


Abstract
In mammalian cells, DNA double-strand breaks are repaired mainly by non-homologous end joining, which modifies and ligates two DNA ends without requiring extensive base pairing interactions for alignment. We investigated the role of DNA polymerases in DNA-PK-dependent end joining of restriction-digested plasmids in vitro and in vivo. Rejoining of DNA blunt ends as well as those with partially complementary 5′ or 3′ overhangs was stimulated by 20–53% in HeLa cell-free extracts when dNTPs were included, indicating that part of the end joining is dependent on DNA synthesis. This DNA synthesis-dependent end joining was sensitive to aphidicolin, an inhibitor of α-like DNA polymerases. Furthermore, antibodies that neutralize the activity of DNA polymerase α were found to strongly inhibit end joining in vitro, whereas neutralizing antibodies directed against DNA polymerases β and ɛ did not. DNA sequence analysis of end joining products revealed two prominent modes of repair, one of which appeared to be dependent on DNA synthesis. Identical products of end joining were recovered from HeLa cells after transfection with one of the model substrates, suggesting that the same end joining mechanisms also operate in vivo. Fractionation of cell extracts to separate PCNA as well as depletion of cell extracts for PCNA resulted in a moderate but significant reduction in end joining activity, suggesting a potential role in a minor repair pathway.
INTRODUCTION
DNA double-strand breaks (DSBs) are generated by endogenously produced radicals and by exogenous agents such as ionizing radiation or radiomimic chemicals such as bleomycin. They can also occur accidentally or intentionally during normal DNA metabolism (1).
Eukaryotes generally repair DSBs either by homologous recombination (HR), a process that requires a repair substrate with extensive homology, or non-homologous end joining (NHEJ), which imposes no particular requirements for homology. In both yeast and mammals, these two pathways appear to compete with each other and whereas budding yeast uses predominantly HR, NHEJ is preferentially utilized in mammalian systems (reviewed in 2). The ratio between NHEJ and HR in higher eukaryotes appears to be modulated during the cell cycle as well as during development (3–5).
In recent years our understanding of the processes underlying NHEJ has been greatly advanced. Analysis of rodent cell lines that are sensitive to DSBs as well as mutants of the yeast Saccharomyces cerevisiae have been crucial for the identification of factors involved in NHEJ (reviewed in 6,7). The heterodimeric DNA end-binding activity Ku plays a key role in NHEJ (reviewed in 6). Ku binds efficiently to DNA ends, protecting them from excessive nucleolytic degradation (reviewed in 8,9). The catalytic subunit of DNA-dependent protein kinase (DNA-PKCS), a large 470 kDa protein of the phosphatidylinositol 3-kinase superfamily, is recruited to the DNA ends by Ku and activated by the DNA ends. The kinase activity, which is essential for efficient NHEJ, is required for translocation of Ku along the DNA helix (10,11).
Another factor identified in genetic studies with rodent mutants is XRCC4 (12). XRCC4 forms a stable complex with DNA ligase IV in both humans and the yeast S.cerevisiae (13,14). It has subsequently been shown that the XRCC4/ligase IV complex is required for the ligation step in NHEJ and cannot be replaced by other cellular ligases (6,15,16). Recent functional and structural studies demonstrate that XRCC4/ligase IV can interact with the Ku and the DNA-PKCS at DNA ends to form a functional complex. Within this complex, XRCC4 may bridge two DNA ends to coordinate rejoining of the broken DNA (17–20). In fact, complementary DNA ends are joined by DNA-PK and XRCC4/ligase IV in the absence of other repair factors, albeit with low efficiency (18,19). A further complex interacting with Ku and participating in NHEJ is composed of the MRE11, RAD50 and the NBS or XRS2 proteins in human and yeast, respectively (21). MRE11 is a double-stranded DNA 3′→5′ exonuclease and single-stranded DNA endonuclease, whereas RAD50 is a coiled coil protein with ATP-dependent DNA binding activity that shares homology with SMC proteins (reviewed in 22). This complex is involved in both HR and NHEJ and has been implicated in trimming of the DNA ends for subsequent repair (22–24). Furthermore, MRE11/RAD50/NBS fulfills a crucial function in the cellular DNA damage response after DSBs (reviewed in 25). Interestingly, the MRE11 complex as well as the Ku proteins have been implicated in maintenance and silencing of telomeres (22,26). The versatility of NHEJ is illustrated by the fact that still another nuclease, the 5′-specific FEN-1, appears to be involved in NHEJ in yeast (27).
Insight into the basic mechanisms of NHEJ have mainly originated from the use of restriction-digested plasmids as defined DSB substrates in cell-free systems and, more recently, the controlled expression of highly specific endonucleases such as HO endonuclease or I-SceI endonuclease (reviewed in 28). Early studies in cell free systems demonstrated that several mechanisms probably representing several pathways of NHEJ operate simultaneously.
Analysis of end joining products indicate that short microhomologies of a few base pairs are preferentially utilized as sites for the rejoining of broken DNA ends (29,30) This has led to the suggestion that NHEJ requires the exposure of single-stranded DNA regions by the action of nucleases and/or helicases (31,32). This process is accompanied by the occurrence of mainly small deletions and, occasionally, larger deletions and insertions, both in vitro (30,33,34) and in vivo (35–37). More rarely, DNA ends with various protruding overhangs are processed to form blunt end DNA intermediates by a combination of nuclease and DNA polymerase (Pol) activities. The blunt end can then either be ligated to a second blunt end (‘blunting’) or serve as a primer for DNA repair synthesis across the DNA break (‘fill-in’) (28,32).
Although there have been striking advances made in the analysis of NHEJ, knowledge of additional factors implicated in this process is very limited. In particular, the potential role of DNA repair synthesis and the DNA polymerases that could be involved in this process remains controversial. In this study we have analyzed the role of DNA repair synthesis in NHEJ of model DSB substrates. Our results indicate that, although DNA repair synthesis is not essential for NHEJ, it is an important factor influencing the outcome of the repair event, thereby counteracting loss of genetic information at the break end. Furthermore, we propose a role for Pol α in this process.
MATERIALS AND METHODS
Materials
All chemicals used were the highest grade available. Aphidicolin was purchased from Sigma, wortmannin from ICN and LY294002 from Promega. Mouse monoclonal antibodies SJK-287-138 and SJK-132-20 against Pol α (38) were purified from hybridoma supernatant using protein G–Sepharose (Pharmacia Sweden). Neutralizing rabbit polyclonal antibodies AHP317 against Ku86 (Serotec) and K18 against Pol ɛ (39) and Pol β (40) were purified by protein A–Sepharose affinity matrix (Pharmacia Sweden).
Cell cultivation and cell extract preparation
HeLa S3 (ATCC CCL 2.2) cells were cultivated in Joklik’s modified Eagle’s medium containing 5% newborn calf serum at 37°C as ‘spinner cells’. Whole cell extracts were prepared in two different ways. First we employed the method of Nishida et al. (41), but found that the NHEJ competence of the extracts varied considerably from batch to batch. Subsequently, we adopted the method of Krude et al. (42) with modifications. All steps were performed at 4°C on ice. Briefly, cells were collected (2000 g, 10 min) and washed twice with PBS and once with hypotonic buffer A (20 mM HEPES–KOH, pH 7.8, 0.5 mM MgCl2, 0.5 mM DTT, 5 mM KCl). After centrifugation (1000 g, 5 min), the cells were resuspended in buffer A at 2 × 108 cells/ml and allowed to swell for 15 min. The cells were then disrupted by 7–15 strokes with a loose fitting pestle in a Dounce homogenizer. Buffer A containing 0.8 M NaCl was added (1 ml per 2 × 108 cells) and proteins were extracted on ice for 90 min. The homogenate was clarified (14 000 g, 30 min) and the supernatant was dialyzed for >4 h against TDEG (50 mM Tris–HCl pH 8.0, 1 mM DTT, 1 mM EDTA, 10% glycerol). The precipitate was removed by centrifugation (20 000 g, 5 min) and the supernatant snap frozen in aliquots and stored at –70°C. Protein concentrations were measured using a standard protein assay (43) and BSA as the standard.
Preparation of the NHEJ substrates
A 23 bp fragment [5′-CACTAGTTGCGCATGGCCTCGCAT, (+)-strand] was introduced into the XmaI and SacI sites of M13mp18 to create M13dsb1, which has a new MluI site. M13dsb2 contained the 599 bp λ MluI–XmaI fragment cloned into the corresponding sites of M13dsb1. M13dsb1 and M13dsb2 resulted in the same substrate after digestion with the above enzymes. M13dsb4 contained a 19 bp fragment [5′-CAACATAAGACGTCTACT, (+)-strand] inserted into the XbaI and EcoRI sites of M13mp18 to create a new AatII site. Plasmids were purified using a Plasmid Maxi Kit (Qiagen) according to the manufacturer’s instructions. The DNA was digested with SmaI (Pharmacia), with KpnI and AatII or sequentially with XmaI (Pharmacia) or its schizoisomer Cfr9I (MBI Fermentas) and MluI (Pharmacia) to give the substrates S, AK and XM, respectively (Fig. 1). Digested DNA was separated from the insert and undigested DNA by agarose gel electrophoresis and purified with a GeneClean II kit (Bio101) according to the manufacturer’s instructions.
Figure 1.

End configuration of the DNA substrates used in this study. The diagram presents the DNA ends created by restriction of M13 mp18 derivatives with XmaI and MluI (XM), SmaI (S) or AatII and KpnI (AK). Sites of homology to the protruding DNA ends are shown in bold.
