

Abstract
Double strand breaks (DSBs) represent highly deleterious DNA damage and need to be accurately repaired. Homology-directed repair and non-homologous end joining (NHEJ) are the two major DSB repair pathways that are highly conserved from yeast to mammals. The choice between these pathways is largely based on 5′ to 3′ DNA resection, and NHEJ proceeds only if resection has not been initiated. In yeast, yKu70/80 rapidly localizes to the break, protecting DNA ends from nuclease accessibility, and recruits additional NHEJ factors, including Nej1 and Lif1. Cells harboring the nej1-V338A mutant exhibit NHEJ-mediated repair deficiencies and hyper-resection 0.15 kb from the DSB that was dependent on the nuclease activity of Dna2–Sgs1. The integrity of Nej1 is also important for inhibiting long-range resection, 4.8 kb from the break, and for preventing the formation of large genomic deletions at sizes >700 bp around the break. Nej1V338A localized to a DSB similarly to WT Nej1, indicating that the Nej1–Lif1 interaction becomes critical for blocking hyper-resection mainly after their recruitment to the DSB. This work highlights that Nej1 inhibits 5′ DNA hyper-resection mediated by Dna2–Sgs1, a function distinct from its previously reported role in supporting Dnl4 ligase activity, and has implications for repair pathway choice and resection regulation upon DSB formation.
Keywords: cell cycle, DNA damage, DNA recombination, DNA repair, genomic instability
Introduction
DNA double strand breaks (DSBs)2 are one of the most deleterious forms of DNA damage. Aberrant repair of these lesions results in mutations, genomic instability, and cell death. In Saccharomyces cerevisiae, DSBs are repaired predominately through homology-directed repair (HDR); however, non-homologous end joining (NHEJ), which requires little or no homology, is conserved and involves direct religation of the DNA ends. yKu70/80 (Ku), Mre11–Rad50–Xrs2 (MRX), Lif1–Dnl4, and Nej1 are the core components of this pathway. It is becoming clear that the functionality of these factors impacts repair pathway choice (for a review, see Ref. 1). Ku and MRX localize first to the break site independently of one another (2). MRX tethers the DNA ends to prevent their separation (3, 4), whereas Ku has high affinity for DSB ends and recruits other factors, including Lif1–Dnl4 and Nej1 (2, 5, 6). Dnl4 interacts with Lif1 and ligates DNA ends to complete repair (7–11). Nej1 has no known enzymatic activity, but it stimulates the ligase activity of Dnl4–Lif1 by interacting with Lif1 and promoting Dnl4 deadenylation (8). Cells lacking NEJ1 are as defective in end joining as ku70Δ and dnl4Δ mutant cells, underscoring the importance of Nej1 in NHEJ (8, 12, 13). Nej1 and Lif1 physically interact via specific residues in the C-terminal region of Nej1 (12, 14–16); however, the importance of their association in repair pathway choice or end resection has not been fully investigated.
The choice between HDR and NHEJ is largely dependent on the regulation of 5′ resection at DSB ends (17). Nej1 and Lif1 enhance Ku stability at the unresected DNA ends (8); however, these observations stem from work in cells carrying full deletions of NEJ1 and LIF1, which could potentially impinge on the stable association of other factors at the break. In G1, NHEJ is the predominant pathway of repair as Mre11 endonuclease activity is inactive and Ku binds the ends of DNA, preventing resection (2, 18–20). In S/G2, when a homologous template is present, Sae2 and Mre11 are activated to initiate resection, which exposes a short track of 3′ ssDNA on each side of the DSB (21). MRX resects 100–300 nucleotides of DNA (22–26). As Ku has reduced affinity for ssDNA ends, it dissociates. The exposed ends are substrates for resection primarily by Exonuclease 1 (Exo1) but also via Dna2–Sgs1 nuclease-helicase as these nucleases share functional redundancy to promote long-range 5′ → 3′ resection (22–24, 26). Once resection is initiated, NHEJ is no longer an option as the ssDNA generated is quickly bound by Rad51, which drives the search for homology and HDR-mediated repair (27). Even in the absence of MRX or Sae2 function, DNA end protection by Ku is not indefinite as resection can be initiated via Exo1 and Dna2-Sgs1 but with delayed timing (26).
Alternative pathways can be utilized to promote survival if cells are unable to repair by NHEJ or HDR. One pathway, microhomology-mediated end joining (MMEJ), depends on a short region of homology (5–25 bp) for repair and is Rad52-independent (1, 28). MMEJ results in small insertions or deletions adjacent to the break site (29–31). Alternatively, single strand annealing (SSA) can be used to repair DSBs when NHEJ and HDR are deficient or no homologous sister chromatid template is located for repair (28, 32). In SSA, homologous ends recognize one another and repair the DSB but with a large deletion of genomic material between the regions of homology on adjacent strands. Yeast cells unable to repair through conventional NHEJ or HDR shift to these alternative pathways to survive, but this comes at the expense of genomic integrity. Thus, functional NHEJ and regulated resection for HDR are essential for proper pathway choice, genomic stability, and cell survival.
Here we characterize Nej1V338A, a mutant we generated previously (16). Our results indicate that the binding between Nej1 and Lif1 subsequent to their localization at the DSB is critical for regulating nuclease function and 5′ resection. We observed that the level of resection in nej1-V338A mutant cells was significantly above the level observed in wild type and similar to that of nej1Δ and ku70Δ mutants. Moreover, we found that Nej1 is important for inhibiting hyper-resection that is mediated by Dna2-Sgs1.
Results
Nej1–Lif1 interactions at DSBs prevent 5′ resection
We previously generated a Val → Ala mutation in Nej1 at amino acid 338 (Nej1V338A) that resulted in a marked reduction in its interaction with Lif1 (16); however, there was >90% efficiency in Dnl4-dependent end-joining activity compared with the ∼2% repair rate in cells where NEJ1 was deleted. Surprisingly, even though nej1-V338A mutant cells were proficient for Dnl4 ligase activity, cell survival significantly decreased when a DSB in the genome was generated by the HO endonuclease (16). These observations suggested that the interaction between Nej1 and Lif1 contributes to repair through a mechanism distinct from DNA ligation (16).
Similar to ku70Δ mutant cells, 5′ resection at a DSB increases when NEJ1, LIF1, or DNL4 has been deleted, underscoring the importance of these factors in stabilizing Ku binding at the DSB (6, 8, 33, 34). Determining the functionality of each individual protein, however, has been challenging because of the interdependency among these factors for their stable association with the DSB. To bring further insight into the role of Nej1 in NHEJ, we characterized the nej1-V338A point mutant at the DSB. We monitored 5′ resection levels in cells blocked in G1 by α-factor because HDR, the central repair pathway, is activated by Cdk1 only when cells go into S phase and is inhibited in G1-arrested cells (35). Moreover, DNA processing was measured in cells with hmlΔ hmrΔ, which lack a homologous template required for canonical HDR repair (Fig. 1A and Ref. 36). Resection levels were determined by a quantitative PCR-based approach after HO-induced DSB formation and relied on two RsaI cut sites located 0.15 and 4.8 kb from the DSB (Fig. 1B and Ref. 37). If 5′ resection has progressed beyond the RsaI recognition site, then the region would not be cleaved and would be amplified by PCR. The experimental design measures 5′ resection at the DSB 6 h after HO induction. The initial kinetics of resection are highly dynamic, and our data do not capture these subtleties; however, the 6-h time point did allow for the accurate determination of stabilized and consistent resection levels across all mutant combinations at both distances from the DSB. Consistent with previous reports and as a control for our experiments, hyper-resection was observed in cells lacking KU70 (Fig. 1, C and D, and Ref. 19). The level of resection in nej1-V338A mutants also increased at both distances from the DSB and was similar to the levels observed in nej1Δ and lif1Δ mutant cells (Fig. 1, C and D). The loss of LIF1 showed less of an impact at 4.8 kb compared with the loss of NEJ1; however, the reason(s) for this difference is currently not clear. Moreover, the increase in resection was not due to NHEJ mutant cells escaping into S phase as flow cytometry indicated all mutants arrested similarly to wild-type cells with > 90% in G1 when resection was monitored (supplemental Table S1). Nej1 was previously shown to promote NHEJ by protecting DSB ends from resection (19), and our findings implicate the association of Nej1–Lif1 in end protection as well (6, 8).
