

Abstract
Background
The MMP (matrix metalloproteinase) family plays diverse and critical roles in directing vascular wall remodeling in atherosclerosis. Unlike secreted‐type MMPs, a member of the membrane‐type MMP family, MT1‐MMP (membrane‐type 1 MMP; MMP14), mediates pericellular extracellular matrix degradation that is indispensable for maintaining physiological extracellular matrix homeostasis. However, given the premature mortality exhibited by MT1‐MMP–null mice, the potential role of the proteinase in atherogenesis remains elusive. We sought to determine the effects of both MT1‐MMP heterozygosity and tissue‐specific gene targeting on atherogenesis in APOE (apolipoprotein E)–null mice.
Methods and Results
MT1‐MMP heterozygosity in the APOE‐null background (Mmp14 +/− Apoe −/−) significantly promoted atherogenesis relative to Mmp14 +/+ Apoe −/− mice. Furthermore, the tissue‐specific deletion of MT1‐MMP from vascular smooth muscle cells (VSMCs) in SM22α‐Cre(+)Mmp14 F/F Apoe −/− (VSMC‐knockout) mice likewise increased the severity of atherosclerotic lesions. Although VSMC‐knockout mice also developed progressive atherosclerotic aneurysms in their iliac arteries, macrophage‐ and adipose‐specific MT1‐MMP–knockout mice did not display this sensitized phenotype. In VSMC‐knockout mice, atherosclerotic lesions were populated by hyperproliferating VSMCs (smooth muscle actin– and Ki67–double‐positive cells) that were characterized by a proinflammatory gene expression profile. Finally, MT1‐MMP–null VSMCs cultured in a 3‐dimensional spheroid model system designed to mimic in vivo–like cell–cell and cell–extracellular matrix interactions, likewise displayed markedly increased proliferative potential.
Conclusions
MT1‐MMP expressed by VSMCs plays a key role in limiting the progression of atherosclerosis in APOE‐null mice by regulating proliferative responses and inhibiting the deterioration of VSMC function in atherogenic vascular walls.
Keywords: aneurysm, atherosclerosis, inflammation, matrix metalloproteinases, muscle, smooth
Subject Categories: Atherosclerosis, Aneurysm, Vascular Biology, Smooth Muscle Proliferation and Differentiation, Genetically Altered and Transgenic Models
Clinical Perspective
What Is New?
The molecular mechanisms underlying proliferative atherosclerosis have not been fully defined.
This animal study suggests that a pericellular collagenase called MT1‐MMP (membrane‐type 1 matrix metalloproteinase), expressed by vascular smooth muscle cells, plays a critical role in limiting the progression of proliferative atherosclerotic lesions.
The loss of vascular smooth muscle cell MT1‐MMP leads to advanced proliferative atherosclerosis and atherosclerotic iliac artery aneurysm formation.
What Are the Clinical Implications?
The functional impairment of vascular smooth muscle cells in regulating vascular wall extracellular matrix remodeling may contribute to the pathogenesis of proliferative atherosclerosis and atherosclerotic iliac artery aneurysm formation.
Introduction
Vascular smooth muscle cells (VSMCs) constitute the major cellular component of the tunica media, where they play key roles in regulating vascular tone and blood flow.1 Under physiological conditions, the number of VSMCs within the arterial wall is tightly controlled, as is arterial wall thickness.1, 2 In marked contrast, during the progression of atherosclerosis, VSMCs proliferate and transition from a contractile to a synthetic phenotype, thereby depositing excess extracellular matrix (ECM) molecules that lead to arterial wall thickening and stiffening, namely, arteriosclerosis.3, 4 However, the molecular mechanisms that underlie VSMC proliferation and arteriosclerosis during the pathological process of atherosclerosis remain largely undefined.
ECM remodeling is mediated by members of the MMP (matrix metalloproteinase) gene family, a group of structurally related proteolytic enzymes that are broadly characterized as either secreted or membrane tethered.5 Consistent with their potential roles in vascular wall pathology, recent studies have characterized the roles of secreted MMPs, particularly MMP‐2, MMP‐8, and MMP‐13, in promoting atherogenesis or plaque rupture.6, 7, 8 With regard to the membrane‐tethered MMPs, MT1‐MMP uniquely serves as a pericellular collagenase that cleaves native, triple‐helical collagens associated with both the basement membrane and interstitial matrix.9 Indeed, in contrast to almost all other MMP family members, in which gene targeting exerts only minor effects on mouse development, MT1‐MMP–knockout animals display severe dwarfism, lipodystrophy, and premature lethality, underscoring the key roles played by MT1‐MMP in maintaining tissue homeostasis.10, 11, 12 Given the premature lethality of MT1‐MMP–knockout mice, however, attempts to characterize the role of MT1‐MMP in atherogenesis have been limited. Schneider et al used a bone marrow transplant system to transfer MT1‐MMP–null bone marrow cells into atherogenic Ldlr −/− mice to assess the role of the proteinase when expressed by myeloid cells in atherogenesis.13 The reconstitution of atherogenic mice with MT1‐MMP–null bone marrow cells did not alter the size of the atheroma or the number of infiltrating macrophages but did result in increased collagen content within the atheromatous lesions.13 Consequently, although myeloid cell–derived MT1‐MMP appears to control collagen turnover, the role of MT1‐MMP in regulating atherogenic responses in non–myeloid cell populations remains unknown.
Despite the paucity of information regarding the role of MT1‐MMP in atherogenesis, MT1‐MMP heterozygous mice, which do not display any of the severe developmental phenotypes observed in the MT1‐MMP–null mice, have been reported to display a protected status from both obesity induced by a high fat diet14 and neointima formation secondary to carotid injury.15 Given the attenuated responses of MT1‐MMP heterozygous animals to pathologic stresses, we initially hypothesized that these mice might likewise be protected from hypercholesterolemic atherogenesis. Contrary to our expectations, we now report that atherogenesis is significantly enhanced in heterozygous Mmp14 +/− Apoe −/− mice. To determine the cellular mechanisms by which MT1‐MMP limits the progression of atherogenesis, we extended our analyses to include newly characterized conditional knockout mice to selectively target MT1‐MMP expression in adipose tissue, macrophages, or VSMCs in APOE (apolipoprotein E)–null mice. Unexpectedly, we found that MT1‐MMP expressed by VSMCs, and not by either adipocytes or macrophages, exerts a profound protective effect against the progression of proliferative atherosclerotic lesions in Apoe −/− mice. Together, these results constitute the first example of MT1‐MMP serving as an antiatherogenic enzyme by directly regulating VSMC function and proliferation in the in vivo setting.
Material and Methods
Animals
Apoe −/− mice16 were purchased from the Jackson Laboratory. MT1‐MMP heterozygous Mmp14 +/− mice were maintained on a C57BL6/J background with >5 generations of backcrossing.14, 17 Mmp14 +/− mice were crossed to Apoe −/− mice to generate Mmp14 +/− Apoe +/− breeders. Mmp14 +/− Apoe +/− mice were used for breeding with Apoe −/− mice to generate Mmp14 +/− Apoe −/− mice and their littermate Mmp14 +/+ Apoe −/− mice for this study. Mmp14 F/F mice were generated as described previously.18 These mice were crossed to SM22α‐Cre (Tg[Tagln‐cre]1Her/J],19 Csf1r‐Cre, or Fabp4‐Cre‐ERT2 transgenic mice (gift from Pierre Chambon, Institute of Genetics and Molecular and Cellular Biology, France),20 and then further crossed to Apoe −/− mice.