NHEJ reactions
The standard repair reaction (25 µl) contained 30 mM HEPES–KOH, pH 7.8, 7 mM MgCl2, 50 µg/ml BSA, 4 mM ATP, 100 µM dNTPs, 40 mM phosphocreatine, 1 µg creatine phosphokinase, 50–100 µg cell extract protein and 100 ng substrate DNA. A reaction mixture containing heat-inactivated cell extract was included as a negative control. Reactions were premixed on ice, started by transfer to 25°C and incubated for 2–4 h. The reactions were terminated by addition of an equal volume of stop buffer (40 mM EDTA, 1% SDS) and 10 µg proteinase K followed by incubation at 37°C for 30 min. DNA was purified by extensive phenol/chloroform extraction, precipitated with ethanol and dissolved in TE (10 mM Tris–HCl, pH 8, 1 mM EDTA). Although covalently closed monomeric repair products were transformed into the open covalent form during the extensive extraction, this method gave higher recoveries and better reproducibility compared to silica matrix-based methods that preserved the covalently closed form.
Measurement of NHEJ in HeLa cells in vivo
With the exception of cell cultivation, all steps were performed at room temperature. Exponentially growing HeLa S3 spinner cells were collected by centrifugation (200 g, 5 min), followed by two washes with fresh medium containing 2.5% fetal calf serum. The cells were resuspended at 5 × 107 cells in the same medium and 400 µl aliquots were mixed with 1 µg DSB substrate and 50–100 ng pBluescript II SK+ (Stratagene) and transferred to electroporation cuvettes with a 4 mm gap (Bio-Rad). After 5 min, the cells were electroporated at 150 V with a 70 ms pulse using an ECM830 square wave electroporator (BTX Genetronics, San Diego, CA). Five minutes after electroporation, the cells were transferred to 100 mm tissue culture dishes and cultured in 20 ml conditioned F13 medium supplemented with 5% fetal calf serum. The NHEJ substrate was re-isolated from the cultured cells after low speed centrifugation and two washes with cold TBS (20 mM Tris–HCl, pH 7.5, 150 mM NaCl) by the method of Sadiev and Taylor (44). Briefly, after alkaline lysis, DNA was sequentially precipitated with 20% polyethylene glycol (PEG) 6000, 2.75 M LiCl (supernatant) and 0.6 vol isopropanol. After a final wash with 70% ethanol, the DNA pellet was resuspended in 50 µl of TE buffer.
Transformation of recovered substrate DNA and sequence analysis of repair products
Appropriate aliquots of recovered substrate DNA were transformed into Escherichia coli XL1blue using standard procedures (45) and plated with X-Gal and IPTG on tetracycline plates. The transformations were repeated at least three times and repair efficiency was measured as the number of plaques formed after transformation. The sequences of the repair joints were determined from transformed repair products by isolation of M13 single-stranded or replicative form DNA according to standard protocols (46) and subsequent sequencing on an ABI Prism 377 automated DNA sequencer (Applied Biosystems).
Southern hybridization
For direct analysis of repair products by Southern hybridization, recovered substrate DNA was resolved by electrophoresis in 0.8% agarose and transferred to a Hybond-N neutral nylon membrane (Amersham) according to standard protocols (46). DNA was detected either using the Rad-free system (Schleicher & Schüll) or DIG chemiluminescent detection and DIG high prime DNA labeling (Roche) with M13mp18 single-stranded DNA as probe according to the manufacturers’ instructions.
Fractionation of cell extract
To separate proliferating cell nuclear antigen (PCNA) into one specific fraction, proteins were fractionated as described previously (47,48) with minor modifications. Briefly, the cell extract was adjusted to 0.1 M NaCl and proteins were first applied to a phosphocellulose (Whatman P11) column in TDEG with 0.1 M NaCl. Bound protein was eluted with TDEG with 1 M NaCl. Peak fractions were combined resulting in fractions I (flow-through) and II (bound and eluted), respectively. Fraction I was adjusted to 0.18 M NaCl and separated on a DEAE–Sephacel column (Pharmacia) with TDEG + 0.18 M NaCl, resulting in fractions IA (flow-through) and IB (bound, eluted as above). When necessary, fractions were concentrated by dialysis against PEG 22 000 and all samples were dialyzed against TDEG. The fractions were used immediately, since they rapidly lost activity upon freezing and thawing.
PCNA depletion
PCNA was removed from cell extracts using a 20 amino acid peptide (KRRQTSMTDFYHSKTTLIFS) from the PCNA interaction site of p21 (amino acids 141–160) and containing biotin on its N-terminus. This peptide strongly interacts with PCNA (49). The peptide was synthesized in a 433A peptide synthesizer (Applied Biosystems). A control peptide in which the critical residues Q, M, F and Y (underlined) were mutated to alanine was unable to precipitate PCNA.
For PCNA removal, 1 µg peptide was linked to 80 µl of streptavidin-coated paramagnetic beads (Dynabeads M-280 Streptavidin; Dynal) in IP buffer (50 mM Tris–HCl, pH 7.5, 100 mM NaCl) at 4°C for 40 min and unbound peptide was washed out with IP buffer. Coupled magnetic beads (16 µl) were then used for depletion of 350 µg cell extract protein in IP buffer in a total volume of 100 µl for 90 min on a rocker at 4°C. Multiple rounds of depletion were required to achieve efficient removal of PCNA. Removal of PCNA was monitored by western analysis. Proteins were separated by SDS–PAGE, transferred to a PVDF membrane (Immobilon-P; Millipore) and PCNA was detected using the monoclonal antibody PC10 (Roche) as described (50).
Purification of PCNA
PCNA was purified from HeLa cells as described (51). Briefly, HeLa cell extract was purified through DEAE–Sephacel, P11 phosphocellulose, phenyl-Sepharose (all from Pharmacia), P11 phosphocellulose, DEAE-Sephacel and hydroxyapatite (Bio-Rad). The purity of the PCNA was >60% after hydroxyapatite chromatography.
RESULTS
Characterization of NHEJ in HeLa cells in vitro
The experiments described here were performed to investigate the role of DNA synthesis in NHEJ. We employed the linear form of M13mp18-derived plasmids cleaved with restriction enzymes to produce DNA substrates that could not be joined by simple ligation of complementary ends (Fig. 1). Treatment with HeLa cell extract resulted in conversion of the substrate into various mono- and multimeric products (Fig. 2). Multimeric products resulted mainly from ligation of complementary restriction ends (Fig. 1). However, for the study of NHEJ, the important products were the circular monomers that arose by self-joining of non-homologous ends. Formation of these circular monomers could be readily detected after transfection of the isolated end joining products into E.coli, since this form is transformed more efficiently than the double-digested substrate by a factor of at least 10 000 (52). Furthermore, the cleavage site of the DNA substrate is located in the α-complementing lacZ reporter gene fragment. Therefore, transfection of end joining products gave rise to either blue or white plaques depending on the reading frame and possible deletions. The plaque color therefore provided additional information on the end joining event (see below).
Figure 2.
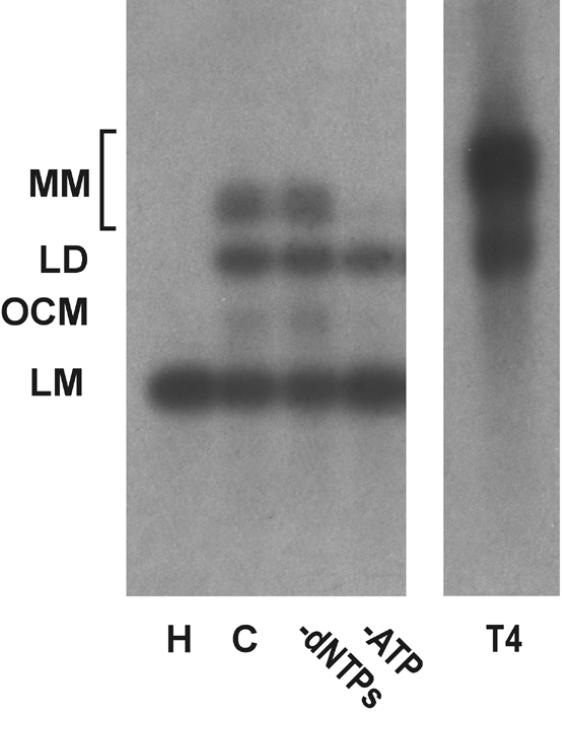
Southern blot analysis of end joining products. DSB substrate XM was incubated with HeLa whole cell extract as described in Materials and Methods. After the end joining reaction, substrate DNA was recovered, resolved by 0.8% agarose gel electrophoresis and analyzed by Southern hybridization using random primed M13mp18 as probe as described in Materials and Methods. H, heated-inactivated whole cell extract control; C, complete reaction; T4, reaction containing 0.5 Weiss U T4 ligase in the absence of cell extract. The DSB substrate linear monomer and various product forms are annotated on the left: LM, linear monomer; OCM, open circular monomer; LD, linear dimer; MM, other multimeric forms.
Except for the blunt end substrate (S), treatment of the DNA substrates with T4 DNA ligase did not result in formation of significant amounts of transformable end joining products, whereas multimeric forms were readily detectable on Southern hybridization (Fig. 2 and data not shown). Notably, treatment of substrate S with calf intestinal alkaline phosphatase did not inhibit end joining in HeLa cell extracts, although it largely prevented ligation by T4 DNA ligase (data not shown). We noticed a near linear relationship between the incubation time with cell extract and DNA substrate and the number of transformants detected for the first 4 h of incubation (data not shown).
