

Abstract
Functional rare variants in drug-related genes are believed to be highly differentiated between ethnic- or racial populations. However, knowledge of population differentiation (PD) of rare single-nucleotide variants (SNVs), remains widely lacking, with the highest fixation indices, (Fst values), from both rare and common variants annotated to specific genes, having only been marginally used to understand PD at the gene level. In this study, we suggest a new, gene-based PD method, PD of Rare and Common variants (PDRC), for analyzing rare variants, as inspired by Generalized Cochran-Mantel-Haenszel (GCMH) statistics, to identify highly population-differentiated drug response-related genes (“pharmacogenes”). Through simulation studies, we reveal that PDRC adequately summarizes rare and common variants, due to PD, over a specific gene. We also applied the proposed method to a real whole-exome sequencing dataset, consisting of 10,000 datasets, from the Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) initiative, and 3,000 datasets from the Genetics of Type 2 diabetes (Go-T2D) repository. Among the 48 genes annotated with Very Important Pharmacogenetic summaries (VIPgenes), in the PharmGKB database, our PD method successfully identified candidate genes with high PD, including ACE, CYP2B6, DPYD, F5, MTHFR, and SCN5A.
Subject terms: Statistical methods, Genetics research
Introduction
Rare variants with large effect sizes have been predicted to exist in the human genome1, 2; also the large effect sizes of these variants have actually been observed, using real data analysis, but without analysis of population differentiation (PD)3, 4. These rare variants tend to be evolutionarily recent alterations, having many functional variants, within drug-related genes (“pharmacogenes”), thought to be highly differentiated between populations5, 6. However, PD approaches, for identifying rare variants, remain quite lacking; to date, the highest frequency indices (i.e., Fst values), from both rare and common variants annotated to specific genes, were used only to nominally understand PD at the gene level7, 8. Considering the fact that Fst values are proportional to minor allele frequencies (MAFs), this gene-level Fst summary is mostly governed by effect sizes (here, the effect sizes are PD) of common variants. Moreover, since previous various established methods (e.g., XP-EHH, and iHS) mainly focus on identifying haplotypes, however, adjacent common variants can strongly affect the results, and thus severely limit the identification of loci in alleles with intermediate frequency9, 10. Furthermore, in those methods, many rare variants in datasets significantly affect their performance by increasing the number of switch errors in the phased haplotypes11–13. Also the method recently proposed by Berg and Coop 14 was mainly for detecting correlations between genetic values and specific environmental variables. As results, these methods are inappropriate for our primary objective, i.e., finding genes with a high level of PD resulting from natural selection, in very recent evolutionary history (based on sequenced data with a large number of rare variants). On the other hand, our proposed method, PD of Rare and Common variants (PDRC), captures PD of both rare and common variants, and summarizes the results at the gene level. In our method, even while a linkage disequilibrium block is not analyzed, we summarize the information in a functional block, and simultaneously focus on very recently selected rare variants in sequenced data, shedding light on inconsistently inherited traits.
Allele frequency differences in pharmacogenes, especially between Africans, Europeans, and Asians, explain the danger of extrapolating therapeutic outcomes from one ethnic group to another15, 16. For instance, while a new drug may be approved by one specific nation’s health regulatory body, many other governments still require their own clinical studies, for their own citizens17. To that end, for ethnic PD differences, the PharmGKB database (www.pharmgkb.org) provides lists of Very Important Pharmacogene (VIPgene) summaries that associate with significant numbers of variant annotations and phenotypes (e.g., metabolism of, or response to, one or several drugs). Thus, although genome-wide variant mapping, to specific genes, was not a focus of our initial research, determining the PD of VIPgenes will enable investigators to find ethnic sensitivities, to therapeutic outcomes, for specific diseases. Additionally, PDRC can be extended to find very recent selection, resulting in considerable numbers of population-specific rare variants, and can even demonstrate associations between a gene and multiple phenotypes (“pleiotropy”), based on sequenced data. For instance, in the future, this approach will enable one to find tissue-specific or cancer-subtype-specific PD in somatic cells, based on data from the emerging technology of single-cell sequencing.
Since the advent of high-throughput (“next generation”) sequencing technology, it has been identified that, throughout the entire human genome, the majority of single-nucleotide variants (SNVs) are rare (86% of the total, with MAFs less than 0.5%)18, and population-specific (53–2%18, 19) (Supplementary Figure S1). Furthermore, rare variants are likely to exert important effects on pharmacogenetically driven phenotypes20–22. Evolutionarily, differences in gene expression, caused by rare variants, contribute to phenotypic diversity23–25. Thus, PD, throughout the human genome, can cause differential ethnic sensitivities to drug responses, through variations that are likely causal for specific genes’ expression and consequently, pathological phenotypes26. Thus, PD considerations are also crucial to drug development, approval, and treatment [PMID: 25669658]; global drug development, or bridging studies, are also important for new drug approval; identification of population-specific rare variants is essential for better understanding of genomic effects on ethnic specific drug responses.
Examination of PD of pharmacogenes requires methods that capture either variant-level or gene-level PD. When the scope of PD is expanded to a gene from a variant, our approach can achieve the following three advantages for population genetics research: (1) overcoming small effect-sizes, which cannot be detected by current variant based identification methods; (2) discovering genes under very recent selection, which also cannot be detected by current selection analyses; and (3) suggestion of additional levels of genetic evidence to explain differences in inherited traits, among populations. In this study, we compare our PDRC method to previously used PD determination algorithms, validating its superior performance at determining PD of numerous SNVs, as related to real whole-exome sequencing (WES) of 10,000 datasets, from the Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) initiative, and 3,000 WES datasets from the Genetics of Type 2 diabetes (Go-T2D) repository.
Herein, we demonstrate that previous PD analysis methods, for common variants7, 27, 28, are not appropriate for analyzing PD of rare variants. We then suggest a new PD analysis method, for rare variants, that is flexible in that it can combine rare and common variants efficiently. Our proposed PD method, Rare and Common variants (PDRC), is based on the Generalized Cochran-Mantel-Haenszel (GCMH) test for conditional independence in three-way contingency tables29, 30. Recently, the CMH test was used for rare variants analysis31. However, it has not been used for developing gene-level statistics, but only for meta-analysis, to summarize the statistics from each study32
Results
The proportion of rare variants in VIP genes
We first procured whole exome sequencing (WES) datasets, the first consisting of 10,000 datasets, from the Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) initiative, and the second consisting of 3,000 datasets from the Genetics of Type 2 diabetes (Go-T2D) repository33. From the PharmGKB data analysis, we selected 48 genes, annotated as “Very Important Pharmacogenes” (VIPs) in PharmGKB (PMID: 11908751). We removed two genes from the original 50 because one was on the sex chromosome and the other was not present in our WES data. Thus, we analyzed 48 VIP genes to determine those with high levels of population differentiation (PD). Since most of the variants in our WES data were less common or rare34, the selected 48 VIP genes mostly consisted of rare variants. The proportions of common variants in VIP genes are shown in Fig. 1. The overall proportion of common variants in VIP genes was 2.80% (max: 8.22%, min: 0%), and most MAFs of variants from the WES dataset were less than 0.05.
