

Abstract
The presence of the dense hydroxyapatite matrix within human bone limits the applicability of conventional protocols for protein extraction. This has hindered the complete and accurate characterization of the human bone proteome thus far, leaving many bone-related disorders poorly understood. We sought to refine an existing method of protein extraction from mouse bone to extract whole proteins of varying molecular weights from human cranial bone. Whole protein was extracted from human cranial suture by mechanically processing samples using a method that limits protein degradation by minimizing heat introduction to proteins. The presence of whole protein was confirmed by western blotting. Mass spectrometry was used to sequence peptides and identify isolated proteins. The data have been deposited to the ProteomeXchange with identifier PXD003215. Extracted proteins were characterized as both intra- and extracellular and had molecular weights ranging from 9.4-629 kDa. High correlation scores among suture protein spectral counts support the reproducibility of the method. Ontology analytics revealed proteins of myriad functions including mediators of metabolic processes and cell organelles. These results demonstrate a reproducible method for isolation of whole protein from human cranial bone, representing a large range of molecular weights, origins and functions.
Keywords: Protein, Extraction, Human, Method, Cranial bone
1. INTRODUCTION
Together, the axial and the appendicular skeleton comprise a dynamic structure essential for structure, protection of vital organs, locomotion, muscle anchoring, maintenance of calcium and phosphate homeostasis, and acid/base regulation. Additionally, the marrow contained in long bones is the site of hematopoiesis. Depending on its composition, skeletal bone can be classified as cortical, compact bone or trabecular, spongy bone.1 Both types have inorganic, organic, water and lipid components. The inorganic or mineral component is predominantly hydroxyapatite while collagens and proteoglycans comprise the organic portion.2 The cellular constituents of bone include osteoblasts, osteoclasts and osteocytes.3,4 These play important roles in bone remodeling that are essential for adaptation to changing biomechanical stresses, and for the turnover of old bone and subsequent replacement with new, stronger bone.
Dysregulation of osteoclasts and osteoblasts can give rise to diseases such as Paget disease of bone,5 osteopetrosis,6 or craniosynostosis.7 However, our understanding of the pathophysiology underlying these and other bone disorders8 is still limited. The development of analytical methods enabling accurate profiling of the bone proteome has immense potential to elucidate these disease mechanisms. Recent methods have shown potential in deriving bone proteins and characterizing them through liquid chromatography-mass spectrometry (LC-MS) platforms. For example, Jiang et al. isolated peptides from canine skull and analyzed them through mass spectrometry by first demineralizing the hydroxyapatite matrix of canine bone using hydrochloric acid.9 While effective at the peptide level, this method was compromised at the protein level by the degradation of protein in strong acid.
Mechanical processing of ancient human bone samples has been previously described by archeologists as an alternative means to extract and analyze proteins.10 In this method, samples were ground by hand with an agate mortar until powder formed. Isolated proteins of this class were identified following mass spectrometry and ranged in molecular weight from 20.1-269.2 kilo-Dalton (kDa). Applicability of this method to current biomedical research is limited by inherent differences between bone obtained from archeological and living specimens.
Recently, Alves et al. extracted protein from femoral trabecular bone of four patients undergoing total hip arthroplasty.11 Collected specimens were mechanically fragmented using a dismembrator and proteins were subsequently homogenized and denatured before being analyzed using nanoflow LC-MS/MS. Notably, 3038 unique proteins were detected with 844 present in all four femoral samples.
The extraction of protein specifically from human skull bone poses additional challenges due to the high density of cortical bone which hinders the extraction of protein from its hydroxyapatite matrix.12 The heat or chemicals needed to break down this matrix compromise protein stability. To date, there has been no method developed for extraction and analysis of proteins from human skull without extensive protein damage. In our laboratory, we are interested in elucidating differential protein expression between patent and prematurely fused human cranial sutures. Therefore, our research objectives call for a protein extraction technique of high yield, resolution and protein integrity. Here we describe a method of protein extraction from human cortical bone that minimizes thermal injury to protein. Using LC-MS/MS platforms, we detected proteins with a wide range of molecular weights that are derived from both intra- and extracellular milieus. Our methods can be utilized for detecting protein-level changes in diseases such as craniosynostosis.
