

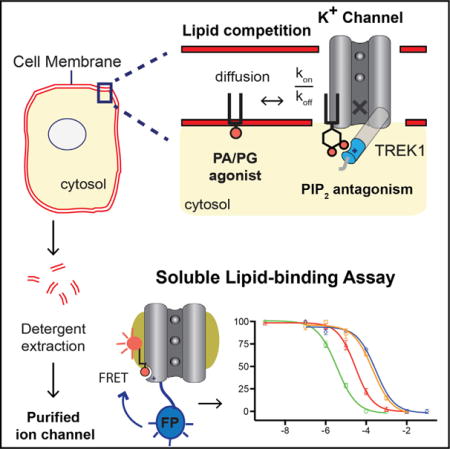
SUMMARY
Lipid regulation of ion channels by low-abundance signaling lipids phosphatidylinositol 4,5-bisphosphate (PIP2) and phosphatidic acid (PA) has emerged as a central cellular mechanism for controlling ion channels and the excitability of nerves. A lack of robust assays suitable for facile detection of a lipid bound to a channel has hampered the probing of the lipid binding sites and measuring the pharmacology of putative lipid agonists for ion channels. Here, we show a fluorescent PIP2 competition assay for detergent-purified potassium channels, including TWIK-1-related K+-channel (TREK-1). Anionic lipids PA and phosphatidylglycerol (PG) bind dose dependently (9.1 and 96 mM, respectively) and agonize the channel. Our assay shows PIP2 binds with high affinity (0.87 mM) but surprisingly can directly antagonize TREK-1 in liposomes. We propose a model for TREK-1 lipid regulation where PIP2 can compete with PA and PG agonism based on the affinity of the lipid for a site within the channel.
Graphical abstract
INTRODUCTION
Tandem of pore domains in a weak inward rectifying K+ channel (TWIK)-1-related K+-channel (TREK-1) is a two-pore domain potassium channel (K2P) expressed throughout the brain and peripheral nervous system (Fink et al., 1996). Small to medium sensory neurons in the dorsal root ganglion (DRG) express high levels of TREK-1, and this expression gives rise to a hyperpolarizing background current that works opposite nociceptive channels (e.g., transient receptor potential cation channel subfamily V member 1 [TRPV1] channels; Alloui et al., 2006) to suppress pain (Li and Toyoda, 2015). Stretch, swelling, poly-unsaturated fatty acids (PUFAs), volatile anesthetics (Enyedi and Czirják, 2010; Honoré et al., 2002; Maingret et al., 2000; Noël et al., 2009; Patel and Honoré, 2001), and the anionic signaling lipids phosphatidylinositol 4,5-bisphosphate (PIP2) and phosphatidic acid (PA) are thought to regulate TREK-1 (Chemin et al., 2005; Comoglio et al., 2014; Lopes et al., 2005). In addition to TREK-1, anionic lipids regulate numerous ion channels that control cell membrane potential, permeability, and excitability (Hansen, 2015; Hille et al., 2015; Suh and Hille, 2008).
Recently, structural and biochemical studies have shown that anionic signaling lipids directly bind to and gate ion channels (Gamper and Shapiro, 2007; Hansen, 2015; Hansen et al., 2011; Svobodova and Groschner, 2016; Whorton and MacKinnon, 2011). For example, PIP2 and PA bind tightly to a lipid binding site located within the transmembrane domain of the inward K+ rectifier Kir2.2, gate the channel in a dose-dependent manner, and define the prototypical “lipid gating” of an ion channel (Hansen, 2015; Hansen et al., 2011). Also, PIP2 inhibits TRPV1 by directly binding to a site in the transmembrane domain of the channel (Gao et al., 2016). Perturbations of anionic lipids in cell membranes regulate TREK-1 similar to Kir and TRPV1, suggesting lipids may bind directly to a site in TREK-1 (Chemin et al., 2005, 2007). Supporting this hypothesis, a recent crystal structure revealed the primary residues that conveyed PIP2 sensitivity are in a structured α helix extending from the fourth transmembrane domain (M4) helix (Dong et al., 2015) (PDB: 4XDK, D subunit) (see Figure S1).
However, which lipids bind and whether they agonize or antagonize the channel remains unclear. PA appears to activate TREK-1 as phospholipase D2 (PLD2) directly binds to TREK-1 and activates the channel through local production of lipid (Comoglio et al., 2014). PIP2 regulation of TREK-1 is more complex; initially, PIP2 was thought to activate the channel, and Gq depletion of PIP2 inactivated the channel (Kang et al., 2006; Lopes et al., 2005). But later studies showed increasing PIP2 after activation inhibits TREK-1 (Chemin et al., 2007). To identify lipid ligands and measure the affinities of lipids directly binding to potassium channels, we developed a simple mix-and-read fluorescent lipid binding assay amenable to TREK-1, TWIK-related arachidonic acid activated K+ channel (TRAAK), Kir2.1, and Kir2.2 channels. We show that PIP2 directly binds to TREK-1 and competes with lipid agonists PA and phosphatidylglycerol (PG) in purified liposomes. These data, combined with previous studies of PLD2 activation of TREK-1 (Comoglio et al., 2014), led us to propose a model where PIP2 can directly antagonize TREK-1.
