

Abstract
Although systematic studies have identified a host of long non-coding RNAs (lncRNAs) which are involved in breast cancer, the knowledge about the methyla-tion-mediated dysregulation of those lncRNAs remains limited. Here, we integrated multi-omics data to analyze the methylated alteration of lncRNAs in breast invasive carcinoma (BRCA). We found that lncRNAs showed diverse methylation patterns on promoter regions in BRCA. LncRNAs were divided into two categories and four subcategories based on their promoter methylation patterns and expression levels be-tween tumor and normal samples. Through cis-regulatory analysis and gene ontology network, abnormally methylated lncRNAs were identified to be associated with can-cer regulation, proliferation or expression of transcription factors. Competing endog-enous RNA network and functional enrichment analysis of abnormally methylated lncRNAs showed that lncRNAs with different methylation patterns were involved in several hallmarks and KEGG pathways of cancers significantly. Finally, survival analysis based on mRNA modules in networks revealed that lncRNAs silenced by high methylation were associated with prognosis significantly in BRCA. This study enhances the understanding of aberrantly methylated patterns of lncRNAs and pro-vides a novel insight for identifying cancer biomarkers and potential therapeutic tar-gets in breast cancer.
Keywords: lncRNA, DNA methylation, network, breast cancer, hallmark
INTRODUCTION
Breast cancer is a genetically malignant tumor caused by a variety of elements involving the accumulation of genetic changes [1–3]. During the past decades, many genetic pathogenic factors have been recognized. With the study developed, besides PCGs large numbers of non-coding RNAs (ncRNAs) which were transcribed by genome sequences but not coding proteins were identified [4, 5]. MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) are crucial directions in the researches of ncRNAs. The miRNAs inhibit the translation or regulation of target genes that carry miRNA binding sites in their 3′ untranslated regions (UTRs) [6]. Many investigations illustrated the importance of miRNAs in diseases [7]. MiRNAs are also implicated in breast cancer and many other cancers [8]. For example, the miR-200 family (miR-200a, miR-200b, miR-200c, miR-141 and miR-429) and miR-205 regulated epithelial to mesenchymal transition by targeting ZEB1 and SIP1 [9]. These two genes were involved in epithelial to mesenchymal transition and tumor metastasis. Yang et al. [10] proved that the overexpression of miR-346 reduced the expression of SRCIN1 and promoted cell proliferation, colony formation, and sensitivity to Docetaxel (Doc) in breast cancer. For lncRNAs, Jadaliha et al. [11] used MALAT1 knockdown/overexpression experiments to confirm the functional significance of MALAT1 as a metastasis driver and a prognostic factor in ER negative, lymph node negative breast cancer. Zhang et al. [12] substantiated that HOTAIR was a biomarker for breast cancer. Although ncRNAs have been described with relatively definite molecular mechanisms in cancers, the regulatory mechanisms of ncRNAs in tumors were unclear especially DNA methylation of lncRNAs.
As one of the most important epigenetic modifications, DNA methylation involves the addition of a methyl group to the C5 position of cytosine residues which is catalyzed by DNA methyltransferases. For PCGs the hypermethylation in promoter inhibits the combination of transcription factors and silences cancer suppressor genes, whereas the hypomethylation in promoter activates oncogenes [2, 13]. For instance, Tang et al. [14] have found that hypomethylation of RPTOR, MGRN1 and RAPSN led to high odds ratios (ORs) in peripheral blood DNA of breast cancer compared with normal tissues and the function of hypomethylation was validated in three independent large sample sets through MassARRAY EpiTyper assays. Yi et al. [15] have verified that overexpression of NSUN2 by DNA hypomethylation facilitated cell proliferation, migration, and invasion in the progress and development of breast cancer. In addition, Luo et al. analyzed the relationship between PTEN hypermethylation and breast cancer. PTEN promoter hypermethylation was increased significantly in ductal carcinoma in situ (DCIS) and invasive ductal carcinoma (IDC) and associated with the risk of DCIS and IDC [16]. Yu et al. [17] found the promoter hypermethylation led to the silence of RASSF2A which was regarded as a tumor suppressor gene. Therefore aberrant DNA methylation in promoter is a prominent feature for the identification of new targets in the pathophysiology of tumors and therapeutic intervention.
Based on the research foundation of PCGs, we assumed that DNA methylation of lncRNA promoters might be an epigenetic regulator of lncRNAs expression. Several lncRNAs have demonstrated the hypothesis. For instance, the hypomethylation of AFAP1-AS1 in Barrett's esophagus and esophageal adenocarcinoma caused the overexpression of AFAP1-AS and affected the proliferation and colony-forming ability [18]. Aberrant DNA hypermethylation downregulated the expression of SOX21-AS1 and low expression of SOX21-AS1 might be an adverse prognostic biomarker in oral cancer [19]. However, these researches only analyzed the DNA methylation of few specific lncRNAs but did not identify the DNA methylation pattern of lncRNAs systematically. Liao et al. [20] analyzed the DNA methylation models of lncRNA promoter in colorectal cancer. They found the hypermethylation of lncRNA promoter offered a new clue for the biological researchers to further understand the function of lncRNAs in colorectal cancer. Zhi et al. [21] created a novel re-annotation strategy to display the DNA methylation patterns of lncRNAs in pan-cancers. But the two studies did not assess how DNA methylation regulated the expression of lncRNAs accurately. They only used few methods to verify the function of lncRNAs.
With the improvement of high-throughput sequencing technology, multitudinous data such as RNA-seq and HumanMethylation450 BeadChip (450K) have been applied for the analysis of cancers [22, 23]. High throughput multi-omics data has facilitated the development of large-scale identification and systemic analysis of novel cancer biomarkers. In this study, a new strategy was raised to observe the DNA methylation patterns of lncRNAs systematically based on genome-wide lncRNA expression, gene expression and DNA methylation profiles in BRCA from The Cancer Genome Atlas (TCGA) project. Firstly, we re-annotated the promoters of lncRNAs and obtained 137 high methylated lncRNAs (HMLncs) and 101 low methylated lncRNAs (LMLncs). Then cis-regulatory function enrichment analysis and gene ontology (GO) term network were carried out to predict the function of lncRNAs. HMLncs and LMLncs mainly influenced regulation (HMLncs: 126, LMLncs: 118). Moreover, LMLncs also influenced proliferation. Next, using the investigation of the competing endogenous RNA (ceRNA) networks, we discovered that lncRNAs classified by DNA methylation were enriched in different types of cancer hallmarks, and validated the impact of DNA methylation for lncRNAs. Finally, through the survival analysis, we identified high-down module associated with prognosis in BRCA. The study presents the methylated regulation mechanism of lncRNAs in BRCA and provided potential cancer biomarkers for diagnosis and treatment.
RESULTS
DNA methylation pattern in lncRNA promoters
To assess patterns of lncRNA, the DNA methylation, lncRNA expression, mRNA expression and clinical data of breast invasive carcinoma (BRCA) were used in this study. The quantity of samples for each profile was in Table 1.
Table 1. The number of samples in each type of data.
After the profiles preprocessed, differentially methylated lncRNAs (DMLs), differentially expressed lncRNAs (DELs) and differentially expressed genes (DEGs) were recognized respectively between tumor and normal samples (Figure 1A). To estimate the importance of DNA methylation for lncRNAs, lncRNAs were divided into two categories based on the mean methylation level in promoter and expression of lncRNAs. Both the categories were differentially expressed lncRNAs. The first category of lncRNAs were the high methylated lncRNAs (HMLncs) that DNA methylation level in promoter was up-regulated compared with normal samples. The other category of lncRNAs were the low methylated lncRNAs (LMLncs) that DNA methylation level in promoter was down-regulated compared with normal samples.
Figure 1. The workflow of our study.