Mammalian NHEJ is largely dependent on the activity of the DNA-dependent protein kinase, DNA-PK, since mutants in the DNA-PKCS as well as the DNA end-binding Ku subunit lead to a gross defect in this process (reviewed in 6). Therefore, we tested whether the NHEJ activity described here is also dependent on DNA-PK activity. Addition of wortmannin and LY294002, chemical inhibitors of DNA-PK and other enzymes of the phosphatidylinositol 3-kinase family, inhibited NHEJ in vitro in a dose-dependent manner. A concentration of 1 µM wortmannin reduced NHEJ by >85% and concentrations of 2 µM inhibited formation of circular end joining products completely (Fig. 3A), as measured by transformation. As revealed by Southern analysis, formation of linear dimers and multimeric forms was also strongly reduced (Fig. 3B). A concentration of 250 µM LY294002 was required to inhibit formation of circular end joining products by 74%. The relatively lower sensitivity of our NHEJ activity compared to other reports (53) is probably due to the higher concentration of ATP and the ATP-regenerating system employed here. LY294002 is competitive with ATP for DNA-PK (54). Furthermore, a polyclonal antibody against the Ku86 subunit of DNA-PK inhibited NHEJ in vitro (Fig. 3D and E), excluding the possibility that inhibition of NHEJ is due to inhibition of another enzyme of the phosphatidylinositol 3-kinase family. This antibody has been previously found to inhibit NHEJ (53). Southern analysis revealed that only formation of circular monomers was inhibited, whereas formation of multimeric forms by direct ligation appeared even to be increased (Fig. 3E). Together, our results indicate that at least formation of circular monomers by the NHEJ activity described here is dependent on DNA-PK.
Figure 3.
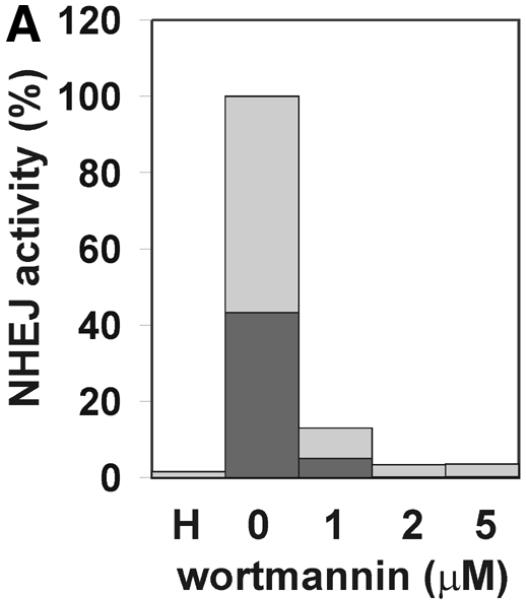
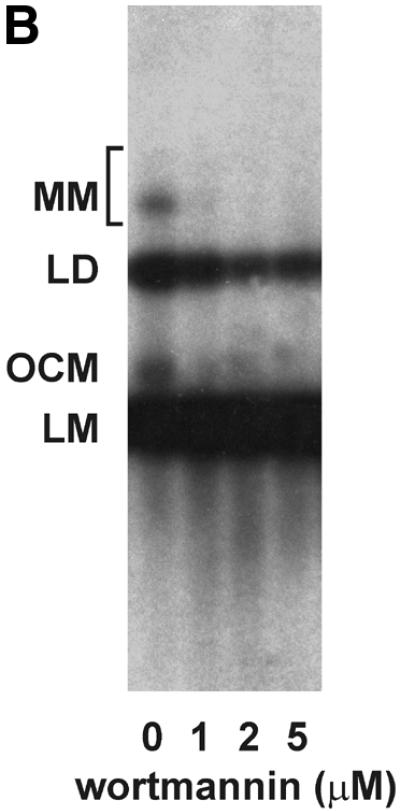
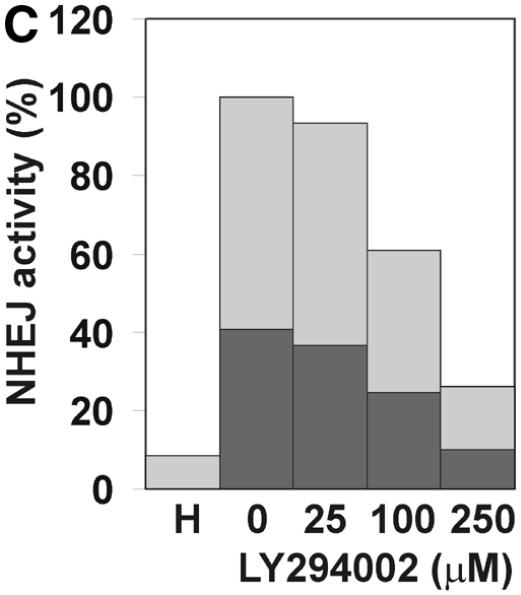
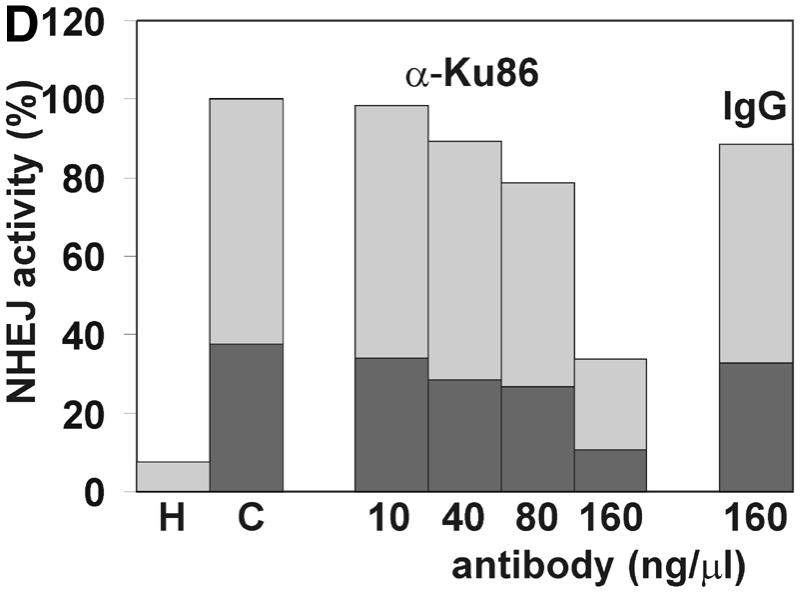
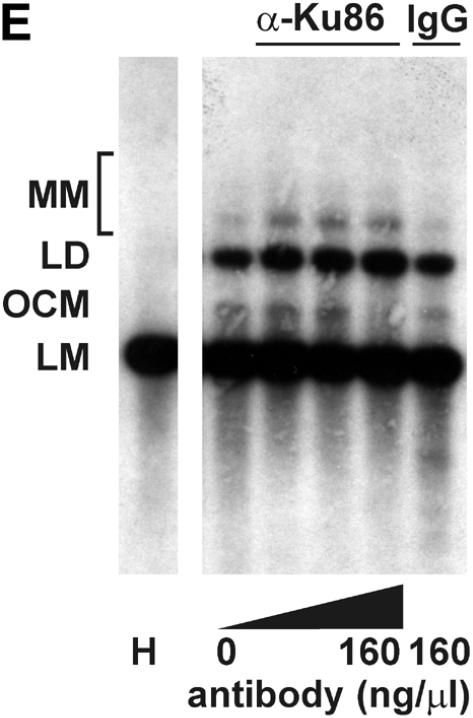
The effect of inhibitors of the DNA-PK on NHEJ in HeLa cell extracts. DSB substrate XM was incubated with HeLa whole cell extract as described in Materials and Methods. The indicated concentrations of wortmannin (A and B), LY294002 (C), both chemical inhibitors of DNA-PK and other enzymes of the phosphatidylinositol 3-kinase family, or antibodies against Ku86 (D and E) were added to each reaction. In (A), (C) and (D), the substrate DNA was recovered after the NHEJ reaction and transfected into E.coli cells to assess formation of circular monomeric end joining products as described in Materials and Methods. The number of plaques formed was measured and they were scored as blue or white. NHEJ activity is represented as the relative percentage of plaques compared to the complete reaction. The fraction of blue plaques is indicated in black and the fraction of white plaques in gray. H, negative control containing heated cell extract. In (B) and (D), the substrate DNA was recovered after the repair reaction, resolved by 0.8% agarose gel electrophoresis and analyzed by Southern hybridization using random primed M13mp18 as probe as described in Materials and Methods. Inhibitor concentrations are indicated under the lanes. H, negative control containing heated cell extract. The DSB substrate linear monomer and various product forms are annotated on the left: LM, linear monomer; OCM, open circular monomer; LD, linear dimer; MM, other multimeric forms.
Nucleotide requirements of the end joining reaction
We next addressed the nucleotide requirement of NHEJ measured in vitro. As shown in Figure 4, NHEJ was absolutely dependent on ATP, as would be expected, since it requires a DNA ligation step. Lower concentrations of dNTPs could substitute for ATP only to a small degree. On the other hand, there was no absolute dependence on the presence of dNTPs in the reaction. This is consistent with earlier reports in which NHEJ was measured in the absence of dNTPs (32,55,56). Nevertheless, we detected a substantial reduction in NHEJ efficiency for all three substrates tested when dNTPs were omitted from the reaction (Fig. 4). The reduction in circular monomer products was more apparent for DNA substrates with 5′ and 3′ protruding ends (XM, 29%; AK, 53%). In particular, for substrate XM, the sub-population of end joining products that gave rise to white plaques was substantially reduced. Circular monomers produced from the blunt end DNA substrate S were reduced only by 20%. Similar results were observed when the blunt end DNA substrate was dephosphorylated by calf intestinal alkaline phosphate to prevent simple ligation (data not shown).
Figure 4.