RESULTS
Development of a Lipid Binding Assay
To test PIP2 binding to TREK-1, we fused a nanoluciferase (Nluc) to the C terminus of the channel and identified a soluble fluorescent PIP2 (FL-PIP2) analog suitable for binding to TREK-1. Figure 1A shows a cartoon of the assay design. Activation of Nluc produced an FL-PIP2-dependent bioluminescence resonance energy transfer (BRET) signal specific to TREK-1 (Figure 1B). When directly titrated, the soluble FL-PIP2 probe bound TREK-1 with a dissociation constant (Kd) of 1.6 μM and a Hill slope of 1.35 (Figure 1C).
Figure 1. Soluble FL-PIP2 Binding Assay to PIP2 Channels.

(A) Cartoon representation showing a single subunit of detergent-purified TREK-1 fused to the bioluminescent protein, Nluc. Excitation of bound FL-PIP2 by Nluc produces a BRET-specific signal. A structure of FL-PIP2 (right) labeled with BODIPY-TMR (red circle) (542/574 nm).
(B) Specific binding of FL-PIP2 to TREK-1, TRAAK, Kir2.1, and Kir2.2 and competition with 100 μM non-fluorescent 8:0 PIP2. No binding was observed for GIRK.
(C) Binding saturation curve of FL-PIP2 to TREK-1 (Kd, 1.6 μM; Hill slope, 1.4).
(D) Dose-dependent competition of bound 3H-PIP2 (200 nM) with FL-PIP2 (Kd, 0.8 μM; Hill slope, −0.9).
(E) 8:0 PIP2 competition FL-PIP2 for TREK-1 (Kd of 0.9 ± 0.1 μM), TRAAK (1.4 ± 0.3 μM), Kir2.1 (0.8 ± 0.2 μM), and Kir2.2 (0.1 ± 0.1 μM).
(F) 8:0 PA competes with Kd of 19.6 ± 2.9 μM for TREK-1, 8.5 ± 0.6 μM for TRAAK, 20.1 ± 5.6 μM for Kir2.1, and 29.1 ± 5.3 μM for Kir2.2.
Error bars represent SEM (n = 3–6). See also Figures S1 and S2 and Table S1.
In addition to TREK-1, FL-PIP2 bound to purified Kir2.2, Kir2.1, and TRAAK channels (Figure 1B). As mentioned, Kir2.2 has known direct binding of PIP2 to the transmembrane domain at the cytoplasmic interface (Hansen et al., 2011). TRAAK is also thought to bind PIP2 (Enyedi and Czirják, 2010). Kir2.2 had the highest affinity for PIP2 at 120 nM followed by Kir2.1, TREK-1, and TRAAK. As a negative control, we tested FL-PIP2 binding to G protein inward-rectifying potassium channel (GIRK/Kir3.2). GIRK is a Kir2 homolog, but only weakly binds PIP2 in the absence of G protein (>30 μM) (Whorton and MacKinnon, 2011). As expected, we saw no significant binding of FL-PIP2 up to 10 μM (Figures 1B and 1C). The assay also worked using a FRET-based assay with GFP (Figure S2A), albeit it required 100 times the protein as the BRET assay (100 nM versus 1.0 nM).
To eliminate the potential artifacts from the luciferase enzyme or the fluorophore attached to the probe, we confirmed PIP2 binding to TREK-1 in a radioactive version of the lipid binding assay using a tritiated (3H) PIP2 and a scintillation proximity assay (SPA). Figure 1D shows binding of 3H-PIP2 to detergent-purified TREK-1 channel; FL-PIP2 dose-dependently competed with 3H-PIP2 (Kd, 0.80 μM; Hill slope, −0.85) (see also Figure S2C).
Pharmacology of Endogenous Membrane Lipids
We rapidly screened the binding of endogenous cellular lipids to four potassium channels and identified high-affinity lipid ligands. PA and PIP2 bound with varying affinity to TREK-1, TRAAK, Kir2.2, and Kir2.1 (Figures 1E and 1F). We determined a Kd for each lipid by fitting the data to a sigmoidal dose-response curve with variable slope (GraphPad Prism). Head group composition drove TREK-1 lipid specificity for both soluble short chain (Figure S2D) and native membrane lipids (Figures 2A and 2B). PA and PIP2 consistently bound the tightest (Table S1). The zwitterionic lipids 8:0 phosphatidylethanolamine (PE) and 8:0 phosphatidylcholine (PC) had no affinity for TREK-1 (Figure S2C).