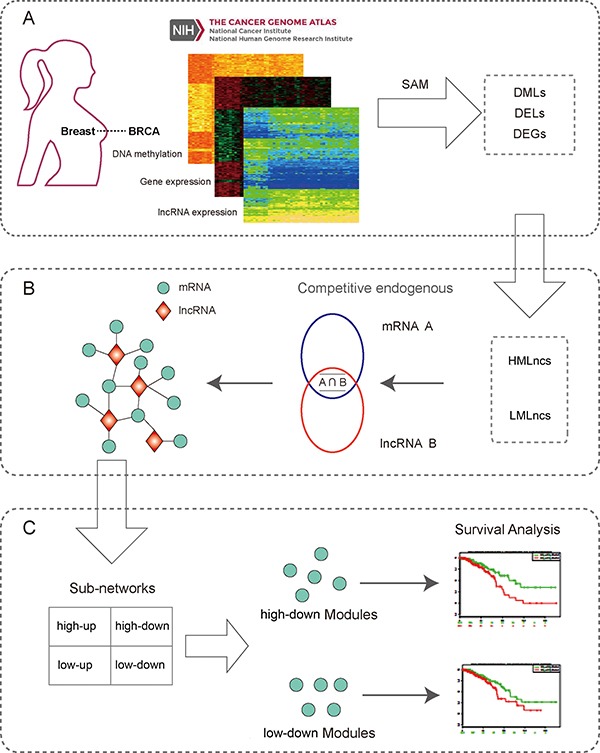
(A) Data source and molecular filter. Expression and methylation data were downloaded from The Cancer Genome Atlas database. Then differential genes and lncRNAs were extracted by SAM method. (B) The construction of lncRNA-mRNA ceRNA network. The intersection of DMLs and DELs were re-classified into HMLncs and LMLncs based on the pattern of methylation. The competitive endogenous mRNAs of these lncRNAs were obtained to construct the ceRNA network. Blue and red circles are the miRNAs interacted with mRNA A and lncRNA B, respectively. (C) The analysis of lncRNAs in different networks. Four sub-networks were extracted from the background network. Sub-networks were analyzed by various methods such as function enrichment analysis, hallmark analysis and survival analysis to validate the effect of DNA methylation to lncRNAs. (DML: Differentially Methylated LncRNA, DEL: Differentially Expressed LncRNA, DEG: Differentially Expressed Gene, HMLnc: high methylated LncRNA, LMLnc: low methylated LncRNA, PCG: Protein Coding Gene).
Next we used GREAT, HMDD and ceRNA networks to identify the influence of lncRNAs in BRCA. Four sub-networks were extracted from the background ceRNA network. Then two modules were got from high-down and low-down network. To evaluate the association of the two modules with patient prognosis we used clinical data from TCGA and three sample sets from GEO to do survival analysis to verify the reliability of our modules (Figure 1B and 1C).
We regarded the region 2kb upstream from TSS as the promoter of lncRNA. In this study, 18,772 probes were located in most lncRNA promoters (87.53%, 3,459). The average value of DNA methylation probes within one lncRNA promoter was computed as the DNA methylation level of the lncRNA. The DNA methylation levels in promoters had a different distribution between tumor and normal samples (Kolmogorov-Smirnov (K-S) test, P = 7.15e-05). By comparing the DNA methylation and gene expression between tumor and normal samples, we identified 743 DMLs and 3206 DELs. This result indicated that lncRNAs exhibited differentially methylated and expressed patterns between tumor and normal samples.
Then 238 lncRNAs (137 HMLncs and 101 LMLncs) that showed both differential methylation and differential expression were picked for the further analysis of lncRNAs. Aberrant DNA methyltransferases might cause the chromosome abnormality and tumor development, and cancer cells usually showed higher methylation than normal cells in promoters of PCGs [24]. We found DNA methylation level in lncRNA promoter showed the similar pattern with that in PCG promoter and was high methylation in tumor samples. Then DNA methylation levels in tumor and normal samples were used to assess the statistical difference with the SAM method based on all 238 lncRNAs, 137 HMLncs and 101 LMLncs, respectively. The three lncRNA sets were all differential (P < =0.01) (Figure 2A, 2B and 2C). The overall DNA methylation level in promoter tended to rise and exceeded 0.5 (Figure 2A). Moreover bidirectional hierarchical clustering analysis using DNA methylation level indicated that the 238 lncRNAs divided the tumor and normal samples obviously. HMLncs and LMLncs both could be clustered into several groups. The result suggested that 238 DMLs in tumors had similar methylation patterns, and HMLncs and LMLncs also displayed the consistency of DNA methylation levels in tumors respectively (Figure 2D, 2E and 2F).
Figure 2. The DNA methylation patterns of tumors and normal samples.
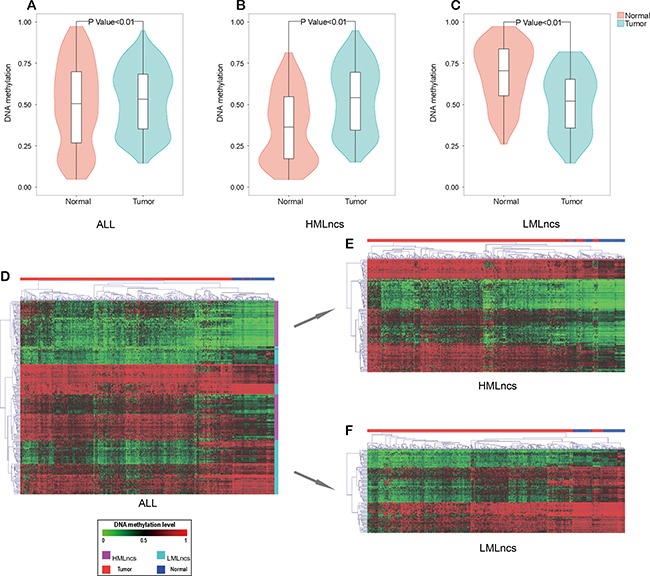
(A) The methylation level of all differential lncRNAs (HMLncs + LMLncs). (B) The methylation level of HMLncs. (C) The methylation level of LMLncs. (D) The bidirectional hierarchical cluster of all differential lncRNAs (HMLncs + LMLncs). (E) The bidirectional hierarchical cluster of HMLncs. (F) The bidirectional hierarchical cluster of LMLncs.
The function interpretation of cis-regulatory regions for lncRNA
Previous studies proved that differentially methylated lncRNAs might be involved in DNA repair, cell apoptosis, cell cycle and many other cancer-related functions [20].
In order to estimate whether lncRNAs classified by DNA methylation influenced tumor progress through cis-regulatory mechanism, a nearest strategy was applied to analyze the function of lncRNAs through the tool GREAT. The tool GREAT integrated annotations from 20 ontologies including gene ontology, Human Phenotype, Disease Ontology, MSigDB Cancer Neighborhood, PANTHER Pathway and so on. Indeed, the lncRNAs were significantly enriched in many ontologies (p < =0.05) (Figure 3). For example, HMLncs were mainly enriched in ameboidal cell migration (GO:0001667), organ development (GO:0048513), positive regulation of cellular process (GO:0048522) and system development (GO:0048731) for gene ontology. LMLncs were mainly enriched in positive regulation of alpha-beta T cell activation (GO:0046635), negative regulation of multicellular organismal metabolic process (GO:0044252), cell activation (GO:0001775) and regulation of multicellular organismal process (GO:0051239) for gene ontology. Cell-cell signaling, cell process, metabolism and angiogenesis were deeply correlated with the progression of cancers [25, 26]. Moreover, GO terms (top 30 rank of p-value) in HMLncs exhibited much stronger relevance than those in LMLncs according to p-value and gene number (Figure 3A and 3B). Besides, we also summarized the functions of disease ontology, MSigDB pathway and PANTHER pathway. HMLncs were enriched in malignant neoplasm of breast and focal adhesion, and LMLncs were enriched in mammary cancer, STAT3 pathway and Wnt signaling pathway [27–30] (Supplementary Table 1).
Figure 3. The cis-regulatory function of HMLncs and LMLncs.

(A and B) The top-30 ranking GO terms of HMLncs and LMLncs based on p-values, respectively. The orange bars represented the GO terms associated with breast cancer. (C and D) The GO term networks. The nodes represented the GO terms enriched significantly. (E and F) The frequency of occurrence of genes in GO terms of HMLncs and LMLncs. Each row represented a gene. And each column was a GO term. The genes were selected if they were regulated by most lncRNAs (top 50 genes). The bar plot at the left side was the count of GO terms of each gene.