Figure 1.
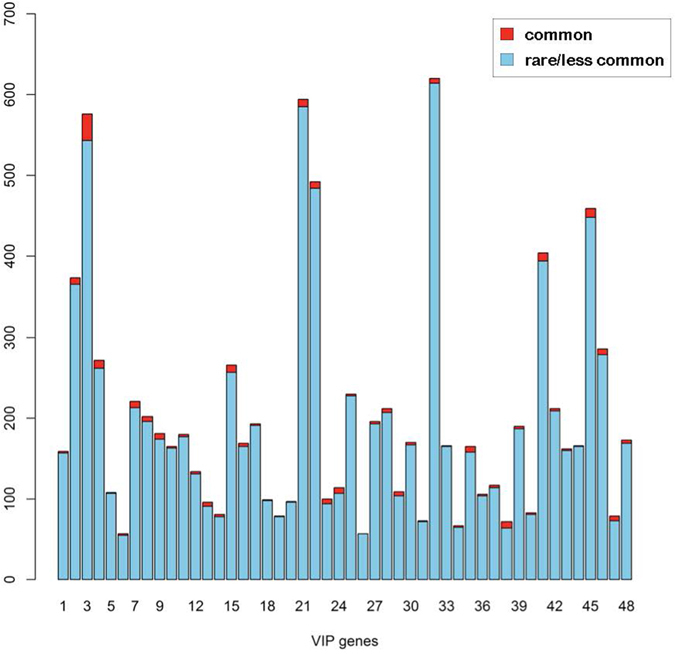
Rare variants in VIP genes. The blue-colored bars depict the number of rare/less common, and the bars are colored red to describe the amount of common variants, in the 48 VIP genes, of our datasets. Only a few of the variants in VIP genes in our datasets were common enough to be detected as highly differentiated variants, via Fst.
Very small Fst from the variants in 48 VIP genes
We first calculated fixation index (Fst) values for all the variants of each VIP gene. For this, we used Weir’s estimate, because it is unbiased, even when the sample sizes are unequal35. Since most of the variants were rare, their Fst values were also very small36. For this reason, the median Fst values for the 48 VIP genes were all less than 0.01, and only six VIP genes had at least one variant with an Fst greater than 0.25. These high Fst values were all from common variants, with MAFs greater than 0.05. For instance, rs1229984(T/C) showed the largest Fst, 0.55, among the variants annotated to the 48 VIP genes, where the minor nucleotide for East Asian is T instead of C, and the frequency of the allele C of East Asian is 0.7445. The nine variants with Fst values greater than 0.25, from the six VIP genes, are summarized in Table 1. Although the MAFs of two variants, rs4846051 in MTHFR and rs6012687, in PTGIS, were not much higher than 0.05 (0.0532 and 0.0628, respectively), the Fst values of the two variants were 0.3000 and 0.2803, respectively. Moreover, racial or ethnic differences were found in the allele frequencies of rs4846051, related to the variation of response to methotrexate, in rheumatoid arthritis37.
Table 1.
List of Variants with Fst values greater than 0.25.
| Gene | SNP | MAFa | Fst |
|---|---|---|---|
| F5 | rs13306334 | 0.1656 | 0.4027 |
| F5 | rs9332658 | 0.1755 | 0.3763 |
| F5 | rs6020 | 0.2105 | 0.3365 |
| MTHFR | rs4846051 | 0.0532 | 0.3000 |
| ADH1B | rs1229984 | 0.1657 | 0.5591 |
| CYP3A4 | rs2687116 | 0.1303 | 0.4819 |
| CYP3A4 | rs2242480 | 0.2959 | 0.2803 |
| PTGIS | rs6012687 | 0.0628 | 0.2581 |
| CYP2D6 | rs1081003 | 0.1346 | 0.3291 |
a Minor Allele Frequency of variants, as measured from our exome sequencing data.
Figure 2 shows that the distribution of Fst in our WES data varied according to the VIP genes’ MAFs. This also shows that the maximum Fst, for all the variants with MAFs < 3%, was less than 0.25. Thus, Fst could not detect the variants of PD when their MAFs were smaller than 3%. In our WES data, 97.5% of the variants had MAFs smaller than 3%. Thus, the majority of variants in our data could not be identified, more specifically, as variants of PD. On the other hand, the common variants with MAFs >5% could be easily identified as variants of PD, by using Fst.
Figure 2.
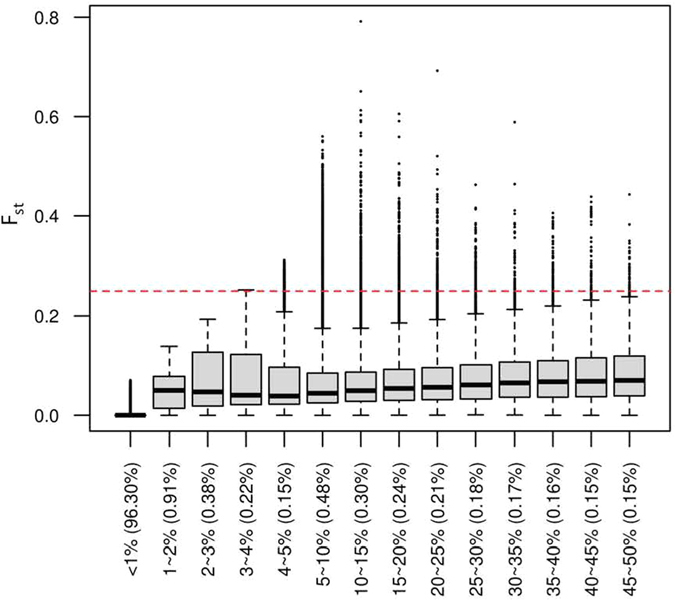
Boxplots of the Fst values of our WES data, according to MAFs of variants. The percentages of the variants which have the range of MAF over the number of all the variants in WES data are shown in x axis with parenthesis; the red line represents Fst cut-off 0.25 according to Wright’s Fst criteria23. Cardoso, et al.23 and Strauss, et al.24 defined a gene with PD if it contains at least one Single Nucleotide Polymorphism (SNP) with Fst value greater than 0.25.
For the VIP genes, 97.14% of variants had MAFs of less than 3%. Thus, the mere use of Fst made it difficult to identify VIP variants related to PD.
SKAT analysis for all VIPgenes
We additionally performed the sequence kernel association test (SKAT)38 for all VIPgenes, even while our objective and the scope of analyses were limited to detecting differences between a pair of populations, at the gene-level. As expected, an association was found for most pairwise comparison of ancestral groups for VIP genes (472 from 480 pairs) after Bonferroni correction39. Specifically, for all VIPgenes, there were large differences in African Americans, due to their distinct genetic history40. Since SKAT can only perform for pairwise comparison of ancestral groups, these results are rather limited. For a more comprehensive comparison of all five ancestral groups, SKAT must be extended, to compare multiple groups.
