

Abstract
Osteogenesis imperfecta (OI) is a collagen-related disorder associated to dominant, recessive or X-linked transmission, mainly caused by mutations in type I collagen genes or in genes involved in type I collagen metabolism.
Among the recessive forms, OI types VII, VIII, and IX are due to mutations in CRTAP, P3H1, and PPIB genes, respectively. They code for the three components of the endoplasmic reticulum complex that catalyzes 3-hydroxylation of type I collagen α1Pro986. Under-hydroxylation of this residue leads to collagen structural abnormalities and results in moderate to lethal OI phenotype, despite the exact molecular mechanisms are still not completely clear.
To shed light on these recessive forms, primary fibroblasts from OI patients with mutations in CRTAP (n = 3), P3H1 (n = 3), PPIB (n = 1) genes and from controls (n = 4) were investigated by a functional proteomic approach. Cytoskeleton and nucleoskeleton asset, protein fate, and metabolism were delineated as mainly affected. While western blot experiments confirmed altered expression of lamin A/C and cofilin-1, immunofluorescence analysis using antibody against lamin A/C and phalloidin showed an aberrant organization of nucleus and cytoskeleton.
This is the first report describing an altered organization of intracellular structural proteins in recessive OI and pointing them as possible novel target for OI treatment.
Significance
OI is a prototype for skeletal dysplasias. It is a highly heterogeneous collagen-related disorder with dominant, recessive and X-linked transmission. There is no definitive cure for this disease, thus a better understanding of the molecular basis of its pathophysiology is expected to contribute in identifying potential targets to develop new treatments.
Based on this concept, we performed a functional proteomic study to delineate affected molecular pathways in primary fibroblasts from recessive OI patients, carrying mutations in CRTAP (OI type VII), P3H1 (OI type VIII), and PPIB (OI type IX) genes. Our analyses demonstrated the occurrence of an altered cytoskeleton and, for the first time in OI, of nuclear lamina organization. Hence, cytoskeleton and nucleoskeleton components may be considered as novel drug targets for clinical management of the disease.
Finally, according to our analyses, OI emerged to share similar deregulated pathways and molecular aberrances, as previously described, with other rare disorders caused by different genetic defects.
Those aberrances may provide common pharmacological targets to support classical clinical approach in treating different diseases.
Keywords: Osteogenesis imperfecta, Cytoskeleton, Nuclear lamina, Cofilin-1, Lamin A/C, Protein network
Graphical abstract
Highlights
-
•
Functional proteomics was applied to characterize recessive OI type VII, VIII and IX.
-
•
Pathway analysis highlighted aberrances in ECM/cytoskeleton/nucleoskeleton signaling.
-
•
Affection of cytoskeleton and nucleoskeleton, with cofilin-1and lamin A/C, was proved.
-
•
Some of the aberrances we observed in OI are common to other rare disorders.
-
•
May those shared aberrances provide common pharmacological targets?
1. Introduction
Osteogenesis imperfecta (OI) is a clinically and molecularly highly heterogeneous group of inherited bone disorders whose phenotype ranges from barely detectable to perinatal lethal. It occurs worldwide without gender preference and with an estimated incidence of 1/10,000 live births. OI, also known as “brittle bone disease”, is prevalently characterized by low bone mass, skeletal deformities, bone fragility and growth deficiency [1].
The majority of OI cases (85–90%) have dominant transmission and are caused by structural or quantitative mutations in the COL1A1 and COL1A2 genes, which respectively encode α1(I) and α2(I) chains of type I collagen: the major structural protein of bone, skin and tendon extracellular matrix (ECM). For many years, OI has been considered a collagen disease and it was classified, according to clinical and radiological criteria, in four different types (I to IV) [2].
Since 2006, also autosomal recessive OI forms (10–15%) have been discovered. These are due to mutations in non-collagenous genes whose protein products are involved in type I collagen post-translational modification, folding, intracellular trafficking and extracellular matrix incorporation [3], [4], [5]. More recently, other mutations causative of recessive and X-linked OI have been described in genes that do not code for collagenous or collagen-handling proteins, but rather for proteins involved in osteoblast differentiation and bone mineralization [6], [7]. OI is now defined as a collagen-related disorder whose classification is based on the altered intracellular or extracellular metabolic pathways important for bone development [5], [7], [8].
Recessive OI types VII, VIII and IX, belonging to the group of OI forms caused by defects in collagen post-translational modification and folding [5], are linked to mutations in CRTAP (coding for cartilage-associated protein [CRTAP]), P3H1 (coding for prolyl 3-hydroxylase 1 [P3H1]), which was until 2014 reported as LEPRE1 (for more details see the “Osteogenesis imperfecta variant database”, https://oi.gene.le.ac.uk/), and PPIB (coding for cyclophilin B [CyPB]) genes, respectively [3], [4], [9]. CRTAP, P3H1 and CyPB associate in 1:1:1 ratio to form, in the rough endoplasmic reticulum (rER), a complex that is responsible for the 3-hydroxylation of α1Pro986 residue in unfolded α1 chains of collagen type I [10], [11].
Up to date, the biological significance of 3-hydroxylated α1Pro986 has not been completely understood: while on one hand it is thought to facilitate the formation or stabilization of collagen trimers [12], on the other one, it has been suggested to destabilize the collagen triple helix [13]. In addition, the complex was also described to function as a chaperone and to prevent type I collagen chains from forming premature aggregates [10].
CRTAP is a helper non-enzymatic protein whose presence is required for the in vivo functionality of the 3-hydroxylation complex [3]. Despite most of this protein is located in the ER, a small amount of CRTAP is secreted in ECM, where its biological function is still unknown [14]. P3H1 is the enzymatic component of the complex that catalyzes the prolyl 3-hydroxylation reaction [15]. An isoform of P3H1 is secreted as leprecan, a chondroitin sulfate proteoglycan of the ECM [16].
The last component of the complex, CyPB, is an ER peptidyl-proyil cis-trans isomerase (PPIase), from the immunophilin family [17], that is responsible for the cis-trans isomerization of peptidyl-prolyl bonds, a rate-limiting step for collagen folding [18]. CyPB also acts as a member of many foldase and chaperone complexes, including BIP, GRP94, PDI, calreticulin, and calnexin, some of which we previously found deregulated in the Brtl−/+ knock-in murine model of dominant OI [19].
Mutations in CRTAP or P3H1 genes cause absence or a severely reduction in α1Pro986 3-hydroxylation levels [20]. In particular, null mutations in either CRTAP or P3H1 result in the absence of both proteins from mutant cells, indicating mutual stabilization of CRTAP and P3H1 in the complex [21]. According to the delay in collagen folding, CRTAP and P3H1 deficiency indirectly causes the over-modification of type I collagen α chains and their consequent intracellular retention [20]. On the contrary, CyPB expression is independent from protein levels of the other two components of the complex. Nonetheless, in PPIB-null cells CRTAP and P3H1 abundance is moderately reduced, as a probable consequence of delay in complex stability [4], [21]. In the absence of CyPB, 3-hydroxylation of α1Pro986 is actually merely reduced [22] or normal [4], and collagen α chains over-modification is not detectable in all cases. These observations suggest that CyPB may not be the only PPIase contributing to proper collagen folding [4].
Although there are distinctive features, OI types VII, VIII and IX phenotypically overlap dominant types II and III. The clinical features of CRTAP and P3H1 mutant patients are consistently similar and include very low bone mineral density, rhizomelia, severe undertubulation of long bones, gracile ribs, neonatal fractures, growth deficiency, near normal head circumference, white or light blue sclerae. OI patients carrying CyPB defects lack rhizomelia [22].
Despite OI mainly affects bone tissue, skin outcomes have been proved both in patients and in murine models [23], [24], [25]. Skin quantitative magnetic resonance microimaging and histological analysis have recently been proposed as tools for less invasive OI diagnosis [26], [27]. Previously, we demonstrated consistent similarities in cellular pathway affection between bone and skin specimens in the Brtl−/+ mouse, model for dominant OI [19], [28]. Such alterations were also detected in primary fibroblasts of dominant OI patients [29].
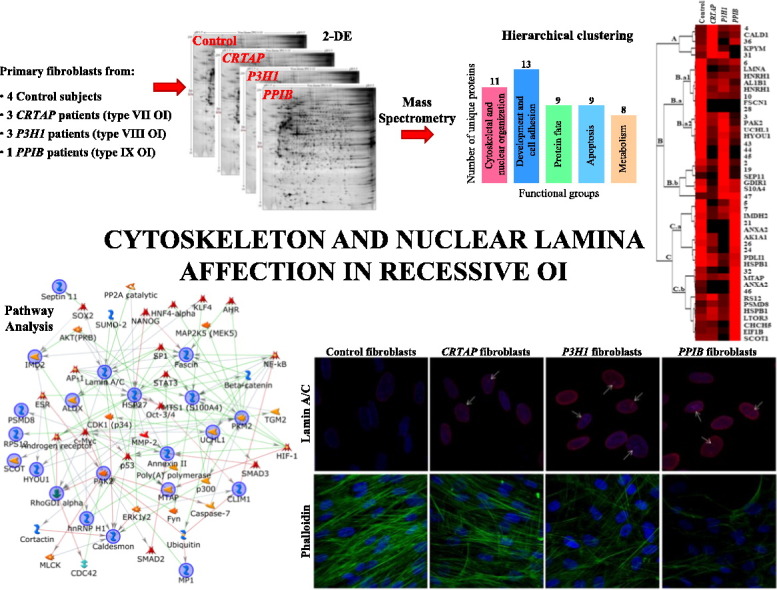
Using a functional proteomic approach, we attempted to gain new insights into biochemical mechanisms and molecular pathways affected by recessive OI type VII, VIII, or IX. Performing 2-DE and image analysis, significant differentially expressed proteins were detected among the three tested forms of recessive OI and among them and control fibroblasts. Mass spectrometry (MS) identified protein-differences resulted principally involved in cytoskeletal and nuclear organization and development, protein fate, and metabolism. To interpret the functional molecular meaning of such aberrances in protein abundance, we processed the identified proteins by the MetaCore pathway analysis software. The overwhelming majority of MS identified proteins were tightly functionally cross-linked thus stressing the high probative value of our data and suggesting their actual involvement in OI. Particularly, heat shock protein beta-1 (HSP27), pyruvate kinase PKM (PKM2), lamin A/C, serine/threonine-protein kinase PAK 2 (PAK2), and protein S100A4 (MTS1), which are known to be involved in cell proliferation, cytoskeleton and nuclear lamina organization, and in osteogenesis, resulted the main central hubs of the net.
Based on our previous findings concerning deregulation of cytoskeleton in dominant OI, we focused our attention on nuclear lamina and actin cytoskeleton organization and we proved their consistent disorganization in CRTAP, P3H1 and PPIB mutant fibroblasts.
According to our data, cytoskeleton and nucleoskeleton emerged as possible novel therapeutic targets in OI.
2. Material and methods
2.1. Human fibroblasts
Human fibroblasts from skin biopsy of four controls, three OI patients with mutations in the CRTAP gene, three OI patients with mutations in the P3H1 gene and one patient with a mutation in the PPIB gene were obtained after informed consent and used up to passage P10.
All patients were diagnosed with severe/lethal OI and their mutations were previously reported in [3], [30], [31], [32]. The detailed clinical outcome of one P3H1 mutant patient, which was compound heterozygous for two causative mutations previously identified in different OI cases [33], [34], will be described elsewhere.
Cells were grown at 37 °C in humidified atmosphere containing 5% CO2 and cultured in Dulbecco Modified Eagle's Medium (D-MEM) supplemented with 10% (v/v) Fetal Bovine Serum (FBS), 4 mM glutamine, 100 μg/ml penicillin and 0.1 mg/ml streptomycin. For each experiment 2.5 × 104 cells/cm2 were plated and harvested after 5 days.
