

Abstract
The interaction of the intrinsically disordered polypeptide islet amyloid polypeptide (IAPP), which is associated with type 2 diabetes (T2D), with the Alzheimer's disease amyloid-β (Aβ) peptide modulates their self-assembly into amyloid fibrils and may link the pathogeneses of these two cell-degenerative diseases. However, the molecular determinants of this interaction remain elusive. Using a systematic alanine scan approach, fluorescence spectroscopy, and other biophysical methods, including heterocomplex pulldown assays, far-UV CD spectroscopy, the thioflavin T binding assay, transmission EM, and molecular dynamics simulations, here we identified single aromatic/hydrophobic residues within the amyloid core IAPP region as hot spots or key residues of its cross-interaction with Aβ40(42) peptide. Importantly, we also find that none of these residues in isolation plays a key role in IAPP self-assembly, whereas simultaneous substitution of four aromatic/hydrophobic residues with Ala dramatically impairs both IAPP self-assembly and hetero-assembly with Aβ40(42). Furthermore, our experiments yielded several novel IAPP analogs, whose sequences are highly similar to that of IAPP but have distinct amyloid self- or cross-interaction potentials. The identified similarities and major differences controlling IAPP cross-peptide interaction with Aβ40(42) versus its amyloid self-assembly offer a molecular basis for understanding the underlying mechanisms. We propose that these insights will aid in designing intervention strategies and novel IAPP analogs for the management of type 2 diabetes, Alzheimer's disease, or other diseases related to IAPP dysfunction or cross-amyloid interactions.
Keywords: Alzheimer disease, amyloid, amyloid-beta (AB), diabetes, peptide conformation, peptide interaction, protein aggregation, protein misfolding, protein-protein interaction, islet amyloid polypeptide
Introduction
Amyloid self-assembly underlies the pathogenesis of an increasing number of cell-degenerative diseases, including AD3 and T2D (1). Increasing evidence suggests that cross-interactions between different amyloidogenic proteins or polypeptides, for which we here use the term “cross-amyloid interactions,” modulate their self-assembly into amyloid fibrils and thus may link different diseases to each other (1–4). However, underlying mechanisms have not been yet understood.
In the case of AD and T2D, epidemiological and pathophysiological evidence suggests that the two diseases are linked to each other (3, 5, 6). A possible molecular link could be the interaction between their key amyloidogenic polypeptides Aβ (AD) and IAPP (T2D) (3, 7, 8). In fact, a nanomolar affinity interaction between early prefibrillar Aβ40(42) and IAPP species has been shown in vitro to suppress amyloidogenesis, whereas seed amounts of Aβ40(42) fibrils are able to cross-seed IAPP amyloidogenesis in vitro and in animal models in vivo (3, 7–9). Importantly, the 37-residue-long IAPP, which in soluble form functions as a glucose regulatory neuropeptide and, similar to Aβ40(42), is present both in blood and CSF, has recently been found to co-localize with Aβ plaques in the brains of AD patients, suggesting a possible (patho-)physiological role for the cross-interaction between the two polypeptides (3, 5, 10, 11).
The two intrinsically disordered but highly amyloidogenic polypeptides Aβ40(42) and IAPP share a sequence similarity of ∼50% and an identity of ∼25% (7). Notably, the highest sequence similarities are within β-sheet-forming regions, and IAPP uses the same binding sites, “hot segments” IAPP(8–18) and IAPP(22–28), in both its self-assembly and its hetero-assembly with Aβ40(42) (Fig. 1) (12–15). The involved IAPP interaction surfaces may thus share similarities; however, yet unidentified differences should also exist (12).
Figure 1.

Aβ40(42), IAPP, and synthetic IAPP mutants (left, short names); C-terminal amides and Cys2–Cys7 disulfide bridge not shown. Similar/identical residues between Aβ and IAPP are underlined (7). Previously identified “hot segments” of IAPP self-assembly and its hetero-assembly with Aβ are in boldface type (12), and introduced Ala substituents are shown in red.
Due to the highly dynamic nature and strong self- and co-aggregation propensities of IAPP and Aβ40(42), characterization of their hetero-assemblies at an atomic level via NMR or X-ray crystallography has not been yet possible. Uncovering the molecular determinants of their cross-interaction is, however, of high importance both for elucidating its potential role in disease pathogenesis and for designing possible intervention strategies and novel therapeutic IAPP analogs. In fact, the IAPP–Aβ40(42) interaction has already been successfully applied to design bioactive IAPP analogs or IAPP-derived peptides as inhibitors of amyloidogenesis of IAPP and/or Aβ40(42) (5, 8, 16, 17). In addition, pramlintide, a soluble and bioactive IAPP analog, which has been used in T2D treatment for several years, has recently been suggested to be a promising candidate for the treatment of AD and certain cancers as well (5, 18, 19).
By using a systematic alanine scan approach, fluorescence and far-UV CD spectroscopy, heterocomplex pulldown assays combined with NuPAGE gel electrophoresis and Western blotting, sedimentation assays, the thioflavin T (ThT) binding assay, transmission electron microscopy (TEM), and molecular dynamics (MD) simulations, we here uncover key molecular determinants of the IAPP cross-interaction with Aβ40(42) and characterize their role in IAPP solubility, conformation, and amyloid self-assembly. Our results provide a molecular basis for the interaction of IAPP with Aβ40(42) versus its amyloidogenic self-assembly and for the design of intervention strategies and IAPP analogs with tailored self-/cross-interactions.
Results
Interactions of IAPP “hot segments” with IAPP and Aβ40
First, we asked which are the key residues of the interaction of the partial IAPP “hot segments” IAPP(8–18) and IAPP(22–28) with IAPP or Aβ40. We addressed this question by using alanine scanning and a fluorescence spectroscopic titration assay developed by us earlier (8, 12, 20, 21). This assay is based on the principle that changes in fluorescence of fluorescently labeled proteins or polypeptides are very sensitive indicators of their interactions and can be used to determine their binding affinities (22).
Our previous studies have shown that N-terminal fluorescein-labeled IAPP(8–18) and IAPP(22–28) bind Aβ40 and IAPP with apparent dissociation constants (Kd(app)) ranging from 200 to 400 nm (12). Here, we titrated a series of synthetic N-terminal fluorescently labeled single-point mutants of the above “hot segments” (at 5 nm) with Aβ40 and IAPP (pH 7.4) (Table 1) (data not shown). The results showed that individual substitution of each of the four residues of segment IAPP(9–12) and of residues Phe15, Leu16, Phe23, and Ile26 by Ala strongly reduced the binding affinities toward both Aβ40 and IAPP (Table 1). By contrast, no significant effects were found when all other residues of IAPP(8–18) or IAPP(22–28) were substituted by Ala (Table 1). These results indicated that the four residues of IAPP(9–12) and residues Phe15, Leu16, Phe23, and Ile26 might comprise key residues of the IAPP interaction surface with full-length IAPP and Aβ40(42).
Table 1.
Kd(app) values of interactions of IAPP(8–18), IAPP(22–28), and their Ala mutants with IAPP or Aβ40 as determined by fluorescence titration assays
N-terminal fluorescein-labeled IAPP segments (5 nm) were titrated with IAPP or Aβ40 (pH 7.4). Kd(app) values were determined from one or three binding curves, as indicated; shown are the S.D. values from three binding curves. NB, no binding up to 2 μm IAPP or Aβ40 (mutants in boldface type).
| IAPP segment | Kd(app) interaction with IAPP | Kd(app) interaction with Aβ40 |
|---|---|---|
| nm | nm | |
| IAPP(8–18) | 233 ±102a | 275 ±48a |
| [Ala9]IAPP(8–18) | NB | NB |
| [Ala10]IAPP(8–18) | NB | NB |
| [Ala11]IAPP(8–18) | NB | NB |
| [Ala12]IAPP(8–18) | NB | NB |
| [Ala14]IAPP(8–18) | 478 | 257 ±16 |
| [Ala15]IAPP(8–18) | NB | NB |
| [Ala16]IAPP(8–18) | NB | NB |
| [Ala17]IAPP(8–18) | 274 | 99 ±14 |
| [Ala18]IAPP(8–18) | 169 | 358 ±16 |
| IAPP(22–28) | 398 ±143a | 363 ±62a |
| [Ala22]IAPP(22–28) | 215 | 218 |
| [Ala23]IAPP(22–28) | NB | NB |
| [Ala24]IAPP(22–28) | 324 | 331 |
| [Ala26]IAPP(22–28) | NB | NB |
| [Ala27]IAPP(22–28) | 277 | 335 |
| [Ala28]IAPP(22–28) | 260 | 138 |
a Values derived from Ref. 12.
Interactions of 8-Ala and 4-Ala-containing IAPP mutants with IAPP and Aβ40(42)
To investigate this hypothesis, we synthesized and studied 17 mutants of full-length IAPP comprising one mutant with substitutions of all eight of the above identified residues by Ala, two quadruple-point mutants, four triple-point mutants, six double-point mutants, and four single-point Ala mutants (Fig. 1). The affinities of their interactions with prefibrillar IAPP, Aβ40, and Aβ42 species were then quantified by fluorescence spectroscopic titrations (Fig. 2, supplemental Figs. S1 and S2, and Tables 2 and 3). Synthetic Nα-terminal fluorescein-labeled IAPP (Fluos-IAPP) or its mutants (5 nm) were titrated with IAPP, Aβ40, and Aβ42 (pH 7.4), and fluorescence spectra were recorded (8, 12, 20). Of note, nanomolar affinities (Kd(app) values) have been determined previously for interactions of Fluos-IAPP with IAPP and Aβ40(42) by using this methodology and were confirmed in the context of the current studies (Table 2, Fig. 2, and supplemental Figs. S1 and S2) (8, 12, 20).
Figure 2.

Determination of Kd(app) values of interactions of IAPP and selected Ala mutants with Aβ40 by fluorescence spectroscopic titrations. Fluorescence emission spectra of N-terminal fluorescein-labeled IAPP or mutants (Fluos-peptide, 5 nm) alone and after titration with Aβ40 (molar ratios of Fluos-peptide/Aβ40 as indicated) are shown for the following peptides: IAPP (A), 8A (B), 4A (C), A15,23,26 (D), A15,23 (E), A23,26 (F), A23 (G), A26 (H), and A16 (I). In the insets, the binding curves are shown; data are means ± S.D. (error bars) from three binding curves. Calculated Kd(app) values are shown in Table 2. a.u., arbitrary units.
Table 2.