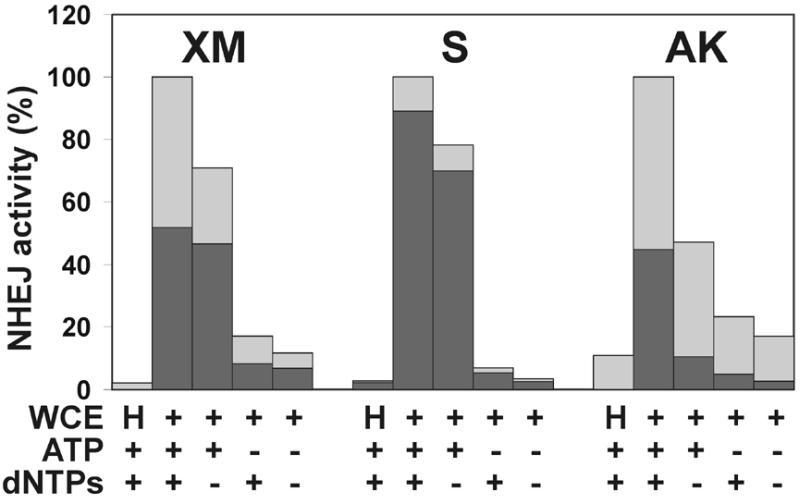
Requirement for nucleotides for NHEJ in vitro. DSB substrates were incubated with HeLa whole cell extract as described in Materials and Methods. ATP and/or dNTP were added to the reactions to 4 mM and 100 µM concentrations, respectively, as indicated. Substrate DNA was recovered and transfected into E.coli to assess formation of circular monomeric end joining products as described in Materials and Methods. The number of plaques formed was measured and they were scored as blue or white. NHEJ activity is presented as the percentage of plaques compared to the complete reaction. The fraction of blue plaques is indicated in black and the fraction of white plaques in gray. H, negative control containing heated cell extract.
Aphidicolin inhibits NHEJ in vitro
The stimulatory effect of dNTPs suggested that DNA synthesis may play a role in NHEJ. To study this further, and in order to identify the types of Pols that might be involved in this process, we tested the effect of chemical inhibitors of Pols on NHEJ in vitro. In the presence of aphidicolin, an inhibitor of the Pol α-like Pols α, δ and ɛ, formation of transformable end joining products was reduced. The inhibition was not complete, but reached levels similar to those found when dNTPs were omitted, a 37% compared to 29% reduction (Fig. 5). End joining of substrate AK was an exception, showing only a 20% reduction compared to a 53% reduction in the absence of dNTPs. Notably, the sub-population of end joining products giving rise to white plaques was again particularly affected with substrate XM. The differential effect of aphidicolin on the three substrates can be best explained by the complex mode of inhibition by aphidicolin (57). Aphidicolin is a competitive inhibitor with respect to dCTP and, to a lesser extend, with respect to dTTP. The inhibition therefore corresponds well with the need for incorporation of dCMP into the different substrates (Fig. 6).
Figure 5.
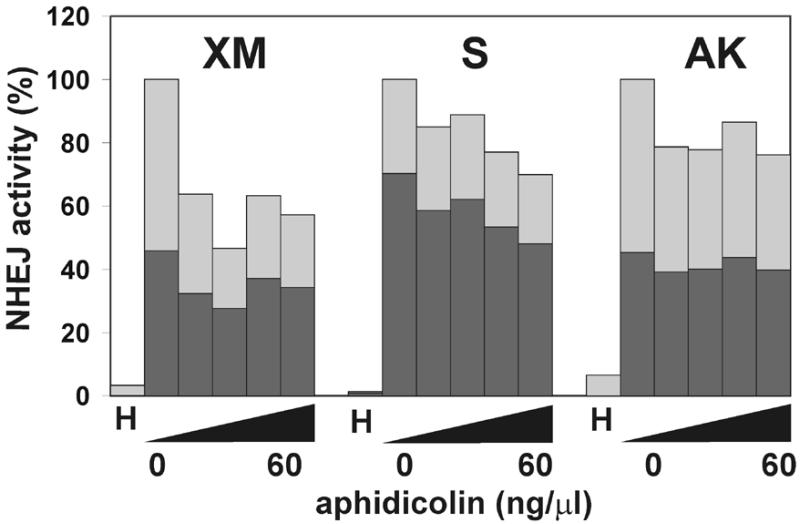
The effect of aphidicolin on NHEJ in vitro. DSB substrates were incubated with HeLa whole cell extract as described in Materials and Methods in the presence of increasing concentrations of aphidicolin (10, 20, 40 and 60 ng/µl). Substrate DNA was recovered to assess the formation of circular monomeric end joining products as described in Materials and Methods. The fraction of blue plaques is indicated in black and the fraction of white plaques in gray. H, negative control containing heated cell extract.
Figure 6.
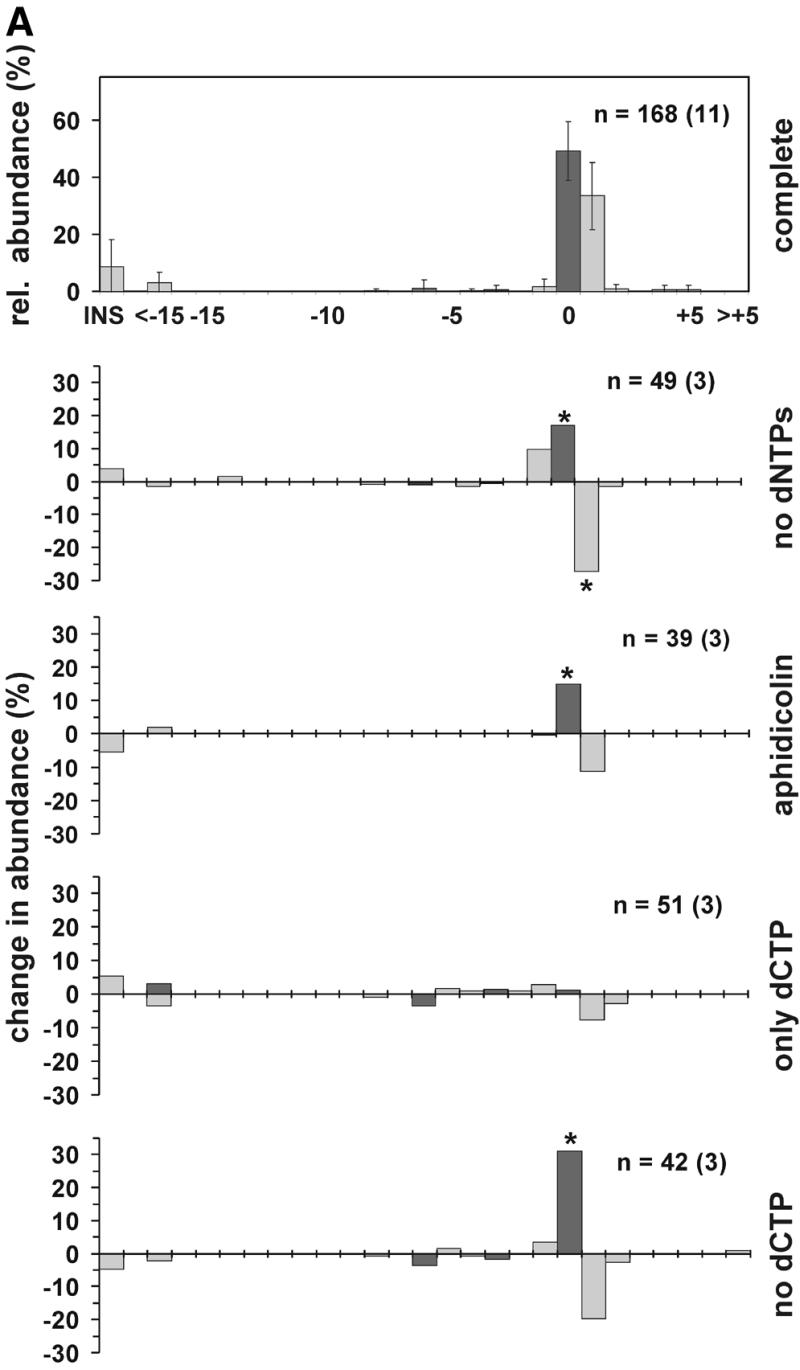

The effect of DNA repair synthesis on the distribution of NHEJ products. (A) DSB substrate XM was incubated with HeLa whole cell extract as described in Materials and Methods. After end joining, the reaction substrate DNA was recovered and transfected into E.coli to assess the formation of circular monomeric end joining products as described in Materials and Methods. The DNA sequence at the break site was determined for an appropriate number of products giving rise to white and blue plaques. The abundance of a particular NHEJ product was then determined as a fraction of this product configuration among the blue or white plaques. The NHEJ products are shown as a distribution of product length and marked in black or gray, corresponding to blue or white plaque phenotype, respectively. The topmost panel presents the relative abundance of end joining products in the complete reaction, the error bar indicating the standard deviation. Lower panels indicate the change in abundance of end joining products when compared to the complete reaction and in response to omission or inclusion of compounds as shown on the right. Aphidicolin was present at 60 ng/µl. n, number of individual products analyzed. The numbers in independent experiments are indicated in parentheses. Statistically significant changes (P < 0.05, unpaired Student’s t-test) in product distribution, when compared with the complete reaction, are indicated by an asterisk. (B) The diagram presents the most abundant configurations of NHEJ joining intermediates representing different repair modes. See text for more details.
The effect of DNA repair synthesis on end joining products
Omission of dNTPs or addition of aphidicolin not only influenced the overall rate of NHEJ but also appeared to influence the type of end joining products formed, as indicated by a change in the ratio of white to blue plaques. We therefore isolated and sequenced end joining products in order to study them more closely. Since substrate XM appeared to be joined most efficiently and we also noticed a clear effect on plaque phenotype distribution, we concentrated on analysis of end joining products from this substrate. In several independent experiments, two end joining products appeared to be predominant (Fig. 6A). Most abundant was the product with length 0, i.e. the two protruding ends were joined without deletion or insertion of nucleotides, utilizing a microhomology of two of the four nucleotides between the overhangs (Fig. 6B, direct mode). The sequence corresponded to either the (+)- or (–)-strand or, in rare cases, both, when the 2 bp mismatch was not corrected. The product with length +1 was also regularly recovered. Here, the protruding ends formed a 3 bp overhang leaving a 1 nt gap on each side (Fig. 6B, overlap mode). Again, a 2 bp microhomology was utilized with either strand serving as template. Joining products that contained mismatches at the ends were also found. On the other hand, deletions at the DNA ends were rare, accounting for <5% of joining products. Even a 4 bp sequence of the protruding MluI overhang paired very inefficiently with a homologous sequence in the immediate proximity of the other end (Fig. 1), indicating that the end joining activity described here minimizes loss of DNA.