Atherosclerosis Study
The atherogenic Western diet, composed of 17% (kcal/kcal) protein, 43% carbohydrate, and 41% fat with 1.5 g/kg cholesterol, was purchased from Research Diets. Male and female mice were fed a Western diet for 12 weeks beginning at 8 weeks of age. All procedures were approved by the University of Michigan committee on the use and care of animals, conforming to the guidelines of the International Association for Assessment and Accreditation of Laboratory Animal Care.
Morphometric Analysis of Atherosclerosis
After euthanasia by CO2 asphyxiation, blood was collected through portal veins. Animals were perfused with PBS and then 10% formalin in PBS through their left ventricles at a rate of 1 mL/min, as described previously.21 Arterial trees were carefully dissected to include the brachiocephalic, left common carotid, and subclavian arteries, as well as the descending thoracic and abdominal aortas with the bilateral iliac arteries. Adipose tissues attached to the arteries were carefully removed under a dissecting microscope. After Oil Red O staining and repeated washing in ethanol and water, aortic trees were pinned against a black background, and the aortic trees were digitally photographed under a dissecting microscope.22 Oil Red O–positive areas were quantified as the percentage of plaque area per total arterial tree or abdominal aorta.
Collagen Staining and Histologic Analysis
Sections were stained with hematoxylin and eosin and sirius red.23 The atheroma area and vascular wall area were quantified in cross‐sections using ImageJ software (National Institutes of Health), and the average plaque and vascular wall areas were determined. Immunostaining was performed using the ABC system. The primary antibodies used were rabbit polyclonal anti–α‐smooth muscle actin (anti–α‐SMA) antibody, anti‐F4/80 antibody, and anti‐Ki67 antibody.
Vascular Smooth Muscle Cells
Mouse aortic smooth muscle cells were isolated with collagenase digestion, as described by others.24 Isolated primary mouse VSMCs were cultured in high‐glucose DMEM with 10% FBS and passed twice before experiments. Mouse primary VSMCs immortalized with the SV40 large T antigen were purchased from ATCC.25
3‐Dimensional Spheroid Culture
Cells (2×104) were cultured as hanging droplets in a 384‐well Perfecta3D hanging droplet plate (3D Biomatrix) for 48 hours before assays.26 Cells were cultured in high‐glucose DMEM with 10% FBS medium supplemented with 0.24% Methocel A4M (Dow).
Whole‐Genome Expression Analysis
The total RNA was extracted from cultured primary VSMCs. Two independent pairs of samples were used for the DNA microarray experiments. Labeled cRNA was hybridized to Affymetrix Mouse Gene ST 2.1 strips at the University of Michigan DNA Sequencing Core. Expression values were calculated using a robust multiarray average. Probe sets that showed >2‐fold difference between the groups with a minimal expression value >24 in at least 1 of the samples were chosen for further statistical analyses. Data analysis was performed with R version 3.1.1, and a heat map was created using the gplots package (R package version 2.17.0, http://CRAN.R-project.org/package=gplots). Significant biological pathways represented by the differentially expressed genes were determined using PANTHER (http://pantherdb.org/).
Statistical Analyses
Values were expressed as mean±SEM. The distribution of weight, fat mass, cholesterol profile, fasting insulin, and glucose levels between 2 groups were analyzed using a 2‐tailed, unpaired Student t test. To assess the differences in atherogenesis between the multiple groups, 2‐way ANOVA was used, followed by a post hoc pairwise comparison using the Tukey procedure. P values <0.05 were considered significant.
Results
The Effect of the MT1‐MMP Gene Dose on Fat Mass and Dyslipidemia in APOE‐Null Mice
We previously demonstrated that MT1‐MMP heterozygosity renders mice resistant to obesity induced by a high fat diet.14 Based on this finding, we hypothesized that the allelic reduction of the MT1‐MMP gene would protect mice from hypercholesterolemic atherosclerosis, a disease process that is often associated with obesity in humans.27 As such, we crossbred MT1‐MMP heterozygous (Mmp14 +/−) mice with APOE‐null mice to generate Mmp14 +/− Apoe −/− mice and examined the effects of MT1‐MMP heterozygosity on weight, fat mass, blood glucose, and insulin levels as well as cholesterol profiles. As expected, heterozygous (HT) Mmp14 +/− Apoe −/− male mice on a Western diet were leaner than littermate wild‐type (WT) Mmp14 +/+ Apoe −/− mice (WT 33.0±1.0 g, HT 29.0±0.9 g, n=9 each, P=0.009; Figure 1A). When fat mass was assessed at the end of the 12‐week Western diet, the total and epididymal fat masses were significantly smaller in the Mmp14 +/− Apoe −/− than Mmp14 +/+ Apoe −/− male mice (percentage of total fat mass per weight: WT 5.1±0.3%, HT 3.6±0.6%, n=9 each, P=0.03; percentage of epididymal fat per weight: WT 3.4±0.2%, HT 2.2±0.3%, n=9 each, P=0.007; Figure 1B). Fasting blood glucose levels were similar between the groups (WT 162±43 [n=8] versus HT 161±27 mg/dL [n=9], P=0.5); however, the fasting insulin concentration was substantially lower in the MT1‐MMP heterozygous mice (WT 11.8±4.3 [n=8] versus HT 3.8±0.6 mU/L [n=9], P=0.04), suggesting an increased insulin sensitivity of Mmp14 +/− Apoe −/− mice that occurs in parallel with their leaner phenotype. Of note, MT1‐MMP heterozygosity did not change the blood cholesterol levels in APOE‐null mice fed a Western diet (total cholesterol: WT 1021±59 [n=8] versus HT 1012±52 mg/dL [n=9], P=0.9; direct low‐density lipoprotein: WT 359±27 versus HT 349±23 mg/dL, P=0.8; Figure 1C). Consistent with their leaner phenotype and relative insulin sensitivity, the blood triglyceride content tended to be lower in HT mice, but this difference did not reach statistical significance (WT 142±10 mg/dL [n=8], HT 118±11 mg/dL [n=9], P=0.1).
Figure 1.
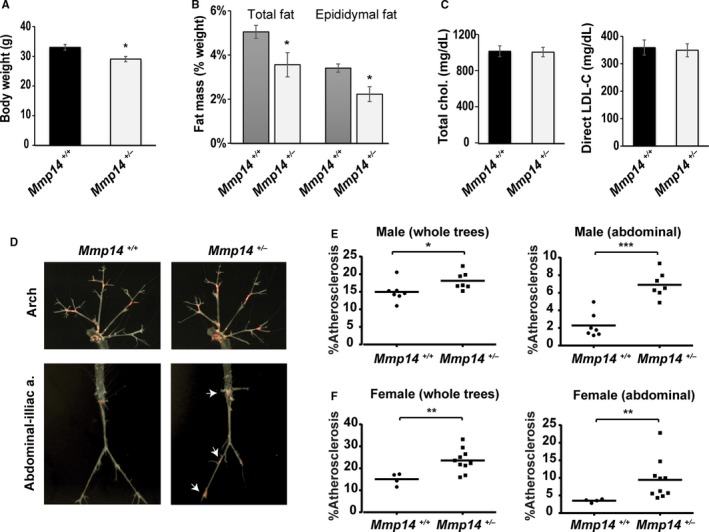
MT1‐MMP (membrane‐type 1 matrix metalloproteinase) heterozygosity promotes atherosclerosis. A, Mmp14 +/+ Apoe −/− mice and Mmp14 +/− Apoe −/− mice were fed a Western diet for 12 weeks beginning at 8 weeks of age. Body weight (g) at the end of study. B, Percentage of fat mass (wt/wt) of total (epididymal plus inguinal) and epididymal fat pads. C, Fasting serum cholesterol (total and direct low‐density lipoprotein) levels. Mean±SEM, n=8 and n=10, respectively; *P<0.05. D, Atherosclerotic lesions detected with Oil Red O staining in Mmp14 +/+ Apoe −/−and Mmp14 +/− Apoe −/− male mice. Arrows point to the increased atherogenesis distributed in abdominal aorta and iliac arteries specifically, as found in Mmp14 +/− Apoe −/− mice. E and F, Oil Red O–positive atherosclerotic plaque areas quantified in all aortic trees and abdominal aortas in male mice (n=7 each) and female mice (n=4 and n=10). Mean±SEM, *P<0.05, **P<0.005, ***P<0.0005. a. indicates artery.