To illustrate the relationship among GO terms enriched by lncRNAs, the GO terms were inputted into GO term network. The HMLncs network had three large connected components (nodes > 20). The largest one included 66 GO terms and 63 of them were related to regulation (Figure 3C). The LMLncs network had two large connected components (nodes > 20). They included 38 and 22 GO terms, respectively. And all of the GO terms in two large connected components were related to regulation. Besides, 13 GO terms (59.09% of second large connected component) were related to proliferation (Figure 3D). The results suggested that lncRNAs mainly interfered in the regulation and proliferation of BRCA, and these biological processes with similar functions often kept the strong connectedness with each other.
To evaluate which PCGs were cis-regulated by lncRNAs frequently, we counted the number of PCGs in the result of GREAT enrichment analysis (biological process) for HMLncs and LMLncs. The genes were ranked by the frequency of enrichments in biological process. Of the top 10 genes in HMLncs, nine were demonstrated to be associated with BRCA: FOXJ1 [31], HDAC2 [32], WNT1 [33], SOX18 [34], NOTCH4 [35], SP3 [36], HOXA11 [37], HOXC11 [38] and FOXD3 [39]. In addition, there were six genes coding transcription factors in the top 10 genes (FOXJ1, SOX18, SP3, HOXA11, HOXC11 and FOXD3) (Figure 3E). FOXJ1 and FOXD3 were the members of forkhead box (FOX) and genes in FOX family played key roles in many cancer-related biological processes, such as metastasis, development, organization differentiation, cell proliferation, cell apoptosis, cell migration, invasion, and longevity [40]. HOXA11, and HOXC11 were the members of homeobox family and the two genes were validated to be correlated with the survival of BRCA [37, 38].
In conclusion, these HMLncs tended to cis-regulate genes which coded transcription factors and lead to the dysregulation of transcription in BRCA.
For LMLncs, Interleukins were a group of cytokines which regulated the immune systems and many autoimmune diseases or immune deficiency were lack of them [41]. Interleukin-6 (IL6, GO term count=103), interleukin-18 (IL18, GO term count=73) and Interleukin-6 receptor (IL6R, GO term count=46) in top 10 genes of LMLncs (Figure 3F) have important roles in inflammation and many biological processes such as enhancing the power of tumor cell apoptosis, inhibiting tumorigenesis and inhibiting tumor angiogenesis [42, 43].
The identification of cancer-related hallmarks in ceRNA network
To analyze how DNA methylation regulated the expression of lncRNAs, we further separated HMLncs and LMLncs into four groups: high-up group (high methylation and up-regulated lncRNA expression), high-down group (high methylation and down-regulated lncRNA expression), low-up group (low methylation and up-regulated lncRNA expression) and low-down group (low methylation and down-regulated lncRNA expression).
Many studies have illustrated that lncRNAs often combined with the target miRNAs to regulate the expression of mRNAs [44, 45]. To measure the potential tumorigenic role of lncRNAs through miR-mediated interaction in BRCA, we used DEGs and DELs to construct a background network and regarded the four groups mentioned above as seeds to mine four sub lncRNA-mRNA ceRNA networks: high-up network, high-down network, low-up network and low-down network (Table 2 and Figure 4). The lncRNAs interacted with many PCGs and had high degrees in these ceRNA networks. Besides, although the number of seed lncRNAs in high-down and low-down network were not as many as in the other two networks, the lncRNAs had the larger average degrees (high-up: 297, high-down: 504, low-up: 300, low-down: 835) (Table 2). This result suggested that down-regulated lncRNAs mediated more genes and were more valuable than up-regulated lncRNAs in BRCA.
Table 2. The interaction information of four groups.
| lncRNAs | miRNAs | mRNAs | interaction pairs | |
|---|---|---|---|---|
| high-up | 13 | 101 | 469 | 558 |
| high-down | 9 | 51 | 728 | 1,231 |
| low-up | 7 | 58 | 277 | 295 |
| low-down | 6 | 83 | 687 | 1,129 |
Figure 4. The lncRNA-mRNA ceRNA networks.

(A and B) The relationship between lncRNAs and miRNAs. The lncRNA names were written in the outer circle. The bar plots in the middle circle represented the methylation level and expression level of lncRNAs, respectively. The inner lines represented the interactions between lncRNAs and miRNAs. The orange color represented the up-regulated lncRNA expression. The blue color represented the down-regulated lncRNA expression. (C, D, E and F) The lncRNA-mRNA ceRNA sub-networks. The node size of lncRNAs was correlated with degree.
In order to evaluate the function of four groups of lncRNAs, the PCGs in four networks were inputted into DAVID (https://david.ncifcrf.gov/) to make the function enrichment analysis (Supplementary Table 2).
Although the biological processes of tumors were especially complex, the intricacy of tumors could be simplified and represented by some cancer-related hallmarks which influenced tumor development and metabolism [46]. These hallmarks offered a new direction for understanding the mechanism of cancers.
Function enrichment analysis revealed that genes in four networks were enriched in the hallmarks of cancers significantly (Figure 5A) [47]. The genes were mainly enriched in six kinds of hallmarks (21 biological processes): “Self Sufficiency in Growth Signals”, “Insensitivity to Antigrowth Signals”, “Evading Apoptosis”, “Sustained Angiogenesis”, “Tissue Invasion and Metastasis” and “Genome Instability and Mutation”. In these hallmarks, “Self Sufficiency in Growth Signals” and “Insensitivity to Antigrowth Signals” covered the most genes and were the most affected hallmarks in BRCA. Another interesting observation was that these two kinds of hallmarks shared four biological processes (“signal transduction”, “cell proliferation”, “intracellular signal transduction” and “regulation of cell cycle”). Besides, there were six biological processes involved in the proliferation functions and this demonstrated their key roles in the development of BRCA.
Figure 5. The function enrichment of mRNAs.
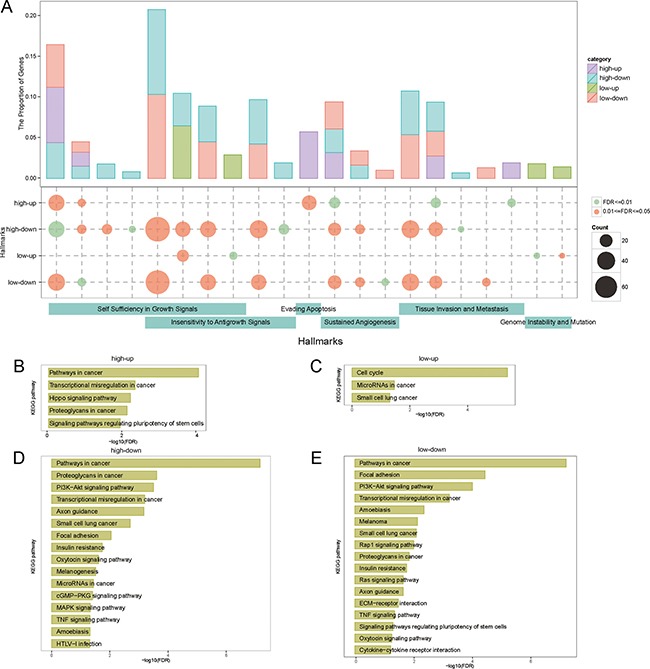
Each column represented a hallmark. (A) The distribution of hallmarks. The top panel was the proportion of genes: (genes enriched in hallmarks in ceRNA network). In the middle panel, the node size was the number of genes enriched in hallmarks. The bottom bar represented the classification of hallmarks. (B, C, D and E) The significant KEGG pathways.
An overall discovery of the function profiles of ceRNA networks also showed some common functions in different networks. For example, three networks (high-up, high-down, low-down) shared four biological processes (“positive regulation of endothelial cell proliferation”, “positive regulation of mesenchymal cell proliferation”, “angiogenesis” and “positive regulation of cell migration”) and these biological processes were the common characteristics of tumors. Most cancer cells owned the capacity of constant proliferation. Normal tissues always mastered the production and release of growth-promoting signals, but cancer cells disturbed the signals and led to the abnormity of tissues [48]. Cancer cells needed nutrients to sustain its growth and evacuate metabolic wastes, so they depended on the process of angiogenesis [49].