Next we addressed how the product distribution is modified when dNTPs are omitted. As can be seen in Figure 6A, there was a significant decrease in the products forming white plaques with length +1 and a concurrent increase in the products forming blue plaques with length 0. Furthermore, shorter products, in particular the –1 product, were found more regularly. We observed a similar effect on the distribution of products in the presence of aphidicolin, although this effect was not as pronounced as in the absence of dNTPs.
The end configuration of the ‘overlap mode’ (Fig. 6B) suggested that dCMP alone could be sufficient to support formation of products with length +1. We therefore analyzed products from reactions to which only dCTP or only the other three nucleotides had been added (Fig. 6A). Indeed, the presence of dCTP was critical for +1 product formation. However, dCTP alone did not support NHEJ efficiently, since end joining activity was only marginally higher than in the absence of dNTPs (data not shown).
Taken together, sequencing indicated that dNTPs and DNA synthesis by an aphidicolin-sensitive Pol are required for the +1 product from substrate XM. For substrate AK, the results from limited sequencing were similar, whereas product lengths from the blunt end substrate S did not seem to be affected by DNA synthesis. In this case, the vast majority of the joining products retained the original sequence.
Anti-DNA polymerase α antibodies inhibit the end joining reaction
To identify the Pol required for the DNA repair synthesis step we studied the effects of antibodies neutralizing Pol activities on NHEJ in vitro (Fig. 7). Antibodies against Pol α strongly inhibited the end joining process in a concentration-dependent manner. As little as 5 ng/µl antibody SJK-287-138 (38) inhibited NHEJ by 12% and at a concentration of 80 ng/µl end joining was reduced to <20% (Fig. 7A). A second antibody against Pol α, SJK-132-20, gave similar results, although inhibition was less pronounced (data not shown). Interestingly, the antibodies inhibited formation of white and blue repair products to the same extent and no effect on the distribution of end joining products was found, as analyzed by sequencing (data not shown). Southern analysis revealed that the antibodies specifically inhibited formation of circular monomers, whereas formation of multimeric forms was stimulated (Fig. 7B). This is distinct from the results obtained in the absence of dNTP or after addition of aphidicolin. Binding of the antibody molecule to Pol α may disrupt the repair complexes and thereby affect all NHEJ products. In contrast, antibodies neutralizing the activity of Pol β or ɛ as well as purified mouse IgG fraction had no effect on NHEJ of substrate XM, even at high concentrations. We have shown previously that a neutralizing antibody against Pol ɛ inhibits replicative DNA synthesis in a similar manner to the antibodies against Pol α that were utilized in this study (39). Furthermore, a polyclonal antibody against Pol β has been shown to efficiently inhibit base excision repair in cell extracts (40). Taken together, these results indicate that Pol α is specifically required for joining of non-complementary DNA ends but not for joining of complementary DNA ends.
Figure 7.
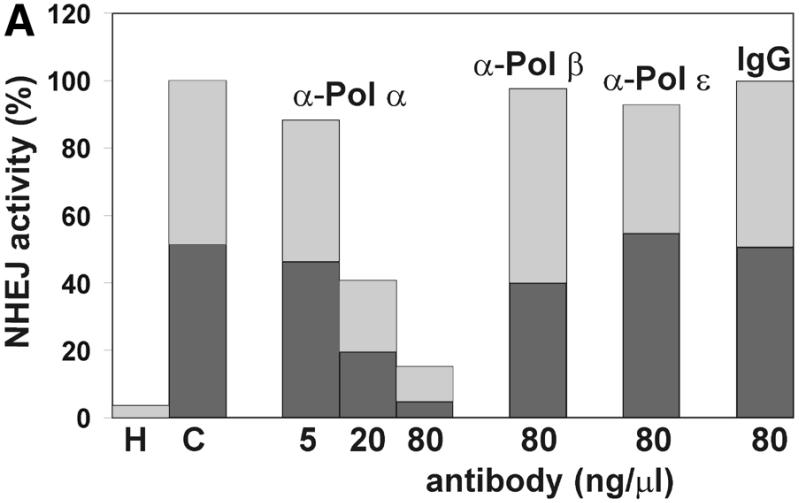
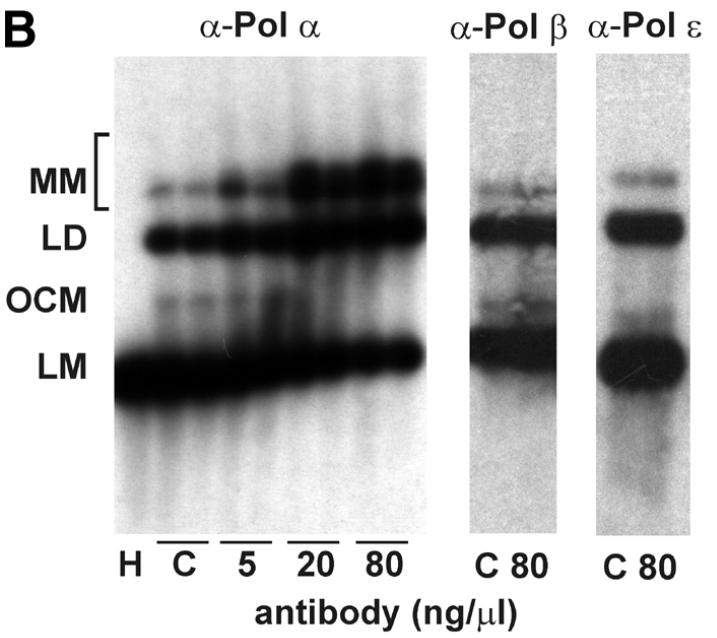
Antibodies against DNA polymerase α inhibit NHEJ. HeLa whole cell extracts were incubated for 1 h on ice with the indicated amounts of antibodies neutralizing the polymerase activity of Pols α, β and ɛ or in the presence of mouse IgG antibody fraction. NHEJ activity of the extracts in the presence of the antibodies was then measured with the DSB substrate XM as described in Materials and Methods. (A) After the end joining reaction, substrate DNA was recovered and transfected into E.coli to assess formation of circular monomeric end joining products as described in Materials and Methods. The fraction of blue plaques is indicated in black and the fraction of white plaques in gray. H, negative control containing heat-inactivated cell extract; C, complete reaction in the absence of antibodies. (B) After the end joining reaction the substrate DNA was recovered and analyzed by Southern hybridization as described in Materials and Methods. Duplicates are presented for the reaction containing antibodies against Pol α. The DSB substrate linear monomer and various product forms are annotated on the left: LM, linear monomer; OCM, open circular monomer; LD, linear dimer; MM, other multimeric forms.
NHEJ of model substrates in HeLa cells in vivo
To study how far NHEJ observed in the cell-free system reflects the situation in vivo, we introduced substrate XM to HeLa cells by electroporation. At the times indicated, plasmid DNA was recovered and analyzed by transformation. Circular pBlueScript vector served as an internal control to assess the efficiency of transformation and recovery. The majority of the NHEJ products from substrate XM in vivo could already be detected 30 min after electroporation (Fig. 8). Only a small further increase was observed up to 2 h of culture, after which a decrease in products occurred, possibly due to instability of the substrate DNA within the cells. The distribution of the end joining products formed in transfected HeLa cells in vivo and in cell extracts in vitro did not vary noticeably, as judged by plaque color distribution and DNA sequencing of products (data not shown). These results indicate that the NHEJ measured in vitro well reflects the conditions in vivo.
Figure 8.

NHEJ of a DSB model substrate in HeLa cells in vivo. Substrate XM was introduced into HeLa cells by electroporation. After transfection, cells were cultured for the indicated times after which substrate DNA was recovered and transfected into E.coli to assess formation of circular monomeric end joining products as described in Materials and Methods. NHEJ activity after 2 h in culture was taken as 100%. Error bars represent the standard deviation of two independent experiments.
The influence of PCNA on NHEJ in vitro
PCNA is a small nuclear protein that forms a trimer of doughnut-shaped structure with a central hole large enough to encircle DNA. It serves as a sliding clamp that mediates contact of proteins with DNA (58). PCNA takes part in replication as well as many DNA repair pathways, particularly by strongly increasing the processivity of Pol δ and, under certain conditions, Pol ɛ. Since our results indicated a role of aphidicolin-sensitive Pol α, δ or ɛ in NHEJ, the involvement of PCNA was studied to aid identification of the Pol associated with NHEJ. We employed two independent methods. First, PCNA was separated from the majority of other cellular factors by passage through two ion exchange columns (Fig. 9A) and NHEJ was then studied in combinations of the resulting fractions. However, the NHEJ activity of the combined fractions decreased rapidly upon fractionation, with ∼30% activity remaining after two columns. The three resulting fractions were all required to obtain efficient end joining (Fig. 9B). Omission of the fraction containing PCNA decreased end joining by 21% and the activity could be fully restored by addition of purified PCNA. Since we detected only a moderate effect of PCNA using fractionation, we depleted HeLa whole cell extract of PCNA using a p21-derived peptide (49) as a second approach to study the effect of PCNA on NHEJ. Removal of PCNA was nearly complete, as indicated by western analysis of the cell extract (Fig. 9C). PCNA-depleted cell extracts showed decreased NHEJ activity in several independent experiments, although the reduction was only 15% (Fig. 9D). Addition of purified PCNA to the depleted cell extract restored the activity. A control peptide mutated in the residues critical for PCNA binding did not precipitate PCNA and had no effect on NHEJ, confirming that the observed effect is specific for PCNA depletion. Taken together, PCNA influences NHEJ in vitro, although the modest effect argues for a minor or redundant role in this process.