Figure 2. TREK-1 Pharmacology of Membrane Lipids.

(A) Single-point competition of soluble lipids (100 μM) with FL-PIP2 (500 nM) shows specificity for anionic lipids. Phosphatidylinositol (PI), PE, and PC do not compete for FL-PIP2 binding.
(B) TREK-1 competition curves comparing head group specificity of full-length anionic lipids. PIP2 binds with the highest affinity (Kd, 0.87 ± 0.11 μM). Kds for 18:0–20:4 PIP2 and 18:1–18:1 PIP2 are indistinguishable.
(C) Single-point competition (100 μM) of FL-PIP2 comparing cellular lipids with free fatty acids. The diacylglycerols (DAGs), 8:0 and 18:1 DAG, sphingosine-1-phosphate, stearic acid (18:0), inositol triphosphate (IP3), and EPA (20:5) fail to compete off FL-PIP2.
***p < 0.0001; **p < 0.001; *p < 0.05, Student’s t test. Error bars represent SEM (n = 3–10). See also Figures S3–S5 and Table S1.
Application of 10 μM arachidonic acid (AA) (free fatty acid) to cells expressing TREK-1 robustly activates TREK-1 (Patel et al., 1998). We tested for direct binding of AA to TREK-1 and found both ω-3 and ω-6 AA (free fatty acid) competed with FL-PIP2 (Figure 2C). Fatty acid binding was highly specific because eicosapentaenoic acid (EPA, 20:5 ω-3) and saturated fatty acids failed to bind (Figure 3C). The Kds for free ω-3 and ω-6 AAs were relatively weak, 67.1 ± 10.8 μM and 73.1 ± 12.1 μM, respectively (Figure 3D; Table S1), suggesting phospholipid incorporation into the membrane may also contribute to the activation of TREK-1.
Figure 3. TREK-1 Lipid Gating by Anionic Lipids.

Ion flux assays of purified TREK-1 (GFP or Nluc tagged) reconstituted into DOPC proteoliposomes with anionic lipid.
(A) DOPG (PG) dose-dependently agonizes TREK-1, 15% > 10% > 5% (0.81 ± 0.07, 0.43 ± 0.19, 0.16 ± 0.02, respectively, normalized fluorescence). A black arrow indicates the initiation of flux with CCCP. No flux was observed with 25% DOPE (PE) (red trace).
(B) Comparison of end fluorescence shown in (A).
(C) DOPA (PA) also agonizes TREK-1 at 15 and 10 mol% (0.43 ± 0.11 and 0.21 ± 0.05, respectively).
(D) Comparison of end fluorescence shown in (C).
(E) DOPIP2 (PIP2) fails to activate TREK-1 (0.5, 1, and 2 mol% tested).
(F) Comparison of end fluorescence shown in (E).
(G) Mixtures of PIP2 and PG (10%) show 5 mol% PIP2 competes with PG to inhibit TREK-1 (0.11 ± 0.08, normalized fluorescence). Low concentration of PIP2 in the presence of 10% PG enhanced TREK-1 agonism (orange trace, 0.79 ± 0.04).
(H) Comparison of end fluorescence shown in (G).
(I) Fluorescence emission from TREK1 with a GFP tag after reconstitution into proteoliposomes.
Error bars represent SEM (n = 3–5); no error bar in (I) is a single measurement. ***p < 0.0001, **p < 0.001, *p < 0.05, ANOVA-Tukey’s multiple comparison test.
Incorporation of AA into nociceptive membrane directly modulates touch through arachidonyl-containing membrane lipids (Vásquez et al., 2014). If the arachidonyl acyl chains of activating lipids bind directly to the channel, this could increase the affinity and further activate the channel. Using our BRET assay, we found arachidonic-acid-containing PA (AA-PA) (18:0–20:4) bound 2-fold tighter compared with 18:1 PA (9.1 μM versus 15.8 μM; p = 0.0423). AA-PA was also statistically a higher affinity than 16:0–18:1 PA (p = 0.0356). This was not true of arachidonyl PIP2 (18:0–20:4) (Figures S3A and S3B; Table S1). Complete removal of an acyl chain had the greater affect. TREK-1 affinity for lyso-PA decreased 3.5-fold compared with diacyl (18:1) PA (56.0 μM versus 15.8 μM) (see also Table S1). Shotgun lipidomics of HEK cells treated with 50 μM AA showed an ~10- and 3-fold increase in arachidonyl PG and PA, respectively, further supporting a role for arachidonyl-containing phospholipid in TREK-1 activation (Figure S4A).