The PDRC tests for VIP genes
Three different weighting schemes, equal weights, inverse of MAF, and inverse of MAF2 (square of MAF) were adopted to compute PDRC test statistics. If we identified highly differentiated pharmacogenes among the 48 VIP genes by p-value, the number of those identified varied substantially in different weighting schemes. The p-values from PDRC tests, with equal weight, were very small, while the weight based on the inverse of MAF mitigated this phenomenon, as we already mentioned, such that the implementation of weight based on the inverse of MAF could reduce the false positive rate. Note that only two genes, BRCA1 and CYP2B6, were identified by all nine of our different analyses (three strategies and three different weighting schemes), when we considered the p-values after Bonferroni correction. Considering that the equal weight is prone to increased false positive rates, the test result from the equal weighting scheme can be easily violated. Additionally, we evaluated the distribution of PDRC test statistics for each selection strategy and weighting scheme (Table 2), using all 18,281 genes in our WES datasets. We then calculated the 95th percentile for all PDRC results. We selected the PD genes among the top 5% of genes and provided a more detailed description. The top 5% percentile of the PD measurement, from all the genes in the datasets, has often been used for PD detection41–44
Table 2.
95th percentile of PDRC statistics for each selection strategy and weighting scheme.
| Weighting Scheme | Selection strategy | |||||
|---|---|---|---|---|---|---|
| All | Rare/Less Common | Protein Altering | ||||
| Statistics | −log10(p-value) | Statistics | −log10(p-value) | Statistics | −log10(p-value) | |
| Equal Weight | 8667.888 | >100 | 6689.85 | >100 | 3791.395 | >100 |
| 1/MAF | 109.9717 | 22.44083 | 108.2756 | 22.07928 | 47.79004 | 9.300198 |
| 1/MAF2 | 54.35791 | 10.67047 | 54.17336 | 10.63187 | 28.91686 | 5.420125 |
Firstly, we obtained the gene-based summary statistics by using an ‘all variant’ selection strategy. Although the impact of synonymous variants on proteins was not confirmed, we assumed that all the SNPs possessed the potential for phenotypic variation. With the weights based on the inverse of MAF, PD values of all 48 genes were identified (p-value [Bonferroni, n = 48] < 0.05), making it possible to claim that all the VIP genes are highly differentiated (Table 3). On the other hand, considering the 95th percentile of the statistics, 6 genes, ACE, CYP2B6, DPYD, F5, MTHFR, and SCN5A, seemed to be specifically differentiated among the VIP genes. With weights based on the inverse of MAF2, PD values for all 48 genes identified (p-value [Bonferroni, n = 48] < 0.05), and the statistics from four genes, CYP2B6, DPYD, F5, and SCN5A, were larger than the 95th percentile.
Table 3.
10 genes from PDRC tests, using ‘all’ or ‘rare/less-common’ variant selection strategies.
| Gene | PDRC statistics (all) | PDRC statistics (rare/less common) | No. of variant | Fst | |||||
|---|---|---|---|---|---|---|---|---|---|
| Weight:1 | Weight: 1/MAF | Weight:1/MAF2 | Weight:1 | Weight:1/MAF | Weight:1/MAF2 | Common | Rare | Max. | |
| DPYD | 685.41 | 129.22 | 62.52 | 1660.63 | 131.13 | 62.52 | 8 | 365 | 0.0731 |
| F5 | 5561.83 | 148.38 | 83.68 | 5766.33 | 149.95 | 83.68 | 33 | 543 | 0.4027 |
| MTHFR | 1752.40 | 117.12 | 52.88 | 2814.08 | 114.40 | 52.88 | 10 | 262 | 0.3000 |
| SCN5A | 5608.12 | 148.77 | 65.16 | 14227.78 | 149.18 | 65.16 | 6 | 614 | 0.2084 |
| CYP3A4 | 13393.42 | 13.15 | 7.88 | 304.07 | 12.15 | 7.88 | 2 | 160 | 0.4819 |
| CYP3A5 | 8674.38 | 51.27 | 15.36 | 5369.90 | 50.29 | 15.36 | 1 | 165 | 0.2125 |
| EGFR | 40.72 | 107.68 | 53.18 | 6729.38 | 108.06 | 53.18 | 11 | 448 | 0.1594 |
| CYP2E1 | 18902.68 | 68.42 | 24.36 | 9627.48 | 65.97 | 24.36 | 3 | 177 | 0.2128 |
| ACE | 3382.44 | 154.36 | 53.90 | 16909.20 | 154.33 | 53.90 | 9 | 585 | 0.1887 |
| CYP2B6 | 96.18 | 204.00 | 66.52 | 1358.99 | 204.16 | 66.52 | 2 | 228 | 0.0728 |
Data are PDRC statistics yielded from the ‘all’ and ‘rare/less common’ strategies. Numbers of common and rare variants in each selected gene are described. Max. Fst is the maximal Fst value estimated from variants of each gene.
Secondly, when the PDRC test uses a ‘rare/less-common’ variant selection strategy, the results are similar to tests using all the variants (Table 3). Due to the fact that the weights, based on the MAFs, enable the PDRC test to put more weight on rare variants, PDRC tests using two different variant selection strategies, ‘all’ and ‘rare/less-common’ yielded the same list of genes.
Thirdly, when the PDRC test uses a ‘protein-altering’ variant selection strategy, a different list of genes was obtained (Table 4). In this case, the PDRC test identified PD in 10 and 12 genes with PD, with weights of the inverse of MAF and the inverse of MAF2, respectively (p-value [Bonferroni, n = 48] < 0.05). By the evaluated 95th percentile, one (CYP2E1) and six genes (BRCA1, CYP2B6, DPYD, F5, MTHFR, and EGFR) were selected with weights of the inverse of MAF and inverse of MAF2, respectively. Since the PDRC test combined all the effects of common and rare variants, while up-weighting rare variants, it successfully selected highly differentiated VIP genes that could not be detected by Fst.
Table 4.
Nine genes from PDRC tests, using ‘protein-altering’ variant selection strategy.
| Gene Sym | PDRC statistics (protein-altering) | No. of variant | Fst | ||||
|---|---|---|---|---|---|---|---|
| Weight:1 | Weight:1/MAF | Weight:1/MAF2 | Non Protein altering | Protein altering | Protein altering common | Max. | |
| DPYD | 724.50 | 5.32 | 32.22 | 229 | 144 | 3 | 0.0731 |
| F5 | 2280.62 | 0.90 | 43.78 | 271 | 305 | 13 | 0.4027 |
| MTHFR | 958.49 | 4.22 | 32.22 | 181 | 91 | 3 | 0.3000 |
| ADH1B | 8207.13 | 7.33 | 2.38 | 78 | 39 | 1 | 0.5591 |
| EGFR | 970.60 | 17.55 | 30.41 | 323 | 136 | 1 | 0.1594 |
| CYP2E1 | 2818.22 | 51.69 | 7.73 | 113 | 67 | 0 | 0.2128 |
| ACE | 6183.58 | 10.44 | 20.50 | 332 | 262 | 0 | 0.1887 |
| BRCA1 | 1403.64 | 20.15 | 29.45 | 248 | 244 | 3 | 0.1377 |
| CYP2B6 | 968.54 | 20.62 | 31.96 | 115 | 115 | 1 | 0.0728 |
Data are PDRC statistics yielded from the ‘protein altering’ strategy. Numbers of protein-altering and common & protein-altering variant in each selected gene are described. Max. Here, Fst is the maximal Fst value estimated from the variants of each gene.
