

Abstract
Epidemiological findings support the hypothesis that type 2 diabetes mellitus (T2DM) is a risk factor for osteoarthritis (OA). Moreover, OA cartilage from patients with T2DM exhibits a greater response to inflammatory stress, but the molecular mechanism is unclear. To investigate whether the antioxidant defense system participates in this response, we examined here the expression of nuclear factor-erythroid 2-related factor (Nrf-2), a master antioxidant transcription factor, and of heme oxygenase-1 (HO-1), one of its main target genes, in OA cartilage from T2DM and non-T2DM patients as well as in murine chondrocytes exposed to high glucose (HG). Ex vivo experiments indicated that Nrf-2 and HO-1 expression is reduced in T2DM versus non-T2DM OA cartilage (0.57-fold Nrf-2 and 0.34-fold HO-1), and prostaglandin E2 (PGE2) release was increased in samples with low HO-1 expression. HG-exposed, IL-1β-stimulated chondrocytes had lower Nrf-2 levels in vitro, particularly in the nuclear fraction, than chondrocytes exposed to normal glucose (NG). Accordingly, HO-1 levels were also decreased (0.49-fold) in these cells. The HO-1 inducer cobalt protoporphyrin IX more efficiently attenuated PGE2 and IL-6 release in HG+IL-1β-treated cells than in NG+IL-1β-treated cells. Greater reductions in HO-1 expression and increase in PGE2/IL-6 production were observed in HG+IL-1β-stimulated chondrocytes from Nrf-2−/− mice than in chondrocytes from wild-type mice. We conclude that the Nrf-2/HO-1 axis is a critical pathway in the hyperglucidic-mediated dysregulation of chondrocytes. Impairments in this antioxidant system may explain the greater inflammatory responsiveness of OA cartilage from T2DM patients and may inform treatments of such patients.
Keywords: diabetes, heme oxygenase, inflammation, Nuclear factor 2 (erythroid-derived 2-like factor) (NFE2L2) (Nrf2), osteoarthritis
Introduction
Type 2 diabetes mellitus (T2DM)5 is a chronic metabolic disorder associated with many adverse complications. Accumulating epidemiological and experimental findings support the hypothesis that T2DM is an independent risk factor for osteoarthritis (OA), the most frequent joint disease (1), or for its severity (2–4). However, the mechanisms underlying the connection between both diseases remain unclear.
Together, oxidative stress and pro-inflammatory mediators, particularly interleukin 1β (IL-1β), actively induce the changes in articular cartilage that predispose this tissue to the development of OA (5, 6) as well as to T2DM and T2DM complications (7–10). Elevated intracellular glucose levels primarily generate oxidative damage as a consequence of glycolytic pathway saturation in the cell and the subsequent production of advanced glycation end products (11–13). This oxidative stress mediated by excess glucose-induced impairments in the antioxidant defense system may also contribute to diabetic complications. Moreover, similar oxidative stress disturbances could occur in OA (14, 15).
Nuclear factor-erythroid 2-related factor-2 (Nrf-2), a master transcription factor involved in antioxidant signaling and the cell survival response, regulates a wide battery of cytoprotective responses and helps attenuate metabolic, neurodegenerative, and other age-related diseases (16–19). Likewise, evidence for altered Nrf-2 signaling in aging and metabolic disorders has been reported (19, 20).
As shown in recent studies, Nrf-2 is a pivotal target for the prevention and attenuation of diabetes mellitus (17, 21) and for controlling bone and cartilage destruction induced by oxidative stress (22, 23). Under physiological conditions, Nrf-2 is generally located in the cytoplasm and binds to its inhibitor, Kelch-like ECH-associated protein 1 (Keap1), leading to its degradation. However, in response to oxidative or electrophilic stress, Nrf-2 dissociates from Keap1 and translocates to the nucleus to bind antioxidant-responsive elements in the promoter regions of its downstream antioxidant genes, including heme oxygenase-1 (HO-1) (24).
HO-1 is a crucial antioxidant enzyme that catalyzes the degradation of heme into iron, carbon monoxide, and biliverdin (25). This enzyme regulates catabolic and anabolic processes in OA chondrocytes (26). HO-1 overexpression in cartilage prevents the pro-inflammatory mediator-induced activation of catabolic, apoptotic, or senescence pathways (27–29). Thus, HO-1 represents an important part of the cellular response to inflammatory and oxidative stress in joints (30). Interestingly, the expression and activity of this enzyme are down-regulated in the vasculature and retinal tissue from patients with diabetes (31–33) as well as in in vitro and in vivo cellular models of high glucose exposure (34, 35). However, the expression and involvement of the Nrf-2/HO-1 antioxidant axis in type 2 diabetes-related OA has not yet been studied.
For a better understanding of the link between OA and T2DM, we recently reported the higher responsiveness of OA cartilage from patients with T2DM to IL-1β-induced inflammatory stress (36). Similarly, the exposure of IL-1β-stimulated chondrocytes to high glucose exacerbates the activation of pathological pathways, which is blocked by ROS scavengers (36). These findings further support the critical role of oxidative stress generated under hyperglucidic conditions in the activation of catabolic responses in cartilage. However, the pathological mechanisms triggered by a high glucose environment that ultimately establishes the redox imbalance in cartilage remain evasive. We studied the Nrf-2/HO-1 axis in human OA cartilage from patients with or without diabetes and in chondrocytes in a high-glucose environment to determine whether the antioxidant defense system is impaired in this excess glucose- and low-grade inflammation-induced phenomenon in cartilage.
Results
Nrf-2/HO-1 expression was decreased in OA cartilage from patients with diabetes and was inversely correlated with the production of inflammatory mediators
We recently observed a more pronounced inflammatory phenotype of OA cartilage from patients with diabetes, based on IL-6 and prostaglandin E2 (PGE2) release (36). We evaluated the expression of the Nrf-2 and HO-1 proteins in 8 cartilage explants from patients with T2DM and 8 explants from non-diabetic patients matched for age and body mass index (BMI) to elucidate whether impairments to the antioxidant system underlie this altered phenotype (age: 64.8 ± 11.1 versus 68.0 ± 8.7; BMI: 30.1 ± 4.9 versus 29.9 ± 4.7; gender: 6 versus 3 females for T2DM and non-T2DM patients, respectively). Additionally, the other clinical characteristics of all patients were similar.
The Nrf-2 and HO-1 expression levels were reduced in OA cartilage from patients with T2DM compared with the expression levels in non-diabetic patients (0.57-fold and 0.34-fold for Nrf-2 and HO-1, respectively, p < 0.05) (Fig. 1, A and B). Likewise, the HO-1 and Nrf-2 expression levels varied similarly; Nrf-2 expression was higher in patients with HO-1 expression levels above the median (Fig. 1C). Conversely, PGE2 release and HO-1 expression varied in opposite ways; PGE2 release was higher in patients with HO-1 expression levels below the median (Fig. 1D).
Figure 1.

Reduced Nrf-2/HO-1 expression in OA cartilage from patients with T2DM was associated with increased production of pro-inflammatory mediators. The HO-1 (A) and Nrf-2 (B) levels in explants of OA cartilage from patients with or without T2DM were evaluated by Western blotting. The values were normalized to the β-actin levels. Each symbol represents an OA patient without (OA, □) or with T2DM (OA-T2DM, ●) (n = 8 per condition). The dependence between Nrf-2 levels (C) or PGE2 production (D) and HO-1 expression in all groups of patients (▵) was assayed by dichotomizing the Nrf-2/PGE2 values as a function of the median HO-1 levels. The bars represent the means ± S.D. for each condition. *, p ≤ 0.05.