Moreover we found that the high-down and low-down network regulated 14 (66.67%) and 12 (57.14%) biological processes and almost covered all kinds of hallmarks. This implied the importance and high coverage of the down-regulated lncRNAs in the development of BRCA. In addition, the low-up network regulated “Genome Instability and Mutation” specifically, but other three networks were not related to this hallmark.
The four networks were also enriched in many cancer-related KEGG pathways (Figure 5B–5E). Some common KEGG pathways located in three of the four networks were identified: “Pathways in cancer”, “Transcriptional misregulation in cancer”, “Proteoglycans in cancer” and “Small cell lung cancer”. “Pathways in cancer” was the most significant pathway (FDR from 2.01e-06 to 8.32e-03) and was consisted of many other cancer-related pathways including “Wnt signaling pathway”, “mapk signaling pathway” and “vega signaling pathway” [50]. Proteoglycans in the tumor microenvironment bound to numerous matrix molecules, growth factors and inflammatory mediators thus influencing the development of cancer [51]. “Small cell lung cancer” also had high frequency in different networks. Our previous study has demonstrated that BRCA and lung cancer kept the strong methylation correlation [13]. We found high-down and low-down network were enriched in more KEGG pathways than the other two networks, and the results of FDR were more significantly as well (Figure 5D and 5E). Most of these pathways were associated with cancers such as “PI3K-Akt signaling pathway”, “Axon guidance” and “TNF signaling pathway” [52–54]. Furthermore, high-down and low-up network controlled the pathway “microRNAs in cancer”. The networks were filtered based on the ceRNA relationship and this result verified the reliability of our ceRNA networks.
In total, 93 genes in high-up network were enriched in hallmarks and KEGG pathways. The lncRNA LEMD1-AS1 interacted with 122 genes and 28 genes were enriched in hallmarks and KEGG pathways, and overlapped 30.11% of 93 genes. 289 genes in high-down network were enriched in hallmarks and KEGG pathways. The lncRNA MAGI2-AS3 interacted with 335 genes and 135 genes were enriched in hallmarks and KEGG pathways, and overlapped 46.71% of 289 genes. 39 genes in low-up network were enriched in hallmarks and KEGG pathways. The lncRNA LINC00707 interacted with 154 genes and 28 genes were enriched in hallmarks and KEGG pathways, and overlapped 71.8% of 39 genes. 246 genes in low-down network were enriched in hallmarks and KEGG pathways. The lncRNA RP11-73M18 interacted with 399 genes and 161 genes were enriched in hallmarks and KEGG pathways, and overlapped 40.35% of 246 genes. The lncRNAs with high degrees tended to interact with cancer-related genes, furthermore lncRNAs in high-down and low-up network had more significant coverage of cancer genes compared with other networks.
As the core link between mRNAs and lncRNAs, miRNAs played key role in development of BRCA. Therefore, miRNA enrichment analysis was used to confirm the function of ceRNA networks. Then the hypergeometric test (Materials and Methods) was used to estimate the function of miRNAs which were interacted with four lncRNA groups, respectively. The miRNAs in high-down group was involved in “Breast Neoplasms” significantly (p = 0.0366), but the other three groups were not (Supplementary Table 3). Previous studies have demonstrated that the high methylation in promoter inhibited downstream expression of lncRNA, and the silence of lncRNAs often played an important role in the progress of cancers [19]. In high-down group, 28 miRNAs were annotated in the breast neoplasms. Then the 28 miRNAs was mapped into the miRNA family information. Nine miRNAs (hsa-mir-302a, hsa-mir-302b, hsa-mir-302c, hsa-mir-302d, hsa-mir-373, hsa-mir-520a, hsa-mir-520b, hsa-mir-520c and hsa-mir-520d) were belonged to “miR-302-3p/372-3p/373” and “miR-520” families, respectively. All miRNAs have been demonstrated to be associated with BRCA through literatures by HMDD.
The identification of clinically relevant modules in ceRNA networks
Wu et al. [55] discovered that it might be possible to improve the survival quality of cancer patients by finding and repairing survival-related gene signatures other than a single gene, and hub genes also played important roles in networks. Therefore, we extracted hub PCGs of the four networks as module to do the survival analysis. However, only high-down and low-down network had the hub PCGs (high-down: seven hub PCGs (NFAT5, QKI, SCHIP1, PRRG1, FOXN3, SACS and NFIB), low-down: six hub PCGs (FOXP2, QKI, ARHGEF10, PTPN14, SPRY2 and DPYSL2)). Next, a multivariate survival analysis based on the two classes of hub PCG modules was performed to estimate the power of predicting survival status in BRCA.
The high-down module was associated with the survival time in BRCA and distinguished patients into high-risk and low-risk group significantly (Log-rank test, p = 0.01474), but the low-down module could not (Log-rank test, p = 0.06716) (Figure 6A and 6B). However, it was unclear whether the high-down module had stable power of classifying the patient subtypes in BRCA significantly.
Figure 6. The survival analysis of high-down and low-down modules.
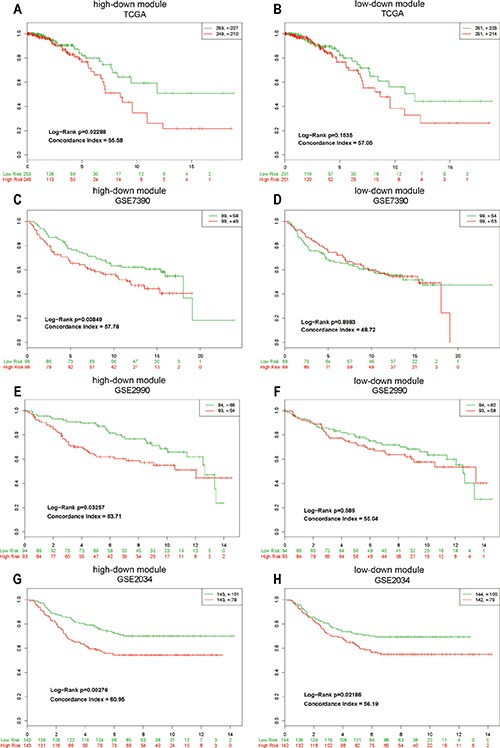
“+” represented lost to follow-up patients. The legend in the top right corner represented the total number and lost to follow-up number of patients. The green and red digitals on the bottom represented number of living patients at that time. (A and B) The survival analysis in TCGA data set. (C–H) The survival analysis in GEO data sets.
To validate the predictive power of high-down module, we did survival analysis in another three independent data sets. As a result, the high-down module kept stable separating capacity for patient survival (GSE2034: p = 2.76e-03, GSE2990: p =3.26e-02, GSE7390: p = 3.85e-02), but the low-down module was only significant in GSE2034 (p = 2.19e-02) (Figure 6C–6H). Our observation might be rewarding to separate high-risk from low-risk patients and provide a new clinical application for the prognostic and surgical management of BRCA.
DISCUSSION
LncRNAs have been demonstrated to be associated with the development and progress of breast cancer. For instance, Xue et al. [56] has found that the overexpression of HOTAIR increased the breast cancer cell proliferation, and its depletion could contribute to the treatment of breast cancer. DNA methylation in promoter always suppresses the combination of transcription factors with DNA chain and decreases the transcription of downstream, so the expression of genes is reduced [2]. It has been recognized that changes in DNA methylation may in part be disruption of the regulatory control of specific promoter usage in cancers. Therefore the dysregulation of lncRNAs in breast cancer may be regulated by DNA methylation in promoter.
