

ABSTRACT
Cancer is driven by mutations in genes whose products participate in major signaling pathways that fuel cell proliferation and survival. It is easy to assume that the more of these so-called driver mutations a tumor accumulates, the faster it progresses. However, this does not appear to be the case: Data from large-scale genome sequencing studies indicate that mutations in driver oncogenes often are mutually exclusive. The mechanisms underlying the mutual exclusivity of oncogenes are not completely understood, but recent reports suggest that the mechanisms may depend on the tumor type, and the nature of interacting oncogenes. Here we discuss our recent findings that the oncogenes KRASG12D and BRAFV600E are mutually exclusive in lung cancer in mouse models because their coexpression leads to oncogene-induced senescence.
Keywords: mutual exclusivity, oncogene, RAF, RAS, senescence
Major driving oncogenes are commonly mutually exclusive
Tumor development is driven by mutations that stimulate intracellular pathways that regulate cell proliferation, survival, and invasion. Additional mutations that synergistically increase tumor growth are retained; but mutations that counteract each other, are selected against in tumor evolution. Data from cancer genome sequencing efforts have revealed that mutations in major cancer driving oncogenes (e.g., RAS, RAF, and EGFR) are often mutually exclusive, especially if the oncogenes participate in the same signal transduction pathway.1-10 Why should 2 activating mutations in the same pathway that exert similar effects be disadvantageous for a tumor cell? One potential and frequently cited explanation is that 2 activating mutations do not occur in the same tumor cell because they are functionally redundant; i.e., that their coexistence does not provide an additional benefit to the cell.11-13 If that was the case, their coexpression should not bring any negative consequences to the cell. Another potential explanation is that, coexpression of 2 oncogenes is harmful and causes cell cycle exit, senescence, or death (Fig. 1). Until recently, those 2 possibilities have not been addressed experimentally under physiological conditions in vivo.
Figure 1.
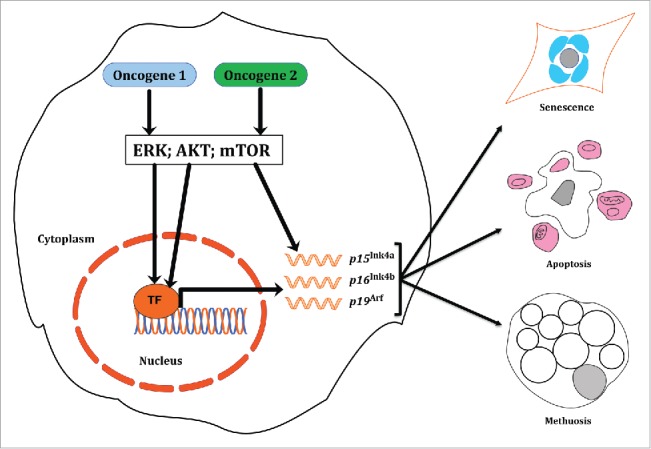
Co-expression of 2 potent oncogenes in the same cell may result in different outcomes. Expression of 2 strong oncogenes (KRAS and BRAF; or KRAS and EGFR) hyperactivates their downstream signaling pathways (ERK, AKT, mTOR etc.), and leads to transcriptional activation of target proteins. The most frequently reported suit of proteins encompasses cyclin-dependent kinase inhibitors, p15Ink4b,p16Ink4a, and p19Arf. Those proteins may, in turn, drive cells toward senescence, or death through either apoptosis, or the recently described form of cell death, methuosis. In our recent paper22 we suggest that induction of senescence underlies the mutual exclusivity of KRASG12D and BRAFV600E in lung cancer.
