., van der Vliet,P. and Kaptein,R. (1993) Solution structure of the POU-specific DNA-binding domain of Oct-1. Nature, 362, 852–855. [DOI] [PubMed] [Google Scholar]
- 6.Endo T., Ohta,K., Saito,T., Haraguchi,K., Nakazato,M., Kogai,T. and Onaya,T. (1994) Structure of the rat thyroid transcription factor-1 (TTF-1) gene. Biochem. Biophys. Res. Commun., 204, 1358–1363. [DOI] [PubMed] [Google Scholar]
- 7.Gruschus J., Tsao,D., Wang,L., Nirenberg,M. and Ferretti,J. (1997) Interactions of the vnd/NK-2 homeodomain with DNA by nuclear magnetic resonance spectroscopy: basis of binding specificity. Biochemistry, 36, 5372–5380. [DOI] [PubMed] [Google Scholar]
- 8.Kissinger C., Liu,B., Martin-Blanco,E., Kornberg,T. and Pabo,C. (1990) Crystal structure of an engrailed homeodomain-DNA complex at 2.8 Å resolution: a framework for understanding homeodomain-DNA interactions. Cell, 63, 579–590. [DOI] [PubMed] [Google Scholar]
- 9.Liu B., Kissinger,C. and Pabo,C. (1990) Crystallization and preliminary X-ray diffraction studies of the engrailed homeodomain and of an engrailed homeodomain/DNA complex. Biochem. Biophys. Res. Commun., 171, 257–259. [DOI] [PubMed] [Google Scholar]
- 10.Qian Y., Billeter,M., Otting,G., Muller,M., Gehring,W. and Wuthrich,K. (1989) The structure of the Antennapedia homeodomain determined by NMR spectroscopy in solution: comparison with prokaryotic repressors. Cell, 59, 573–580. [DOI] [PubMed] [Google Scholar]
- 11.Qian Y., Furukubo-Tokunaga,K., Resendez-Perez,D., Muller,M., Gehring,W. and Wuthrich,K. (1994) Nuclear magnetic resonance solution structure of the fushi tarazu homeodomain from Drosophila and comparison with the Antennapedia homeodomain. J. Mol. Biol., 238, 333–345. [DOI] [PubMed] [Google Scholar]
- 12.Wolberger C., Vershon,A.K., Liu,B., Johnson,A.D. and Pabo,C.O. (1991) Crystal structure of a MAT alpha 2 homeodomain-operator complex suggests a general model for homeodomain-DNA interactions. Cell, 67, 517–528. [DOI] [PubMed] [Google Scholar]
- 13.Gehring W.J., Qian,Y.Q., Billeter,M., Furukubo-Tokunaga,K., Schier,A.F., Resendez-Perez,D., Affolter,M., Otting,G. and Wuthrich,K. (1994) Homeodomain-DNA recognition. Cell, 78, 211–223. [DOI] [PubMed] [Google Scholar]
- 14.Burglin T. (1994) Comprehensive classification of homeobox genes. In Duboule,D. (ed.), Guidebook to the Homeobox Genes. Oxford University Press, Oxford, UK.
- 15.Banerjee-Basu S., Landsman,D. and Baxevanis,A.D. (1999) Threading analysis of Prospero-type homeodomains. In Silico Biol., 1, 163–173. [PubMed] [Google Scholar]
- 16.Breitling R. and Gerber,J.K. (2000) Origin of the paired domain. Dev. Genes Evol., 210, 644–650. [DOI] [PubMed] [Google Scholar]
- 17.Brooke N.M., Garcia-Fernandez,J. and Holland,P.W. (1998) The ParaHox gene cluster is an evolutionary sister of the Hox gene cluster. Nature, 392, 920–922. [DOI] [PubMed] [Google Scholar]
- 18.Davidson D. (1995) The function and evolution of Msx genes: pointers and paradoxes. Trends Genet., 11, 405–411. [DOI] [PubMed] [Google Scholar]
- 19.Ferrier D.E.K. and Holland,P.W. (2001) Ancient origin of the HOX gene cluster. Nature Rev., 2, 33. [DOI] [PubMed] [Google Scholar]
- 20.Kappen C. (2000) Analysis of a complete homeobox gene repertoire: implications for the evolution of diversity. Proc. Natl Acad. Sci. USA, 97, 4481–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Felsenstein J. (1993) PHYLIP Phylogeny Inference Package 3.5. Department of Genetics, The University of Washington, Seattle, WA.