A high-glucose environment reduced Nrf-2/HO-1 expression in IL-1β-stimulated murine chondrocytes
Glucose has been shown to down-regulate HO-1 promoter activity and HO-1 levels (37). Consequently, a high glucose environment in T2DM may be responsible for the reduced Nrf-2/HO-1 expression, as observed in OA cartilage from patients with T2DM. Murine chondrocytes were stimulated with or without IL-1β (5 ng/ml) for the indicated times in the presence of normal glucose (5.5 mm) or high glucose (25 mm) to explore this possibility. As shown in Fig. 2 and Fig. 3, reduced HO-1 and Nrf-2 mRNA expression levels were observed at 72 h of incubation with high glucose compared with cells treated with normal glucose; however, these differences in expression were not significant at the protein level. The IL-1β treatment further enhanced the impact of high glucose on the Nrf-2 and HO-1 expression levels at both the mRNA (Fig. 2A and Fig. 3A) and protein levels (Fig. 2D and Fig. 3B). This effect was significant at 48 h and particularly at 72 h (0.81-fold Nrf-2 and 0.48-fold HO-1 reductions at the protein level, p < 0.05). Additionally, we did not observe a simultaneous regulation of Kelch-like ECH-associated protein 1 (Keap-1) expression (Nrf-2 cytoplasmic inhibitor) that counteracted the variations in the Nrf-2 levels (Fig. 2B). Subsequently, the Nrf-2/Keap-1 ratio confirmed the modulation of Nrf-2 expression by high glucose (Fig. 2C).
Figure 2.

Nrf-2 expression was reduced in murine chondrocytes incubated with high glucose. Chondrocytes were incubated with normal glucose (5 mm; NG) or high glucose (25 mm; HG) in the presence or absence of IL-1β (5 ng/ml) for the indicated times. The expression levels of the Nrf-2 (A) and Keap-1 (B) genes were evaluated by qRT-PCR. The values were normalized to HPRT expression (n = 6). C, The Nrf-2/Keap-1 expression ratio was calculated. The total (D) and nuclear (E) Nrf-2 protein expression levels were measured by Western blotting. The values were normalized to β-actin expression (n = 6). Each symbol represents an experiment from one litter of mice in normal glucose (○) and high glucose (●) conditions. The bars represent the means ± S.D. for each condition. *, p ≤ 0.05.
Figure 3.

HO-1 expression was reduced in murine chondrocytes incubated with high glucose and is associated with Nrf-2 levels and negatively associated with the production of inflammatory mediators. Chondrocytes were incubated with NG or HG in the presence or absence of IL-1β for the indicated times. Expression of the HO-1 gene (A) or protein (B) was evaluated by qRT-PCR or Western blotting, respectively. The values were normalized to the HPRT levels for gene expression or to the β-actin levels for protein quantification (n = 6 per condition). Each symbol represents an experiment from one litter of mice in normal glucose (○) and high glucose (●) conditions. The bars represent the means ± S.D. for each condition. *, p ≤ 0.05. Dependence analysis between Nrf-2 expression (C) or IL-1β-induced PGE2 production (D) and HO-1 expression was assayed by dichotomizing Nrf-2/PGE2 values as a function of the median HO-1 levels. Each symbol (▵) represents the Nrf-2/PGE2 value obtained from chondrocytes incubated in both normal and high glucose and whose HO-1 expression is lower or higher than the median level (n = 72 for the Nrf-2/HO-1 analysis; n = 10 for the PGE2/HO-1 analysis). The bars represent the means ± S.D. for each condition. *, p ≤ 0.05; **, p ≤ 0.01.
We assayed the variations in the nuclear levels of this transcription factor 30 min after treatment to determine whether Nrf-2 nuclear translocation was impaired (Fig. 2E). Nuclear levels of Nrf-2 were reduced in chondrocytes grown under high-glucose conditions in both the presence and absence of IL-1β compared with that of cells grown under normal glucose conditions (0.73-fold and 0.61-fold, respectively). Consistent with these findings and similar to our observations with human OA cartilage, we observed a significant dependence between the Nrf-2 and HO-1 expression levels in cultured murine chondrocytes, as the levels of these proteins varied in the same way (Fig. 3C).
Reduced HO-1 expression favored the increased responsiveness of chondrocytes to IL-1β in a high-glucose environment
Consistent with our observations in OA cartilage, HO-1 expression was inversely correlated with PGE2 production in IL-1β-stimulated murine chondrocytes (Fig. 3D). Based on these findings, HO-1 expression may regulate chondrocyte activation by pro-inflammatory cytokines. Chondrocytes were co-incubated with cobalt protoporphyrin IX (CoPP), an inducer of HO-1 activity, for 72 h, and IL-6 and PGE2 release was measured to further confirm this hypothesis (Fig. 4, A and B). CoPP attenuated both IL-1β-induced IL-6 and PGE2 production. Interestingly, this reduction was significantly stronger in cells grown in high glucose, with a 49% decrease in IL-6 release and a 98% decrease in PGE2 release compared with a 25 and 91% release, respectively, from cells grown in normal glucose. Moreover, because HO-1 is an antioxidant enzyme, we also determined whether CoPP reduced the previously observed high glucose-stimulated increase in IL-1β-induced ROS production (36). As expected, CoPP, through HO-1, strongly reduced the ROS levels, returning them to the control values (Fig. 4C).
Figure 4.
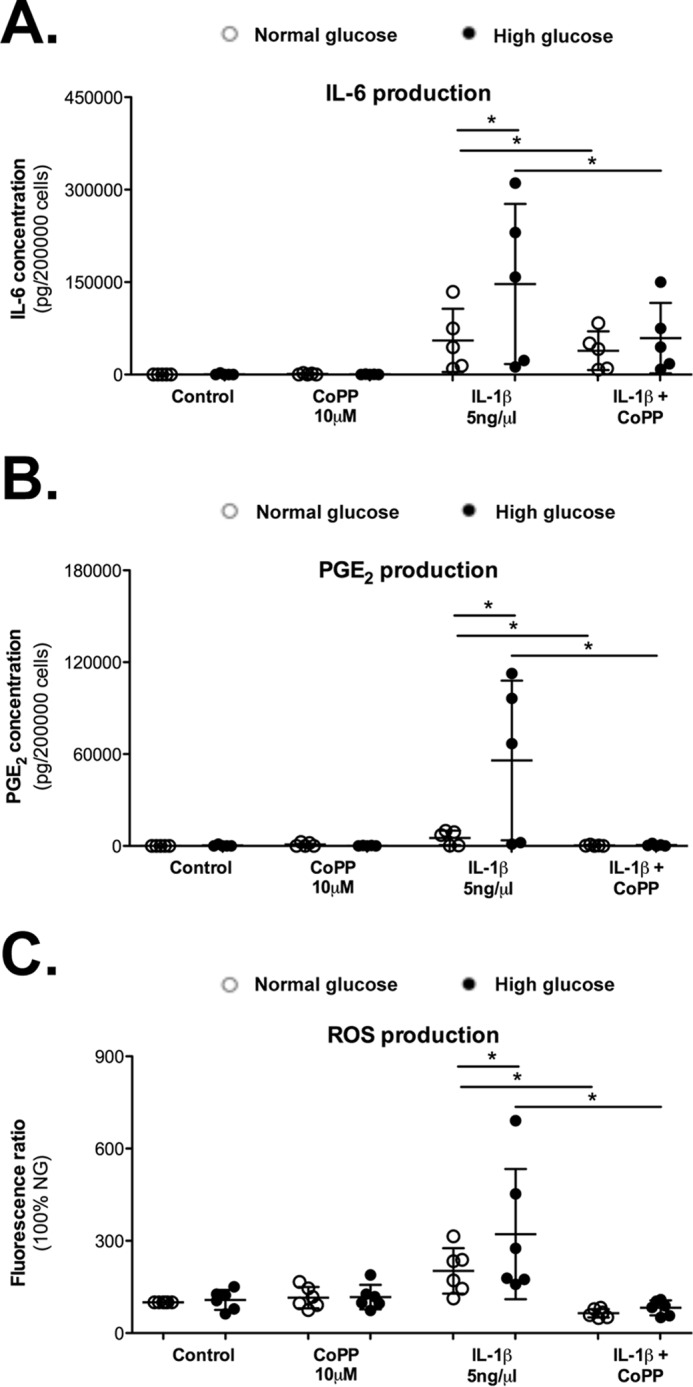
HO-1 protects chondrocytes against increased responsiveness to IL-1β in a high glucose environment. Chondrocytes were co-incubated with an inducer of HO-1, CoPP (10 μm), for 72 h. Release of the IL-6 (A) and PGE2 (B) was assayed. C, ROS production was evaluated using a fluorometric assay with DCFDA. Represented data are -fold induction compared with the control condition without IL-1β and normalized to intracellular protein quantity. Each symbol represents an experiment from one litter of mice in normal glucose (○) and high glucose (●) conditions. The bars represent the means ± S.D. for each condition (n = 6). *, p ≤ 0.05.