2.2. Sample preparation for 2-DE analysis
Cells were lysed in the following 2-DE lysis buffer: 7 M urea, 2 M thiourea, 4% (w/v) CHAPS, 1% (w/v) dithioerythritol (DTE). Samples were sonicated on ice for 2 min, vortexed and then centrifuged at 15,000 × g for 5 min at 4 °C. The supernatants were recovered and, after estimation of protein concentration by Bradford's assay [35], they were aliquoted and stored at − 80 °C, until use.
2.3. 2-DE
2-DE was performed using the Immobiline-polyacrylamide system, as previously described [36], [37]. IEF was carried out on non-linear wide-range immobilized pH gradients (IPG) (pH 3–10; 18 cm long IPG strips; GE Healthcare, Uppsala, Sweden) and performed using the Ettan™ IPGphor system (GE Healthcare). Strips for analytical runs were rehydrated with 350 μl of lysis buffer containing bromophenol blue in trace and 0.2% (v/v) carrier ampholyte, for 12 h at room temperature. Sixty micrograms protein, diluted in 100 μl of lysis buffer with bromophenol blue in trace, was loaded at the cathodic end of the strip by dedicated cups, and the run was performed using the Manifold Tray for Ettan™ IPGphor™ (GE Healthcare). Proteins were focused, at 16 °C, according to the following voltage steps: 200 V for 8 h, from 200 V to 3500 V in 2 h, 3500 V for 2 h, from 3500 V to 5000 V in 2 h, 5000 V for 3 h, from 5000 V to 8000 V in 1 h, 8000 V for 3 h, from 8000 V to 10,000 V in 1 h, 10,000 V until a total of 93,000 Vh was reached.
MS-preparative strips were rehydrated with 350 μl of lysis buffer containing traces of bromophenol blue and 2% (v/v) carrier ampholyte, for 12 h at room temperature. Sample load, 600 μg protein per strip, was performed by cup-loading at the cathodic end of the strip, and IEF was achieved in the Manifold Tray for Ettan™ IPGphor™ (GE Healthcare) by applying the same voltage protocol of analytical runs and by reaching a total of 100.000 Vh.
After IEF, analytical and MS-preparative strips were equilibrated for 12 min in 6 M urea, 30% (v/v) glycerol, 2% (w/v) SDS, 0.05 M Tris–HCl pH 6.8, 2% (w/v) DTE; and for further 5 min in the same equilibration buffer except for 2% (w/v) DTE that was substituted with 2.5% (w/v) iodoacetamide. Bromophenol blue was used as tracking dye.
The second dimension was carried out, at 10 °C, on house-made 9–16% polyacrylamide linear gradient gels (18 cm × 20 cm × 1.5 mm) at 40 mA/gel constant current, until the dye front reached the bottom of the gel.
Analytical and MS-preparative gels were stained with ammoniacal silver nitrate [38], [39] and with a MS-compatible silver staining procedure [40], respectively. All gels were scanned using the ImageScanner III (GE Healthcare).
2.4. Image analysis and statistics
A 2-D spot map was obtained for each fibroblast sample, and the corresponding image analysis was carried out using the ImageMaster 2D Platinum v. 6.0 software (GE Healthcare). An intra-class analysis was performed by comparing the gels from the same condition. In order to detect any statistically significant quantitative and qualitative difference occurring among them, a differential inter-class analysis was done comparing, at the same time, all the 4 classes. Protein spots that showed at least ± 2 fold change in relative volume (%V) ratio, among the four conditions, at a 95% confidence level (1-way ANOVA: p ≤ 0.05), were considered significant differences. Pair-wise comparison of experimental groups was also performed for the significant differences that were selected according to above applied parameters by the Tukey's post-hoc multiple comparison test using the Excel Template inerSTAT-a v. 2.0.
The distribution of differentially expressed proteins among the six performed pair comparisons was visualized by a six-way Venn diagram that was obtained using the freely accessible jvenn software (http://bioinfo.genotoul.fr/jvenn/example.html) [41].
2.5. Protein identification
Differentially expressed spots were manually excised from MS-preparative gels, destained [42], and dehydrated in acetonitrile. After spot rehydration in trypsin solution (Sigma Aldrich, St. Louis, MO), in-gel protein digestion was performed by an overnight incubation at 37 °C. From each protein digest, 1.25 μl of tryptic-peptide solution was directly spotted onto the MALDI target and air-dried. Then, 0.75 μl of matrix solution (5 mg/ml alpha-cyano-4-hydroxycynnamic acid in 50% (v/v) acetonitrile and 0.5% (v/v) trifluoroacetic acid) was added to the dried samples and allowed to dry again. Mass spectra were acquired using an Ultraflex III MALDI-TOF/TOF mass spectrometer (Bruker Daltonics, Billerica, MA), equipped with a 200 Hz smartbeam™ I laser, in reflector positive mode with a laser frequency set to 100 Hz. Spectra were analyzed by Flex Analysis software v. 3.0. Mascot (Matrix Science Ltd., London, UK, http://www.matrixscience.com) PMF database searching was carried out in Swiss-Prot database (version 2016_11; 553,231 sequences; 197,953,409 residues) set for Homo sapiens. The experimental and theoretical peptide fingerprinting patterns Δmass was < 100 ppm, and trypsin was selected as the digestion enzyme with one allowed missed cleavage. Alkylation of cysteine by carbamidomethylation was assumed as fixed modification and oxidation of methionine as a possible one. The parameters used to accept identifications were the number of matched peptides, the extent of sequence coverage, and the probabilistic score.
Peptide digests that did not give unambiguous identifications were further analyzed performing MS/MS, using the same mass spectrometer. Two/three PMF peaks showing a high intensity were CID fragmented using Argon as collision gas, and MALDI-TOF/TOF tandem MS was performed in LIFT mode by software controlled data acquisition. Fragmented ions were analyzed using the Flex Analysis software v.3.0. The MS/MS database searching was carried out in the Swiss-Prot database (version 2016_11; 553,231 sequences; 197,953,409 residues) using the on-line available MASCOT MS/MS ion search software. The parameters applied for database searching were: Homo sapiens for taxonomy, trypsin specificity, one missed cleavage allowed, ± 100 ppm as peptide precursor mass tolerance, ± 0.6 Da allowed fragment mass tolerance, + 1 as peptide precursor charge state, carbamidomethylation of cysteine and oxidation of methionine as fixed and possible modifications, respectively. Mascot ion score, peptide coverage by “b” and “y” ions, and expected value were considered for protein identification.
2.6. Functional analysis
Differentially expressed proteins were subjected to unsupervised hierarchical clustering (HC): firstly, according to their expression levels in the 4 different tested conditions, and then, basing on their functional description by Gene Ontology (GO) terms. The former HC analysis was done using Cluster 3.0, an open source clustering software (http://bonsai.hgc.jp/~mdehoon/software/cluster/index.html) [43], that clusters proteins of interest according to their expression similarities. Relative volume (%V) mean values of significant differentially expressed proteins, that were computed in spot maps from control cells, CRTAP, P3H1, and PPIB fibroblasts, were normalized and loaded into Cluster 3.0 software. HC expression matrix was computed using Absolute value of centered correlation distance (Pearson) and complete linkage. The clustering results were indeed visualized by the open source tool Java TreeView [44].
Functional HC was performed using the DAVID (Database for Annotation, Visualization and Integrated Discovery) bioinformatics resource v. 6.7 (http://david.abcc.ncifcrf.gov) [45]. The list of UniProtKB accession numbers of unique MS identified differentially expressed proteins was processed through the DAVID functional annotation tool according to Gene Ontology (GO) terms. Protein functional classes were obtained by DAVID clustering and according to literature text mining. The main resulting significant classes were then visualized using an histogram from a five-way Venn diagram obtained by the freely accessible jvenn software [41].
To proceed in functional investigation, MS identified proteins were analyzed by pathway analysis using the MetaCore network building tool (GeneGo Inc., St. Joseph, MI), which operates basing on an in-house manually annotated database of protein interactions and metabolic reactions from scientific literature. Proteins of interest were imported into MetaCore and processed using the shortest path algorithm that allowed to introduce into the net a maximum of one protein, not included among the experimental proteins, to interconnect experimental factors. Hypothetical generated networks were indeed prioritized according to their statistical significance (p < 0.001) and graphically visualized by hubs (proteins) and edges (the relationships between proteins).
2.7. Western blotting
For 1-D western blots, protein extracts from all control and pathological fibroblasts were obtained in presence of the Laemmli buffer: 100 mM Tris–HCl pH 6.8, 2% (w/v) SDS, 20% (v/v) glycerol, 4% (v/v) β-mercaptoethanol, and heated at 95 °C for 5 min [46]. For each sample, 25 μg of proteins was subjected to SDS-PAGE and electroblotting, as previously described [19]. For 2-D western blots, whole fibroblast lysates from control subjects and from CRTAP, P3H1, and PPIB mutant patients were obtained using a conventional 2-DE lysis buffer: 7 M urea, 2 M thiourea, 4% (w/v) CHAPS, 1% (w/v) DTE. For each tested condition, 120 μg of proteins was subjected to IEF and SDS-PAGE according to the same procedure used for the 2-D analytical runs. 2-D electroblotting experimental procedure was the same described in [19].
The immunodetection was achieved applying the protocol previously reported [19] and using the following antibodies: rabbit polyclonal anti-lamin A/C (sc-20681) from Santa Cruz Biotechnology (San Jose, CA, USA), 1:1000 diluted; rabbit monoclonal anti-cofilin (D3F9) from Cell Signaling Technology (Danvers, MA, USA), 1:1000 diluted; and polyclonal goat anti-rabbit IgG secondary antibody (A0545) from Sigma-Aldrich (Saint Louis, MO, USA), 1:7000 diluted. Immunoreactive bands were visualized by chemiluminescence using ECL detection reagents (GE Healthcare). Chemiluminescent signals were captured by ChemiDoc™ imaging system (Bio-Rad). The obtained 1-D and 2-D western blot images were then analyzed with the ImageQuant v. 3.0 software (Molecular Dynamics World Headquarters, Sunnyvale, CA) and the ImageMaster 2D Platinum v. 6.0 software, respectively. For 1-D western blot, β-tubulin immunoblotting ensured equal loading of samples in each lane. Rabbit polyclonal anti-β-tubulin (sc-9104) from Santa Cruz Biotechnology (San Jose, CA, USA), 1:1000 diluted, was used.
2.8. Immunofluorescence analyses
For lamin staining, cells were fixed with cold 100% CH₃OH for 15 min, washed with PBS and blocked 1 h in 1% (w/v) BSA in PBS, 0.3% (v/v) Triton X-100. Then, cells were incubated with 1:200 lamin antibody (Santa Cruz Biotechnology) in 1.5% (w/v) BSA in PBS, 0.3% (v/v) Triton X-100 overnight at 4 °C. AlexaFluor 633 anti-rabbit IgG (Thermo Scientific, Waltham, MA, USA) was used as secondary antibody diluted 1:500 in PBS, 1% (w/v) BSA.
For phalloidin staining, cells were fixed in 3.7% (v/v) PFA in PBS for 10 min at room temperature and blocked in 1% (w/v) BSA, 0.05% (v/v) Triton X-100 in PBS for 30 min. Then, cells were incubated with Alexa 488-phalloidin (Molecular Probes, Invitrogen, Carlsbad, CA, USA) diluted 1:40 in 1% (w/v) BSA in PBS for 1 h at room temperature. Nuclei were counterstained by 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). Samples were examined by TCS SP2-Leica confocal microscope (Leica, Wetzlar, Germany).