- 24.Dayhoff M. (1978) Atlas of Protein Sequence and Structure. National Biomedical Research Foundation, Washington, DC.
- 25.Saitou N. and Nei,M. (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4, 406–425. [DOI] [PubMed] [Google Scholar]
- 26.Tucker-Kellogg L., Rould,M.A., Chambers,K.A., Ades,S.E., Sauer,R.T. and Pabo,C.O. (1997) Engrailed (Gln50→Lys) homeodomain-DNA complex at 1.9 Å resolution: structural basis for enhanced affinity and altered specificity. Structure, 5, 1047–1054. [DOI] [PubMed] [Google Scholar]
- 27.Ouweneel W.J. (1976) Developmental genetics of homoeosis. Adv. Genet., 18, 179–248. [DOI] [PubMed] [Google Scholar]
- 28.Lewis E.B. (1978) A gene complex controlling segmentation in Drosophila. Nature, 276, 565–570. [DOI] [PubMed] [Google Scholar]
- 29.Kappen C., Schughart,K. and Ruddle,F.H. (1989) Two steps in the evolution of Antennapedia-class vertebrate homeobox genes. Proc. Natl Acad. Sci. USA, 86, 5459–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey W.J., Kim,J., Wagner,G.P. and Ruddle,F.H. (1997) Phylogenetic reconstruction of vertebrate Hox cluster duplications. Mol. Biol. Evol., 14, 843–853. [DOI] [PubMed] [Google Scholar]
- 31.Scott M.P. (1993) A rational nomenclature for vertebrate homeobox (HOX) genes. Nucleic Acids Res., 21, 1687–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duboule D. and Dolle,P. (1989) The structural and functional organization of the murine HOX gene family resembles that of Drosophila homeotic genes. EMBO J., 8, 1497–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laughon A. (1991) DNA binding specificity of homeodomains. Biochemistry, 30, 11357–11367. [DOI] [PubMed] [Google Scholar]
- 34.Piper D.E., Batchelor,A.H., Chang,C.P., Cleary,M.L. and Wolberger,C. (1999) Structure of a HoxB1-Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell, 96, 587–597. [DOI] [PubMed] [Google Scholar]
- 35.Davis A.P., Witte,D.P., Hsieh-Li,H.M., Potter,S.S. and Capecchi,M.R. (1995) Absence of radius and ulna in mice lacking hoxa-11 and hoxd-11. Nature, 375, 791–795. [DOI] [PubMed] [Google Scholar]
- 36.Thompson A.A. and Nguyen,L.T. (2000) Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nature Genet., 26, 397–398. [DOI] [PubMed] [Google Scholar]
- 37.Davis A.P. and Capecchi,M.R. (1994) Axial homeosis and appendicular skeleton defects in mice with a targeted disruption of hoxd-11. Development, 120, 2187–2198. [DOI] [PubMed] [Google Scholar]
- 38.Goodman F., Giovannucci-Uzielli,M.L., Hall,C., Reardon,W., Winter,R. and Scambler,P. (1998) Deletions in HOXD13 segregate with an identical, novel foot malformation in two unrelated families. Am. J. Hum. Genet., 63, 992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortlock D.P. and Innis,J.W. (1997) Mutation of HOXA13 in hand-foot-genital syndrome. Nature Genet., 15, 179–180. [DOI] [PubMed] [Google Scholar]
- 40.Goodman F.R., Bacchelli,C., Brady,A.F., Brueton,L.A., Fryns,J.P., Mortlock,D.P., Innis,J.W., Holmes,L.B., Donnenfeld,A.E., Feingold,M., Beemer,F.A., Hennekam,R.C. and Scambler,P.J. (2000) Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am. J. Hum. Genet., 67, 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian V., Meyer,B.I. and Gruss,P. (1995) Disruption of the murine homeobox gene Cdx1 affects axial skeletal identities by altering the mesodermal expression domains of Hox genes. Cell, 83, 641–653. [DOI] [PubMed] [Google Scholar]