Nrf-2 knock-out chondrocytes showed reduced HO-1 expression and higher responsiveness to IL-1β in a high glucose environment
Nrf-2 is recognized as the main inducer of HO-1 gene expression (38). Here, Nrf-2 and HO-1 expression were shown to vary in the same way in OA cartilage and in murine chondrocytes exposed to a high glucose environment. Additionally, sulforaphane, a known natural inducer of Nrf-2, increased HO-1 expression, reduced ROS accumulation, and attenuated the production of IL-6 in chondrocytes exposed to IL-1β (Fig. 5), suggesting a role for Nrf-2 in controlling the catabolic response. Therefore, we conducted studies in chondrocytes from Nrf-2 knock-out (Nrf-2−/−) mice using the same experimental approach to further address the dependence of IL-1β-induced HO-1 expression in response to high glucose on Nrf-2 (Fig. 6). As expected, HO-1 expression was drastically reduced in Nrf-2−/− chondrocytes at 72 h compared with that in the wild-type cells (Fig. 6A). IL-6 and PGE2 production were measured to determine whether the Nrf-2 knock-out also had an impact on the pro-inflammatory phenotype of chondrocytes. We observed a strengthening of PGE2 production induced by IL-1β in chondrocytes from Nrf-2−/− mice compared with wild-type mice, achieving significant differences in those cells incubated in high glucose (Fig. 6B); moreover, similar results were obtained for IL-6 levels.
Figure 5.
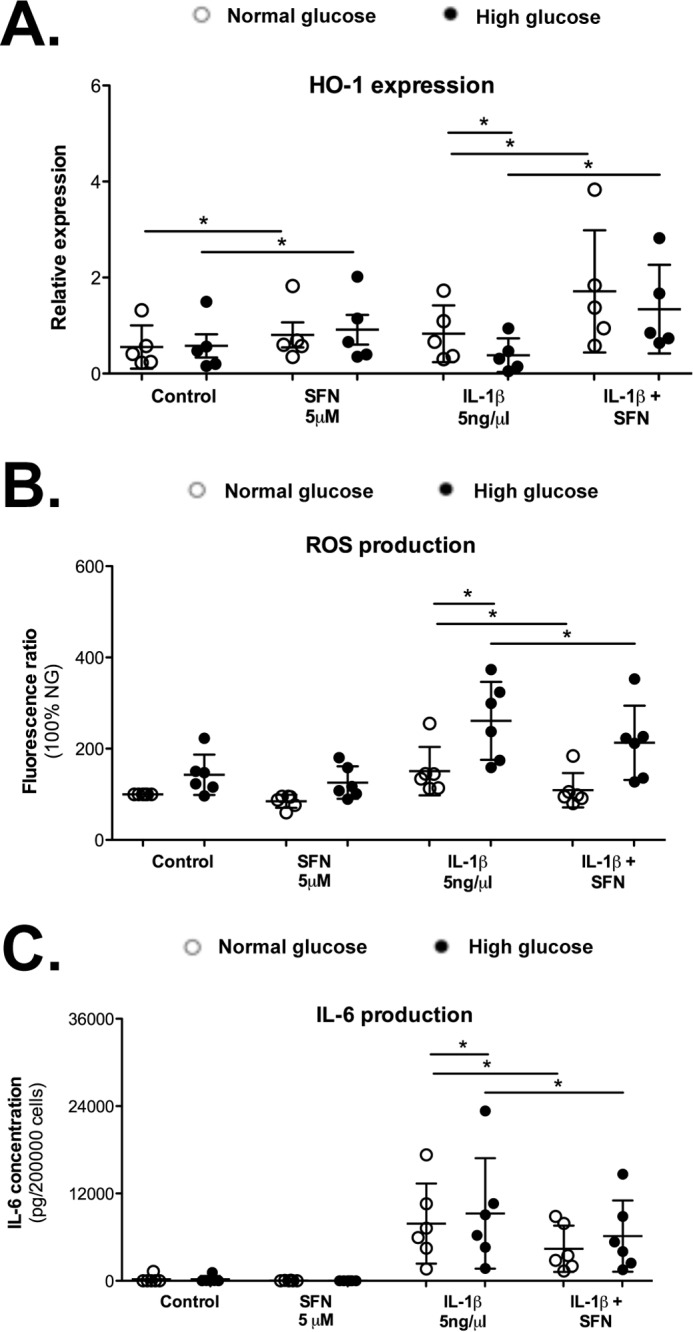
Sulforaphane increased HO-1 expression and attenuated ROS and inflammatory production. Chondrocytes were preincubated with SFN (5 μm) for 30 min before stimulation in normal glucose (5 mm) or high glucose (25 mm) with/without IL-1β (5 ng/ml). HO-1 expression (A) and ROS (B) and IL-6 production (C) were assayed after 72 h treatment. Each symbol represents an experiment from one litter of mice in normal glucose (○) and high glucose (●) conditions. The bars represent the means ± S.D. for each condition. *, p ≤ 0.05.
Figure 6.