Genes identified via PDRC test with supporting evidence
From our PDRC test results using an ‘all’ or ‘rare/less-common’ variant selection strategy, PDs for all 48 VIP genes were identified, and supported by known PD values in many SNPs of VIP genes, based on microarray-derived previous research45. When we additionally evaluated the 95th percentile of PDRC statistics, with weights based on either inverse of MAF or MAF2 (square of MAF) six genes, BRCA1, CYP2E1, ACE, CYP2B6, SCN5A, and EGFR, were selected, with no variants having Fst values greater than 0.25. Therefore, we propose these six genes to be specifically differentiated pharmacogenes, among other VIP genes. Besides, we found supporting evidence for high PD levels in these six genes, from either ethnic variation at the genetic46–52, or epigenetic, level53–57. The ethnic variations in genetic sequences of six genes have been reported, but the PDs of these genes could not be identified via Fst. Genetic polymorphisms can be combined at the gene level, and their synergic variability could potentially affect the PD of either gene expression or phenotypes. For this reason, the gene-based statistic, PDRC, is advantageous for finding potential PDs at the epigenetic level. In fact, we specifically found evidence for PD at epigenetic level of three genes CYP2B6, ACE, and SCN5A; we will describe them in the following paragraph.
Hepatic CYP2B6 expression is variable by ethnicity.53 Note that the maximum Fst among the SNPs in CYP2B6 was less than 0.08, but the combined effect of the rare variants seemed to affect the variation in gene expression. In fact, there were only two common variants in CYP2B6 from our WES data. This result shows that although a gene does not contain any SNPs, with large Fst values, it can be a gene with a high level of PD having large effects on expression levels. The Mantel-Haenszel odds ratios from each pair of ancestry groups are summarized in Table 5. Here, the European ancestry group is used as a baseline ancestry. African Americans tended to have more minor alleles than Europeans in our WES data (Mantel-Haenszel odds ratio, 2.33; 95% CI, 2.23 to 2.42). If we suppose that the minor allele potentially reduces a gene’s fitness, defined as the availability of a gene to perform a particular function, this may play a role in female African Americans having the lowest CYP2B6 expression53.
Table 5.
Mantel-Haenszel log odds ratios and confidence intervals of 3 genes.
| CYP2B6 | |
|---|---|
| MHi and 95% confidence interval | |
| African American and European | 2.3266 (2.2333, 2.4237) |
| East Asian and European | 1.4618 (1.3844, 1.5434) |
| American Hispanic and European | 1.2242 (1.1358, 1.3196) |
| South Asian and European | 1.4167 (1.3406, 1.4972) |
| ACE | |
| African American and European | 1.9047 (1.8543, 1.9562) |
| East Asian and European | 1.5142 (1.4702, 1.5597) |
| American Hispanic and European | 0.9461 (0.8972, 0.9977) |
| South Asian and European | 1.6006 (1.5525, 1.6500) |
| SCN5A | |
| African American and European | 2.9808 (2.8671, 3.0991) |
| East Asian and European | 2.3238 (2.2309, 2.4203) |
| American Hispanic and European | 1.3359 (1.2425, 1.4363) |
| South Asian and European | 2.3298 (2.2336, 2.4302) |
European are used as baseline to estimate the Mantel-Haenszel log odds ratios.
ACE, angiotensin-converting enzyme, yielded the second largest test statistic when ‘all’ variants were used, with the weight of inverse of MAF. The maximum Fst value among the SNPs in ACE was less than 0.2. ACE also was the predominant enzyme for bradykinin metabolism in human, where bradykinin is a potent endogenous, endothelium-dependent vasodilator. Consequently, reduced bradykinin expression could affect hypertension58. Since angiotensin is a vasoconstrictor, ACE inhibitors are widely prescribed for the treatment of hypertension, although their efficacy has been reported to vary among different ethnic groups48. In addition, another previous study reported an interactive effect of ethnicity and an ACE gene insertion/deletion polymorphism associated with vascular reactivity48, and our current analysis result also showed ethnic sensitivity to ACE inhibitors. In our WES data, African Americans tended to have more minor alleles than Europeans (Mantel-Haenszel odds ratio = 1.90; 95% CI, 1.85 to 1.96), which is concordant with the phenotype of significantly attenuated vasodilation in Africans, when compared to Europeans48. The Mantel-Haenszel odds ratios and their confidence intervals are summarized in Table 5.
SCN5A, the sodium channel (voltage-gated) type V alpha subunit, includes variants with Fst values less than 0.14. However, the rate of cardiovascular disease (CVD), a SCN5A-related pathological phenotype, is likely related to ethnicity49, 54, 55. For instance, the prevalence of CVD is higher in rural southeastern regions of the US, with the largest African American population, compared to other regions54, 55. Similarly, Hispanics and African Americans have different genetic backgrounds, and patterns of linkage disequilibrium (LD), compared to populations of European descent49–51. Also, it is possible that genetic variation in SCN5A associates with electrocardiography (ECG), and cardiac traits that can vary, depending on the ancestral populations55, and this possibility is supported by the results of our analysis. African Americans, South Asians, and East Asians tended to have more minor alleles than Europeans in our WES data (Table 5). For instance, the Mantel-Haenszel odds of SCN5A from African Americans are 2.98 times higher than those from Europeans, at a 95% confidence level between 2.87 times higher and 3.10 times higher. This result also supports that PD of SCN5A potentially affecting ethnic variation of CVD prevalence, which is higher in African Americans than Europeans.
Discussion
With the advent of low-cost, high-throughput sequencing technologies, a large number of rare variants have been identified in the human genome34, and it has been widely accepted that recent positive selection could result in between-group population differentiation (PD), in the human genome59, 60. Most rare variants are assumed to be driven by very recent positive selection, but have not yet reached fixation60, implying adaptation of modern-day humans to localized evolutionary pressure60. Similarly, when we identified pharmacogenes from whole-exome sequencing (WES) data, most rare variants were also population-specific. However, although most rare variants were identified as population-specific61, methods for measuring PD, using rare variant datasets, have not been well developed. In this study, we proposed a new PD analysis method, PD of Rare and Common variants (PDRC), based on the Generalized Cochran-Mantel-Haenszel (GCMH) test, for gene level analysis of PD in rare and common variants. Because PDRC can put more weight on rare variants, it enables us to avoid the minor allele frequency (MAF) dependency problem of chi-square statistics. Thus, PDRC can find significant genes, according to their PD, by placing more weight on rare variants. Since PD could potentially be used to identify distinct ethnic sensitivities in drug responses, we sought to find pharmacogenes, associated with PD, using our WES data. In addition to gene-level analysis, our PDRC method can be extended to find very recently selected genes. Such analyses will result in considerable identification of genes with population-specific rare variants, and even associations between genes and multiple phenotypes, based on sequencing data.
