

Abstract
Cardiovascular physiology exhibits time-of-day-dependent oscillations, which are mediated by both extrinsic (e.g., environment/behavior) and intrinsic (e.g., circadian clock) factors. Disruption of circadian rhythms negatively affects multiple cardiometabolic parameters. Recent studies suggest that the cardiomyocyte circadian clock directly modulates responsiveness of the heart to metabolic stimuli (e.g., fatty acids) and stresses (e.g., ischemia/reperfusion). The aim of this study was to determine whether genetic disruption of the cardiomyocyte circadian clock impacts insulin-regulated pathways in the heart. Genetic disruption of the circadian clock in cardiomyocyte-specific Bmal1 knockout (CBK) and cardiomyocyte-specific Clock mutant (CCM) mice altered expression (gene and protein) of multiple insulin signaling components in the heart, including p85α and Akt. Both baseline and insulin-mediated Akt activation was augmented in CBK and CCM hearts (relative to littermate controls). However, insulin-mediated glucose utilization (both oxidative and non-oxidative) and AS160 phosphorylation were attenuated in CBK hearts, potentially secondary to decreased Inhibitor-1. Consistent with increased Akt activation in CBK hearts, mTOR signaling was persistently increased, which was associated with attenuation of autophagy, augmented rates of protein synthesis, and hypertrophy. Importantly, pharmacological inhibition of mTOR (rapamycin; 10 days) normalized cardiac size in CBK mice. These data suggest that disruption of cardiomyocyte circadian clock differentially influences insulin-regulated processes, and provide new insights into potential pathologic mediators following circadian disruption.
Keywords: circadian rhythm, hypertrophy, insulin signaling, metabolism
Introduction
Cardiovascular disease (CVD) is the leading cause of death in the United States and industrialized countries world-wide.(1) Diabetes mellitus increases the incidence of CVD, and worsens outcomes.(2) The most common form of diabetes is type 2 diabetes, which is characterized by both insulin resistance and insulin insufficiency.(3) Peripheral tissues exhibit differential insulin resistance, in terms of the impact on both distinct organs and intracellular signaling branches, which contributes to pathogenesis of the disease. In the case of the heart, insulin influences a host of processes central to cardiac function, ranging from glucose uptake and utilization, to the turnover of cellular constituents (e.g., protein, organelles).(4,5) Despite hyperactivation of the insulin signaling pathway in type 2 diabetes, insulin-mediated glucose uptake is repressed. The contribution of aberrant cardiac insulin signaling toward the development of heart failure in diabetes has recently been reviewed.(6)
Twenty-four-hour cycles have been observed for various biological functions, in essentially all species.(7,8) These oscillations are driven by both extrinsic and intrinsic factors. The latter includes the cell autonomous circadian clock, a sophisticated transcription-translation feedback loop mechanism that temporally partitions cellular processes. Two critical clock components include brain and muscle arnt-like 1 (Bmal1) and circadian locomotor output cycles kaput (Clock), which bind to conserved E-boxes in the promoter region of clock controlled genes (as a heterodimer), resulting in 24-hour oscillations in gene expression (and ultimately biologic processes).(9,10) Although originally identified in the suprachiasmatic nucleus (SCN), this transcriptional network has been described in virtually all mammalian cells/organs, influencing processes ranging from the whole body (e.g., behaviors) to cellular levels (e.g., metabolism/signaling).(7,8) In doing so, circadian clocks temporally orchestrate biologic/cellular functions, ensuring appropriate responses to predicted environmental stimuli/stresses.
Circadian disruption, whether environmental (e.g., shift work) or genetic (e.g., polymorphisms), increases the risk of obesity, diabetes, and CVD.(11–14) The clinical impact of abnormal 24-hr rhythms is underscored by observations such as non-dipping hypertension, which results in greater risk of adverse cardiac events relative to dipping hypertension.(15,16) Consistent with these reports, animal studies indicate that germline knockout of circadian clock components increases adiposity, alters insulin secretion/sensitivity, impacts blood pressure, and impairs contractile function of the heart.(17–20) Moreover, circadian clocks are altered during various pathologies, including obesity, diabetes, and hypertension.(21–23) Collectively, these observations suggest that disruption of circadian clocks may play a causal role in the development of cardiometabolic diseases.
Recent studies suggest that the cardiomyocyte circadian clock influences numerous processes essential for maintenance of cardiac function, including metabolism, electrical activity, and cell signaling.(24–26) In the latter case, we have reported that the cardiomyocyte circadian clock influences responsiveness of the heart to both physiologic (e.g., fatty acids) and pathologic (e.g., ischemia/reperfusion) stimuli/stresses.(27–29) We now show that genetic disruption of the cardiomyocyte circadian clock leads to alterations in gene/protein expression of multiple insulin signaling components. This results in chronic activation of the Akt/mTOR/S6 signaling axis, increased protein synthesis, and increased cardiac size; the latter are normalized by pharmacologic inhibition of mTOR. Chronic mTOR activation is also associated with inhibition of autophagy. Conversely, despite chronic activation of Akt, insulin-mediated glucose utilization was attenuated following circadian disruption. These studies highlight a critical role of the cardiomyocyte circadian clock in regulating myocardial insulin signaling/sensitivity, and suggest that circadian disruption differentially influences distinct branches of the insulin signaling cascade known to regulate critical cardiac processes.