3. Results and discussion
Fibroblasts are, along with osteoblasts and other derivatives of mesenchymal cells (e.g. chondroblasts, cementoblasts and odontoblasts), the major producers of collagen, and, as a consequence, they are among the main affected cells in collagen-related disorders. Since fibroblasts are the most abundant mesenchymal cells in the stroma of most tissues and organs, several tissues may be affected in OI.
Despite bone presents the main aberrances, OI extra-skeletal manifestations have been described. Beyond skin affections, blue sclerae and dentinogenesis imperfecta, also cardiovascular diseases, pulmonary defects, gastrointestinal manifestations, and skeletal muscle complications are reported in OI [8], [47], [48], [49].
According to these statements and to our previous analyses, we took advantage of fibroblast cells to investigate OI molecular basis. Protein 2-D profiles of skin primary fibroblasts obtained from control subjects and from patients carrying mutations in CRTAP (OI type VII), P3H1 (OI type VIII), and PPIB (OI type IX) genes were compared applying a functional proteomic approach, consisting of 2-DE, mass spectrometry and dedicated bioinformatics tools for functional analysis.
We investigated fibroblast cells from 4 controls, 3 patients affected by OI type VII, 3 OI type VIII patients, and 1 OI type IX patient. OI type IX is quite rare, thus we were able to obtain fibroblasts from only one case [5], [31]. Fibroblasts from this unique type IX OI proband were anyway run in triplicate.
Representative silver stained electropherograms of control, CRTAP, P3H1, and PPIB fibroblast samples are reported in Fig. 1. Approximately 2000 spots were well detected in each gel from control and disease conditions with consistent experimental reproducibility. According to applied statistics, as described in Material and methods, 47 protein spots were found to differ in expression when control and pathological groups were compared. Supplementary Table S1 and Venn diagram, Fig. S1 (Supplementary material), detailed the six different performed pair comparisons and the corresponding detected (Supp. Venn diagram) and identified differences (Suppl. Table S1). Overall, 27 differentially expressed protein spots, which consists in 24 unique proteins, were successfully identified by MS, as summarized in Supplementary Table S1 and specified in Table 1.
Fig. 1.

Silver stained electropherograms of fibroblasts from control (A), CRTAP (B), P3H1 (C), and PPIB (D) subjects. Numbers and circles visualize quantitative and/or qualitative significant differences detected among the analyzed conditions.
Table 1.
Differentially expressed protein spots identified by MS.
| Spot n. | Protein description | MetaCore namea | UniProt ID/nameb | Mascot search results |
Mean %V ± SD × 10− 4c |
1-Way ANOVA p-value | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | N. of matched peptides | Sequence coverage (%) | Control | CRTAP | P3H1 | PPIB | |||||
| Cytoskeletal and nuclear organization, and development | |||||||||||
| 8 | Caldesmon | Caldesmon | Q05682/CALD1 | 199 | 25/46 | 29 | 214 ± 203§d | 664 ± 200§¥# | 120 ± 160¥ | 241 ± 49# | 0.012326 |
| 9 | Prelamin-A/C | Lamin A/C | P02545/LMNA | 136 | 15/25 | 22 | 119 ± 68$ | – | 20 ± 20$ | 35 ± 10 | 0.012354 |
| 12 | Serine/threonine-protein kinase PAK 2 | PAK2 | Q13177/PAK2 | 143 | 12/17 | 33 | 424 ± 97⁎ | 228 ± 68 | 426 ± 127& | 113 ± 59⁎& | 0.007873 |
| 16 | Fascin | Fascin | Q16658/FSCN1 | 214 | 21/48 | 47 | 380 ± 161 | – | – | – | 0.00067 |
| 20 | Septin-11 | Septin 11 | Q9NVA2/SEP11 | 62 | 5/7 | 10 | 71 ± 69$ | – | 462 ± 218$ | – | 0.002164 |
| 25 | Annexin A2 | Annexin II | P07355/ANXA2 | 216 | 18/24 | 43 | 101 ± 145⁎ | – | – | 402 ± 69⁎ | 0.002210 |
| 27 | Annexin A2 | Annexin II | P07355/ANXA2 | 227 | 20/30 | 48 | 63 ± 18 | – | – | 86 ± 41 | 0.000326 |
| 34 | Rho GDP-dissociation inhibitor 1 | RhoGDI alpha | P52565/GDIR1 | 169 | 15/33 | 50 | 1437 ± 443§ | 216 ± 58§#¥ | 1822 ± 393¥ | 1345 ± 94# | 0.000456 |
| 41 | Ragulator complex protein LAMTOR3 | MP1 | Q9UHA4/LTOR3 | 152 | 7/8 | 66 | 52 ± 25 | 66 ± 18 | 41 ± 7& | 101 ± 5& | 0.029419 |
| 42 | Protein S100-A4 | MTS1 (S100A4) | P26447/S10A4 | 115 | 7/11 | 45 | 3909 ± 572§ | 1234 ± 646§¥ | 3774 ± 1387¥ | 3155 ± 367 | 0.005465 |
| Protein fate (synthesis, folding, proteolysis) | |||||||||||
| 1 | Hypoxia up-regulated protein 1 | HYOU1 | Q9Y4L1/HYOU1 | 255 | 35/63 | 42 | 561 ± 110⁎§ | 187 ± 162§ | 419 ± 71 | 186 ± 151⁎ | 0.009483 |
| 17 | Heterogeneous nuclear ribonucleoprotein H | hnRNP H1 | P31943/HNRH1 | 156 | 13/21 | 37 | 428 ± 92§ | 1155 ± 96§¥# | 175 ± 168¥ | 176 ± 110# | 0.020274 |
| 18 | Heterogeneous nuclear ribonucleoprotein H | hnRNP H1 | P31943/HNRH1 | 198 | 22/55 | 59 | 394 ± 154§ | 1424 ± 75§¥# | 238 ± 31¥ | 141 ± 72# | 0.029005 |
| 23 | PDZ and LIM domain protein 1 | CLIM1 | O00151/PDLI1 | 197 | 17/31 | 54 | 412 ± 115$ | 313 ± 87 | 148 ± 29$ | 325 ± 87 | 0.025707 |
| 30 | 26S proteasome non-ATPase regulatory subunit 8 | PSMD8 | P48556/PSMD8 | 104 | 7/9 | 16 | 71 ± 54⁎ | 133 ± 79 | 79 ± 117 | 332 ± 134⁎ | 0.039994 |
| 33 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 | UCHL1 | P09936/UCHL1 | 173 | 11/20 | 65 | 237 ± 75§ | 95 ± 16§ | 230 ± 92 | 116 ± 18 | 0.02802 |
| 35 | Heat shock protein beta-1 | HSP27 | P04792/HSPB1 | 131 | 9/22 | 57 | 192 ± 198⁎ | 361 ± 15 | 185 ± 20& | 557 ± 97&⁎ | 0.020487 |
| 37 | Heat shock protein beta-1 | HSP27 | P04792/HSPB1 | 82 | 5/8 | 28 | 89 ± 17$ | 77 ± 23¥ | 29 ± 8$¥& | 81 ± 10& | 0.006612 |
| LATQSNEITIPVTFESR | |||||||||||
| 38 | 40S ribosomal protein S12 | RPS12 | P25398/RS12 | 96 | 6/11 | 43 | 48 ± 16 | 70 ± 19¥ | 27 ± 6&¥ | 79 ± 5& | 0.010931 |
| 40 | Eukaryotic translation initiation factor 1b | – | O60739/EIF1B | 83 | 5/15 | 49 | 59 ± 42⁎ | 68 ± 64# | – | 190 ± 9⁎# | 0.017554 |
| Metabolism | |||||||||||
| 11 | Pyruvate kinase PKM | PKM2 | P14618/KPYM | 107 | 9/14 | 19 | 487 ± 120§ | 1175 ± 319§¥ | 227 ± 207¥ | – | 0.000451 |
| 13 | Inosine-5′-monophosphate dehydrogenase 2 | IMD2 | P12268/IMDH2 | 157 | 13/19 | 27 | 797 ± 97 | 328 ± 301# | 534 ± 306 | 1027 ± 19# | 0.025709 |
| GKLPIVNEDDELVAIIAR | |||||||||||
| 14 | Succinyl-CoA:3-ketoacid coenzyme A transferase 1 | SCOT | P55809/SCOT1 | 128 | 11/19 | 24 | 154 ± 112 | 151 ± 178 | – | 400 ± 117 | 0.047791 |
| 15 | Aldehyde dehydrogenase X, mitochondrial | – | P30837/AL1B1 | 166 | 16/26 | 30 | 326 ± 61⁎§$ | 143 ± 97§ | 172 ± 37$ | 63 ± 4⁎ | 0.00606 |
| 22 | Alcohol dehydrogenase [NADP(+)] | ALDX | P14550/AK1A1 | 195 | 16/30 | 52 | 382 ± 81 | 210 ± 72 | – | 383 ± 76 | 0.000186 |
| 29 | S-methyl-5′-thioadenosine phosphorylase | MTAP | Q13126/MTAP | 163 | 15/25 | 48 | 159 ± 41 | 106 ± 39# | 138 ± 49& | 264 ± 40#& | 0.013231 |
| 39 | Coiled-coil-helix-coiled-coil-helix domain-containing protein 5 | – | Q9BSY4/CHCH5 | 96 | 5/9 | 65 | 32 ± 23⁎ | 27 ± 23# | – | 107 ± 53⁎# | 0.009274 |
Protein name in MetaCore network. Proteins excluded from the net according to the set parameters (–).
UniProt ID and entry name.
Each value represents the mean ± SD of individually computed %V in spot maps from control fibroblasts, and from fibroblasts obtained from OI patients carrying mutations in CRTAP, P3H1, or PPIB genes.
Pair-wise comparison was performed using the Tukey's post hoc test (p ≤ 0.05). Only proteins showing both statistical reliability and, at least, 2 fold change in expression are listed as significant differences: Control vs CRTAP (§); control vs P3H1 ($); control vs PPIB (*); CRTAP vs P3H1 (¥); CRTAP vs PPIB (#); P3H1 vs PPIB (&).
3.1. Clusterization of differentially expressed proteins: hierarchical clustering
Significant differentially expressed protein spots, independently on their successful identification by MS, were clustered by unsupervised hierarchical clustering (HC) based on their expression profiles in the 4 different tested conditions. The normalized mean-relative-volume (%V) values of all the proteins of interest were plotted against every analyzed sample set in order to highlight differences and similarities occurring among such protein spots, in relation to each tested condition (see Material and methods). HC results were visualized by expression matrix and dendrogram, and three main clusters were delineated: clusters A, B, and C, as shown in Fig. 2.
Fig. 2.

Unsupervised Hierarchical Clustering (HC) analysis. Normalized %V mean values of significant differences, as computed in spot maps from control, CRTAP, P3H1, and PPIB cells, are visualized by expression matrix, where each represented %V value is proportional to color intensity, from black = zero expression to red = maximum positive expression. Spot identity (protein name, as listed in Table 1, or number, for MS not identified spots) is annotated on the right of the expression matrix, while dendrogram of complete linkage clustering, based on centered correlation distance, is reported on the left. Main clusters are named from top to bottom and the corresponding expression profiles of their included proteins are shown on the right.
Cluster A is the smaller obtained group containing only 5 significant protein differences whose expression profiles are similar in control, P3H1, and PPIB and different in CRTAP cells, where such proteins result up-regulated.
Cluster B includes 21 proteins that were further sub-clustered into 2 sub-groups: B.a and B.b. Mainly according to protein differential abundances in P3H1 fibroblasts, the sub-cluster B.a was additionally divided into B.a1 and B.a2. The 8 proteins from B.a1 show expression profiles similar in the three pathological conditions, with an evident down-regulation in respect to the control. B.a2 consists of 7 proteins under-expressed in CRTAP and PPIB patients. Sub-cluster B.b comprises 6 differences whose expression profiles are similar in control, CRTAP, and PPIB but different in P3H1 cells, where the highest expression was detected.