2. MATERIALS & METHODS
2.1 Materials
This study was approved by the University of Chicago Institutional Review Board (IRB #15-0539). Both patent and pathologically fused (craniosynostotic) human cranial suture samples stripped of periosteum were extracted from patients undergoing cranial vault reconstruction at the University of Chicago Medicine Hospital from January 2013 through September 2014. As a pair of suture types (patent/fused) was harvested during surgery, each patient served as their own internal control. Samples were immediately labeled, frozen in liquid nitrogen and then stored at −80°C until use. Ethylenediaminetetraacetic acid (EDTA), aprotinin, leupeptin and pepstatin, sodium orthovanadate (Na3VO4), ammonium bicarbonate (NH4HCO3), and dithiothreitol (DTT) were acquired from Sigma Aldrich (St Louis, MO).
2.2 Lysis buffer preparation
A solution of 2 ml of 1.2 M tris-acetate (pH 6.8), 1 ml 10% SDS, 50 μl glycerol, 100 μl 100 mM EDTA, and 7 ml H2O was mixed and placed on ice. Immediately before use, 10 μl each of 1 mg/ml aprotinin, leupeptin and pepstatin were added. Additionally, 100 μl 1 M β-glycerol phosphate, 50 μl 200 mM Na3VO4, and 500 μl 1 M DTT were added. The solution was mixed in a conical tube by hand for approximately 30 seconds, placed on ice, and subsequently aliquoted into 500 μl Eppendorf tubes (each) and kept on ice.
2.3 Sample preparation
The suture samples were removed from the freezer and wrapped in several layers of autoclaved aluminum foil. A hammer was used to break up the sample and the fragments were placed into a mortar and pestle kept on dry ice. The fragments were ground into a fine powder before being placed into Eppendorf tubes. The tubes were vortexed for one minute or until the ground suture went into solution. If necessary, more lysis buffer was added to solubilize the ground suture.
The solution was boiled for ten minutes (ensuring that the eppendorf tops were screwed on tightly) and then frozen at −80°C. The samples were thawed and then centrifuged at 14000 rpm for 10 minutes. The supernatant was removed and placed into two ml tubes. The supernatant was sonicated for a total of ten seconds and then needle-sheared using a 25G needle. It was then aliquoted and frozen at −80°C.
2.4 Protein digestion
1D gel electrophoresis was performed on sample eluates using 20 ug for a gel plug digest. IP eluates were loaded onto a 12% MOPS buffered SDS-PAGE gel (Invitrogen, Thermo Scientific, San Jose, CA), and run for 10 minutes at 200 V resulting in a ~2 cm “gel plug.” The gel was stained with 25 ml Imperial Stain (Thermo Scientific, San Jose, CA) at room temperature, and de-stained overnight in deionized H2O at 4°C.
The gel plugs for each sample to be analyzed were excised by sterile razor blade and chopped into ~ 1 mm3 pieces. Each section was washed in deionized H2O and destained using 100 mM NH4HCO3 pH 7.5 in 50% acetonitrile. A reduction step was performed by addition of 100 μl 50 mM NH4HCO3 pH 7.5 and 10 μl of 200 mM tris(2-carboxyethyl)phosphine HCl at 37 °C for 30 minutes. The proteins were alkylated by addition of 100 μl of 50 mM iodoacetamide prepared fresh in 50 mM NH4HCO3 pH 7.5 buffer and allowed to react in the dark at 20 °C for 30 minutes. Gel sections were washed in water, then in acetonitrile, and vacuum dried. Trypsin digestion was carried out overnight at 37 °C with 1:50-1:100 enzyme–protein ratio of sequencing grade-modified trypsin (Promega, Madison WI) in 50 mM NH4HCO3 pH 7.5, and 20 mM CaCl2. Peptides were sequentially extracted with 5% formic acid, then 75% acetonitrile with 5% formic acid, combined and vacuum dried.