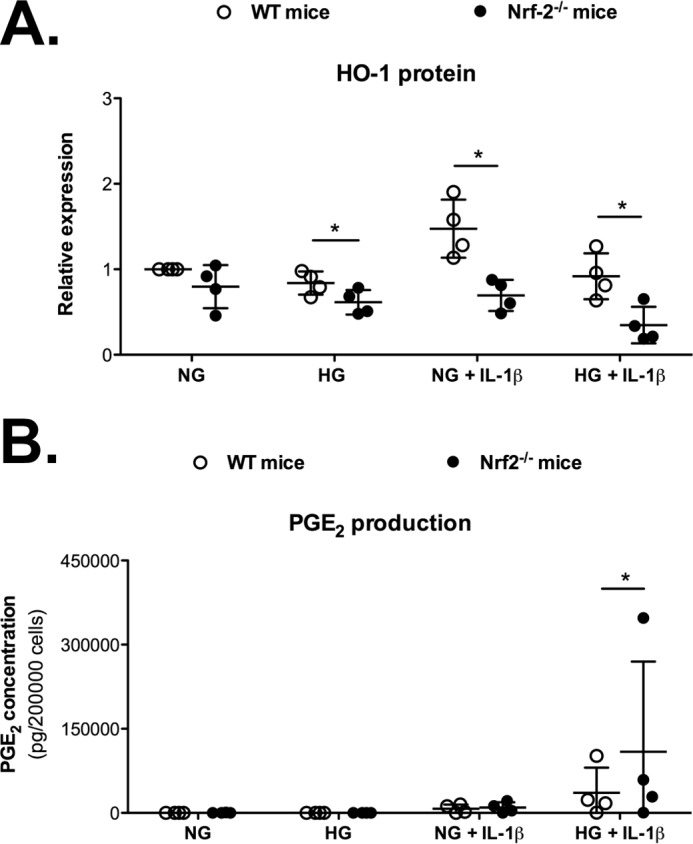
Nrf-2 knock-out (Nrf-2−/−) chondrocytes exposed to high glucose exhibited lower HO-1 expression and increased responsiveness to IL-1β. A, the expression of the HO-1 protein was evaluated in chondrocytes from wild-type (WT) or Nrf-2−/− mice that were stimulated as described above for 72 h. The values were normalized to β-actin expression. Represented data are -fold induction compared with the control condition without IL-1β. B, PGE2 production was also measured. Each symbol represents an experiment from one litter of wild-type (○) and Nrf-2−/− (●) mice. The bars represent the means ± S.D. for each condition. *, p ≤ 0.05.(n = 4).
Discussion
T2DM is currently considered an additional risk factor for OA occurrence and OA severity, delineating the T2DM-associated OA phenotype (2, 3, 39). In addition to insulin resistance, chronic hyperglycemia is one of the main biological features involved in diabetic complications (10, 13). Moreover, we recently reported a more pronounced inflammatory phenotype in OA cartilage from patients with diabetes and enhanced IL-1β-induced inflammation in cultured chondrocytes exposed to excess glucose (36). To further elucidate these findings, we show in this study the expression of Nrf-2, the master transcriptional regulator of antioxidant responses, and one of its main target genes, HO-1, is reduced in OA cartilage from patients with T2DM. These observations are mimicked in vitro by exposing IL-1β-stimulated chondrocytes to high glucose. Linking our findings, impaired Nrf-2/HO-1 signaling is responsible for the increased responsiveness to IL-1β, as shown by the increased IL-6 and PGE2 release and ROS production by chondrocytes.
The Nrf-2/HO-1 axis is a crucial cell survival mechanism that counteracts oxidative stress and inflammation (24, 40). Deficiencies in this axis have been identified in some systemic diabetic complications, such as retinopathy or cardiopathy (33–35, 41). In the present study we investigated whether the Nrf-2/HO-1 pathway was also impaired in OA joints from patients with T2DM and whether it participates in the pathological mechanisms predisposing cartilage to diabetes-associated OA. For this purpose, we selected osteoarthritic cartilage explants from patients with and without diabetes and matched them based on age, BMI, gender, and other metabolic co-morbidities, similar to our previous report (36). Both Nrf-2 and HO-1 expression were decreased in diabetic OA cartilage. The HO-1 promoter (HMOX1) contains binding sites for several transcription factors, notably the antioxidant-responsive element site for Nrf-2; however, activator protein-1 (AP-1), cAMP response element-binding protein (CREB), and nuclear factor- κB (NF-κB) can also activate its expression (38). In our study HO-1 expression in OA cartilage at least partially depended on the Nrf-2 levels. Nrf-2/HO-1 signaling protects the joint against the activation of pathological pathways (42–44). Here, we revealed a negative correlation between PGE2 production and the HO-1 levels, suggesting a deleterious effect of a reduction in Nrf-2/HO-1 expression on the inflammatory profile of cartilage.
A decrease in Nrf-2/HO-1 signaling and a subsequent increase in ROS release are associated with exposure to a high glucose environment in different cell types. Retinal endothelial cells incubated with high glucose exhibit reduced Nrf-2 transcriptional activity (45). Similar results were observed in human microvessel endothelial cells (46). Interestingly, decreased HO-1 levels were also detected in these in vitro models (46) as well as in animal models of diabetes mellitus (31, 35). Therefore, we used an in vitro approach to evaluate whether a diabetes-related high glucose environment may participate in reducing Nrf-2/HO-1 signaling in T2DM OA cartilage. Murine chondrocytes were stimulated with IL-1β, a cytokine known to be involved in the pathophysiology of OA and T2DM, and were exposed to high glucose. The hyperglucidic environment reduced the early nuclear translocation of Nrf-2 as well as its total protein level. As expected, HO-1 expression was also reduced and was positively correlated with the Nrf-2 levels. This correlation between the expression levels of both proteins was further confirmed in chondrocytes from Nrf-2−/− mice. The genetic invalidation of Nrf-2 expression drastically reduced HO-1 expression.
The transcriptional activity of Nrf-2 is mainly regulated by its cytoplasmic repressor, Keap-1, although other inhibitors also block its activity in the nucleus (24). We failed to detect any modulation of Keap-1 expression under high glucose conditions and/or IL-1β stimulation, suggesting that repressors other than Keap-1 participate in the alterations in the Nrf-2 pathway observed in our model. For instance, the glycogen synthase kinase 3β (GSK3β)/Fyn pathway and BTB domain and CNC homolog 1 (bach1) (47) impair Nrf-2 signaling in diabetic complications and have been involved in pathological processes in joints (48–52). Based on these findings, hyperglycemia/diabetes modulates the Nrf-2/HO-1 axis through different repressors; however, additional studies are required to elucidate the specific pathways activated in chondrocytes.
The beneficial effect of activation of the Nrf-2/HO-1 axis on diabetic conditions is widely accepted (17, 35). Moreover, accumulating evidence has revealed a protective role for the Nrf-2/HO-1 axis in joint diseases (22, 23, 26, 43, 53). As shown in the present study, the reduction of the Nrf-2/HO-1 levels contributes to a pro-inflammatory imbalance in IL-1β-stimulated chondrocytes exposed to a high glucose environment. HO-1 expression was negatively correlated with the production of inflammatory mediators. Likewise, treatment with sulforaphane, a known natural inductor of Nrf-2, and subsequent HO-1 up-regulation protected chondrocytes against ROS accumulation and inflammatory production. Accordingly, the chemical induction of HO-1 activity reduced IL-6 and PGE2 release in a more significant manner in those chondrocytes exposed to high glucose. Finally, HO-1 expression was reduced in chondrocytes from Nrf-2 knock-out mice incubated with high glucose and the inflammatory response to IL-1β was further exacerbated. Our results are corroborated by Cai et al. (43), who observed that an Nrf-2 deletion results in increased disease severity in different animal models of OA. Additionally, the recovery of Nrf-2 activity inducing HO-1 expression decreased OA pathogenesis (43). Accordingly, deficiency of bach1 favoring Nrf-2 transcriptional activity protects against development of two different types of OA: aging- and post-traumatic-associated OA pathogenesis (51). Thereby, these findings suggest anti-catabolic roles of Nrf-2 in different OA subsets. Here, we provide for the first time strong evidence that Nrf2 is also pivotal to counteract the pathological pathways activated by high-glucose stress in a diabetes-related OA context. However, HO-1 activation independent of Nrf-2 may also protect against the development of this disease (51, 54). In this sense, Nrf-2 knock-out mice failed to completely block the effect of high glucose on HO-1 expression, suggesting that pathways other than Nrf-2 signaling may also be involved in regulating HO-1 expression.