Also cluster C was divided into sub-clusters: C.a and C.b. While the 10 C.a proteins show down-regulation in CRTAP and P3H1 patients, the 11 C.b differences display approximately similar expression in control, CRTAP, and P3H1 and an up-regulation in PPIB fibroblasts.
As clearly represented by the Fig. 2 expression matrix, HC highlighted a general down-regulation trend of protein spot differences in CRTAP mutants. Despite each pathological investigated condition showed a peculiar expression profile of protein differences, CRTAP and P3H1 cells presented more similarities, well resembling CRTAP/P3H1 known mutual stabilization in rER [21].
The MS identified unique proteins were also classified according to DAVID HC (as detailed in Material and methods) and based on their main biological functions, as supported by scientific literature, into three main functional groups: i) cytoskeletal and nuclear organization and development, ii) protein fate (synthesis, folding, and proteolysis), and iii) metabolism, as indicated in Table 1. Those functional groups were also delineated in our previous work on dominant OI to cluster proteins changing in expression among Brtl+/− mice and wild-type littermates [19].
Reported findings interestingly indicate that, despite the majority of deregulated proteins identified in dominant and recessive OI forms are different, specific biological processes and molecular functions are characteristically affected in OI, independently of Mendelian mode of transmission and of mutated gene.
3.2. Clusterization of differentially expressed proteins: functional groups
Identified differentially expressed proteins were clustered based on all their characterizing GO terms. Besides “biological process”, also “molecular function” and “cellular component” GO terms, along with literature text mining, were taken into account and 5 functional groups were delineated: i) cytoskeletal and nuclear organization, ii) development and cell adhesion, iii) protein fate (synthesis, folding, proteolysis), iv) apoptosis, and v) metabolism, as shown in Fig. 3. i) and ii) were the main represented functional groups including 11 (Fig. 3: dark pink bar) and 13 (Fig. 3: blue bar) proteins, respectively. iii) grouped 9 proteins (Fig. 3: green bar), as well as iv) (Fig. 3: light blue bar), and v) comprised 8 proteins (Fig. 3: orange bar). More than half of the identified unique proteins resulted involved in multiple biological functions, cytoskeleton and nucleoskeleton, and development and cell adhesion appeared as the most affected structures and processes in recessive OI.
Fig. 3.

Histogram from a five-way Venn diagram of functional clustering of the MS identified unique proteins. The histogram displays the number of unique proteins, listed below each bar, in every delineated functional cluster, according to all the GO terms characterizing differentially expressed proteins (i.e. biological process, molecular function, and cellular component).
This functional clustering, despite the small number of proteins found differentially expressed, may account for relevant pathways altered in OI. Indeed, the few aberrantly expressed multifunctional factors that we identified may lead to a simultaneous affection of several pathways determining loss of cell and tissue functionality and homeostasis. Moreover, those “moonlight” proteins act key roles in pathways that are known, or at least hypothesized, to be relevant in OI pathogenesis, as later discussed.
3.3. Pathway analysis
In order to functionally correlate the identified proteins, MetaCore pathway analysis was performed applying the shortest path algorithm [50]. Only proteins closely related in function were indeed included into the generated networks. About 90% of MS identified proteins clustered into the most relevant of the generated nets (Fig. 4: blue circles; Table 1), thus demonstrating the significance of produced data and, particularly, of the biological meaning of their altered expression.
Fig. 4.

MetaCore protein network. MetaCore analysis was used to map identified proteins (blue circles) on existing mammalian pathways and networks, according to known protein-protein interactions and other features established in the literature. Network proteins are visualized by specific symbols, which define the functional nature of the protein (network caption). The edges indicate the relationships existing between individual proteins, and the arrowheads represent the direction of the interaction. The following line colors designate the nature of the interaction: red = negative effect; green = positive effect; gray = unspecified effect. Based on selected parameters, only eukaryotic translation initiation factor 1b (EIF1B), coiled-coil-helix-coiled-coil-helix domain-containing protein 5 (CHCHD5), and aldehyde dehydrogenase X, mitochondrial (AL1B1) were excluded from the net.
Studying the network, identified multifunctional proteins that here represent the main central hubs, i.e. heat shock protein beta-1 (HSP27), pyruvate kinase PKM (PKM2), lamin A/C, serine/threonine-protein kinase PAK 2 (PAK2), and protein S100A4 (MTS1), focused the attention mainly on cytoskeleton and nucleoskeleton organization. A higher level of complexity in interpreting this result was related to the MetaCore-added and not-experimentally-detected factors. Their tight crosstalk with experimental hubs delineated complex pathways that unambiguously correlate cytoskeleton and nucleoskeleton to development, differentiation and functionality of organs and apparatus usually affected in OI. Actually, several of those not-experimental-detected factors directly control or are controlled by the above reported central hubs and they have key functions in regulating signal transduction and transcription during development, cell proliferation, differentiation, survival, and apoptosis. Moreover, most of them are transcription factors whose detection is usually difficult in 2-D gels since their levels relay under the limits of detection by conventional staining protocols.
More in detail, besides the almost pleiotropic proteins c-Myc, p53, Oct-3/4, all described in Bianchi et al. [19], Nanog, and Sox2, whose altered expression affect development in several tissues, the transcriptional factors NF-kB and KLF4 operate in signaling pathways whose proper and balanced regulation is essential for normal bone and tooth development and remodeling [51], [52], [53], [54], [55], [56]. Of relevance, altered NF-kB function has been described in genetic disorders, such as ectodermal dysplasias (ED), that impair, as well as OI, skin, eye, inner ear, limbs and bone [57]. NF-kB signaling is known to depend or to intersect signal transduction of the two fundamental bone regulators: TGF-β and BMPs, being it activated by TGF-β/BMP non-canonical pathway or interfering with Smad signaling, respectively [58], [59], [60]. Moreover, Smad2/3, which are two other transcription factors added by the software, were previously proved, along with TGF-β, as altered in dominant OI [29], [61].
Smad and NF-kB interestingly emerged as critical factors for osteoblast differentiation and for bone mass regulation by intersecting Wnt/β-catenin pathway [62], [63]. Once again, β-catenin is a MetaCore-added factor interconnected with the main central hubs of the net. Wnt/β-catenin and TGF-β/BMP signaling are known to regulate each other and to cooperate in controlling fibroblast and osteoblast collagen production [64], [65], [66], [67]. Dysfunctions of such pathways are related to fibrosis and aberrances in bone formation and wound healing [66], [68], [69], [70], with this latter being one of the main cause of skeletal deformity in OI. Interestingly, mutations in WNT1, which is a regulator of canonical Wnt-signaling [71], [72], are causative of recessive OI [73], [74].
TGF-β/BMP non-canonical pathway also controls another MetaCore-added protein: AP-1, which is known to operate in bone disruption/formation [75] and that is linked to all the central hubs of the generated net, except for PKM2.
Still in line with TGF-β/BMP and Wnt/β-catenin but also with integrin and cadherin cascades, which are all fundamental in skeletal and skin development and that are reported to crosstalk each other [76], the software generated the net including also AKT, ERK1/2 and p300. They properly fit into the MetaCore-delineated signaling interconnection and into the induced responses by exerting key roles in the above listed pathways [64], [65], [76], [77].
Finally, the not-experimentally detected HIF-1 protein directly controls the experimentally detected fascin, S100A4, and annexin II, all factors operating in cytoskeleton dynamics and bone metabolism. HIF-1 is necessary for embryogenesis, physiological development of several organs (e.g. bone, lung and heart), and tissue remodeling/healing. Defects in hypoxia/HIF-1 signaling lead to anomalies in skeletal, cardio-vascular and respiratory systems [78], [79], [80]. Accordingly, OI probands develop, in addition to bone aberrances, cardio-vascular defects and pulmonary affections [8], [47], [81].
3.4. MetaCore central hubs: nucleoskeleton and cytoskeleton affection
Despite several of the main protein-actors on the stage of OI morbidity differ between dominant and recessive forms, shared clinical features in different OI types suggest common molecular pathomechanisms at the bases of this disorder. Our functional proteomic study has evidenced, once more, that cytoskeletal anomalies characterized OI cells and that they may contribute to OI pathogenesis. Here we also proved, for the first time, the occurrence of altered nucleoskeleton in recessive OI by having detected aberrances in lamin A/C expression and organization in the nucleus. Interestingly lamin A/C was included in the above described cluster B.a1, where proteins sharing similar expression in all patients were grouped.
Lamins are nuclear type V intermediate filaments [82]. They are the principal component of the nucleoskeleton and their main function is to provide shape, stiffness and architecture to the nucleus. Lamins are deeply involved in chromatin organization and gene expression, as well as in DNA replication and repair, thus concurring to cell signaling, proliferation, migration and differentiation [83], [84], [85]. Lamin A and C are A-type lamins derived by alternative splicing of the LMNA gene. Both these isoforms are equally present in mammalian lamina, the fibrous protein meshwork located just underneath the inner nuclear membrane [86]. Lamin A/C is developmentally regulated and mainly expressed in differentiated cells [84]. Of relevance, several studies demonstrated the crucial role of lamin A/C in osteoblastogenesis and bone formation, in vitro and in vivo. While in osteogenic differentiating human mesenchymal stem cells (MSCs) the loss of lamin A/C inhibits osteoblast differentiation and mineralization [87], [88], in mature osteoblast it leads to a reduced mineralization and alkaline phosphatase expression, thus suggesting that lamin A/C is required for both MSC commitment and for mature osteoblast physiological functions [87]. On the other hand, inhibition of lamin A/C facilitates the differentiation of human MSCs into adipocytes [87]. Interestingly, MSCs from adult Brtl+/− mice have a reduced osteogenic potential and an increased ability to differentiate toward adipocytes [89], as well as it was observed in PEDF knock-out mouse, a model of recessive OI that showed enhanced adiposity and decreased bone mineral content [90].
Of great relevance to correlate the lamin aberrancy that we detected to OI traits, mice lacking lamin A/C or enzymes involved in its processing present a bone phenotype very similar to those observed in dominant and recessive OI patients and murine models. Lmna−/− mice actually display a severe osteopenic phenotype with significant decrease in bone mass and bone apposition, and with poor bone microarchitecture [91]. Also Zmpste24−/− mice, which lack zmpste24 enzyme for lamin A/C processing, show an osteopenic phenotype with brittle bones and spontaneous bone fractures [92]. In transgenic mice, which resemble the Hutchinson-Gilford progeria syndrome (HGPS), the bone phenotype is reverted by completely silencing endogenous mutated Lmna gene and inducing transgenic expression of human lamin A/C [93].
LMNA mutations or incorrect processing of lamin A/C are causative of a wide spectrum of human heterogeneous diseases termed laminopathies [94], several of which share some features with OI. Certain forms of laminopathies affect different adipose and bone tissues [95], with MAD (mandibuloacral dysplasia) and HPGS presenting the main bone and dental affections (i.e. osteoporosis, osteolysis, bone and dental deformities, and spontaneous fractures) [96], [97], [98], [99].
After its identification among protein spots that significantly change in abundance (Table 1: spot n. 9), we proved lamin A/C aberrant protein-expression in recessive OI performing 2-D western blot. As shown in Fig. 5A, fibroblasts from CRTAP and P3H1 patients show an evident increased immunoreactive signal of lamin C protein species (isoelectric series at lower MW) when compared to the control cells. On the other hand, P3H1 fibroblasts present depleted signals of lamin A protein species (isoelectric series at higher MW; Fig. 5A) with respect to control and CRTAP cells, which instead have a similar intensity. Fibroblasts from the PPIB proband are discordant with the other investigated cells displaying a dramatic reduction of both lamin A and C.