Coexpression of mutant forms of KRAS and BRAF induces senescence
A significant body of evidence pointed to induction of a permanent cell cycle arrest, often referred to as senescence, after activation of more than one oncogene.14,15 Senescence is a stress response of a cell to different insults, including suboptimal growth conditions, toxins, reactive oxygen species, and radiation. Oncogene-induced senescence (OIS) results from oncogene activation that leads to the generation of unusually high number of origins of replication and subsequent DNA damage response (DDR) and cell cycle arrest. A common denominator of diverse pathways leading to senescence is expression of cell cycle inhibitory proteins (CDK inhibitors).16 Among them, p16Ink4a and p19Arf play a pivotal role, and together with p15Ink4b are encoded by the INK4 locus.17,18
Could senescence explain the mutual exclusive pattern of oncogenic RAS and RAF mutations? In melanoma, the most frequently mutated driving oncogenes, NRASQ61R and BRAFV600E, activate the mitogen-activated protein kinase (MAPK) pathway.19 Those NRAS and BRAF mutations are mutually exclusive to a point that even when both mutations are found in the same tumor, they can be traced to different clones, each with a single mutation.20 Consistent with the senescence hypothesis of mutual exclusivity, forced expression of NRASQ61R in an endogenous BRAFV600E-expressing melanoma cell line resulted in a flattened cell morphology, accumulation of cells in G1/G0 phase of the cell cycle, and increased levels of senescence-associated (SA)-β-galactosidase (SA-β-gal) activity.21 Those changes were accompanied, and potentially induced by, hyperactivation of the MAPK pathway as judged by high levels of phosphorylated (p)-MEK and pERK. However, those studies did not address whether the crucial CDK inhibitors p16INK4a and p14ARF, and also p53, were involved. Moreover, NRASQ61R was ectopically overexpressed rather than expressed at physiologic levels from the endogenous promoter; it is well known that overexpression of oncogenes can cause senescence.
We recently studied the mutual exclusive nature of KRASG12D and BRAFV600E – 2 potent oncogenes and activators of MAPK signaling, in mouse models of lung cancer.22 In these models, inhalation of a Cre-adenovirus induces expression of KRASG12D or BRAFV600E from the endogenous promoters which results in physiological levels of oncogene expression in the lung.23,24 Under these conditions, BRAFV600E produced more tumors and an overall higher tumor burden than KRASG12D. On the other hand, even though KRASG12D produced fewer tumors, the tumors were larger and of a more advanced grade, suggesting a faster progression to established advanced tumors. Those results are consistent with previous studies showing that BRAFV600E has a stronger tumor-initiating capacity than KRASG12D, whereas KRASG12D can generate bigger and more advanced lesions.25-27 Indeed, the cell proliferation index was higher in KRASG12D than in BRAFV600E tumors.
We then determined the impact of expressing both oncogenes simultaneously. To approach this issue, we intercrossed BRAFV600E and KRASG12D mice to produce offspring where both oncogenes could be induced in the lung following Cre-adenovirus inhalation. Strikingly, tumor burden, number, and individual tumor area were markedly lower in double-mutant mice than in BRAFV600E mice, indicating that activation of KRASG12D expression in the setting of BRAFV600E expression is disadvantageous and reduces tumor formation. This result suggests that functional redundancy of BRAFV600E and KRASG12D is not a likely explanation for their mutual exclusivity: If the mutations were functionally redundant, we should not have observed reduced tumor burden caused by BRAFV600E. More importantly, the number of proliferating cells was lower in double mutant tumors, raising the possibility of growth arrest due to senescence induction. To investigate the molecular mechanisms behind the oncogene mutual exclusivity more closely, we isolated mouse embryonic fibroblasts (MEFs) from 12.5-day-old BRAFV600E, KRASG12D, and double-mutant embryos. We first evaluated proliferation of MEFs after in vitro transduction with the Cre-adenovirus. Consistent with the in vivo lung tumor data, proliferation of BRAFV600E/KRASG12D double-mutant MEFs was lower than in MEFs expressing either oncogene alone; and they expressed higher levels of pERKs, showed strong SA-β-galactosidase staining, and expressed higher levels of cell cycle inhibitors p16Ink4a, p15Ink4b, and p19Arf. The p21Cip1 tumor suppressor was not involved in the senescence response, as its mRNA and protein levels were unaltered. Moreover, expression of the MAPK pathway negative regulators of the Dusp, Sprouty, or Spread families was unaltered. The lack of activation of p21Cip1 was surprising given that p19Arf is a positive regulator of p53, the main transcriptional activator of p21Cip1. One possibility is that the levels of p19Arf were not sufficiently high to stabilize the p53 protein to a point where it obtains robust transcriptional activity. To further elucidate the requirement for both KRASG12D and BRAFV600E expression for the induction of senescence, we knocked down the expression of the KRASG12D oncogene in the double-mutant MEFs using retrovirally–delivered shRNAs. Knock down of KRASG12D expression increased proliferation of BRAFV600E/KRASG12D MEFs, suggesting that oncogenic KRAS is functionally involved in senescent response induced by both oncogenes. Similarly, knockdown of either p16Ink4a or p15Ink4b significantly increased BRAFV600E/KRASG12D MEFs proliferation.