2.5 LC-MS/MS analysis
All fused and patent samples were re-suspended in Burdick & Jackson HPLC-grade water containing 0.2% formic acid (Fluka), 0.1% trifluoroacetic acid (TFA, Pierce, Waltham, MA), and 0.002% Zwittergent 3–16 (Calbiochem, Billerica, MA), a sulfobetaine detergent that contributes the following distinct peaks at the end of chromatograms: MH+ at 392, and in-source dimer [2M+H+] at 783, and some minor impurities of Zwittergent 3–12 seen as MH+ at 336. The peptide samples were loaded to a 0.25 μl C8 OptiPak trapping cartridge custom-packed with Michrom Magic (Optimize Technologies, Oregon City, OR) C8, washed with Mobile phase A solution, then switched in-line with a 20 cm by 75 μm C18 packed spray tip nano column packed with Michrom Magic C18AQ, for a 2-step gradient. Mobile phase A was water/acetonitrile/formic acid (98/2/0.2) and mobile phase B was acetonitrile/isopropanol/water/formic acid (80/10/10/0.2). Using a flow rate of 350 nl/min, a 90 minute, 2-step LC gradient was run from 5% B to 50% B in 60 minutes, followed by 50%–95% B over the next 10 minutes, hold 10 minutes at 95% B, back to starting conditions and re-equilibrated.
The samples were analyzed via electrospray tandem mass spectrometry (MS/MS) on a Thermo Q-Exactive Orbitrap mass spectrometer, using a 70,000 reversed phase survey scan in profile mode, m/z 360–2000 Da, using lock masses, followed by 10 MS/MS higher-energy collision dissociation fragmentation scans at 17,500 resolution on doubly and triply charged precursors. Single charged ions were excluded, and ions selected for MS/MS were placed on an exclusion list for 60 seconds.
2.6 Data analysis
Tandem mass spectra were extracted and charge states deconvoluted and deisotoped by Proteo Wizard version 3.0.6447. All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.3.02) and X! Tandem13 (The GPM, thegpm.org; version CYCLONE (2010.12.01.1)). Mascot was set up to search the 140530_SPROT_HUMAN database (Uniprot 5-30-2014, 88698 entries) with the enzyme set to trypsin. X! Tandem was set up to search the 140117_SPROT_HUMAN database (Uniprot 1-17-2014, 69025 entries) also with enzymatic cleavage set as trypsin. Mascot and X! Tandem were searched with a fragment ion mass tolerance of 0.60 Da and a parent ion tolerance of 20 ppm. Carbamidomethylation of cysteine was specified as a fixed modification. Other variable modifications that were specified were modification from Glu to pyro-Glu at the N-terminus, ammonia-loss at the N-terminus, oxidation of methionine, formylation of the N-terminus, phosphorylation of serine, threonine and tyrosine and glyGly of lysine.
Scaffold (version Scaffold_4.2.1, Proteome Software Inc., Portland, OR) was used to validate MS/MS-based peptide and protein identifications. The following rules were used to derive a list of confident peptides and proteins: a minimum observance of 2 peptides per protein, 95% peptide and 99% protein probabilities based on PeptideProphet14 and ProteinProphet15 algorithms within Scaffold. Gene ontology (GO) analysis was performed on the list of all identified proteins using Panther gene analysis tool.16 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository17 with the dataset identifier PXD003215 and DOI 10.6019/PXD003215.
2.7 Western Blot
Cranial suture samples were collected with a target size of 1 cm2. Protein was extracted from suture samples using two different methods of mechanical sample processing. The sample in lane 1 was processed with a mortar and pestle, and the sample in lane 2 was processed with a hammer. 30 μl of each lysate were loaded onto a NuPage gel (200V, 115 mA/gel, 1hr) although this was not normalized for protein concentration. Separated proteins were transferred to a nitrocellulose membrane (0.2 μm pore size) using a tank transfer system (200 V, 230 mA, 1 hour). Transfer was confirmed with Ponceau S staining. Actin was probed for using mouse anti-actin (cat#69100, clone C4) from MP Biomedical (Santa Ana, CA). Actin band intensity was quantified with densitometry using ImageJ18.
3. RESULTS
3.1 Suture Collection
In total, ten unique sutures were collected from five patients (ages 3-12 months). Patent sutures included four coronal sutures and one metopic suture. Fused sutures included three sagittal, one coronal and one metopic suture.
3.2 Protein Characterization
A total of 664 proteins were identified in our analysis (Supplementary Table 1). Molecular weights of proteins isolated (as reported by UniProt) ranged from 0-300+ kDa with a majority of proteins (83.3%) between 0-75 kDa in weight (Figure 1). A total of 624 proteins were identified from the patent samples, with 275 proteins (44.1%) common to all patent samples. Similarly, 634 proteins were identified in fused samples with 199 (31.3%) common to all fused samples. Further details on protein sequence coverage, spectral counts, and identification probabilities are given in Supplementary Table 1.