By evaluating measures for PD, we showed that both the chi-square test and Fst are dependent on MAF. For a given MAF, the maximum chi-square test statistic is shown to be proportional to MAF (S1 text62). The motivation of introducing the weight of MAF was to reduce the effect of MAF on the chi-square statistics. Without using the weight, the PDRC test statistic L 2 is also highly dependent on MAF62. However, by introducing weights, based on the inverse of MAF or MAF2, B k (n k −μ k) becomes less affected by MAF, as does the PDRC test statistic L 2. Similarly, this MAF-based weighting scheme has been widely used for genetic association tests that assign more weight to rare variants, and less weight to common variants38, 63. In our PD analysis, the introduction of weight tends to avoid reporting too many genes, with extremely small p-values, when the sample size is large.
Furthermore, if we assume that rare variants are driven by positive selection, the introduction of MAF weight is biologically meaningful, having only recently been introduced (but not yet fixed) in the genome. Under positive selection, MAF could be regarded as a rough measure of the age of the variant38. In this sense, the observable PD of rare and young variants is likely to be smaller than others, because the recent mutation could not have had sufficient time to be fixed in the population. Therefore, when we calculate a gene-level summary, it is biologically convincing to multiply a bigger weight to a more recently made variant.
In summary, we propose a new test (PDRC) for identifying genes having PD, based on next-generation sequencing (NGS) data. Our PDRC test provides a gene-based statistic for summarizing the effects of both rare and common variants. The possible impact of linkage- disequilibrium (LD), among rare variants in real WES datasets, on PDRC statistics, was investigated through permutation; it was also controlled by the implementation of weights, based on the inverse of MAF or MAF2. Also, through simulation studies, we demonstrated that PDRC tests well preserved type I error, which was not affected by the MAF distribution of genes, when the variants were considered independent of each other. Through an application to a real 13 K exome sequencing dataset, the PDRC test successfully identified pharmacogenes, with high levels of PD, from 48 Very Important Pharmacogenes (VIPs), according to different weighting schemes and selection strategies. To compare our results with known findings, at both the genetic and/or epigenetic levels, we specifically selected six genes, whose statistics were larger than the 95th percentile, and simultaneously, without any variants, with Fst >0.25. Although the PD in these six genes could not be identified by Fst values, earlier studies have claimed the existence of PD at the genetic46–52, or epigenetic levels53–57, also supporting our findings46–58, 64, 65.
The gene-level-PD captured by our PDRC method can be used for the identification of recent adaptations by humans from sequencing data. For decades, genomewide research of natural selection has found that very recent beneficial genetic adaptation is often fixed in the human genome66–68. To that end, notable numbers of population-specific rare variants in our data also support recent adaptation that could provide selection of PD throughout the human genome.
Furthermore, If the ADME of a drug is closely related to a PD of pharmacogenes, we will be able to identify the scope of further investigation, and also devise ways researchers can screen drugs targeting genes with high PD, which can potentially be prone to be sensitive to ethnic factors, and also to suggest distinct pairs of ancestral allele groups, based on Mantel-Haenszel odds ratios29, 69. While it is hard to obtain whole genome sequencing data, at such large sample sizes, from the human genome, our method can be simply applied to WGS data. Longer computing time could be the only challenge to the application of our PDRC method to WGS data analysis. Besides, our method can be applied to other organisms, including viruses and bacteria, and more specifically the human microbiome. Specifically, RNA viruses tend to rapidly mutate, because of the lack of proofreading by their polymerase70. Therefore, when a target for vaccination is explored, our method potentially enables discovery of the most efficient ways to immunize the targeted host against the pathogen; and, considering that our PDRC method can capture the PD in bacterial genes, it could also be potentially applied to antibiotic resistance research.
In conclusion, our PDRC method precisely detects associations between multiple phenotypes and specific genes, based on sequencing data, and also facilitates interpretation of the possible biological impact, of rare variants, on specific traits of interest. This approach can accurately identify specific genes with high levels of PD, under very recent evolutionary selection. Here, we effectively identified highly population-differentiated pharmacogenes, by summarizing the effects of both rare and common variants, at the gene level. Such knowledge will greatly improve the design of therapeutic strategies for patients of distinct ethnicities, or even finding cancer-subtype-specific or tissue-specific somatic mutations, based on the emerging technology of single-cell sequencing. These results will also improve understanding of the recent evolution of SNVs, in specific genes, and possibly even indicate the selective pressure responsible for their respective associated phenotypes.
Materials and Methods
WES Datasets
We obtained whole exome sequencing (WES) data set from two consortia, T2D-GENES (Type 2 Diabetes Genetic Exploration by Next-generation sequencing in Ethnic Samples) and Go-T2D (the Genetics of Type 2 Diabetes Consortium)33. T2D-GENES is a NIDDK-funded research consortium that seeks to identify genetic variants for Type 2 Diabetes (T2D) through multiethnic sequencing studies. Go-T2D is a high-resolution study of type 2 diabetes genetic architecture through whole-genome sequencing of 2850 Europeans. T2D-GENES comprises three projects, from which we used data from Project 1. Project 1 seeks to assess whether less common variants play roles in T2D risk, in addition to similarities and differences in the distribution of T2D risk variants across ancestry groups. Presently, the T2D-GENES and Go-T2D initiatives are carrying out deep whole-exome sequencing (WES) of 13 K individuals, from which we used 12,844 unrelated individuals (6,474 cases, 6,370 controls) for our analysis. The total numbers of the five ancestry groups were as follows: 2025 African Americans, 2164 East Asians, 1938 American Hispanics, 2199 South Asians and 4518 Europeans. Among the five populations, cases and controls were well balanced for each ethnic group. From the European population, about 2000 samples were collected for case and control groups, respectively. Similarly, about 1000 samples were selected for case and control groups, respectively, from other ethnic groups. A more detailed description is given in the main paper33. Sequencing was completed at the Broad Institute using the Agilent (Santa Clara, CA) v2 capture reagent on HiSeq machines. After quality control, 3,130,381 variants matches to the datasets. In total, there were 62,489 common variants (MAF > 0.05) and 2,951,589 rare variants (0 < MAF < 0.01).
