

Abstract
Mitochondrial biogenesis (MB) is an adaptive response to maintain metabolic homeostasis after mitochondrial dysfunction. Induction of MB during APAP hepatotoxicity has not been studied. To investigate this, mice were treated with toxic doses of APAP and euthanized between 0 and 96h. At early time points, APAP caused both mitochondrial dysfunction and reduction of mitochondrial mass, indicated by reduced activity of electron transport chain (ETC) complexes I and IV and depletion of mitochondrial DNA (mtDNA), respectively. Both ETC activity and mtDNA gradually recovered after 12 h, suggesting that MB occurs at late time points after APAP overdose. Immunofluorescent staining of mitochondria with mitochondrial outer membrane protein Tom20 further demonstrated that MB occurs selectively in hepatocytes surrounding necrotic areas. MB signaling mediators including PPARγ co-activator 1-α (Pgc-1α), nuclear respiratory factor-1 (Nrf-1) and mitochondrial fission protein dynamin-related protein-1 (Drp-1) were induced. Pgc-1α was selectively increased in hepatocytes surrounding necrotic areas. In addition, the time course of MB induction coincides with increased liver regeneration. Post-treatment with the known MB inducer SRT1720 increased Pgc-1α expression and liver regeneration, resulting in protection against late liver injury after APAP overdose. Thus, induction of MB is an important feature during APAP hepatotoxicity and liver regeneration.
Keywords: Acetaminophen, hepatotoxicity, mitochondria biogenesis, regeneration, PPARγ coactivator 1-α, SRT1720
1. INTRODUCTION
Acetaminophen (APAP)-induced liver injury is the leading cause of acute liver failure in the United States and many other Western countries (Lee, 2008). Numerous studies have established the critical role of mitochondria in the initiation and progression of APAP hepatotoxicity in both mice and humans (Placke et al., 1987; Meyers et al., 1988; Jaeschke, 1990; Kon et al., 2004; LoGuidice and Boelsterli, 2011; Ramachandran et al., 2011; McGill et al., 2012a; 2014; Du et al., 2017). There is considerable evidence that the reactive metabolite of APAP binds to mitochondrial proteins (Tirmenstein and Nelson, 1989; McGill et al., 2012b; Xie et al., 2014, 2015) leading to altered mitochondrial morphology (Placke et al., 1987), inhibition of mitochondrial respiration (Meyers et al., 1988), mitochondrial oxidative stress and peroxynitrite formation (Jaeschke, 1990; Cover et al., 2005), loss of mitochondrial membrane potential (Kon et al., 2004; McGill et al., 2011 ; Xie et al., 2014) and release of mitochondrial proteins into the cytosol and plasma (Bajt et al., 2006; McGill et al., 2012a; 2014). In addition, several interventions aimed at preventing or reducing mitochondrial dysfunction have been shown to protect against APAP-induced liver injury, including post-treatment with the antidote GSH or N-acetylcysteine (NAC) (James et al., 2003; Knight et al., 2002; Saito et al., 2010) or SOD-mimetic Mito-Tempo (Du et al., 2017) to scavenge ROS, inhibition of the mitochondrial membrane permeability transition (MPT) (Kon et al., 2004; Ramachandran et al., 2011) and activation of autophagy to remove damaged mitochondria (Ni et al., 2012).
Mitochondrial biogenesis (MB) is the growth and division of existing mitochondria, resulting in increased mitochondrial mass within cells. The primary purpose of MB is to maintain or restore energy homeostasis during energy deprivation or following a mitochondrial insult. Several signaling mediators control this process, but PPARγ co-activator-1α (Pgc-1α) is thought to be the master regulator (Scarpulla, 2008). Although Pgc-1α itself does not bind to DNA, it interacts with other transcription factors in the nucleus to induce expression of genes that are important for MB (Scarpulla, 2008). In particular, induction of nuclear respiratory factor (Nrf) 1 by Pgc-1α controls the coordinate expression of other genes involved in MB in the nucleus and the mitochondria, especially those encoding subunits of the electron transport chain (ETC) complexes (Baker et al., 2007; Scarpulla, 2008). Importantly, Pgc-1α itself can be activated by the AMP-activated protein kinase (Ampk) and Sirtuin-1 (Sirt-1). It is also important to note that mitochondrial dynamics, involving mitochondrial fission and fusion are also carefully coordinated in order to ensure proper organization of the mitochondrial network (Chan, 2006a,b). In some cases, MB may determine whether a cell survives or dies (Jornayvaz et al., 2010; Scarpulla, 2008), and in fact, impairment of MB is thought to contribute to several forms of tissue injury. In the liver, there is some evidence that the stress caused by chronic ethanol feeding can induce MB (Han et al., 2012), though it is not yet clear what role this plays in alcohol-induced liver injury. In extrahepatic tissues, kidney cells treated with pro-oxidants showed increased MB after the initial stress, and overexpressing Pgc-1α enhanced recovery of mitochondrial function (Rasbach et al., 2007). Pharmacological induction of MB enhances regeneration and recovery in various rodent models of acute kidney injury (Rehman et al., 2013; Whitaker et al., 2013; Funk and Schnellmann, 2013; Jesinkey et al., 2014; Garrett et al., 2014; Khader et al., 2014) as well as other models of tissue injury (Finck and Kelly, 2007; St-Pierre et al., 2005; Funk et al., 2010).
Since mitochondrial dysfunction is a key factor in APAP-induced liver injury (Jaeschke et al., 2012), we hypothesized that MB could be affecting APAP hepatotoxicity. Therefore, the major objective of the present study was to characterize the time course of MB following APAP overdose and to determine whether or not induction of MB could be beneficial during APAP hepatotoxicity.
2. METHODS
2.1. Animals
Male C57BL/6J mice (8–12 weeks old) were purchased from Jackson Laboratories (Bar Harbor, ME) and kept in an environmentally controlled room with a 12 h light/dark cycle and ad libitum access to food (LabDiet® PicoLab® Rodent Diet 20, #5053 Purina, Missouri, USA) and water. Mice were i.p. treated with 200 or 300 mg/kg APAP (Sigma-Aldrich) dissolved in warm saline after overnight fasting, and euthanized at the indicated time points between 0 and 96h after APAP injection for collection of blood and liver samples. SRT1720 (EMD Millipore) was dissolved in 10% DMSO plus 2% Tween 20 and was i.p. administered at either 1.5h or 12h and 36h post-APAP. All vehicle control mice received the same volume of DMSO (1 mL/kg) and Tween 20 (0.2 mL/kg). Blood was drawn from the caudal vena cava using a heparinized syringe. The liver was divided into several pieces some of which were used for mitochondrial isolation (Du et al., 2015), others for embedding in OCT medium for immunofluorescent staining or fixing in 10% phosphate-buffered formalin for histological analysis. The remaining pieces were snap-frozen in liquid nitrogen and stored at −80°C for later analyses. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University Kansas Medical Center and followed the criteria of the National Research Council for the care and use of laboratory animals.
2.2. Biochemical assays
Plasma ALT activity was determined using an ALT kit (Pointe Scientific, MI). Hepatic mitochondria were isolated, and submitochondrial particles were prepared by two cycles of freezing/thawing. Mitochondrial respiratory complex I & IV enzyme activity was assayed as described (Larosche et al. 2007; Kwong and Sohal, 2000). For complex I activity, the reaction mixture contained 25mM potassium phosphate buffer (pH 7.4), 5 mM MgCl2, 2mM KCN, 2.5 mg bovine serum albumin, 100mM NADH, 100mM ubiquinone, and 2mg antimycin. The reaction was initiated by addition of submitochondrial particles (20 −50mg of protein), and complex I activity measured by following the decrease in absorbance due to oxidation of NADH to NAD at 340 for 2 min. Complex I activity was then calculated as the difference between the total enzymatic rates and that obtained with the addition of rotenone (5mg). For complex IV enzyme activity, the assay mixture had 10 mM potassium phosphate and 15 mM ferrocytochrome c, to which submitochondrial sample (1–5mg protein) was added and the decrease in absorbance due to the oxidation of ferrocytochrome c followed at 550nm for 30s. Complex IV activity was then calculated from the initial rate. GSH and GSSG levels were measured using a modified method of the Tietze assay as described (McGill and Jaeschke, 2015).