Whole‐Body MT1‐MMP Heterozygosity Accelerates Atherogenesis
MT1‐MMP WT and HT mice were fed a Western diet for 12 weeks beginning at 8 weeks of age. Contrary to our prediction, atherogenesis was more advanced in Mmp14 +/− Apoe −/− mice compared with Mmp14 +/+ Apoe −/− littermate controls (Figure 1D). Particularly, advanced atherogenesis was notable in the abdominal aorta and iliac/femoral arteries of Mmp14 +/− Apoe −/− mice (Figure 1D arrows). The percentage of total plaque area was increased by 21% in male mice (Mmp14 +/+ Apoe −/− 14.9±0.9% versus Mmp14 +/− Apoe −/− 18.1±0.8%, n=7 each, P=0.04; Figure 1E, left), whereas the abdominal percentage of plaque area was increased by 200% (2.3±0.5% versus 6.9±0.5%, n=7 each, P<0.0001; Figure 1E, right). In female mice, the total plaque area in Mmp14 +/− Apoe −/− animals was increased by 56% (15.1±1.3% [n=4] versus 23.6±1.6% [n=10], P=0.002; Figure 1F, left), with the abdominal plaque area increasing by 171% relative to the Mmp14 +/+ Apoe −/− female controls (3.5±0.2% [n=4] versus 9.5±1.8% [n=10], P=0.002; Figure 1F, right).
We next examined left carotid artery sections to assess the structure of the atherosclerotic plaque and the associated level of vascular wall remodeling. Interestingly, the average plaque area of the Mmp14 +/− Apoe −/− mice was 3 times as large as that of the Mmp14 +/+ Apoe −/− mice, but this was not statistically significant given the variable severity of atherosclerosis in the heterozygous group (WT 59±7×103 μm2 versus HT 189±84×103 μm2, n=6 each, P=0.15; Figure 2A). Concurrent with a trend of increased atherosclerosis, we observed a 1.7‐fold increase in the vascular wall area, underscoring the presence of outward vascular wall remodeling in MT1‐MMP heterozygous APOE‐null mice (133±16×103 μm2 versus 234±19×103 μm2, n=6 each, P=0.004). Furthermore, both the atherosclerotic plaques and the vascular walls of MT1‐MMP HT mice displayed an increased collagen fiber content relative to WT mice, as assessed by sirius red staining (sirius red–positive area: WT 26.6±3.3% versus HT 44.0±4.7%, n=6 each, P=0.009; Figure 2B), suggesting that MT1‐MMP heterozygosity promoted vascular wall arteriosclerosis along with plaque formation in the APOE‐null background. MT1‐MMP HT mice also displayed an apparent loss of a contractile phenotype (ie, loss of SMA staining) coupled with disrupted elastic lamina, which in turn elicited more complex and advanced atherosclerotic lesions (Figure 2C and 2D).
Figure 2.
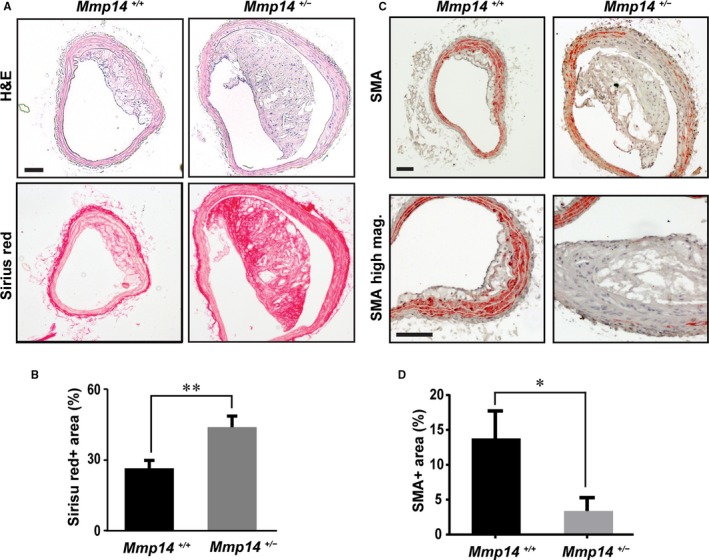
MT1‐MMP (membrane‐type 1 matrix metalloproteinase) heterozygosity promotes plaque formation and outward vascular remodeling. A, Representative histology sections of left common carotid arteries. Hematoxylin and eosin (H&E) and sirius red staining. Scale=100 μm. B, Sirius red–positive area (%). C, Immunostaining of smooth muscle actin (SMA) in vascular walls and atherosclerotic plaques (orange). Nuclei were counterstained (blue). Lower panels are of higher magnification. Scale=100 μm. D, SMA‐positive area (percentage). Mag indicates magnification. *P<0.05, **P<0.005
MT1‐MMP Gene Targeting in VSMCs Accelerates Atherosclerosis Progression
Although MT1‐MMP heterozygosity was found to promote atherogenesis, the cellular mechanisms underlying the aggravated atherogenesis were unclear. In obesity, perivascular adipose tissues play a key role in atherogenesis.28 In contrast, dysfunctional VSMCs might also be responsible for inducing inflammatory atherogenesis.4 Because MT1‐MMP is highly expressed in both adipocytes9, 10 and VSMCs,15 we sought to use tissue‐specific knockout mice in an effort to determine the cell type responsible for the MT1‐MMP–dependent regulation of atherogenesis. When male mice in each group were fed a Western diet, we observed markedly augmented atherogenesis and aneurysm formation only with VSMC‐specific MT1‐MMP gene targeting (ie, SM22α‐Cre[+]Mmp14 F/F Apoe −/− mice), but not following adipocyte MT1‐MMP gene targeting (ie, aP2‐Cre‐ERT2[+]Mmp14 F/F Apoe −/− mice; Figure 3A). Atherogenesis in SM22α‐Cre(+)Mmp14 F/F Apoe −/− mice frequently extended to femoral arteries with significant outward remodeling having been noted, in tandem with aneurysm formation in the iliac arteries (Figure 3A and 3B). Importantly, total plaque area was increased by 190% in SM22α‐Cre(+)Mmp14 F/F Apoe −/− male mice compared with Cre(−)Mmp14 F/F Apoe −/− male mice (8.0±1.4% [n=8] versus 23.5±1.4% [n=13], P<0.0001) and 210% in female mice (4.9±0.9% [n=5] versus 15.5±3.5% [n=7], P=0.03). In contrast, significant differences were not observed between Cre‐negative controls and the adipose‐specific MT1‐MMP deletion model (Figure 3C and Figure S1), suggesting that the VSMCs are the primary cell type that mediates MT1‐MMP–dependent modulation of atherogenesis progression.
Figure 3.