Lastly, we tested the contribution of the glycerol backbone to TREK-1 affinity. Ceramide-1-phophate (C1P) is often a saturated lipid with an identical head group as PA and differs only in the acyl chain linkage (Figures S3E–S3G). We found C1P binding to TREK-1 was 4-fold weaker than PA (79.4 μM versus 19.6 μM; 8:0 and 16:0 lipids were similar). The Hill coefficients were near 1.0 for each lipid. Furthermore, lyso-C1P, i.e., sphingosine-1-phosphate (S1P), had no affinity at 100 μM (Table S1). These results suggest the acyl chain and backbone combine to give high-affinity lipid binding in TREK-1. Also, the chemical specificity suggests a structurally well-defined lipid binding site.
Lipid Gating of TREK-1
To determine whether PG, PA, and PIP2 are TREK-1 agonists or antagonists, we tested ion conductance in a flux assay (Brohawn et al., 2012). PA and PG are both products of the enzyme PLD2 (Yang et al., 1967), and we expect these lipids to agonize the channel as PLD2 binds directly to the C terminus of TREK-1 and activates the channel (Comoglio et al., 2014) (see also Figure S5). 5–15 mol% PG dose-dependently activated TREK-1 in 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) liposomes, as determined by potassium ion flux (Figures 3A and 3B). PG is relatively abundant in the plasma membrane, and the low affinity is appropriate for agonizing the channel. The flux assay assumes equal incorporation of the channel between lipid types. We confirmed TREK-1 incorporation into the liposome using fluorescence from a GFP tag on TREK-1 (Figure 3I). In PG liposomes, the fluorescence was similar, suggesting the effect of PG on TREK-1 is a lipid-specific interaction. PA likewise activated the channel, albeit with less flux (Figure 3C). Incorporation of TREK-1 appeared to increase with 15 mol% PA compared with 10 mol% (the GFP fluorescence increased to 17,800 RU; p = 0.02, unpaired Student’s t test). Hence, some of the increased PA activation may be an artifact of increased TREK-1 incorporation. In contrast with PG and PA, increasing concentrations of PIP2 (0.5, 1, and 2 mol%) failed to activate TREK-1: the highest concentration of PIP2, 2 mol%, induced the least flux (Figures 3E and 3F). Given that our soluble binding assay shows that PIP2 binds the channel, we reasoned PIP2 could bind and antagonize TREK-1.
To determine antagonistic properties of PIP2 on TREK-1, we combined increasing concentration of PIP2 with 10% PG, a concentration of PG that opens the channel (Figure 3A, blue trace). Consistent with antagonism, TREK-1 currents were completely inhibited in 5 mol% PIP2 (Figure 3C), as were channels in 2 mol%. Unexpectedly, sub-saturating PIP2 (0.5 mol%) in the presence of 10 mol% PG enhanced TREK-1 activity (Figures 3G and 3H). Reconstitution efficiency of TREK-1 into the liposomes does not account for the increased flux because the incorporation in 0.5 mol% differed from 5 mol% by less than 20% (Figure 3I), but flux increased 7-fold.
To further confirm that the lipid Kd arises from the signaling lipid directly binding to structured protein, we mutated residues in the distal end of the M4 helix, a site previously implicated in lipid binding based on electrophysiology experiments (Chemin et al., 2005). Consistent with direct lipid binding, we found mutating R297 to cysteine completely blocked 500 nM 3H-PIP2 binding to TREK-1 (Figure 4A). To confirm the functionality of this cysteine mutant, we tested channel conductance in our ion flux assay. To do this, we made a partner double-cysteine mutation, F171C + R297C (cysTREK), to trap the channel in a disulfide bonded or “lipid-bound” (active) conformation when oxidized, analogous to a pair of cys mutants previously characterized in TRAAK channels using whole-cell patch clamp (Brohawn et al., 2014a) (see Figure S1). Like patch clamp, cysTREK in liposomes rescued flux activity when oxidized; but when the disulfides were reduced, the channel’s flux significantly decreased, consistent with low-affinity agonist binding (Figure 4C). We confirmed a 10-fold loss in lipid binding affinity using our BRET assay (Figure 4B).
Figure 4. Characterization of the TREK-1 Lipid Binding Site.

(A) Cysteine mutants of TREK-1 directly block 500 nM PIP2 binding. R297 contributes to a known lipid binding site (Chemin et al., 2005), and mutant R297C/F171C (cysTREK) traps an open conformation when disulfide bonded (Brohawn et al., 2014a).
(B) Titration of mutant TREK-1 with FL-PIP2 results in an ~10-fold decrease in FL-PIP2 affinity.
(C) Ion flux assay of cysTREK-1 rescues activation in the absence of lipid binding when oxidized (purple trace), but not when reduced with DTT (orange circles); control vesicles are shown as gray circles.
(D) Small molecules fluphenazine (FLZ), thioridazine (TRZ), and norfluoxetine (NFX) dose-dependently compete with FL-PIP2. FLZ inhibition is incomplete (red arrow and dashed marker) despite a high affinity, Kd of 6.5 ± 2.0 μM. Error bars represent SEM (n = 3–6).