Methods
Mice
The cardiomyocyte circadian clock was genetically ablated by targeting BMAL1 and CLOCK, as described previously.(24,25) Twelve to sixteen week old male cardiomyocyte-specific Bmal1 knockout (CBK; BMAL1flox/flox/α-MHC-CRE+/−) and littermate control mice (CON; BMAL1flox/flox/MHC-CRE−/−) on the C57BL/J6 background were utilized, as well as cardiomyocyte-specific Clock mutant (CCM; α-MHC-dnCLOCK+/−) and littermate control mice (WT; α-MHC-dnCLOCK−/−) on the FVB/N background. Mice were kept in temperature, humidity, and light-controlled rooms on a strictly enforced 12-hour light/12-hour dark schedule, where ZT0 refers to the beginning of the light phase and ZT12 refers to the beginning of the dark phase. All experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. It is noteworthy that the current study utilized CCM mice as a secondary model of cardiomyocyte-specific circadian clock disruption, and as such the data obtained for this model is located in supplemental figures.
Rapamycin Feeding
In select experiments, mice were provided food supplemented with encapsulated rapamycin at 42ppm (eRAPA, Rapamycin Holdings Inc., San Antonio, TX); diet containing encapsulation material was used as a control.
In vivo Insulin Challenge
Mice were fasted for 6 hours prior to anesthetization (400 mg/kg chloral hydrate), followed by administration of insulin (0.167 U/kg; a sub-maximal insulin level, determined by initial dose-response studies [data not shown]) or vehicle (saline) via the inferior vena cava (at ZT0 or ZT12). Hearts were isolated and snap frozen in liquid nitrogen 5 minutes after insulin/saline administration. Frozen hearts were stored at −80°C, and subsequently utilized for Western blot analysis.
Ex vivo Myocardial Insulin Challenge
Hearts were isolated (at ZT0 or ZT12) and perfused in the non-recirculating working mode as described previously.(24,25) A subset of hearts were challenged with insulin (100 μU/mL) for 5 minutes, and subsequently utilized for Western blot analysis. A second set of hearts were challenged with insulin (0, 10, or 100 μU/mL) for 30 minutes, during which time metabolic fluxes were measured via addition of metabolic tracers: D-[U-14C]-glucose (0.20 mCi/L; oxidative and non-oxidative glucose utilization) and L-[2,3,4,5,6-3H]-phenylalanine (0.20 mCi/L; protein synthesis).(30) More specifically, the following was assessed: 1) full oxidation of exogenous glucose by following the release of [14C]-CO2 into the perfusate (i.e., exogenous glucose oxidation; (31)); 2) the anaerobic catabolism of exogenous glucose to lactate by following the release of [14C]-lactate into the perfusate (i.e., a non-absolute measure that correlates with glucose uptake plus glycolysis; (32)); 3) the net incorporation of exogenous glucose into glycogen by following the generation of [14C]-glycogen (i.e., net glycogen synthesis; (33)); and 4) the net incorporation of exogenous phenylalanine into protein by following the generation of [3H]-protein (i.e., net protein synthesis; (34)). In the case of [14C]-CO2 and [14C]-lactate release, coronary effluent was collected at 5-minute intervals during the perfusion, and data are presented as steady state values (i.e., the last 10 minutes of the perfusion protocol). In the case of net glycogen and protein synthesis, hearts were freeze clamped at the end of the perfusion protocol, followed by extraction of [14C]-glycogen and [3H]-protein, as described previously.(33,35)
In vivo Protein Synthesis
Mice were injected via tail vein with 150 mM Phenylalanine plus L-[2,3,4,5,6-3H] Phenylalanine (0.18 mCi/kg), according to the flooding dose technique.(35) Myocardial protein was precipitated and washed with 10% trichloroacetic acid, neutralized with 1 N NaOH, and counted in a scintillation counter (Beckman Coulter).
Quantitative RT-PCR
RNA was extracted from hearts followed by assessment of gene expression using standard procedures as described previously.(36,37) Gene expression analysis of insulin signaling and autophagic components (Insr, Irs1, Irs2, Pik3ca, Pik3r1, Pdpk1, Akt1, Akt2, As160, Gsk3β, I-1, Mtor, Ulk1, Lc3, P62) was performed using conventional methods with SYBR Green; primer sequences are included in Supplemental Table 1. All RT-PCR data were normalized to the average expression of three different housekeeping genes: acidic ribosomal phosphoprotein P0 (36B4), hypoxanthine phosphoribosyltransferase 1 (Hprt), and TATA-binding protein (Tbp), as described previously.(38) ΔΔCt values are represented as fold change from a designated control group.