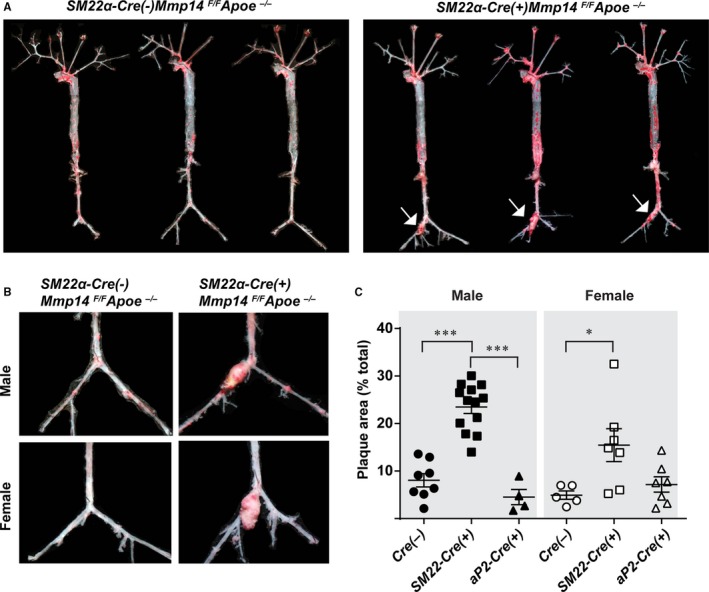
Vascular smooth muscle cell MT1‐MMP (membrane‐type 1 matrix metalloproteinase) gene targeting promotes atherosclerosis and aneurysm formation. A, Whole arterial trees assessed for Oil Red O–positive atherosclerotic plaque area (red). Arrows point to atherosclerotic aneurysms. Representative arterial trees from SM22α‐Cre(−)MMP14 F/ FA poe −/− and SM22α‐Cre(+)MMP14 F/ FA poe −/− mice are shown; n=13 and n=20, respectively. B, Higher magnification of aortic aneurysms found in SM22α‐Cre(+)MMP14 F/ FA poe −/− mice. C, Percentage of plaque area in Cre(−)Mmp14 F/ FA poe −/−, SM22α‐Cre(+)Mmp14 F/ FA poe −/− , aP2‐Cre‐ERT2(+)Mmp14 F/ FA poe −/− mice, males and females (n=8, n=13, and n=4 in male mice, and n=5, n=7, and n=7 in female mice, respectively). *P<0.05, ***P<0.0005.
The Loss of VSMC MT1‐MMP Leads to Proliferative Atherosclerotic Lesions and Aneurysm Formation
After 12‐week Western diet feeding, SM22α‐Cre(+)Mmp14 F/F Apoe −/− mice developed extensive atherosclerosis in their common iliac arteries followed by aneurysm formation (Figure 4A and 4B). No aneurysm formation was observed in the Cre(−)Mmp14 F/F Apoe −/− mice (8 male and 5 female mice), whereas 11 of 12 male and 6 of 7 female SM22α‐Cre(+)Mmp14 F/F Apoe −/− mice developed strikingly enlarged aneurysms that were readily observable on dissection (Figure 3B). These dysmorphic lesions displayed significant vascular wall thickening as well as atheroma formation (Figure 4A). Masson's Trichrome and Verhoeff–Van Gieson staining demonstrated the disruption and loss of elastic laminae (Figure 4A). SMA staining showed an increased number of SMA+ cells in vascular walls as well as within atheroma, where Ki67 staining confirmed that large numbers of SMA+ cells existed in a proliferative state (Figure 4A and 4D). Infiltration of F4/80‐positive cells was observed in the atheroma and the vascular walls of affected vessels (Figure 4A, F4/80 staining) but to a lesser extent compared with VSMCs. Image quantification of the lesions confirmed an increased atheroma area (Figure 4B) and outward remodeling (Figure 4C), which were coupled with an increased number of proliferating VSMCs (Figure 4D). Because macrophages are potentially targeted by SM22α promoter‐driven Cre expression,29 we specifically targeted these cells using Csf1r‐Cre(+)Mmp14 F/F Apoe −/− mice and examined atherosclerosis and aneurysm formation. Neither littermate control Csf1r‐Cre(−)Mmp14 F/F Apoe −/− mice nor Csf1r‐Cre(+)Mmp14 F/F Apoe −/− mice developed atherosclerotic iliac aneurysm after 12 weeks of Western diet (n=7 for each group tested), and no significant differences in the sizes of atherosclerosis lesions were observed (n=4 each).
Figure 4.
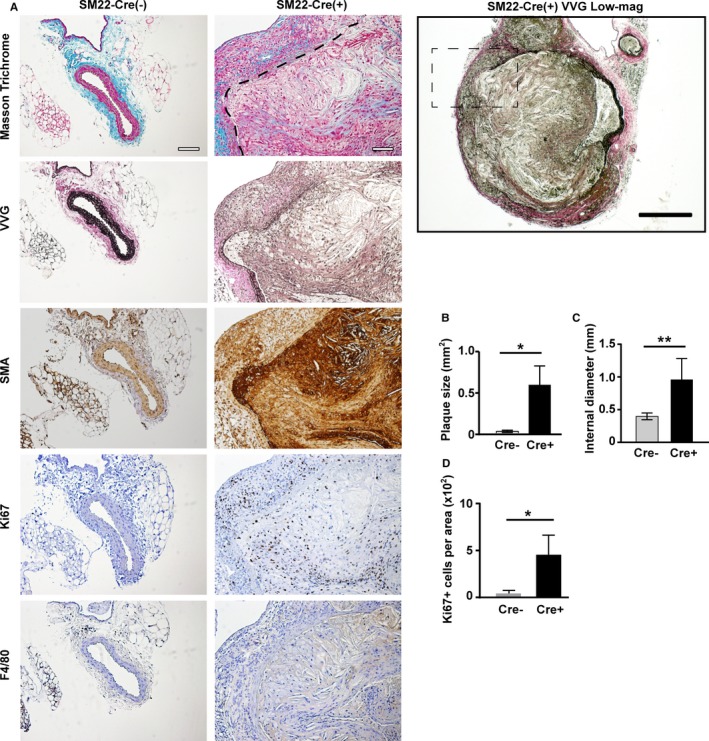
The loss of vascular wall integrity and vascular smooth muscle cell (VSMC) proliferation in VSMC MT1‐MMP (membrane‐type 1 matrix metalloproteinase) knockout mice. A, Atherosclerotic aneurysms found in iliac arteries of SM22α‐Cre(+)MMP14 F/ FA poe −/− mice. Masson's Trichrome staining; the border between the vascular wall and the atheroma is demarcated with a dashed line. Verhoeff–Van Gieson staining (VVG), smooth muscle actin (SMA) staining, Ki67 staining, and F4/80 (a macrophage marker) staining. Scale=100 μm. The lower magnification (×4) micrograph of SM22α‐Cre(+)MMP14 F/ FA poe −/− VVG staining on the right. Scale=200 μm. The lesion in the dashed square is shown on the left. B, Plaque area. C, The internal diameter of iliac arteries. D, Ki67‐positive area. Mean±SEM; n=5 and n=7 for each group. One‐way ANOVA. *P<0.05, **P<0.005.