Kd(app) values of interactions of IAPP and its Ala mutants with IAPP, Aβ40, and Aβ42 as determined by fluorescence spectroscopic titrations
Nα-amino-terminal fluorescein-labeled IAPP or IAPP mutants (5 nm) were titrated with IAPP, Aβ40, or Aβ42 (pH 7.4). Kd(app) values are from three binding curves; shown are S.D. values from three binding curves except for the values of IAPP-IAPP/-Aβ40 interactions, for which S.E. values are shown (see Footnote a). NB, no binding at IAPP or Aβ40(42) concentrations ≤ 5 μm. Mutants/values in boldface type indicate ≥19-fold weaker binding than IAPP toward IAPP and/or Aβ40 and/or Aβ42.
| IAPP and mutants | Kd(app) with IAPP | Kd(app) with Aβ40 | Kd(app) with Aβ42 |
|---|---|---|---|
| nm | nm | nm | |
| IAPP | 9.7 ± 0.9a | 48.5 ± 4.2a | 219 ± 17b |
| 8A | 8400 ± 1900c | NB | NB |
| 4A | 4500 ± 400c | NB | NB |
| A(9–12) | 28.1 ± 2.2 | 501.7 ± 26.8 | 241 ± 1.2 |
| A15,23,26 | 14.3 ± 0.9 | 210.2 ± 2.2 | 190 ± 16 |
| A16,23,26 | 12.1 ± 0.4 | 154.8 ± 10.8 | 175.1 ± 16.8 |
| A15,16,26 | 55.4 ± 29.4 | 273.4 ± 4.2 | 360.3 ± 1.4 |
| A15,16,23 | 20.1 ± 1.1 | 180.3 ± 1.8 | 109.7 ± 8.1 |
| A15,16 | 11.5 ± 0.8 | 369.8 ± 66.7 | 75.8 ± 1.4 |
| A23,26 | 47.7 ± 6.6 | 927.3 ± 45.6 | 162 ± 41 |
| A15,23 | 19.4 ± 3.8 | NB | NB |
| A16,23 | 15.3 ± 1.7 | 205.7 ± 4.5 | 195.1 ± 6.4 |
| A15,26 | 19.5 ± 2.1 | 405.8 ± 28.6 | 751.4 ± 64 |
| A16,26 | 21.2 ± 0.4 | 102.7 ± 2.4 | 161.2 ± 12.1 |
| A23 | 58 ± 11 | NB | 299 ± 8 |
| A26 | 44 ± 4 | 2150 ± 100 | 329 ± 9 |
| A15 | 12 ± 0.3 | 399.3 ± 27.1 | 359.7 ± 14.9 |
| A16 | 40.2 ± 3.9 | 184.3 ± 6.6 | 469.4 ± 22.5 |
Table 3.
Factorial decrease of apparent binding affinity (Kd(mut)/Kd(wt)) of mutants to IAPP, Aβ40, and Aβ42 as compared with wild-type IAPP
Apparent Kd values determined by fluorescence spectroscopic titrations (Table 2): Nα-terminal fluorescein-labeled IAPP mutants (5 nm) were titrated with IAPP, Aβ40, or Aβ42 (pH 7.4). Mutants/values in boldface type indicate ≥19-fold weaker binding than IAPP.
| IAPP and mutants | Kd(mut)/Kd(wt) (with IAPP) | Kd(mut)/Kd(wt) (with Aβ40) | Kd(mut)/Kd(wt) (with Aβ42) |
|---|---|---|---|
| IAPP | 1 | 1 | 1 |
| 8A | 850 | >200a | >45a |
| 4A | 450 | >200a | >45a |
| A(9–12) | 3 | 10 | 1 |
| A15,23,26 | 1 | 4 | 1 |
| A16,23,26 | 1 | 3 | 1 |
| A15,16,26 | 6 | 6 | 2 |
| A15,16,23 | 2 | 4 | 0.5 |
| A15,16 | 1 | 8 | 0.5 |
| A23,26 | 5 | 19 | 1 |
| A15,23 | 2 | >200a | >45a |
| A16,23 | 2 | 4 | 1 |
| A15,26 | 2 | 8 | 3 |
| A16,26 | 2 | 2 | 1 |
| A23 | 6 | >200a | 1 |
| A26 | 5 | 44 | 1 |
| A15 | 1 | 8 | 2 |
| A16 | 4 | 4 | 2 |
a No binding at ≤5 μm.
Dramatically decreased binding affinities to both Aβ40(42) and IAPP were found for 8A and 4A as compared with IAPP (Fig. 2, supplemental Figs. S1 and S2, and Tables 2 and 3). For instance, titrations of Fluos-IAPP (5 nm) with different molar ratios of Aβ40 resulted in significant enhancement of its fluorescence emission already at low nanomolar Aβ40 concentrations, as reported previously (8) (Fig. 2). By contrast, no fluorescence changes were observed when Fluos-8A or Fluos-4A (5 nm) was titrated with Aβ40 up to a 1000-fold molar excess, and qualitatively similar results were obtained in the titrations with Aβ42 and IAPP (Fig. 2 and supplemental Figs. S1 and S2). The affinities of the interactions of Fluos-8A or Fluos-4A with Aβ40 (estimated Kd(app) > 10 μm) were thus >200-fold weaker than the affinity of the Fluos-IAPP–Aβ40 interaction (Kd(app) = 48.5 nm (8)). Similarly, the affinities of the interactions of Fluos-8A and Fluos-4A with IAPP were 850- and 450-fold weaker, respectively, than the affinity of the interaction of Fluos-IAPP with IAPP (Kd(app) = 9.7 nm (20)), whereas affinities of the interactions of Fluos-8A or Fluos-4A with Aβ42 were >45-fold weaker than the affinity of the Fluos-IAPP–Aβ42 interaction (Kd(app) = 219 nm) (supplemental Figs. S1 and S2 and Tables 2 and 3).
The determined Kd(app) values were used for the calculation of the differences between binding free energies of the Ala mutants and IAPP regarding their interaction with IAPP or Aβ40(42) (ΔΔG values) (Fig. 3). In the case of the 8A and 4A mutants, the ΔΔG values were higher than 2 or 3 kcal/mol (Fig. 3). Thus, the simultaneous substitution of the eight residues within IAPP(9–12) and residues Phe15, Leu16, Phe23, and Ile26 (in 8A) or of the four residues Phe15, Leu16, Phe23, and Ile26 (in 4A) by Ala caused a strong destabilization of the IAPP–Aβ40(42) and IAPP–IAPP complexes. By contrast, Fluos-A(9–12) was found to bind IAPP and Aβ40(42) with only ≤10-fold lower affinities than Fluos-IAPP (Tables 2 and 3). These results suggested that the “core region” of both IAPP self-assembly and its hetero-assembly with Aβ40(42) lies within IAPP(15–26) and that the four residues Phe15, Leu16, Phe23, and Ile26 might comprise key residues of these processes.
Figure 3.

Identification of hot spots and key residues of IAPP self-assembly and its hetero-assembly with Aβ40(42) via fluorescence spectroscopic titrations (A–C). Differences in binding free energy of Ala mutants (mut) and wild-type (wt) IAPP (ΔΔGmut − wt or ΔΔG) toward wild-type IAPP (A), Aβ40 (B), or Aβ42 (C) (means ± S.D. (error bars); n = 3 assays). Mutants in boldface type are those with strongly diminished binding affinity to IAPP, Aβ40, or Aβ42 as compared with IAPP.
Interactions of single-point mutants of IAPP with IAPP and Aβ40(42)
To directly address the above hypothesis, interactions of single mutants A15, A16, A23, and A26 with Aβ40(42) and IAPP were studied next (Fig. 2, supplemental Figs. S1 and S2, and Tables 2 and 3). All four mutants bound IAPP with only slightly weaker affinities than the affinity of IAPP self-assembly, suggesting that none of the four residues Phe15, Leu16, Phe23, and Ile26 in isolation plays a key role in IAPP self-assembly. By contrast, strongly reduced affinities were found for the interactions of A23 and A26 with Aβ40; the affinity of A23 was >200-fold (ΔΔG > 3 kcal/mol) and that of A26 44-fold decreased (ΔΔG = 2.2 kcal/mol) as compared with the affinity of IAPP (Fig. 3). However, substitution of Phe15 or Leu16 by Ala did not strongly affect IAPP affinity to Aβ40 (ΔΔG < 1 kcal/mol) (Fig. 3). These findings suggested that IAPP residues Phe23 and Ile26 (ΔΔG > 2.0 kcal/mol) are hot spots of the IAPP interaction surface with Aβ40 (Fig. 3). Importantly, the binding affinities of the above four single Ala mutants for Aβ42 were very similar to the affinity of IAPP (supplemental Fig. S1 and Tables 2 and 3). Thus, none of the above mutations in isolation was able to affect the interaction of IAPP with Aβ42.
Heterocomplex pulldown assays
To obtain more support for the above findings, mixtures of IAPP, 4A, and A23 with Aβ40 or Aβ42 were subjected to heterocomplex pulldown assays combined with NuPAGE gel electrophoresis and Western blotting (Fig. 4 and supplemental Fig. S3) (8, 20). Thereby, synthetic Nα-terminal biotin-labeled IAPP (biotin-IAPP), 4A (biotin-4A), or A23 (biotin-A23) (2.5 μm) were incubated (1 h) with Aβ40(42) (pH 7.4); heterocomplexes were isolated by streptavidin-coated magnetic beads (Fig. 4). Heterocomplex components were revealed after dissociation and release from the beads via NuPAGE and Western blotting with an anti-Aβ antibody and streptavidin-peroxidase (POD) (labeled anti-Biotin in Fig. 4 and supplemental Fig. S3). The Western blot with the anti-Aβ antibody revealed the presence of significant amounts of Aβ40 or Aβ42 bound to biotin-IAPP (Fig. 4 and supplemental Fig. S3) (8, 12). By contrast, no Aβ40 was found in lanes corresponding to Aβ40 bound to biotin-4A or biotin-A23 (Fig. 4), whereas the amounts of Aβ42 bound to biotin-4A were strongly reduced as compared with Aβ42 bound to biotin-IAPP (supplemental Fig. S3). These findings were thus in very good agreement with the results of the fluorescence titration studies.
Figure 4.

Identification of hot spots and key residues of IAPP hetero-assembly with Aβ40 via pulldown assays. A, pulldown assays of biotin-4A– and biotin-IAPP–Aβ40 hetero-assemblies. Top, anti-Aβ40 Western blot analysis (WB) of mixtures or their components as indicated (Aβ40, 5 μm; biotin-peptide, 2.5 μm) following biotin pulldown and peptide dissociation from beads. Bottom, anti-biotin WB of the same incubations as above. B, biotin-A23- versus biotin-IAPP–Aβ40 pull-down assay. Top, anti-Aβ40 WB of mixtures or their components as indicated (Aβ40, 5 μm; biotin-peptide, 2.5 μm) following biotin pulldown and peptide dissociation from beads. Bottom, anti-biotin Western blotting of the above incubations. Aβ40 control lane (A and B), Aβ40 not incubated with beads. Results shown are representative of three assays.