2.3. mtDNA levels
mtDNA was measured as previously described (Cover et al., 2005). Briefly, total hepatic DNA was isolated with Genomic-tip 100/G columns (QIAGEN GmbH, Hilden, Germany) then blotted onto Hybond-N nylon membranes (GE Healthcare). Membranes were first hybridized with a 10.9-kilobase mtDNA probe (nucleotides 4964 −15,896) generated by long PCR and labeled by random priming, then stripped and hybridized with a mouse Cot-1 nDNA probe (Invitrogen, Cergy Pontoise, France). The levels were determined by densitometry analysis of autoradiographs and normalized to nuclear DNA levels.
2.4. Histology
Formalin-fixed tissue samples were embedded in paraffin and 5 μm thick sections were cut and transferred to glass slides. The slides were then stained with hematoxylin and eosin (H&E) for evaluation of tissue necrosis. Necrosis was quantified by a pathologist (A.F.) who was blinded to the sample identities. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay was performed for assessment of DNA strand breaks with the In Situ Cell Death Detection Kit, AP (Roche Diagnostics, Indianapolis, IN) following manufacturer’s instructions. Sections were also stained for proliferating cell nuclear antigen (PCNA) using a rabbit polyclonal anti-PCNA antibody, according to the manufacturer’s instructions (Santa Cruz Biotechnology, Dallas, TX). Immunofluorescence staining was performed with OCT-embedded tissue. Cryosections were cut 6 μm thick and fixed with 5% paraformaldehyde for 10 mins. After washing with 100D7 PBS, tissues were blocked with 5% normal goat serum followed by overnight incubation with the rabbit anti-Tom20 antibody (1:250 dilution) (sc11415 Santa Cruz, Dallas, TX) or anti-Pgc-1α antibody (1:250 dilution) (PA5-38021, Pierce, Rockford, IL). The secondary antibody was Alexa Fluor 594-conjugated goat anti-rabbit antibody (A11037, Life Technologies, Eugene, OR). Nuclei were stained with DAPI containing mounting medium (Life Technologies) when placing coverslips, and images were obtained using a Zeiss Axiovert inverted fluorescence microscope (Carl Zeiss AG, Jena, Germany).
2.5. Western blotting
Western blotting was performed as previously described (Bajt et al., 2000). The primary antibodies used in this study were cyclin D1 (sc717), PCNA (sc7907) and Nrf-1 (sc33771) from Santa Cruz Biotechnology (Dallas, TX); phosphorylated-Ampk (#2535), Drp-1 (#8570) and beta-actin (#4970) from Cell Signaling Technology (Danvers, MA); the Pgc-1α (PA5-38021) antibody was purchased from Pierce (Rockford, IL). The Mitoprofile Total Oxphos Rodent WB antibody cocktail (Cat#ab110413) was from Abcam. Horseradish peroxidase-coupled anti-mouse or anti-rabbit IgG was used as the secondary antibody. Proteins were visualized by enhanced chemiluminescence (Amersham Biosciences Inc., Piscataway, NJ).
2.6. Statistics
All data were expressed as mean ± SEM. For two groups with normally distributed data, the Student’s t-test was used. For comparison of more than two groups, statistical significance was evaluated by one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls test for multiple comparisons. For non-normally distributed data, ANOVA was performed on ranks, followed by Dunn’s multiple comparisons. P<0.05 was considered significant.
3. RESULTS
3.1. Induction of mitochondrial biogenesis during APAP hepatotoxicity
To investigate the time course of mitochondrial biogenesis following APAP overdose, C57BL/6J mice were treated with 200 or 300 mg/kg APAP and sacrificed at multiple time points between 0 and 96h post-APAP. Two doses of APAP were chosen to provide a range of injury, since the activation of recovery processes such as mitochondrial biogenesis, are typically commensurate with initial liver injury. The 300mg/kg dose typically replicates an APAP overdose and produced significant liver injury, which peaked at 12h (Figure 1), and then began to normalize, as indicated by plasma ALT and centrilobular necrosis in H&E-stained liver sections (Fig. 1A–C). The 200mg/kg dose produced still substantial liver injury as seen by plasma ALT release and extent of centrilobular necrosis (Fig 1A&B). Consistent with mitochondrial dysfunction, activities of the ETC complexes I and IV were dose-dependently impaired by more than 50% (300 mg/kg dose) at 24h post-APAP compared to baseline for the 300mg/kg dose, slightly less impairment with the 200mg/kg dose (Fig. 2A,B). mtDNA levels in the liver also decreased during this time (Fig. 2C,D). Interestingly, however, both ETC activity and mtDNA increased after 24h, and were mostly restored by 72h (Fig. 2). It is interesting to note that despite the 2–3 fold difference in liver injury between the two doses (Figure 1 A & B) decrease in respiratory complex activity as well as rate of recovery is not very different between the doses (Figure 2 A & B). This suggests that the magnitude of injury at 200mg/kg APAP is sufficient to cause maximal induction of mitochondrial biogenesis, suggesting a threshold response for this phenomenon. All these data suggest that APAP overdose induces MB in mouse livers at later time points after injury.
Figure 1. Time course and dose response of liver injury after acetaminophen treatment.
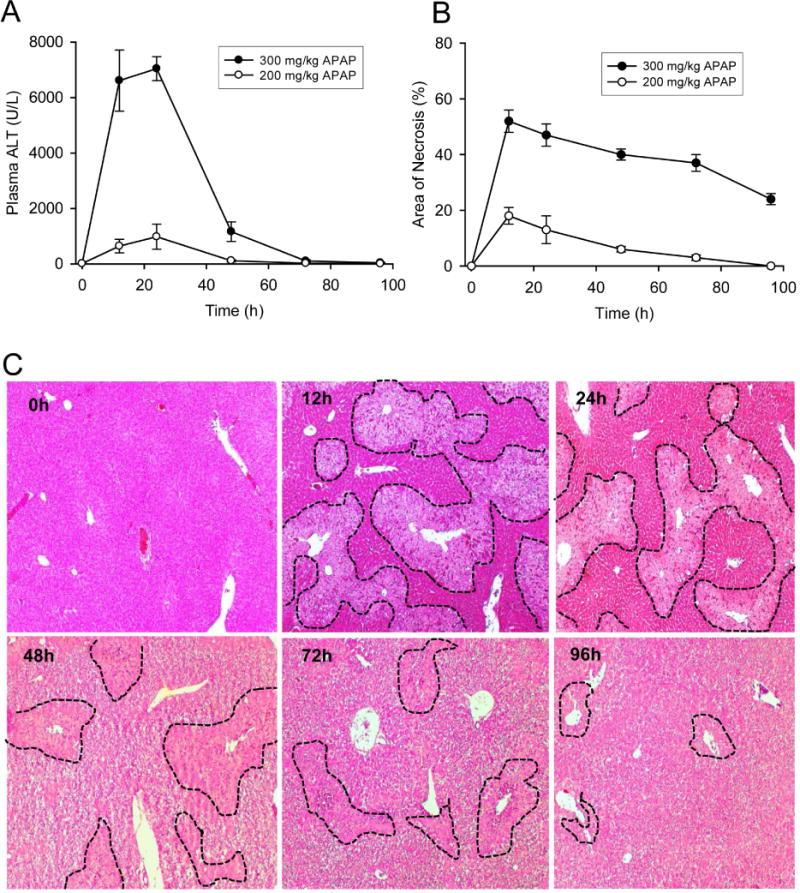
Mice were treated with 200 or 300 mg/kg acetaminophen (APAP) and sacrificed at various time points between 0 and 96h. (A) Plasma alanine aminotransferase (ALT) activity. (B) Areas of necrosis (%). (C) Representative H&E-stained liver sections (original magnification 50×) after 300 mg/kg APAP with necrotic areas outlined. Necrotic areas were identified by lack of nuclear staining or pyknotic nuclei and lines were marked along the boundaries. Data are expressed as mean ± SEM for n = 4–6 animals per group and time point.