Proinflammatory and Metabolically Dysfunctional MT1‐MMP–Null VSMCs
To assess the role of MT1‐MMP in VSMC function, we isolated primary VSMCs from descending aortas. As expected, VSMCs isolated from SM22‐Cre(+)Mmp14 F/F Apoe −/− mice demonstrated specific Cre expression along with the suppression MT1‐MMP gene expression (Figure S2). In vitro, MT1‐MMP–null VSMCs displayed a flattened, spread shape with higher stress fiber formation (Figure 5A), whereas the expression of other MMPs (eg, MMP‐2, MMP‐8, MMP‐9, MMP‐13, and MT2‐MMP [MMP15]), were not significantly different between control and MT1‐MMP–deleted cells (Figure S2). The Acta2 gene, which encodes α‐SMA, was expressed equally in primary VSMCs isolated from SM22‐Cre(−)Mmp14 F/F Apoe −/− and SM22‐Cre(+)Mmp14 F/F Apoe −/− mice (Figure S2). To gain further insight into the effects of MT1‐MMP gene targeting in VSMCs, whole‐genome transcriptome analysis was performed. Using a minimum 2‐fold difference as a cutoff, 414 genes were found to be differentially expressed between the 2 groups (the top 70 genes are shown in Figure 5B, and all differentially expressed genes are listed in Table S1; the microarray data is available at NCBI Gene Expression Omnibus [GSE] as GSE100661). Interestingly, the genes upregulated in SM22α‐Cre(+)Mmp14 F/F Apoe −/− VSMCs (>2.3‐fold change) were aggregated in the pathways of inflammation and cell killing (Figure 5C), suggesting the acquisition of a proinflammatory phenotype in MT1‐MMP–null VSMCs.
Figure 5.
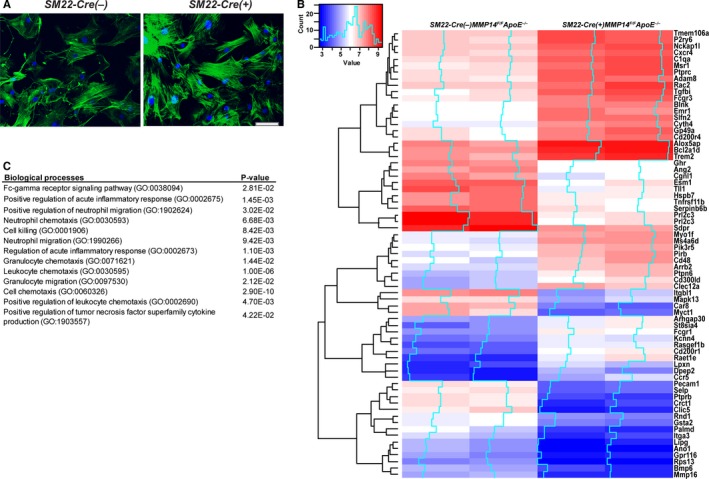
Proinflammatory gene expression in MT1‐MMP (membrane‐type 1 matrix metalloproteinase)–null vascular smooth muscle cells (VSMCs). A, Primary aortic VSMCs isolated from Cre(−)Mmp14 F/ FA poe −/− and SM22α‐Cre(+)/Mmp14 F/ FA poe −/− mice. F‐actin (green), nuclei (blue). B, Representative genes differentially expressed in VSMCs from 2 independent pairs of Cre(−)Mmp14 F/ FA poe −/− and SM22α‐Cre(+)/Mmp14 F/ FA poe −/− mice. C, Gene Ontology (GO) biological processes represented by the genes upregulated in VSMCs isolated from the SM22α‐Cre(+)/Mmp14 F/ FA poe −/− mice.
MT1‐MMP Gene Targeting Promotes VSMC Proliferation in 3‐Dimensional Cell–Cell and Cell–ECM Contexts
To gain insights into the mechanism by which MT1‐MMP–null VSMCs engage in the formation of proliferative atherosclerotic lesions, we developed an in vivo–like 3‐dimensional (3‐D) spheroid culture system in which in vivo–like cell–cell and cell–ECM interactions could be recapitulated. Using Ki67 as an index of proliferative responses, MT1‐MMP–null primary VSMCs displayed markedly higher activity relative to WT cells (Figure 6A and 6B). To determine whether the observed differences in cell proliferation were a direct consequence of MT1‐MMP gene targeting (ie, as opposed to a secondary response of VSMCs recovered from the advanced atherogenic lesions found in VSMC‐knockout mice), we used immortalized mouse primary VSMCs to further define the role of MT1‐MMP in regulating VSMC proliferation. In 3‐D spheroid culture, similar to the findings obtained with freshly isolated VSMCs, small interfering RNA–mediated MT1‐MMP gene silencing in mouse VSMCs induced a marked increase in the number of Ki67‐positive VSMCs (Figure 6C and 6D). Of note, under 2‐D culture conditions, MT1‐MMP silencing exerted minimal effects on mouse VSMC proliferation (Figure 6E and 6F). Taken together, these results support a model in which MT1‐MMP plays a required role in regulating VSMC proliferative activity but only under 3‐D conditions that more closely recapitulate the in vivo environment.
Figure 6.
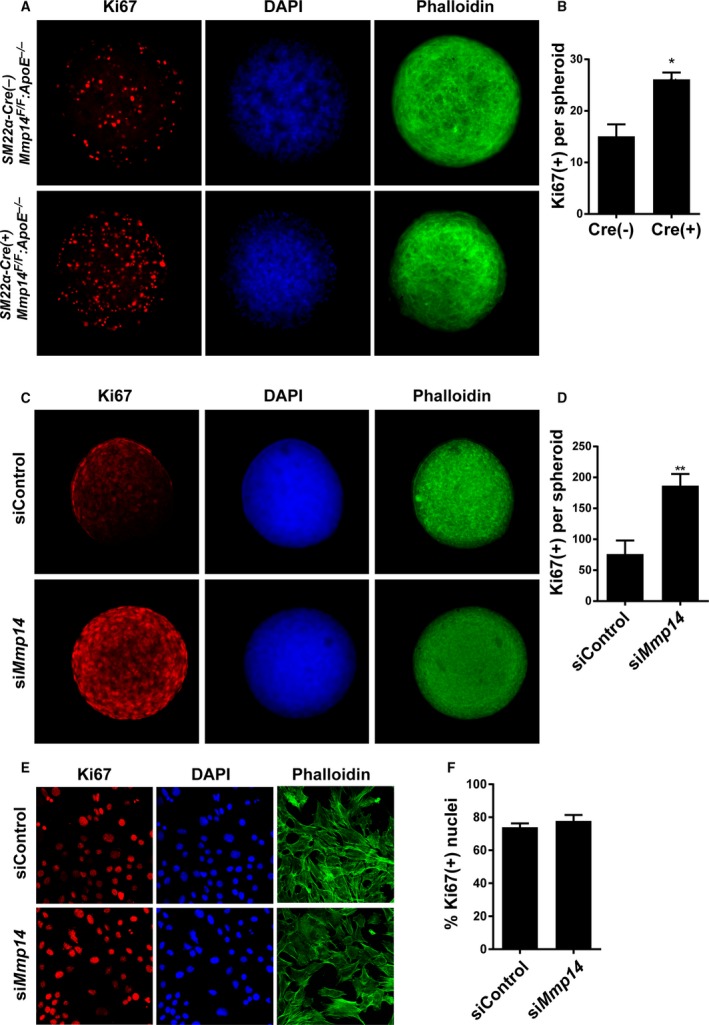
MT1‐MMP (membrane‐type 1 matrix metalloproteinase) limits vascular smooth muscle cell (VSMC) proliferation in 3‐dimensional (3‐D) organoids. A, Primary VSMCs isolated from Cre(−)Mmp14 F/ FA poe −/− mice and Cre(+)Mmp14 F/ FA poe −/− mice were cultured as a 3‐D spheroids for 48 hours and stained for Ki67 (red), nuclei (DAPI, blue), and actin (phalloidin, green). B, Quantified intensity of Ki67 staining per spheroid. n=5 to 7. *P<0.05. C, Immortalized mouse VSMCs (MOVAS) transiently transfected with small interfering RNA (siRNA) control (siControl) and MT1‐MMP siRNA (siMmp14). Ki67 (red), nuclei (blue), actin (green). D, Ki67 staining intensity per spheroid of MOVAS. **P<0.005. E, 2‐D cultured MOVAS transfected with control and MT1‐MMP siRNA. Ki67 (red), nucleus (blue), and actin (green). F, Ki67‐positive nuclei per total nuclei count.