In conclusion, the Nrf-2/HO-1 axis is dysfunctional in diabetic osteoarthritic cartilage and represents a critical pathway involved in the hyperglucidic-mediated dysregulation of articular chondrocytes (Fig. 7). The impairment of this antioxidant system may explain the greater inflammatory responsiveness of cartilage from patients with T2DM and may provide new targeted therapeutic avenues for treating patients with the diabetes-related OA phenotype.
Figure 7.
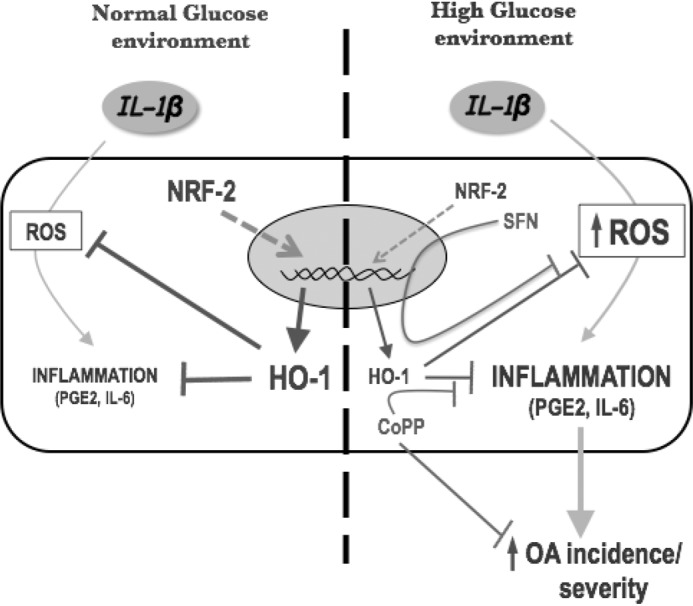
Hypothetical mechanism by which impairments in the Nrf-2/HO-1 axis favors catabolic responsiveness to IL-1β in chondrocytes exposed to a high glucose environment. The Nrf-2/HO-1 axis is one of the most important anti-inflammatory and antioxidant protective systems. Under stress conditions Nrf-2 is translocated to the nucleus to activate antioxidant gene expression, including one of its main target HO-1. Next, HO-1 counteracts the catabolic pathways induced by inflammatory stimuli such as IL-1β. However, Nrf-2 signaling is impaired in a high glucose environment, which is manifested as dysfunction in Nrf-2 synthesis and translocation and a subsequent reduction in the HO-1 levels. Thus, chondrocytes lost the capacity to control ROS production and inflammation (i.e. PGE2 and IL-6 release). This event further favors the activation of pathological pathways, leading to an increase in the susceptibility to OA and disease severity. SFN, a Nrf-2 activator, and CoPP, an inducer of HO-1, attenuate this phenomenon.
Experimental procedures
Collection of OA human cartilage
Human knee explants were obtained from patients with OA who were undergoing total joint replacement at Saint-Antoine Hospital (Paris, France). The diagnosis of OA of the knee was based on criteria from the American College of Rheumatology (55). Patients were screened for diabetes using their medical files, drug prescriptions, and patient interviews. For each patient with diabetes who was included in the study, we matched a non-diabetic patient undergoing total knee joint replacement due to OA by age and BMI to avoid confounding factors. The explants from each patient were manually dissected from all remaining cartilage zones (i.e. tibial plateaus and femoral condyles), mixed to obtain homogeneous isolated cartilage samples and managed using previously described methods (36). Briefly, the cartilage explants were cut into small pieces (∼5 mm3), washed several times with PBS, and incubated in DMEM (25 mm) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 4 mm glutamine for 24 h at 37 °C. After incubation, the explants were frozen, and protein extracts were obtained using previously established methods (56). Briefly, the tissues were ground in liquid nitrogen using a mortar and pestle, after which the proteins were extracted with lysis buffer and used for the Western blotting experiments. In parallel, conditioned media (CM) were also collected, centrifuged (1600 × g for 6 min), and stored at −20 °C. Each volume of medium was normalized to the wet weight of the explants (6 ml/g tissue) (36). Informed consent was obtained from each patient for the use of their tissues and clinical data. All experiments using human samples were approved by a French Institutional Review Board (Comité de Protection des Personnes, Paris Ile de France 5, April 2012).
Primary culture and treatment of murine articular chondrocytes
Mouse primary chondrocytes were isolated from the articular cartilage of 5–6-day-old newborn C57BL/6 mice from Janvier (St. Berthevin, France) and were seeded at a density of 8 × 103 cells per cm as previously described (57). Articular chondrocytes obtained from newborn mice using this protocol were validated as cells presenting characteristics similar to fully mature murine chondrocytes (58). Chondrocytes were cultured in complete DMEM for a week and were then incubated in serum-free DMEM containing 0.1% bovine serum albumin (BSA) for 24 h before treatment (basal medium). Subsequently, the murine chondrocytes were incubated with normal glucose (5.5 mm) or high glucose (25 mm) in the presence or absence of IL-1β (5 ng/ml) (PeproTech, Rocky Hill, NJ) for the indicated times. Cell lysates were collected for mRNA or protein extraction, and supernatants were collected for the assays.
For mechanistic studies, chondrocytes that had been cultured with normal or high glucose in the presence or absence of IL-1β (5 ng/ml) for 72 h were co-treated with CoPP, an inducer of HO-1 activity (10 μm) (Enzo Life Sciences, Villeurbanne, France) whose non-toxic effect at this concentration had been checked using an LDH assay and its specificity described in previous publications (29, 59, 60). In additional experiments, cells were pretreated for 30 min with the Nrf2 activator compound sulforaphane (SFN; 5 μm) (Sigma). Moreover, experiments were also performed using Nrf-2 knock-out (Nrf-2−/−) (61) mice generated from inbred Nrf-2 heterozygous mice on a C57BL/6J background, as described by El Ali et al. (62). The mice were housed in a pathogen-free facility and were handled in accordance with the principles and procedures outlined in Council Directive 86/609/EEC.
The efficiency of the IL-1β dose employed was assayed previously, and the cytotoxic effects of the treatments and an osmotic effect of high glucose were excluded (36). The CoPP dose was chosen based on dose-effect experiments (data not shown) and literature data. All experiments with murine articular chondrocytes were performed using protocols approved by the French and European ethics committees (Comité Régional d'Ethique en Expérimentation Animale N°3 de la région Ile de France).
RNA extraction and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from the chondrocytes using the ReliaPrep RNA Cell Miniprep System kit (Promega, Madison, WI), and the concentrations were determined by spectrophotometry (Eppendorf, Le Pecq, France). Reverse transcription utilized 500 ng of total RNA and the Omniscript RT kit (Qiagen). The levels of the IL-6, HO-1, Nrf-2, and Keap-1 mRNAs were quantified using a Light Cycler LC480 (Roche Diagnostics). The PCR amplification conditions were: initial denaturation for 5 min at 95 °C followed by 40 cycles consisting of 10 s at 95 °C, 15 s at 60 °C, and 10 s at 72 °C. Product formation was detected at 72 °C in the fluorescein isothiocyanate channel. The relative mRNA expression levels were calculated and normalized to the levels of the murine hypoxanthine guanine phosphoribosyltransferase (HPRT) mRNA using the 2 ΔΔCT method (specific mouse primer sequences are shown in Table 1). All measurements were performed in duplicate.
Table 1.