(E) A cartoon showing a proposed model for lipid gating of TREK-1 channels. Anionic lipids PG, PA, and PIP2 diffuse laterally in the plasma membrane and bind to the channel (a single subunit of TREK is shown). A lipid binding motif (cyan cylinder) extends from the last transmembrane helix of the M4 helix near the inner leaflet of the plasma membrane. (Left) Binding of PA (or PG) to the lipid binding motif causes the tip of the helix to translocate up, activating the channel. A solid red sphere represents the negatively charged head group phosphate. Purple spheres represent ions in the conduction pathway. (Right) PIP2 displaces PA from the lipid binding site. The geometry of the PIP2 head group positions the two negatively charged 4′- and 5′-phosphates of PIP2 (solid red circles) ~8Å away from the membrane compared with PA (the distance of the inositol ring). With PIP2 bound, the helix adopts a conformation favoring a more closed state of the channel. An X indicates a blocked conduction pathway.
See also Figures S6 and S7.
Lastly, we investigated the ability of TREK-1 small-molecule antagonists norfluoxetine (Dong et al., 2015) (NFX), fluphenazine (Thümmler et al., 2007) (FLZ), and thioridazine (Thümmler et al., 2007) (TRZ) to compete with FL-PIP2 binding. FLZ bound tightly to TREK-1 with a Kd of 6.5 ± 2.0 μM, followed by TRZ of 52.7 ± 5.5 μM, and NFX of 68.2 ± 14.0 μM (Figure 4D). Also, binding was channel specific because PIP2 binding to TRAAK was unaffected by TRZ (Figure S4B), in agreement with previous studies (Thümmler et al., 2007). Because PIP2 appears to antagonize TREK-1, it is unclear whether binding of small molecules is directly competing or if there are multiple conformations that lead to antagonism. Interestingly, FLZ appeared to bind non-competitively because 40% of FL-PIP2 remained bound at what appeared to be near-saturating concentrations (Figure 4D).
DISCUSSION
Taken together, the nanomolar affinity for PIP2, the specificity for the glycerol backbone and acyl chains, and the agonism by anionic lipids (PA or PG) solidify a high-affinity lipid binding site within TREK-1. Figure 4E shows a proposed model for lipid gating in TREK-1. The PIP2 affinity for TREK-1 (860 nM) is only slightly weaker than Kir2.1 (450 nM) and suggests that structural determinants like Kir2.2 exist (Hansen, 2015; Hansen et al., 2011).
We directly measured the functional effect of PIP2 on TREK-1 channels in a purified reconstituted system (Figure 3A). The purified system is critical to determining the direct effect of a lipid, because whole cells are confounded by indirect effects of PIP2. For example, PIP2 can activate PLD2 (Lopez et al., 1998), which would indirectly agonize TREK-1 through the generation of PA (see Figure S5A), yet appear as activation by PIP2. The lateral distribution of lipids varies dramatically (Simons and Sampaio, 2011) in a membrane, and measuring the precise concentration of lipids near a channel is difficult (Hansen, 2015). This limits our ability to correlate the antagonism in our purified system with exact relevant lipid concentrations in a biological membrane. However, crude increases in PIP2 concentration after initial activation have been shown to inactivate TREK-1 in cell culture (Chemin et al., 2007), consistent with our direct PIP2 antagonism playing a role in vivo.
The opposite effect of low and high levels of PIP2 in the presence of PG (Figures 3G and 3H) may suggest multiple sites for PIP2 binding to TREK-1. TREK-1 is a pseudotetramer (dimer of dimer) and could have two distinct lipid binding sites (one agonizing and one antagonizing). If so, agonizing lipids PA and PG could compete with PIP2 through an allosteric mechanism at distinct sites. Alternatively, the dimer could have only one PIP2 site (two per dimer) and only antagonize when both sites of the dimer are bound. At sub-saturating concentrations, the presence of PIP2 would give the appearance of higher anionic lipid concentration but lack a sufficient concentration to occupy both PIP2 sites, thus failing to antagonize the channel. This effect resembles Kir, a homotetramer, which is activated when sub-saturating PA is added to agonizing PIP2 (Cheng et al., 2011; Hansen, 2015). At high concentrations, PA competes off PIP2 and antagonizes Kir (Hansen et al., 2011; Lee et al., 2013). As mentioned, PIP2 could also indirectly activate TREK-1 through the enzyme PLD2. We recently showed PLD2 necessarily translocates to PIP2 during activation (Petersen et al., 2016). These findings combined with Comoglio’s model of localization of PLD2 in the activation of TREK-1 (Comoglio et al., 2014) suggest PIP2 could indirectly activate TREK-1 through increasing PLD2 activation and production of PA and PG agonists (see Figure S5A). This later mode of action assumes PIP2 must bind PLD2 with higher affinity than TREK-1, which is not known. A comparison of these two modes of activation is shown in Figure S5A, (i) and (ii). Distinguishing their relative contributions will require further investigation. The expected changes in cellular lipid concentration relative to TREK-1 activity are modeled in Figure S5B.