In this study, we characterized the changes in DNA methylation of lncRNA promoters in BRCA. We integrated the DNA methylation, lncRNA expression and mRNA expression profiles from TCGA and TANRIC to investigate the methylation of lncRNAs. Our research showed that a large number of lncRNAs were epigenetically deregulated in promoters. We divided the lncRNAs into different groups based on the high methylation and low methylation, and explored their biological and clinical relationship with BRCA. The result indicated that HMLncs and LMLncs cis-regulated cancer-related biological processes (Figure 3). HMLncs were enriched in 126 biological processes associated with regulation function and LMLncs were enriched in 118 biological processes associated with regulation function. Besides LMLncs were enriched in 18 biological processes associated with proliferation. In GO term networks, regulation functions and proliferation functions had the largest connected component and strong combination with each other. Moreover previous studies have shown that lncRNAs regulated PCG activity through the cis-regulation in two modes. The first mode was that the lncRNA products regulated the activity of chromosome through recruiting epigenetic modifiers. The second mode was that the transcription of lncRNAs through PCG promoters or a cis-regulatory element (RE) could influence the expression of PCGs [57–60]. The result showed that epigenetically dysregulated lncRNAs were always located near the genes correlated with the regulation and proliferation function. Moreover we found many high methylated lncRNAs cis-regulated PCGs which coded transcription factors such as FOXJ1, SOX18, SP3, HOXA11, HOXC11 and FOXD3. In molecular biology, transcription factor could regulate the rate transcription of genetic information from DNA to mRNA by biding to a specific DNA sequence. The dysregulation of transcription factor will lead to the abnormal expression of genes near that sequence[61]. This observation suggests that high methylated lncRNAs modulate BRCA through influencing the expression of transcription factors, which provides a foundation for the further analysis of lncRNA dysregulation in BRCA.
Although we have confirmed the role of lncRNAs through cis-regulation, lncRNAs have other ways regulating cancers. Competing endogenesis relationship is another mode that lncRNAs influence the expression of mRNAs by binding to the target miRNAs of mRNAs. To predict the function of lncRNAs, we constructed ceRNA networks through four groups which were classified based on the patterns of DNA methylation and lncRNA expression. The structure of ceRNA networks showed that although high-down and low-down network consisted of less lncRNAs, they attracted more mRNAs (high-down: 728, low-down: 687) than the other two networks (Figure 4) (Table 2). Because of the positive correlation between mRNA and lncRNA, the expression of mRNAs was always inhibited. This result suggests that the majority of differentially expressed mRNA was down-regulated in BRCA.
Tumorigenesis is a complex and dynamic biological process that is controlled by multiple elements of genetic and epigenetic variations. In this study, we found that competing endogenesis relationship regulated key components of cancer-associated hallmarks and KEGG pathways [46]. Four biological processes (“positive regulation of endothelial cell proliferation”, “positive regulation of mesenchymal cell proliferation”, “angiogenesis” and “positive regulation of cell migration”) in hallmarks and three KEGG pathways (“Pathways in cancer”, “Transcriptional misregulation in cancer”, “Proteoglycans in cancer” and “Small cell lung cancer”) were identified in three lncRNA networks as the common characteristics of tumors, and previous studies have demonstrated the relationship between these functions and cancers [13, 47, 50, 51]. These results suggest the mRNAs regulated by lncRNAs could modulate the progress and development of BRCA. Moreover the high-down and low-down network also controlled more hallmarks and KEGG pathways (approximately 10 hallmarks and KEGG pathways) than high-up and low-up network and had the larger coverage of genes enriched in hallmarks and KEGG pathways (Figure 5). Moreover most of these hallmarks and KEGG pathways were associated with BRCA such as “PI3K-Akt signaling pathway”, “Transcriptional misregulation in cancer”, “Proteoglycans in cancer” and so on. These finding further validated the importance of down-regulated lncRNAs in BRCA. “MicroRNAs in cancer” was also enriched significantly. MiRNAs were regarded as the important element connecting the function between mRNA and lncRNA and the dysregulation of miRNAs might lead to abnormal expression of mRNAs and lncRNAs in diseases [62]. Next the miRNAs interacted with lncRNAs were used to assess the function of lncRNAs. Only high-down network was enriched in “Breast Neoplasms” significantly. We thought the lncRNAs down-regulated by high methylation may be more important, and also demonstrate the role of miRNA regulators in BRCA.
Finally, in order to estimate the relationship between clinical prognosis and mRNA modules, the survival analysis was used to evaluate the function of different lncRNA networks. The hub PCG module in high-down network divided patients into high-risk group and low-risk group significantly (P < = 0.01), but low-down PCG module could not. Next we applied three sample sets of BRCA from GEO database to verify the reliability of our finding. The result showed that high-down module divided the patients significantly in three sets (GSE2034: p = 2.76e-03, GSE2990: p = 3.26e-02, GSE7390: p = 3.85e-02), but low-down module was only working in one set (GSE2034: p = 2.19e-02) (Figure 6). From the survival analysis, the lncRNAs silenced by high methylation in promoter have been demonstrated to be potential roles in the prognosis of BRCA, and might be further evaluated for use as cancer biomarkers and potential therapeutic targets. At last, because of lack of hub PCGs in low-up module and high-up module we couldn't demonstrate the function of them for survival status.
In summary, our integration analysis of multiple omics data sets is demonstrated to identify epigenetically abnormal lncRNAs and assess the importance of high methylation for function of lncRNAs in BRCA.
The present study only analyzes the epigenetically dysregulated lncRNAs based on DNA methylation in BRCA, but recent studies have demonstrated that the N6-methyladenosine (m6A) as the RNA post-transcriptional modifications also influences the function of RNAs. Because of the lack of data for m6A in breast cancer, we didn't analyze the relationship between m6A and lncRNA [63]. In the future we hope that growing omics data in different cancers will be analyzed and biological experiments will be performed to demonstrate our study.
MATERIALS AND METHODS
Data source
Patient clinical data, methylation data and RNA-seq data were downloaded from TCGA (http://cancergenome.nih.gov/). Molecular data from the following platforms were used in our study. DNA methylation data was Infinium 450k arrays (Level 3) and RNA-seq data (Level 3) was RNASeqV2. The lncRNA data were obtained from TANRIC database (http://ibl.mdanderson.org/tanric/design/basic/index.html) [64]. We selected 529 tumor samples of which DNA methylation data, RNA-seq data and lncRNA data were all available.
The annotation file (gencode.v19.long_noncoding_RNAs) for lncRNAs was derived from GENCODE (http://www.gencodegenes.org/). The interaction pairs of mRNA-miRNA (386 miRNAs, 13,802 mRNAs and 423,975 interaction pairs) and lncRNA-miRNA (277 miRNAs, 1,127 lncRNAs and 10,212 interaction pairs) were downloaded from the starBase V2.0 database (http://starbase.sysu.edu.cn/) [65].
The information of transcription factors was downloaded from the Animal Transcription Factor Database (AnimalTFDB, http://www.bioguo.org/AnimalTFDB/index.php) [66]. The miRNA family information was from TargetScan (http://www.targetscan.org/vert71/).
Three validation sets of survival analysis were downloaded from Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) [67]. In total, 286 samples in GSE2034 [68], 187 samples in GSE2990 [69] and 198 samples in GSE7390 [70] were analyzed to confirm our results in this study.
Data filtering and normalization
In order to estimate the quality of data, we counted the number of missing values in DNA methylation data, RNA-seq data and lncRNA data. The sites with missing values in more than 30% samples were removed. Then the average value of the site was calculated to impute the other missing values. In total, 393,983 DNA methylation probes, 3,952 lncRNAs and 16,826 mRNAs were retained.
DNA methylation of lncRNA
To estimate the methylation level of a given probe, the beta-value was used as the ratio Methy/(Methy + Unmethy), where “Methy” represented the intensity of methylation of the probe and “Unmethy” represented the intensity of unmethylation of the probe [71]. The beta-value “0” stood for unmethylation, and “1” stood for methylation.
We mapped 393,983 450K probes to lncRNA annotation file and extracted the lncRNA promoter information. Since the regulatory mechanism of lncRNA was similar to the PCG in promoter, the region 2kb upstream from the transcription start site (TSS) of lncRNA was regarded as the promoter and the DNA methylation probes in promoter were obtained for further analysis [21, 57]. Then the average value of DNA methylation probes in promoter of one lncRNA was calculated as the DNA methylation level of the lncRNA. The intersection of lncRNAs between the lncRNA methylation profile and the lncRNA expression profile were selected for our further analysis.