Western Blotting
Qualitative analysis of protein expression and phosphorylation status was performed via standard western blotting procedures as described previously.(39) Briefly, 20 μg protein lysate was separated on polyacrylamide gels and transferred to PVDF membranes. Membranes were probed for the following targets: IRS1 (Cell Signaling 3194), p-IRS1Tyr612 (Abcam ab66153), p110α (Cell Signaling 4255), p85α (Abcam ab22653), p-PDPK1Ser241 (Cell Signaling 3438), PDPK1 (Cell Signaling 3062), p-AktSer473 (Cell Signaling 9271), p-AktThr308 (Cell Signaling 9275), Akt (Cell Signaling 9272), p-AS160Thr642 (Cell Signaling 8881), AS160 (Millipore 07-741), p-GSK3βSer9 (Cell Signaling 9336), GSK3α/β (Santa Cruz 7291), Inhibitor-1 (Abcam 40877), p-mTORSer2448 (Cell Signaling 2971), mTOR (Cell Signaling 2983), p-Ulk1Ser757 (Cell Signaling 6888), Ulk1 (Sigma Aldrich A7481), LC3A/B (Cell Signaling 12741), p62 (Abnova H00008878-MO1), p-S6Ser240/244 (Cell Signaling 2215), S6 (Cell Signaling 2317), p-4EBP1Thr37/46 (Cell Signaling 2855), 4EBP1 (Cell Signaling 9452), GLUT1 (Abcam ab40084), GLUT4 (Millipore 07-1404), Hexokinase II (Cell Signaling 2867), Calsequestrin (Abcam ab3516). Rabbit and mouse HRP-conjugated secondary antibodies (Santa Cruz sc-2004 and sc-2005, respectively) were used for chemiluminescent detection with Luminata Forte Western Blotting substrate (Millipore). Importantly, due to the nature of time course studies, in order to minimize the contribution that position on the gel might have on outcomes, samples were randomized on gels; samples were re-ordered post-imaging, only for the sake of illustration of representative images (note, all bands for representative images for an individual experiment were from the same gel).
Electron Microscopy
Myocardial apex samples harvested from 16-hr fasted mice were fixed in a modified Karnovsky’s fixative (2% Paraformaldehyde/2% Glutaraldehyde in 0.1M Phosphate Buffer), embedded, and sectioned at 70–90 nm using standard EM techniques, followed by imaging at the High Resolution Imaging Facility at the University of Alabama at Birmingham using a Tecnai T12 Spirit (FEI, Hillsboro, OR).(40)
Statistical analysis
Statistical analyses were performed using student t-tests (Excel 2010) and two-way ANOVA (SPSS, IBM Software) followed by LSD post hoc analyses for pair-wise comparisons, as described previously. In all analyses, the null hypothesis of no model effects was rejected at p < 0.05.
Results
Genetic disruption of the cardiomyocyte circadian clock alters expression of key insulin signaling components in the heart
To investigate whether disruption of the cardiomyocyte circadian clock might influence insulin signaling in the heart, mRNA levels of insulin signaling components were measured in hearts isolated from CBK and littermate CON mice at the beginning of the light phase (ZT0), the middle of the light phase (ZT6), the beginning of the dark phase (ZT12), and the middle of the dark phase (ZT18). This analysis revealed significant time main effects for Irs1, Akt1, and Akt2 (and a trend for Pik3r1; p=0.079), as well as significant genotype main effects for Insr, Irs1, Pik3r1, Akt1, and Akt2; of the latter genes, Insr, Pik3r1, Akt1, and Akt2 were significantly lower in CBK than in CON hearts (Figure 1A). A similar analysis of CCM (a second model of cardiomyocyte-specific circadian clock disruption) and CON hearts showed significant time main effects for Irs1, in addition to significant genotype main effects for irs2, pik3r1, and akt1; CCM hearts exhibit decreased expression of Irs2, Pik3r1, and Akt1 (Supplemental Figure 1A). Collectively, these data suggest that genetic disruption of the cardiomyocyte circadian clock attenuates gene expression of distinct insulin signaling components in the heart, particularly Pik3r1 and Akt1 (i.e., decreased in both CBK and CCM hearts).
Figure 1.
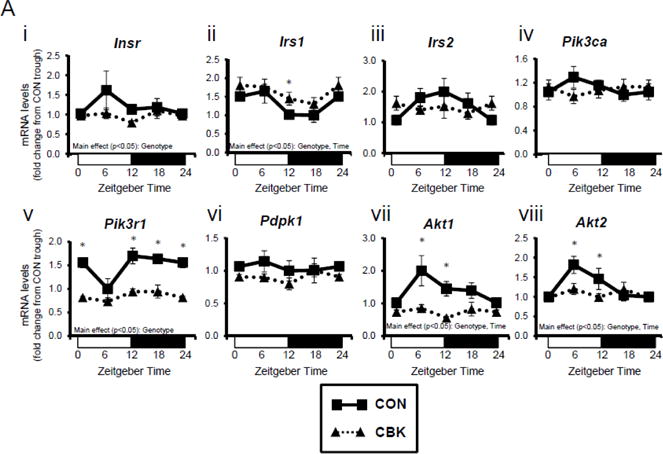
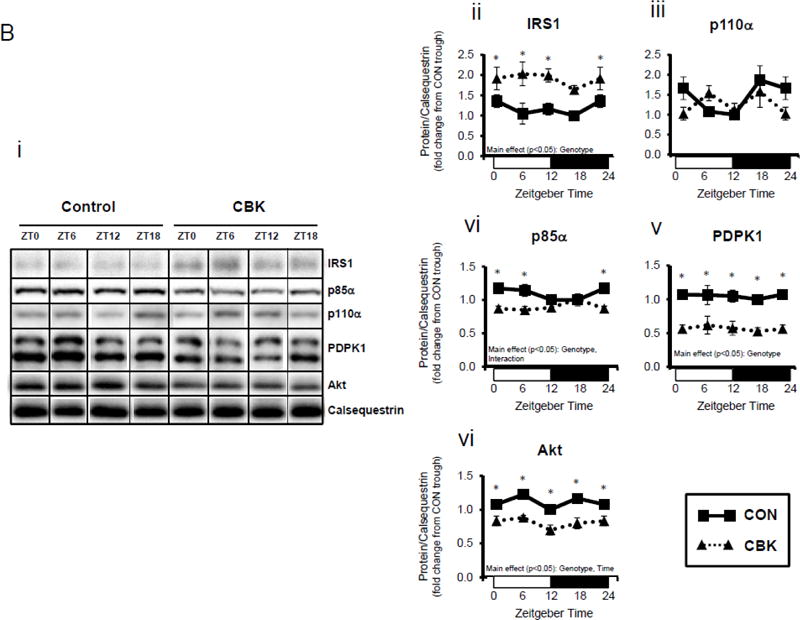
Genetic ablation of the cardiomyocyte circadian clock influences diurnal variations in mRNA (A) and protein (B) levels of key insulin-signaling components in the heart. CBK and littermate control (CON) mouse hearts were collected at distinct times of day, followed by RT-PCR (A; 4–7 independent observations) and Western blot (B; n=4–7 independent observations) analysis. Data are represented as fold change from the trough of CON hearts. Data for ZT0 is duplicated as ZT24 for visualization purposes only. All data are shown as mean ± SEM. *, p<0.05 for CBK versus CON hearts within a ZT.