Cell death may underlie the mutual exclusivity of other oncogenes
Interestingly, tumor burden in the double-mutant mice was similar to the tumor burden observed in KRASG12D mice. This finding raised the possibility that KRASG12D expression inhibits efficient BRAFV600E-induced tumorigenesis. Mutant KRAS was previously shown to induce apoptosis, primarily by stimulating the RASSF1/Nore/Mst1 tumor suppressor pathway,28 or by being phosphorylated by protein kinase C (PKC), and translocated to mitochondria.29 We stained lung sections for cleaved caspase-3 and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) – well-established markers of apoptosis. We reasoned that any proapoptotic effect of KRASG12D expression should be rapid and therefore evaluated apoptosis in lungs of mice, 4 and 10 d after Cre-adenovirus inhalation. However, the number of apoptotic cells in BRAFV600E, KRASG12D, and double-mutant lungs did not differ suggesting that apoptosis does not contribute to the mutual exclusive nature of the oncogenes. We cannot, however, exclude the possibility that double-mutant cells underwent another form of cell death. Along those lines, a recent study by Varmus and co-workers30 showed that lung cancer cells coexpressing mutant KRAS and mutant epidermal growth factor receptor (EGFR) – which are mutually exclusive oncogenes – undergo methuosis, a cell death pathway characterized by vacuolization and ruffled cell membrane.31-33 Consistent with our findings, lung cancer cells expressing both KRAS and EGFR displayed higher pERK levels.30
Ras mutations may coexist with mutations in some of its downstream pathways
Mutations in KRAS are not mutually exclusive with mutations in all of its downstream protein mediators. Activating mutations in PI3KCA frequently coexist with mutant KRAS in lung cancer.34,35 This may be related to the fact that KRAS is a relatively poor activator of the PI3K-AKT pathway,36 so it may benefit from activation of this pathway without the risk of inducing senescence. Accordingly, it has been proposed that activation of PI3K-AKT pathway in mutant KRAS tumor cells may stimulate tumor growth by reducing KRAS-induced senescence.37 In keeping with this notion, KRAS mutations frequently co-occur with mutations in HRAS, a much stronger PI3K activator,36 in soft tissue sarcomas;13 but HRAS mutations are mutually exclusive with loss of the PTEN tumor suppressor in skin cancer.38 On the contrary, HRAS is recruited to the plasma membrane and activates CRAF less efficiently than KRAS.36 It is possible that RAF-MEK-ERK activation by HRAS is suboptimal and may benefit from additional activation by e.g., mutant BRAF.