Figure 9.

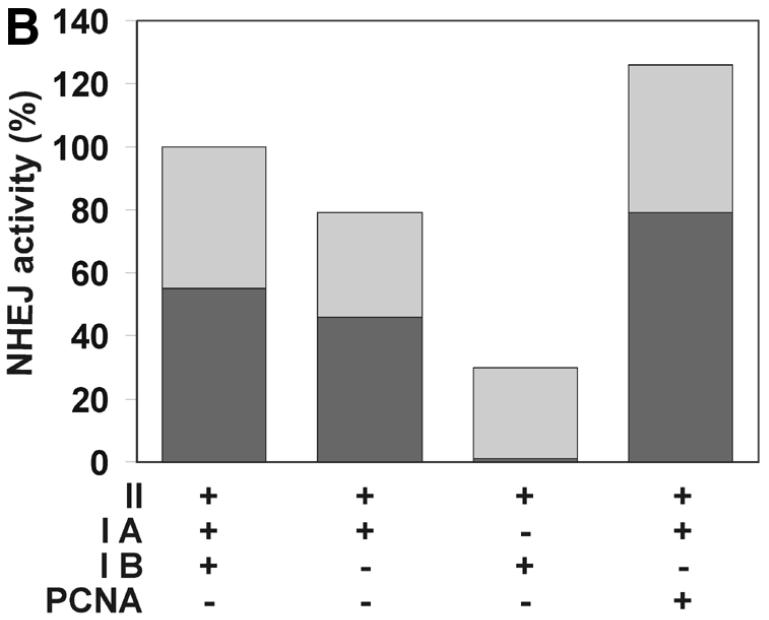
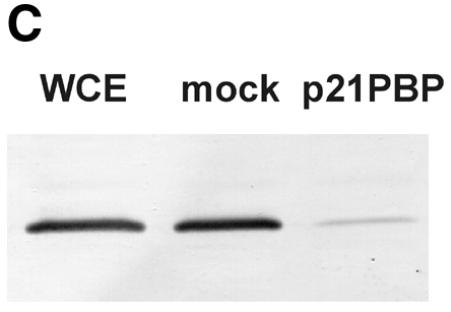
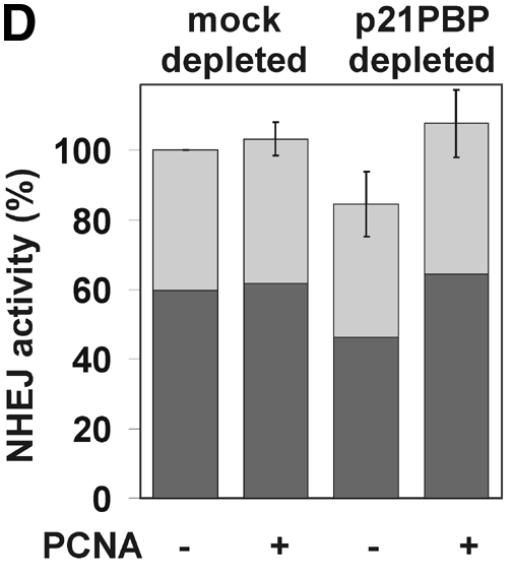
The effect of PCNA on NHEJ. (A) Flow diagram of fractionation of the HeLa cell extract to separate PCNA. As described in Materials and Methods, HeLa whole cell extract was passed through a phosphocellulose column, resulting in fractions I (flow-through) and II (bound). PCNA-containing fraction I was further separated through DEAE–Sephacel to generate fractions IA and IB, the latter containing PCNA. (B) DSB substrate XM was incubated with combinations of different fractions from the above fractionation scheme as indicated. Substrate DNA was recovered to assess formation of circular monomeric end joining products as described in Materials and Methods, defining the NHEJ activity of the combined fractions IA, IB and II as 100%. The fraction of blue plaques is indicated in black and the fraction of white plaques in gray. Note that substitution of fraction IB with purified PCNA restores NHEJ to >100%. (C) Western analysis indicating the removal of PCNA from HeLa cell extract. PCNA was depleted from HeLa whole cell extract with a p21-derived peptide (PBP for PCNA-binding peptide) or mock-depleted with a mutated p21-derived peptide (mock). The mutated p21-derived peptide was incapable of precipitating PCNA and served as a negative control. PCNA was detected in cell extracts by western analysis as described in Materials and Methods. (D) Depletion of PCNA causes a small but significant reduction in NHEJ activity in HeLa cell extracts. NHEJ activity of PCNA-depleted and mock-depleted HeLa cell extract was determined as described in Materials and Methods. NHEJ activity of the mock-depleted cell extract was arbitrarily taken as 100%. Error bars indicate the standard deviation of total NHEJ activity. The reduction in NHEJ in PCNA-depleted cell extracts was highly significant in eight independent experiments (P = 0.0012, one-sided Student’s t-test).
DISCUSSION
In this paper we describe an NHEJ activity that apparently depends on the activity of DNA-PK, since it is inhibited by chemical inhibitors of the phosphatidylinositol 3-kinase family (11,53,54,59) and by antibodies against Ku86. The importance of DNA-PK and the DNA end binding activity Ku for efficient NHEJ has been long recognized based on genetic studies in rodent cell lines and yeast and by studying NHEJ in vivo (reviewed in 6). Binding of Ku to the DNA ends appears to be a prerequisite for successful repair (9,53,60). Ku, as part of DNA-PK, facilitates transient bridging of the two ends (61) and may play an important role in recruitment of additional end joining factors, such as nucleases and ligases, to the break site (17,21,61,62). End joining is accompanied by extensive loss of DNA at the break site in the absence of Ku (9), consistent with a protective role against nucleolytic degradation of DNA ends. Our results indicate that DNA synthesis constitutes a yet unrecognized factor contributing to stability of the DNA ends. The shorter end joining products that we detected in the absence of dNTPs (Fig. 6) could reflect a disturbance in the balance between nuclease and polymerase activity.
In fact, DNA repair synthesis influences not only the outcome, but also the efficiency of the repair event in NHEJ in vitro, since omission of dNTPs reduced the amount of NHEJ products. In reality, the effect may be even stronger, because low levels of dNTPs may be provided during the NHEJ reaction from degraded DNA via the ATP-driven salvage pathway. This would explain the low levels of overlap mode end joining products that are dependent on DNA synthesis even in the absence of externally added dNTPs (Fig. 6).
Several conflicting reports on the role of DNA synthesis in NHEJ have been published. They can be explained by the influence of varying DNA structures at the DNA break ends. The configuration of the DNA ends determines the quality of joining products (9,28,63). In some studies joining of non-complementary DNA ends occurred in the absence of dNTPs and, consequently, in the absence of DNA synthesis (32,56). Using partially purified calf thymus extracts, the same group found that dNTPs were only required for fill-in DNA synthesis opposite a 5′ overhang to allow subsequent blunt end ligation (32). This reaction was inhibited by ddTTP, but not by aphidicolin. These results do not contradict our results, since we noticed that repair via blunt end ligation occurred extremely rarely in our experimental system (Fig. 6A). On the other hand, Lehmann et al. found that aphidicolin inhibited end joining in activated Xenopus germinal vesicle extracts (34). As in our study, small homologies were utilized for end joining, but the results of the latter study differed from those of our study in that deletions were generated. Pfeiffer and co-workers reported that ‘overlap mode’ NHEJ by partial alignment of non-identical DNA overhangs was inhibited by ddNTPs in Xenopus egg extracts (29,64), an effect that we also observed in HeLa cell extract (data not shown). The authors proposed that the DNA could not be ligated after incorporation of the chain terminator into gaps resulting from overlaps. These results emphasize the importance of DNA repair synthesis, but do not address directly which type of Pols are required during the process.
Considering the very specific role of Pol α in producing RNA/DNA primers during DNA replication (reviewed in 65), the apparent involvement of this enzyme in NHEJ is surprising. When compared to other Pols, there are only a few studies suggesting a role for Pol α in DNA repair processes (66,67). On the other hand, Pol α seems to fit well with this process. It is an aphidicolin-sensitive enzyme with moderate processivity independent of auxiliary factors (68), which could be expected from a Pol needed for synthesis of relatively short DNA repair patches (69). Furthermore, Pol α was isolated from HeLa cells as an activity capable of performing DNA synthesis on discontinuous, unlinked DNA templates (70), a process resembling NHEJ. In contrast, nuclear Pols β, δ and ɛ showed only very little DNA synthesis on the same templates.
Few studies have thus far addressed the involvement of Pols in NHEJ directly. Sensitivity of NHEJ activities to ddTTP has been taken as an indication that Pol β is involved in this process. The Pol β-related POL IV in budding yeast was indeed found to be required for NHEJ of non-complementary DNA ends (71). In contrast, deficiency in Pol β in mouse fibroblasts does not render the cells more sensitive to γ-irradiation (72) and V(D)J recombination proceeds normally in the absence of Pol β in mouse cells (73), arguing that Pol β is not involved in mammalian NHEJ. Göttlich et al. (74) described the partial purification of a NHEJ activity from Xenopus egg extracts. Characterization of this activity revealed the presence of Pol ɛ, ligase III and FEN-1, in addition to 3′ and 5′ directional exonuclease activities. This activity differs from NHEJ of Xenopus whole egg extract in that it generates deletions during the joining process. Since neither Ku nor DNA-PKCS could be detected, this activity probably represents a minor pathway of NHEJ (Fig. 10).
Figure 10.