Figure 1.

The Nej1–Lif1 interaction prevents end resection at an HO-induced DSB. A and B, schematic showing regions around the HO cut site on chromosome (Chr) III. MATα1 and MATα2 loci are labeled and relevant for the mating type assay. CEN is the centromere of Chr III. The ChIP probe used in this study is 0.6 kb from the DSB. The RsaI sites used in the qPCR resection assays, 0.15 and 4.8 kb from the DSB, are also indicated. C–F, resection of DNA 0.15 and 4.8 kb away from the HO DSB as measured by percentage of ssDNA 0 and 6 h post-DSB induction in G1 cells in wild type (JC3585), nej1Δ (JC3884), nej1-V338A (JC3896), yku70Δ (JC3632) lif1Δ (JC3906), and nej1Δ yku70Δ (JC3928). Error bars represent the standard error of three replicates. Significance was determined using one-tailed, unpaired Student's t test. All mutants were compared with wild-type cells (*, p < 0.05; **, p < 0.01). The enrichment of Nej1-13Myc (JC1687) and Nej1V338A-13Myc (JC3160) (G) or Lif1-6HA in wild type (JC2665), nej1Δ (JC2884), and nej1-V338A (JC3828) cells (H) was determined at 0.6 kb from the DSB. The -fold enrichment is normalized to recovery at the SMC2 locus. Error bars represent the standard deviation of three replicates. Significance was determined using one-tailed, unpaired Student's t test (*, p < 0.05). The non-tagged control wild type (JC727), nej1Δ (JC1342), and nej1-V338A (JC2659) cells are shown in supplemental Fig. 1, A and B.
The strongest evidence that Nej1 contributes to Ku functionality comes from the increase in Rad51 levels at the DSB in nej1Δ mutant cells (8). Previous reports showed that direct measurements of Ku were similar in wild type and nej1Δ mutants by standard ChIP methods (8), and it was only when ChIP was performed under non-cross-linking conditions that a decrease in DNA-bound Ku was detected in cells lacking NEJ1. To address whether there is a Ku-independent role for Nej1 in resection, NEJ1 and KU70 were both disrupted. Resection was indistinguishable between ku70Δ single and ku70Δ nej1Δ double mutant cells (Fig. 1, E and F), supporting a model whereby Nej1 does indeed prevent resection via a pathway epistatic with Ku.
To determine whether the mutant protein is properly recruited to the break, we performed ChIP at the DSB after HO induction. The level of Nej1V338A-Myc recovery was similar to wild-type Nej1-Myc, suggesting that the resection defect with nej1-V338A is not attributable to failed recruitment but rather its functionality after localization (Fig. 1G and supplemental Fig. S1A). In vitro work previously showed that Lif1 binding to Ku-bound DNA was not dependent on Nej1, but the interaction was enhanced with the addition of Nej1 (8). The importance of Nej1 for Lif1 recovery at a DSB in vivo has not been reported. Upon HO-induced DSB formation, there was no significant difference in the recovery of Lif1-HA in nej1Δ mutants compared with wild-type cells, and there was no difference in Lif1-HA levels at the break in cells in which NEJ1 was deleted compared with the point mutant nej1-V338A (Fig. 1H and supplemental Fig. S1B). However, compared with wild type, there was a ∼50% decrease of Lif1-HA recovery in nej1-V338A mutant cells (Fig. 1H). We conclude that Nej1 is enriched at the DSB independently of its interaction with Lif1 but that through their association Nej1 may maximize Lif1 retention at the break.
MRX-dependent resection events and interactions between MRX and Nej1
In the absence of KU70, end protection is lost, and resection increases at both 0.15 and 4.8 kb from the DSB (6, 8) (Fig. 1, C and D). The initial resection of 100–300 nucleotides is regulated by MRX, and consistent with previous reports, we observed that rad50Δ mutant cells exhibited a decrease in resection that was restored back to wild-type levels in ku70Δ rad50Δ double mutants (Fig. 2, A and B, and Refs. 20, 22–25, 27, 35, and 36). However, when the deletion of RAD50 was combined with either nej1Δ or nej1-V338A, the resection levels in double mutant cells remained indistinguishable from rad50Δ (Fig. 2, A–D). A similar decrease in resection was also observed when rad50Δ was combined with lif1Δ (Fig. 2, E and F). Consistent with previous literature, the level of resection in ku70Δ rad50Δ above that of rad50Δ was Exo1-dependent (supplemental Fig. S2, A and B, and Ref. 20). Thus, there is a distinction when combining rad50Δ with the loss of KU70 compared with the loss of factors that support Ku stability. The DNA ends of nej1Δ rad50Δ and nej1-V338A rad50Δ mutant cells are not substrates for Exo1, suggesting that Ku still provides a level of DNA end protection when NEJ1 is deleted or when Nej1–Lif1 interactions are disrupted.
Figure 2.
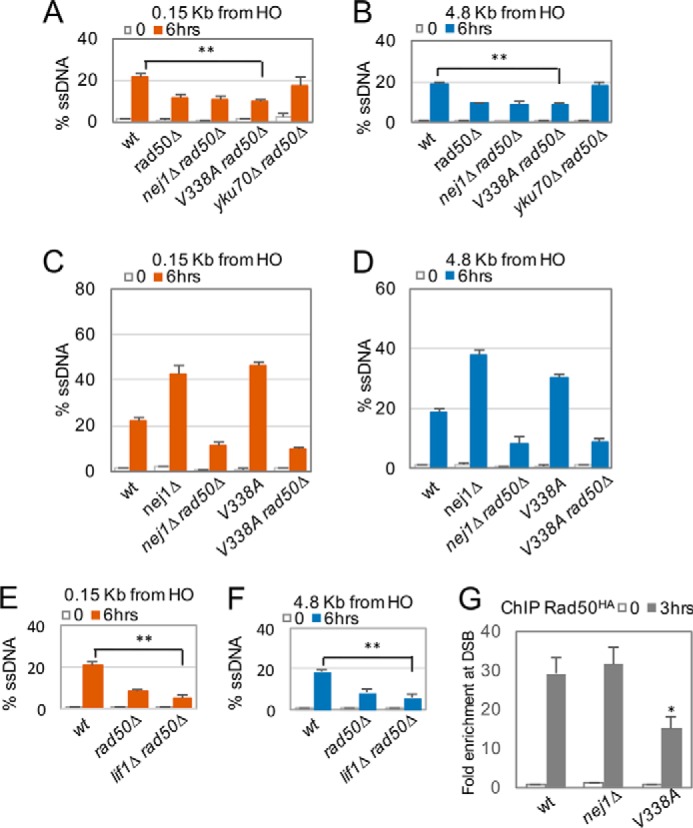
Neither nej1Δ nor nej1-V338A rescues the resection defect in rad50Δ mutant cells. Resection of DNA 0.15 and 4.8 kb away from the HO DSB was measured by percentage of ssDNA 0 and 6 h after DSB induction in wild type (JC3585), rad50Δ (JC3882), nej1Δ rad50Δ (JC3887), nej1-V338A rad50Δ (JC3897), and yku70Δ rad50Δ (JC3878) (A and B) and in nej1Δ (JC3884), nej1-V338A (JC3896), and lif1Δ rad50Δ (JC3907) cells (C–F) in G1 stage of the cell cycle. Error bars represent standard error of three replicates. Significance was determined using one-tailed, unpaired Student's t test where mutants were compared with wild-type cells for changes in resection levels (**, p < 0.01). G, ChIP assay as described in Fig. 1, E and F, was performed to measure the enrichment of Rad50-3HA at a DSB in wild type (JC3306), nej1Δ (JC3307), and nej1-V338A cells (JC3347). Error bars represent the standard deviation of three replicates. Significance was determined using one-tailed, unpaired Student's t test. All mutants were compared with wild-type cells (*, p < 0.05).