Next, we assessed whether transcriptional alterations following genetic disruption of the cardiomyocyte circadian clock were associated with parallel changes in protein levels. This analysis revealed a significant time main effect for Akt, as well as genotype main effects for IRS1, p85α (encoded by Pik3r1), PDPK1, and Akt (Figure 1B). Consistent with alterations in mRNA levels (Figure 1A), p85α, PDPK1, and Akt were decreased in CBK hearts (by 20%, 50%, and 30%, respectively). In contrast, IRS1 was increased almost 2-fold in CBK hearts (consistent with increased mRNA levels; Figures 1A–B). Similar to findings in CBK mice, we observed decreased p85α, PDPK1, and Akt protein levels in CCM hearts (versus CON hearts; Supplemental Figure 1B). Collectively, these data suggest that genetic disruption of the cardiomyocyte clock decreases protein levels of multiple insulin signaling components in the heart, including p85α, PDPK1, and Akt.
Differential myocardial insulin signaling/sensitivity following cardiomyocyte clock disruption
Next, we evaluated whether myocardial insulin signaling/sensitivity is altered following genetic disruption of the cardiomyocyte circadian clock. Initially, the phosphorylation status of insulin signaling components was assessed in hearts isolated from ad libitum fed CBK, CCM, and littermate CON mice, at distinct times of the day. Due to observed genotype-specific alterations in total protein levels for distinct insulin signaling components, phospho-proteins were normalized to calsequestrin; normalization to total protein levels can be found in Supplemental Figure 3. Regarding CBK versus CON hearts, there were significant time main effects for p-AktSer473 and p-AktThr308, and a significant genotype main effect for p-AktSer473 (as well as a trend for p-AktThr308; p=0.087) (Figure 2A). p-AktSer473 was chronically increased at all times of day in CBK hearts (by approximately 2-fold). Although no significant genotype main effects were observed for phospho-proteins in CCM hearts, a time main effect was observed for p-AktThr308 (Supplemental Figure 2A).
Figure 2.
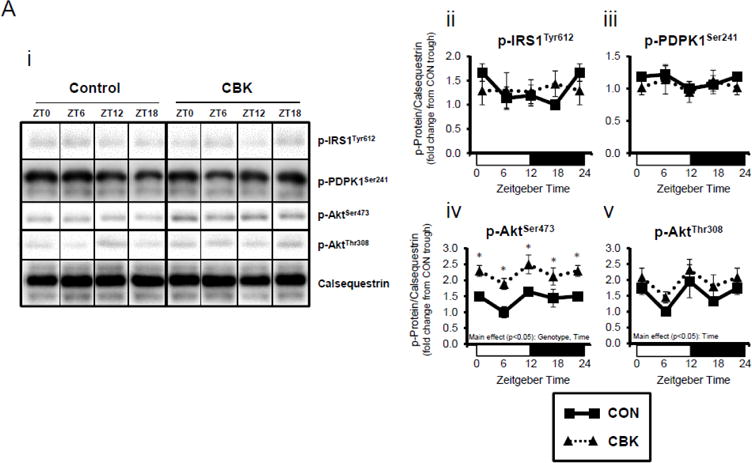
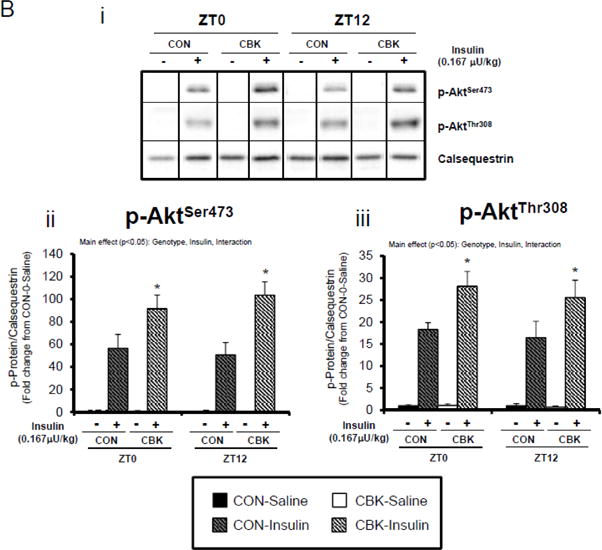
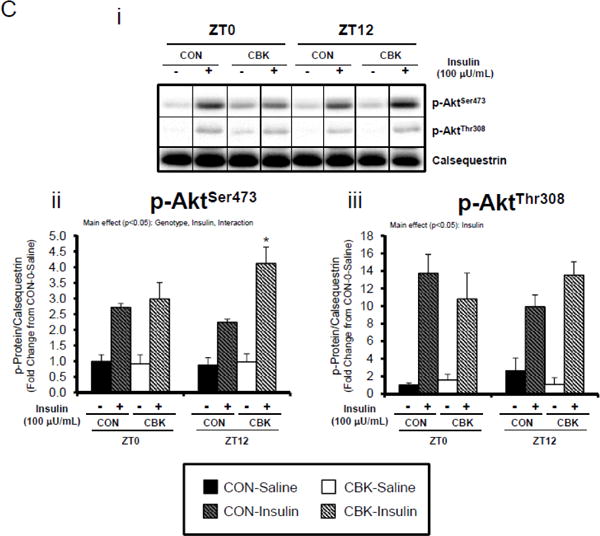
Perturbations in baseline and insulin-mediated phosphorylation status of key insulin-signaling components in CBK hearts in vivo (A-B) and ex vivo (B). Analysis of the phosphorylation status of insulin signaling components in CBK and CON hearts isolated at distinct times of day from ad libitum fed mice (A; 4–7 independent observations). Effects of challenging CBK and CON hearts with insulin (5 minutes) in vivo (0.167μU/kg; B; 3–7 independent observations) or ex vivo (100μU/mL; C; 3–7 independent observations) on Akt phosphorylation at distinct times of the day; saline challenge serves as vehicle control. Data are represented as fold change from the trough of CON hearts (A), or as fold change from CON hearts challenged with saline at ZT0 (B, C). Data for ZT0 is duplicated as ZT24 for visualization purposes only (A). All data are shown as mean ± SEM. *, p<0.05 for CBK versus CON hearts within a ZT (A), or CBK versus CON hearts within a ZT plus saline/insulin experimental group (B).
We next evaluated whether disruption of the cardiomyocyte circadian clock influences myocardial insulin sensitivity by investigating insulin-mediated Akt phosphorylation in vivo. After a 6-hr fast, CBK, CCM, and littermate CON mice were challenged with insulin or saline (vehicle) at either ZT0 or ZT12. In CON mice, insulin increased p-AktSer473 and p-AktThr308 independent of the time-of-day (Figure 2B). Insulin-mediated activation of p-AktSer473 and p-AktThr308 was significantly higher in CBK hearts (relative to CON hearts), independent of time-of-day (Figure 2B). Similarly, an augmented response to insulin was observed in CCM (compared to CON) hearts, particularly at ZT12 (Supplemental Figure 2B). In an attempt to eliminate potential neurohumoral factors influencing insulin sensitivity in vivo, CBK and littermate CON hearts were then challenged ex vivo with insulin (100 μU/mL) for 5-min at ZT0 and ZT12; CCM hearts were not investigated ex vivo. Similar to observations in vivo, isolated perfused CBK hearts showed increased insulin-mediated activation of p-AktSer473, particularly at ZT12 (Figure 2C). Collectively, these data suggest that genetic disruption of the cardiomyocyte circadian clock enhances insulin-mediated Akt phosphorylation in the heart.