Primer sequences used for real-time PCR
| Gene | Forward | Reverse |
|---|---|---|
| Nrf-2 | 5′-CATGATGGACTTGGAGTTGC-3′ | 5′-CCTCCAAAGGATGTCAATCAA-3′ |
| Keap-1 | 5′-CACAGCAGCGTGGAGAGA-3′ | 5′-CAACATTGGCGCGACTAGA-3′ |
| HO-1 | 5′-AGGCTAAGACCGCCTTCCT-3′ | 5′-TGTGTTCCTCTGTCAGCATCA-3′ |
| HPRT | 5′-AGGACCTCTCGAAGTGT-3′ | 5′-ATTCAAATCCCTGAAGTACTCAT-3′ |
IL-6 and PGE2 assessments
The IL-6 concentrations in the murine cell supernatants were measured using the Quantikine enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Lille, France). The concentrations of human IL-6 were measured in human CM using the Pelikine compact kit (Sanquin, Amsterdam, The Netherlands). The concentrations of the murine and human pro-inflammatory bioactive lipid PGE2 in the cell supernatants and CM were measured using the enzymatic immunoassay (EIA) kit (Cayman Chemical, Ann Arbor, MI). The limits of detection were 7.8, 0.6, and 7.8 pg/ml for the murine/human IL-6 and PGE2 assessments, respectively. Duplicate measurements were performed.
Protein extraction and Western blotting
Protein extracts from human cartilage explants were prepared using previously described methods (56). Murine chondrocytes were cultured in 12-well plates (4 × 104 cells/well) in duplicate and were treated as indicated. Subsequently, the total intracellular proteins were obtained as previously described (63). In some experiments, cytosolic and nuclear fractions were obtained from the murine chondrocytes using commercially available NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Waltham, MA) according to the manufacturer's recommendations. Protein extracts from human cartilage (30 μg) and total proteins (30 μg) or nuclear fractions (20 μg) from murine chondrocytes were resolved by SDS-PAGE and immunoblotted with the indicated antibodies: Nrf-2 antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA; sc-722) (80–100 kDa) (64), HO-1 antibody (1:1,000; Enzo Life Sciences, Lausen, Switzerland; SPA-895) (32 kDa), or actin antibody (1:5,000; Sigma; A5316). Signals were detected using enhanced chemiluminescence (ECL), and the blots were exposed to Fujifilm LAS-300 (Fujifilm Medical Systems, Stamford, CT). The relative levels of protein expression were calculated using densitometry and were normalized to the actin levels. We used Image-Gauge software (Science Lab 2004; Fujifilm) for the densitometry analysis.
Cellular ROS production
Chondrocytes were seeded and cultured in 96-well plates at a density of 1 × 104 cells per well, as described above. After 72-h of treatment, ROS production was measured using a fluorometric assay with dichlorodihydrofluorescein diacetate (DCFDA) (Molecular Probes, Life Technologies, Saint Aubin, France). Briefly, chondrocytes were incubated with 17 μm DCFDA diluted in the fasting medium for 60 min at 37 °C in the dark. Subsequently, the chondrocytes were washed with PBS, and fluorescence was measured using the Fluostar Galaxy reader (BMG Labtech, Ortenberg, Germany) at an excitation wavelength of 485 nm and an emission wavelength of 520 nm and then analyzed using the Biolise system (Labsystems, Helsinki, Finland). The intracellular proteins were collected with NaOH (0.5 m), and the concentrations were measured using a spectrophotometer and a protein assay kit (Bio-Rad) to normalize the results. The ROS production is represented as -fold induction from that of the control and by micrograms of protein. All measurements were performed in triplicate.
Statistical analysis
All data are reported as points representing one single experiment from one litter of mice or one patient with standard deviation to represent error. All tests were analyzed using GraphPad Prism 5 (GraphPad Software, San Diego, CA) with the Wilcoxon test for paired variables and the Mann-Whitney test for unpaired variables. Additionally, analysis of data from Nrf-2−/− experiments was performed by paired t test. p ≤ 0.05 was considered statistically significant.
Author contributions
C. V.-G., A. C., M.-C. L., X. H., S. K.-R., R. M., F. B., and J. S. were responsible for the study design, manuscript preparation, and data interpretation. A. S. organized and collected the human tissue samples and participated in designing the experiments with human tissue and in data interpretation. C. V.-G., A. C., and A. P. performed the experiments. S. K.-R. was responsible for generating the Nrf-2−/− mice and was involved in data interpretation. All authors reviewed and approved the final manuscript.
Acknowledgments
We thank Valérie Domergue, Ayma Galland, and the Unité Mixte de Service–Institut Fédératif de Recherche (UMS-IPSIT) for excellent technical assistance for animal testing.
Note added in proof
Panels A and B of Fig. 2 were duplicated in the version of this article that was published as a Paper in Press on July 6, 2017. This error has now been corrected and does not affect the results or conclusions of this work.
This work was supported by the French State Transimmunom Funds managed by the ANR within the Investissements d'Avenir Program (ANR-11-IDEX-0004-02) and Fondation Arthritis Jacques Courtin. The authors declare that they have no conflicts of interest with the contents of this article.
- T2DM
- type 2 diabetes mellitus
- Nrf-2
- nuclear factor-erythroid 2-related factor-2
- PGE2
- prostaglandin E2
- CoPP
- cobalt protoporphyrin IX
- DCFDA
- 2′,7′-dichlorofluorescein diacetate
- HG
- high glucose
- HO-1
- heme oxygenase-1
- HPRT
- hypoxanthine guanine phosphoribosyltransferase
- NG
- normal glucose
- OA
- osteoarthritis
- ROS
- reactive oxygen species
- BMI
- body mass index
- CM
- conditioned media
- qRT-PCR
- quantitative RT-PCR
- SFN
- sulforaphane.