The large selective increase of arachidonyl-PG (Figure S4A) with AA treatment may indicate PG-specific regulation of TREK-1. However, the addition of AA to cells can activate TREK-1 in seconds (Patel et al., 1998), and our lipidomics study incorporated AA over 15 min. Furthermore, free AA increases TRAAK currents in liposomes (Brohawn et al., 2014b). Additional experiments with sufficient temporal resolution are needed to better define the role of free AA in TREK-1 activation.
So why does PIP2 antagonize TREK-1 in liposomes when PA and PG are activating? All three lipids are anionic. Inhibition by a cellular feedback mechanism is impossible in our purified (cell-free) liposomes. Rather, a structural or biophysical feature must explain the functional selectivity. Recent crystal structures of TREK-1 and TRAAK suggested that the lipid binding site, i.e., the distal end of the M4 helix21, can move toward the membrane in the active conformation (Brohawn et al., 2014a; Dong et al., 2015; Lolicato et al., 2014) (Figure S1). The span of the inositol head group is substantial, 8.1 Å (see PIP2 in Figure S2B), and could displace the most distal charges by positioning the anionic phosphate ~8 Å away from the membrane (Figure 4E; Figure S2B, PIP2 chemical structure). This model is consistent with the previous up/down activating models established through double-cysteine mutants of TRAAK (Brohawn et al., 2014a). Conceivably, a thinner membrane could allow PIP2 to shift up relative to the TREK-1 and allow the lipid binding site to sit closer to the active conformation. Gain-of-function mutations in TRAAK appeared to destabilize the TM4 helix prior to the lipid binding motif (Lolicato et al., 2014), which could also allow the lipid binding motif to associate with the membrane (move up), although the key residues were not visible in the pertinent structures (Lolicato et al., 2014; Miller and Long, 2012).
We conclude that lipid agonists and antagonists dually and directly contribute to gating of TREK-1 by binding with high affinity to the channel. We also demonstrate a simple mix-and-read fluorescent assay that accurately characterizes lipid specificity and affinities of a lipid bound to an ion channel. The state-dependent selectivity combined with the small requirement for protein make our soluble assay amenable to ultrahigh-throughput drug screening (2–3 million compounds). Our assay could be combined with high-throughput functional screens (200,000–300,000 compounds) (Su et al., 2016), which would allow a complete strategy for screening of ion channel in a purified system using techniques analogous to soluble proteins.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
TREK-1, TRAAK, Kir2.1, Kir2.2, or Kir3.2 (GIRK) containing a C-terminal fluorescent protein (either GFP or Nluc) with 103-His tag was expressed in Pichia yeast. After cryo-milling, the proteins were extracted in dodecyl-β-D-maltoside (DDM) with protease inhibitors and purified to homogeneity on a cobalt affinity column followed by size exclusion chromatography (SEC). The final SEC buffer contained 20 mM Tris (pH 8.0), 150 mM KCl, 1 mM EDTA, and 2 mM DDM. All proteins were expressed in milligram quantities with a predominant monodispersed peak (Figure S4C) corresponding to the expected molecular weight (MW) of an assembled channel with a fluorescent tag in detergent.
BRET Lipid Binding Assay
The BRET was done in a 384-well plate, with each well having a total of 50 μL of total reaction volume consisting of 1 nM (binding sites) purified ion channel tagged with Nluc, 500 nM BODIPY-TMR phosphatidylinositol 4,5-bisphosphate (FL-PIP2) (Echelon Biosciences), various concentrations of competing ligand, and 1:2,000 furimazine (NanoGlo; Promega), the substrate for Nluc, in the SEC buffer with 7 mM DDM final concentration. BRET signal brought about by the binding of FL-PIP2 to TREK-1-Nluc and competition with a ligand was calculated by subtracting signal from Nluc-only control enzyme (purified from bacteria) and measured using Envision Multilabel 2104 plate reader (PerkinElmer) set for dual-emission detection (540 nm for Nluc and 574 nm for FL-PIP2) and automatic BRET ratio calculation. Nluc control enzyme was purified from BL1 bacteria cells. Although not used here, background fluorescence can be determined using excess non-FL-PIP2 (e.g., C8-PIP2) rather than the Nluc control enzyme. Data points were fit to a sigmoidal dose-response curve with variable slope using GraphPad Prism. A FRET version of the assay employed a GFP tag rather than an Nluc tag. See also Figure S2A and Supplemental Experimental Procedures.