Insulin-mediated glucose metabolism is attenuated in CBK hearts
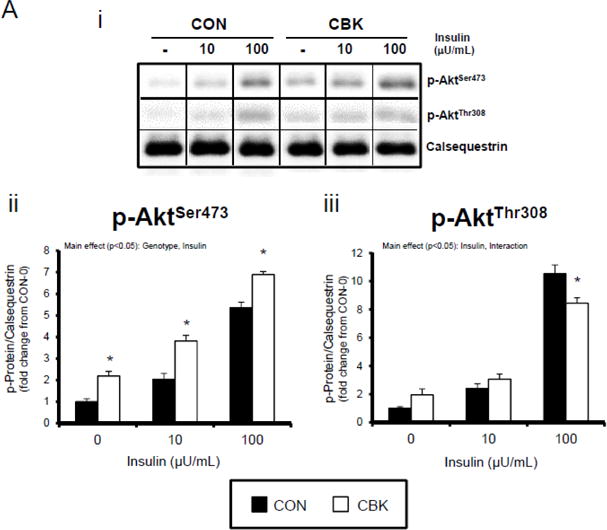
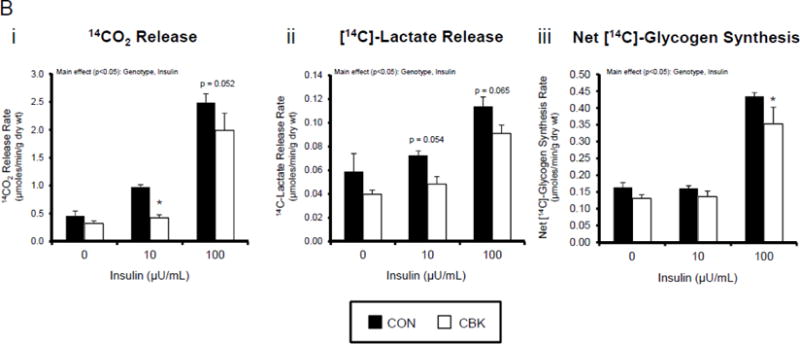
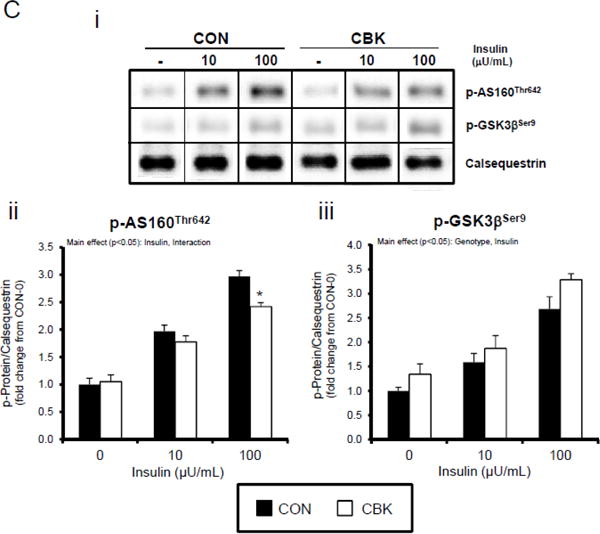
We next investigated whether perturbations in insulin signaling/sensitivity following genetic disruption of the cardiomyocyte circadian clock influenced insulin-regulated processes. First, we assessed glucose utilization. Accordingly, CBK and littermate CON hearts were perfused ex vivo in the working mode for 30-min under baseline, sub-maximal (10 μU/mL), and maximal (100 μU/ml) insulin stimulation conditions; only a single time of day (ZT12) was investigated. Insulin dose-dependently increased both p-AktSer473 and p- AktThr308 (Figure 3A). Moreover, p-AktSer473 levels were augmented in CBK (relative to CON) hearts, at all insulin concentration. Although p-AktThr308 tended to be higher in CBK hearts at basal and sub-maximal conditions, it was significantly lower following maximal insulin stimulation (Figure 3A). In marked contrast, insulin-mediated glucose utilization (both oxidative ([14C]-CO2 release) and non-oxidative ([14C]-lactate release and net [14C]-glycogen synthesis)) was significantly attenuated in CBK (compared to CON) hearts (Figure 3B). To address the apparent disconnect between Akt signaling and glucose utilization, the Akt targets AS160 (involved in GLUT4 trafficking and glucose uptake) and GSK3β (involved in regulation of glycogen synthesis) were assessed in CBK and littermate CON hearts. Consistent with attenuated glucose utilization, CBK hearts exhibited decreased insulin-mediated p-AS160Thr642 relative to CON hearts (Figure 3C). Similarly, p-AS160Thr642 