Ku and MRX are believed to function antagonistically in repair pathway choice (2, 33). MRX promotes Ku removal from unrepaired DSBs (2, 38, 39), and although Ku stabilizes MRX at the break (6), Ku inhibits 5′ end degradation by MRX. Interestingly, interactions between Lif1 and Xrs2 actually enhance the binding of core NHEJ factors to Ku-bound DNA (4, 5, 40). Previous work showed that the level of Rad50 at the break site decreased in cells lacking LIF1 (6). We wanted to investigate whether Nej1 influenced MRX binding at the DSB; therefore, we performed ChIP on Rad50-HA. In nej1-V338A mutant cells, Rad50-HA recovery was ∼30% lower relative to wild type and nej1Δ, which were indistinguishable from one another (Fig. 2G and supplemental Fig. S1B). In all, the presence of Nej1V338A at the DSB results in a decrease in the association of Lif1 and MRX with the DSB that is statistically significant (Figs. 1, G and H, and 2G), but it is unclear why their recovery is decreased in nej1-V338A mutant cells but not in cells with the full NEJ1 deletion.
Nej1–Lif1 interactions inhibit Dna2–Sgs1-mediated resection at the DSB site
MRX activity is the central nuclease for initiating resection; however, two redundant nucleases, Exo1 and Dna2, also localize via distinct mechanisms to the DSB and function in long-range resection after MRX starts the process (25, 26, 41). Dna2 requires Sgs1 helicase; as such, the disruption of SGS1 has been used to characterize the role of Dna2-dependent 5′ resection at the DSB because DNA2 is an essential gene involved in Okazaki fragment processing (26, 42–44). In line with previous reports and in contrast to rad50Δ mutant cells, we observed no defects in the initiation of resection at the 0.15 kb site in sgs1Δ or exo1Δ single mutant cells (Fig. 3A).
Figure 3.

Hyper-resection in nej1 mutants at 0.15 kb is Dna2–Sgs1-dependent. A–E, resection of DNA 0.15 kb away from the HO DSB as measured by percentage of ssDNA at 0 and 6 h after DSB induction in wild type (JC3585), rad50Δ (JC3882), sgs1Δ (JC3754), exo1Δ (JC3755), nej1-V338A (JC3896), nej1-V338A exo1Δ (JC3899), nej1Δ (JC3884), nej1Δ exo1Δ (JC3886), yku70Δ (JC3632), yku70Δ exo1Δ (JC3877), nej1-V338A sgs1Δ (JC3898), nej1Δ sgs1Δ (JC3885), and yku70Δ sgs1Δ (JC3850) in G1 stage of the cell cycle. Error bars represent standard error of three replicates. Significance was determined using one-tailed, paired Student's t test (ns, not significant; *, p < 0.05; **, p < 0.01).
We wanted to determine resection dynamics in RAD50+ cells and the contribution of Exo1 and Dna2 nucleases to hyper-resection at 0.15 kb in the nej1 mutants. Resection at 0.15 kb in exo1Δ nej1-V338A double mutants was slightly higher than that measured in nej1-V338A single mutant cells (Fig. 3B). Similar trends were observed in double mutants when exo1Δ was combined with nej1Δ or ku70Δ (Fig. 3C). In contrast, the hyper-resection levels at 0.15 kb in nej1-V338A, nej1Δ, and ku70Δ single mutants were significantly reduced in all double mutants carrying sgs1Δ and similar to the level measured in wild-type cells (Fig. 3, D and E). In all, these data suggest that Nej1 prevents unregulated resection 0.15 kb from the break that is mediated by Dna2–Sgs1 but not Exo1.
Resection at 0.15 kb was dependent on MRX as hyper-resection in all nej1 mutants was reduced to below wild-type levels upon the further deletion of RAD50 (Table 1 and Fig. 4). MRX is critical for Dna2–Sgs1 recruitment to the DSB (21); therefore, to understand whether the physical presence of MRX or its nuclease activity influenced Nej1 inhibition of Dna2–Sgs1, we utilized a nuclease-dead mutant of MRE11, mre11-3. This allele was designed to disrupt the conserved phosphoesterase motif III of Mre11 (18, 45). Cells harboring mre11-3 show DSB repair defects but no disruption in the ability of Mre11 to interact with Rad50 (18, 45). We compared resection in rad50Δ and mre11-3 in combination with the nej1 mutants and with the loss of either SGS1 or EXO1 (Table 1 and Fig. 4). One notable difference in resection at 0.15 kb was observed when comparing nej1Δ exo1Δ mre11-3 (26.4 ± 0.94%) with nej1Δ exo1Δ rad50Δ (4.24 ± 0.92%) (Table 1 and Fig. 4). The level of resection in nej1Δ exo1Δ mre11-3 triple mutants when Dna2–Sgs1 is the only functional nuclease remained elevated above wild-type levels (21.9 ± 1.7%). Thus, the presence of MRX, independent of its nuclease activity, promotes resection that is Dna2–Sgs1-dependent. Surprisingly and in contrast to the triple mutants with nej1Δ, the level of resection in nej1-V338A exo1Δ mre11-3 mutants remained low (10.4 ± 0.9%) (Table 1 and Fig. 4). The full reason(s) for the difference between nej1Δ and nej1-V338A is unclear (see “Discussion”). Nevertheless, the physical presence of Nej1 at the DSB correlates with the inhibition of Dna2–Sgs1-dependent 5′ resection in this mutant combination, which is also highlighted when comparing the level of resection in exo1Δ mre11-3 double mutant cells (9.8 ± 0.2%) with nej1Δ exo1Δ mre11-3 triple mutant cells (26.4 ± 0.94%) (Table 1 and Fig. 4).
Table 1.