Interactions of double/triple mutants of IAPP with IAPP and Aβ40(42)
To identify possible direct or indirect interactions/coupling between the identified key residues and their effects on the stabilities of IAPP homo-assemblies or their hetero-assemblies with Aβ40(42), interactions of double and triple mutants were studied next (Figs. 2 and 3, supplemental Figs. S1 and S2, and Tables 2 and 3). All hetero-assemblies between IAPP and double/triple mutants exhibited only weakly reduced stabilities (<10-fold) as compared with IAPP–IAPP assemblies (Tables 2 and 3). Also, most of the double/triple mutants were found to bind Aβ40 with only weakly reduced (<10-fold) affinity as compared with IAPP with two exceptions: A15,23, which exhibited a >200-fold weaker affinity, and A23,26, which exhibited a 19-fold weaker affinity for Aβ40 as compared with IAPP (Tables 2 and 3).
The dramatically impaired binding of A15,23 to Aβ40 was consistent with our finding that Phe23 is a hot spot of the IAPP interaction surface with Aβ40. On the other hand, the finding that binding affinities of several triple/double point mutants to Aβ40 were only <10- or 20-fold lower than the affinity of IAPP suggested that the presence of two or three Ala-substituted hot spots/key residues compensates for the detrimental effects of mutating a hot spot to Ala. Cooperative, direct/indirect interactions between key residues and/or structural perturbations caused by the mutations probably account for these findings (23). Importantly, in the case of the hetero-assemblies of Aβ42 with double/triple Ala mutants, a strong destabilization (ΔΔG > 2) was found only for the A15,23-Aβ42 hetero-assemblies (Fig. 3, supplemental Fig. S1, and Tables 2 and 3). These latter results suggested that the presence of either Phe15 or Phe23 is required for the interaction of IAPP with Aβ42.
Effects of IAPP-GI mutants on amyloidogenesis of Aβ40(42) and IAPP
We have reported previously that IAPP-GI, a double N-methylated analog of full-length IAPP, is a potent inhibitor of amyloidogenesis of Aβ40, Aβ42, and IAPP (8, 20). Three mutants of IAPP-GI were synthesized, and their effects on the kinetics of amyloidogenesis of Aβ40, Aβ42, and IAPP were studied by using the ThT binding assay (supplemental Fig. S4). Mutants comprised 4A-GI, an IAPP-GI analog with all four key residues substituted by Ala, the IAPP-GI analog A23-GI with Phe23 substituted by Ala, and the IAPP analog A15,23-GI, containing substitutions of both Phe15 and Phe23 by Ala. In strong contrast to IAPP-GI, the Ala-substituted IAPP-GI analogs were unable to block Aβ40(42) and/or IAPP amyloidogenesis (supplemental Fig. S4). These results supported the suggestion that the four residues Phe15, Leu16, Phe23, and Ile26 play an important role in IAPP–Aβ40(42) and IAPP–IAPP interactions.
Self-assembly propensities of IAPP mutants
The self-assembly propensities of the mutants were studied next (Table 4 and data not shown). Fluorescence spectroscopic titrations showed that 8A and 4A exhibited a >1000-fold weaker self-assembly propensity than IAPP. By contrast, the Kd(app) values of self-assembly of most other mutants were very similar to the Kd(app) of IAPP self-assembly and to the Kd(app) values of their interactions with IAPP (Tables 3 and 4) (data not shown). Only in the case of the self-assembly of A15,16 and of A23,26 were markedly reduced (10–15-fold weaker) affinities found (Table 4). This latter result was consistent with an important role for homotypic interactions in early steps of IAPP self-assembly, as suggested for IAPP fibrils as well (Table 4) (14, 15). Taken together, the studies on the self-assembly potency of the mutants provided additional evidence for a crucial, concerted role of residues Phe15, Leu16, Phe23, and Ile26 in IAPP self-assembly.
Table 4.
Self-assembly potentials (Kd(app) values) and factorial decrease of self-assembly potentials (Kd(mut)/Kd(wt)), increase of solubilities, and decrease of amyloidogenicities of mutants in comparison with IAPP
| IAPP and mutantsa | Kd(app) self-assemblyb,c,d,e | Kd(mut)/Kd(wt) (self-assembly)b,c,d,e | -Fold higher solubility than IAPPf | -Fold lower amyloidogenicity than IAPPg |
|---|---|---|---|---|
| nm | ||||
| IAPP | 9.7 ± 0.9d | 1 | 1 | 1 |
| 8A | NB | >1000 | >100 | >100 |
| 4A | NB | >1000 | >100 | >100 |
| A(9–12) | 87.8 ± 4.8 | 9 | <100 | >100 |
| A15,23,26 | 10.4 ± 1.1 | 2 | >100 | >100 |
| A16,23,26 | 22.3 ± 2.3 | 2 | <100 | >100 |
| A15,16,26 | 16.9 ± 5.8 | 2 | <100 | <20 |
| A15,16,23 | 17.8 ± 1.4 | 2 | <100 | <20 |
| A15,16 | 132.7 ± 26.6 | 14 | <100 | 20–99 |
| A23,26 | 108.3 ± 3.8 | 11 | >100 | 20–99 |
| A15,23 | 21.7 ± 11.8 | 2 | 50–99 | 20–99 |
| A16,23 | 5.9 ± 1.6 | 1 | <100 | <20 |
| A15,26 | 21.5 ± 3.5 | 2 | <100 | <20 |
| A16,26 | 19 ± 1 | 2 | <100 | <20 |
| A23 | 24.4 ± 4.8 | 3 | <20 | 20–99 |
| A26 | 32 ± 4 | 3 | <20 | <20 |
| A15 | 21.8 ± 1.8 | 2 | <20 | <20 |
| A16 | 58 ± 8 | 6 | <20 | 20–99 |
a Mutants in boldface type are those with strongly diminished binding affinity to Aβ40(42) (Tables 2 and 3). Boldface values indicate strong or medium effects of mutations on IAPP self-assembly potency, solubility, or amyloidogenicity.
b Determined via titrations of Nα-amino-terminal fluorescein-labeled IAPP mutants (5 nm) with non-labeled mutants (pH 7.4).
c Kd(app) values are from three binding curves; numbers in parentheses are S.D. values from three binding curves except for the value of the Fluos-IAPP–IAPP interaction (see Footnote d).
d Kd(app) ± S.E. from Ref. 20.
e NB, no binding at mutant concentrations ≤5 μm.
f Solubilities (aqueous buffer, pH 7.4) determined via sedimentation assays at 100 μm (Fig. 5A); solubilities of A15,23 and single mutants were determined also at 50 and 20 μm, respectively (supplemental Fig. S5); IAPP is insoluble at 1 μm (Fig. 5A) (20).
Solubility of IAPP mutants
We next addressed the question whether mutations affected IAPP solubility by using a sedimentation assay (Fig. 5A). The focus was on mutants that exhibited impaired bindings to Aβ40(42) and/or IAPP because such mutants could become leads for soluble IAPP analogs with tailored self-/cross-interaction properties. Peptides were incubated at 100 μm in aqueous buffer (pH 7.4) for 7 days, and, following centrifugation, peptide amounts in supernatants and pellets were quantified (Fig. 5A). Of note, IAPP has previously been found to precipitate within a few days already at 1 μm by using the same methodology (8, 20). A dramatic increase of solubility (>100-fold) as compared with IAPP was found for four of the 17 mutants (i.e. 8A, 4A, A15,23,26, and A23,26) (Fig. 5A and Table 4). Additional sedimentation assays at 50 or 20 μm revealed that A15,23 was >50-fold more soluble than IAPP and that the solubilities of the four single mutants were similar to the solubility of IAPP (Table 4 and supplemental Fig. S5). Thus, four of the six mutants identified here exhibiting impaired binding to IAPP or Aβ40(42) (i.e. 8A, 4A, A23,26, and A15,23) had strongly improved solubilities compared with IAPP.
Figure 5.

Effects of Ala mutations on IAPP solubility and conformation as determined by a sedimentation assay (A) and CD spectroscopy (B–F). A, solubilities of Ala mutants at 100 μm versus IAPP at 100 μm (7-day-aged) and 1 μm (IAPP*, 5-day-aged) (aqueous buffer, pH 7.4) as determined by a sedimentation assay (means ± S.D. (error bars), three assays). Peptide amounts (percentage of total) in supernatants after 7 days of incubation are shown; a mutant is judged as soluble when ≥80% is found in supernatant (indicated by a dotted red line). Different colors are used for each of the four different groups of mutants (8-Ala- and 4-Ala-containing mutants, triple mutants, double mutants, and single mutants). B–E, CD spectra (10 μm, pH 7.4) of 8-Ala- and 4-Ala-containing mutants (B), triple mutants (C), double mutants (D), and single mutants (E) versus IAPP (CD spectrum of IAPP (in boldface type) shown in each graph for comparison).
Secondary structure of IAPP mutants
Effects of Ala mutations on the conformation of IAPP were then investigated by far-UV CD spectroscopy (Fig. 5 (B–E) and supplemental Fig. S6). The spectra of freshly dissolved IAPP and the mutants (10 μm; pH 7.4) were of similar shapes and indicative of mixtures of random coil and β-sheet/β-turn structural elements (Fig. 5, B–E). Also, no strong differences were observed between the magnitudes of the CD spectra of IAPP and most of the mutants (Fig. 5, B–E) (24). Only in the case of Ala(9–12) and A16 were the magnitudes of the CD spectra strongly reduced compared with IAPP (Fig. 5, B and E). As expected for peptides with high oligomerization propensity, a clear concentration dependence was observed in the CD spectra even at low micromolar concentrations (data not shown) (24). However, oligomerization did not result in precipitation or strong changes of secondary structure contents of IAPP and most mutants over at least a 10-fold concentration range (5–50 μm) (supplemental Fig. S6). Determination of secondary structure contents via deconvolutions of CD spectra obtained between 5 and 50 μm suggested 30–50% β-turn/β-sheet, 40–60% random coil and 5–15% α-helical contents for most mutants (supplemental Fig. S6) (25–27). For IAPP, 30–40% β-turn/β-sheet, 50–60% random coil, and 10% α-helix were determined, consistent with earlier reported values (supplemental Fig. S6) (24). Together, the CD studies provided evidence that (a) mutations did not cause large structural changes in IAPP and (b) β-strand–loop–β-strand conformers are major populations of folded species in IAPP and the mutants. The strongly impaired binding of A23, A26, A23,26, and A15,23 to Aβ40(42) could thus be due to the substitution of IAPP residues making direct contacts with Aβ40(42) by Ala, whereas the restored high-affinity binding of many double and all triple Ala mutants could be due to local structural changes related to the introduced substitutions. Such changes could be mediated for instance via local fold (de)stabilization and/or registry shifts in β-strands, yielding alternate Aβ40(42) binding surfaces. The high conformational flexibility of both Aβ40(42) and IAPP strongly supports this suggestion.