- 42.Silberg D.G., Swain,G.P., Suh,E.R. and Traber,P.G. (2000) Cdx1 and cdx2 expression during intestinal development. Gastroenterology, 119, 961–971. [DOI] [PubMed] [Google Scholar]
- 43.Kourakis M.J. and Martindale,M.Q. (2000) Combined-method phylogenetic analysis of Hox and ParaHox genes of the metazoa. J. Exp. Zool., 288, 175–191. [DOI] [PubMed] [Google Scholar]
- 44.Faiella A., D’Esposito,M., Rambaldi,M., Acampora,D., Balsofiore,S., Stornaiuolo,A., Mallamaci,A., Migliaccio,E., Gulisano,M. and Simeone,A. (1991) Isolation and mapping of EVX1, a human homeobox gene homologous to even-skipped, localized at the 5′ end of HOX1 locus on chromosome 7. Nucleic Acids Res., 19, 6541–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Esposito M., Morelli,F., Acampora,D., Migliaccio,E., Simeone,A. and Boncinelli,E. (1991) EVX2, a human homeobox gene homologous to the even-skipped segmentation gene, is localized at the 5′ end of HOX4 locus on chromosome 2. Genomics, 10, 43–50. [DOI] [PubMed] [Google Scholar]
- 46.Pollard S.L. and Holland,P.W. (2000) Evidence for 14 homeobox gene clusters in human genome ancestry. Curr. Biol., 10, 1059–1062. [DOI] [PubMed] [Google Scholar]
- 47.Matsui T., Hirai,M., Hirano,M. and Kurosawa,Y. (1993) The HOX complex neighbored by the EVX gene, as well as two other homeobox-containing genes, the GBX-class and the EN-class, are located on the same chromosomes 2 and 7 in humans. FEBS Lett., 336, 107–110. [DOI] [PubMed] [Google Scholar]
- 48.Xiang B., Weiler,S., Nirenberg,M. and Ferretti,J.A. (1998) Structural basis of an embryonically lethal single Ala → Thr mutation in the vnd/NK-2 homeodomain. Proc. Natl Acad. Sci. USA, 95, 7412–7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanaka M., Chen,Z., Bartunkova,S., Yamasaki,N. and Izumo,S. (1999) The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development, 126, 1269–1280. [DOI] [PubMed] [Google Scholar]
- 50.Tanaka M., Kasahara,H., Bartunkova,S., Schinke,M., Komuro,I., Inagaki,H., Lee,Y., Lyons,G.E. and Izumo,S. (1998) Vertebrate homologs of tinman and bagpipe: roles of the homeobox genes in cardiovascular development. Dev. Genet., 22, 239–249. [DOI] [PubMed] [Google Scholar]
- 51.Schott J.J., Benson,D.W., Basson,C.T., Pease,W., Silberbach,G.M., Moak,J.P., Maron,B.J., Seidman,C.E. and Seidman,J.G. (1998) Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science, 281, 108–111. [DOI] [PubMed] [Google Scholar]
- 52.Zhu W., Shiojima,I., Hiroi,Y., Zou,Y., Akazawa,H., Mizukami,M., Toko,H., Yazaki,Y., Nagai,R. and Komuro,I. (2000) Functional analyses of three Csx/Nkx-2.5 mutations that cause human congenital heart disease. J. Biol. Chem., 275, 35291–35296. [DOI] [PubMed] [Google Scholar]
- 53.Bodmer R. (1993) The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development, 118, 719–729. [DOI] [PubMed] [Google Scholar]
- 54.Jowett A.K., Vainio,S., Ferguson,M.W., Sharpe,P.T. and Thesleff,I. (1993) Epithelial-mesenchymal interactions are required for msx 1 and msx 2 gene expression in the developing murine molar tooth. Development, 117, 461–470. [DOI] [PubMed] [Google Scholar]