References
- 1. Guillemin F., Rat A. C., Mazieres B., Pouchot J., Fautrel B., Euller-Ziegler L., Fardellone P., Morvan J., Roux C. H., Verrouil E., Saraux A., Coste J., and 3000 Osteoarthritis group (2011) Prevalence of symptomatic hip and knee osteoarthritis: a two-phase population-based survey. Osteoarthritis Cartilage 19, 1314–1322 [DOI] [PubMed] [Google Scholar]
- 2. Louati K., Vidal C., Berenbaum F., and Sellam J. (2015) Association between diabetes mellitus and osteoarthritis: systematic literature review and meta-analysis. RMD Open 1, e000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schett G., Kleyer A., Perricone C., Sahinbegovic E., Iagnocco A., Zwerina J., Lorenzini R., Aschenbrenner F., Berenbaum F., D'Agostino M. A., Willeit J., and Kiechl S. (2013) Diabetes is an independent predictor for severe osteoarthritis: results from a longitudinal cohort study. Diabetes Care 36, 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hart D. J., Doyle D. V., and Spector T. D. (1995) Association between metabolic factors and knee osteoarthritis in women: the Chingford Study. J. Rheumatol. 22, 1118–1123 [PubMed] [Google Scholar]
- 5. Hui W., Young D. A., Rowan A. D., Xu X., Cawston T. E., and Proctor C. J. (2016) Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum Dis. 75, 449–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu-Bryan R., and Terkeltaub R. (2015) Emerging regulators of the inflammatory process in osteoarthritis. Nat. Rev. Rheumatol. 11, 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Donath M. Y., and Mandrup-Poulsen T. (2008) The use of interleukin-1-receptor antagonists in the treatment of diabetes mellitus. Nat. Clin. Pract. Endocrinol. Metab 4, 240–241 [DOI] [PubMed] [Google Scholar]
- 8. Donath M. Y., and Shoelson S. E. (2011) Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107 [DOI] [PubMed] [Google Scholar]
- 9. Herder C., Dalmas E., Böni-Schnetzler M., and Donath M. Y. (2015) The IL-1 pathway in type 2 diabetes and cardiovascular complications. Trends Endocrinol. Metab. 26, 551–563 [DOI] [PubMed] [Google Scholar]
- 10. Li J., Huang M., and Shen X. (2014) The association of oxidative stress and pro-inflammatory cytokines in diabetic patients with hyperglycemic crisis. J. Diabetes Complications 28, 662–666 [DOI] [PubMed] [Google Scholar]
- 11. Giacco F., and Brownlee M. (2010) Oxidative stress and diabetic complications. Circ. Res. 107, 1058–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhuo Q., Yang W., Chen J., and Wang Y. (2012) Metabolic syndrome meets osteoarthritis. Nat. Rev. Rheumatol. 8, 729–737 [DOI] [PubMed] [Google Scholar]
- 13. Yan L. J. (2014) Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J. Diabetes Res. 2014, 137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X., Hunter D., Xu J., and Ding C. (2015) Metabolic triggered inflammation in osteoarthritis. Osteoarthritis Cartilage 23, 22–30 [DOI] [PubMed] [Google Scholar]
- 15. Courties A., Gualillo O., Berenbaum F., and Sellam J. (2015) Metabolic stress-induced joint inflammation and osteoarthritis. Osteoarthritis Cartilage 23, 1955–1965 [DOI] [PubMed] [Google Scholar]
- 16. Denzer I., Münch G., and Friedland K. (2016) Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds. Pharmacol. Res. 103, 80–94 [DOI] [PubMed] [Google Scholar]
- 17. Uruno A., Furusawa Y., Yagishita Y., Fukutomi T., Muramatsu H., Negishi T., Sugawara A., Kensler T. W., and Yamamoto M. (2013) The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 33, 2996–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soares M. P., and Ribeiro A. M. (2015) Nrf2 as a master regulator of tissue damage control and disease tolerance to infection. Biochem. Soc. Trans. 43, 663–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bruns D. R., Drake J. C., Biela L. M., Peelor F. F. 3rd, Miller B. F., and Hamilton K. L. (2015) Nrf2 signaling and the slowed aging phenotype: evidence from long-lived models. Oxid. Med. Cell Longev. 2015, 732596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seo H. A., and Lee I. K. (2013) The role of Nrf2: adipocyte differentiation, obesity, and insulin resistance. Oxid Med. Cell Longev. 2013, 184598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schneider K. S., and Chan J. Y. (2013) Emerging role of Nrf2 in adipocytes and adipose biology. Adv. Nutr. 4, 62–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wruck C. J., Fragoulis A., Gurzynski A., Brandenburg L. O., Kan Y. W., Chan K., Hassenpflug J., Freitag-Wolf S., Varoga D., Lippross S., and Pufe T. (2011) Role of oxidative stress in rheumatoid arthritis: insights from the Nrf2-knockout mice. Ann. Rheum. Dis. 70, 844–850 [DOI] [PubMed] [Google Scholar]
- 23. Maicas N., Ferrándiz M. L., Brines R., Ibáñez L., Cuadrado A., Koenders M. I., van den Berg W. B., and Alcaraz M. J. (2011) Deficiency of Nrf2 accelerates the effector phase of arthritis and aggravates joint disease. Antioxid. Redox. Signal 15, 889–901 [DOI] [PubMed] [Google Scholar]
- 24. Niture S. K., Khatri R., and Jaiswal A. K. (2014) Regulation of Nrf2-an update. Free Radic. Biol. Med. 66, 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mawatari T., Nakamichi I., Suenaga E., Maloney W. J., and Smith R. L. (2013) Effects of heme oxygenase-1 on bacterial antigen-induced articular chondrocyte catabolism in vitro. J. Orthop. Res. 31, 1943–1949 [DOI] [PubMed] [Google Scholar]
- 26. Guillén M., Megías J., Gomar F., and Alcaraz M. (2008) Haem oxygenase-1 regulates catabolic and anabolic processes in osteoarthritic chondrocytes. J. Pathol. 214, 515–522 [DOI] [PubMed] [Google Scholar]
- 27. Clérigues V., Guillén M. I., Castejón M. A., Gomar F., Mirabet V., and Alcaraz M. J. (2012) Heme oxygenase-1 mediates protective effects on inflammatory, catabolic, and senescence responses induced by interleukin-1β in osteoarthritic osteoblasts. Biochem. Pharmacol. 83, 395–405 [DOI] [PubMed] [Google Scholar]
- 28. Clérigues V., Murphy C. L., Guillén M. I., and Alcaraz M. J. (2013) Haem oxygenase-1 induction reverses the actions of interleukin-1β on hypoxia-inducible transcription factors and human chondrocyte metabolism in hypoxia. Clin. Sci. 125, 99–108 [DOI] [PubMed] [Google Scholar]
- 29. Kim H. A., Lee K. B., and Bae S. C. (2005) The mechanism of low-concentration sodium nitroprusside-mediated protection of chondrocyte death. Arthritis Res. Ther. 7, R526–R535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benallaoua M., François M., Batteux F., Thelier N., Shyy J. Y., Fitting C., Tsagris L., Boczkowski J., Savouret J. F., Corvol M. T., Poiraudeau S., and Rannou F. (2007) Pharmacologic induction of heme oxygenase 1 reduces acute inflammatory arthritis in mice. Arthritis Rheum. 56, 2585–2594 [DOI] [PubMed] [Google Scholar]
- 31. He M., Pan H., Xiao C., and Pu M. (2013) Roles for redox signaling by NADPH oxidase in hyperglycemia-induced heme oxygenase-1 expression in the diabetic retina. Invest. Ophthalmol. Vis. Sci. 54, 4092–4101 [DOI] [PubMed] [Google Scholar]
- 32. Adaikalakoteswari A., Balasubramanyam M., Rema M., and Mohan V. (2006) Differential gene expression of NADPH oxidase (p22phox) and hemoxygenase-1 in patients with type 2 diabetes and microangiopathy. Diabet. Med. 23, 666–674 [DOI] [PubMed] [Google Scholar]
- 33. Nowak W. N., Borys S., Kusińska K., Bukowska-Strakova K., Witek P., Koblik T., Józkowicz A., Małecki M. T., and Dulak J. (2014) Number of circulating pro-angiogenic cells, growth factor and anti-oxidative gene profiles might be altered in type 2 diabetes with and without diabetic foot syndrome. J. Diabetes Investig. 5, 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barbagallo I., Vanella A., Peterson S. J., Kim D. H., Tibullo D., Giallongo C., Vanella L., Parrinello N., Palumbo G. A., Di Raimondo F., Abraham N. G., and Asprinio D. (2010) Overexpression of heme oxygenase-1 increases human osteoblast stem cell differentiation. J. Bone Miner. Metab. 28, 276–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li M., Kim D. H., Tsenovoy P. L., Peterson S. J., Rezzani R., Rodella L. F., Aronow W. S., Ikehara S., and Abraham N. G. (2008) Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes 57, 1526–1535 [DOI] [PubMed] [Google Scholar]