Proposed roles for Pol α and PNCA in NHEJ. The figure presents a schematic overview of different NHEJ pathways described in the literature or identified in this study and characteristic intermediates of these pathways. A Ku-dependent pathway relying on the activities of DNA-PK, ligase IV/XRCC4 and MRE11 exonuclease is prominent in mammalian cells (53,60). This pathway appears to be branched, based on the requirement for DNA repair synthesis. A DNA synthesis-dependent pathway apparently requires Pol α activity to complete end joining whereas a DNA-independent pathway does not need any Pol activity. A Ku-independent pathway can be detected in many organisms in cases where the major pathway is defective or overloaded. This pathway may rely on the activities of Pol ɛ, FEN-1 and ligase III (74). Our results are consistent with a role of PCNA in this alternative pathway. See Discussion for details.
In cells, several alternative NHEJ pathways with different requirements for DNA repair synthesis may operate simultaneously, as has been proposed by Moore and Haber (75). These authors found that the predominant end joining products contained either short insertions or short deletions when studying HO endonuclease-induced NHEJ in S.cerevisiae. The authors could show that the deletion, but not the insertion, mechanism depended on the RAD50/MRE11/XRS2 gene products. When breaks were induced in G1 phase of the cell cycle, end joining became less efficient and, in particular, formation of the insertion products was affected. In this case, additional products with large deletions appeared. Taken together, the mechanistic similarities between our study and the studies described above suggests that multiple alternative NHEJ pathways are conserved in different organisms (Fig. 10).
The DNA sliding clamp PCNA is of central importance for DNA replication as well as for multiple DNA repair pathways. It can interact with several proteins, thereby mediating a stable contact with the DNA (see 76 for a review). In Drosophila, the repair of DSBs created by P-element excision and X-ray damage requires PCNA as well as Ku and DmRAD54 in a homology-dependent process (77–80). Since removal of PCNA decreased NHEJ activity only moderately, it may not be critical for repair. However, PCNA appears to facilitate the process. Furthermore, PCNA depletion did not affect the product distribution as judged by analysis of plaque phenotype and analysis of DNA sequences of end joining products (data not shown). Therefore, PCNA probably does not serve as an auxiliary factor for a Pol. The relatively weak effect of PCNA suggests that it is involved in a minor pathway of NHEJ (Fig. 10), possibly in association with the PCNA-interacting nuclease FEN-1 (27,74). To our knowledge, this is the first report where a factor involved in NHEJ has been identified by biochemical fractionation and reconstitution of repair activity with the purified factor.
In conclusion, our results indicate that Pol activity represents an important factor affecting NHEJ, marking a branch point of the repair pathway and guaranteeing a minimum loss of DNA sequence information during this process (Fig. 10). The results suggest that Pol α is responsible for DNA repair synthesis in vitro. PCNA plays a less important role in NHEJ, probably being involved in a minor alternative pathway. Altogether, the data presented here help to explain the molecular basis for several modes of NHEJ that have been regularly observed.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr Samuel Wilson for kindly providing us with the antibodies against Pol β. We are grateful to Dr Deborah Kaska for careful reading and valuable comments on the manuscript. This study was supported by a grant from the Research Council for the Environment and Natural Resources, Academy of Finland and by grants D11-6 E417-4 F111-9 from the Center for International Mobility, Finland, and D/95/05708 from the Deutscher Akademischer Austauschdienst, Germany, to H.P.
References
- 1.Friedberg E., Walker,G. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 2.Haber J.E. (2000) Recombination: a frank view of exchanges and vice versa. Curr. Opin. Cell Biol., 12, 286–292. [DOI] [PubMed] [Google Scholar]
- 3.Takata M., Sasaki,M.S., Sonoda,E., Morrison,C., Hashimoto,M., Utsumi,H., Yamaguchi-Iwai,Y., Shinohara,A. and Takeda,S. (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J., 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee S.E., Mitchell,R.A., Cheng,A. and Hendrickson,E.A. (1997) Evidence for DNA-PK-dependent and -independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Mol. Cell. Biol., 17, 1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Essers J., van Steeg,H., de Wit,J., Swagemakers,S.M.A., Vermeij,M., Hoeijmakers,J.H.J. and Kanaar,R. (2000) Homologous and non-homologous recombination differentially affect DNA damage repair in mice. EMBO J. 19, 1703–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Critchlow S.E. and Jackson,S.P. (1998) DNA end-joining: from yeast to man. Trends Biochem. Sci., 23, 394–398. [DOI] [PubMed] [Google Scholar]
- 7.Kanaar R., Hoeijmakers,J.H.J. and van Gent,D.C. (1998) Molecular mechanisms of DNA double strand break repair. Trends Cell Biol., 8, 483–489. [DOI] [PubMed] [Google Scholar]
- 8.Featherstone C. and Jackson,S.P. (1999) Ku, a DNA repair protein with multiple cellular functions? Mutat. Res., 434, 3–15. [DOI] [PubMed] [Google Scholar]
- 9.Feldmann E., Schmiemann,V., Goedecke,W., Reichenberger,S. and Pfeiffer,P. (2000) DNA double-strand break repair in cell-free extracts from Ku80-deficient cells: implications for Ku serving as an alignment factor in non-homologous DNA end joining. Nucleic Acids Res., 28, 2585–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurimasa A., Kumano,S., Boubnov,N.V., Story,M.D., Tung,C.-S., Peterson,S.R. and Chen,D.J. (1999) Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol. Cell. Biol., 19, 3877–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calsou P., Frit,P., Humbert,O., Muller,C., Chen,D.J. and Salles,B. (1999) The DNA-dependent protein kinase catalytic activity regulates DNA end processing by means of Ku entry into DNA. J. Biol. Chem., 274, 7848–7856. [DOI] [PubMed] [Google Scholar]
- 12.Li Z., Otevrel,T., Gao,Y., Cheng,H.L., Seed,B., Stamato,T.D., Tacclioli,G.E. and Alt,F.W. (1995) The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell, 83, 1079–1089. [DOI] [PubMed] [Google Scholar]
- 13.Critchlow S.E., Bowater,R.P. and Jackson,S.P. (1997) Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol., 7, 588–598. [DOI] [PubMed] [Google Scholar]
- 14.Hermann G., Lindahl,T. and Schär,P. (1998) Saccharomyces cerevisiae LIF1: a function involved in DNA double-strand break repair related to mammalian XRCC4. EMBO J., 17, 4188–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank K.M., Sekuguchi,J.M., Seidl,K.J., Swat,W., Rathbun,G.A., Cheng,H.-L., Davidson,L., Kangaloo,L. and Alt,F.W. (1998) Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature, 396, 173–177. [DOI] [PubMed] [Google Scholar]
- 16.Gao Y., Ferguson,D.O., Xie,W., Manis,J.P., Sekiguchi,J., Frank,K.M., Chaudhuri,J., Horner,J., DePinho,R.A. and Alt,F.W. (2000) Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature, 404, 897–900. [DOI] [PubMed] [Google Scholar]
- 17.McElhinny S.A.N., Snowden,C.M., McCarville,J. and Ramsden,D.A. (2000) Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol. Cell. Biol., 20, 2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L., Trujillo,K., Sung,P. and Tomkinson,A.E. (2000) Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J. Biol. Chem., 275, 26196–26205. [DOI] [PubMed] [Google Scholar]
- 19.Lee K.-J., Huang,J., Takeda,Y. and Dynan,W.S. (2000) DNA ligase IV and XRCC4 form a stable mixed tetramer that functions synergistically with other repair factors in a cell-free end-joining system. J. Biol. Chem., 275, 34787–34796. [DOI] [PubMed] [Google Scholar]
- 20.Junop M.S., Modesti,M., Guarné,A., Ghirlando,R., Gellert,M. and Yang,W. (2000) Crystal structure of the XRCC4 DNA repair protein and implications for end joining. EMBO J., 19, 5962–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goedecke W., Eijpe,M., Offenberg,H.H., van Aalderen,M. and Heyting,C. (1999) Mre11 and Ku70 interact in somatic cells, but are differentially expressed in early meiosis. Nature Genet., 23, 194–198. [DOI] [PubMed] [Google Scholar]
- 22.Haber J.E. (1998) The many interfaces of Mre11. Cell, 95, 583–586. [DOI] [PubMed] [Google Scholar]
- 23.Paull T.T. and Gellert,M. (1998) The 3′ to 5′ exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol. Cell, 1, 969–979. [DOI] [PubMed] [Google Scholar]
- 24.Paull T.T. and Gellert,M. (2000) A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc. Natl Acad. Sci. USA, 97, 6409–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petrini J.H. (1999) The mammalian Mre11-Rad50-nbs1 protein complex: integration of functions in the cellular DNA-damage response. Am. J. Hum. Genet., 64, 1264–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin S.G., Laroche,T., Suka,N., Grunstein,M. and Gasser,S.M. (1999) Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell, 97, 621–633. [DOI] [PubMed] [Google Scholar]
- 27.Wu X., Wilson,T.E. and Lieber,M.R. (1999) A role for FEN-1 in nonhomologous DNA end joining: the order of strand annealing and nucleolytic processing events. Proc. Natl Acad. Sci. USA, 96, 1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Labhart P. (1999) Nonhomologous DNA end joining in cell-free systems. Eur. J. Biochem., 265, 849–861. [DOI] [PubMed] [Google Scholar]
- 29.Thode S., Schäfer,A., Pfeiffer,P. and Vielmetter,W. (1990) A novel pathway of DNA end-to-end joining. Cell, 60, 921–928. [DOI] [PubMed] [Google Scholar]
- 30.Thacker J., Chalk,J., Ganesh,A. and North,P. (1992) A mechanism for deletion formation in DNA by human cell extracts: the involvement of short sequence repeats. Nucleic Acids Res., 20, 6183–6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicolás A.L., Munz,P.L. and Young,C.S.H. (1995) A modified single-strand annealing model best explains the joining of DNA double-strand breaks in mammalian cells and cell extracts. Nucleic Acids Res., 23, 1036–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mason R.M., Thacker,J. and Fairmann,M.P. (1996) The joining of non-complementary DNA double-strand breaks by mammalian extracts. Nucleic Acids Res., 24, 4946–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicolás A.L. and Young,C.S.H (1994) Characterization of DNA end joining in a mammalian cell nuclear extract: junction formation is accompanied by nucleotide loss, which is limited and uniform but not site specific. Mol. Cell. Biol., 14, 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehmann C.W., Trautman,J.K. and Caroll,D. (1994) Illegitimate recombination in Xenopus: characterization of end-joined junctions. Nucleic Acids Res., 22, 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips J.W. and Morgan,W.F. (1994) Illegitimate recombination induced by DNA double-strand breaks in a mammalian chromosome. Mol. Cell. Biol., 14, 5794–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sargent R.G., Brenneman,M.A. and Wilson,J.H. (1997) Repair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol. Cell. Biol., 17, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang F., Han,M., Romnienko,P.J. and Jasin,M. (1998) Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl Acad. Sci. USA, 95, 5172–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka S., Hu,S.Z., Wang,T.S.-F. and Korn,D. (1982) Preparation and preliminary characterization of monoclonal antibodies against human DNA polymerase α. J. Biol. Chem., 257, 8386–8390. [PubMed] [Google Scholar]
- 39.Pospiech H., Kursula,I., Abdel-Aziz,W., Malkas,L., Uitto,L., Kastelli,M., Vihinen-Ranta,M., Eskelinen,S. and Syvaoja,J.E. (1999) A neutralizing antibody against human DNA polymerase ɛ inhibits cellular but not SV40 DNA replication. Nucleic Acids Res., 27, 3799–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singhal R.K., Prasad,R. and Wilson,S.H. (1995) DNA polymerase β conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J. Biol. Chem., 270, 949–957. [DOI] [PubMed] [Google Scholar]
- 41.Nishida C., Reinhard,P. and Linn,S. (1988) DNA repair synthesis in human fibroblasts requires DNA polymerase δ. J. Biol. Chem., 263, 501–510. [PubMed] [Google Scholar]
- 42.Krude T., Jackman,M., Pines,J. and Laskey,R.A. (1997) Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell, 88, 109–119. [DOI] [PubMed] [Google Scholar]
- 43.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 44.Sadiev S. and Taylor,A. (1996) Alternative method for isolation of double-stranded template for DNA sequencing. Biotechniques, 21, 233–235. [DOI] [PubMed] [Google Scholar]
- 45.Inoue H., Nojima,H. and Okayama,H. (1990) High efficiency transformation of Escherichia coli with plasmids. Gene, 96, 23–28. [DOI] [PubMed] [Google Scholar]
- 46.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1989) Current Protocols in Molecular Biology. Wiley Interscience, New York, NY.