Amyloidogenicity of IAPP mutants
Next, amyloidogenicities of IAPP and the 17 Ala mutants were studied by using the ThT binding assay in combination with TEM (Fig. 6). First, kinetics of amyloid formation were studied by incubating the peptides at 12 μm (pH 7.4) for 7 days (Fig. 6, A–D). Of note, IAPP has previously been found to be amyloidogenic at a concentration 19-fold lower than the above one (625 nm) (8). Seven of the 17 mutants comprising the two triple mutants A15,16,23 and A15,16,26; the three double mutants A15,26, A16,23, and A16,26; and the two single mutants A15 and A26 were found to aggregate into ThT binding assemblies (Fig. 6, A–D). The presence of significant amounts of amyloid fibrils in their incubations was confirmed by TEM (Fig. 6, A–D). The above seven mutants were thus <20-fold less amyloidogenic than IAPP, which is consistent with the corresponding mutations having only weakly affected IAPP amyloidogenicity.
Figure 6.

Effects of mutations on IAPP amyloidogenicity as determined by the ThT binding assay and TEM. A–D, identification of weak effects (<20-fold lower amyloidogenicity than IAPP (20)) by following kinetics of fibrillogenesis at 12 μm (pH 7.4): 8-Ala- and 4-Ala-containing (A), triple-Ala (B), double-Ala (C), and single-Ala (D) mutants. Left panels, ThT binding; right panels, representative TEM images from 7-day-aged incubations. E–H, identification of medium (20–99-fold weaker amyloidogenicity than IAPP) or strong (>100-fold weaker amyloidogenicity than IAPP) effects of mutations on IAPP amyloidogenicity. The kinetics of fibrillogenesis of IAPP and mutants that did not form fibrils at 12 μm according to both ThT binding and TEM (A–D) were studied at 62.5 μm (pH 7.4) (20): 8-Ala- and 4-Ala-containing (E), triple-Ala (F), double-Ala (G), and single-Ala (H) mutants. Left panels, ThT binding; right panels, representative TEM images of 14-day-aged incubations except for A15,16 (aged 7 days) and A16,26 (aged 24 h). Colored circles in the right-hand corners of TEM pictures have the same colors as the symbols of the data shown in the graphs of the corresponding ThT binding assays and indicate the mutant whose TEM picture is shown. Data shown in the ThT binding assays are means ± S.D. (error bars) from three assays; scale bars in TEM micrographs represent 100 nm; a.u., arbitrary units.
To identify mutations that strongly affected IAPP amyloidogenicity, the 10 mutants that did not form fibrils at 12 μm were studied with regard to their amyloidogenicity at 62.5 μm, corresponding to a 100-fold higher concentration than the concentration under which IAPP already forms fibrils (8) (Fig. 6, E–H). No ThT binding was found in 14-day-aged incubations of the five mutants 8A, 4A, A(9–12), A15,23,26, and A16,23,26, and the absence of fibrils was confirmed by TEM (Fig. 6, E and F). By contrast, the three double mutants A15,16, A23,26, and A15,23 were amyloidogenic according to both the ThT binding assay and TEM (Fig. 6G). Notably, in the case of A16 and A23, weak ThT binding was observed, and TEM indicated the presence of both fibrils and significant amounts of amorphous aggregates (Fig. 6H).
Taken together, the above studies showed that mutations caused strong (>100-fold reduced amyloidogenicity; 5 mutants), medium (20–99-fold reduced amyloidogenicity; 5 mutants), and weak/no (<20-fold reduced amyloidogenicity; 7 mutants) effects on IAPP amyloidogenicity (results summarized in Table 4). Their effects depended both on the nature of the substituted residues and on their position within the IAPP sequence. In this context, amyloidogenicities of A23, A26, A23,26, and A15,23, which exhibited strongly impaired binding to Aβ40(42), were affected but not strongly reduced as compared with IAPP. Thus, the presence of Phe23 and Ile26 or Phe15 and Phe23 in isolation or combination with each other does not strongly affect IAPP amyloidogenesis. Importantly, the strongly reduced amyloidogenicity of the 4A mutant and the pronounced differences between the effects of triple/double mutations on IAPP amyloidogenicity suggested that cooperative interactions between Phe15, Leu16, Phe23, and Ile26 underlie their effects on IAPP amyloidogenesis.
Effects of mutations on IAPP protofilament stability studied by MD simulations
To obtain more information about the mechanistic basis of the above-observed effects of mutations on IAPP amyloidogenicity, we next performed all-atom MD simulations in explicit water. The stability of a native IAPP protofilament model was compared with the stability of protofilament models of selected mutants. We focused on the four mutants A23, A26, A15,23, and 4A, which exhibited dramatically impaired binding to Aβ40(42) but similar (A26), significantly reduced (A23 and A15,23), or strongly reduced amyloidogenicities (4A) as compared with IAPP (Table 4). We constructed hexameric protofilament models of IAPP and the mutants based on the IAPP fibril structure suggested by Eisenberg et al. (14) and recent results by some of us (28) (Fig. 7). Notably, the Eisenberg IAPP fibril structure has already been used in MD simulations by various groups to study both IAPP self-assembly and its hetero-assembly with Aβ40(42) (29–31). Our simulations addressed the stability of the native IAPP protofilament as compared with the constructed protofilament models containing substitutions of Phe23 by Ala (A23), of Ile26 by Ala (A26), of Phe15 and Phe23 by Ala (A15,23), and of Phe15, Leu16, Phe23, and Ile26 by Ala (4A) (Fig. 7).
Figure 7.

MD simulation of a hexameric protofilament model of native IAPP and mutants. A, Cα backbone RMSD of the amyloid core region Phe15–Ser29 versus simulation time for native IAPP (black) and mutants A26 (light blue), A23 (dark blue), A15,23 (purple), and 4A (red). B, conformation of native IAPP after ∼3 μs of simulation time in top view (left) and side view (right). The top view illustrates that the mutation sites Leu16 and Ile26 form an internal hydrophobic amyloid core, whereas Phe15 and Phe23 are oriented toward the outer quaternary contact level. The side view shows ordered strand-loop-strand stacking and disordered terminal parts of the β-sheets. The dashed blue rectangle illustrates the region considered for RMSD calculation. The bottom row shows representative conformations after ∼3 μs of simulation time for A26 (C), A23 (D), A15,23 (E), and 4A (F). The conformations of A23 and A15,23 show no major difference as compared with native IAPP, indicating that Phe15 and Phe23 are less important for internal protofilament stability. In contrast, mutations within the hydrophobic amyloid core (such as in A26 and 4A) result in formation of internal water channels, and the combination of the four mutations in 4A causes a significant destabilization of the native IAPP structure.
Overall, the results of the MD simulations (Fig. 7) were in good agreement with the results of our ThT binding and TEM studies (Fig. 6 and Table 4) and with the results of previous studies (12–14, 30, 32–39). During the 3 μs of simulation, the native IAPP protofilament model remained stable and close to the starting structure as quantified by Cα backbone root mean square deviation (RMSD) of the amyloid core region Phe15–Ser29 (Fig. 7A). The MD simulations suggested that a major factor contributing to the stability of the U-shaped, β-strand–loop–β-strand motif of the IAPP protofilament is the hydrophobic core formed by residues located close to the loop region IAPP(18–22) (14, 30, 31) (Fig. 7B). Thereby, interactions between hydrophobic and/or aromatic residues probably play an important role (14, 30, 31).
In the case of the A26 protofilament model, substitution of Ile26 by Ala resulted in only a weak destabilization of the hydrophobic core, as evident by the fact that only few water molecules were found in the inner side of the strands after ∼3 μs of simulation time (Fig. 7, A and C). In addition, no significant changes in the Phe23–Phe23 stacking interactions and the U-shaped structure of the A26 fibrils were observed. Thus, based on the MD simulations, the Ile26 → Ala mutation was not expected to cause a significant effect on IAPP amyloidogenicity, which was in good agreement with the findings of the amyloidogenicity assays.
In the case of the A23 mutation, both the tight hydrophobic core of the IAPP protofilament and the starting protofilament structure were retained despite the lack of Phe23–Phe23 stacking interactions (Fig. 7, A and D). Thus, a not strongly reduced amyloidogenicity would be expected for A23 as compared with IAPP. Phe23–Phe23 interactions could be more important on the quaternary structure level in fibrillar double/multilayer IAPP assemblies than for the single protofilament layer used here in the MD simulations. This hypothesis was in good agreement with the observed weak ThT binding and the significant amounts of amorphous aggregates found in A23 incubations in addition to the fibrillar assemblies.
For the A15,23 protofilament model, both the tight hydrophobic core and the protofilament structure were mostly retained; few water molecules were observed only in the peripheral region (Fig. 7, A and E). Thus, mutating both F15 and F23 by Ala was not expected to strongly affect IAPP amyloidogenicity as found above by the ThT binding and TEM studies.
Importantly, the MD simulations also suggested that the simultaneous substitution of Phe15, Leu16, Phe23, and Ile26 by Ala (in 4A) causes dramatic effects on IAPP protofilament structure and stability (Fig. 7, A and F) as found above by the amyloidogenicity studies as well. This is reflected in the large RMSD with respect to the native IAPP protofilament structure (Fig. 7A). The MD simulations indicated formation of a water channel within the loop region, which was most likely due to gaps in the hydrophobic core. Furthermore, a markedly increased disorder in loop stacking was observed, possibly due to both the lack of Phe23–Phe23 stacking interactions and the simultaneous destabilization of the hydrophobic core.
More quantitative support for the above suggested effects of mutations on the stability of the hydrophobic core of IAPP amyloid protofilaments was obtained by calculating average values of the structural RMSD from the native IAPP protofilament model and solvent-accessible surface area (SASA) of the IAPP amyloid core region IAPP(15–29) (supplemental Table S1) (14, 30, 39, 40). A strongly increased SASA was observed in sequence 4A(15–29) as compared with IAPP(15–29) (568 ± 145 Å2 versus 205 ± 57 Å2). In strong contrast to 4A, the SASA values of A23, A26, and A15,23 were comparable with the SASA of IAPP(15–29) (supplemental Table S1). Similar to the SASA findings, a strong RMSD deviation from the native IAPP protofilament model was observed for the sampled structures of 4A, whereas the average RMSD of all other mutants remained close to native IAPP (supplemental Table S1).