Percentage of ssDNA 0.15 kb from the DSB
The percentage of ssDNA after 6-h HO induction (±S.E.) is shown.
| 0.15 kb |
|||
|---|---|---|---|
| RAD50+ MRE11+ | rad50Δ | mre11-3 | |
| WT | 21.9 ± 1.7 | 11.4 ± 1.7 | 7.7 ± 1.7 |
| nej1Δ | 42.8 ± 3.6 | 10.9 ± 1.7 | 14.6 ± 4.1 |
| nej1Δ sgs1Δ | 30.4 ± 0.5 | 7.9 ± 1.4 | 11.8 ± 3.0 |
| nej1Δ exo1Δ | 50.1 ± 0.7 | 4.2 ± 0.9 | 26.4 ± 0.9 |
| nej1-V338A | 46.0 ± 1.8 | 9.4 ± 0.8 | 31.1 ± 2.7 |
| nej1-V338A sgs1Δ | 25.9 ± 8.8 | 10.3 ± 1.2 | 16.9 ± 4.1 |
| nej1-V338A exo1Δ | 55.0 ± 1.9 | 5.5 ± 0.6 | 10.4 ± 0.9 |
| sgs1Δ | 21.0 ± 3.0 | 10.8 ± 2.4 | 15.4 ± 1.4 |
| exo1Δ | 27.1 ± 6.6 | 6.6 ± 1.2 | 9.8 ± 0.2 |
Figure 4.

Comparing hyper-resection between rad50 and mre11-3 mutants. Resection of DNA 0.15kb away from the HO DSB was measured by percentage of ssDNA at 6 h post-DSB induction in G1 cells in WT (JC3585), rad50Δ (JC3882), mre11-3 (JC4010), nej1Δ (JC3884), nej1Δ sgs1Δ (JC3885), nej1Δ exo1Δ (JC3886), nej1-V338A (JC3896), nej1-V338A sgs1Δ (JC3898), nej1-V338A exo1Δ (JC3899), sgs1Δ (JC3754), exo1Δ (JC3755), nej1Δ rad50Δ (JC3887), nej1Δ sgs1Δ rad50Δ (JC3888), nej1Δ exo1Δ rad50Δ (JC3889), nej1-V338A rad50Δ (JC3897), nej1-V338A sgs1Δ rad50Δ (JC3900), nej1-V338A exo1Δ rad50Δ (JC3901), sgs1Δ rad50Δ (JC3883), exo1Δ rad50Δ (JC3881), nej1Δ mre11-3 (JC4049), nej1Δ sgs1Δ mre11-3 (JC4051), nej1Δ exo1Δ mre11-3 (JC4052), nej1-V338A mre11-3 (JC4179), nej1-V338A sgs1Δ mre11-3 (JC4180), nej1-V338A exo1Δ mre11-3 (JC4181), mre11-3 sgs1Δ (JC4047), and exo1Δ mre11-3 (JC4048). Error bars represent standard error of three replicates. Significance was determined using one-tailed, paired Student's t test (ns, not significant; *, p < 0.05; **, p < 0.01); # denotes statistical significance as compared with wild type at p < 0.01.
Long-range resection defects in nej1 mutants
We next wanted to understand whether the differences in short-range resection (0.15 kb) extended to long-range resection (4.8 kb). The levels of resection decreased at a distance 4.8 kb from the DSB in both sgs1Δ and exo1Δ single mutant cells, which is consistent with previous reports (Fig. 5A and Refs. 25, 26, and 41). Similar to resection levels at 0.15 kb, the loss of SGS1 partially reversed hyper-resection at 4.8 kb in nej1Δ mutants to an intermediate level between nej1Δ and sgs1Δ single mutant cells and similar to wild-type cells (Fig. 5, B and C). The level of resection in nej1-V338A sgs1Δ double mutant cells was indistinguishable from wild type and above the level measured in sgs1Δ mutants (Fig. 5, B and C).
Figure 5.

Hyper-resection in nej1 mutants at 4.8 kb is suppressed by deletion of SGS1 or Exo1. A–E, resection of DNA 0.15 and 4.8 kb away from the HO DSB as measured by percentage of ssDNA at 0 and 6 h post-DSB induction in G1 cells in wild type (JC3585), rad50Δ (JC3882), sgs1Δ (JC3754), exo1Δ (JC3755), nej1-V338A (JC3896), nej1-V338A exo1Δ (JC3899), nej1Δ (JC3884), nej1Δ exo1Δ (JC3886), exo1Δ yku70Δ (JC3877), nej1-V338A sgs1Δ (JC3898), nej1Δ sgs1Δ (JC3885), and yku70Δ sgs1Δ (JC3850). Error bars represent standard error of three replicates. Significance was determined using one-tailed, unpaired Student's t test. Double mutants in B and D were compared with nej1 mutant cells, and double mutants in C and E were compared with sgs1Δ or exo1Δ single mutants, respectively (ns, not significant; *, p < 0.05; **, p < 0.01).
A different pattern emerged with the loss of EXO1 (Fig. 5, D and E). The resection levels in nej1Δ exo1Δ and nej1-V338A exo1Δ double mutant strains were reduced to less than 50% of wild-type levels and indistinguishable from exo1Δ single mutants at the 4.8-kb site (Fig. 5, D and E). Long-range resection was decreased in all cells lacking EXO1 to below wild-type levels and in marked contrast to the levels of resection in these double mutants 0.15 kb from the DSB where hyper-resection was observed (Tables 1 and 2 and Fig. 3, B and C). Similar resection trends were observed when exo1Δ or sgs1Δ was combined with ku70Δ (Fig. 5, C and E). In all, our data support Exo1 as the central nuclease for extended long-range resection.
Table 2.
Percentage of ssDNA 4.8 kb from the DSB
The percentage of ssDNA after 6-h HO induction (±S.E.) is shown.
| 4.8 kb |
|||
|---|---|---|---|
| RAD50+ MRE11+ | rad50Δ | mre11-3 | |
| WT | 18.7 ± 1.3 | 9.5 ± 0.4 | 5.6 ± 0.4 |
| nej1Δ | 37.6 ± 2.1 | 8.6 ± 1.8 | 10.9 ± 3.0 |
| nej1Δ sgs1Δ | 26.6 ± 1.3 | 8.0 ± 1.5 | 9.4 ± 4.3 |
| nej1Δ exo1Δ | 6.9 ± 0.7 | 6.0 ± 1.7 | 3.0 ± 0.4 |
| nej1-V338A | 30.1 ± 1.6 | 7.8 ± 1.3 | 29.7 ± 3.3 |
| nej1-V338A sgs1Δ | 18.6 ± 1.3 | 12.7 ± 1.4 | 18.4 ± 3.3 |
| nej1-V338A exo1Δ | 4.4 ± 0.4 | 8.1 ± 1.0 | 8.5 ± 1.2 |
| sgs1Δ | 9.8 ± 4.0 | 10.7 ± 1.3 | 17.9 ± 2.7 |
| exo1Δ | 6.2 ± 3.8 | 8.8 ± 1.2 | 2.9 ± 0.2 |
Nej1–Lif1 prevents Sgs1- and Exo1-dependent large deletions during DSB repair
We next wanted to determine whether the interplay between Nej1 and the nucleases impacts genome stability specifically in the vicinity of a DSB where we had observed aberrant resection and in the same system where one DSB was generated by the HO endonuclease (29). Cells that survive continuous HO cutting (survivors) arise from an imprecise repair event where the HO recognition site becomes disrupted and no DSB is generated. The extent of genomic alterations during repair can be measured by determining the mating type of survivors. Two genes that regulate mating type, MATα1 and MATα2, are located adjacent to the HO-induced DSB (Figs. 1A and 6A). Their expression activates α-type genes and inhibits a-type genes. Conversely, large deletions (>700 bp) around the DSB where both α1 and α2 have been disrupted result in an a-like mating type (Fig. 6A). From three independent experiments, we determined the mating type of >300 independent survivors in each mutant background.
Figure 6.