- 47.Prelich G., Tan,C.K., Kostura,M., Mathews,M.B., So,A.G., Downey,K.M. and Stillman,B. (1987) Functional identity of proliferating cell nuclear antigen and a DNA polymerase-δ auxiliary protein. Nature, 326, 517–520. [DOI] [PubMed] [Google Scholar]
- 48.Shivji M.K., Kenny,M.K. and Wood,R.D. (1992) Proliferating cell nuclear antigen is required for DNA excision repair. Cell, 69, 367–374. [DOI] [PubMed] [Google Scholar]
- 49.Warbrick E., Lane,D.P., Glover,D.M. and Cox,L.S. (1995) A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21WAF1 and proliferating cell nuclear antigen. Curr. Biol., 5, 275–282. [DOI] [PubMed] [Google Scholar]
- 50.Uitto L., Halleen,J., Hentunen,T., Höyhtyä,M. and Syväoja,J.E. (1995) Structural relationship between DNA polymerases ɛ and ɛ* and their occurrence in eukaryotic cells. Nucleic Acids Res., 23, 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Downey K.M. and So,A.G. (1995) Purification of mammalian polymerases: DNA polymerase δ. Methods Enzymol., 262, 84–92. [DOI] [PubMed] [Google Scholar]
- 52.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 53.Baumann P. and West,S.C. (1998) DNA end-joining catalyzed by human cell-free extracts. Proc. Natl Acad. Sci. USA, 95, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Izzard R.A., Jackson,S.P. and Smith,G.C.M. (1999) Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res., 59, 2581–2586. [PubMed] [Google Scholar]
- 55.North P., Ganesh,A. and Thacker,J. (1990) The rejoining of double-strand breaks in DNA by human cell extracts. Nucleic Acids Res., 18, 6205–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fairman M.P., Johnson,A.P. and Thacker,J. (1992) Multiple components are involved in the efficient joining of double strand DNA breaks in human cell extracts. Nucleic Acids Res., 20, 4145–4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fry M. and Loeb,L. (1986) Animal Cell DNA Polymerases. CRC Press, Boca Raton, FL.
- 58.Warbrick E. (2000) The puzzle of PCNA’s many partners. Bioessays, 22, 997–1006. [DOI] [PubMed] [Google Scholar]
- 59.Gu X.-Y., Bennett,R.A.O. and Povrik,L.F. (1996) End-joining of free radical-mediated DNA double-strand breaksin vitro is blocked by the kinase inhibitor wortmannin at a step preceding removal of damaged 3′ termini. J. Biol. Chem., 271, 19660–19663. [DOI] [PubMed] [Google Scholar]
- 60.Labhart P. (1999) Ku-dependent nonhomologous DNA end joining in Xenopus egg extracts. Mol. Cell. Biol., 19, 2585–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramsden D.A. and Gellert,M. (1998) Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J., 17, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kabotyanski E.B., Gomelsky,L., Han,J.-O., Stamato,T.D. and Roth,D.B. (1998) Double strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res., 26, 5333–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson A.P. and Fairmann,M.P. (1996) The identification and characterization of mammalian proteins involved in the rejoining of DNA double-strand breaks in vitro. Mutat. Res., 364, 103–116. [DOI] [PubMed] [Google Scholar]
- 64.Pfeiffer P. and Vielmetter,W. (1988) Joining of nonhomologous DNA double strand breaks in vitro. Nucleic Acids Res., 16, 907–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waga S. and Stillman,B. (1998) The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem., 67, 721–751. [DOI] [PubMed] [Google Scholar]
- 66.Oda N., Saxena,J.K., Jenkins,T.M., Prasad,R., Wilson,S.H. and Ackerman,E.J. (1996) DNA polymerases α and β are required for DNA repair in an efficient nuclear extract from Xenopus oocytes. J. Biol. Chem., 271, 13816–13820. [DOI] [PubMed] [Google Scholar]
- 67.Holmes A.M. and Haber,J.E. (1999) Double-strand break repair in yeast requires both leading and lagging strand DNA polymerases. Cell, 96, 415–424. [DOI] [PubMed] [Google Scholar]
- 68.Wang T.S.-F. (1996) Cellular DNA polymerases. In DePamphlis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 461–494.
- 69.Whisnant-Hurst N. and Leadon,S.A. (1999) Induced repair of DNA double-strand breaks at the G1/S-phase border. Radiat. Res., 151, 257–262. [PubMed] [Google Scholar]
- 70.Islas Á,.L., Fairley,C.F. and Morgan,W.F. (1998) DNA synthesis on discontinuous templates by human DNA polymerases: implications for non-homologous DNA recombination. Nucleic Acids Res., 26, 3729–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilson T.E. and Lieber,M.R. (1999) Efficient processing of DNA ends during yeast nonhomologous end joining. Evidence for a DNA polymerase β (Pol4)-dependent pathway. J. Biol. Chem., 274, 23599–23609. [DOI] [PubMed] [Google Scholar]
- 72.Sobol R.W., Horton,J.K., Kühn,R., Gu,H., Singhal,R.K., Prasad,R., Rajewsky,K. and Wilson,S.H. (1996) Requirement of mammalian DNA polymerase-β in base-excision repair. Nature, 379, 183–186. [DOI] [PubMed] [Google Scholar]
- 73.Esposito G., Texido,G., Betz,U.A.K., Gu,H., Müller,W., Klein,U. and Rajewsky,K. (2000) Mice reconstituted with DNA polymerase β-deficient fetal liver cells are able to mount a T cell-dependent immune response and mutate their Ig genes normally. Proc. Natl Acad. Sci. USA, 97, 1166–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Göttlich B., Reichenberger,S., Feldmann,E. and Pfeiffer,P. (1998) Rejoining of DNA double-strand breaks in vitro by single-strand annealing. Eur. J. Biochem., 258, 387–395. [DOI] [PubMed] [Google Scholar]
- 75.Moore J.K. and Haber,J.E. (1996) Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 2164–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jónsson Z.O. and Hübscher,U. (1997) Proliferating cell nuclear antigen: more than a clamp for DNA polymerases. Bioessays, 19, 967–975. [DOI] [PubMed] [Google Scholar]
- 77.Henderson D.S., Banga,S.S., Grigliatti,T.A. and Boyd,J.B. (1994) Mutagen sensitivity and suppression of position-effect variegation result from mutations in mus209, the Drosophila gene encoding PCNA. EMBO J., 13, 1450–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beall E.L. and Rio,D.C. (1996) Drosophila IRBP/Ku p70 corresponds to the mutagen-sensitive mus309 gene and is involved in P-element excision in vivo. Genes Dev., 10, 921–933. [DOI] [PubMed] [Google Scholar]
- 79.Henderson D.S. and Glover,D.M. (1998) Chromosome fragmentation resulting from an inability to repair transposase-induced DNA double-strand breaks in PCNA mutants of Drosophila. Mutagenesis, 13, 57–60. [DOI] [PubMed] [Google Scholar]
- 80.Kooistra R., Pastink,A., Zonneveld,J.B.M., Lohman,P.H.M. and Eeken,J.C.J. (1999) The Drosophila melanogaster DmRAD54 gene plays a crucial role in double-strand break repair after P-element excision and acts synergistically with Ku70 in the repair of X-ray damage. Mol. Cell. Biol., 19, 6269–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]