Radioactive Lipid Binding Assay
Binding of 3H-PIP2 to TREK-1 was detected in a SPA (Perkin Elmer). 100 nM TREK-1 binding sites was mixed in 0.6 mg/mL polyvinyltoluene (PVT) anti-mouse SPA beads, 0.8 μg/ml anti-His antibody, SEC buffer with 7 mM DDM, and a signal measured in a scintillation counter.
Potassium Flux Assay
Lipids of a desired ratio were mixed, dried, rehydrated, sonicated, and solubilized with 3 mM DDM. Proteoliposomes were formed by mixing 1:100 TREK-1/lipids and removal of detergent with BioBeads (150 mM KCl, 20 mM HEPES [pH 7.4]). Sonicated proteoliposomes (5 μL) were added to 195 μL of flux assay buffer (150 mM NaCl, 20 mM HEPES [pH 7.4], 2 μM 9-amino-6-chloro-2-methoxyacridine [ACMA]) in a 96-well plate at 25°C. Flux was initiated by the addition of the protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (1 μM final concentration).
Shotgun Lipidomics
HEK293 cells were treated in 10 cm plates with 50 μM AA for 15 min. The cells were scraped and frozen for lipid extraction. The lipids were extracted using a modified Bligh and Dyer procedure (Han et al., 2012) and analyzed by electrospray ionization (ESI) direct infusion analysis (Yang et al., 2009).
Statistical Methods
All statistical calculations were performed in GraphPad Prism v6.0. For the Student’s t test, significance was calculated using a two-tailed unpaired parametric test with significance defined as p < 0.05. For the multiple comparison test, significance was calculated using an ordinary one-way ANOVA and Tukey’s multiple comparison test with a single pooled variance.
Supplementary Material
Highlights.
A soluble fluorescent assay directly measures lipid binding to potassium channels
Lipids are direct ligands for two-pore domain potassium channels TREK and TRAAK
Phosphatidic acid (PA) and phosphatidylglycerol (PG) agonize TREK-1
Phosphatidylinositol 4,5 bisphosphate (PIP2) can directly antagonize TREK-1
Acknowledgments
We thank Arman Nayebosadri for help with flux assays and Andrew Hansen for discussion and comments on the manuscript. This work was supported by a Director’s New Innovator Award (1DP2NS087943-01 to S.B.H.) from the NIH.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, five figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.07.034.
AUTHOR CONTRIBUTIONS
S.B.H. and C.C. designed the experiments and wrote the manuscript. C.C. performed all the experiments except for the shotgun lipidomics, which was performed by M.W. and analyzed by X.H. and S.B.H.
References
- Alloui A, Zimmermann K, Mamet J, Duprat F, Noël J, Chemin J, Guy N, Blondeau N, Voilley N, Rubat-Coudert C, et al. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 2006;25:2368–2376. doi: 10.1038/sj.emboj.7601116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn SG, del Mármol J, MacKinnon R. Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science. 2012;335:436–441. doi: 10.1126/science.1213808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn SG, Campbell EB, MacKinnon R. Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature. 2014a;516:126–130. doi: 10.1038/nature14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn SG, Su Z, MacKinnon R. Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci USA. 2014b;111:3614–3619. doi: 10.1073/pnas.1320768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honoré E. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 2005;24:44–53. doi: 10.1038/sj.emboj.7600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Patel AJ, Duprat F, Sachs F, Lazdunski M, Honore E. Up- and down-regulation of the mechano-gated K(2P) channel TREK-1 by PIP (2) and other membrane phospholipids. Pflugers Arch. 2007;455:97–103. doi: 10.1007/s00424-007-0250-2. [DOI] [PubMed] [Google Scholar]
- Cheng WWL, D’Avanzo N, Doyle DA, Nichols CG. Dual-mode phospholipid regulation of human inward rectifying potassium channels. Biophys J. 2011;100:620–628. doi: 10.1016/j.bpj.2010.12.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comoglio Y, Levitz J, Kienzler MA, Lesage F, Isacoff EY, Sandoz G. Phospholipase D2 specifically regulates TREK potassium channels via direct interaction and local production of phosphatidic acid. Proc Natl Acad Sci USA. 2014;111:13547–13552. doi: 10.1073/pnas.1407160111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong YY, Pike ACW, Mackenzie A, McClenaghan C, Aryal P, Dong L, Quigley A, Grieben M, Goubin S, Mukhopadhyay S, et al. K2P channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science. 2015;347:1256–1259. doi: 10.1126/science.1261512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90:559–605. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M. Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J. 1996;15:6854–6862. [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci. 2007;8:921–934. doi: 10.1038/nrn2257. [DOI] [PubMed] [Google Scholar]