Consistent with the mutual complementation of the MAPK-ERK and PI3K-AKT pathways, mutations in BRAF have been found together with mutations in PI3K or with PTEN deletions in human tumors, and were shown experimentally to increase tumor aggressiveness in mice.39-42 Mechanistically, activation of both pathways seems to enable escape from senescence.43 However, another study revealed that expression of KRASG12V, or expression of either of 3 KRAS mutants that specifically activate one of the 3 major KRAS downstream pathways MAPK-ERK, PI3K-AKT, and RalGDS, markedly reduces endogenous BRAFV600E-induced lung cancer, suggesting that activation of canonical KRAS pathways is incompatible with BRAFV600E mutation.25 This interesting result, should, however, be interpreted with caution as high level of transgene expression of the 3 KRAS mutants was used. Regardless, it seems clear that the mechanism that underlies the mutual exclusive nature of oncogenic RAS and RAF is potent, and effectively eliminates double-mutant cells in tumor development and progression. Intriguingly, mutations in KRAS and BRAF may sporadically co-occur in advanced, metastatic disease44 likely due to inactivation of several tumor suppressive mechanisms.
A key pathway downstream of both ERK and AKT is the mTOR pathway which is responsible for new biomass synthesis required for cell growth and proliferation. It has been proposed that excessively strong mTOR signaling may stimulate OIS, especially when the cell cycle is inhibited by CDKs.45 Our data support this scenario. Lung tumors that developed in BRAFV600E/KRASG12D mice showed higher levels of phosphorylated AKTSer437, a known downstream “feed-back-target” of mTORC2 and pS6Ser235/236 protein which is downstream of mTORC1. Why does expression of both oncogenes synergistically increase mTOR activation is unclear, but may involve cumulative activation of Akt pathway directly by KRASG12D, and indirectly by BRAFV600E through activation of p90 ribosomal S6 kinase (RSK),46 inhibition of PTEN expression via AP-1 transcription factor,47 negative regulation of LKB1/AMPK signaling,48,49 or through a yet another mechanism. However, those possibilities require an experimental verification.
Summary
Our paper aimed at elucidating the mechanisms behind the mutual exclusive nature of 2 major human oncogenes, KRASG12D and BRAFV600E using a mouse model of lung cancer.22 In this paper we propose that cell cycle arrest and senescence are causally involved in this response. Our results imply that if a BRAFV600E-mutant cell acquires an additional KRASG12D mutation, or vice versa, it will hyperactivate MAPK-ERK pathway and senesce, and be outcompeted by single mutant cells. This concept fits well in the so-called “Goldilocks Principle," the idea that certain biological processes require precise levels in order to promote fitness, where either too little or too much is detrimental.50 The evidence that ERK signaling obeys this principle was nicely illustrated in a mouse model of breast cancer in which mutant KRAS expression levels were doxycycline-regulated. Low levels of KRAS activity promoted tumor formation, whereas high levels induced growth arrest and senescence.51
Presently, it is unclear whether the mutual exclusive nature of oncogenes may be exploited therapeutically. Some light on this matter was shed by an intermittent use of kinase inhibitors like BRAFV600E and MEK inhibitors. When both inhibitors were withdrawn from addicted cells, the pERK levels in those cells rebound, and their growth was inhibited.52 Moreover, pharmacologic inhibition of CDK4/6, which de facto mimics the function of the endogenous CDK inhibitors, p16INK4a and p15INK4b, is undergoing clinical trials in a number of cancer types.53