Figure 2. Electron transport chain activity and mitochondrial DNA levels in the liver after acetaminophen treatment.
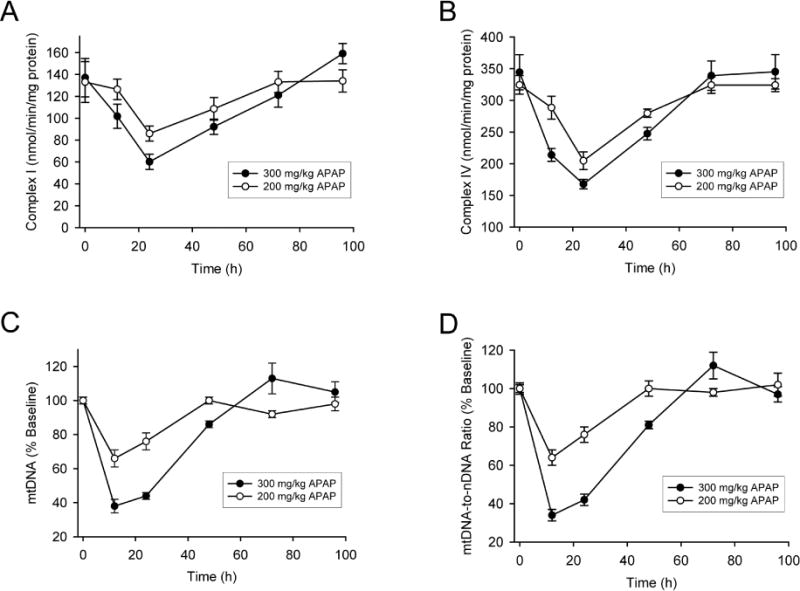
Mice were treated with 200 or 300 mg/kg acetaminophen (APAP) and sacrificed at various time points between 0 and 96h. After isolation of liver mitochondrial by subcellular fractionation, enzyme activity of mitochondrial complex I (A) and complex IV (B) were then measured over time. Mitochondrial DNA (mtDNA) levels in the liver were also measured over time and expressed as absolute content (C) or normalized to nuclear DNA (D). Data are expressed as mean ± SEM for n = 4–6 animals per group and time point.
3.2. Mitochondrial biogenesis occurs selectively in hepatocytes surrounding necrotic areas
To localize the area of MB in liver tissues after APAP overdose, mitochondria were stained with Tom20, the central component of the TOM (translocase of outer membrane) receptor complex. Consistent with the time course of ETC activity and mtDNA content, Tom20 staining began to increase in a subset of hepatocytes a few cells away from the necrotic area boundary by 24h post-APAP (Fig. 3D, arrows). By 48h after APAP, intense Tom20 staining was evident exclusively in hepatocytes surrounding necrotic areas (Fig. 3E&F). Interestingly, we also noticed a less-stained zonal area sandwiched between the necrotic area and healthy hepatocytes at 12h (Fig. 3C), in which the hepatocytes might be in severe stress, with excessive mitophagy taking place to remove damaged mitochondrial, thus decreasing the amount of mitochondria in those hepatocytes.
Figure 3. Immunofluorescent staining of Tom20 in the liver after APAP treatment.
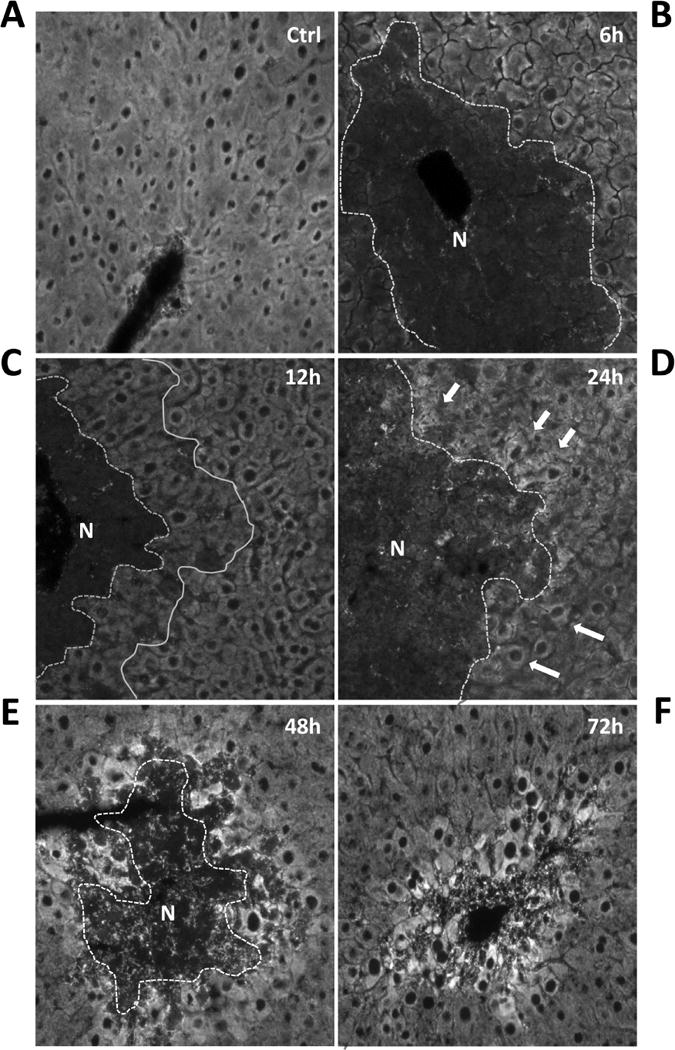
Mice were treated with 300 mg/kg acetaminophen (APAP) and sacrificed at various time points between 0 and 72h. Liver cryosections were stained with the mitochondrial protein Tom20 as described in the methods section and immunofluorescence images acquired on an inverted fluorescence microscope (Original magnification 200×). “N” indicates the necrotic area, which was identified based on absence of nuclear staining and cell contents, and is outlined with a dotted line.
3.3. Mitochondrial biogenesis signaling during APAP hepatotoxicity
To further investigate signaling events involved in MB after APAP-induced liver injury, we performed immunoblotting for several major MB signaling mediators. Pgc-1α is considered the master regulator of MB and activates the transcription factors that coordinate the expression of nuclear and mitochondrial genes necessary for MB, particularly for expression of ETC subunits (Baker et al., 2007; Scarpulla, 2008). Pgc-1α can itself be activated by Ampk. Phosphorylation of Ampk was observed as early as 6 h after APAP treatment and this was sustained until at least 48 h (Fig. 4A). Though expression of Pgc-1α was diminished during early liver injury, protein levels were restored at 24h and appeared to increase at 48h (Fig. 4A). Nrf-1, which is important for Pgc-1α function and helps to coordinate the expression of MB genes (Scarpulla, 2012), also showed increased expression (Fig. 4A). These changes were accompanied by a dramatic induction of the mitochondrial fission protein Drp-1 (Fig. 4A), suggesting not only growth of existing mitochondria but also division to form new healthy mitochondria. To examine areas of induction of biogenesis signaling within the liver, sections were stained for the central regulator Pgc-1α. We found that Pgc-1α was evenly expressed in hepatocytes in all areas of the liver in control mice (Fig. 4B). However, at 48h after APAP treatment, Pgc-1α expression was exclusively increased in hepatocytes surrounding necrotic areas (Fig. 4B). In addition, Pgc-1α staining colocalized with the nucleus in these areas (Fig. 4C). Together with the selective increase in staining of Tom20 in Figure 3, these data suggested that Pgc-1α translocated to the nucleus and activated MB in hepatocytes surrounding necrotic areas.