Fig. 5.

Lamin A/C protein abundance (A) and nuclear organization in human dermal fibroblasts from control subjects and OI patients with mutations in CRTAP, P3H1, and PPIB genes (B). A: Representative 2-D western blot images are shown. Histograms visualize the total spot Vol of each isoelectric series: lamin A in dark gray and lamin C in light gray. B: Representative confocal immunofluorescence images are shown. Lamin A/C signal is shown in red. Counterstaining of the nuclei was performed using DAPI (blue). An altered lamin distribution was detected in all pathological primary cells. Scale bar 10 μm.
These data suggested the occurring of nuclear lamina and nucleoskeleton disorganization. To evaluate eventual correlated nuclear aberrances, a confocal immunofluorescence analysis for lamin A/C was indeed performed on fibroblasts from all the four analyzed conditions. As illustrated in Fig. 5B, lamin A/C homogenously distributed at the nuclear periphery of control cells. On the contrary, recessive OI fibroblasts displayed lamin A/C staining not only peripherally but also throughout the nucleoplasm where lamin A/C foci were also detected (Fig. 5B: arrows), thus suggesting the occurrence of lamin fibers accumulation in some restricted areas. Interestingly, aberrant lamin A/C organization and intranuclear foci presence were described in fibroblasts from patients affected by different laminopathies, as well as in human transfected fibroblasts and myoblasts expressing mutant lamin A/C [100], [101], [102]. Furthermore, CRTAP, P3H1, and PPIB fibroblasts presented a lamin A/C “granular aspect” resembling the lamin A/C “honeycomb” pattern observed in nuclei from laminophatic cells [100], [103], [104], [105].
Why different genetic disorders, caused by different mutated genes whose protein products act in different and (apparently) not overlapping pathways, present similar aberrances is still puzzling. On the bases of our data, nucleoskeleton and cytoskeleton affection may intriguingly represent a molecular bridge between OI and other apparently not related diseases, such as laminopathies. Alterations in nucleoskeleton, actually, negatively affect the cytoskeleton organization and cytoskeleton aberrances determine nucleoskeleton, and chromatin, affection [106], [107], [108].
A-type lamins anchor nucleoskeleton to cytoskeleton and indirectly regulate cytosolic actin assembly [109], [110]. Such cytoskeleton/nucleoskeleton interactions act as sensors and transducers of signals from the cytoplasm to the nucleus, and vice versa, to regulate nuclear position, gene expression, and cell functions [111]. In turn, the cytoskeleton is connected to the ECM via focal adhesion (FA) integrins [112]. Of note, abnormal ECM composition, which is a hallmark of OI [8], was reported in lipodystrophy and in model cell lines of Hutchinson-Gilford Progeria (HGPS), as linked, respectively, to altered TGF-β and Wnt signaling [113], [114]. To close the circle, lamins indirectly interfere with TGF-β and Wnt signaling by regulating the inner nuclear membrane integral proteins MAN1, which inactivate Smads, and emerin that mediates the nuclear export of β-catenin [115], [116], [117], [118].
Our data suggest an alteration of the tight interconnection among nucleus, cytoskeleton and ECM in OI cells, and their strong cooperation in sensing, integrating, and transducing extracellular stimuli for cell response [119] may represent a molecular link between OI and other disorders, such as laminopathies.
Based on the here proved aberrant organization of nuclear lamina, on the high percentage (46%) of differentially expressed proteins involved in cytoskeleton asset, on the MetaCore built network, and, finally, on previous data from dominant forms of the disease [29], we hypothesized that also cytoskeleton was affected in investigated fibroblast cells. Cofilin-1 protein abundance levels were evaluated to assess cytoskeleton “health status” in CRTAP, P3H1, and PPIB fibroblast samples by using 1-D western blot. Cofilin-1 was chosen as it is an essential regulator of actin dynamics, by inducing F-actin depolymerization and inhibiting G-actin polymerization [120], and on the bases of its proved deregulation in a murine model of dominant OI [19], [29]. Very recently, Wiggan et al. have also described cofilin-1 to indirectly regulate nuclear architecture and as its deregulation, along with ADF (actin-depolymerizing factor or destrin), provokes consistent nuclear lamina disruption and aberrant chromatin organization [121]. In addition, the MetaCore processing of cofilin-1 with the differentially expressed proteins generated, by shortest path, a significant network where this G- and F-actin modulator was perfectly integrated, Supplementary Fig. S2. Consistent with previously described data in dominant OI [29], we found a statistically significant increase of cofilin-1 in fibroblasts from CRTAP and P3H1 patients, Fig. 6A. Despite it was not statistically significant, an increase of cofilin-1 was detected also in PPIB cells.
Fig. 6.

Cofilin-1 protein abundance (A) and cytoskeletal organization in human dermal fibroblasts from OI patients (B). A: Representative 1-D western blot images are shown. Histograms visualize normalized mean relative-integrated density ± SD values of detected bands. * and § symbols indicate the significance of expression changes occurring between control and CRTAP, and control and P3H1 fibroblasts, respectively. B: Representative confocal immunofluorescence images are shown. Fluorescently labeled phalloidin (green), specifically interacting with actin, was used. Counterstaining of the nuclei was performed using DAPI (blue). A disorganized cytoskeletal network was evident in all patients primary cells. Scale bar 10 μm.
Then, to verify the presence of cytoskeletal anomalies, a confocal immunofluorescence analysis was performed on fibroblasts from controls and probands by using the fluorescently labeled phalloidin, which specifically targets actin. A disorganized cytoskeleton, with an abnormal distribution of actin fibers, was revealed, Fig. 6B. This finding maybe reflects the tight structural/functional coupling of nucleoskeleton to cytoskeleton.
The relevance of actin cytoskeleton in osteoblast differentiation and bone development has been largely discussed in our previous work on dominant OI [29]. It has been even demonstrated that cell viability and differentiation of human MSCs into osteoblasts are enhanced by stabilizing actin filaments through cofilin-1 and destrin inhibition, while the opposite effect was achieved silencing the cofilin-1 inhibitor LIMK1 [122], [123]. Noteworthy, we identified two differentially expressed proteins involved in LIMK1 activity control: p21-activated kinase 2 (PAK2) and Rho GDP-dissociation inhibitor 1 (RHOGDI1 or RHOGDIα). PAK2, one of the main central hubs in the MetaCore generated network, is a downstream effector of Rho small GTPases that phosphorylates and activates LIMK1 [124] and that is known to operate in many cellular processes depending on cytoskeleton dynamics [125]. RHOGDI1, instead, regulates several Rho GTPases, such as Cdc42, preventing the release of GDP [126]. PAK2 controls, by a positive feedback, its own activation inhibiting RHOGDI1 and maintaining the active state of Cdc42, which in turn activates PAK2 [127], as highlighted in Fig. 4. PAK2 decreased in CRTAP and PPIB fibroblasts, Table 1, spot 12. In these patients, PAK2 down-regulation may lead to a reduced LIMK1 activity and to altered cytoskeleton dynamics.
4. Conclusions
In this work, we reported for the first time nucleoskeleton and cytoskeleton aberrances in recessive OI forms characterized by collagen structural defects. We also highlighted, according to our previous analyses, that specific biological processes and molecular functions, mainly related to cytoskeleton and nucleoskeleton, are similarly affected in dominant and recessive OI, independently of their different protein abundance profiles and mutated genes. We indeed believe that nucleus and cytoskeleton organization may represent novel targets to develop innovative treatment for OI.
The tight interconnection among nucleus, cytoskeleton, and ECM and their strong cooperation in sensing, integrating, and transducing stimuli for cell response [119] may represent a molecular bridge among different diseases.
In line with this concept, some of the molecular pathways that we hypothesized, according to our data, to be aberrant in OI have been previously described as affected also in laminopathies. Despite their distinctive etiology and phenotypic features, OI and laminopathies present affection of nuclear lamina and cytoskeleton, and of TGF-β, integrin and Wnt signaling, even though such aberrances occur in response of different trigger events.
In conclusion, the comparison of aberrant molecular pathways that characterize different forms of the same disorder or different disorders, caused by different mutated genes, may pave the way to a molecular reviewing of diseases based on similarities in molecular affection. Such perspective may flank conventional clinical practice as well as pharmacological approach by suggesting novel drug targets.
The following are the supplementary data related to this article.
Detected quantitative and qualitative differences in the six different performed pair comparisons.
Six-way Venn diagram (A), displayed by Edwards' mode, and histogram (B) of the quantitative and qualitative differentially expressed protein-spots detected by crossed comparisons among control subjects and OI patients carrying mutations in CRTAP, P3H1, and PPIB genes. A and B show distribution of common and peculiar differences in the six performed pair-comparisons.
MetaCore protein network obtained by processing cofilin-1 with the differentially expressed proteins in primary fibroblasts from CRTAP, P3H1, and PPIB patients and from control subjects. MetaCore analysis was used to map identified proteins (blue circles) on existing mammalian pathways and networks, according to known protein-protein interactions and other features established in the literature. Network proteins are visualized by specific symbols, which define the functional nature of the protein (network caption). The edges indicate the relationships existing between individual proteins, and the arrowheads represent the direction of the interaction. The following line colors designate the nature of the interaction: red = negative effect; green = positive effect; gray = unspecified effect. Based on selected parameters, only eukaryotic translation initiation factor 1b (EIF1B), coiled-coil-helix-coiled-coil-helix domain-containing protein 5 (CHCHD5), and aldehyde dehydrogenase X, mitochondrial (AL1B1) were excluded from the net.
Supplementary material
Transparency document
Transparency document.
Acknowledgments
Acknowledgment
We thank Prof. Maurizia Valli and Dr. Simona Viglio, Dept. of Molecular Medicine, Biochemistry Unit, University of Pavia, Pavia, Italy and Prof. Roy Morello, Dept. of Physiology and Biophysics, UAMS, Little Rock, AR, USA for providing some of the primary fibroblasts used in this research. We are grateful to Dr. Patrizia Vaghi, Centro Grandi Strumenti, University of Pavia, Italy for technical assistance with confocal microscopy.
Acknowledgments
Author contributions
Conceived and designed the project: Laura Bianchi, Luca Bini, AF; designed experiments: Laura Bianchi, Luca Bini, AF; performed experiments: AG, CC, RB, AA; analyzed data: AG, Laura Bianchi, RB, CC, CL; clinical contribution: MA, GO, SAT; wrote manuscript: AG, Laura Bianchi, Luca Bini, AF; critical review: Laura Bianchi, Luca Bini, AF, AG, RB; approval of the manuscript: all authors.
Funding
This work was supported by Telethon Foundation [grant n. GGP13098] to AF and Laura Bianchi, and by Fondazione Cariplo [grant n. 2013-0612] to AF and Luca Bini.
Conflict of interest
The authors have declared no conflict of interest.
Footnotes
The Transparency document associated with this article can be found, in online version.