Large deletions around the DSB are reduced upon deletion of long-range nucleases. A, mating type analysis of survivors from persistent DSB induction assays. Disruption of the MATα1 gene results in a sterile phenotype, and disruption of the MATα2 gene (∼700 bp upstream of HO cut site) results in an a-like phenotype in the mating type assays. The mating phenotype is a readout for the type of repair: mutated HO endonuclease, α survivors; small insertions and deletions, sterile survivors; and >700-bp deletions, a-like survivors. B–F, the mating phenotype of survivors was determined in wild type (JC727), lif1Δ (JC1343), nej1Δ (JC1342), nej1-V338A (JC2659), yku70Δ (JC3848), rad50Δ (JC3313), nej1Δ rad50Δ (JC3314), nej1-V338A rad50Δ (JC3833), sgs1Δ (JC3757), nej1Δ sgs1Δ rad50Δ (JC3761), rad50Δ yku70Δ (JC3835), yku70Δ sgs1Δ rad50Δ (JC3840), nej1-V338A sgs1Δ rad50Δ (JC3846), exo1Δ (JC3767), nej1Δ exo1Δ rad50Δ (JC3770), yku70Δ exo1Δ rad50Δ (JC3841), and nej1-V338A exo1Δ rad50Δ (JC3847). All mating type measurements are in supplemental Fig. S3. Error bars represent standard deviation of at least three replicates. Significance was determined using two-tailed, unpaired Student's t test (*, p < 0.05; **, p < 0.01). Chr, chromosome.
The majority of repair events in wild-type survivors arise from a small 2-bp insertion that alters HO and expression of MATα1 (Fig. 6A), resulting in a yeast strain with a sterile mating type (29). Survivors with mating type α result from mutations inactivating the HO endonuclease and not from mutations at the HO cut site (confirmed by sequencing the HO cut site of six nej1Δ survivors) (29). We determine the mating types in all survivors (Fig. 6B and supplemental Fig. S3, A–C) and were particularly interested in the level of large deletions (a-like survivors) in the various mutant backgrounds. Compared with wild type (1.3%), the percentage of NHEJ mutant survivors exhibiting an increase in large deletions (>700 bp) increased in ku70Δ (9.0%), nej1Δ (12.5%), and lif1Δ (19.1%); however, this did not hold true for nej1-V338A (0.33%) (Fig. 6, B and C, and supplemental Fig. S3D). In rad50Δ (5.1%), the percentage of survivors with large deletions was above that of wild type (Fig. 6D) but less than when other factors central to NHEJ were deleted. The level of large deletions in nej1-V338A rad50Δ double mutant survivors was above the level of the rad50Δ single mutant (Fig. 6D and Ref. (29) and similar to the level measured in cells lacking NEJ1 and nej1Δ rad50Δ (Fig. 6D and supplemental Fig. S3A). In contrast, no large deletions were observed when either of the two long-range resection nucleases, SGS1 or EXO1, was disrupted (Fig. 6, E and F). Moreover, in all mutants containing sgs1Δ, we observed a decrease in the percentage of a-like survivors containing large genome deletions (Fig. 6E and supplemental Fig. S3B). Similarly, all survivors in which EXO1 was further disrupted showed a decrease in the percentage of large deletions around the DSB (Fig. 6F and supplemental Fig. S3C). Importantly, in nej1Δ rad50Δ, yku70Δ rad50Δ, and nej1-V338A rad50Δ double mutants, the increase in large deletions was not overamplified as a result of reduced cell viability because the viability in triple mutants in which SGS1 or EXO1 was further disrupted still remained <2.5% of wild type even though the percentage of large deletion significantly decreased (Table 3 and Fig. 6, E and F). Taken together, our data support a model whereby the increase in large genomic deletions around a DSB is primarily a consequence of aberrant long-range nuclease activity.
Table 3.
Viability upon continuous HO induction
| Strain | Genotype | Survival | % of survival compared with WT |
|---|---|---|---|
| JC727 | Wild type | 3.56 × 10−3 | 100 |
| JC3313 | rad50Δ | 5.77 × 10−5 | 1.62 |
| JC1342 | nej1Δ | 8.21 × 10−5 | 2.31 |
| JC3848 | yku70Δ | 2.38 × 10−5 | 0.67 |
| JC2659 | nej1-V338A | 9.28 × 10−4 | 26.1 |
| JC1343 | lif1Δ | 1.67 × 10−5 | 0.47 |
| JC3314 | nej1Δ rad50Δ | 1.82 × 10−5 | 0.51 |
| JC3835 | yku70Δ rad50Δ | 7.36 × 10−6 | 0.21 |
| JC3833 | nej1-V338A rad50Δ | 2.54 × 10−5 | 0.71 |
| JC3315 | lif1Δ rad50Δ | 5.79 × 10−6 | 0.16 |
| JC3757 | sgs1Δ | 3.81 × 10−3 | 107 |
| JC3761 | nej1Δ sgs1Δ rad50Δ | 3.76 × 10−5 | 1.06 |
| JC3840 | yku70Δ sgs1Δ rad50Δ | 3.29 × 10−5 | 0.92 |
| JC3846 | nej1-V338A sgs1Δ rad50Δ | 6.73 × 10−5 | 1.89 |
| JC3767 | exo1Δ | 3.68 × 10−3 | 103 |
| JC3770 | nej1Δ exo1Δ rad50Δ | 9.72 × 10−5 | 2.54 |
| JC3841 | yku70Δ exo1Δ rad50Δ | 2.01 × 10−5 | 0.63 |
| JC3847 | nej1-V338A exo1Δ rad50Δ | 1.91 × 10−5 | 0.54 |
Discussion
5′–3′ DNA end resection is a critical event in DSB repair. If resection is initiated, then NHEJ is no longer a repair pathway option. Sae2 binds the MRX complex, stimulating Mre11 to initiate resection, and Exo1 is the central nuclease in long-range resection. Dna2–Sgs1 serves as backup to both, and its regulation in DSB resection has remained somewhat obscure, particularly under conditions where MRX and Exo1 are present. Although Exo1 and Dna2–Sgs1 normally perform long-range resection, both can initiate resection at the break site in the absence of RAD50, albeit with delayed kinetics and low efficiency (26). Nucleases are under CDK1 and cell cycle control. In S phase, CDK1 activity increases, and Sae2 and MRX are phosphorylated to initiate resection. This regulatory step promotes HDR-mediated repair during a cell cycle stage when a homolog is present for recombination (35, 46–48). In G1 when CDK1 activity is low or when CDK1 is knocked down, resection is heavily reduced (35, 46–48). It is under these conditions in G1 when NHEJ is the major repair pathway that we observed the impact of Nej1 on 5′ resection.
Our work presents two novel findings for Nej1 at the DSB. First, using a point mutant, nej1-V338A, we showed that the loss of Nej1–Lif1 interactions subsequent to their recruitment to the DSB site leads to 5′ hyper-resection, which offers one explanation for why this allele shows NHEJ defects when it is capable of promoting Dnl4-dependent ligation reactions (16). These data provide an element of mechanistic insight. Nej1 not only needs to be recruited to the DSB, but it must interact with Lif1 through important contacts such as Val-338 to promote NHEJ-mediated repair (Fig. 7). Second, short-range hyper-resection in the nej1 mutants can be attributed to a loss in regulating the activity of Dna2–Sgs1 (Fig. 7B). The hyper-resection level at 0.15 kb in nej1 mutants is restored to levels similar to wild type when SGS1 is additionally disrupted, supporting a role for the NHEJ factors in inhibiting Dna2–Sgs1 that might be distinct from DNA end protection per se. Indeed, Exo1 has a very high affinity for DNA ends not protected by Ku; however, the deletion of EXO1 did not restore resection to wild type like the loss of SGS1. In contrast to Exo1, Dna2–Sgs1 can initiate resection in the presence of Ku (19), and in nej1 mutants, our data suggest that a level of Ku remains bound to the DNA ends (Fig. 2A). The physical presence of Nej1 and its integrity at the DSB are important to inhibit Dna2 activity, and this, by extension, is another way that Nej1 promotes NHEJ.