- 55.Qiu M., Bulfone,A., Ghattas,I., Meneses,J.J., Christensen,L., Sharpe,P.T., Presley,R., Pedersen,R.A. and Rubenstein,J.L. (1997) Role of the Dlx homeobox genes in proximodistal patterning of the branchial arches: mutations of Dlx-1, Dlx-2 and Dlx-1 and -2 alter morphogenesis of proximal skeletal and soft tissue structures derived from the first and second arches. Dev. Biol., 185, 165–184. [DOI] [PubMed] [Google Scholar]
- 56.Satokata I., Ma,L., Ohshima,H., Bei,M., Woo,I., Nishizawa,K., Maeda,T., Takano,Y., Uchiyama,M., Heaney,S., Peters,H., Tang,Z., Maxson,R. and Maas,R. (2000) Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nature Genet., 24, 391–395. [DOI] [PubMed] [Google Scholar]
- 57.Satokata I. and Maas,R. (1994) Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nature Genet., 6, 348–356. [DOI] [PubMed] [Google Scholar]
- 58.van den Boogaard M.J., Dorland,M., Beemer,F.A. and van Amstel,H.K. (2000) MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nature Genet., 24, 342–343. [DOI] [PubMed] [Google Scholar]
- 59.Jabs E.W., Muller,U., Li,X., Ma,L., Luo,W., Haworth,I.S., Klisak,I., Sparkes,R., Warman,M.L., Mulliken,J.B. et al. (1993) A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell, 75, 443–450. [DOI] [PubMed] [Google Scholar]
- 60.Wilkie A.O., Tang,Z., Elanko,N., Walsh,S., Twigg,S.R., Hurst,J.A., Wall,S.A., Chrzanowska,K.H. and Maxson,R.E. (2000) Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nature Genet., 24, 387–390. [DOI] [PubMed] [Google Scholar]
- 61.Price J.A., Bowden,D.W., Wright,J.T., Pettenati,M.J. and Hart,T.C. (1998) Identification of a mutation in DLX3 associated with tricho- dento-osseous (TDO) syndrome. Hum. Mol. Genet., 7, 563–569. [DOI] [PubMed] [Google Scholar]
- 62.Simeone A., Acampora,D., Pannese,M., D’Esposito,M., Stornaiuolo,A., Gulisano,M., Mallamaci,A., Kastury,K., Druck,T., Huebner,K. et al. (1994) Cloning and characterization of two members of the vertebrate Dlx gene family. Proc. Natl Acad. Sci. USA, 91, 2250–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scherer S.W., Heng,H.H., Robinson,G.W., Mahon,K.A., Evans,J.P. and Tsui,L.C. (1995) Assignment of the human homolog of mouse Dlx3 to chromosome 17q21.3-q22 by analysis of somatic cell hybrids and fluorescence in situ hybridization. Mamm. Genome, 6, 310–311. [DOI] [PubMed] [Google Scholar]
- 64.Nakamura S., Stock,D.W., Wydner,K.L., Bollekens,J.A., Takeshita,K., Nagai,B.M., Chiba,S., Kitamura,T., Freeland,T.M., Zhao,Z., Minowada,J., Lawrence,J.B., Weiss,K.M. and Ruddle,F.H. (1996) Genomic analysis of a new mammalian distal-less gene: Dlx7. Genomics, 38, 314–324. [DOI] [PubMed] [Google Scholar]
- 65.Hanson I.M., Fletcher,J.M., Jordan,T., Brown,A., Taylor,D., Adams,R.J., Punnett,H.H. and van Heyningen,V. (1994) Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nature Genet., 6, 168–173. [DOI] [PubMed] [Google Scholar]
- 66.Wawersik S., Purcell,P. and Maas,R.L. (2000) Pax6 and the genetic control of early eye development. Results Probl. Cell Differ., 31, 15–36. [DOI] [PubMed] [Google Scholar]
- 67.Sheng H.Z., Zhadanov,A.B., Mosinger,B.,Jr, Fujii,T., Bertuzzi,S., Grinberg,A., Lee,E.J., Huang,S.P., Mahon,K.A. and Westphal,H. (1996) Specification of pituitary cell lineages by the LIM homeobox gene Lhx3. Science, 272, 1004–1007. [DOI] [PubMed] [Google Scholar]