Figure 4. Activation or expression of mitochondrial biogenesis signaling mediators after acetaminophen treatment.
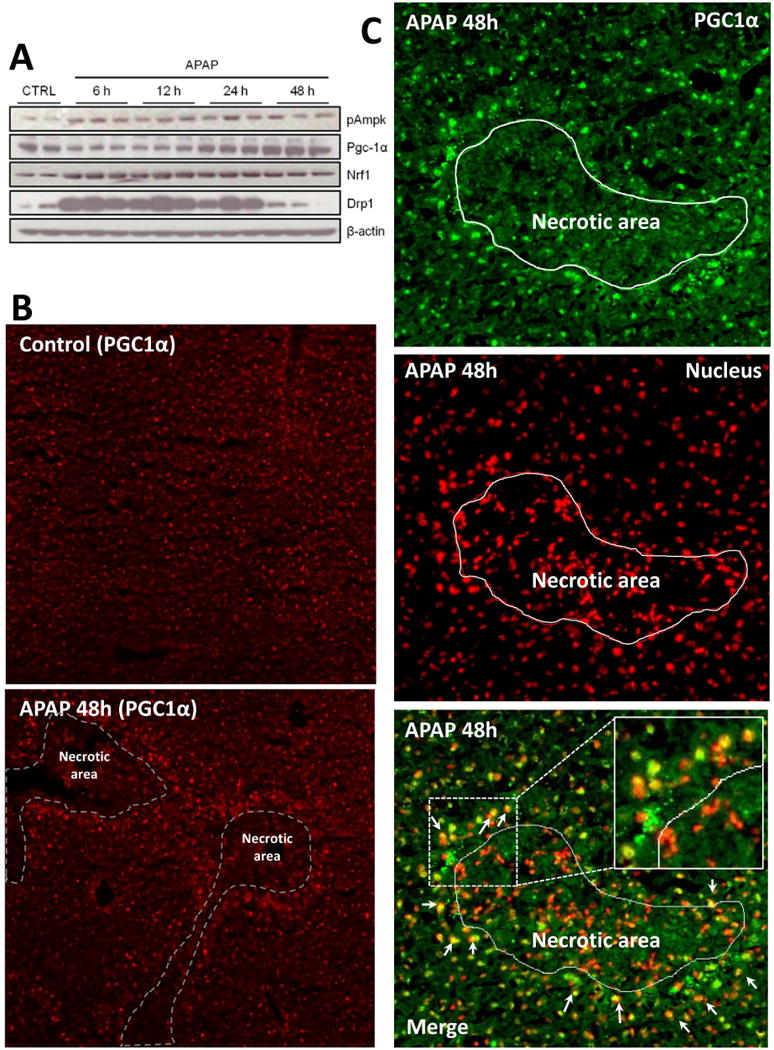
Mice were treated with 300 mg/kg acetaminophen (APAP) and sacrificed at various time points between 0 and 48h. (A) Liver homogenates were separated by SDS-PAGE and expression of several major mitochondrial biogenesis (MB) markers was assessed by immunoblotting. (B) Liver cryosections from controls and animals after 48h APAP were subjected to immunofluorescent staining of Pgc-1α (red). (C) For co-localization studies, in some liver sections 48h after APAP, Pgc-1α and nuclear DAPI signals were false colored green and red, respectively, to observe the merged signal as yellow (Original magnification 100×). Inset shows a magnification of the indicated area to show yellow merged signal in cell nuclei outside necrotic area. Necrotic areas were identified by pyknotic nuclei in DAPI images and marked as indicated.
3.4. Induction of mitochondrial biogenesis protects against APAP hepatotoxicity
Stimulation of MB through Pgc-1α signaling has been reported to rescue mitochondrial function and improve outcome in several pathologies, including cardiovascular diseases, nephrotoxicity, neurodegenerative diseases and also chronic liver injury (Finck and Kelly, 2007; St-Pierre et al., 2006; Funk et al., 2010; Rehman et al., 2013). We hypothesized that induction of MB would then protect against APAP-induced liver injury. To evaluate this, we treated mice with APAP, followed by SRT1720, an established inducer of MB signaling (Milne et al., 2007; Cameron et al., 2016). SRT1720 was given at 1.5h post-APAP to avoid any relevant effect on metabolic activation of APAP. In support of the late induction of MB (after 12h post-APAP), SRT1720 did not affect the early injury at 6h but significantly decreased the late injury at 24h post-APAP, as indicated by the 38% reduction in plasma ALT, decrease in the area of necrosis and of TUNEL-positive cells (Fig. 5A, B). This was also supported by the timely and advanced Tom20 staining in hepatocytes surrounding necrotic areas in the SRT1720-treated mice, while it was still mainly absent in the vehicle-treated mice (Fig. 5C). In addition, total GSH levels were significantly increased while the GSSG/GSH ratio was decreased (Fig. 5D, E), indicating a lower oxidant stress in these mice. SRT1720 treatment also blunted the decrease in mitochondrial respiratory complex proteins seen after APAP by 24 hours (Fig. 6A–C), which could facilitate faster recovery of mitochondrial function. Nrf 1 levels were not further increased by SRT1720 treatment (Fig. 6A, D); however, this may be due to their already significant induction after APAP, which is probably the maximum levels which could be attained under these conditions.
Figure 5. Induction of mitochondrial biogenesis protected against acetaminophen hepatotoxicity.
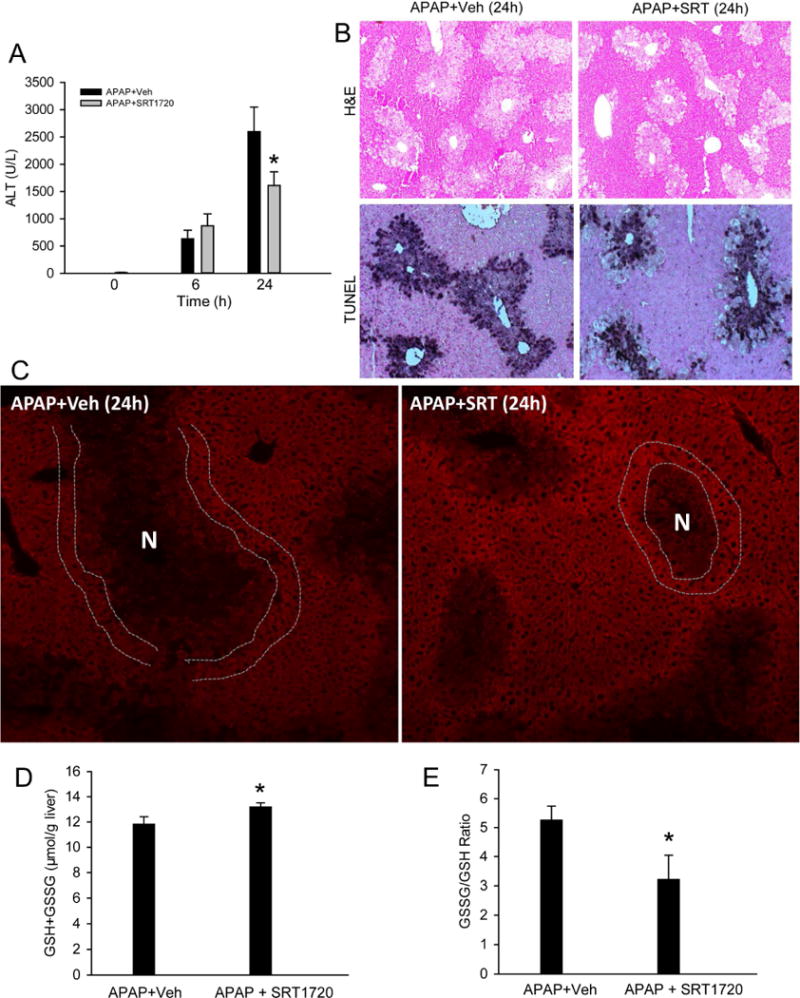
Mice were treated with 300 mg/kg acetaminophen (APAP) followed by 10 mg/kg SRT1720 or its vehicle 1.5h later, and sacrificed at 6h or 24h post-APAP. (A) Plasma alanine aminotransferase (ALT) activity. (B) H&E- or TUNEL-stained liver sections at 24h (Original magnification 50×). (C) Immunofluorescent staining of Tom20 (red) in liver tissue at 24h (Original magnification 100×). Necrotic areas were identified by lack of nuclear staining or pyknotic nuclei in corresponding DAPI images and marked as indicated. Double white lines highlight cells with enhanced Tom20 staining around the necrotic area. (D) Total GSH levels and (E) GSSG/GSH ratio at 24h. All measurements were carried out as described in the materials and methods section. Data are expressed as mean ± SEM for n = 4–6 animals per group and time point. *P< 0.05 vs. APAP+Veh.