Discussion
Our studies uncovered similarities and major differences between IAPP interactions mediating its hetero-assembly with Aβ40(42) versus its self-assembly (Fig. 8). In particular, we (a) identified the aromatic/hydrophobic residues Phe23 and Ile26 within the well-known IAPP self-assembly–mediating segment FGAIL as hot spots of the IAPP interaction with Aβ40 but not with IAPP (14, 15, 32, 37) (notably, a very strong energetic contribution was found for Phe23 and a weaker one for Ile26); (b) showed that Phe15 or Phe23 is required for IAPP interaction with Aβ42 but not with IAPP; and (c) showed that the four aromatic/hydrophobic residues Phe15, Leu16, Phe23, and Ile26 within the IAPP amyloid core IAPP(15–29) act in concert to function as key molecular determinants of both IAPP self-assembly and its hetero-assembly with Aβ40(42).
Figure 8.
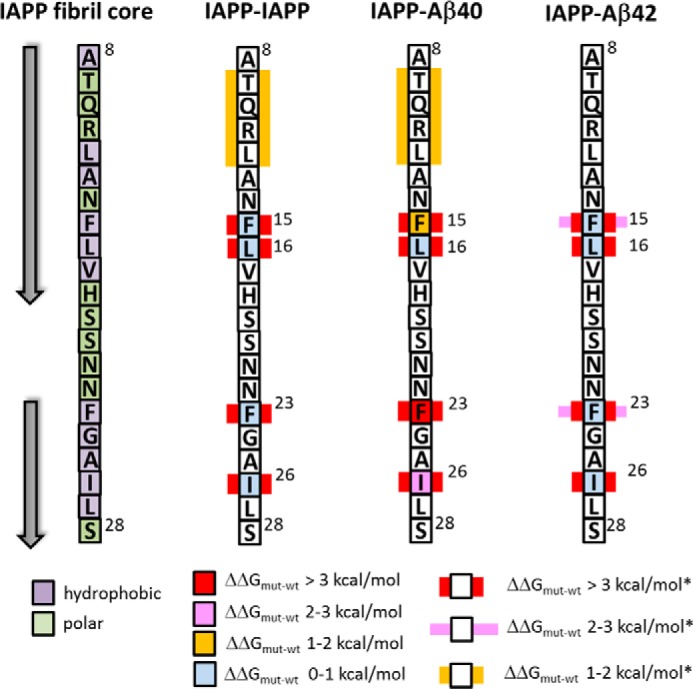
Summary of identified IAPP hot spots/key residues within amyloid core segment IAPP(8–28) mediating interactions with IAPP, Aβ40, or Aβ42. Hot spots (ΔΔGmut − wt (ΔΔG) > 2 kcal/mol) are shown as red (ΔΔG > 3 kcal/mol) or pink (ΔΔG, 2–3 kcal/mol) squares. Key residues (i.e. residues whose substitution by Ala results in ΔΔG > 2 kcal/mol only in the presence of all other residues substituted by Ala marked in the same way) are shown by using, in addition to the square, a red (ΔΔG > 3 kcal/mol*) or pink (ΔΔG = 2–3 kcal/mol*) rectangle. Additional yellow rectangles indicate ΔΔG of 1–2 kcal/mol* when all residues marked in this way are substituted by Ala (mutant A(9–12)). Left, IAPP(8–28); arrows, β-strands of reported amyloidogenic conformers (12, 13, 15, 32, 33, 37).
In this context, our studies suggest that the above four residues and their interactions play a key role in the stability of the hydrophobic core of the IAPP amyloid core sequence IAPP(15–29), which probably strongly contributes to both IAPP amyloid self-assembly and its cross-interactions with Aβ40(42). We also identified combinations of two or three of the above key residues whose substitution by Ala restores binding to Aβ40(42) even in the presence of hot spots that are substituted by Ala. Furthermore, our findings provided the first evidence for differences between the IAPP–Aβ40 versus IAPP–Aβ42 interaction interfaces. Structural differences between Aβ40 and Aβ42 and the highly dynamic and polymorphic nature of IAPP–Aβ40(42) hetero-assemblies probably account for this latter finding (9, 12, 29, 41, 42).
The finding that each of the two aromatic residues Phe15 and Phe23 in isolation affects but is not of crucial importance for IAPP amyloidogenesis is in very good agreement with previous results (35, 38, 43). In fact, substitution of Phe23 or Phe15 by leucine has been reported to only weakly suppress (F23L mutant) or accelerate (F15L mutant) IAPP amyloidogenesis (35, 38, 43). Furthermore, non-additive effects on IAPP amyloidogenesis have been reported for various different multiple substitutions within the IAPP sequence, including F15L and F23L, which is also consistent with our findings (38, 43). Regarding the two hydrophobic residues Leu16 and Ile26, strongly reduced amyloidogenicities have been previously reported for mutants L16Q, I26P, and I26D (43–45). However, our studies showed that the conservative mutations L16A and I26A did not strongly affect IAPP self-assembly, suggesting that Leu16 and Ile26 in isolation are not key determinants of this process. Effects of Pro, Gln, or Asp substituents on peptide structure and amyloidogenicity most likely account for the apparent discrepancy between our findings and the previous ones.
The finding that specific aromatic/hydrophobic residues within the amyloid core of IAPP are able to control its cross-interaction with Aβ40(42) but do not play a crucial role in its amyloid self-assembly is of high importance because it offers a molecular basis for deciphering mechanisms underlying the above and possibly cross-interactions between other amyloidogenic polypeptides as well (1, 2, 23, 46). In fact, interactions of aromatic/hydrophobic residues play a variety of roles in protein–protein interactions, including amyloid self-assembly, and common molecular recognition rules are often involved (23, 34, 47). However, their roles in cross-interactions of intrinsically disordered proteins/polypeptides, including amyloidogenic ones, are largely unknown (47).
Based on the above, we suggest that residues Phe23 and Ile26 and either residue Phe23 or Phe15 of early prefibrillar IAPP conformers make direct and energetically important contacts with early prefibrillar Aβ40 and Aβ42 species, respectively (Fig. 9). In particular, the Phe residues could function as anchor residues, as often observed in interactions between disordered proteins/polypeptides (47, 48). We also suggest that the high plasticity of the IAPP–Aβ40(42) interaction interface and direct/indirect interactions between identified key residues Phe15, Leu16, Phe23, and Ile26, probably in the context of previously proposed β-strand–loop–β-strand IAPP conformers, play a crucial role in stability of prefibrillar Aβ40(42)-IAPP hetero-assemblies (Fig. 9) (13, 15). In fact, various polymorphic amyloidogenic IAPP conformers enabling such interactions have been proposed with IAPP(23–27) or FGAIL and IAPP(13–18) or ANFLVH sequences being parts of β-strands connected to each other by a loop formed by IAPP(19–22) (13, 15, 29, 30, 32, 33, 39, 40). Because (a) formation of a β-strand by FGAIL is probably an early required step in IAPP amyloid self-assembly (15, 32, 33, 37), (b) the ANFLVH segment is the “core” of the second β-strand of amyloidogenic IAPP conformers (13, 15, 33, 36, 39, 40), (c) mutations of IAPP residues Phe15, Phe23, and Ile26 can affect the rate of IAPP amyloidogenesis (38, 43, 44), and (d) Aβ40(42) uses the same binding sites for both its self-assembly and its cross-interaction with IAPP (12), the above suggested interactions would be expected to modulate amyloidogenesis of both IAPP and Aβ40(42) variably and with strong dependence on peptide folds/assemblies present, as found by us and others (Fig. 9) (3, 7–9, 42, 46, 49, 50).
Figure 9.

Schematic presentation of proposed IAPP residues and folds mediating cross-interactions between early prefibrillar IAPP and Aβ40(42) species (top) versus IAPP amyloid self-assembly (bottom) based on our current results and previous findings (8, 9, 12–15, 29, 30, 32, 33, 38–40, 42, 63). Here identified IAPP hot spots/key residues are indicated by red/orange symbols in early disordered conformers only. The IAPP self-assembly-mediating sequence FGAIL is shown in yellow. The disulfide bridge between Cys2 and Cys7 in IAPP is indicated by a continuous line. Only a few heterodimers under the various possible hetero-assemblies are shown; previously identified IAPP binding sequences within Aβ40(42) are shown in purple (12, 42).
Finally, our studies generated a number of novel IAPP analogs that have very high sequence similarity to IAPP but strongly differ from it with regard to their amyloid self-assembly potency and/or their ability to cross-interact with Aβ40(42). For instance, we identified (a) the four IAPP mutants 8A, 4A, A15,23, and A23,26, which exhibit both strongly impaired binding to IAPP and/or Aβ40(42) and improved solubility and/or amyloidogenicity, as compared with IAPP, and (b) the three IAPP mutants A23, A26, and A23,26, which are unable to interact with Aβ40 but are still able to bind IAPP with high affinity. Thus, these peptides are suitable templates for designing novel IAPP analogs with tailored self-/cross-amyloid interactions and related functions (6, 11).
In summary, here we identified single aromatic/hydrophobic residues within the IAPP amyloid core region that are able to control its interaction with Aβ40(42) but not IAPP self-assembly. We also identified four aromatic/hydrophobic residues, which, in combination, are able to control both IAPP amyloid self-assembly and its cross-interaction with Aβ40(42). Furthermore, we devised a number of different full-length IAPP analogs that have a high sequence similarity to IAPP but distinct profiles regarding their amyloid self-assembly and/or their cross-interaction with Aβ40(42). Our results provide a molecular basis for deciphering IAPP–Aβ40(42) interactions and designing intervention strategies and novel IAPP analogs with optimized self-/cross-interactions as leads for therapeutics in T2D, AD, and possibly other devastating diseases linked to IAPP dysfunction and/or its cross-amyloid interactions (1, 3, 5, 6, 10, 11, 18, 19).