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1]. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502:333-9; PMID:24132290; https://doi.org/ 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MD, Niu B, McLellan MD, Uzunangelov V, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014; 158:929-44; PMID:25109877; https://doi.org/ 10.1016/j.cell.2014.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]. Maitra A, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Best Pract Res Clin Gastroenterol 2006; 20:211-26; PMID:16549325; https://doi.org/ 10.1016/j.bpg.2005.10.002 [DOI] [PubMed] [Google Scholar]
- [4]. Goydos JS, Mann B, Kim HJ, Gabriel EM, Alsina J, Germino FJ, Shih W, Gorski DH. Detection of B-RAF and N-RAS mutations in human melanoma. J Am Coll Surg 2005; 200:362-70; PMID:15737846; https://doi.org/ 10.1016/j.jamcollsurg.2004.10.032 [DOI] [PubMed] [Google Scholar]
- [5]. Alsina J, Gorsk DH, Germino FJ, Shih W, Lu SE, Zhang ZG, Yang JM, Hait WN, Goydos JS. Detection of mutations in the mitogen-activated protein kinase pathway in human melanoma. Clin Cancer Res 2003; 9:6419-25; PMID:14695143 [PubMed] [Google Scholar]
- [6]. Borras E, Jurado I, Hernan I, Gamundi MJ, Dias M, Marti I, Mane B, Arcusa A, Agundez JA, Blanca M, et al. Clinical pharmacogenomic testing of KRAS, BRAF and EGFR mutations by high resolution melting analysis and ultra-deep pyrosequencing. BMC Cancer 2011; 11:406; PMID:21943394; https://doi.org/ 10.1186/1471-2407-11-406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]. Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol 2011; 12:175-80; PMID:21277552; https://doi.org/ 10.1016/S1470-2045(10)70087-5 [DOI] [PubMed] [Google Scholar]
- [8]. Kinno T, Tsuta K, Shiraishi K, Mizukami T, Suzuki M, Yoshida A, Suzuki K, Asamura H, Furuta K, Kohno T, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014; 25:138-42; PMID:24297085; https://doi.org/ 10.1093/annonc/mdt495 [DOI] [PubMed] [Google Scholar]
- [9]. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002; 418:934; PMID:12198537; https://doi.org/ 10.1038/418934a [DOI] [PubMed] [Google Scholar]
- [10]. Seth R, Crook S, Ibrahem S, Fadhil W, Jackson D, Ilyas M. Concomitant mutations and splice variants in KRAS and BRAF demonstrate complex perturbation of the Ras/Raf signalling pathway in advanced colorectal cancer. Gut 2009; 58:1234-41; PMID:19474002; https://doi.org/ 10.1136/gut.2008.159137 [DOI] [PubMed] [Google Scholar]
- [11]. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011; 11:761-74; PMID:21993244; https://doi.org/ 10.1038/nrc3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008; 9:517-31; PMID:18568040; https://doi.org/ 10.1038/nrm2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Yeang CH, McCormick F, Levine A. Combinatorial patterns of somatic gene mutations in cancer. FASEB J 2008; 22:2605-22; PMID:18434431; https://doi.org/ 10.1096/fj.08-108985 [DOI] [PubMed] [Google Scholar]
- [14]. Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol 2005; 37:961-76; PMID:15743671; https://doi.org/ 10.1016/j.biocel.2004.10.013 [DOI] [PubMed] [Google Scholar]
- [15]. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15:482-96; PMID:24954210; https://doi.org/ 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- [16]. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell 2007; 130:223-33; PMID:17662938; https://doi.org/ 10.1016/j.cell.2007.07.003 [DOI] [PubMed] [Google Scholar]
- [17]. Sherr CJ. Ink4-Arf locus in cancer and aging. Wiley Interdisciplinary Rev 2012; 1:731-41; https://doi.org/ 10.1002/wdev.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Lanigan F, Geraghty JG, Bracken AP. Transcriptional regulation of cellular senescence. Oncogene 2011; 30:2901-11; PMID:21383691; https://doi.org/ 10.1038/onc.2011.34 [DOI] [PubMed] [Google Scholar]
- [19]. Zhang T, Dutton-Regester K, Brown KM, Hayward NK. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res 2016; 29(3):266-83; PMID:26833684 [DOI] [PubMed] [Google Scholar]
- [20]. Sensi M, Nicolini G, Petti C, Bersani I, Lozupone F, Molla A, Vegetti C, Nonaka D, Mortarini R, Parmiani G, et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene 2006; 25:3357-64; PMID:16462768; https://doi.org/ 10.1038/sj.onc.1209379 [DOI] [PubMed] [Google Scholar]