was lower in CBK hearts isolated from ad libitum fed mice, independent of the time-of-day; AS160 total protein was also reduced in CBK mice (as well as a trend for decreased mRNA levels in CBK mice [p = 0.10]; Figure 3D). Although p-GSK3βSer9 is lower in CBK hearts independent of the time-of-day (in the absence of alterations in total protein or mRNA levels; Figure 3D), ex vivo insulin stimulation normalized p-GSK3βSer9 levels (Figure 3C). Both p-AS160Thr642 and p-GSK3βSer9 were attenuated in CCM (versus CON) hearts (Supplemental Figure 4).
Figure 3.
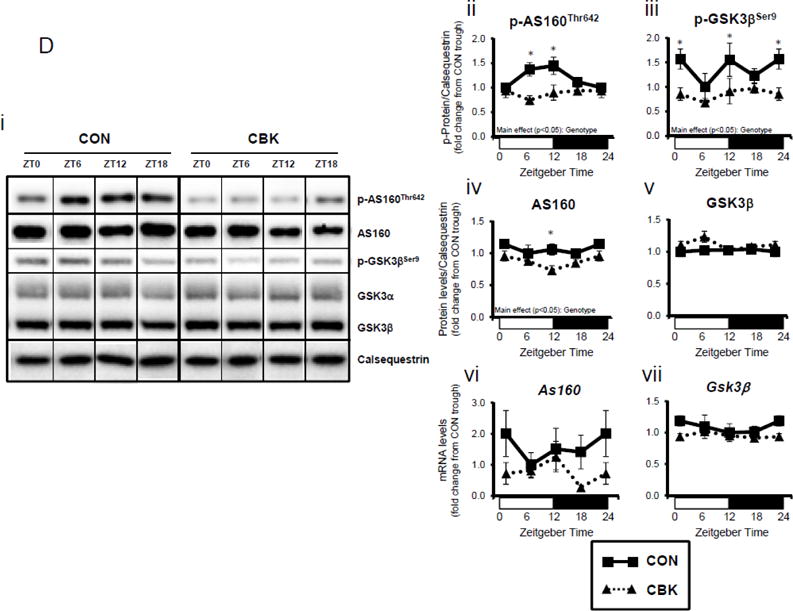
Disruption of the cardiomyocyte circadian clock alters insulin-mediated signaling (A, C, D, E) and glucose utilization (B). Effects of challenging CBK and CON hearts with insulin (0, 10, or 100μU/mL; 30 minutes) ex vivo at ZT12 on Akt phosphorylation (A; 5 independent observations), glucose utilization (B; 5 independent observations), as well as on AS160 and GSK3β phosphorylation (C; 5 independent observations). CBK and littermate CON hearts were collected at distinct times of day, followed by RT-PCR and Western blot analysis for AS160 (D; 3–7 independent observations), GSK3β (D; 3–7 independent observations), and I-1 (E; 3–7 independent observations). Data are represented as fold change from CON hearts challenged with saline (A, C), as absolute rates (B), or as fold change from the trough of CON hearts (D, E). Data for ZT0 is duplicated as ZT24 for visualization purposes only (A). All data are shown as mean ± SEM. *, p<0.05 for CBK versus CON hearts within a ZT (D, E), or CBK versus CON hearts within a ZT plus saline/insulin experimental group (A, B, C).