Discussion
MMP family members play a key role in ECM turnover in a wide variety of developmental and disease processes.30 Among the collagen‐degrading MMPs, secreted (MMP‐3, MMP‐8, MMP‐13) and membrane‐type MMPs (MT1‐ and MT2‐MMP) display distinct temporospatial differences in their patterns of expression and activity.9 Unlike other MMPs, MT1‐MMP (MMP14) is the only family member whose activity is indispensable for postnatal development.10, 11 Because of the premature morbidity and mortality displayed by MT1‐MMP–null mice, the role of the proteinase in cardiovascular disease has remained elusive. To date, the only MT1‐MMP–expressing cellular compartment tested in a mouse atherogenesis model was bone marrow–derived myeloid cells; however, no substantial impact on atheroma size was observed following bone marrow reconstitution with Mmp14 −/− cells.13 In our study, we identified the critical role played by VSMC‐derived MT1‐MMP in regulating the progression of atherosclerotic lesions and the associated formation of vascular aneurysms.
Previously, we demonstrated that MT1‐MMP heterozygosity protects C57BL/6 mice from diet‐induced adipose tissue expansion.14 Similarly, in this study, heterozygous Mmp14 +/− mice displayed a leaner phenotype relative to Mmp14 +/+ mice in an APOE‐null background. Consistent with their leaner phenotype, MT1‐MMP heterozygous mice displayed higher insulin sensitivity than WT mice. Given the metabolically improved status of Mmp14 +/− mice, we initially hypothesized that MT1‐MMP heterozygosity would protect mice from hypercholesterolemic atherosclerosis, which is often associated with increased adiposity. Contrary to our expectations, MT1‐MMP heterozygous APOE‐null mice developed more extensive atherosclerotic lesions than MT1‐MMP–sufficient APOE‐null mice, suggesting a potentially beneficial role for MT1‐MMP in limiting disease progression. The results also suggest that the inverse relationship observed between fat mass and atherosclerosis—the so‐called obesity paradox in humans—may reflect the independent biological effects exerted by a cohort of modifier genes, including MT1‐MMP, on adipose tissue and vascular function.
To define the cellular mechanisms by which MT1‐MMP exerts its antiatherogenic effects, we embarked on a series of studies aimed at tissue‐specific MT1‐MMP gene targeting in APOE‐null mice. In the early stages of our efforts, we focused on 2 cell types: adipocytes and VSMCs. We initially hypothesized that the loss of adipocyte MT1‐MMP would modify atherogenesis via the potentially causal links that exist between adipose tissue and the vascular wall.31 In APOE‐null mice, however, we were unable to observe a significant impact on atherogenesis following gene targeting of adipocyte‐derived MT1‐MMP. In contrast, the SM22α‐Cre‐mediated loss of VSMC MT1‐MMP strikingly aggravated atherosclerosis progression in APOE‐null mice. Whole‐genome transcriptome analyses indicated that a series of proinflammatory genes were upregulated in MT1‐MMP–null VSMCs, a finding consistent with the proatherogenic phenotype of SM22α‐Cre(+)Mmp14 F/F Apoe −/− mice. Furthermore, our in vitro studies demonstrated that MT1‐MMP targeting in either primary or immortalized VSMCs promotes cell proliferation under 3‐D spheroid culture conditions.26 As such, 3‐D spheroid culture appears to reflect a set of conditions that better reflect our in vivo findings and, as such, can be used to more faithfully address cellular behavior in tissue‐like contexts by re‐creating cell–cell and cell–ECM interactions ex vivo. Indeed, previous work has demonstrated that VSMC proliferative activity can be regulated by cell–cell adhesion32, 33, 34 and cell–ECM interactions.35, 36 Of note, the enhanced proliferative responses displayed by MT1‐MMP–null VSMCs were not observed under conventional 2‐D culture conditions in which the cell–cell and cell–ECM interactions that are encountered in vivo are replaced by cell culture atop a nonphysiologic, planar, and rigid substratum. Finally, it is interesting to note that the proliferative effects of MT1‐MMP gene silencing were not observed in other cell types, for example, 3T3‐L1 preadipocytes (data not shown), reinforcing the unique role played by MT1‐MMP in VSMC biology.
In considering the mechanisms by which MT1‐MMP might control proliferative activity, efforts are complicated by the proteinase's broad substrate repertoire, ranging from type I collagen37 to CD4438 and cadherins.39 As such, MT1‐MMP can potentially regulate VSMC proliferation by degrading any number of membrane‐associated protein targets as well as pericellular ECM molecules, thereby modifying both cell–cell adhesion and cell–ECM interaction. In atherosclerosis, the expression of adhesion molecules and ECM proteins is highly upregulated40; as such, a decrease in MT1‐MMP activity would be predicted to trigger the excess accumulation of ECM macromolecules as well as cell‐surface adhesion molecules. At this juncture, we posit that changes in the dynamics of ECM turnover and cell‐surface molecule expression occurring within the vascular wall lead to the unregulated proliferation of VSMCs and the development of a proinflammatory phenotype. Interestingly, at least in terms of proliferative responses, the use of a 3‐D spheroid model allowed us to recapitulate the MT1‐MMP–dependent biological processes ex vivo. Nevertheless, it remains unclear how the cleavage of MT1‐MMP substrate(s) accelerates VSMC proliferation in APOE‐null mice. This caveat notwithstanding, our data clearly highlight the role played by VSMC‐derived MT1‐MMP in limiting the progression of proliferative atherosclerotic lesions. Because MT1‐MMP is a membrane‐bound proteinase, the physical proximity of substrates with the enzyme is likely critical for the protective effects exerted by VSMCs in limiting the expansion of atherosclerotic lesions. Our study also suggests that other atherosclerosis‐associated MMPs (eg, MMP‐2, MMP‐8, MMP‐13) do not compensate for the genetic loss of MT1‐MMP and are unable to limit atherosclerosis progression. Although MT2‐MMP (MMP15) was also expressed in mouse VSMCs (Figure S2), as reported previously in rat VSMCs,41 the biological phenotypes conferred by MT1‐MMP gene loss were not rescued by the presence of MT2‐MMP. Differences in the hemopexin domain structure, posttranslational modification, or protein trafficking may underlie the specific effects mediated by MT1‐MMP versus MT2‐MMP.42
Although increased collagen content within atheroma might be predicted to play a protective role against plaque rupture, collagen accumulation in arterial walls could also accentuate vascular sclerosis and stiffening. In turn, stiff and sclerotic blood vessels could increase luminal sheer stress, thereby increasing the chance of plaque rupture. In future studies, an assessment of hemodynamic changes and plaque instability in our model will be required to more accurately define the role of MT1‐MMP in cardiovascular disease. Interestingly, in APOE‐null mice, arterial wall stiffness is known to be increased through VSMC lysyl oxidase activity.43 In turn, increased tissue stiffness could control VSMC proliferation through the activation of a mechanotransduction pathway, for example, YAP/TAZ transcription activity.44 As such, we posit that the pathologic accumulation of ECM macromolecules secondary to the loss of MT1‐MMP activity may further promote vascular wall rigidity in the APOE‐null mice, leading to hyperproliferative vascular lesions and aneurysm formation. Given that we observed severe atherosclerosis and aneurysm formation in iliac and femoral arteries, site‐specific increases in vascular wall thickening and arteriosclerosis are likely related to the distinct mechanical properties of vessel walls observed along the arterial tree.45 Indeed, femoral arteries as well as the abdominal aorta are known to display higher wall thickness with lower content of elastic lamina compared with carotid arteries and thoracic aortas.45 Together, differences in mechanical stress and ECM composition may render VSMCs in abdominal aortae and femoral arteries more vulnerable to atherogenic proliferation.