- [21]. Petti C, Molla A, Vegetti C, Ferrone S, Anichini A, Sensi M. Coexpression of NRASQ61R and BRAFV600E in human melanoma cells activates senescence and increases susceptibility to cell-mediated cytotoxicity. Cancer Res 2006; 66:6503-11; PMID:16818621; https://doi.org/ 10.1158/0008-5472.CAN-05-4671 [DOI] [PubMed] [Google Scholar]
- [22]. Cisowski J, Sayin VI, Liu M, Karlsson C, Bergo MO. Oncogene-induced senescence underlies the mutual exclusive nature of oncogenic KRAS and BRAF. Oncogene 2016; 35:1328-33; PMID:26028035; https://doi.org/ 10.1038/onc.2015.186 [DOI] [PubMed] [Google Scholar]
- [23]. Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001; 15:3243-8; PMID:11751630; https://doi.org/ 10.1101/gad.943001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev 2007; 21:379-84; PMID:17299132; https://doi.org/ 10.1101/gad.1516407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Vandal G, Geiling B, Dankort D. Ras effector mutant expression suggest a negative regulator inhibits lung tumor formation. PLoS One 2014; 9:e84745; PMID:24489653; https://doi.org/ 10.1371/journal.pone.0084745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]. Trejo CL, Juan J, Vicent S, Sweet-Cordero A, McMahon M. MEK1/2 inhibition elicits regression of autochthonous lung tumors induced by KRASG12D or BRAFV600E. Cancer Res 2012; 72:3048-59; PMID:22511580; https://doi.org/ 10.1158/0008-5472.CAN-11-3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Andreadi C, Cheung LK, Giblett S, Patel B, Jin H, Mercer K, Kamata T, Lee P, Williams A, McMahon M, et al. The intermediate-activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev 2012; 26:1945-58; PMID:22892241; https://doi.org/ 10.1101/gad.193458.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene 2003; 22:8999-9006; PMID:14663478; https://doi.org/ 10.1038/sj.onc.1207111 [DOI] [PubMed] [Google Scholar]
- [29]. Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, Miura J, Wiener HH, Wright L, Saba SG, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell 2006; 21:481-93; PMID:16483930; https://doi.org/ 10.1016/j.molcel.2006.01.012 [DOI] [PubMed] [Google Scholar]
- [30]. Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. Elife 2015; 4:e06907; PMID:26047463; https://doi.org/ 10.7554/eLife.06907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Chi S, Kitanaka C, Noguchi K, Mochizuki T, Nagashima Y, Shirouzu M, Fujita H, Yoshida M, Chen W, Asai A, et al. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene 1999; 18:2281-90; PMID:10327074; https://doi.org/ 10.1038/sj.onc.1202538 [DOI] [PubMed] [Google Scholar]
- [32]. Overmeyer JH, Kaul A, Johnson EE, Maltese WA. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol Cancer Res 2008; 6:965-77; PMID:18567800; https://doi.org/ 10.1158/1541-7786.MCR-07-2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]. Maltese WA, Overmeyer JH. Methuosis: nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am J Pathol 2014; 184:1630-42; PMID:24726643; https://doi.org/ 10.1016/j.ajpath.2014.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Kawano O, Sasaki H, Endo K, Suzuki E, Haneda H, Yukiue H, Kobayashi Y, Yano M, Fujii Y. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer 2006; 54:209-15; PMID:16930767; https://doi.org/ 10.1016/j.lungcan.2006.07.006 [DOI] [PubMed] [Google Scholar]
- [35]. Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, Zakowski MF, Rusch V, Sima CS, Ladanyi M, et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Therapeutics 2012; 11:485-91; PMID:22135231; https://doi.org/ 10.1158/1535-7163.MCT-11-0692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Yan J, Roy S, Apolloni A, Lane A, Hancock JF. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem 1998; 273:24052-6; PMID:9727023; https://doi.org/ 10.1074/jbc.273.37.24052 [DOI] [PubMed] [Google Scholar]