Figure 7.

Model for Nej1 at the DSB. A, Ku and MRX are the first components to arrive at a DSB and function antagonistically to each other in repair pathway choice (2, 36). Ku has high affinity for DNA ends and recruits Nej1 and Lif1–Dnl4 to the DSB to promote NHEJ repair (2–6). Lif1 also interacts with Xrs2 of the MRX complex, stabilizing the complex at the DSB (4–6, 42). MRX is also required for the recruitment of Dna2–Sgs1 to the DSB (21). Ku binds DNA ends to protect them from 5′ DNA resection initiation mediated by the short-range nuclease Mre11 and subsequent resection elongation by the long-range nucleases Exo1 and Dna2–Sgs1, which have functionally redundant roles. MRX is the central short-range nuclease, and Exo1 is the central long-range nuclease with Dna2–Sgs1 performing a backup function. B, our data indicate that the interaction between Lif1 and Nej1 is critical to inhibit resection initiation by the Dna2 nuclease at DSBs. In nej1-V338A mutant cells, Dna2–Sgs1 is misregulated, and 5′ hyper-resection ensues. The regulation of Dna2–Sgs1 at the DSB is dependent on Ku, but not solely through DNA end protection. All observations with nej1Δ mutants were epistatic with yku70Δ mutants. Our results provide insight to previous observations that Nej1 and Lif1 are believed to enhance Ku stability at unresected DNA ends and that direct measurements of Ku were similar in wild-type and nej1Δ mutant cells, but there was an increase in Rad51 levels at the DSB in nej1Δ cells (8).
In RAD50+ cells, deletion of either SGS1 or EXO1 reversed hyper-resection 4.8 kb from the break site in nej1-V338A mutant cells (Fig. 5, B–E, and Table 2). In all mutant combinations where EXO1 was deleted, there was a marked reduction in long-range resection to below the levels in wild-type cells (Fig. 5, D and E). In nej1-V338A sgs1Δ and nej1Δ sgs1Δ double mutant cells, Exo1 is the functional nuclease 4.8 kb from the DSB, and resection was similar to levels in wild-type cells. Alternatively, in nej1-V338A exo1Δ and nej1Δ exo1Δ double mutant cells where Dna2-Sgs1 is the only functional long-range nuclease available, resection levels were reduced to less than half that measured in wild-type cells (Fig. 5, D and E, and Table 2). These data demonstrate that Exo1 is the primary nuclease 4.8 kb from the break with Dna2–Sgs1 being less efficient. However, the level of resection at 4.8 kb was not merely an extension of resection dynamics closer to the break site in cells in which EXO1 was deleted. For example, in nej1-V338A exo1Δ and nej1Δ exo1Δ double mutant cells, resection levels at 0.15 kb remained elevated above wild type and similar to levels in nej1-V338A and nej1Δ single mutants (Tables 1 and 2). Taken together, our data support a model where in an otherwise wild-type background Dna2–Sgs1 has robust nuclease activity 0.15 kb from the DSB and must be actively inhibited by the core NHEJ factors, including the Nej1–Lif1 interaction, but Exo1 is less efficient and most active in the absence of KU and MRX.
Despite the marked similarities in resection between nej1Δ and nej1-V338A (Figs. 2–4), differences were observed depending on the presence or absence of EXO1 together with mre11-3. For example, resection at both 0.15 and 4.8 kb in nej1Δ mre11-3 double mutant cells was below wild type; however, in nej1-V338A mre11-3 mutants, hyper-resection was observed at both distances (Tables 1 and 2). In contrast, with the further loss of EXO1, nej1-V338A exo1Δ mre11-3 triple mutant cells had a defect in resection at 0.15 and 4.8kb, and the nej1Δ exo1Δ mre11-3 triple mutant cells exhibited hyper-resection at 0.15 kb but a resection defect at 4.8 kb (Tables 1 and 2). One contributing factor might involve the decrease in Lif1 and Rad50 associated with the DSB in nej1-V338A but not nej1Δ mutant cells (Figs. 1H and 2G). This might result in an overall “null-like” phenotype for MRX functionality at the break site. Indeed, nej1-V338A exo1Δ mre11-3 and nej1-V338A exo1Δ rad50Δ triple mutant cells both show similar defects in resection at 0.15 kb (Table 1). However, the situation is likely more complex, involving the dynamic interplay of factors at the DSB, because in EXO1+ cells hyper-resection was observed in nej1-V338A mre11-3 but not nej1-V338A rad50Δ (Table 1).
MRX stabilizes Dna2–Sgs1 recruitment to the break through direct interactions (19, 49). When Sgs1 was the sole nuclease present, resection at 0.15 kb was reduced in exo1Δ mre11-3 and exo1Δ rad50Δ double mutants compared with wild-type cells (Table 1). When considering the disruption of NEJ1, the level of resection was restored to near wild-type levels in nej1Δ exo1Δ mre11-3 triple mutant cells but not nej1Δ exo1Δ rad50Δ mutants, revealing that Nej1 integrity is important to inhibit Dna2–Sgs1 functionality at the break site. Furthermore, these data also confirm that, although its nuclease activity is non-functional, the physical presence of MRX at the DSB is important to improve the efficiency of resection initiation by Dna2-Sgs1, likely through its stabilizing effects on Dna2-Sgs1 (19, 49). As discussed above, the level of resection at 0.15 kb in nej1Δ exo1Δ mre11-3 triple mutant cells was not maintained 4.8 kb from the break (Table 2), suggesting that the efficiency of Dna2–Sgs1 decreases between 0.15 and 4.8 kb. The regulation of Dna2–Sgs1 requires further characterization; however, these results underscore the importance of Exo1 in resection at a distance 4.8 kb from the break even when resection is initiated by Dna2–Sgs1.
For cells to survive chronic HO induction (DSB formation), the break must be repaired imprecisely to prevent further cleavage. This can occur through imprecise NHEJ, MMEJ, or SSA, which requires long regions of homology and results in the loss of a large amount of genetic material during repair. Loss of NEJ1 resulted in an increase in a-like survivors, which exhibit repair with large deletions of >700 bp. The most common type of survivors in cells with the nej1-V338A mutation were similar to wild-type and were repaired with small insertions or deletions (Fig. 6B). This may be explained by the fact that Dnl4 ligase activity remains proficient in nej1-V338A cells (16); thus, the vast majority of survivors repair quickly via error-prone direct ligation through Dnl4 rather than an alternative pathway such as SSA. In addition to mating type determination, we also used PCR to confirm that the few nej1-V338A survivors with an a-like mating type contained large deletions (supplemental Fig. S3D). Additionally, upon the further deletion of RAD50 in nej1-V338A mutant cells, an increase in a-like survivors (large deletions) was observed at levels found in nej1Δ and nej1Δ rad50Δ mutant cells (Fig. 6D). The difference between nej1-V338A and nej1-V338A rad50Δ could be attributed to DNA end tethering by MRX (3). One model is that the untethered DNA ends would be unable to repair efficiently via direct ligation, resulting in an increase in repair via SSA and large genomic loss from an overall increase in end resection by the time DSB ends rejoin (50, 51). Lastly, we characterized whether Sgs1 and Exo1 activity contributed to the formation of large genomic deletions during repair. The number of a-like survivors was reduced in mutant cells when either nuclease was deleted (Fig. 6, E and F). Thus, the Nej1–Lif1 interaction and the stable association of NHEJ factors at the DSB promote genome stability and prevent large genomic rearrangements in part by inhibiting Dna2-Sgs1-dependent hyper-resection at the break site.