Finally, it remains to be determined whether the inflammatory gene expression profile observed in MT1‐MMP–null VSMCs is restricted to atherogenic milieu encountered in vivo. Interestingly, in our hands, the increase in inflammatory gene expression of MT1‐MMP–null VSMCs was coupled with decreased mitochondrial activity (T. Akama, PhD, unpublished data, 2016). Furthermore, recent studies have demonstrated a critical role played by a VSMC phenotypic switch to macrophage‐like cells in atherogenesis.4, 46 Because MT1‐MMP also regulates inflammatory responses in macrophages,47 the proteinase may well play a key role in controlling a complex set of metabolic and phenotypic switching programs that are engaged in atherogenic VSMCs. Although further work is needed to delineate MT1‐MMP function during atherogenesis, our work highlights the previously unsuspected vessel wall–protective effects exerted by this membrane‐anchored metalloproteinase in the VSMC compartment.
Sources of Funding
Funding was provided by the McKay Research Grant from University of Michigan Cardiovascular Center, NIH R21HL106332, and R01DK102656 to Chun. NIH R01AI105068‐01 to Weiss. NIH Cancer Biology Training Program Grant T32‐CA009676 supported Bahr.
Disclosures
None.
Supporting information
Table S1. List of Genes Differentially Expressed in Cre(−) and SM22α‐Cre(+)Mmp14 F/F Vascular Smooth Muscle Cells
Figure S1. Arterial trees dissected from Cre(−), SM22α‐Cre(+), and aP2‐Cre(+) MMP14 F/F Apoe −/− male and female mice after a 12‐week Western diet.
Figure S2. Gene expression of Acta2 (α‐SMA), Cre enzyme, MT1‐MMP (membrane‐type 1 matrix metalloproteinase; MMP14), MMP‐2, MMP‐8, MMP‐9, MMP‐13, and MT2‐MMP (MMP15) in primary vascular smooth muscle cells (VSMCs) isolated from Cre(−) and SM22α‐Cre(+) MMP14 F/F Apoe −/− mice. Almost complete suppression of MT1‐MMP expression in primary VSMCs was confirmed coupled with SM22‐dependent Cre expression. Expression of other matrix metalloproteinases was not affected. ***P<0.001.
Acknowledgments
We like to thank Dr Pierre Chambon (Institute of Genetics and Molecular and Cellular Biology, France) for sharing Fabp4‐Cre‐ERT2 transgenic mice. Current affiliation of Öhman is Duke‐Nus Medical School, Singapore.
(J Am Heart Assoc. 2017;6:e003693 DOI: 10.1161/JAHA.116.003693.)28735290
References
- 1. Olivetti G, Anversa P, Melissari M, Loud AV. Morphometric study of early postnatal development of the thoracic aorta in the rat. Circ Res. 1980;47:417–424. [DOI] [PubMed] [Google Scholar]
- 2. Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89:957–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 4. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Page‐McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuzuya M, Nakamura K, Sasaki T, Wu Cheng X, Itohara S, Iguchi A. Effect of MMP‐2 deficiency on atherosclerotic lesion formation in apoE‐deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:1120–1125. [DOI] [PubMed] [Google Scholar]
- 7. Laxton RC, Hu Y, Duchene J, Zhang F, Zhang Z, Leung K‐Y, Xiao Q, Scotland RS, Hodgkinson CP, Smith K, Willeit J, López‐Otín C, Simpson IA, Kiechl S, Ahluwalia A, Xu Q, Ye S. A role of matrix metalloproteinase‐8 in atherosclerosis. Circ Res. 2009;105:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quillard T, Araújo HA, Franck G, Tesmenitsky Y, Libby P. Matrix metalloproteinase‐13 predominates over matrix metalloproteinase‐8 as the functional interstitial collagenase in mouse atheromata. Arterioscler Thromb Vasc Biol. 2014;34:1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sabeh F, Li XY, Saunders TL, Rowe RG, Weiss SJ. Secreted versus membrane‐anchored collagenases: relative roles in fibroblast‐dependent collagenolysis and invasion. J Biol Chem. 2009;284:23001–23011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3‐dimensional development of white adipose tissue. Cell. 2006;125:577–591. [DOI] [PubMed] [Google Scholar]
- 11. Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, Ward JM, Birkedal‐Hansen H. MT1‐MMP‐deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. [DOI] [PubMed] [Google Scholar]
- 12. Zhou Z, Apte SS, Soininen R, Cao R, Baaklini GY, Rauser RW, Wang J, Cao Y, Tryggvason K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane‐type matrix metalloproteinase I. Proc Natl Acad Sci USA. 2000;97:4052–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schneider F, Sukhova GK, Aikawa M, Canner J, Gerdes N, Tang SM, Shi GP, Apte SS, Libby P. Matrix‐metalloproteinase‐14 deficiency in bone‐marrow‐derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008;117:931–939. [DOI] [PubMed] [Google Scholar]
- 14. Chun TH, Inoue M, Morisaki H, Yamanaka I, Miyamoto Y, Okamura T, Sato‐Kusubata K, Weiss SJ. Genetic link between obesity and MMP14‐dependent adipogenic collagen turnover. Diabetes. 2010;59:2484–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Filippov S, Koenig GC, Chun TH, Hotary KB, Ota I, Bugge TH, Roberts JD, Fay WP, Birkedal‐Hansen H, Holmbeck K, Sabeh F, Allen ED, Weiss SJ. MT1‐matrix metalloproteinase directs arterial wall invasion and neointima formation by vascular smooth muscle cells. J Exp Med. 2005;202:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yana I, Sagara H, Takaki S, Takatsu K, Nakamura K, Nakao K, Katsuki M, Taniguchi S, Aoki T, Sato H, Weiss SJ, Seiki M. Crosstalk between neovessels and mural cells directs the site‐specific expression of MT1‐MMP to endothelial tip cells. J Cell Sci. 2007;120:1607–1614. [DOI] [PubMed] [Google Scholar]
- 18. Tang Y, Rowe RG, Botvinick EL, Kurup A, Putnam AJ, Seiki M, Weaver VM, Keller ET, Goldstein S, Dai J, Begun D, Saunders T, Weiss SJ. MT1‐MMP‐dependent control of skeletal stem cell commitment via a beta1‐integrin/YAP/TAZ signaling axis. Dev Cell. 2013;25:402–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. [DOI] [PubMed] [Google Scholar]
- 20. Imai T, Takakuwa R, Marchand S, Dentz E, Bornert J‐M, Messaddeq N, Wendling O, Mark M, Desvergne B, Wahli W, Chambon P, Metzger D. Peroxisome proliferator‐activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc Natl Acad Sci USA. 2004;101:4543–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ohman MK, Wright AP, Wickenheiser KJ, Luo W, Russo HM, Eitzman DT. Monocyte chemoattractant protein‐1 deficiency protects against visceral fat‐induced atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maganto‐Garcia E, Tarrio M, Lichtman AH. Mouse models of atherosclerosis. Curr Protoc Immunol. 2001;Chapter 15:Unit 15.21.1‐23. [DOI] [PubMed] [Google Scholar]