- [37]. Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell 2011; 42:36-49; PMID:21474066; https://doi.org/ 10.1016/j.molcel.2011.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38]. Mao JH, To MD, Perez-Losada J, Wu D, Del Rosario R, Balmain A. Mutually exclusive mutations of the Pten and ras pathways in skin tumor progression. Genes Dev 2004; 18:1800-5; PMID:15289454; https://doi.org/ 10.1101/gad.1213804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]. Janku F, Lee JJ, Tsimberidou AM, Hong DS, Naing A, Falchook GS, Fu S, Luthra R, Garrido-Laguna I, Kurzrock R. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One 2011; 6:e22769; PMID:21829508; https://doi.org/ 10.1371/journal.pone.0022769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40]. Trejo CL, Green S, Marsh V, Collisson EA, Iezza G, Phillips WA, McMahon M. Mutationally activated PIK3CA(H1047R) cooperates with BRAF(V600E) to promote lung cancer progression. Cancer Res 2013; 73:6448-61; PMID:24019382; https://doi.org/ 10.1158/0008-5472.CAN-13-0681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41]. Marsh Durban V, Deuker MM, Bosenberg MW, Phillips W, McMahon M. Differential AKT dependency displayed by mouse models of BRAFV600E-initiated melanoma. J Clin Invest 2013; 123:5104-18; PMID:24200692; https://doi.org/ 10.1172/JCI69619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42]. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 2009; 41:544-52; PMID:19282848; https://doi.org/ 10.1038/ng.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43]. Vredeveld LC, Possik PA, Smit MA, Meissl K, Michaloglou C, Horlings HM, Ajouaou A, Kortman PC, Dankort D, McMahon M, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev 2012; 26:1055-69; PMID:22549727; https://doi.org/ 10.1101/gad.187252.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44]. Oliveira C, Velho S, Moutinho C, Ferreira A, Preto A, Domingo E, Capelinha AF, Duval A, Hamelin R, Machado JC, et al. KRAS and BRAF oncogenic mutations in MSS colorectal carcinoma progression. Oncogene 2007; 26:158-63; PMID:16953233; https://doi.org/ 10.1038/sj.onc.1209758 [DOI] [PubMed] [Google Scholar]
- [45]. Leontieva OV, Blagosklonny MV. Tumor promoter-induced cellular senescence: cell cycle arrest followed by geroconversion. Oncotarget 2014; 5:12715-27; PMID:25587030; https://doi.org/ 10.18632/oncotarget.3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46]. Romeo Y, Moreau J, Zindy PJ, Saba-El-Leil M, Lavoie G, Dandachi F, Baptissart M, Borden KL, Meloche S, Roux PP. RSK regulates activated BRAF signalling to mTORC1 and promotes melanoma growth. Oncogene 2013; 32:2917-26; PMID:22797077; https://doi.org/ 10.1038/onc.2012.312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47]. Hettinger K, Vikhanskaya F, Poh MK, Lee MK, de Belle I, Zhang JT, Reddy SA, Sabapathy K. c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differentiation 2007; 14:218-29; PMID:16676006; https://doi.org/ 10.1038/sj.cdd.4401946 [DOI] [PubMed] [Google Scholar]
- [48]. Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell 2009; 33:237-47; PMID:19187764; https://doi.org/ 10.1016/j.molcel.2008.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49]. Esteve-Puig R, Canals F, Colome N, Merlino G, Recio JA. Uncoupling of the LKB1-AMPKalpha energy sensor pathway by growth factors and oncogenic BRAF. PLoS One 2009; 4:e4771; PMID:19274086; https://doi.org/ 10.1371/journal.pone.0004771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50]. Amin AD, Rajan SS, Groysman MJ, Pongtornpipat P, Schatz JH. oncogene overdose: Too much of a bad thing for oncogene-addicted cancer cells. Biomarkers Cancer 2015; 7:25-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51]. Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol 2007; 9:493-505; PMID:17450133; https://doi.org/ 10.1038/ncb1567 [DOI] [PubMed] [Google Scholar]
- [52]. Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015; 27:240-56; PMID:25600339; https://doi.org/ 10.1016/j.ccell.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53]. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov 2016; 6:353-67; PMID:26658964; https://doi.org/ 10.1158/2159-8290.CD-15-0894 [DOI] [PMC free article] [PubMed] [Google Scholar]