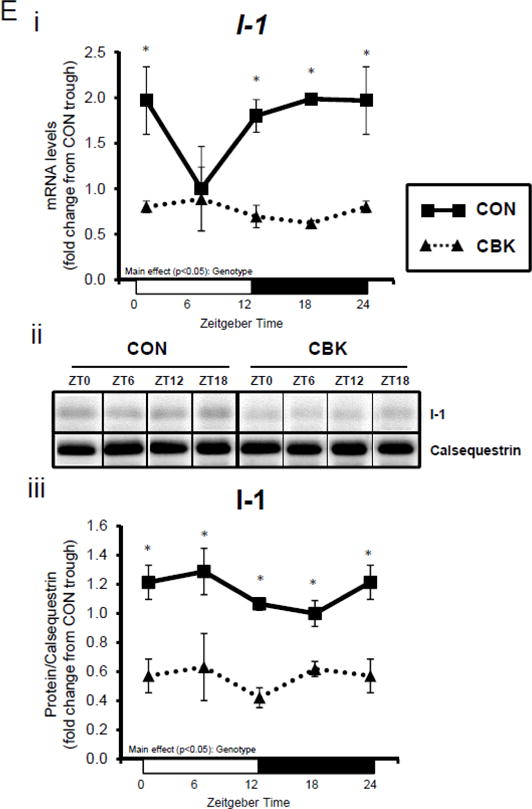
A dissociation appears to exist between Akt activation and its downstream target AS160 and GSK3β (although in the latter case, insulin appears to normalize p-GSK3βSer9) following genetic disruption of the cardiomyocyte circadian clock. Both AS160 and GSK3β are dephosphorylated by protein phosphatase 1 (PP1). Analyses of recent unbiased microarray studies in CBK and CCM hearts suggest that, although PP1 expression is not influenced by circadian clock disruption, inhibitor-1 (I-1; which usually represses PP1 activity) is markedly repressed following cardiomyocyte circadian clock disruption.(24,25) RT-PCR and Western Blot analyses of CBK and CON hearts confirmed 55% and 50% decreases of I-1 in CBK hearts at mRNA and protein levels, respectively (Figure 3E). I-1 was similarly decreased in CCM hearts (data not shown). These observations suggest that chronic activation of PP1, secondary to diminished I-1, may account for attenuation of both p-AS160Thr642 and p-GSK3βSer9, as well as attenuated glucose utilization, following genetic disruption of the cardiomyocyte circadian clock.
Chronic activation of mTOR in CBK hearts is associated with repressed autophagy
Insulin is a potent anabolic hormone, attenuating autophagy via the Akt/mTOR/ULK1 pathway, and stimulating protein synthesis via the Akt/mTOR/S6/4EBP1 pathway. Given increased p-AktSer473 (and p-AktThr308 to a lesser extent) following genetic disruption of the cardiomyocyte circadian clock, we next measured p-mTORSer2448 in CBK and CON hearts isolated from ad libitum fed mice at distinct times of the day. Independent of the time-of-day, p-mTORSer2448 was persistently elevated in CBK hearts (in the absence of alterations in mTOR total protein or mRNA levels; Figure 4A). Interrogation of ULK1, a known mTOR target, revealed elevated p-ULK1Ser757 levels; total ULK1 protein levels were also elevated in CBK hearts (whereas mRNA levels were persistently repressed; Figure 4B). Interestingly, CON hearts exhibited biphasic patterns for p-mTORSer2448 and p-ULK1Ser757, both peaking at ZT0 and ZT12 (Figure 4A–B). Consistent with the inhibitory effect of mTOR/ULK1 signaling on autophagy, LC3 lipidation also exhibited a biphasic pattern in CON hearts, troughing at ZT0 and ZT12 (Figure 4C). However, LC3 lipidation was not altered in CBK hearts (although Lc3 mRNA was chronically elevated in CBK hearts; Figure 4C). Similarly, a second autophagic marker, p62, was chronically elevated in CBK hearts (while P62 gene expression was diminished; Figure 4D). In order to stimulate autophagy, CON and CBK mice were fasted (16-hr). Fasting increased LC3 lipidation and decreased p62 levels, independent of genotype (Figure 4E). In addition, p62 was elevated in CBK hearts independent of feeding status (genotype main effect; Figure 4E). Furthermore, electron micrographs revealed a greater number of autophagosome structures in CBK hearts (versus CON hearts) following fasting (Figure 4E). Collectively, these data suggest impairment of autophagic flux in CBK hearts, thus attenuating p62 and autophagosome degradation.