References
- 1.Byers P.H., Cole W.G. Osteogenesis imperfecta. In: Royce P.M., Steinmann B., editors. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. 2nd ed. Wiley-Liss; New York: 2002. pp. 385–430. [Google Scholar]
- 2.Sillence D.O., Senn A., Danks D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979;16(2):101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J., Bächinger H.P., Pace J.M., Schwarze U., Byers P.H., Weis M., Fernandes R.J., Eyre D.R., Yao Z., Boyce B.F., Lee B. CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127(2):291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 4.Barnes A.M., Chang W., Morello R., Cabral W.A., Weis M., Eyre D.R., Leikin S., Makareeva E., Kuznetsova N., Uveges T.E., Ashok A., Flor A.W., Mulvihill J.J., Wilson P.L., Sundaram U.T., Lee B., Marini J.C. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006;355(26):2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forlino A., Marini J.C. Osteogenesis imperfecta. Lancet. 2016;387(10028):1657–1671. doi: 10.1016/S0140-6736(15)00728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marini J.C., Blissett A.R. New genes in bone development: what's new in osteogenesis imperfecta. J. Clin. Endocrinol. Metab. 2013;98(8):3095–3103. doi: 10.1210/jc.2013-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindert U., Cabral W.A., Ausavarat S., Tongkobpetch S., Ludin K., Barnes A.M., Yeetong P., Weis M., Krabichler B., Srichomthong C., Makareeva E.N., Janecke A.R., Leikin S., Röthlisberger B., Rohrbach M., Kennerknecht I., Eyre D.R., Suphapeetiporn K., Giunta C., Marini J.C., Shotelersuk V. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat. Commun. 2016;7:11920. doi: 10.1038/ncomms11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forlino A., Cabral W.A., Barnes A.M., Marini J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011;7(9):540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes A.M., Carter E.M., Cabral W.A., Weis M., Chang W., Makareeva E., Leikin S., Rotimi C.N., Eyre D.R., Raggio C.L., Marini J.C. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 2010;362(6):521–528. doi: 10.1056/NEJMoa0907705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishikawa Y., Wirz J., Vranka J.A., Nagata K., Bächinger H.P. Biochemical characterization of the prolyl 3-hydroxylase 1·cartilage-associated protein·cyclophilin B complex. J. Biol. Chem. 2009;284:17641–17647. doi: 10.1074/jbc.M109.007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myllyharju J., Kivirikko K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20(1):33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Krane S.M. The importance of proline residues in the structure, stability and susceptibility to proteolytic degradation of collagens. Amino Acids. 2008;35(4):703–710. doi: 10.1007/s00726-008-0073-2. [DOI] [PubMed] [Google Scholar]
- 13.Jenkins C.L., Bretscher L.E., Guzei I.A., Raines R.T. Effect of 3-hydroxyproline residues on collagen stability. J. Am. Chem. Soc. 2003;125(21):6422–6427. doi: 10.1021/ja034015j. [DOI] [PubMed] [Google Scholar]
- 14.Castagnola P., Gennari M., Morello R., Tonachini L., Marin O., Gaggero A., Cancedda R. Cartilage associated protein CASP is a novel developmentally regulated chick embryo protein. J. Cell Sci. 1997;110:1351–1359. doi: 10.1242/jcs.110.12.1351. [DOI] [PubMed] [Google Scholar]
- 15.Vranka J.A., Sakai L.Y., Bächinger H.P. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 2004;279(22):23615–23621. doi: 10.1074/jbc.M312807200. [DOI] [PubMed] [Google Scholar]
- 16.Wassenhove-McCarthy D.J., McCarthy K.J. Molecular characterization of a novel basement membrane-associated proteoglycan, leprecan. J. Biol. Chem. 1999;274:25004–25017. doi: 10.1074/jbc.274.35.25004. [DOI] [PubMed] [Google Scholar]
- 17.Barik S. Immunophilins: for the love of proteins. Cell. Mol. Life Sci. 2006;63(24):2889–2900. doi: 10.1007/s00018-006-6215-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinmann B., Bruckner P., Superti-Furga A. Cyclosporin A slows collagen triple-helix formation in vivo: indirect evidence for a physiologic role of peptidyl-prolyl cis‑trans‑isomerase. J. Biol. Chem. 1991;266(2):1299–1303. [PubMed] [Google Scholar]
- 19.Bianchi L., Gagliardi A., Gioia R., Besio R., Tani C., Landi C., Cipriano M., Gimigliano A., Rossi A., Marini J.C., Forlino A., Bini L. Differential response to intracellular stress in the skin from osteogenesis imperfecta Brtl mice with lethal and non lethal phenotype: a proteomic approach. J. Proteome. 2012;75(15):4717–4733. doi: 10.1016/j.jprot.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marini J.C., Cabral W.A., Barnes A.M. Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. 2010;339:59–70. doi: 10.1007/s00441-009-0872-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang W., Barnes A.M., Cabral W.A., Bodurtha J.N., Marini J.C. Prolyl 3-hydroxylase 1 and CRTAP are mutually stabilizing in the endoplasmic reticulum collagen prolyl 3-hydroxylation complex. Hum. Mol. Genet. 2010;19:223–234. doi: 10.1093/hmg/ddp481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Dijk F.S., Nesbitt I.M., Zwikstra E.H., Nikkels P.G., Piersma S.R., Fratantoni S.A., Jimenez C.R., Huizer M., Morsman A.C., Cobben J.M., van Roij M.H., Elting M.W., Verbeke J.I., Wijnaendts L.C., Shaw N.J., Högler W., McKeown C., Sistermans E.A., Dalton A., Meijers-Heijboer H., Pals G. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009;85(4):521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansen B., Jemec G.B. The mechanical properties of skin in osteogenesis imperfecta. Arch. Dermatol. 2002;138(7):909–911. doi: 10.1001/archderm.138.7.909. [DOI] [PubMed] [Google Scholar]
- 24.Canuto H.C., Fishbein K.W., Huang A., Doty S.B., Herbert R.A., Peckham J., Pleshko N., Spencer R.G. Characterization of skin abnormalities in a mouse model of osteogenesis imperfecta using high resolution magnetic resonance imaging and Fourier transform infrared imaging spectroscopy. NMR Biomed. 2012;25(1):169–176. doi: 10.1002/nbm.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baldridge D., Lennington J., Weis M., Homan E.P., Jiang M.M., Munivez E., Keene D.R., Hogue W.R., Pyott S., Byers P.H., Krakow D., Cohn D.H., Eyre D.R., Lee B., Morello R. Generalized connective tissue disease in Crtap −/− mouse. PLoS One. 2010;5(5):e10560. doi: 10.1371/journal.pone.0010560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashinsky B.G., Fishbein K.W., Carter E.M., Lin P.C., Pleshko N., Raggio C.L., Spencer R.G. Multiparametric classification of skin from osteogenesis imperfecta patients and controls by quantitative magnetic resonance microimaging. PLoS One. 2016;11(7):e0157891. doi: 10.1371/journal.pone.0157891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balasubramanian M., Sobey G.J., Wagner B.E., Peres L.C., Bowen J., Bexon J., Javaid M.K., Arundel P., Bishop N.J. Osteogenesis imperfecta: ultrastructural and histological findings on examination of skin revealing novel insights into genotype-phenotype correlation. Ultrastruct. Pathol. 2016;40(2):71–76. doi: 10.3109/01913123.2016.1140253. [DOI] [PubMed] [Google Scholar]
- 28.Forlino A., Tani C., Rossi A., Lupi A., Campari E., Gualeni B., Bianchi L., Armini A., Cetta G., Bini L., Marini J.C. Differential expression of both extracellular and intracellular proteins is involved in the lethal or nonlethal phenotypic variation of BrtlIV, a murine model for osteogenesis imperfecta. Proteomics. 2007;7(11):1877–1891. doi: 10.1002/pmic.200600919. [DOI] [PubMed] [Google Scholar]
- 29.Bianchi L., Gagliardi A., Maruelli S., Besio R., Landi C., Gioia R., Kozloff K.M., Khoury B.M., Coucke P.J., Symoens S., Marini J.C., Rossi A., Bini L., Forlino A. Altered cytoskeletal organization characterized lethal but not surviving Brtl +/− mice: insight on phenotypic variability in osteogenesis imperfecta. Hum. Mol. Genet. 2015;24(21):6118–6133. doi: 10.1093/hmg/ddv328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valli M., Barnes A.M., Gallanti A., Cabral W.A., Viglio S., Weis M.A., Makareeva E., Eyre D., Leikin S., Antoniazzi F., Marini J.C., Mottes M. Deficiency of CRTAP in non-lethal recessive osteogenesis imperfecta reduces collagen deposition into matrix. Clin. Genet. 2012;82(5):453–459. doi: 10.1111/j.1399-0004.2011.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caparrós-Martin J.A., Valencia M., Pulido V., Martínez-Glez V., Rueda-Arenas I., Amr K., Farra C., Lapunzina P., Ruiz-Perez V.L., Temtamy S., Aglan M. Clinical and molecular analysis in families with autosomal recessive osteogenesis imperfecta identifies mutations in five genes and suggests genotype-phenotype correlations. Am. J. Med. Genet. A. 2013;161A(6):1354–1369. doi: 10.1002/ajmg.a.35938. [DOI] [PubMed] [Google Scholar]
- 32.Takagi M., Ishii T., Barnes A.M., Weis M., Amano N., Tanaka M., Fukuzawa R., Nishimura G., Eyre D.R., Marini J.C., Hasegawa T. A novel mutation in LEPRE1 that eliminates only the KDEL ER- retrieval sequence causes non-lethal osteogenesis imperfecta. PLoS One. 2012;7(5):e36809. doi: 10.1371/journal.pone.0036809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willaert A., Malfait F., Symoens S., Gevaert K., Kayserili H., Megarbane A., Mortier G., Leroy J.G., Coucke P.J., De Paepe A. Recessive osteogenesis imperfecta caused by LEPRE1 mutations: clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J. Med. Genet. 2009;46(4):233–241. doi: 10.1136/jmg.2008.062729. [DOI] [PubMed] [Google Scholar]
- 34.Pepin M.G., Schwarze U., Singh V., Romana M., Jones-Lecointe A., Byers P.H. Allelic background of LEPRE1 mutations that cause recessive forms of osteogenesis imperfecta in different populations. Mol. Genet. Genomic Med. 2013;1(4):194–205. doi: 10.1002/mgg3.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 36.Gorg A., Postel W., Gunther S. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis. 1988;9:531–546. doi: 10.1002/elps.1150090913. [DOI] [PubMed] [Google Scholar]
- 37.Bjellqvist B., Pasquali C., Ravier F., Sanchez J.C., Hochstrasser D. A nonlinear wide-range immobilized pH gradient for two-dimensional electrophoresis and its definition in a relevant pH scale. Electrophoresis. 1993;14:1357–1365. doi: 10.1002/elps.11501401209. [DOI] [PubMed] [Google Scholar]
- 38.Oakley B.R., Kirsch D.R., Morris N.R. A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal. Biochem. 1980;105:361–363. doi: 10.1016/0003-2697(80)90470-4. [DOI] [PubMed] [Google Scholar]
- 39.Hochstrasser D.F., Patchornik A., Merril C.R. Development of polyacrylamide gels that improve the separation of proteins and their detection by silver staining. Anal. Biochem. 1988;173:412–423. doi: 10.1016/0003-2697(88)90208-4. [DOI] [PubMed] [Google Scholar]