- Gao Y, Cao E, Julius D, Cheng Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature. 2016;534:347–351. doi: 10.1038/nature17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Yang K, Gross RW. Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom Rev. 2012;31:134–178. doi: 10.1002/mas.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SB. Lipid agonism: the PIP2 paradigm of ligand-gated ion channels. Biochim Biophys Acta. 2015;1851:620–628. doi: 10.1016/j.bbalip.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. 2011;477:495–498. doi: 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B, Dickson EJ, Kruse M, Vivas O, Suh BC. Phosphoinositides regulate ion channels. Biochim Biophys Acta. 2015;1851:844–856. doi: 10.1016/j.bbalip.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honoré E, Maingret F, Lazdunski M, Patel AJ. An intracellular proton sensor commands lipid- and mechano-gating of the K(+) channel TREK-1. EMBO J. 2002;21:2968–2976. doi: 10.1093/emboj/cdf288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Han J, Kim D. Mechanism of inhibition of TREK-2 (K2P10.1) by the Gq-coupled M3 muscarinic receptor. Am J Physiol Cell Physiol. 2006;291:C649–C656. doi: 10.1152/ajpcell.00047.2006. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Wang S, Borschel W, Heyman S, Gyore J, Nichols CG. Secondary anionic phospholipid binding site and gating mechanism in Kir2.1 inward rectifier channels. Nat Commun. 2013;4:2786. doi: 10.1038/ncomms3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Toyoda H. Role of leak potassium channels in pain signaling. Brain Res Bull. 2015;119(Pt A):73–79. doi: 10.1016/j.brainresbull.2015.08.007. [DOI] [PubMed] [Google Scholar]
- Lolicato M, Riegelhaupt PM, Arrigoni C, Clark KA, Minor DL., Jr Transmembrane helix straightening and buckling underlies activation of mechanosensitive and thermosensitive K(2P) channels. Neuron. 2014;84:1198–1212. doi: 10.1016/j.neuron.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CMB, Rohács T, Czirják G, Balla T, Enyedi P, Logothetis DE. PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J Physiol. 2005;564:117–129. doi: 10.1113/jphysiol.2004.081935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez I, Arnold RS, Lambeth JD. Cloning and initial characterization of a human phospholipase D2 (hPLD2). ADP-ribosylation factor regulates hPLD2. J Biol Chem. 1998;273:12846–12852. doi: 10.1074/jbc.273.21.12846. [DOI] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honore E. Lysophospholipids open the two-pore domain mechano-gated K(+) channels TREK-1 and TRAAK. J Biol Chem. 2000;275:10128–10133. doi: 10.1074/jbc.275.14.10128. [DOI] [PubMed] [Google Scholar]
- Miller AN, Long SB. Crystal structure of the human two-pore domain potassium channel K2P1. Science. 2012;335:432–436. doi: 10.1126/science.1213274. [DOI] [PubMed] [Google Scholar]
- Noël J, Zimmermann K, Busserolles J, Deval E, Alloui A, Diochot S, Guy N, Borsotto M, Reeh P, Eschalier A, Lazdunski M. The mechano-activated K+ channels TRAAK and TREK-1 control both warm and cold perception. EMBO J. 2009;28:1308–1318. doi: 10.1038/emboj.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AJ, Honoré E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001;24:339–346. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998;17:4283–4290. doi: 10.1093/emboj/17.15.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen EN, Chung HW, Nayebosadri A, Hansen SB. Kinetic disruption of lipid rafts is a mechanosensor for phospholipase D. Nat Commun. 2016;7:13873. doi: 10.1038/ncomms13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Sampaio JL. Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol. 2011;3:a004697. doi: 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Brown EC, Wang W, MacKinnon R. Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. Proc Natl Acad Sci USA. 2016;113:5748–5753. doi: 10.1073/pnas.1602815113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svobodova B, Groschner K. Mechanisms of lipid regulation and lipid gating in TRPC channels. Cell Calcium. 2016;59:271–279. doi: 10.1016/j.ceca.2016.03.012. [DOI] [PubMed] [Google Scholar]
- Thümmler S, Duprat F, Lazdunski M. Antipsychotics inhibit TREK but not TRAAK channels. Biochem Biophys Res Commun. 2007;354:284–289. doi: 10.1016/j.bbrc.2006.12.199. [DOI] [PubMed] [Google Scholar]
- Vásquez V, Krieg M, Lockhead D, Goodman MB. Phospholipids that contain polyunsaturated fatty acids enhance neuronal cell mechanics and touch sensation. Cell Rep. 2014;6:70–80. doi: 10.1016/j.celrep.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011;147:199–208. doi: 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SF, Freer S, Benson AA. Transphosphatidylation by phospholipase D. J Biol Chem. 1967;242:477–484. [PubMed] [Google Scholar]
- Yang K, Cheng H, Gross RW, Han X. Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Anal Chem. 2009;81:4356–4368. doi: 10.1021/ac900241u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.