Experimental procedures
Yeast strains and media
All strains used in this study are listed in supplemental Table S2. For experiments involving the induction of an HO DSB, cells were grown in YPLGg medium containing 1% yeast extract, 2% Bacto peptone, 2% lactic acid, 3% glycerol, and 0.05% glucose. For the mating type assays, the YPA plates (1% yeast extract, 2% Bacto peptone, and 0.0025% adenine) were supplemented with 2% glucose or 2% galactose (16).
Antibodies
Primary antibodies used were anti-HA (mouse; Sigma, F-7) and anti-Myc (mouse; Cell Signaling Technology, 9B11). Secondary antibodies were coupled to horseradish peroxidase (Bio-Rad).
Mating type assay
Mating type assays were performed as described previously (29). Surviving colonies from continuous growth on YPA + 2% Gal for 3–4 days were restreaked onto YPA + 2% Glc plates and incubated at 30 °C for 1–2 days. Each plate was replica-plated onto fresh YPA + 2% Glc plates with both JC158 and JC159 to test for α and a-like survivors, respectively. These plates were incubated at 30 °C overnight and then replica-plated onto minimal media (MIN) plates containing 2% glucose and 0.6% yeast extract and incubated at 30 °C for 1 day. Colonies growing on the JC158 plate are “α” survivors, colonies growing on the JC159 plate are “a-like” survivors, and colonies that do not grow on either plate are “sterile” survivors.
Chromatin immunoprecipitation
ChIPs were performed as described (16). Chromatin fractionation was performed by spinning the cell lysate at 13,200 rpm for 15 min after which the pellet was resuspended in lysis buffer and sonicated to yield DNA fragments ∼500 bp in length. The sonicated lysate was then incubated with beads + anti-HA (12CA5) or anti-Myc (9E10) antibody or unconjugated beads (control) for 2 h at 4 °C. Quantitative PCR was performed using the Applied Biosystems QuantStudio 6 Flex machine. The reaction contained PerfeCTa qPCR Supermix (Quanta Biosciences Inc.), primers, and 5′ FAM-labeled/3′ TAMRA-labeled probes specific to 0.6 kb away from the cut site (HO2), the cut site to measure cutting efficiency (HO6), or the SMC2 control region. Enrichment was calculated relative to SMC2 and corrected for cut efficiency, which was determined from the loss of PCR product at HO6 after 3-h induction compared with time 0.
qPCR-based resection assay
The resection assay was performed as described (37). This method was previously verified by Southern blotting with ssDNA-binding probes on both sides of the DSB (37). Cells were grown overnight to 1 × 107 cells/ml in YPLG. Cells were pelleted and resuspended in fresh YPLG ± 2% galactose and α-factor to maintain G1 arrest as verified by flow cytometry. At the t = 0 and t = 6-h time points, genomic DNA was purified using standard genomic preparation methods and resuspended in 100 of ml double distilled H2O. Genomic DNA was treated with 0.005 μg/μl RNase A (Sigma) for 45 min at 37 °C. 2 μl of DNA was added to tubes containing Cut Smart buffer with or without RsaI restriction enzyme (New England Biolabs) and incubated at 37 °C for 2 h. Quantitative PCR was performed using the Applied Biosystems QuantStudio 6 Flex machine. PowerUp SYBR Green Master Mix (Applied Biosystems) was used to quantify resection 0.15 and 4.8 kb from the DSB, and Pre1 was used as a negative control. RsaI-cut DNA was normalized to uncut DNA as described previously to quantify the percentage of ssDNA/total DNA (37). Primers and probes are in supplemental Table S3. The primers are located at optimal sites where resection via Mre11 and the delayed effects of Dna2–Sgs1 and Exo1 on close-range resection can be distinguished from their effects on long-range resection.
Author contributions
K. S. S. designed, performed, and analyzed the experiments and wrote the paper. B. L. M. constructed mutant strains and helped in the design of the experiments. S. P. L.-M. contributed to the interpretation of the data and the preparation of the manuscript. J. A. C. conceived and coordinated the study, analyzed the experiments, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. John Petrini for strains and helpful discussions and Drs. Karine Dubrana and Aaron Goodarzi for helpful suggestions.
This work was supported by operating grants from Canadian Institutes of Health Research (CIHR) Grants MOP-82736 and MOP-137062 and Natural Sciences and Engineering Research Council of Canada Grant 418122 (to J. A. C.) and CIHR Grant MOP-13639 (to S. P. L.-M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S3 and Tables S1–S3.
- DSB
- double strand break
- HDR
- homology-directed repair
- HO
- homothallic switching endonuclease
- NHEJ
- non-homologous end joining
- Ku
- yKu70/80
- MRX
- Mre11–Rad50–Xrs2
- ssDNA
- single-stranded DNA
- MMEJ
- microhomology-mediated end joining
- SSA
- single strand annealing
- qPCR
- quantitative PCR.