Figure 1.

Distribution of molecular weights of proteins extracted from cranial suture samples.
3.3 Method Reproducibility
To further characterize reproducibility of the protein extraction method, correlation measures of protein spectral counts between experiments were calculated. Figure 2A depicts the correlation of spectral counts of all identified proteins between the patent samples. The upper-diagonal portion of the plot depicts correlation scores that indicate variation between samples. High correlation scores (>0.9) are observed when comparing patent samples even across patients and suture type (coronal, metopic), indicative of high experimental reproducibility. The corresponding correlation plots for fused samples are shown in Figure 2B. As shown, there is significant correlation among the fused sagittal samples (>0.8, indicated by large text in Figure 2B) and between sagittal and metopic samples. However, the correlation of protein spectral count between coronal and other types are however low (0.6-0.7, indicated by small text in Figure 2B).
Figure 2.

Correlation measures of protein spectral counts of (A) patent sutures and (B) fused sutures. Samples are labeled with three identifiers. P indicates a patent suture, F indicates a fused suture, and S, C, M correspond to sagittal, coronal and metopic sutures, respectively. The numbers 1-5 indicate the patient from which the suture was taken.
3.4 Functional Characteristics and Biology
Proteins were identified as deriving from both the intracellular and extracellular compartments (Figure 3A) using a search of the UniProt database. While all cellular compartments were represented by the intracellular proteins, the majority (40.6%) was located in the cytoplasm, while the nucleus (17.4%) and mitochondria (5.8%) represented the second and third most common sites of origin, respectively (Figure 3B).
Figure 3A.

Intra- vs. extracellular distribution of protein origin as recorded in UniprotKB.
Figure 3B.

Localization of isolated proteins of intracellular origin as recorded in UniProtKB.
The results of gene ontology analysis are illustrated in Figure 4. Within biological processes, metabolic process (24.2%), cellular process (18.5%) and localization (9.5%) were the top identified GO categories. Within molecular function, catalytic activity (32.5%), binding (25.2%) and structural activity (13.8%) were the top protein functions associated with suture proteins. Finally, within the cellular component category, cell parts (32.3%), organelle (21.6%) and extracellular (19.0%) components were the most associated with identified suture proteins.
Figure 4.

Protein Ontology as generated by PANTHER. The top three functions represented in the “Biological Categories,” “Molecular Function” and “Cellular Component” categories are represented by bold text.
3.5 Whole Protein Isolation
Isolated (not degraded) actin was detected by Western blot. Actin bands were present in suture samples processed using two different mechanical processing methods. The band intensity of the sample processed using a mortar and pestle was 12% of the band intensity of the sample processed using a hammer (Figure 5).
Figure 5.
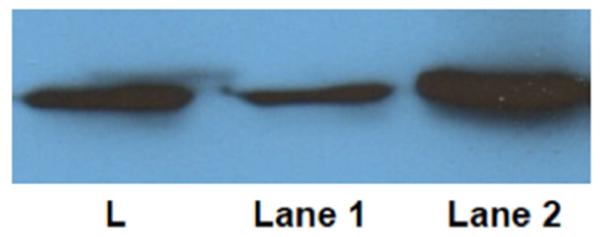
Western blot probing for actin (42 kDa). Actin was probed in protein extracts of suture mechanically processed with a mortar and pestle (lane 1) and with a hammer (lane 2). L indicates ladder.
4. Discussion
No method has yet described a means to isolate whole protein from human cranial bone. Protein extraction from human bone is challenging due to the fragility and sensitivity of proteins to heat and acidic degradation – factors that are necessary for the disruption of the rigid hydroxyapatite matrix. This has led to previous investigation of bone biology using in vitro studies of osteoblasts and osteoclasts – a technique inherently limited by its inability to replicate the in vivo environment of bone.19 We hypothesized that we could devise a method that minimized protein loss due to acidic or thermal degradation. To this end, we fragmented cranial bone samples first using a hammer to increase the surface area-to-volume ratio before subjecting the bone to the friction and heat generated by the mortar and pestle. This limited heat introduction into samples and minimized thermal protein degradation. Protein was then stabilized in a lysis buffer consisting of several protease inhibitors, adapted from Ciaccio et al.20 This was followed by brief boiling, freeze-thaw cycles and sonication to further disrupt cell membranes and to facilitate isolation of intracellular proteins.21 Centrifugation was used to remove mineral matrix debris from samples. Of note, no protein analysis of this matrix debris was performed, but will be the focus of future experimentation to further characterize the efficiency of our technique.