Experimental procedures
Peptides and peptide synthesis
Aβ40 (TFA salt) was synthesized by Fmoc solid-phase synthesis on Tentagel R PHB resin (Rapp Polymere), purified by RP-HPLC, and treated as described (12, 51, 52). Aβ40 stocks were freshly prepared in 1,1,3,3,3,3-hexafluoro-2-isopropanol (HFIP) (4 °C) and treated as described; their concentrations were determined by the bicinchoninic acid (BCA) assay (Pierce) (8, 12). IAPP and IAPP-GI were obtained by Fmoc solid-phase peptide synthesis (SPPS), subjected to air oxidation, and purified by RP-HPLC as described previously (20). For the preparation of IAPP stocks, IAPP was dissolved in HFIP (200–500 μm) and filtered over 0.2-μm filters (Millipore) (4 °C), and concentrations were determined by UV spectroscopy (8, 24). Nα-Terminal fluorescein-labeled IAPP (Fluos-IAPP) was synthesized by SPPS and purified via RP-HPLC, and its HFIP stock was made as described (12, 20). Synthetic Aβ42 (TFA salt) was from Bachem and PSL, and Aβ42 stocks in HFIP were freshly prepared as described (12). The Ala mutants of the hot segments IAPP(8–18) and IAPP(22–28) and their N-terminal fluorescein-labeled analogs (C-terminal acids) were synthesized by Fmoc SPPS using Wang resin (0.6 mmol/g), cleaved from the resin, and purified by RP-HPLC using previously published protocols (12, 20, 52). All Ala mutants of full-length IAPP, IAPP-GI, and their Nα-terminal fluorescein-/biotin-labeled analogs (C-terminal amides) were synthesized by Fmoc SPPS using RINK-resin (0.2 mmol/g), cleaved from the resin, and purified by RP-HPLC using previously published protocols (12, 20, 52). Briefly, couplings were performed using Fmoc-protected amino acids and O-benzotriazole-N,N,N′,N′-tetramethyluronium tetrafluoroborate mostly in a 3-fold molar excess and N,N-diisopropylethylamine (DIEA) in a 4.5-fold molar excess in N,N-dimethylformamide except for Cys (3-fold molar excess) which was coupled using 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) (3-fold molar excess) and DIEA (3.4-fold molar excess). Pseudoproline dipeptides (3-fold molar excess) were used for the dipeptide sequences Ala5-Thr6, Ala8-Thr9, Ser19-Ser20, and Ser28-Ser29 and were coupled using HATU (3-fold molar excess) and DIEA (4.5-fold molar excess) in N,N-dimethylformamide. Fluorescein- and biotin-labeled peptides were synthesized by coupling 5(6)-carboxyfluorescein or Fmoc-protected ϵ-aminocaproic acid followed by biotin to the peptide resins as described (20). Most of the amino acids, including 5(6)-carboxyfluorescein and biotin, were coupled twice except for Gln10, Arg11, Leu12, and Ala13, for which triple couplings were used; for pseudoproline dipeptides, mostly single (IAPP analogs) or double (IAPP-GI analogs) couplings were applied. Peptide cleavage from the resin was performed by treatment with a mixture of TFA/water/thioanisole/ethanedithiol/phenol (83/4.5/4.5/2/6 (v/v/v/v/w)) (3 h), and disulfide bridge formation was performed by air oxidation in the presence of 3 m guanidine HCl in 0.1 m NH4HCO3 containing 20% DMSO as described (20, 52). Stock solutions of all Ala mutants, including their fluorescently labeled or biotin-labeled analogs, were made in HFIP (4 °C) as described for IAPP, and concentrations were determined by UV spectroscopy (8, 12). All synthetic peptides were characterized by MALDI-MS.
Determination of binding affinities and ΔΔG values by fluorescence spectroscopic titrations
Fluorescence measurements were performed with a JASCO FP-6500 fluorescence spectrophotometer using an assay system that we have previously developed and applied for the determination of the affinity (Kd(app)) of the interaction of IAPP or its segments with IAPP, Aβ40, and Aβ42 (8, 12, 20). Briefly, excitation was at 492 nm, and fluorescence emission spectra were recorded between 500 and 600 nm within 2–5 min following solution preparation at 25 °C.
Apparent affinities (Kd(app) values) of the interactions of the partial IAPP segments (hot segments) with IAPP or Aβ40 were determined by titration of freshly made solutions of synthetic N-terminal fluorescein-labeled IAPP segments and their Ala mutants (5 nm) with IAPP or Aβ40 (up to 2 μm) as described (12). Kd(app) values of interactions of full-length IAPP or its Ala mutants with IAPP, Aβ40, Aβ42, or Ala mutants were determined by titration of freshly made solutions of synthetic Nα-terminal fluorescein-labeled IAPP (Fluos-IAPP) or mutant (Fluos-mutant) (5 nm) with IAPP, Aβ40, Aβ42, or the Ala mutant (up to 5 μm; exception: Fluos-8A (or Fluos-4A) + IAPP, up to 15 (or 10) μm) as described (8, 12, 20).
Briefly, freshly made stock solutions of unlabeled peptides and their fluorescently labeled analogs in HFIP were used (8, 12, 20). All assays were performed in 10 mm sodium phosphate buffer, pH 7.4 (1% HFIP). Under these experimental conditions, freshly made solutions have been shown previously to contain fluorescently labeled IAPP (5 nm) mainly in a monomeric state, whereas titrations of fluorescently labeled mutants with non-labeled ones suggest that at 5 nm, the mutants are also mainly present as monomers (see Kd(app) values of self-association of mutants in Table 4) (12, 20). The above experimental set-up allows for addressing interactions between early prefibrillar, mostly monomeric species of IAPP or its mutants with early prefibrillar species of Aβ40(42), IAPP, or the mutants (8, 12, 20). Reversibility of binding of Fluos-IAPP/mutants to IAPP or Aβ40(42) was verified. Of note, the presence of 1% HFIP has been shown previously to not affect binding affinities (12). Binding affinities were estimated by using 1:1 binding models; however, more complicated binding models may also apply due to the strong self-assembly potentials of these peptides (8, 12, 20). Determined Kd(app) values are means ± S.D. from three binding curves. The changes in binding free energy were calculated via the equation, ΔΔG = RTln(Kd(mut)/Kd(wt)), where R is the gas constant and T is absolute temperature). Determined Kd(app) values of Fluos-IAPP interaction with IAPP, Aβ40, and Aβ42 were 9.7, 48.5, and 219 nm, corresponding to binding free energies, ΔG values, of −10.9 kcal/mol (IAPP–IAPP), −9.9 kcal/mol (IAPP–Aβ40), and −9.7 kcal/mol (IAPP–Aβ42) (8, 20). Of note, the Kd(app) values of these latter interactions were determined in the context of the current fluorescence titration studies as well as internal controls and were very similar to the ones determined previously (8, 12, 20).
Biotin pulldown assays, NuPAGE, and Western blot analysis
A previously developed assay system for the pulldown of IAPP–Aβ heterocomplexes was applied (8, 20). Briefly, synthetic Nα-terminal biotinylated IAPP (biotin-IAPP) or Ala mutants (2.5 μm) were incubated (1 h) alone or in a mixture with freshly dissolved Aβ40 or Aβ42 (5 μm) in 200 μl of 10 mm sodium phosphate buffer (pH 7.4) at room temperature. Streptavidin-coated magnetic beads were washed, blocked, and incubated with assay buffer, and the preincubated peptide solutions (see above) were added to the beads as described (8, 20). Nonspecific binding of Aβ40(42) was determined by incubating Aβ40(42) alone (5 μm) with beads. After incubation (4 h), bead-bound complexes were isolated by magnetic affinity, beads were washed, and reducing NuPAGE sample loading buffer was added. The beads were boiled (5 min), and supernatants containing complexes or peptides alone were subjected to NuPAGE electrophoresis in 4–12% BisTris gels with MES running buffer according to the manufacturer's (Invitrogen) recommendations. Equal amounts of peptides were loaded in each lane; loaded amounts for Aβ40(42) controls (freshly dissolved peptides without incubation with beads) were 0.2 μg (Aβ40) and 0.3 μg (Aβ42). Thereafter, peptides were blotted onto nitrocellulose using an XCell S Blot Module blotting system (Invitrogen). Aβ40(42) bound to biotin-IAPP or biotin-mutants was revealed by Western blotting using a polyclonal rabbit anti-Aβ antibody (Sigma) in combination with a POD-coupled secondary antibody and the SuperSignal West Dura Extended Duration Substrate staining solution (Thermo Scientific). Negligible nonspecific binding to the beads was found for Aβ40(42) under the applied conditions. Control blots were also performed to detect biotinylated peptides and were developed with streptavidin-POD (anti-Biotin in the figures). A molecular weight marker ranging from 3.5 to 260 kDa (Novex) was electrophoresed in the same gels.
Sedimentation assays
A previously developed sedimentation assay for the determination of solubility of IAPP and its analogs was applied (16, 20). Briefly, IAPP and all mutants were incubated at 100 μm in 10 mm sodium phosphate buffer, pH 7.4, at room temperature. Of note, under these conditions, IAPP has been previously shown to immediately precipitate (16, 20). IAPP solubility was also tested at 1 μm, where it has previously been found to precipitate within a few days (20). In addition, solubility of A15,23 and single Ala mutants was studied also at 50 and 20 μm, respectively. At the time points 0 h and 7 days, solutions were centrifuged (20,000 × g for 20 min), and peptide amounts in supernatants and pellets were quantified by the BCA protein assay (Pierce) (16, 20). The amount of peptide present in a non-centrifuged sample at 0 h was also quantified in each experimental set-up and corresponded to the total peptide amount. A mutant was judged as soluble when >80% of mutant was present in the supernatant at the 7-day time point. Results in Fig. 5A and supplemental Fig. S5 show amounts (percentage of total) of soluble peptides present in supernatants at the end of the incubation process (7 days) except for 1 μm IAPP (end of incubation at 5 days) (20).
Far-UV CD spectroscopy and determination of secondary structure contents
Far-UV CD measurements were carried out using a Jasco 715 spectropolarimeter. Spectra were collected between 195 and 250 nm at 0.1-nm intervals with a response time of 2 s and at room temperature; each spectrum is the average of three spectra. CD measurements were performed using freshly made solutions containing IAPP or Ala mutants at 5–50 μm (as indicated in the figure legends) in aqueous 10 mm sodium phosphate buffer, pH 7.4, containing 1% HFIP, as described previously for IAPP (20, 24). Briefly, CD solutions were obtained by diluting peptides from their freshly made HFIP stocks, and IAPP was in the lag time of its fibrillization process under these conditions (data not shown and results of previous studies) (8, 16, 20, 24, 53). The spectrum of the buffer was subtracted from the CD spectra of the peptide solutions. Secondary structure contents (supplemental Fig. S6) were calculated by deconvolutions of CD spectra of IAPP and the mutants performed with ContinLL at DichroWeb using the reference spectra set 7 (25–27, 54).