- 23. Chun TH, Inoue M. 3‐D adipocyte differentiation and peri‐adipocyte collagen turnover. Methods Enzymol. 2014;538:15–34. [DOI] [PubMed] [Google Scholar]
- 24. Lynn Ray J, Leach R, Herbert J‐M, Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23:185–188. [DOI] [PubMed] [Google Scholar]
- 25. Afroze T, Husain M. C‐Myb‐binding sites mediate G1/S‐associated repression of the plasma membrane Ca2+‐ATPase‐1 promoter. J Biol Chem. 2000;275:9062–9069. [DOI] [PubMed] [Google Scholar]
- 26. Moraes C, Labuz JM, Leung BM, Inoue M, Chun TH, Takayama S. On being the right size: scaling effects in designing a human‐on‐a‐chip. Integr Biol (Camb). 2013;5:1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. [DOI] [PubMed] [Google Scholar]
- 28. Chang L, Villacorta L, Li R, Hamblin M, Xu W, Dou C, Zhang J, Wu J, Zeng R, Chen YE. Loss of perivascular adipose tissue on peroxisome proliferator‐activated receptor‐gamma deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation. 2012;126:1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen Z, Li C, Frieler RA, Gerasimova AS, Lee SJ, Wu J, Wang MM, Lumeng CN, Brosius FC, Duan SZ, Mortensen RM. Smooth muscle protein 22 alpha‐Cre is expressed in myeloid cells in mice. Biochem Biophys Res Commun. 2012;422:639–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gustafson B. Adipose tissue, inflammation and atherosclerosis. J Atheroscler Thromb. 2010;17:332–341. [DOI] [PubMed] [Google Scholar]
- 32. Uglow EB, Slater S, Sala‐Newby GB, Aguilera‐Garcia CM, Angelini GD, Newby AC, George SJ. Dismantling of cadherin‐mediated cell‐cell contacts modulates smooth muscle cell proliferation. Circ Res. 2003;92:1314–1321. [DOI] [PubMed] [Google Scholar]
- 33. Koutsouki E, Beeching CA, Slater SC, Blaschuk OW, Sala‐Newby GB, George SJ. N‐cadherin‐dependent cell‐cell contacts promote human saphenous vein smooth muscle cell survival. Arterioscler Thromb Vasc Biol. 2005;25:982–988. [DOI] [PubMed] [Google Scholar]
- 34. Hou R, Liu L, Anees S, Hiroyasu S, Sibinga NES. The FAT1 cadherin integrates vascular smooth muscle cell growth and migration signals. J Cell Biol. 2006;173:417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schlosser A, Pilecki B, Hemstra LE, Kejling K, Kristmannsdottir GB, Wulf‐Johansson H, Moeller JB, Fuchtbauer EM, Nielsen O, Kirketerp‐Moller K, Dubey LK, Hansen PB, Stubbe J, Wrede C, Hegermann J, Ochs M, Rathkolb B, Schrewe A, Bekeredjian R, Wolf E, Gailus‐Durner V, Fuchs H, Hrabe de Angelis M, Lindholt JS, Holmskov U, Sorensen GL. MFAP4 promotes vascular smooth muscle migration, proliferation and accelerates neointima formation. Arterioscler Thromb Vasc Biol. 2016;36:122–133. [DOI] [PubMed] [Google Scholar]
- 36. Ikesue M, Matsui Y, Ohta D, Danzaki K, Ito K, Kanayama M, Kurotaki D, Morimoto J, Kojima T, Tsutsui H, Uede T. Syndecan‐4 deficiency limits neointimal formation after vascular injury by regulating vascular smooth muscle cell proliferation and vascular progenitor cell mobilization. Arterioscler Thromb Vasc Biol. 2011;31:1066–1074. [DOI] [PubMed] [Google Scholar]
- 37. Ohuchi E, Imai K, Fujii Y, Sato H, Seiki M, Okada Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem. 1997;272:2446–2451. [DOI] [PubMed] [Google Scholar]
- 38. Kajita M, Itoh Y, Chiba T, Mori H, Okada A, Kinoh H, Seiki M. Membrane‐type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol. 2001;153:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Covington MD, Burghardt RC, Parrish AR. Ischemia‐induced cleavage of cadherins in NRK cells requires MT1‐MMP (MMP‐14). Am J Physiol Renal Physiol. 2006;290:F43–F51. [DOI] [PubMed] [Google Scholar]
- 40. Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001;52:372–386. [DOI] [PubMed] [Google Scholar]
- 41. Shofuda K, Yasumitsu H, Nishihashi A, Miki K, Miyazaki K. Expression of three membrane‐type matrix metalloproteinases (MT‐MMPs) in rat vascular smooth muscle cells and characterization of MT3‐MMP with and without transmembrane domain. J Biol Chem. 1997;272:9749–9754. [DOI] [PubMed] [Google Scholar]
- 42. Hotary K, Allen E, Punturieri A, Yana I, Weiss SJ. Regulation of cell invasion and morphogenesis in a three‐dimensional type I collagen matrix by membrane‐type matrix metalloproteinases 1, 2, and 3. J Cell Biol. 2000;149:1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kothapalli D, Liu SL, Bae YH, Monslow J, Xu T, Hawthorne EA, Byfield FJ, Castagnino P, Rao S, Rader DJ, Puré E, Phillips MC, Lund‐Katz S, Janmey PA, Assoian RK. Cardiovascular protection by ApoE and ApoE‐HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep. 2012;2:1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hong J‐H, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, Mueller E, Benjamin T, Spiegelman BM, Sharp PA, Hopkins N, Yaffe MB. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309:1074–1078. [DOI] [PubMed] [Google Scholar]
- 45. Dinardo CL, Venturini G, Zhou EH, Watanabe IS, Campos LC, Dariolli R, da Motta‐Leal‐Filho JM, Carvalho VM, Cardozo KH, Krieger JE, Alencar AM, Pereira AC. Variation of mechanical properties and quantitative proteomics of VSMC along the arterial tree. Am J Physiol Heart Circ Physiol. 2014;306:H505–H516. [DOI] [PubMed] [Google Scholar]
- 46. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AAC, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4‐dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shimizu‐Hirota R, Xiong W, Baxter BT, Kunkel SL, Maillard I, Chen X‐Y, Sabeh F, Liu R, Li X‐Y, Weiss SJ. MT1‐MMP‐dependent regulation of a PI3Kδ‐nucleosome remodeling axis controls macrophage immune function. Genes Dev. 2012;26:395–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of Genes Differentially Expressed in Cre(−) and SM22α‐Cre(+)Mmp14 F/F Vascular Smooth Muscle Cells
Figure S1. Arterial trees dissected from Cre(−), SM22α‐Cre(+), and aP2‐Cre(+) MMP14 F/F Apoe −/− male and female mice after a 12‐week Western diet.
Figure S2. Gene expression of Acta2 (α‐SMA), Cre enzyme, MT1‐MMP (membrane‐type 1 matrix metalloproteinase; MMP14), MMP‐2, MMP‐8, MMP‐9, MMP‐13, and MT2‐MMP (MMP15) in primary vascular smooth muscle cells (VSMCs) isolated from Cre(−) and SM22α‐Cre(+) MMP14 F/F Apoe −/− mice. Almost complete suppression of MT1‐MMP expression in primary VSMCs was confirmed coupled with SM22‐dependent Cre expression. Expression of other matrix metalloproteinases was not affected. ***P<0.001.