The reproducibility of this method is demonstrated by the protein profiles of two types of suture-associated human cranial bone. On comparison of biological replicates, both sample groups had high correlation scores. To our knowledge, no other method has been as extensively tested to show reproducibility. By using two separate suture types (fused and patent), and demonstrating consistent inter-group protein extraction profiles, the consistency of the method is supported.
A heterogeneous group of proteins was isolated, consisting of both intra- and extracellular proteins with molecular weights ranging from 9-629 kDa. The majority of intracellular proteins were located in the cytoplasm, although all intracellular compartments were represented.
The isolation of whole protein was demonstrated with Western blotting. Of the methods detailed above that have been used to isolate peptides for analysis with mass spectrometry, only one described whole protein extraction. In this study, we probed for actin because of its ubiquitous nature. Since the method of using a hammer to mechanically process samples yielded a more intense actin band signal than the sample processed with the mortar and pestle, it appears more protein is preserved when less friction is used to morcellate samples.
The presence of proteins such as osteomodulin, an osteoblast maturation marker induced by osteoclast activity,22 periostin, a protein secreted by osteoblasts and osteoblast cell lines,23 and asporin, a protein that contributes to osteoblast-driven collagen biomineralization activity and has been implicated in the development of osteoarthritis,24,25 suggest that our isolate is representative of in vivo protein expression in bone. Additionally, as bone is largely composed of collagen, it was expected and accordingly reflected in our results that there were many collagen proteins isolated including Col6A1. Col6A1 has been linked to the RANK-RANKL osteoclast activation pathway and has been linked to the development of rheumatoid arthritis.26
Further characterizing the bone proteome can have enormous implications for research in several bone-related disorders. For instance our current focus is to identify differential expression of proteins in prematurely fused versus patent human cranial sutures in an attempt to unravel candidate genes in nonsyndromic craniosynostosis.
This method is limited due to the use of pediatric cranial bone – this bone is known to be more malleable and elastic than adult cranial bone, which has more completely ossified.27 This feature of pediatric bone may have facilitated the breakdown of the hydroxyapatite matrix. We are planning to study this method for protein extraction in the adult setting.
While the protein yield was relatively low, the application of this method to human cranial bone represents a first pass at this technique. Refinement of this method to standardize lysate protein concentrations and to produce a higher protein recovery will be the goal of future experiments.
Additional limitations include the application of this method to different bone types including trabecular. As trabecular bone lacks the rigidity and density inherent to cortical bone, we suspect overall protein yield would be higher as less mechanical force (and heat) would be necessary to interrupt the hydroxyapatite matrix thereby minimizing protein degradation.
Conclusions
Here we describe a reproducible method for isolating whole protein, with a wide range of molecular weights and of several intra- and extracellular origins, from human cranial bone. By mechanically processing cranial suture samples in a manner that minimized the introduction of heat, protein degradation was minimized. This can be instrumental in the characterization of the human bone proteome, which can shed light on poorly understood pathophysiologic conditions ranging from malignancy to trauma as well as facilitate the development of therapeutic interventions.
Supplementary Material
Supplementary Table 1: Table of proteins detected by mass spectrometry with their spectral count abundances
Acknowledgements
This project was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health through Grant Number UL1 TR000430.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure
The authors declare no competing financial interest.