Identification of differentially methylated and differentially expressed genes
Significance analysis of microarrays (SAM) was applied to identify the differential genes between tumor and normal samples for DNA methylation, RNA-seq and lncRNA data, respectively. SAM was developed based on t-test and adjusted the p-value to assess the statistically significant changes for genes [72]. The differentially methylated lncRNAs were identified with q-value < = 0.05 and difference value of DNA methylation level > = 0.1 between tumor and normal samples. The differentially expressed genes were identified with q-value <= 0.05 and fold-change > = 2 or < = 0.5 between tumor and normal samples. The differentially expressed lncRNAs were identified with q-value < = 0.05 [13].
The prediction of the cis-regulation of lncRNAs
In order to compute the effect of the HMLncs and LMLncs, we used the Genomic Regions Enrichment of Annotations Tool (GREAT: http://bejerano.stanford.edu/great/public/html/index.php) to assess the function of target lncRNAs [73]. The web application used the bed format file of the target lncRNAs to predict the function of HMLncs and LMLncs and tested the false positives of results through the hypergeometric test. The result of annotation consisted of about 20 kinds of ontologies including gene ontology, Human Phenotype, Disease Ontology, MSigDB Cancer Neighborhood, PANTHER Pathway and so on. We extracted the bed format file of lncRNAs from lncRNA annotation file to make the enrichment analysis.
The construction of the GO term network
The GO term network was picked out from gene2go (18 March 2015 updated), which was downloaded from NCBI [74]. Only considering ‘is_a’ relationships, the final network included 218 terms and 267 edges for HMLncs, 186 terms and 172 edges for LMLncs. The Go term network was visualized through the software Cytoscape [75].
The prediction of miRNAs targeting lncRNAs
The relationship between miRNAs and diseases was downloaded from the Human MicroRNA Disease Database (HMDD: http://cmbi.bjmu.edu.cn/hmdd), which collected the experimentally supported human miRNA-disease associations.
The hypergeometric test was used to filter the significant functions of the miRNAs with p-values less than 0.05:
| (1) |
Where N was the total number of human genome miRNAs in HMDD, M was the size of miRNAs annotated in one disease, n was the size of miRNAs interacted with HMLncs or LMLncs and m was the intersection of n and M.
The construction of ceRNA network
The interaction pairs of lncRNA-miRNA and mRNA-miRNA were 10,212 and 423,975, respectively. We got 4,563,164 ceRNA pairs between mRNAs and lncRNAs which shared with the same miRNA.
The lncRNA-mRNA ceRNA network was constructed as follows: First, expression correlation between DEGs and DELs was calculated using Pearson correlation coefficient (PCC) between matched mRNA and lncRNA expression profiles:
| (2) |
Where n was defined as the number of samples in BRCA, xik was the expression level of gene i in sample k, xjk was the expression level of gene j in sample k.
Because of the competing endogenous interaction between lncRNAs and mRNAs, the lncRNA-mRNA pairs with PCC > 0 and p-value < = 0.01 were chosen for further analysis. Second, an lncRNA-mRNA pair which interacted with more than one same miRNA and whose hypergeometric test based on lncRNA-miRNA pair and mRNA-miRNA pair was significant (p-value < = 0.01) was considered as a candidate interaction pair (Figure 1). Finally the lncRNA-mRNA pairs satisfying the two conditions were used to construct the lncRNA-mRNA ceRNA network. The ceRNA network was visualized through the software Cytoscape [75].
Functional enrichment analysis
Function enrichment analysis was performed through the DAVID Bioinformatics Resources (https://david.ncifcrf.gov/) [76]. The DAVID enrichment result only checked KEGG pathways and GO biological process (BP) terms and chose the human genome as the background. The terms with FDR < = 0.05 were considered as the statistically significant result.
The identification of clinically related mRNAs correlated with lncRNAs
To measure the clinical effect of mRNA modules in ceRNA networks, we used the prognostic index (PI), also named as risk score, to classify the risk groups. The module was consisted of hub mRNAs (degree > = 5) in ceRNA networks. PI was known as the liner Cox model, PI=β1x1+β2x2+…+βpxp, where p was the number of prognostic mRNAs in the module of ceRNA networks, βp was regarded as the risk coefficient which was calculated in the multivariate Cox regression analysis and xp was regarded as the expression level of mRNAp. The R package “survival” was used to calculate the risk score of each sample and generated the risk groups. Then the patients were divided into high-risk group and low-risk group by the median value of risk score. Kaplan-Meier method and log-rank test was used to assess the survival difference between two patient groups [13].
SUPPLEMENTARY MATERIALS TABLES
Acknowledgments
We acknowledge the help of Chunquan Li from Harbin Medical University-Daqing for his comments. We thank Jianmei Zhao, Jiazi Ma, Xi Han and Chao Wang for modifying the pictures and manuscript.
Abbreviations
- (lncRNAs)
long non-coding RNAs
- (PCGs)
protein coding genes
- (CH3)
covalent addition of a methyl group
- (ORs)
odds ratios
- (DCIS)
ductal carcinoma in situ
- (IDC)
invasive ductal carcinoma
- (ncRNAs)
non-coding RNAs
- (miRNAs)
microRNAs
- (UTRs)
untranslated regions
- (Doc)
Docetaxel
- (BRCA)
breast invasive carcinoma
- (TCGA)
The Cancer Genome Atlas
- (HMLncs)
high methylated lncRNAs
- (LMLncs)
low methylated lncRNAs
- (GO)
gene ontology
- (ceRNA)
competing endogenous RNA
- (GEO)
Gene Expression Omnibus
- (SAM)
Significance analysis of microarrays
- (DMLs)
differentially methylated lncRNAs
- (DELs)
differentially methylated lncRNAs
- (DEGs)
differentially expressed genes
- (GREAT)
Genomic Regions Enrichment of Annotations Tool
- (HMDD)
Human MicroRNA Disease Database
- (PCC)
Pearson correlation coefficient
- (K-S)
Kolmogorov-Smirnov
- (FOX)
forkhead box
- (IL)
Interleukin
- (MAGI2)
membrane associated guanylate kinase
- (RE)
regulatory element
Authors’ contributions
Conceived and designed the experiments: YZ CLZ XYW. Performed the experiments: CLZ XCL NZ YHW. Analyzed the data: CLZ XYW XCL NZ YHW XLH CC. Contributed reagents/materials/analysis tools: JZ ML. Wrote the paper: YZ CLZ NZ XCL.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest to disclose.
FUNDING
This work was supported by National Natural Science Foundation of China [grant number 31371334, 81573021 to Y Zhang], the Innovation Research Funds for Graduate Student of Harbin Medical University [grant number YJSCX2015-45HYD to XY Wang] and Harbin Medical University-Daqing Science Fund [DQXN201609 to CL Zhang].