Figure 6.
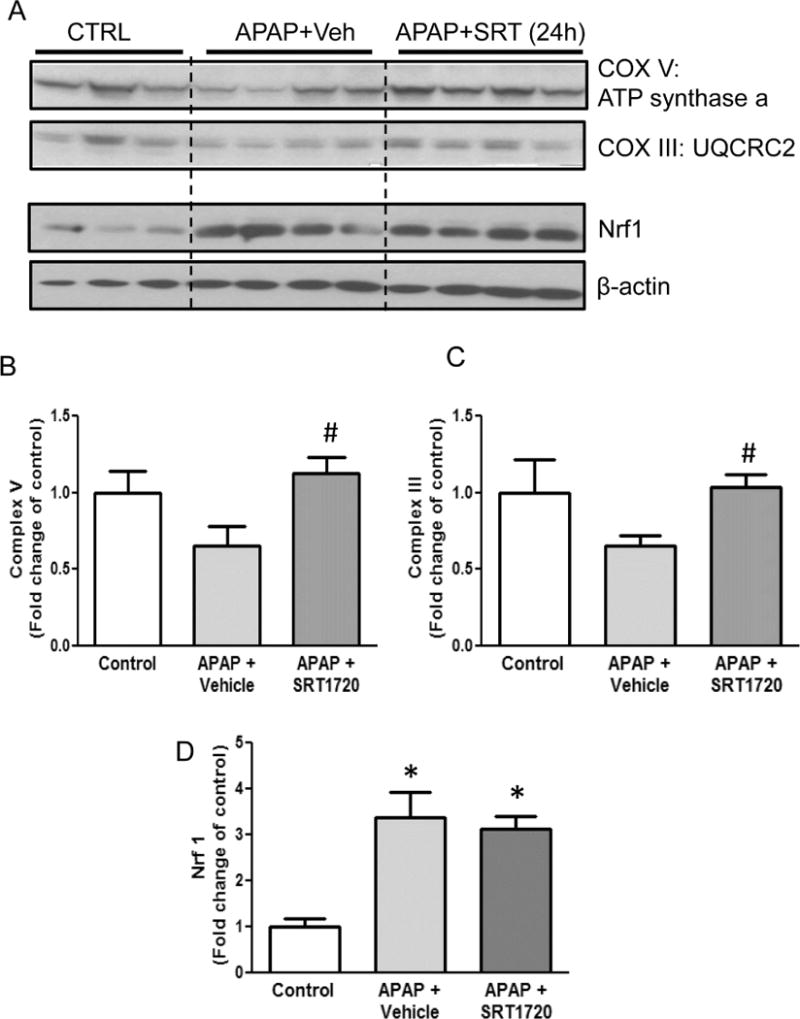
Induction of mitochondrial biogenesis blunted APAP-induced loss of mitochondrial protein. Mice were treated with 300 mg/kg acetaminophen (APAP) followed by 10 mg/kg SRT1720 or its vehicle 1.5h later, and sacrificed at 24h post-APAP. Following sacrifice, expression of mitochondrial complex III Core protein 2 (48 kD) and complex V alpha (55 kD) subunits, as well as Nrf 1 (69 kD) levels were analyzed in liver homogenates by western blotting (A), followed by densitometry for complex V (B), complex III (C) and Nrf 1 (D). Data represent means ± SEM for n = 4–6 animals per group. *P< 0.05 vs. Ctrl. #P < 0.05 vs. APAP + Veh.
3.5. Induction of mitochondrial biogenesis promotes liver regeneration after APAP hepatotoxicity
Promoting liver regeneration may be a promising approach to the treatment of APAP-induced liver injury because of the relatively late presentation of most patients who overdose on APAP (Larson, 2007). MB has been reported to coincide with the tissue injury process, and its induction has been shown to accelerate recovery and regeneration after injury in other organs (Wagatsuma et al., 2011 ; Yin et al., 2008; Rasbach et al., 2007; Tran et al., 2011). Since SRT1720 protected against late APAP-induced injury, we hypothesized that MB may be important for liver regeneration after APAP hepatotoxicity. Consistent with our hypothesis, liver regeneration, as indicated by PCNA expression, began at 24h post-APAP (Fig. 7A), the same time point at which we first observed an increase in mtDNA. PCNA levels continued to increase until at least 48h (Fig. 7A), similar to the time course for MB. Interestingly, the regenerating cells, as indicated by PCNA-positivity, were also observed selectively in hepatocytes surrounding necrotic areas (Fig. 7B). To further test this idea, we treated mice with APAP followed by SRT1720 at 12h and 36h post-APAP. The time points were chosen to avoid any effect on the early liver injury which may give the appearance of improved recovery.
Figure 7. Induction of mitochondrial biogenesis promoted liver regeneration after acetaminophen treatment.