- 40.Sinha P., Poland J., Schnolzer M., Rabilloud T. A new silver staining apparatus and procedure for matrix-assisted laser desorption/ionization-time of flight analysis of proteins after two-dimensional electrophoresis. Proteomics. 2001;1:835–840. doi: 10.1002/1615-9861(200107)1:7<835::AID-PROT835>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 41.Bardou P., Mariette J., Escudié F., Djemiel C., Klopp C. jvenn: an interactive Venn diagram viewer. BMC Bioinf. 2014;15:293. doi: 10.1186/1471-2105-15-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gharahdaghi F., Weinberg C.R., Meagher D.A., Imai B.S., Mische S.M. Mass spectrometric identification of proteins from silver-stained polyacrylamide gel: a method for the removal of silver ions to enhance sensitivity. Electrophoresis. 1999;20:601–605. doi: 10.1002/(SICI)1522-2683(19990301)20:3<601::AID-ELPS601>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 43.de Hoon M.J., Imoto S., Nolan J., Miyano S. Open source clustering software. Bioinformatics. 2004;20(9):1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 44.Saldanha A.J. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 45.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 46.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 47.Folkestad L., Hald J.D., Gram J., Langdahl B.L., Hermann A.P., Diederichsen A.C., Abrahamsen B., Brixen K. Cardiovascular disease in patients with osteogenesis imperfecta - a nationwide, register-based cohort study. Int. J. Cardiol. 2016;225:250–257. doi: 10.1016/j.ijcard.2016.09.107. [DOI] [PubMed] [Google Scholar]
- 48.Folkestad L., Hald J.D., Canudas-Romo V., Gram J., Hermann A.P., Langdahl B., Abrahamsen B., Brixen K. Mortality and causes of death in patients with osteogenesis imperfecta: a register-based nationwide cohort study. J. Bone Miner. Res. 2016;31(12):2159–2166. doi: 10.1002/jbmr.2895. [DOI] [PubMed] [Google Scholar]
- 49.DiGirolamo D.J., Singhal V., Chang X., Lee S.J., Germain-Lee E.L. Administration of soluble activin receptor 2B increases bone and muscle mass in a mouse model of osteogenesis imperfecta. Bone Res. 2015;3:14042. doi: 10.1038/boneres.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bianchi L., Bruzzese F., Leone A., Gagliardi A., Puglia M., Di Gennaro E., Rocco M., Gimigliano A., Pucci B., Armini A., Bini L., Budillon A. Proteomic analysis identifies differentially expressed proteins after HDAC vorinostat and EGFR inhibitor gefitinib treatments in Hep-2 cancer cells. Proteomics. 2011;11(18):3725–3742. doi: 10.1002/pmic.201100092. [DOI] [PubMed] [Google Scholar]
- 51.Chang J., Wang Z., Tang E., Fan Z., McCauley L., Franceschi R., Guan K., Krebsbach P.H., Wang C.Y. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat. Med. 2009;15(6):682–689. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abu-Amer Y. NF-κB signaling and bone resorption. Osteoporos. Int. 2013;24(9):2377–2386. doi: 10.1007/s00198-013-2313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyce B.F., Yao Z., Xing L. Functions of nuclear factor kappaB in bone. Ann. N. Y. Acad. Sci. 2010;1192:367–375. doi: 10.1111/j.1749-6632.2009.05315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohazama A., Sharpe P.T. TNF signalling in tooth development. Curr. Opin. Genet. Dev. 2004;14(5):513–519. doi: 10.1016/j.gde.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 55.Kim J.H., Kim K., Youn B.U., Lee J., Kim I., Shin H.I., Akiyama H., Choi Y., Kim N. Kruppel-like factor 4 attenuates osteoblast formation, function, and cross talk with osteoclasts. J. Cell Biol. 2014;204(6):1063–1074. doi: 10.1083/jcb.201308102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee H.K., Lee D.S., Park S.J., Cho K.H., Bae H.S., Park J.C. Nuclear factor I-C (NFIC) regulates dentin sialophosphoprotein (DSPP) and E-cadherin via control of Krüppel-like factor 4 (KLF4) during dentinogenesis. J. Biol. Chem. 2014;289(41):28225–28236. doi: 10.1074/jbc.M114.568691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aradhya S., Nelson D.L. NF-kappaB signaling and human disease. Curr. Opin. Genet. Dev. 2001;11(3):300–306. doi: 10.1016/s0959-437x(00)00194-5. [DOI] [PubMed] [Google Scholar]
- 58.Dai L., Aye Thu C., Liu X.Y., Xi J., Cheung P.C. TAK1, more than just innate immunity. IUBMB Life. 2012;64(10):825–834. doi: 10.1002/iub.1078. [DOI] [PubMed] [Google Scholar]
- 59.Li Y., Li A., Strait K., Zhang H., Nanes M.S., Weitzmann M.N. Endogenous TNFalpha lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-kappaB. J. Bone Miner. Res. 2007;22(5):646–655. doi: 10.1359/jbmr.070121. [DOI] [PubMed] [Google Scholar]
- 60.Yamazaki M., Fukushima H., Shin M., Katagiri T., Doi T., Takahashi T., Jimi E. Tumor necrosis factor alpha represses bone morphogenetic protein (BMP) signaling by interfering with the DNA binding of Smads through the activation of NF-kappaB. J. Biol. Chem. 2009;284(51):35987–35995. doi: 10.1074/jbc.M109.070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grafe I., Yang T., Alexander S., Homan E.P., Lietman C., Jiang M.M., Bertin T., Munivez E., Chen Y., Dawson B., Ishikawa Y., Weis M.A., Sampath T.K., Ambrose C., Eyre D., Bächinger H.P., Lee B. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 2014;20(6):670–675. doi: 10.1038/nm.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Labbé E., Letamendia A., Attisano L. Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-beta and wnt pathways. Proc. Natl. Acad. Sci. U. S. A. 2000;97(15):8358–8363. doi: 10.1073/pnas.150152697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Silva I., Branco J.C. Rank/Rankl/opg: literature review. Acta Reumatol. Port. 2011;36(3):209–218. [PubMed] [Google Scholar]
- 64.Rodríguez-Carballo E., Ulsamer A., Susperregui A.R., Manzanares-Céspedes C., Sánchez-García E., Bartrons R., Rosa J.L., Ventura F. Conserved regulatory motifs in osteogenic gene promoters integrate cooperative effects of canonical Wnt and BMP pathways. J. Bone Miner. Res. 2011;26(4):718–729. doi: 10.1002/jbmr.260. [DOI] [PubMed] [Google Scholar]
- 65.Guo X., Wang X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19(1):71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheon S.S., Wei Q., Gurung A., Youn A., Bright T., Poon R., Whetstone H., Guha A., Alman B.A. Beta-catenin regulates wound size and mediates the effect of TGF-beta in cutaneous healing. FASEB J. 2006;20(6):692–701. doi: 10.1096/fj.05-4759com. [DOI] [PubMed] [Google Scholar]
- 67.Klapholz-Brown Z., Walmsley G.G., Nusse Y.M., Nusse R., Brown P.O. Transcriptional program induced by Wnt protein in human fibroblasts suggests mechanisms for cell cooperativity in defining tissue microenvironments. PLoS One. 2007;2(9):e945. doi: 10.1371/journal.pone.0000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varga J., Whitfield M.L. Transforming growth factor-beta in systemic sclerosis (scleroderma) Front. Biosci. (Schol. Ed.) 2009;1:226–235. doi: 10.2741/s22. [DOI] [PubMed] [Google Scholar]
- 69.Dimitriou R., Tsiridis E., Giannoudis P.V. Current concepts of molecular aspects of bone healing. Injury. 2005;36(12):1392–1404. doi: 10.1016/j.injury.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 70.Wei J., Fang F., Lam A.P., Sargent J.L., Hamburg E., Hinchcliff M.E., Gottardi C.J., Atit R., Whitfield M.L., Varga J. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012;64(8):2734–2745. doi: 10.1002/art.34424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Willert K., Nusse R. Wnt proteins. Cold Spring Harb. Perspect. Biol. 2012;4(9):a007864. doi: 10.1101/cshperspect.a007864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Noort M., Meeldijk J., van der Zee R., Destree O., Clevers H. Wnt signaling controls the phosphorylation status of beta-catenin. J. Biol. Chem. 2002;277(20):17901–17905. doi: 10.1074/jbc.M111635200. [DOI] [PubMed] [Google Scholar]
- 73.Faqeih E., Shaheen R., Alkuraya F.S. WNT1 mutation with recessive osteogenesis imperfecta and profound neurological phenotype. J. Med. Genet. Jul 2013;50(7):491–492. doi: 10.1136/jmedgenet-2013-101750. [DOI] [PubMed] [Google Scholar]
- 74.Pyott S.M., Tran T.T., Leistritz D.F., Pepin M.G., Mendelsohn N.J., Temme R.T., Fernandez B.A., Elsayed S.M., Elsobky E., Verma I., Nair S., Turner E.H., Smith J.D., Jarvik G.P., Byers P.H. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2013;92(4):590–597. doi: 10.1016/j.ajhg.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wagner E.F., Eferl R. Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 2005;208:126–140. doi: 10.1111/j.0105-2896.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 76.Marie P.J., Haÿ E., Saidak Z. Integrin and cadherin signaling in bone: role and potential therapeutic targets. Trends Endocrinol. Metab. 2014;25(11):567–575. doi: 10.1016/j.tem.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 77.Lim X., Nusse R. Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb. Perspect. Biol. 2013;5(2) doi: 10.1101/cshperspect.a008029. (pii: a008029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang Y., Wan C., Gilbert S.R., Clemens T.L. Oxygen sensing and osteogenesis. Ann. N. Y. Acad. Sci. 2007;1117:1–11. doi: 10.1196/annals.1402.049. [DOI] [PubMed] [Google Scholar]
- 79.Semenza G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014;76:39–56. doi: 10.1146/annurev-physiol-021113-170322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shimoda L.A., Semenza G.L. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011;183(2):152–156. doi: 10.1164/rccm.201009-1393PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Soma K., Abe H., Takeda N., Shintani Y., Takazawa Y., Kojima T., Fujiu K., Semba H., Yamashita H., Hirata Y., Fukayama M., Nagai R. Myocardial involvement in patients with osteogenesis imperfecta. Int. Heart J. 2012;53(1):75–77. doi: 10.1536/ihj.53.75. [DOI] [PubMed] [Google Scholar]
- 82.Parry D.A., Conway J.F., Steinert P.M. Structural studies on lamin. Similarities and differences between lamin and intermediate-filament proteins. Biochem. J. 1986;238:305–308. doi: 10.1042/bj2380305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Prokocimer M., Davidovich M., Nissim-Rafinia M., Wiesel-Motiuk N., Bar D.Z., Barkan R., Meshorer E., Gruenbaum Y. Nuclear lamins: key regulators of nuclear structure and activities. J. Cell. Mol. Med. 2009;13(6):1059–1085. doi: 10.1111/j.1582-4934.2008.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dechat T., Adam S.A., Taimen P., Shimi T., Goldman R.D. Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2010;2(11):a000547. doi: 10.1101/cshperspect.a000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Osmanagic-Myers S., Dechat T., Foisner R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015;29(3):225–237. doi: 10.1101/gad.255968.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stuurman N., Heins S., Aebi U. Nuclear lamins: their structure, assembly, and interactions. J. Struct. Biol. 1998;122:42–66. doi: 10.1006/jsbi.1998.3987. [DOI] [PubMed] [Google Scholar]
- 87.Akter R., Rivas D., Geneau G., Drissi H., Duque G. Effect of lamin A/C knockdown on osteoblast differentiation and function. J. Bone Miner. Res. 2009;24(2):283–293. doi: 10.1359/jbmr.081010. [DOI] [PubMed] [Google Scholar]
- 88.Rauner M., Sipos W., Goettsch C., Wutzl A., Foisner R., Pietschmann P., Hofbauer L.C. Inhibition of lamin A/C attenuates osteoblast differentiation and enhances RANKL-dependent osteoclastogenesis. J. Bone Miner. Res. 2009;24(1):78–86. doi: 10.1359/jbmr.080902. [DOI] [PubMed] [Google Scholar]