ThT binding assays
Kinetics of amyloidogenesis of IAPP and the Ala mutants were determined at room temperature using previously established ThT binding assay systems (8, 12, 17, 20). The effects of Ala mutations on IAPP amyloidogenicity were determined by two rounds of screening. First, kinetics of amyloidogenesis of IAPP and all Ala mutants were studied at 12 μm (in 50 mm aqueous sodium phosphate buffer, pH 7.4, containing 100 mm NaCl and 0.5% HFIP), which corresponds to a 19-fold higher concentration than the concentration (625 nm) under which IAPP has been previously found to form fibrils within 24 h (20). ThT binding was measured at various time points up to 7 days. Mutants that did not bind ThT under the above conditions were subjected to a second round of screening at 62.5 μm (in aqueous 10 mm Tris-HCl buffer, pH 7.4, containing 2% HFIP), corresponding to a 100-fold higher concentration than the concentration under which IAPP aggregates into fibrils, and amyloidogenesis was followed up to 14 days by the ThT binding assay (20). IAPP was included in each experimental set as control, and results of ThT assays were confirmed by TEM (Fig. 7).
The effects of IAPP-GI and its Ala mutants (at 1:1) on the kinetics of fibrillogenesis of IAPP (12 μm), Aβ40 (16.5 μm), and Aβ42 (16.5 μm) (supplemental Fig. S4) were studied in aqueous 50 mm sodium phosphate buffer, pH 7.4, containing 100 mm NaCl and 1% HFIP (Aβ40(42)-related studies) or 0.5% HFIP (IAPP-related studies) as described (8, 17, 20). At various time points, aliquots of the incubations were gently mixed with a 20 μm solution of ThT in 0.05 m glycine/NaOH, pH 8.5 (17, 20). ThT binding was determined immediately by measuring fluorescence emission at 486 nm, following excitation at 450 nm using a 2030 Multilabel Reader VictorX3 (PerkinElmer Life Sciences) (17, 20).
TEM
TEM was applied to confirm the results of the ThT assays as described previously (8, 20). Briefly, 10-μl aliquots of solutions used for ThT assays were applied on carbon-coated grids at the end of the incubation process. Following washing with distilled water, grids were stained with aqueous 2% (w/v) uranyl acetate and examined with a JEOL JEM 100CX electron microscope at 100 kV (8, 20).
Molecular dynamics simulation
Simulation models
The IAPP hexamer structure was extracted from the full-length, all-atom IAPP fibril model suggested by Eisenberg et al. (14), which is based on crystal fibril structures of IAPP segments NNFGAIL and SSTNVG. Within the amyloid fibril hierarchy, the IAPP hexamer corresponds to a so-called protofilament consisting of two antiparallel β-sheets connected by a loop region. This tertiary structure results from six IAPP monomers stacked on top of each other in β-hairpin conformation with an interpeptide spacing of 4.8 Å. In each monomer, residues Cys2 and Cys7 were connected by a disulfide bridge, and the relevant alanine mutations were inserted into an energy-minimized native IAPP hexamer structure.
Simulation protocol
The simulations were performed using the Amber14 simulation package with the all-atom force field ff14SB and explicit TIP3P water (55, 56). 12 neutralizing chloride ions were added, and standard ionization states at pH 7 were used for ionizable side chains. The IAPP hexamers were placed into an octahedral simulation box with a minimum distance of 12 Å to the box boundaries, resulting in system sizes of ∼3,200 solute and ∼11,000 solvent atoms. Before starting the integration in time, the energy of each model system was minimized using 10,000 steps of steepest-descent algorithm. The systems were then heated up to 300 K in steps of 50 K and 100 ps each. A subsequent equilibration phase was divided into 2 ns in the NVT ensemble and a further 4 ns in the NPT ensemble using the Berendsen weak-coupling scheme with a reference temperature of T = 300 K, a reference pressure of p = 1 bar, and coupling time constants of τT = 0.1 ps and τp = 1.0 ps (57). During the equilibration, the positions of heavy backbone atoms were restrained using a harmonic potential with a force constant of krestr = 2.4 kcal/(mol·Å2). In a subsequent relaxation phase, the position restraints were gradually removed by reducing krestr in steps of 0.5 kcal/(mol·Å2) and 10 ps each.
The unrestrained production simulations were performed for ∼3 μs in the NPT ensemble at T = 300 K and pressure of 1 bar using the standard leapfrog algorithm. The combination of the SETTLE algorithm for bond constraining and hydrogen mass repartitioning allowed the use of an integration time step of 4 fs (58).
Long-range Coulomb interactions were treated using periodic boundary conditions and the particle mesh Ewald method with a 12-Å real-space cut-off (59). Simulation frames were saved every 80 ps.
Simulation analysis
The resulting simulation trajectories were analyzed using the program Cpptraj version 14.05 (60).
To compare the stability of mutants with respect to the native IAPP hexamer, a Cα backbone RMSD was calculated within the amyloid core segment Phe15–Ser29, which includes the loop region and the mutation-relevant parts of the β-sheets. Both peripheral monomers at the hexamer tips were excluded from RMSD calculation due to considerable conformational fluctuations inherent to small amyloid protofilament models (28).
The SASA within the hexamer's internal amyloid core was quantified using the LCPO algorithm of Weiser et al. (61). Only amino acids pointing toward the internal amyloid core within the mutation-relevant region were included in the calculation: Leu16, His18, Asn22, Gly24, Ile26, and Ser28. Mean and S.D. values of RMSD and SASA were calculated, discarding the first 200 ns of each trajectory, based on the structural equilibration time of mutant 4A. Representative conformations were visualized in VMD version 1.9.2 (62).
Author contributions
M. B., K. H., M. K., A. S., E. M., L.-M. Y., and A. C. performed experiments and analyzed data. C. V. F. performed and analyzed MD simulations. M. Z. designed and analyzed simulations. A. K. designed experiments, analyzed data, and wrote the manuscript. All authors have approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank V. Ecker for technical assistance, K. Tas for help with cell toxicity assays, and E. Andreetto for helpful advice in peptide synthesis and valuable discussions. We are grateful to S. Weinkauf, M. Hanzlik, and C. Peters for help with TEM and to J. Bernhagen for valuable discussions and support.
This work was supported by Deutsche Forschungsgemeinschaft Grants SFB 1035 B06 (to A. K.) and B02 (to M. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Table 1 and Figs. S1–S6.
- AD
- Alzheimer's disease
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- T2D
- type 2 diabetes
- Aβ
- amyloid-β
- IAPP
- islet amyloid polypeptide
- ThT
- thioflavin T
- TEM
- transmission electron microscopy
- MD
- molecular dynamics
- Fluos-IAPP
- Nα-amino-terminal fluorescein-labeled IAPP
- SASA
- solvent-accessible surface area
- HFIP
- 1,1,3,3,3,3-hexafluoro-2-isopropanol
- SPPS
- solid-phase peptide synthesis
- RP
- reverse phase
- DIEA
- N,N-diisopropylethylamine
- HATU
- 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- POD
- peroxidase
- WB
- Western blot analysis
- A15
- A16, A23, and A26, alanine single mutants F15A, L16A, F23A, and I26A, respectively
- 8A
- simultaneous substitution of the eight residues within IAPP(9–12) and residues Phe15, Leu16, Phe23, and Ile26
- 4A
- substitution of the four residues Phe15, Leu16, Phe23, and Ile26.
References
- 1. Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Luo J., Wärmländer S. K., Gräslund A., and Abrahams J. P. (2016) Cross-interactions between the Alzheimer disease amyloid-β peptide and other amyloid proteins: a further aspect of the amyloid cascade hypothesis. J. Biol. Chem. 291, 16485–16493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oskarsson M. E., Paulsson J. F., Schultz S. W., Ingelsson M., Westermark P., and Westermark G. T. (2015) In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease. Am. J. Pathol. 185, 834–846 [DOI] [PubMed] [Google Scholar]
- 4. Horvath I., and Wittung-Stafshede P. (2016) Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 113, 12473–12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lutz T. A., and Meyer U. (2015) Amylin at the interface between metabolic and neurodegenerative disorders. Front. Neurosci. 9, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verdile G., Fuller S. J., and Martins R. N. (2015) The role of type 2 diabetes in neurodegeneration. Neurobiol. Dis. 84, 22–38 [DOI] [PubMed] [Google Scholar]
- 7. O'Nuallain B., Williams A. D., Westermark P., and Wetzel R. (2004) Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 279, 17490–17499 [DOI] [PubMed] [Google Scholar]
- 8. Yan L. M., Velkova A., Tatarek-Nossol M., Andreetto E., and Kapurniotu A. (2007) IAPP mimic blocks Aβ cytotoxic self-assembly: cross-suppression of amyloid toxicity of Aβ and IAPP suggests a molecular link between Alzheimer's disease and type II diabetes. Angew. Chem. Int. Ed. Engl. 46, 1246–1252 [DOI] [PubMed] [Google Scholar]
- 9. Hu R., Zhang M., Chen H., Jiang B., and Zheng J. (2015) Cross-seeding interaction between β-amyloid and human islet amyloid polypeptide. ACS Chem. Neurosci. 6, 1759–1768 [DOI] [PubMed] [Google Scholar]
- 10. Jackson K., Barisone G. A., Diaz E., Jin L. W., DeCarli C., and Despa F. (2013) Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 74, 517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Westermark P., Andersson A., and Westermark G. T. (2011) Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 91, 795–826 [DOI] [PubMed] [Google Scholar]
- 12. Andreetto E., Yan L. M., Tatarek-Nossol M., Velkova A., Frank R., and Kapurniotu A. (2010) Identification of hot regions of the Aβ-IAPP interaction interface as high-affinity binding sites in both cross- and self-association. Angew. Chem. Int. Ed. Engl. 49, 3081–3085 [DOI] [PubMed] [Google Scholar]
- 13. Dupuis N. F., Wu C., Shea J. E., and Bowers M. T. (2011) The amyloid formation mechanism in human IAPP: dimers have β-strand monomer-monomer interfaces. J. Am. Chem. Soc. 133, 7240–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wiltzius J. J., Sievers S. A., Sawaya M. R., Cascio D., Popov D., Riekel C., and Eisenberg D. (2008) Atomic structure of the cross-beta spine of islet amyloid polypeptide (amylin). Protein Sci. 17, 1467–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wiltzius J. J., Sievers S. A., Sawaya M. R., and Eisenberg D. (2009) Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 18, 1521–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan L. M., Velkova A., Tatarek-Nossol M., Rammes G., Sibaev A., Andreetto E., Kracklauer M., Bakou M., Malideli E., Goeke B., Schirra J., Storr M., and Kapurniotu A. (2013) Selectively N-methylated soluble IAPP mimics as potent IAPP receptor agonists and nanomolar inhibitors of cytotoxic self-assembly of both IAPP and Aβ40. Angew. Chem. Int. Ed. Engl. 52 10378–10383 [DOI] [PubMed] [Google Scholar]