- 36. Laiguillon M. C., Courties A., Houard X., Auclair M., Sautet A., Capeau J., Fève B., Berenbaum F., and Sellam J. (2015) Characterization of diabetic osteoarthritic cartilage and role of high glucose environment on chondrocyte activation: toward pathophysiological delineation of diabetes mellitus-related osteoarthritis. Osteoarthritis Cartilage 23, 1513–1522 [DOI] [PubMed] [Google Scholar]
- 37. Chang S. H., Barbosa-Tessmann I., Chen C., Kilberg M. S., and Agarwal A. (2002) Glucose deprivation induces heme oxygenase-1 gene expression by a pathway independent of the unfolded protein response. J. Biol. Chem. 277, 1933–1940 [DOI] [PubMed] [Google Scholar]
- 38. Ozen M., Zhao H., Lewis D. B., Wong R. J., and Stevenson D. K. (2015) Heme oxygenase and the immune system in normal and pathological pregnancies. Front. Pharmacol. 6, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berenbaum F. (2011) Diabetes-induced osteoarthritis: from a new paradigm to a new phenotype. Ann. Rheum. Dis. 70, 1354–1356 [DOI] [PubMed] [Google Scholar]
- 40. O'Connell M. A., and Hayes J. D. (2015) The Keap1/Nrf2 pathway in health and disease: from the bench to the clinic. Biochem. Soc. Trans. 43, 687–689 [DOI] [PubMed] [Google Scholar]
- 41. Mishra M., Zhong Q., and Kowluru R. A. (2014) Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic. Biol. Med. 75, 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim H. A., Yeo Y., Jung H. A., Jung Y. O., Park S. J., and Kim S. J. (2012) Phase 2 enzyme inducer sulphoraphane blocks prostaglandin and nitric oxide synthesis in human articular chondrocytes and inhibits cartilage matrix degradation. Rheumatology 51, 1006–1016 [DOI] [PubMed] [Google Scholar]
- 43. Cai D., Yin S., Yang J., Jiang Q., and Cao W. (2015) Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Arthritis Res. Ther. 17, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moon S. J., Park J. S., Woo Y. J., Lim M. A., Kim S. M., Lee S. Y., Kim E. K., Lee H. J., Lee W. S., Park S. H., Jeong J. H., Park S. H., Kim H. Y., Cho M. L., and Min J. K. (2014) Rebamipide suppresses collagen-induced arthritis through reciprocal regulation of th17/treg cell differentiation and heme oxygenase 1 induction. Arthritis Rheumatol. 66, 874–885 [DOI] [PubMed] [Google Scholar]
- 45. Zhong Q., Mishra M., and Kowluru R. A. (2013) Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Invest Ophthalmol. Vis. Sci. 54, 3941–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abraham N. G., Kushida T., McClung J., Weiss M., Quan S., Lafaro R., Darzynkiewicz Z., and Wolin M. (2003) Heme oxygenase-1 attenuates glucose-mediated cell growth arrest and apoptosis in human microvessel endothelial cells. Circ. Res. 93, 507–514 [DOI] [PubMed] [Google Scholar]
- 47. Kaspar J. W., and Jaiswal A. K. (2010) Antioxidant-induced phosphorylation of tyrosine 486 leads to rapid nuclear export of Bach1 that allows Nrf2 to bind to the antioxidant response element and activate defensive gene expression. J. Biol. Chem. 285, 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bitar M. S., and Al-Mulla F. (2011) A defect in Nrf2 signaling constitutes a mechanism for cellular stress hypersensitivity in a genetic rat model of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 301, E1119–E1129 [DOI] [PubMed] [Google Scholar]
- 49. Cheng X., Chapple S. J., Patel B., Puszyk W., Sugden D., Yin X., Mayr M., Siow R. C., and Mann G. E. (2013) Gestational diabetes mellitus impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero. Diabetes 62, 4088–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ochiai S., Mizuno T., Deie M., Igarashi K., Hamada Y., and Ochi M. (2008) Oxidative stress reaction in the meniscus of Bach 1-deficient mice: potential prevention of meniscal degeneration. J. Orthop. Res. 26, 894–898 [DOI] [PubMed] [Google Scholar]
- 51. Takada T., Miyaki S., Ishitobi H., Hirai Y., Nakasa T., Igarashi K., Lotz M. K., and Ochi M. (2015) Bach1 deficiency reduces severity of osteoarthritis through upregulation of heme oxygenase-1. Arthritis Res. Ther. 17, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hama M., Kirino Y., Takeno M., Takase K., Miyazaki T., Yoshimi R., Ueda A., Itoh-Nakadai A., Muto A., Igarashi K., and Ishigatsubo Y. (2012) Bach1 regulates osteoclastogenesis in a mouse model via both heme oxygenase 1-dependent and heme oxygenase 1-independent pathways. Arthritis Rheum. 64, 1518–1528 [DOI] [PubMed] [Google Scholar]
- 53. Berenbaum F. (2014) Does broccoli protect from osteoarthritis? Joint Bone Spine 81, 284–286 [DOI] [PubMed] [Google Scholar]
- 54. Davidson R. K., Jupp O., de Ferrars R., Kay C. D., Culley K. L., Norton R., Driscoll C., Vincent T. L., Donell S. T., Bao Y., and Clark I. M. (2013) Sulforaphane represses matrix-degrading proteases and protects cartilage from destruction in vitro and in vivo. Arthritis Rheum. 65, 3130–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Altman R., Asch E., Bloch D., Bole G., Borenstein D., Brandt K., Christy W., Cooke T. D., Greenwald R., and Hochberg M. (1986) Development of criteria for the classification and reporting of osteoarthritis: classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 29, 1039–1049 [DOI] [PubMed] [Google Scholar]
- 56. Laiguillon M. C., Houard X., Bougault C., Gosset M., Nourissat G., Sautet A., Jacques C., Berenbaum F., and Sellam J. (2014) Expression and function of visfatin (Nampt), an adipokine-enzyme involved in inflammatory pathways of osteoarthritis. Arthritis Res. Ther. 16, R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gosset M., Berenbaum F., Thirion S., and Jacques C. (2008) Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 3, 1253–1260 [DOI] [PubMed] [Google Scholar]
- 58. Salvat C., Pigenet A., Humbert L., Berenbaum F., and Thirion S. (2005) Immature murine articular chondrocytes in primary culture: a new tool for investigating cartilage. Osteoarthritis Cartilage 13, 243–249 [DOI] [PubMed] [Google Scholar]
- 59. Rousset F., Nguyen M. V., Grange L., Morel F., and Lardy B. (2013) Heme oxygenase-1 regulates matrix metalloproteinase MMP-1 secretion and chondrocyte cell death via Nox4 NADPH oxidase activity in chondrocytes. PLoS ONE 8, e66478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Megías J., Guillén M. I., Clérigues V., Rojo A. I., Cuadrado A., Castejón M. A., Gomar F., and Alcaraz M. J. (2009) Heme oxygenase-1 induction modulates microsomal prostaglandin E synthase-1 expression and prostaglandin E2 production in osteoarthritic chondrocytes. Biochem. Pharmacol. 77, 1806–1813 [DOI] [PubMed] [Google Scholar]
- 61. Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., and Nabeshima Y. (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322 [DOI] [PubMed] [Google Scholar]
- 62. El Ali Z., Gerbeix C., Hemon P., Esser P. R., Martin S. F., Pallardy M., and Kerdine-Römer S. (2013) Allergic skin inflammation induced by chemical sensitizers is controlled by the transcription factor Nrf2. Toxicol. Sci. 134, 39–48 [DOI] [PubMed] [Google Scholar]
- 63. Masuko-Hongo K., Berenbaum F., Humbert L., Salvat C., Goldring M. B., and Thirion S. (2004) Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 50, 2829–2838 [DOI] [PubMed] [Google Scholar]
- 64. Lau A., Tian W., Whitman S. A., and Zhang D. D. (2013) The predicted molecular weight of Nrf2: it is what it is not. Antioxid. Redox Signal. 18, 91–93 [DOI] [PMC free article] [PubMed] [Google Scholar]