References
- 1.Dempster DW. In: Principles of Bone Biology. Third Martin JPBGRJ, editor. Academic Press; 2008. pp. 447–463. [Google Scholar]
- 2.Lammi MJ, Häyrinen J, Mahonen A. Proteomic analysis of cartilage- and bone-associated samples. Electrophoresis. 2006;27:2687–2701. doi: 10.1002/elps.200600004. [DOI] [PubMed] [Google Scholar]
- 3.Eriksen EF, Axelrod DW, Melsen F. Bone Histomorphometry. Raven Press; 1994. [Google Scholar]
- 4.Nijweide PJ, Burger EH, Feyen JH. Cells of bone: proliferation, differentiation, and hormonal regulation. Physiol. Rev. 1986;66:855–886. doi: 10.1152/physrev.1986.66.4.855. [DOI] [PubMed] [Google Scholar]
- 5.Ralston SH. Pathogenesis of Paget’s disease of bone. Bone. 2008;43:819–825. doi: 10.1016/j.bone.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Teti A, et al. Mechanisms of Osteoclast Dysfunction in Human Osteopetrosis: Abnormal Osteoclastogenesis and Lack of Osteoclast-Specific Adhesion Structures. J. Bone Miner. Res. 1999;14:2107–2117. doi: 10.1359/jbmr.1999.14.12.2107. [DOI] [PubMed] [Google Scholar]
- 7.Lee JC, et al. Role of RANK-RANKL-OPG axis in cranial suture homeostasis. J. Craniofac. Surg. 2011;22:699–705. doi: 10.1097/SCS.0b013e3182077fbd. [DOI] [PubMed] [Google Scholar]
- 8.Asher MA, Burton DC. Adolescent idiopathic scoliosis: natural history and long term treatment effects. Scoliosis. 2006;1:2. doi: 10.1186/1748-7161-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang X, et al. Method development of efficient protein extraction in bone tissue for proteome analysis. J. Proteome Res. 2007;6:2287–2294. doi: 10.1021/pr070056t. [DOI] [PubMed] [Google Scholar]
- 10.Boros-Major A, et al. New perspectives in biomolecular paleopathology of ancient tuberculosis: a proteomic approach. J. Archaeol. Sci. 2011;38:197–201. [Google Scholar]
- 11.Alves RDAM, et al. Unraveling the human bone microenvironment beyond the classical extracellular matrix proteins: a human bone protein library. J. Proteome Res. 2011;10:4725–4733. doi: 10.1021/pr200522n. [DOI] [PubMed] [Google Scholar]
- 12.Peterson J, Dechow PC. Material properties of the inner and outer cortical tables of the human parietal bone. Anat. Rec. 2002;268:7–15. doi: 10.1002/ar.10131. [DOI] [PubMed] [Google Scholar]
- 13.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinforma. Oxf. Engl. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 14.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical Statistical Model To Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 15.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 16.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vizcaíno JA, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simonet WS, et al. Osteoprotegerin: A Novel Secreted Protein Involved in the Regulation of Bone Density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 20.Ciaccio MF, Wagner JP, Chuu C-P, Lauffenburger DA, Jones RB. Systems analysis of EGF receptor signaling dynamics with microwestern arrays. Nat. Methods. 2010;7:148–155. doi: 10.1038/nmeth.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Traditional Methods of Cell Lysis. Available at: https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/traditional-methods-cell-lysis.html. (Accessed: 18th August 2015)
- 22.Ninomiya K, et al. Osteoclastic activity induces osteomodulin expression in osteoblasts. Biochem. Biophys. Res. Commun. 2007;362:460–466. doi: 10.1016/j.bbrc.2007.07.193. [DOI] [PubMed] [Google Scholar]
- 23.Horiuchi K, et al. Identification and Characterization of a Novel Protein, Periostin, with Restricted Expression to Periosteum and Periodontal Ligament and Increased Expression by Transforming Growth Factor β. J. Bone Miner. Res. 1999;14:1239–1249. doi: 10.1359/jbmr.1999.14.7.1239. [DOI] [PubMed] [Google Scholar]
- 24.Sakao K, et al. Asporin and transforming growth factor-beta gene expression in osteoblasts from subchondral bone and osteophytes in osteoarthritis. J. Orthop. Sci. Off. J. Jpn. Orthop. Assoc. 2009;14:738–747. doi: 10.1007/s00776-009-1401-4. [DOI] [PubMed] [Google Scholar]
- 25.Kalamajski S, Aspberg A, Lindblom K, Heinegård D, Oldberg A. Asporin competes with decorin for collagen binding, binds calcium and promotes osteoblast collagen mineralization. Biochem. J. 2009;423:53–59. doi: 10.1042/BJ20090542. [DOI] [PubMed] [Google Scholar]
- 26.Danks L, et al. Extended report: RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann. Rheum. Dis. 2015;75:1187–1195. doi: 10.1136/annrheumdis-2014-207137. [DOI] [PubMed] [Google Scholar]
- 27.Coats B, Margulies SS. Material properties of human infant skull and suture at high rates. J. Neurotrauma. 2006;23:1222–1232. doi: 10.1089/neu.2006.23.1222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Table of proteins detected by mass spectrometry with their spectral count abundances