REFERENCES
- 1.Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, Absher DM, Wold BJ, Myers RM. Dynamic DNA methylation across diverse human cell lines and tissues. Genome research. 2013;23:555–567. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, Camargo AA, Stevenson BJ, Ecker JR, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome research. 2012;22:246–258. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wutz A. Noncoding roX RNA remodeling triggers fly dosage compensation complex assembly. Mol Cell. 2013;51:131–132. doi: 10.1016/j.molcel.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Izzo A, Regnard C, Morales V, Kremmer E, Becker PB. Structure-function analysis of the RNA helicase maleless. Nucleic acids research. 2008;36:950–962. doi: 10.1093/nar/gkm1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalmay T, Edwards DR. MicroRNAs and the hallmarks of cancer. Oncogene. 2006;25:6170–6175. doi: 10.1038/sj.onc.1209911. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Zeng X, He Z, Zou Q. Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources. IEEE/ACM transactions on computational biology and bioinformatics. 2016. [DOI] [PubMed]
- 8.Tang W, Liao Z, Zou Q. Which statistical significance test best detects oncomiRNAs in cancer tissues? An exploratory analysis. Oncotarget. 2016;7:85613–85623. doi: 10.18632/oncotarget.12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 10.Yang F, Luo LJ, Zhang L, Wang DD, Yang SJ, Ding L, Li J, Chen D, Ma R, Wu JZ, Tang JH. MiR-346 promotes the biological function of breast cancer cells by targeting SRCIN1 and reduces chemosensitivity to docetaxel. Gene. 2017;600:21–28. doi: 10.1016/j.gene.2016.11.037. [DOI] [PubMed] [Google Scholar]
- 11.Jadaliha M, Zong X, Malakar P, Ray T, Singh DK, Freier SM, Jensen T, Prasanth SG, Karni R, Ray PS, Prasanth KV. Functional and prognostic significance of long non-coding RNA MALAT1 as a metastasis driver in ER negative lymph node negative breast cancer. Oncotarget. 2016;7:40418–40436. doi: 10.18632/oncotarget.9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, Song X, Wang X, Xie Y, Wang Z, Xu Y, You X, Liang Z, Cao H. Circulating DNA of HOTAIR in serum is a novel biomarker for breast cancer. Breast Cancer Res Treat. 2015;152:199–208. doi: 10.1007/s10549-015-3431-2. [DOI] [PubMed] [Google Scholar]
- 13.Zhang C, Zhao H, Li J, Liu H, Wang F, Wei Y, Su J, Zhang D, Liu T, Zhang Y. The identification of specific methylation patterns across different cancers. PLoS One. 2015;10:e0120361. doi: 10.1371/journal.pone.0120361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Q, Holland-Letz T, Slynko A, Cuk K, Marme F, Schott S, Heil J, Qu B, Golatta M, Bewerunge-Hudler M, Sutter C, Surowy H, Wappenschmidt B, et al. DNA methylation array analysis identifies breast cancer associated RPTOR, MGRN1 and RAPSN hypomethylation in peripheral blood DNA. Oncotarget. 2016;7:64191–64202. doi: 10.18632/oncotarget.11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi J, Gao R, Chen Y, Yang Z, Han P, Zhang H, Dou Y, Liu W, Wang W, Du G, Xu Y, Wang J. Overexpression of NSUN2 by DNA hypomethylation is associated with metastatic progression in human breast cancer. Oncotarget. 2017;8:20751–20765. doi: 10.18632/oncotarget.10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo S, Chen J, Mo X. The association of PTEN hypermethylation and breast cancer: a meta-analysis. Onco Targets Ther. 2016;9:5643–5650. doi: 10.2147/OTT.S111684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu P, Guo Y, Yusufu M, Liu Z, Wang S, Yin X, Peng G, Wang L, Zhao X, Guo H, Huang T, Liu C. Decreased expression of EZH2 reactivates RASSF2A by reversal of promoter methylation in breast cancer cells. Cell Biol Int. 2016;40:1062–1070. doi: 10.1002/cbin.10646. [DOI] [PubMed] [Google Scholar]
- 18.Wu W, Bhagat TD, Yang X, Song JH, Cheng Y, Agarwal R, Abraham JM, Ibrahim S, Bartenstein M, Hussain Z, Suzuki M, Yu Y, Chen W, et al. Hypomethylation of noncoding DNA regions and overexpression of the long noncoding RNA, AFAP1-AS1, in Barrett's esophagus and esophageal adenocarcinoma. Gastroenterology. 2013;144:956–966.e954. doi: 10.1053/j.gastro.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang CM, Wang TH, Chen HC, Li SC, Lee MC, Liou HH, Liu PF, Tseng YK, Shiue YL, Ger LP, Tsai KW. Aberrant DNA hypermethylation-silenced SOX21-AS1 gene expression and its clinical importance in oral cancer. Clin Epigenetics. 2016;8:129. doi: 10.1186/s13148-016-0291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao Q, He W, Liu J, Cen Y, Luo L, Yu C, Li Y, Chen S, Duan S. Identification and functional annotation of lncRNA genes with hypermethylation in colorectal cancer. Gene. 2015;572:259–265. doi: 10.1016/j.gene.2015.07.028. [DOI] [PubMed] [Google Scholar]
- 21.Zhi H, Ning S, Li X, Li Y, Wu W. A novel reannotation strategy for dissecting DNA methylation patterns of human long intergenic non-coding RNAs in cancers. Nucleic acids research. 2014;42:8258–8270. doi: 10.1093/nar/gku575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6:692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- 23.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gill Super HJ. A role for epigenetics in the formation of chromosome translocations in acute leukemia. Cancer Genet. 2015;208:230–236. doi: 10.1016/j.cancergen.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maria RM, Altei WF, Selistre-de-Araujo HS, Colnago LA. Effects of doxorubicin, cisplatin, and tamoxifen on the metabolic profile of human breast cancer MCF-7 cells as determined by 1H HR-MAS NMR. Biochemistry. 2017. [DOI] [PubMed]
- 26.Weigand A, Tasbihi K, Strissel PL, Strick R, Horch RE, Boos AM. [Development of an Innovative Cell Isolation Method for the Investigation of Breast Cancer Pathogenesis and Angiogenesis for Experimental In Vitro And In Vivo Assays]. [Article in German; Abstract available in German from the publisher] Handchir Mikrochir Plast Chir. 2017;49:111–122. doi: 10.1055/s-0042-123706. [DOI] [PubMed] [Google Scholar]
- 27.Kim YJ, Kim K, Lee R, Kim J, Jung W, Paik NS, Moon BI, Lim W, Lee J. Two-year Follow-up of Volumetric-modulated Arc Therapy for Treating Internal Mammary Nodes in Locally Advanced Breast Cancer. Anticancer Res. 2016;36:4847–4851. doi: 10.21873/anticanres.11047. [DOI] [PubMed] [Google Scholar]
- 28.Harrington KM, Clevenger CV. Identification of NEK3 Kinase Threonine 165 as a Novel Regulatory Phosphorylation Site That Modulates Focal Adhesion Remodeling Necessary for Breast Cancer Cell Migration. J Biol Chem. 2016;291:21388–21406. doi: 10.1074/jbc.M116.726190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aghazadeh S, Yazdanparast R. Mycophenolic acid potentiates HER2-overexpressing SKBR3 breast cancer cell line to induce apoptosis: involvement of AKT/FOXO1 and JAK2/STAT3 pathways. Apoptosis. 2016;21:1302–1314. doi: 10.1007/s10495-016-1288-4. [DOI] [PubMed] [Google Scholar]
- 30.Bayerlova M, Klemm F, Kramer F, Pukrop T, Beissbarth T, Bleckmann A. Newly Constructed Network Models of Different WNT Signaling Cascades Applied to Breast Cancer Expression Data. PLoS One. 2015;10:e0144014. doi: 10.1371/journal.pone.0144014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Q, Jackson-Cook C, Lyon D, Perera R, Archer KJ. Identifying molecular features associated with psychoneurological symptoms in women with breast cancer using multivariate mixed models. Cancer Inform. 2015;14:139–145. doi: 10.4137/CIN.S17276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao H, Yu Z, Zhao L, He M, Ren J, Wu H, Chen Q, Yao W, Wei M. HDAC2 overexpression is a poor prognostic factor of breast cancer patients with increased multidrug resistance-associated protein expression who received anthracyclines therapy. Jpn J Clin Oncol. 2016;46:893–902. doi: 10.1093/jjco/hyw096. [DOI] [PubMed] [Google Scholar]
- 33.Si W, Li Y, Shao H, Hu R, Wang W, Zhang K, Yang Q. MiR-34a Inhibits Breast Cancer Proliferation and Progression by Targeting Wnt1 in Wnt/beta-Catenin Signaling Pathway. Am J Med Sci. 2016;352:191–199. doi: 10.1016/j.amjms.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Ma Y, Wang S, Chen F, Gu Y. Suppression of SOX18 by siRNA inhibits cell growth and invasion of breast cancer cells. Oncol Rep. 2016;35:3721–3727. doi: 10.3892/or.2016.4746. [DOI] [PubMed] [Google Scholar]