References
- 1. Daley J. M., Palmbos P. L., Wu D., and Wilson T. E. (2005) Nonhomologous end joining in yeast. Annu. Rev. Genet. 39, 431–451 [DOI] [PubMed] [Google Scholar]
- 2. Wu D., Topper L. M., and Wilson T. E. (2008) Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics 178, 1237–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hopfner K.-P., Craig L., Moncalian G., Zinkel R. A., Usui T., Owen B. A., Karcher A., Henderson B., Bodmer J.-L., McMurray C. T., Carney J. P., Petrini J. H., and Tainer J. A. (2002) The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 418, 562–566 [DOI] [PubMed] [Google Scholar]
- 4. Chen L., Trujillo K., Ramos W., Sung P., and Tomkinson A. E. (2001) Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol. Cell 8, 1105–1115 [DOI] [PubMed] [Google Scholar]
- 5. Palmbos P. L., Wu D., Daley J. M., and Wilson T. E. (2008) Recruitment of Saccharomyces cerevisiae Dnl4-Lif1 complex to a double-strand break requires interactions with Yku80 and the Xrs2 FHA domain. Genetics 180, 1809–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Y., Hefferin M. L., Chen L., Shim E. Y., Tseng H.-M., Kwon Y., Sung P., Lee S. E., and Tomkinson A. E. (2007) Role of Dnl4-Lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat. Struct. Mol. Biol. 14, 639–646 [DOI] [PubMed] [Google Scholar]
- 7. Teo S. H., and Jackson S. P. (1997) Identification of Saccharomyces cerevisiae DNA ligase IV: involvement in DNA double-strand break repair. EMBO J. 16, 4788–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen X., and Tomkinson A. E. (2011) Yeast Nej1 is a key participant in the initial end binding and final ligation steps of nonhomologous end joining. J. Biol. Chem. 286, 4931–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schär P., Herrmann G., Daly G., and Lindahl T. (1997) A newly identified DNA ligase of Saccharomyces cerevisiae involved in RAD52-independent repair of DNA double-strand breaks. Genes Dev. 11, 1912–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilson T. E., Grawunder U., and Lieber M. R. (1997) Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature 388, 495–498 [DOI] [PubMed] [Google Scholar]
- 11. Herrmann G., Lindahl T., and Schär P. (1998) Saccharomyces cerevisiae LIF1: a function involved in DNA double-strand break repair related to mammalian XRCC4. EMBO J. 17, 4188–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frank-Vaillant M., and Marcand S. (2001) NHEJ regulation by mating type is exercised through a novel protein, Lif2p, essential to the ligase IV pathway. Genes Dev. 15, 3005–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valencia M., Bentele M., Vaze M. B., Herrmann G., Kraus E., Lee S. E., Schär P., and Haber J. E. (2001) NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature 414, 666–669 [DOI] [PubMed] [Google Scholar]
- 14. Deshpande R. A., and Wilson T. E. (2007) Modes of interaction among yeast Nej1, Lif1 and Dnl4 proteins and comparison to human XLF, XRCC4 and Lig4. DNA Repair 6, 1507–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ooi S. L., Shoemaker D. D., and Boeke J. D. (2001) A DNA microarray-based genetic screen for nonhomologous end-joining mutants in Saccharomyces cerevisiae. Science 294, 2552–2556 [DOI] [PubMed] [Google Scholar]
- 16. Mahaney B. L., Lees-Miller S. P., and Cobb J. A. (2014) The C-terminus of Nej1 is critical for nuclear localization and non-homologous end-joining. DNA Repair 14, 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Symington L. S. (2016) Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 51, 195–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Foster S. S., Balestrini A., and Petrini J. H. (2011) Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol. Cell. Biol. 31, 4379–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mimitou E. P., and Symington L. S. (2010) Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 29, 3358–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shim E. Y., Chung W.-H., Nicolette M. L., Zhang Y., Davis M., Zhu Z., Paull T. T., Ira G., and Lee S. E. (2010) Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 29, 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cannavo E., and Cejka P. (2014) Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125 [DOI] [PubMed] [Google Scholar]
- 22. Cejka P. (2015) DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J. Biol. Chem. 290, 22931–22938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chung W.-H., Zhu Z., Papusha A., Malkova A., and Ira G. (2010) Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 6, e1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zakharyevich K., Ma Y., Tang S., Hwang P. Y., Boiteux S., and Hunter N. (2010) Temporally and biochemically distinct activities of Exo1 during meiosis: double-strand break resection and resolution of double Holliday junctions. Mol. Cell 40, 1001–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mimitou E. P., and Symington L. S. (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu Z., Chung W.-H., Shim E. Y., Lee S. E., and Ira G. (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krogh B. O., and Symington L. S. (2004) Recombination proteins in yeast. Annu. Rev. Genet. 38, 233–271 [DOI] [PubMed] [Google Scholar]
- 28. McVey M., and Lee S. E. (2008) MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 24, 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moore J. K., and Haber J. E. (1996) Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 16, 2164–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen C., Umezu K., and Kolodner R. D. (1998) Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol. Cell 2, 9–22 [DOI] [PubMed] [Google Scholar]
- 31. Yu X., and Gabriel A. (2003) Ku-dependent and Ku-independent end-joining pathways lead to chromosomal rearrangements during double-strand break repair in Saccharomyces cerevisiae. Genetics 163, 843–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jain S., Sugawara N., Mehta A., Ryu T., and Haber J. E. (2016) Sgs1 and Mph1 helicases enforce the recombination execution checkpoint during DNA double-strand break repair in Saccharomyces cerevisiae. Genetics 203, 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clerici M., Mantiero D., Guerini I., Lucchini G., and Longhese M. P. (2008) The Yku70-Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep. 9, 810–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zierhut C., and Diffley J. F. (2008) Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 27, 1875–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ira G., Pellicioli A., Balijja A., Wang X., Fiorani S., Carotenuto W., Liberi G., Bressan D., Wan L., Hollingsworth N. M., Haber J. E., and Foiani M. (2004) DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431, 1011–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee S. E., Moore J. K., Holmes A., Umezu K., Kolodner R. D., and Haber J. E. (1998) Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94, 399–409 [DOI] [PubMed] [Google Scholar]
- 37. Ferrari M., Dibitetto D., De Gregorio G., Eapen V. V., Rawal C. C., Lazzaro F., Tsabar M., Marini F., Haber J. E., and Pellicioli A. (2015) Functional interplay between the 53BP1-ortholog Rad9 and the Mre11 complex regulates resection, end-tethering and repair of a double-strand break. PLoS Genet. 11, e1004928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wasko B. M., Holland C. L., Resnick M. A., and Lewis L. K. (2009) Inhibition of DNA double-strand break repair by the Ku heterodimer in mrx mutants of Saccharomyces cerevisiae. DNA Repair 8, 162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Balestrini A., Ristic D., Dionne I., Liu X. Z., Wyman C., Wellinger R. J., and Petrini J. H. (2013) The Ku heterodimer and the metabolism of single-ended DNA double-strand breaks. Cell Rep. 3, 2033–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matsuzaki K., Shinohara A., and Shinohara M. (2008) Forkhead-associated domain of yeast Xrs2, a homolog of human Nbs1, promotes nonhomologous end joining through interaction with a ligase IV partner protein, Lif1. Genetics 179, 213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gravel S., Chapman J. R., Magill C., and Jackson S. P. (2008) DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22, 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cejka P., Cannavo E., Polaczek P., Masuda-Sasa T., Pokharel S., Campbell J. L., and Kowalczykowski S. C. (2010) DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nimonkar A. V., Genschel J., Kinoshita E., Polaczek P., Campbell J. L., Wyman C., Modrich P., and Kowalczykowski S. C. (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Niu H., Chung W.-H., Zhu Z., Kwon Y., Zhao W., Chi P., Prakash R., Seong C., Liu D., Lu L., Ira G., and Sung P. (2010) Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 467, 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bressan D. A., Olivares H. A., Nelms B. E., and Petrini J. H. (1998) Alteration of N-terminal phosphoesterase signature motifs inactivates Saccharomyces cerevisiae Mre11. Genetics 150, 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Y., Shim E. Y., Davis M., and Lee S. E. (2009) Regulation of repair choice: Cdk1 suppresses recruitment of end joining factors at DNA breaks. DNA Repair 8, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen X., Niu H., Chung W.-H., Zhu Z., Papusha A., Shim E. Y., Lee S. E., Sung P., and Ira G. (2011) Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol. 18, 1015–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen X., Niu H., Yu Y., Wang J., Zhu S., Zhou J., Papusha A., Cui D., Pan X., Kwon Y., Sung P., and Ira G. (2016) Enrichment of Cdk1-cyclins at DNA double-strand breaks stimulates Fun30 phosphorylation and DNA end resection. Nucleic Acids Res. 44, 2742–2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chiolo I., Carotenuto W., Maffioletti G., Petrini J. H., Foiani M., and Liberi G. (2005) Srs2 and Sgs1 DNA helicases associate with Mre11 in different subcomplexes following checkpoint activation and CDK1-mediated Srs2 phosphorylation. Mol. Cell. Biol. 25, 5738–5751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oza P., Jaspersen S. L., Miele A., Dekker J., and Peterson C. L. (2009) Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 23, 912–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Horigome C., Bustard D. E., Marcomini I., Delgoshaie N., Tsai-Pflugfelder M., Cobb J. A., and Gasser S. M. (2016) PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev. 30, 931–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.