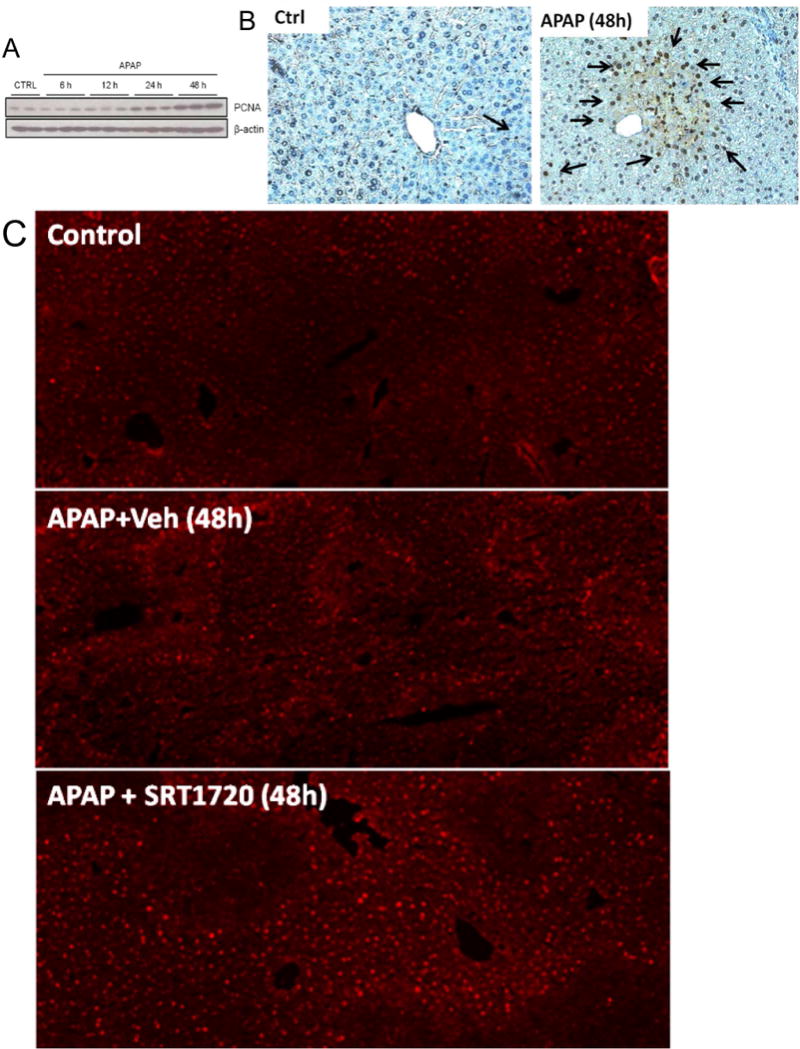
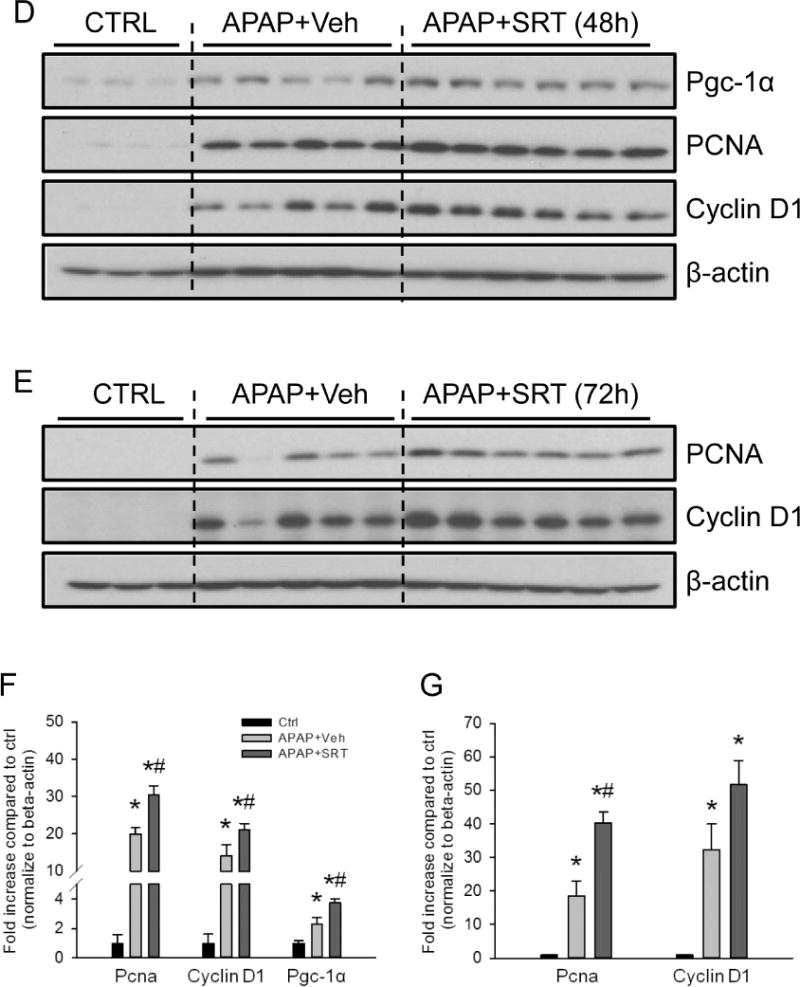
Mice were treated with 300 mg/kg acetaminophen (APAP). Some animals were treated with either SRT1720 (SRT) or its vehicle (Veh) as described in the materials and methods section, 12h and 36h post-APAP and sacrificed at 48h or 72h post-APAP. Following sacrifice, temporal expression of proliferating cell nuclear antigen (PCNA) was analyzed in liver homogenates by (A) western blotting, and (B) by immunohistochemistry on liver sections at 48h post-APAP (Original magnification 50×). Liver cryo-sections were also used for immunofluorescent staining of Pgc-1α in controls and animals treated with APAP ± SRT720 (Original magnification 100×). Pgc-1α (92 kD), PCNA (36 kD) and cyclin D1 (37 kD) expression were also examined by western blotting in livers from mice treated with SRT1720 (SRT) or its vehicle (Veh) at 48h (D, with densitometry in F) and 72h (E, with densitometry in G). Data are expressed as mean ± SEM for n = 3–5 animals per group and time point. *P< 0.05 vs. Ctrl. #P<0.05 vs. APAP + Veh.
Interestingly, we observed an increased expression of Pgc-1α at 48h (Fig. 7C) as well as both PCNA and cyclin D1 at both 48h and 72h (Fig. 7D, E) in the livers from the APAP + SRT1720-treated animals compared with APAP + vehicle-treated mice, and this was further confirmed by densitometric analysis of the blots (Fig. 7F, G). Overall, these data suggest that MB plays a role in liver regeneration and may be important for recovery after liver injury.
4. DISCUSSION
There is considerable evidence that mitochondrial damage plays a major role in the mechanisms of APAP-induced liver injury in both mice and humans (Placke et al., 1987; Meyers et al., 1988; Jaeschke, 1990; Kon et al., 2004; LoGuidice and Boelsterli, 2011; Ramachandran et al., 2011; McGill et al., 2012a, 2014). Recently it was also shown that the removal of these damaged mitochondria by autophagy (mitophagy) can limit APAP-induced cell death in vivo and in vitro (Ni et al., 2012). However, little is known about the mechanisms of recovery of mitochondrial mass and function or the role of MB in liver regeneration during injury resolution. Our data suggest that MB signaling begins early after APAP overdose, but that mitochondrial mass and function are not fully restored until late time points. In addition, MB occurs selectively in hepatocytes surrounding necrotic areas. Furthermore, induction of MB with a known MB-inducer protects against APAP hepatotoxicity and promotes liver regeneration.
4.1. Mitochondrial biogenesis mechanisms
Pgc-1α is generally thought to be the central mediator of MB signaling (Komen and Thorburn, 2014). Activation of Pgc-1α signaling can occur through both increased expression of the protein and post-translational modification, including phosphorylation by active Ampk and deacetylation by Sirt1 (Komen and Thorburn, 2014). However, activation of Pgc-1α is not enough to induce MB. As a co-activator, Pgc-1α must interact with other transcription factors to transactivate expression of MB genes. One such transcription factor is Nrf1 (Scarpulla, 2012). Upon activation, Pgc-1α and Nrf1 induce expression of nuclear genes that are important for MB, including mitochondrial transcription factor A (Tfam). Tfam is a key transcription factor for expression of genes encoded in mtDNA and is also involved in mtDNA replication (Scarpulla, 2012). The fact that we observed phosphorylation of Ampk and increased expression of Nrf1 and Pgc-1α in our samples suggests that all of the key signaling steps required for MB are activated during APAP hepatotoxicity. The reason for the delayed Pgc-1α induction in our experiments is not clear; however, it is important to remember that induced expression is only one of the ways in which Pgc-1α activity can be increased. It is possible that post-translational modifications of Pgc-1α could occur at earlier time points prior to induction of protein expression. Additional studies are needed to assess these mechanisms.
We have previously suggested that APAP overdose induces zonated histological changes in the centrilobular areas of the mouse liver; ranging from inner to outer areas includes necrosis (zone 1), mitochondrial spheroid formation (zone 2), autophagy (zone 3) and mitochondrial biogenesis (zone 4) (Ni et al., 2013). By staining mitochondria with mitochondrial protein Tom20, we noticed a minimally-stained zonal area sandwiched between the necrotic area and healthy hepatocytes at 12h post APAP (Fig. 3C). Although these cells were still surviving, their mitochondrial function may have been severely impaired due to the close proximity to the centrilobular areas, and excessive mitophagy could be taking place to remove damaged mitochondria, thus decreasing mitochondrial staining in those hepatocytes. Interestingly, we also observed that Tom20 staining started to increase from 24h post-APAP in a subset of cells, and the increased Tom20 staining was exclusively in hepatocytes surrounding necrotic areas by 48h (Fig. 3D, E, F). The fact that we also observed a selective increase of Pgc-1α at 48h in these areas (Fig. 4B) further demonstrates that activation of Pgc-1α signaling may promote MB in these hepatocytes. Coincidently, most of the cells surrounding necrotic areas at 48h were positive for the hepatocyte proliferation marker PCNA (Fig. 6B). This suggested that induction of MB may predispose these hepatocytes towards cell regeneration.