- 89.Gioia R., Panaroni C., Besio R., Palladini G., Merlini G., Giansanti V., Scovassi I.A., Villani S., Villa I., Villa A., Vezzoni P., Tenni R., Rossi A., Marini J.C., Forlino A. Impaired osteoblastogenesis in a murine model of dominant osteogenesis imperfecta: a new target for osteogenesis imperfecta pharmacological therapy. Stem Cells. 2012;30:1465–1476. doi: 10.1002/stem.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gattu A.K., Swenson E.S., Iwakiri Y., Samuel V.T., Troiano N., Berry R., Church C.D., Rodeheffer M.S., Carpenter TO, Chung C. Determination of mesenchymal stem cell fate by pigment epithelium derived factor (PEDF) results in increased adiposity and reduced bone mineral content. FASEB J. 2013;27(11):4384–4394. doi: 10.1096/fj.13-232900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li W., Yeo L.S., Vidal C., McCorquodale T., Herrmann M., Fatkin D., Duque G. Decreased bone formation and osteopenia in lamin a/c-deficient mice. PLoS One. 2011;6(4):e19313. doi: 10.1371/journal.pone.0019313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bergo M.O., Gavino B., Ross J., Schmidt W.K., Hong C., Kendall L.V., Mohr A., Meta M., Genant H., Jiang Y., Wisner E.R., Van Bruggen N., Carano R.A., Michaelis S., Griffey S.M., Young S.G. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. U. S. A. 2002;99(20):13049–13054. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Strandgren C., Nasser H.A., McKenna T., Koskela A., Tuukkanen J., Ohlsson C., Rozell B., Eriksson M. Transgene silencing of the Hutchinson-Gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J. 2015;29(8):3193–3205. doi: 10.1096/fj.14-269217. [DOI] [PubMed] [Google Scholar]
- 94.Dobrzynska A., Gonzalo S., Shanahan C., Askjaer P. The nuclear lamina in health and disease. Nucleus. 2016;9:1–16. doi: 10.1080/19491034.2016.1183848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schreiber K.H., Kennedy B.K. When lamins go bad: nuclear structure and disease. Cell. 2013;152(6):1365–1375. doi: 10.1016/j.cell.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cunningham V.J., D'Apice M.R., Licata N., Novelli G., Cundy T. Skeletal phenotype of mandibuloacral dysplasia associated with mutations in ZMPSTE24. Bone. 2010;47(3):591–597. doi: 10.1016/j.bone.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 97.Tanyeri H., Kurklu E., Ak G., Ozturk S., Koray M., Palanduz S. Maxillofacial and dental manifestations in a patient with mandibulo-acral dysplasia. Cranio. 2005;23(1):74–78. doi: 10.1179/crn.2005.011. [DOI] [PubMed] [Google Scholar]
- 98.Gordon C.M., Gordon L.B., Snyder B.D., Nazarian A., Quinn N., Huh S., Giobbie-Hurder A., Neuberg D., Cleveland R., Kleinman M., Miller D.T., Kieran M.W. Hutchinson-Gilford progeria is a skeletal dysplasia. J. Bone Miner. Res. 2011;26(7):1670–1679. doi: 10.1002/jbmr.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.de Paula Rodrigues G.H., das Eiras Tâmega I., Duque G., Spinola Dias Neto V. Severe bone changes in a case of Hutchinson-Gilford syndrome. Ann. Genet. 2002;45(3):151–155. doi: 10.1016/s0003-3995(02)01119-x. [DOI] [PubMed] [Google Scholar]
- 100.Muchir A., Medioni J., Laluc M., Massart C., Arimura T., van der Kooi A.J., Desguerre I., Mayer M., Ferrer X., Briault S., Hirano M., Worman H.J., Mallet A., Wehnert M., Schwartz K., Bonne G. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve. 2004;30(4):444–450. doi: 10.1002/mus.20122. [DOI] [PubMed] [Google Scholar]
- 101.Holt I., Ostlund C., Stewart C.L., Man Nt, Worman H.J., Morris G.E. Effect of pathogenic mis-sense mutations in lamin A on its interaction with emerin in vivo. J. Cell Sci. 2003;116(Pt 14):3027–3035. doi: 10.1242/jcs.00599. [DOI] [PubMed] [Google Scholar]
- 102.Ostlund C., Bonne G., Schwartz K., Worman H.J. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J. Cell Sci. 2001;114(Pt 24):4435–4445. doi: 10.1242/jcs.114.24.4435. [DOI] [PubMed] [Google Scholar]
- 103.Vigouroux C., Auclair M., Dubosclard E., Pouchelet M., Capeau J., Courvalin J.C., Buendia B. Nuclear envelope disorganization in fibroblasts from lipodystrophic patients with heterozygous R482Q/W mutations in the lamin A/C gene. J. Cell Sci. 2001;114(Pt 24):4459–4468. doi: 10.1242/jcs.114.24.4459. [DOI] [PubMed] [Google Scholar]
- 104.Novelli G., Muchir A., Sangiuolo F., Helbling-Leclerc A., D'Apice M.R., Massart C., Capon F., Sbraccia P., Federici M., Lauro R., Tudisco C., Pallotta R., Scarano G., Dallapiccola B., Merlini L., Bonne G. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002;71(2):426–431. doi: 10.1086/341908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goldman R.D., Shumaker D.K., Erdos M.R., Eriksson M., Goldman A.E., Gordon L.B., Gruenbaum Y., Khuon S., Mendez M., Varga R., Collins F.S. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. U. S. A. 2004;101(24):8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Broers J.L., Peeters E.A., Kuijpers H.J., Endert J., Bouten C.V., Oomens C.W., Baaijens F.P., Ramaekers F.C. Decreased mechanical stiffness in LMNA −/− cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum. Mol. Genet. 2004;13(21):2567–2580. doi: 10.1093/hmg/ddh295. [DOI] [PubMed] [Google Scholar]
- 107.Houben F., Ramaekers F.C., Snoeckx L.H., Broers J.L. Role of nuclear lamina-cytoskeleton interactions in the maintenance of cellular strength. Biochim. Biophys. Acta. 2007;1773(5):675–686. doi: 10.1016/j.bbamcr.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 108.Ramdas N.M., Shivashankar G.V. Cytoskeletal control of nuclear morphology and chromatin organization. J. Mol. Biol. 2015;427(3):695–706. doi: 10.1016/j.jmb.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 109.Ho C.Y., Jaalouk D.E., Vartiainen M.K., Lammerding J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature. 2013;497(7450):507–511. doi: 10.1038/nature12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Crisp M., Liu Q., Roux K., Rattner J.B., Shanahan C., Burke B., Stahl P.D., Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol. 2006;172(1):41–53. doi: 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tapley E.C., Starr D.A. Connecting the nucleus to the cytoskeleton by SUN-KASH bridges across the nuclear envelope. Curr. Opin. Cell Biol. 2013;25(1):57–62. doi: 10.1016/j.ceb.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Katsumi A., Orr A.W., Tzima E., Schwartz M.A. Integrins in mechanotransduction. J. Biol. Chem. 2004;279(13):12001–12004. doi: 10.1074/jbc.R300038200. [DOI] [PubMed] [Google Scholar]
- 113.Le Dour C., Wu W., Béréziat V., Capeau J., Vigouroux C., Worman H.J. Extracellular matrix remodeling and transforming growth factor-β signaling abnormalities induced by lamin A/C variants that cause lipodystrophy. J. Lipid Res. 2016;58(1):151–163. doi: 10.1194/jlr.M071381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hernandez L., Roux K.J., Wong E.S., Mounkes L.C., Mutalif R., Navasankari R., Rai B., Cool S., Jeong J.W., Wang H., Lee H.S., Kozlov S., Grunert M., Keeble T., Jones C.M., Meta M.D., Young S.G., Daar I.O., Burke B., Perantoni A.O., Stewart C.L. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev. Cell. 2010;19(3):413–425. doi: 10.1016/j.devcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brachner A., Reipert S., Foisner R., Gotzmann J. LEM2 is a novel MAN1-related inner nuclear membrane protein associated with A-type lamins. J. Cell Sci. 2005;118(Pt 24):5797–5810. doi: 10.1242/jcs.02701. [DOI] [PubMed] [Google Scholar]
- 116.Bourgeois B., Gilquin B., Tellier-Lebègue C., Östlund C., Wu W., Pérez J., El Hage P., Lallemand F., Worman H.J., Zinn-Justin S. Inhibition of TGF-β signaling at the nuclear envelope: characterization of interactions between MAN1, Smad2 and Smad3, and PPM1A. Sci. Signal. 2013;6(280):ra49. doi: 10.1126/scisignal.2003411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vaughan A., Alvarez-Reyes M., Bridger J.M., Broers J.L., Ramaekers F.C., Wehnert M., Morris G.E., Whitfield W.G.F., Hutchison C.J. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J. Cell Sci. 2001;114(Pt 14):2577–2590. doi: 10.1242/jcs.114.14.2577. [DOI] [PubMed] [Google Scholar]
- 118.Markiewicz E., Tilgner K., Barker N., van de Wetering M., Clevers H., Dorobek M., Hausmanowa-Petrusewicz I., Ramaekers F.C., Broers J.L., Blankesteijn W.M., Salpingidou G., Wilson R.G., Ellis J.A., Hutchison C.J. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006;25(14):3275–3285. doi: 10.1038/sj.emboj.7601230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fedorchak G.R., Kaminski A., Lammerding J. Cellular mechanosensing: getting to the nucleus of it all. Prog. Biophys. Mol. Biol. 2014;115(2–3):76–92. doi: 10.1016/j.pbiomolbio.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bamburg J.R. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 121.Wiggan O., Schroder B., Krapf D., Bamburg J.R., DeLuca J.G. Cofilin regulates nuclear architecture through a myosin-II dependent mechanotransduction module. Sci Rep. 2017;7:40953. doi: 10.1038/srep40953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen L., Shi K., Frary C.E., Ditzel N., Hu H., Qiu W., Kassem M. Inhibiting actin depolymerization enhances osteoblast differentiation and bone formation in human stromal stem cells. Stem Cell Res. 2015;15(2):281–289. doi: 10.1016/j.scr.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 123.Van Troys M., Huyck L., Leyman S., Dhaese S., Vandekerkhove J., Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur. J. Cell Biol. 2008;87(8–9):649–667. doi: 10.1016/j.ejcb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 124.Misra U.K., Deedwania R., Pizzo S.V. Binding of activated alpha2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J. Biol. Chem. 2005;280(28):26278–26286. doi: 10.1074/jbc.M414467200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hall A. Rho family GTPases. Biochem. Soc. Trans. 2012;40(6):1378–1382. doi: 10.1042/BST20120103. [DOI] [PubMed] [Google Scholar]
- 126.Garcia-Mata R., Boulter E., Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011;12(8):493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shin E.Y., Shim E.S., Lee C.S., Kim H.K., Kim E.G. Phosphorylation of RhoGDI1 by p21-activated kinase 2 mediates basic fibroblast growth factor-stimulated neurite outgrowth in PC12 cells. Biochem. Biophys. Res. Commun. 2009;379(2):384–389. doi: 10.1016/j.bbrc.2008.12.066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detected quantitative and qualitative differences in the six different performed pair comparisons.
Six-way Venn diagram (A), displayed by Edwards' mode, and histogram (B) of the quantitative and qualitative differentially expressed protein-spots detected by crossed comparisons among control subjects and OI patients carrying mutations in CRTAP, P3H1, and PPIB genes. A and B show distribution of common and peculiar differences in the six performed pair-comparisons.
MetaCore protein network obtained by processing cofilin-1 with the differentially expressed proteins in primary fibroblasts from CRTAP, P3H1, and PPIB patients and from control subjects. MetaCore analysis was used to map identified proteins (blue circles) on existing mammalian pathways and networks, according to known protein-protein interactions and other features established in the literature. Network proteins are visualized by specific symbols, which define the functional nature of the protein (network caption). The edges indicate the relationships existing between individual proteins, and the arrowheads represent the direction of the interaction. The following line colors designate the nature of the interaction: red = negative effect; green = positive effect; gray = unspecified effect. Based on selected parameters, only eukaryotic translation initiation factor 1b (EIF1B), coiled-coil-helix-coiled-coil-helix domain-containing protein 5 (CHCHD5), and aldehyde dehydrogenase X, mitochondrial (AL1B1) were excluded from the net.
Supplementary material
Transparency document.