- 68.Netchine I., Sobrier,M.L., Krude,H., Schnabel,D., Maghnie,M., Marcos,E., Duriez,B., Cacheux,V., Moers,A., Goossens,M., Gruters,A. and Amselem,S. (2000) Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nature Genet., 25, 182–186. [DOI] [PubMed] [Google Scholar]
- 69.Dreyer S.D., Zhou,G., Baldini,A., Winterpacht,A., Zabel,B., Cole,W., Johnson,R.L. and Lee,B. (1998) Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nature Genet., 19, 47–50. [DOI] [PubMed] [Google Scholar]
- 70.Klemm J.D., Rould,M.A., Aurora,R., Herr,W. and Pabo,C.O. (1994) Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA-binding modules. Cell, 77, 21–32. [DOI] [PubMed] [Google Scholar]
- 71.Vahava O., Morell,R., Lynch,E.D., Weiss,S., Kagan,M.E., Ahituv,N., Morrow,J.E., Lee,M.K., Skvorak,A.B., Morton,C.C., Blumenfeld,A., Frydman,M., Friedman,T.B., King,M.C. and Avraham,K.B. (1998) Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science, 279, 1950–1954. [DOI] [PubMed] [Google Scholar]
- 72.de Kok Y.J., van der Maarel,S.M., Bitner-Glindzicz,M., Huber,I., Monaco,A.P., Malcolm,S., Pembrey,M.E., Ropers,H.H. and Cremers,F.P. (1995) Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science, 267, 685–688. [DOI] [PubMed] [Google Scholar]
- 73.Burglin T.R. (1997) Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res., 25, 4173–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wallis D.E., Roessler,E., Hehr,U., Nanni,L., Wiltshire,T., Richieri-Costa,A., Gillessen-Kaesbach,G., Zackai,E.H., Rommens,J. and Muenke,M. (1999) Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nature Genet., 22, 196–198. [DOI] [PubMed] [Google Scholar]
- 75.Bertolino E., Wildt,S., Richards,G. and Clerc,R.G. (1996) Expression of a novel murine homeobox gene in the developing cerebellar external granular layer during its proliferation. Dev. Dyn., 205, 410–420. [DOI] [PubMed] [Google Scholar]
- 76.Gripp K.W., Wotton,D., Edwards,M.C., Roessler,E., Ades,L., Meinecke,P., Richieri-Costa,A., Zackai,E.H., Massague,J., Muenke,M. and Elledge,S.J. (2000) Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nature Genet., 25, 205–208. [DOI] [PubMed] [Google Scholar]
- 77.The International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 78.Ji Y., Eichler,E.E., Schwartz,S. and Nicholls,R.D. (2000) Structure of chromosomal duplicons and their role in mediating human genomic disorders. Genome Res., 10, 597–610. [DOI] [PubMed] [Google Scholar]
- 79.Eichler E.E. (1998) Masquerading repeats: paralogous pitfalls of the human genome. Genome Res., 8, 758–762. [DOI] [PubMed] [Google Scholar]
- 80.Mazzarella R. and Schlessinger,D. (1998) Pathological consequences of sequence duplications in the human genome. Genome Res., 8, 1007–1021. [DOI] [PubMed] [Google Scholar]
- 81.Ohno S. (1970) Evolution by Gene Duplication. George Allen and Unwin, London, UK.
- 82.Sidow A. and Bowman,B.H. (1991) Molecular phylogeny. Curr. Opin. Genet. Dev., 1, 451–456. [DOI] [PubMed] [Google Scholar]
- 83.Sidow A. and Thomas,W.K. (1994) A molecular evolutionary framework for eukaryotic model organisms. Curr. Biol., 4, 596–603. [DOI] [PubMed] [Google Scholar]
- 84.Sidow A. (1996) Gen(om)e duplications in the evolution of early vertebrates. Curr. Opin. Genet. Dev. 6, 715–722. [DOI] [PubMed] [Google Scholar]
- 85.Barton G. (1993) ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng., 10, 37–40. [DOI] [PubMed] [Google Scholar]