PharmGKB Database
We used the PharmGKB (http://www.pharmgkb.org) database (PMID: 11908751) to select pharmacogenes for our PD study. This database is publicly available and encompasses clinical information, including dosing guidelines and drug labels, potentially clinically actionable gene-drug associations and genotype-phenotype relationships. Specifically, the very important pharmacogenes (VIP genes) represent the genes that greatly impact drug responses. These VIP genes have been widely used to decode the genomic effect on drug responses71, 72. VIP genes in PharmGKB were written by Scientific Curators, through extensive literature review, to provide a concise summary of key genes involved in drug responses, and whether these genes have been used for understanding pharmacogenomics15. Among 50 VIP genes in PharmGKB, one is on the sex chromosome and another was not included in our WES dataset. Thus, we used 48 genes for our analysis to identify pharmacogenes with PD.
Test for Population differentiation for rare and common variants (PDRC)
For identifying genes with PD from the WES data, we proposed a new method, PDRC. The PDRC test is a gene-level summary test based on generalized Cochran-Mantel-Haenszel (GCMH) statistics29, 30. The main motivation of using the PDRC test was for extracting gene-based summary statistics that infer an average partial association between ancestral groups and genotypes. Initially, Cochran (1954) proposed a test, ‘average partial association,’ for a set of 2 × 2 tables, using a mean difference weighted across q tables, as determined by levels of the covariates29, 30. In particular, we considered detecting PD through the analysis of q s × r (s ≥ 2, r ≥ 2)) contingency tables under the multiple hypergeometric model assumption30. Here, q is the number of SNPs in a gene; s (=5) represents five ancestry groups, (1 for African Americans, 2 for East Asians, 3 for American Hispanics, 4 for South Asians and 5 for Europeans); r (=2) represents whether an allele is minor or major. For our analysis, we constructed a contingency table for each variant in a gene, and then combined them within a gene. Let index a set of () contingency tables, which correspond to the number of SNPs in a gene. Let index five ancestry groups and j (=1, 2) index minor or major alleles, respectively. Let , where denotes the number of subjects in the sample jointly classified as belonging to the i th ancestry group, the j th allele category and the k th SNP table. In addition, let denote the marginal total number of subjects in the i th ancestry group. In that case, is the marginal total number of subjects with the j th allele category, and is the overall marginal total sample size in the k th SNP table.
Using the GCMH test by Landis et al.30, we introduced the weight (i.e. the inverse of the MAF) into the PDRC test, for handling both rare and common variants, as follows:
| 1 |
| 2 |
| 3 |
| 4 |
with
| 5 |
| 6 |
where is a weight. Then under the null hypothesis of conditional independence. Note that although rare variants often did not exhibit strong linkage disequilibium (LD)73, 74, but independence was also assessed via permutation (S2 text). From our permutation results, weights based on the inverse of MAF reduced the rate of false positives. Therefore, implementation of this type of weights is recommended for analyzing WES datasets like ours.
We reported the weighted Mantel-Haenszel odds ratio69, , to show which ancestral alleles are distinctly different from European alleles (S3 text). We introduced the weight into the Mantel-Haenszel odds ratio and estimated variance69, 75, as follows, using the last cells as a baseline.
| 7 |
where, i = 1, 2, 3 and 4. For the estimation of variance of , we extended the methodology by Robins et al.75 Landis et al. showed that , where is a vector of row scores and is a vector of column scores30. When and , L 2 is the generalized CMH statistic for two nominal variables. In the PDRC test, three types of weights are used: equal weight, inverse of MAF and inverse of MAF2. More details on these weights are given in the Discussion.
Simulation
We conducted a simulation to analyze the type-1-error rate of PDRC test for the analysis of population differentiation (PD) in genes. The null distributions (i.e., no PD in the genome), were generated from several scenarios (S4 Text).
VIP gene analysis through PDRC test
According to the 1000 Genomes Project (PMID: 21030618), 17% of low-frequency variants with MAF ranges of 0.5–5% were observed in a single ancestry group, and 53% of rare variants (MAF < 0.5%) were observed in a single ancestry group19 and similarly, half of the variants in VIP genes in our datasets were population-specific rare variants (Figs 2 and 3). Therefore, our studies of population-specific rare variants are also important for studying pharmacogenes with high PD. Therefore, we investigated the VIP genes in our WES data through the proposed PDRC test. Since the PDRC test statistics could summarize PD information from both rare and common variants, VIP gene analysis, via the PDRC test, has some flexibility in choosing variants in gene analysis, in order to identify genes with high PD.
Figure 3.
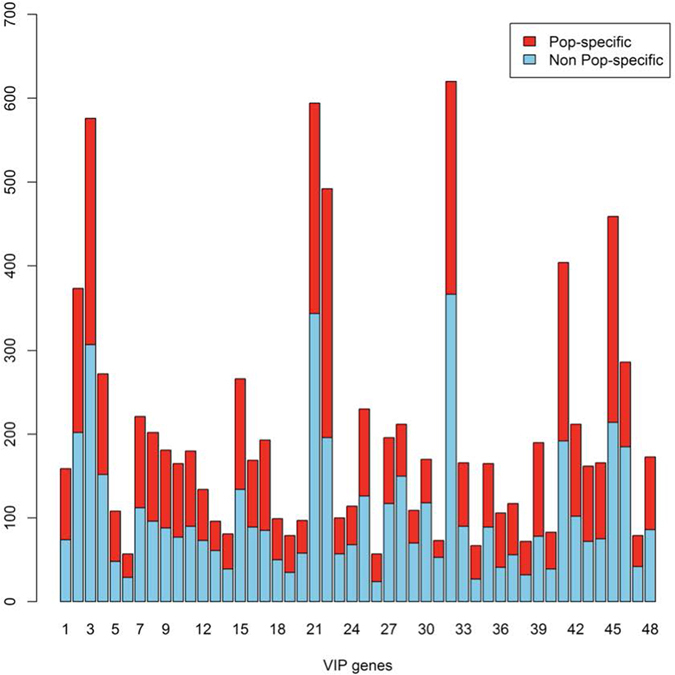
Population-specific variants in VIP genes. The bars are colored red to describe the numbers of population-specific variants, observed only from a single population, in our 48 identified VIP genes. Half of the variants in VIP genes in our datasets were population-specific rare variants.
Variant selection strategy for specific genes
The process of selecting variants representing a specific gene is not straightforward. In our analysis, we considered the following three strategies for choosing variants: (1) all variants, including common and rare ones; (2) less common or rare variants; and (3) protein-altering variants. Since some of the variants do not alter the encoded protein, the phenotypic variation caused by genotypic variation might be summarized only by protein-altering variants76. However, non-protein-altering variants might also cause variation of gene expression, with phenotypic consequences77, 78. Lastly, less common and rare variants are expected to have larger effects than common variants79. Thus, these three strategies are used in the PDRC test for summarizing effects at the gene level.