- 17. Andreetto E., Malideli E., Yan L. M., Kracklauer M., Farbiarz K., Tatarek-Nossol M., Rammes G., Prade E., Neumüller T., Caporale A., Spanopoulou A., Bakou M., Reif B., and Kapurniotu A. (2015) A hot-segment-based approach for the design of cross-amyloid interaction surface mimics as inhibitors of amyloid self-assembly. Angew. Chem. Int. Ed. Engl. 54, 13095–13100 [DOI] [PubMed] [Google Scholar]
- 18. Adler B. L., Yarchoan M., Hwang H. M., Louneva N., Blair J. A., Palm R., Smith M. A., Lee H. G., Arnold S. E., and Casadesus G. (2014) Neuroprotective effects of the amylin analogue pramlintide on Alzheimer's disease pathogenesis and cognition. Neurobiol. Aging 35, 793–801 [DOI] [PubMed] [Google Scholar]
- 19. Venkatanarayan A., Raulji P., Norton W., Chakravarti D., Coarfa C., Su X., Sandur S. K., Ramirez M. S., Lee J., Kingsley C. V., Sananikone E. F., Rajapakshe K., Naff K., Parker-Thornburg J., Bankson J. A., et al. (2015) IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 517, 626–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan L. M., Tatarek-Nossol M., Velkova A., Kazantzis A., and Kapurniotu A. (2006) Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 2046–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tatarek-Nossol M., Yan L. M., Schmauder A., Tenidis K., Westermark G., and Kapurniotu A. (2005) Inhibition of IAPP amyloid fibril formation and apoptotic cell death by a designed IAPP amyloid core-containing hexapeptide. Chem. Biol. 12, 797–809 [DOI] [PubMed] [Google Scholar]
- 22. Eftink M. R. (1997) Fluorescence methods for studying equilibrium macromolecule-ligand interactions. Methods Enzymol. 278, 221–257 [DOI] [PubMed] [Google Scholar]
- 23. Keskin O., Gursoy A., Ma B., and Nussinov R. (2008) Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chem. Rev. 108, 1225–1244 [DOI] [PubMed] [Google Scholar]
- 24. Kayed R., Bernhagen J., Greenfield N., Sweimeh K., Brunner H., Voelter W., and Kapurniotu A. (1999) Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J. Mol. Biol. 287, 781–796 [DOI] [PubMed] [Google Scholar]
- 25. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Whitmore L., and Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 28. Schwierz N., Frost C. V., Geissler P. L., and Zacharias M. (2016) Dynamics of seeded Aβ40-fibril growth from atomistic molecular dynamics simulations: kinetic trapping and reduced water mobility in the locking step. J. Am. Chem. Soc. 138, 527–539 [DOI] [PubMed] [Google Scholar]
- 29. Baram M., Atsmon-Raz Y., Ma B., Nussinov R., and Miller Y. (2016) Amylin-Aβ oligomers at atomic resolution using molecular dynamics simulations: a link between Type 2 diabetes and Alzheimer's disease. Phys. Chem. Chem. Phys. 18, 2330–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wineman-Fisher V., Atsmon-Raz Y., and Miller Y. (2015) Orientations of residues along the β-arch of self-assembled amylin fibril-like structures lead to polymorphism. Biomacromolecules 16, 156–165 [DOI] [PubMed] [Google Scholar]
- 31. Berhanu W. M., Yaşar F., and Hansmann U. H. (2013) In silico cross seeding of Aβ and amylin fibril-like oligomers. ACS Chem. Neurosci. 4, 1488–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Buchanan L. E., Dunkelberger E. B., Tran H. Q., Cheng P. N., Chiu C. C., Cao P., Raleigh D. P., de Pablo J. J., Nowick J. S., and Zanni M. T. (2013) Mechanism of IAPP amyloid fibril formation involves an intermediate with a transient β-sheet. Proc. Natl. Acad. Sci. U.S.A. 110, 19285–19290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dupuis N. F., Wu C., Shea J. E., and Bowers M. T. (2009) Human islet amyloid polypeptide monomers form ordered β-hairpins: a possible direct amyloidogenic precursor. J. Am. Chem. Soc. 131, 18283–18292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gazit E. (2002) A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 16, 77–83 [DOI] [PubMed] [Google Scholar]
- 35. Marek P., Abedini A., Song B., Kanungo M., Johnson M. E., Gupta R., Zaman W., Wong S. S., and Raleigh D. P. (2007) Aromatic interactions are not required for amyloid fibril formation by islet amyloid polypeptide but do influence the rate of fibril formation and fibril morphology. Biochemistry 46, 3255–3261 [DOI] [PubMed] [Google Scholar]
- 36. Mazor Y., Gilead S., Benhar I., and Gazit E. (2002) Identification and characterization of a novel molecular-recognition and self-assembly domain within the islet amyloid polypeptide. J. Mol. Biol. 322, 1013–1024 [DOI] [PubMed] [Google Scholar]
- 37. Tenidis K., Waldner M., Bernhagen J., Fischle W., Bergmann M., Weber M., Merkle M.-L., Voelter W., Brunner H., and Kapurniotu A. (2000) Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J. Mol. Biol. 295, 1055–1071 [DOI] [PubMed] [Google Scholar]
- 38. Tu L. H., and Raleigh D. P. (2013) Role of aromatic interactions in amyloid formation by islet amyloid polypeptide. Biochemistry 52, 333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weirich F., Gremer L., Mirecka E. A., Schiefer S., Hoyer W., and Heise H. (2016) Structural characterization of fibrils from recombinant human islet amyloid polypeptide by solid-state NMR: the central FGAILS segment is part of the β-sheet core. PLoS One 11, e0161243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mirecka E. A., Feuerstein S., Gremer L., Schröder G. F., Stoldt M., Willbold D., and Hoyer W. (2016) β-Hairpin of islet amyloid polypeptide bound to an aggregation inhibitor. Sci. Rep. 6, 33474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Colvin M. T., Silvers R., Ni Q. Z., Can T. V., Sergeyev I., Rosay M., Donovan K. J., Michael B., Wall J., Linse S., and Griffin R. G. (2016) Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 138, 9663–9674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan L. M., Velkova A., and Kapurniotu A. (2014) Molecular characterization of the hetero-assembly of β-amyloid peptide with islet amyloid polypeptide. Curr. Pharm. Des. 20, 1182–1191 [DOI] [PubMed] [Google Scholar]
- 43. Cao P., Marek P., Noor H., Patsalo V., Tu L. H., Wang H., Abedini A., and Raleigh D. P. (2013) Islet amyloid: from fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 587, 1106–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abedini A., Meng F., and Raleigh D. P. (2007) A single-point mutation converts the highly amyloidogenic human islet amyloid polypeptide into a potent fibrillization inhibitor. J. Am. Chem. Soc. 129, 11300–11301 [DOI] [PubMed] [Google Scholar]
- 45. Fox A., Snollaerts T., Errecart Casanova C., Calciano A., Nogaj L. A., and Moffet D. A. (2010) Selection for nonamyloidogenic mutants of islet amyloid polypeptide (IAPP) identifies an extended region for amyloidogenicity. Biochemistry 49, 7783–7789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ma B., and Nussinov R. (2012) Selective molecular recognition in amyloid growth and transmission and cross-species barriers. J. Mol. Biol. 421, 172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Espinoza-Fonseca L. M. (2012) Aromatic residues link binding and function of intrinsically disordered proteins. Mol. Biosyst. 8, 237–246 [DOI] [PubMed] [Google Scholar]
- 48. Rajamani D., Thiel S., Vajda S., and Camacho C. J. (2004) Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 101, 11287–11292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Young L. M., Mahood R. A., Saunders J. C., Tu L. H., Raleigh D. P., Radford S. E., and Ashcroft A. E. (2015) Insights into the consequences of co-polymerisation in the early stages of IAPP and Aβ peptide assembly from mass spectrometry. Analyst 140, 6990–6999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seeliger J., Evers F., Jeworrek C., Kapoor S., Weise K., Andreetto E., Tolan M., Kapurniotu A., and Winter R. (2012) Cross-amyloid interaction of Abeta and IAPP at lipid membranes. Angew. Chem. Int. Ed. Engl. 51, 679–683 [DOI] [PubMed] [Google Scholar]
- 51. Kapurniotu A., Buck A., Weber M., Schmauder A., Hirsch T., Bernhagen J., and Tatarek-Nossol M. (2003) Conformational restriction via cyclization in β-amyloid peptide Aβ(1–28) leads to an inhibitor of Aβ(1–28) amyloidogenesis and cytotoxicity. Chem. Biol. 10, 149–159 [DOI] [PubMed] [Google Scholar]
- 52. Kazantzis A., Waldner M., Taylor J. W., and Kapurniotu A. (2002) Conformationally constrained human calcitonin (hCt) analogues reveal a critical role of sequence 17–21 for the oligomerization state and bioactivity of hCt. Eur. J. Biochem. 269, 780–791 [DOI] [PubMed] [Google Scholar]
- 53. Kapurniotu A., Schmauder A., and Tenidis K. (2002) Structure-based design and study of non-amyloidogenic, double N-methylated IAPP amyloid core sequences as inhibitors of IAPP amyloid formation and cytotoxicity. J. Mol. Biol. 315, 339–350 [DOI] [PubMed] [Google Scholar]
- 54. Provencher S. W., and Glöckner J. (1981) Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20, 33–37 [DOI] [PubMed] [Google Scholar]
- 55. Case D. A., Babin V., Berryman J. T., Betz R. M., Cai Q., Cerutti D. S., Cheatham T. E. 3rd, Darden T. A., Duke R. E., Gohlke H., Goetz A. W., Gusarov S., Homeyer N., Janowski P., Kaus J., et al. (2014) AMBER 14, University of California, San Francisco [Google Scholar]
- 56. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 57. Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., and Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 58. Hopkins C. W., Le Grand S., Walker R. C., and Roitberg A. E. (2015) Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 [DOI] [PubMed] [Google Scholar]
- 59. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 60. Roe D. R., and Cheatham T. E. 3rd (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 [DOI] [PubMed] [Google Scholar]
- 61. Weiser J., Shenkin P. S., and Still W. C. (1999) Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 20, 217–230 [Google Scholar]
- 62. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 63. Petkova A. T., Ishii Y., Balbach J. J., Antzutkin O. N., Leapman R. D., Delaglio F., and Tycko R. (2002) A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.