- 35.Abu-Tayeh H, Weidenfeld K, Zhilin-Roth A, Schif-Zuck S, Thaler S, Cotarelo C, Tan TZ, Thiery JP, Green JE, Klorin G, Sabo E, Sleeman JP, Tzukerman M, et al. ‘Normalizing’ the malignant phenotype of luminal breast cancer cells via alpha(v)beta(3)-integrin. Cell Death Dis. 2016;7:e2491. doi: 10.1038/cddis.2016.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hedrick E, Li X, Safe S. Penfluridol Represses Integrin Expression in Breast Cancer through Induction of Reactive Oxygen Species and Downregulation of Sp Transcription Factors. Mol Cancer Ther. 2017;16:205–216. doi: 10.1158/1535-7163.MCT-16-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia B, Shan M, Wang J, Zhong Z, Geng J, He X, Vu T, Zhang D, Pang D. Homeobox A11 hypermethylation indicates unfavorable prognosis in breast cancer. Oncotarget. 2017;8:9794–9805. doi: 10.18632/oncotarget.14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ali A, Creevey L, Hao Y, McCartan D, O’Gaora P, Hill A, Young L, McIlroy M. Prosaposin activates the androgen receptor and potentiates resistance to endocrine treatment in breast cancer. Breast Cancer Res. 2015;17:123. doi: 10.1186/s13058-015-0636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chu TL, Zhao HM, Li Y, Chen AX, Sun X, Ge J. FoxD3 deficiency promotes breast cancer progression by induction of epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2014;446:580–584. doi: 10.1016/j.bbrc.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 40.Chen HW, Huang XD, Li HC, He S, Ni RZ, Chen CH, Peng C, Wu G, Wang GH, Wang YY, Zhao YH, Zhang YX, Shen AG, et al. Expression of FOXJ1 in hepatocellular carcinoma: correlation with patients’ prognosis and tumor cell proliferation. Mol Carcinog. 2013;52:647–659. doi: 10.1002/mc.21904. [DOI] [PubMed] [Google Scholar]
- 41.Ben Menachem-Zidon O, Avital A, Ben-Menahem Y, Goshen I, Kreisel T, Shmueli EM, Segal M, Ben Hur T, Yirmiya R. Astrocytes support hippocampal-dependent memory and long-term potentiation via interleukin-1 signaling. Brain Behav Immun. 2011;25:1008–1016. doi: 10.1016/j.bbi.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15:79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanchez-Mejias A, Tay Y. Competing endogenous RNA networks: tying the essential knots for cancer biology and therapeutics. J Hematol Oncol. 2015;8:30. doi: 10.1186/s13045-015-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Xu J, Li Y, Zhang J, Chen H, Lu J, Wang Z, Zhao X, Xu K, Li X, Zhang Y. Competing endogenous RNA network analysis identifies critical genes among the different breast cancer subtypes. Oncotarget. 2017;8:10171–10184. doi: 10.18632/oncotarget.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Plaisier CL, Pan M, Baliga NS. A miRNA-regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers. Genome research. 2012;22:2302–2314. doi: 10.1101/gr.133991.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pu SY, Yu Q, Wu H, Jiang JJ, Chen XQ, He YH, Kong QP. ERCC6L, a DNA helicase, is involved in cell proliferation and associated with survival and progress in breast and kidney cancers. Oncotarget. 2017;8:42116–42124. doi: 10.18632/oncotarget.14998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andrade SS, Sumikawa JT, Castro ED, Batista FP, Paredes-Gamero E, Oliveira LC, Guerra IM, Peres GB, Cavalheiro RP, Juliano L, Nazario AP, Facina G, Tsai SM, et al. Interface between breast cancer cells and the tumor microenvironment using platelet-rich plasma to promote tumor angiogenesis - influence of platelets and fibrin bundles on the behavior of breast tumor cells. Oncotarget. 2017;8:16851–16874. doi: 10.18632/oncotarget.15170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abdel-Magid AF. Wnt/beta-Catenin Signaling Pathway Inhibitors: A Promising Cancer Therapy. ACS Med Chem Lett. 2014;5:956–957. doi: 10.1021/ml500276a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Afratis NA, Karamanou K, Piperigkou Z, Vynios DH, Theocharis AD. The role of heparins and nano-heparins as therapeutic tool in breast cancer. Glycoconj J. 2016. [DOI] [PubMed]
- 52.Yang L, Huang F, Mei J, Wang X, Zhang Q, Wang H, Xi M, You Z. Posttranscriptional Control of PD-L1 Expression by 17beta-Estradiol via PI3K/Akt Signaling Pathway in ERalpha-Positive Cancer Cell Lines. Int J Gynecol Cancer. 2017;27:196–205. doi: 10.1097/IGC.0000000000000875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson C, Segovia B, Kandpal RP. EPHA7 and EPHA10 Physically Interact and Differentially Co-localize in Normal Breast and Breast Carcinoma Cell Lines, and the Co-localization Pattern Is Altered in EPHB6-expressing MDA-MB-231 Cells. Cancer Genomics Proteomics. 2016;13:359–368. [PMC free article] [PubMed] [Google Scholar]
- 54.Dasgupta A, Sawant MA, Kavishwar G, Lavhale M, Sitasawad S. AECHL-1 targets breast cancer progression via inhibition of metastasis, prevention of EMT and suppression of Cancer Stem Cell characteristics. Scientific reports. 2016;6:38045. doi: 10.1038/srep38045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu G, Stein L. A network module-based method for identifying cancer prognostic signatures. Genome Biol. 2012;13:R112. doi: 10.1186/gb-2012-13-12-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xue X, Yang YA, Zhang A, Fong KW, Kim J, Song B, Li S, Zhao JC, Yu J. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene. 2016;35:2746–2755. doi: 10.1038/onc.2015.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun M, Kraus WL. Minireview: Long noncoding RNAs: new “links” between gene expression and cellular outcomes in endocrinology. Mol Endocrinol. 2013;27:1390–1402. doi: 10.1210/me.2013-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59. doi: 10.1186/1741-7007-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tesarchik Ia F. [Infection of carp with Neoechinorhynchus rutili acanthocephala]. [Article in Russian] Parazitologiia. 1972;6:190–191. [PubMed] [Google Scholar]
- 61.Karin M. Too many transcription factors: positive and negative interactions. The New biologist. 1990;2:126–131. [PubMed] [Google Scholar]
- 62.Zeng X, Zhang X, Zou Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Briefings in bioinformatics. 2016;17:193–203. doi: 10.1093/bib/bbv033. [DOI] [PubMed] [Google Scholar]
- 63.Chen W, Xing P, Zou Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Scientific reports. 2017;7:40242. doi: 10.1038/srep40242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J, Han L, Roebuck P, Diao L, Liu L, Yuan Y, Weinstein JN, Liang H. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer Res. 2015;75:3728–3737. doi: 10.1158/0008-5472.CAN-15-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic acids research. 2014;42:D92–97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang HM, Liu T, Liu CJ, Song S, Zhang X, Liu W, Jia H, Xue Y, Guo AY. AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic acids research. 2015;43:D76–81. doi: 10.1093/nar/gku887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic acids research. 2013;41:D991–995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 69.Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, Desmedt C, Larsimont D, Cardoso F, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006;98:262–272. doi: 10.1093/jnci/djj052. [DOI] [PubMed] [Google Scholar]
- 70.Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, Viale G, Delorenzi M, Zhang Y, d’Assignies MS, Bergh J, Lidereau R, Ellis P, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007;13:3207–3214. doi: 10.1158/1078-0432.CCR-06-2765. [DOI] [PubMed] [Google Scholar]
- 71.Kobayashi Y, Absher DM, Gulzar ZG, Young SR, McKenney JK, Peehl DM, Brooks JD, Myers RM, Sherlock G. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome research. 2011;21:1017–1027. doi: 10.1101/gr.119487.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Powell JA. GO2MSIG, an automated GO based multi-species gene set generator for gene set enrichment analysis. BMC bioinformatics. 2014;15:146. doi: 10.1186/1471-2105-15-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.