Electronic supplementary material
Acknowledgements
We would like to thank Dr. Hosun Lee for providing valuable comments on this manuscript and Dr. Curt Balch for English editing. This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI15C2165) and by the Bio-Synergy Research Project (2013M3A9C4078158) of the Ministry of Science, ICT and Future Planning through the NRF. Sequencing data from the T2D-GENES Consortium was supported by NIH/NIDDK U01’s DK085501, DK085524, DK085526, DK085545 and DK085584.
Author Contributions
Eunyong Ahn performed the simulations and analyses. Eunyong Ahn and Taesung Park developed the methodology and wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-08468-y
References
- 1.Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park, J.-H. et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet42, 570–575, http://www.nature.com/ng/journal/v42/n7/suppinfo/ng.610_S1.html (2010). [DOI] [PMC free article] [PubMed]
- 3.Del-Aguila JL, et al. Alzheimer’s disease: rare variants with large effect sizes. Curr Opin Genet Dev. 2015;33:49–55. doi: 10.1016/j.gde.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Ramsey LB, et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Research. 2012;22:1–8. doi: 10.1101/gr.129668.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson MR, et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathieson I, Reich D. Differences in the rare variant spectrum among human populations. Plos Genet. 2017;13:e1006581. doi: 10.1371/journal.pgen.1006581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu DD, Zhang YP. Different level of population differentiation among human genes. Bmc Evol Biol. 2011;11:16. doi: 10.1186/1471-2148-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A Map of Recent Positive Selection in the Human Genome. Plos Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sabeti, P. C. et al. Detecting recent positive selection in the human genome from haplotype structure. Nature419, 832–837, http://www.nature.com/nature/journal/v419/n6909/suppinfo/nature01140_S1.html (2002). [DOI] [PubMed]
- 11.Liu X, et al. Detecting and Characterizing Genomic Signatures of Positive Selection in Global Populations. American journal of human genetics. 2013;92:866–881. doi: 10.1016/j.ajhg.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, et al. Genotype calling and haplotyping in parent-offspring trios. Genome Res. 2013;23:142–151. doi: 10.1101/gr.142455.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menelaou A, Marchini J. Genotype calling and phasing using next-generation sequencing reads and a haplotype scaffold. Bioinformatics (Oxford, England) 2013;29:84–91. doi: 10.1093/bioinformatics/bts632. [DOI] [PubMed] [Google Scholar]
- 14.Berg JJ, Coop G. A Population Genetic Signal of Polygenic Adaptation. Plos Genet. 2014;10:e1004412. doi: 10.1371/journal.pgen.1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whirl-Carrillo M, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92:414–417. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramos E, et al. Pharmacogenomics, ancestry and clinical decision making for global populations. Pharmacogenomics J. 2014;14:217–222. doi: 10.1038/tpj.2013.24. [DOI] [PubMed] [Google Scholar]
- 17.Liu JP, Chow SC. Bridging studies in clinical development. Journal of biopharmaceutical statistics. 2002;12:359–367. doi: 10.1081/BIP-120014564. [DOI] [PubMed] [Google Scholar]
- 18.Tennessen JA, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genomes Project C, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramsey LB, et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012;22:1–8. doi: 10.1101/gr.129668.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smeraldi E, Serretti A, Artioli P, Lorenzi C, Catalano M. Serotonin transporter gene-linked polymorphic region: possible pharmacogenetic implications of rare variants. Psychiatric genetics. 2006;16:153–158. doi: 10.1097/01.ypg.0000218611.53064.a0. [DOI] [PubMed] [Google Scholar]
- 22.Zuo L, et al. Rare SERINC2 variants are specific for alcohol dependence in individuals of European descent. Pharmacogenetics and genomics. 2013;23:395–402. doi: 10.1097/FPC.0b013e328362f9f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 24.Kukurba KR, et al. Allelic Expression of Deleterious Protein-Coding Variants across Human Tissues. Plos Genet. 2014;10:e1004304. doi: 10.1371/journal.pgen.1004304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cruchaga, C. et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer/‘s disease. Nature505, 550–554, doi:10.1038/nature12825, http://www.nature.com/nature/journal/v505/n7484/abs/nature12825.html#supplementary-information (2014). [DOI] [PMC free article] [PubMed]
- 26.Wilson JF, et al. Population genetic structure of variable drug response. Nat Genet. 2001;29:265–269. doi: 10.1038/ng761. [DOI] [PubMed] [Google Scholar]
- 27.Wright S. The Genetical Structure of Populations. Annals of Eugenics. 1949;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 28.Weir BS, Cockerham CC. Estimating F-Statistics for the Analysis of Population Structure. Evolution. 1984;38:1358. doi: 10.2307/2408641. [DOI] [PubMed] [Google Scholar]
- 29.Cochran WG. Some Methods for Strengthening the Common χ 2 Tests. Biometrics. 1954;10:417. doi: 10.2307/3001616. [DOI] [Google Scholar]
- 30.Landis JR, Heyman ER, Koch GG. Average Partial Association in Three-Way Contingency Tables: A Review and Discussion of Alternative Tests. International Statistical Review / Revue Internationale de Statistique. 1978;46:237. doi: 10.2307/1402373. [DOI] [Google Scholar]
- 31.Liu, D. J. et al. Meta-analysis of gene-level tests for rare variant association. Nat Genet46, 200–204, doi:10.1038/ng.2852, http://www.nature.com/ng/journal/v46/n2/abs/ng.2852.html#supplementary-information (2014). [DOI] [PMC free article] [PubMed]
- 32.Liu DJ, et al. Meta-analysis of gene-level tests for rare variant association. Nat Genet. 2014;46:200–204. doi: 10.1038/ng.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuchsberger C, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536:41–47. doi: 10.1038/nature18642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Genomes Project C, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weir BS, Hill WG. Estimating F-statistics. Annu Rev Genet. 2002;36:721–750. doi: 10.1146/annurev.genet.36.050802.093940. [DOI] [PubMed] [Google Scholar]
- 36.Jakobsson M, Edge MD, Rosenberg NA. The relationship between F(ST) and the frequency of the most frequent allele. Genetics. 2013;193:515–528. doi: 10.1534/genetics.112.144758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hughes LB, et al. Racial or ethnic differences in allele frequencies of single-nucleotide polymorphisms in the methylenetetrahydrofolate reductase gene and their influence on response to methotrexate in rheumatoid arthritis. Annals of the rheumatic diseases. 2006;65:1213–1218. doi: 10.1136/ard.2005.046797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. American journal of human genetics. 2013;92:841–853. doi: 10.1016/j.ajhg.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonferroni, C. E. In Studi in Onore del Professore Salvatore Ortu Carboni. 13–60 (1935).