SRT1720 is a derivative of the natural product resveratrol that is thought to be a potent activator of Sirt1 and Pgc-1α (Milne et al., 2007; Cameron et al., 2016), although it has been suggested that it may act on Sirt1 indirectly or even have off-target effects that lead to MB (Pacholec et al., 2010; Huber et al., 2010; Komen and Thorburne, 2014). In fact, the effect of resveratrol and its derivatives also seems to involve activation of Ampk (Komen and Thorburne, 2014). In any case, SRT1720 is widely accepted as a useful inducer of Pgc-1α signaling and MB. Our data provide preliminary evidence that post-treatment with SRT1720 to induce Pgc-1α signaling protects against APAP-induced late liver injury and enhances liver regeneration after APAP overdose (Fig. 5, 6). It has previously been shown that knockdown of Pgc-1α in mice exacerbates APAP-induced liver injury (Ye et al., 2014). Although the authors of the latter study concluded that the protective effects of Pgc-1α are due to upregulation of antioxidant genes, it is possible that impairment of MB also played a role. Together, the data suggest that MB is important for recovery of liver function after APAP treatment.
4.2. Mitochondrial biogenesis and mitochondrial dynamics in APAP hepatotoxicity
Recent studies have just begun to explore mitochondrial dynamics (fission, fusion, and mitophagy) during APAP hepatotoxicity (Dara et al., 2015; Kang et al., 2016; Ni et al., 2012; 2013; Ramachandran et al., 2013). In particular, APAP-induced the translocation of Drp-1 to mitochondria in mouse livers and appeared to cause mitochondrial fission in primary mouse hepatocytes (Ramachandran et al., 2013). The Drp-1 activation was mediated by receptorinteracting protein (Rip) kinase signaling, as indicated by the fact that knockdown of Rip3 expression in the liver prevented Drp-1 translocation and reduced the liver injury (Ramachandran et al., 2013). Other studies have also shown protection against APAP toxicity by inhibition of Rip1 in vivo (Dara et al., 2015; Zhang et al., 2014; Takemoto et al., 2014) and in vitro (Ramachandran et al., 2013). Although these data suggest that mitochondrial fission is detrimental during APAP hepatotoxicity, it is known that mitochondrial fission is necessary for MB. Thus, while excessive fission may be harmful at early time points during APAP hepatotoxicity, it is likely to be beneficial at late time points. The latter idea is supported by our data and by the fact that increased MB has been observed by electron microscopy in healthy regenerating hepatocytes surrounding areas of necrosis after APAP treatment (Ni et al., 2013). There is also increasing evidence that autophagy is important for the removal of damaged mitochondria at early time points after APAP overdose (Ni et al., 2012; 2013). Autophagy is activated in hepatocytes after APAP treatment, and N-acetylcysteine (NAC) prevents both autophagy induction and injury suggesting that preventing mitochondrial damage by scavenging the reactive metabolite of APAP eliminates the need for autophagy (Ni et al., 2012).
4.3. Conclusions
We demonstrated that MB occurs selectively in hepatocytes surrounding necrotic areas in APAP hepatotoxicity and that its induction attenuates the injury and promotes liver regeneration. We found that, after an initial reduction due to mitochondrial damage and possibly mitophagy, mitochondrial mass and function increase and return to normal levels after APAP-induced liver injury. We also observed the activation of MB signaling in hepatocytes surrounding necrotic areas. Furthermore, post-treatment with the Sirt1 activator SRT1720 protects against APAP-induced liver injury and induced PCNA and cyclin D1 expression in the liver, suggesting that MB is important in both injury and liver regeneration after APAP toxicity. Induction of MB may be a promising therapeutic approach for patients presenting late after APAP overdose.
Highlights.
A delayed induction of mitochondrial biogenesis occurs after acetaminophen overdose
Mitochondrial biogenesis is prominent in hepatocytes surrounding areas of necrosis
Regions with enhanced mitochondrial biogenesis also upregulate liver regeneration
Pharmacological induction of mitochondrial biogenesis promotes liver regeneration
Acknowledgments
This work was supported in part by the U.S. National Institutes of Health grants R01 DK102142 (W.X.D/HJ.), P20 GM103549 (H.J.), P30 GM118247 (H.J.), and doctoral and postdoctoral fellowships (to M.R.M. and B.L.W.) from the National Institute of Environmental Health Sciences “Training Program in Environmental Toxicology” grant T32 ES007079. Additional support came from a Biomedical Research Training Program (BRTP) award from the University of Kansas Medical Center (to K.D.).
Abbreviations
- ALT
alanine aminotransferase
- APAP
acetaminophen
- Drp-1
dynamin-related protein-1
- ETC
electron transport chain
- GSH
reduced glutathione
- GSSG
glutathione disulfide
- JNK
c-jun N-terminal kinase
- MB
mitochondrial biogenesis
- MPT
mitochondrial permeability transition
- mtDNA
mitochondrial DNA
- NAPQI
N-acetyl-p-benzoquinone imine
- Nrf-1
nuclear respiratory factor-1
- PCNA
proliferating nuclear antigen
- Pgc-1α
PPARγ co-activator 1-α
- RIP3
receptor-interacting protein kinase-3
- Tom20
the central component of the TOM (translocase of outer membrane) receptor complex
- ROS
reactive oxygen species
- TUNEL
terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling assay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST DISCLOSURE
The authors declare no competing financial interest.