- 40.Jorde, L. B. & Wooding, S. P. Genetic variation, classification and ‘race’. Nat Genet (2004). [DOI] [PubMed]
- 41.Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet. 2010;11:17–30. doi: 10.1038/nrg2698. [DOI] [PubMed] [Google Scholar]
- 42.Casto AM, Feldman MW. Genome-wide association study SNPs in the human genome diversity project populations: does selection affect unlinked SNPs with shared trait associations? PLoS genetics. 2011;7:e1001266. doi: 10.1371/journal.pgen.1001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rawlings-Goss RA, Campbell MC, Tishkoff SA. Global population-specific variation in miRNA associated with cancer risk and clinical biomarkers. BMC medical genomics. 2014;7:53. doi: 10.1186/1755-8794-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue Y, et al. Population differentiation as an indicator of recent positive selection in humans: an empirical evaluation. Genetics. 2009;183:1065–1077. doi: 10.1534/genetics.109.107722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yasuda SU, Zhang L, Huang SM. The Role of Ethnicity in Variability in Response to Drugs: Focus on Clinical Pharmacology Studies. Clinical Pharmacology & Therapeutics. 2008;84:417–423. doi: 10.1038/clpt.2008.141. [DOI] [PubMed] [Google Scholar]
- 46.Shanmughapriya S, Nachiappan V, Natarajaseenivasan K. BRCA1 and BRCA2 mutations in the ovarian cancer population across race and ethnicity: special reference to Asia. Oncology. 2013;84:226–232. doi: 10.1159/000346593. [DOI] [PubMed] [Google Scholar]
- 47.Levy-Lahad E, et al. Founder BRCA1 and BRCA2 mutations in Ashkenazi Jews in Israel: frequency and differential penetrance in ovarian cancer and in breast-ovarian cancer families. American journal of human genetics. 1997;60:1059–1067. [PMC free article] [PubMed] [Google Scholar]
- 48.Gainer JV, Stein CM, Neal T, Vaughan DE, Brown NJ. Interactive Effect of Ethnicity and ACE Insertion/Deletion Polymorphism on Vascular Reactivity. Hypertension. 2001;37:46–51. doi: 10.1161/01.HYP.37.1.46. [DOI] [PubMed] [Google Scholar]
- 49.Bush WS, et al. Genetic variation in the rhythmonome: ethnic variation and haplotype structure in candidate genes for arrhythmias. Pharmacogenomics. 2009;10:1043–1053. doi: 10.2217/pgs.09.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reich DE, et al. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- 51.Lohmueller, K. E. et al. Proportionally more deleterious genetic variation in European than in African populations. Nature451, 994–997, http://www.nature.com/nature/journal/v451/n7181/suppinfo/nature06611_S1.html (2008). [DOI] [PMC free article] [PubMed]
- 52.Stephens EA, et al. Ethnic variation in the CYP2E1 gene: polymorphism analysis of 695 African-Americans, European-Americans and Taiwanese. Pharmacogenetics. 1994;4:185–192. doi: 10.1097/00008571-199408000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Lamba V, et al. Hepatic CYP2B6 expression: gender and ethnic differences and relationship to CYP2B6 genotype and CAR (constitutive androstane receptor) expression. The Journal of pharmacology and experimental therapeutics. 2003;307:906–922. doi: 10.1124/jpet.103.054866. [DOI] [PubMed] [Google Scholar]
- 54.Crook ED, Taylor H. Traditional and nontraditional risk factors for cardiovascular and renal disease in African Americans (Part 2): a project of the Jackson Heart Study investigators. The American journal of the medical sciences. 2003;325:305–306. doi: 10.1097/00000441-200306000-00001. [DOI] [PubMed] [Google Scholar]
- 55.Jeff JM, et al. SCN5A variation is associated with electrocardiographic traits in the Jackson Heart Study. Circulation. Cardiovascular genetics. 2011;4:139–144. doi: 10.1161/CIRCGENETICS.110.958124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Calvo E, Baselga J. Ethnic differences in response to epidermal growth factor receptor tyrosine kinase inhibitors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:2158–2163. doi: 10.1200/jco.2006.06.5961. [DOI] [PubMed] [Google Scholar]
- 57.Loong HH, Wei J, Mok TS. Ethnic variation in response to EGFR inhibitors. Drug Discovery Today: Therapeutic Strategies. 2012;9:e61–e66. doi: 10.1016/j.ddstr.2011.04.003. [DOI] [Google Scholar]
- 58.Margolius HS. Theodore Cooper Memorial Lecture. Kallikreins and kinins. Some unanswered questions about system characteristics and roles in human disease. Hypertension. 1995;26:221–229. doi: 10.1161/01.HYP.26.2.221. [DOI] [PubMed] [Google Scholar]
- 59.Sabeti PC, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 60.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. Plos Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Casals F, et al. Whole-exome sequencing reveals a rapid change in the frequency of rare functional variants in a founding population of humans. Plos Genet. 2013;9:e1003815. doi: 10.1371/journal.pgen.1003815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Workman PL, Niswander JD. Population studies on southwestern Indian tribes. II. Local genetic differentiation in the Papago. American journal of human genetics. 1970;22:24–49. [PMC free article] [PubMed] [Google Scholar]
- 63.Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. Plos Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thorn CF, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for angiotensin-converting enzyme. Pharmacogenetics and genomics. 2010;20:143–146. doi: 10.1097/FPC.0b013e3283339bf3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thorn CF, Lamba JK, Lamba V, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for CYP2B6. Pharmacogenetics and genomics. 2010;20:520–523. doi: 10.1097/FPC.0b013e32833947c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akey JM. Constructing genomic maps of positive selection in humans: where do we go from here? Genome Res. 2009;19:711–722. doi: 10.1101/gr.086652.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grossman SR, et al. Identifying Recent Adaptations in Large-scale Genomic Data. Cell. 2013;152:703–713. doi: 10.1016/j.cell.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sabeti PC, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22:719–748. doi: 10.1016/0021-9681(79)90031-6. [DOI] [PubMed] [Google Scholar]
- 70.Elena SF, Sanjuán R. Adaptive Value of High Mutation Rates of RNA Viruses: Separating Causes from Consequences. Journal of Virology. 2005;79:11555–11558. doi: 10.1128/JVI.79.18.11555-11558.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rukov JL, Wilentzik R, Jaffe I, Vinther J, Shomron N. Pharmaco-miR: linking microRNAs and drug effects. Brief Bioinform. 2014;15:648–659. doi: 10.1093/bib/bbs082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J, et al. Genetic polymorphisms of VIP variants in the Tajik ethnic group of northwest China. BMC Genet. 2014;15:102. doi: 10.1186/s12863-014-0102-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? American journal of human genetics. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robins J, Greenland S, Breslow NE. A general estimator for the variance of the Mantel-Haenszel odds ratio. American journal of epidemiology. 1986;124:719–723. doi: 10.1093/oxfordjournals.aje.a114447. [DOI] [PubMed] [Google Scholar]
- 76.Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annual review of genomics and human genetics. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 77.de Coulgeans CD, et al. Synonymous nucleotide polymorphisms influence Dombrock blood group protein expression in K562 cells. Br J Haematol. 2014;164:131–141. doi: 10.1111/bjh.12597. [DOI] [PubMed] [Google Scholar]
- 78.Brest P, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 79.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.