References
- Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-induced factor during acetaminophen-induced liver cell injury. Toxicol Sci. 2006;94:217–225. doi: 10.1093/toxsci/kfl077. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Lawson JA, Vonderfecht SL, Gujral JS, Jaeschke H. Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: evidence for a postmitochondrial processing of caspase-8. Toxicol Sci. 2000;58:109–117. doi: 10.1093/toxsci/58.1.109. [DOI] [PubMed] [Google Scholar]
- Baker MJ, Frazier AE, Gulbis JM, Ryan MT. Mitochondrial protein-import machinery: correlating structure with function. Trends Cell Biol. 2007;17:456–464. doi: 10.1016/j.tcb.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Cameron RB, Beeson CC, Schnellmann RG. Development of therapeutics that induce mitochondrial biogenesis for the treatment of acute and chronic degenerative diseases. J Med Chem. 2016;59:10411–10434. doi: 10.1021/acs.jmedchem.6b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006a;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006b;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2005;315:879–887. doi: 10.1124/jpet.105.088898. [DOI] [PubMed] [Google Scholar]
- Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, Kaplowitz N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology. 2015;62:1847–1857. doi: 10.1002/hep.27939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Farhood A, Jaeschke H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch Toxicol. 2017;91:761–773. doi: 10.1007/s00204-016-1692-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, McGill MR, Xie Y, Jaeschke H. Benzyl alcohol protects against acetaminophen hepatotoxicity by inhibiting cytochrome P450 enzymes but causes mitochondrial dysfunction and cell death at higher doses. Food Chem Toxicol. 2015;86:253261. doi: 10.1016/j.fct.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1 (Pgc-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–2548. doi: 10.1161/CIRCULATIONAHA.107.670588. [DOI] [PubMed] [Google Scholar]
- Funk JA, Odejinmi S, Schnellmann RG. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J Pharmacol Exp Ther. 2010;333:593–601. doi: 10.1124/jpet.109.161992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk JA, Schnellmann RG. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/Pgc-1α activation following ischemia-reperfusion injury. Toxicol Appl Pharmacol. 2013;273:345–354. doi: 10.1016/j.taap.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett SM, Whitaker RM, Beeson CC, Schnellmann RG. Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J Pharmacol Exp Ther. 2014;350:257–264. doi: 10.1124/jpet.114.214700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Ybanez MD, Johnson HS, McDonald JN, Mesropyan L, Sancheti H, Martin G, Martin A, Lim AM, Dara L, Cadenas E, Tsukamoto H, Kaplowitz N. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: biogenesis, remodeling, and functional alterations. J Biol Chem. 2012;287:42165–42179. doi: 10.1074/jbc.M112.377374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber JL, McBurney MW, Distefanos PS, McDonagh T. SIRT1-independent mechanisms of the putative sirtuin enzyme activators SRT1720 and SRT2183. Future Med Chem. 2010;2:1751–1759. doi: 10.4155/fmc.10.257. [DOI] [PubMed] [Google Scholar]
- Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther. 1990;255:935–941. [PubMed] [Google Scholar]
- Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev. 2012;44:88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James LP, McCullough SS, Lamps LW, Hinson JA. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol Sci. 2003;75:458–467. doi: 10.1093/toxsci/kfg181. [DOI] [PubMed] [Google Scholar]
- Jesinkey SR, Funk JA, Stallons LJ, Wills LP, Megyesi JK, Beeson CC, Schnellmann RG. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol. 2014;25:1157–1162. doi: 10.1681/ASN.2013090952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. doi: 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SW, Haydar G, Taniane C, Farrell G, Arias IM, Lippincott-Schwartz J, Fu D. AMPK Activation prevents and reverses drug-induced mitochondrial and hepatocyte injury by promoting mitochondrial fusion and function. PLoS One. 2016;11:e0165638. doi: 10.1371/journal.pone.0165638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khader A, Yang WL, Kuncewitch M, Jacob A, Prince JM, Asirvatham JR, Nicastro J, Coppa GF, Wang P. Sirtuin 1 activation stimulates mitochondrial biogenesis and attenuates renal injury after ischemia-reperfusion. Transplantation. 2014;98:148–156. doi: 10.1097/TP.0000000000000194. [DOI] [PubMed] [Google Scholar]
- Knight TR, Ho YS, Farhood A, Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 2002;303:468–475. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- Komen JC, Thorburn DR. Turn up the power – pharmacological activation of mitochondrial biogenesis in mouse models. Br J Pharmacol. 2014;171:1818–1836. doi: 10.1111/bph.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Sohal RS. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch Biochem Biophys. 2000;373:1622. doi: 10.1006/abbi.1999.1495. [DOI] [PubMed] [Google Scholar]
- Larosche I, Lettéron P, Fromenty B, Vadrot N, Abbey-Toby A, Feldmann G, Pessayre D, Mansouri A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J Pharmacol Exp Ther. 2007;321:526–535. doi: 10.1124/jpet.106.114546. [DOI] [PubMed] [Google Scholar]
- Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis. 2007;11:525–548. doi: 10.1016/j.cld.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Lee WM. Etiologies of acute liver failure. Semin Liver Dis. 2008;28:142–152. doi: 10.1055/s-2008-1073114. [DOI] [PubMed] [Google Scholar]
- LoGuidice A, Boelsterli UA. Acetaminophen overdose-induced liver injury in mice is mediated by peroxynitrite independently of the cyclophilin D-regulated permeability transition. Hepatology. 2011;54:969–978. doi: 10.1002/hep.24464. [DOI] [PubMed] [Google Scholar]
- McGill MR, Jaeschke H. A direct comparison of methods used to measure oxidized glutathione in biological samples: 2-vinylpyridine and N-ethylmaleimide. Toxicol Mech Methods. 2015;25:589–595. doi: 10.3109/15376516.2015.1094844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012a;122:1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Staggs VS, Sharpe MR, Lee WM, Jaeschke H, Acute Liver Failure Study Group Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology. 2014;60:13361345. doi: 10.1002/hep.27265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol. 2012b;264:387–394. doi: 10.1016/j.taap.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H. HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology. 2011;53:974–982. doi: 10.1002/hep.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers LL, Beierschmitt WP, Khairallah EA, Cohen SD. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol Appl Pharmacol. 1988;93:378–387. doi: 10.1016/0041-008x(88)90040-3. [DOI] [PubMed] [Google Scholar]
- Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Williams JA, Jaeschke H, Ding WX. Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biol. 2013;1:427–432. doi: 10.1016/j.redox.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, et al. SRT1720. SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placke ME, Ginsberg GL, Wyand DS, Cohen SD. Ultrastructural changes during acute acetaminophen-induced hepatotoxicity in the mouse: a time and dose study. Toxicol Pathol. 1987;15:431–438. doi: 10.1177/019262338701500407. [DOI] [PubMed] [Google Scholar]
- Ramachandran A, Lebofsky M, Baines CP, Lemasters JJ, Jaeschke H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic Res. 2011;45:156–164. doi: 10.3109/10715762.2010.520319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013;58:2099–2108. doi: 10.1002/hep.26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasbach KA, Schnellmann RG. Pgc-1α over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem Biophys Res Commun. 2007;355:734739. doi: 10.1016/j.bbrc.2007.02.023. [DOI] [PubMed] [Google Scholar]
- Rehman H, Krishnasamy Y, Haque K, Thurman RG, Lemasters JJ, Schnellmann RG, Zhong Z. Green tea polyphenols stimulate mitochondrial biogenesis and improve renal function after chronic cyclosporin a treatment in rats. PloS One. 2013;8:e65029. doi: 10.1371/journal.pone.0065029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 2010;51:246–254. doi: 10.1002/hep.23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Nucleus-encoded regulators of mitochondrial function: integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim Biophys Acta. 2012;1819:1088–1087. doi: 10.1016/j.bbagrm.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the Pgc-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Takemoto K, Hatano E, Iwaisako K, Takeiri M, Noma M, Ohmae S, Toriguchi K, Tanabe K, Tanaka H, Seo S, et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio. 2014;4:777–787. doi: 10.1016/j.fob.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirmenstein MA, Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J Biol Chem. 1989;264:9814–9819. [PubMed] [Google Scholar]
- Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, David S, Burstein D, Karumanchi SA, Stillman IE, Arany Z, Parikh SM. Pgc-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121:4003–4014. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagatsuma A, Kotake N, Yamada S. Muscle regeneration occurs to coincide with mitochondrial biogenesis. Mol Cell Biochem. 2011;349:139–147. doi: 10.1007/s11010-010-0668-2. [DOI] [PubMed] [Google Scholar]
- Whitaker RM, Wills LP, Stallons LJ, Schnellmann RG. cGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J Pharm Exp Ther. 2013;347:626–634. doi: 10.1124/jpet.113.208017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol. 2014;279:266–274. doi: 10.1016/j.taap.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Du K, Dorko K, Kumer SC, Schmitt TM, Ding WX, Jaeschke H. Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol Appl Pharmacol. 2015;289:213–222. doi: 10.1016/j.taap.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D, Wang Y, Li H, Jia W, Man K, Lo CM, Wang Y, Lam KS, Xu A. Fibroblast growth factor 21 protects against acetaminophen-induced hepatotoxicity by potentiating peroxisome proliferator-activated receptor coactivator protein-1α-mediated antioxidant capacity in mice. Hepatology. 2014;60:977–989. doi: 10.1002/hep.27060. [DOI] [PubMed] [Google Scholar]
- Yin W, Signore AP, Iwai M, Cao G, Gao Y, Chen J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke. 2008;39:3057–3063. doi: 10.1161/STROKEAHA.108.520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YF, He W, Zhang C, Liu XJ, Lu Y, Wang H, Zhang ZH, Chen X, Xu DX. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett. 2014;225:445–453. doi: 10.1016/j.toxlet.2014.01.005. [DOI] [PubMed] [Google Scholar]