Figure 4.
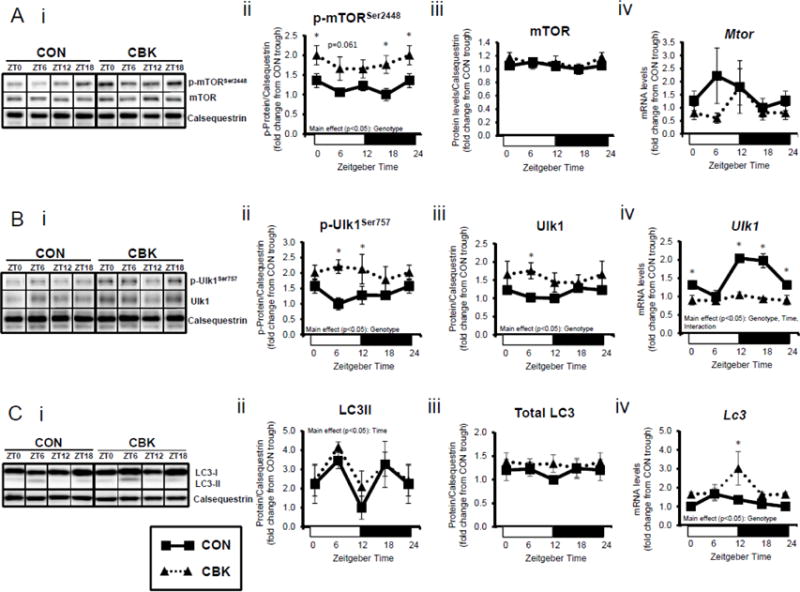
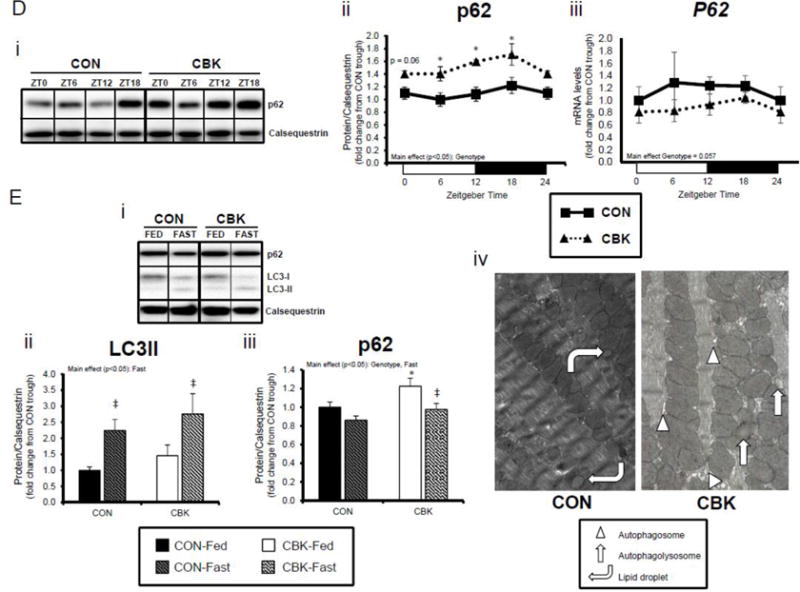
Cardiomyocyte-specific Bmal1 deletion alters autophagy markers in the heart. CBK and littermate control (CON) mouse hearts were collected at distinct times of day, followed by Western blot and RT-PCR analysis for mTOR (A; 3–7 independent observations), ULK1 (B; 3–7 independent observations), LC3 (C; 3–7 independent observations), and p62 (D; 3–7 independent observations). In a separate set of studies (E; 3–7 independent observations), CBK and CON mice were either fed ad libitum or fasted for 16 hours (ZT4 to ZT20), followed by collection of hearts (at ZT20), and subsequent assessment of LC3 lipidation, p62, and accumulation of autophagosomes. Data are represented as fold change from the trough of CON hearts (A, B, C, D), or as fold change from CON hearts from ad libitum fed mice (E). Data for ZT0 is duplicated as ZT24 for visualization purposes only (A, B, C, D). All data are shown as mean ± SEM. *, p<0.05 for CBK versus CON hearts within a ZT (A, B, C, D), or CBK versus CON hearts within a fed/fasted experimental group (E).
Chronic activation of mTOR in CBK hearts is associated with protein synthesis augmentation
We next investigated the effects of chronic activation of mTOR following circadian disruption on modulators of protein synthesis. Both p-S6Ser240/244 and p-4EBP1Thr37/46 exhibited a trend for a time-of-day-dependent oscillations in CON hearts (1-way ANOVA), with a peak at ZT0 (beginning of the light phase) and trough at ZT18 (middle of the dark phase; Figure 5A); these oscillations were mirrored by time-of-day-dependent fluctuations in net [3H]-protein synthesis in CON hearts (Figure 5A). Importantly, time-of-day-dependent oscillations in p-S6Ser240/244, p-4EBP1Thr37/46, and net [3H]-protein synthesis were absent in CBK hearts (Figure 5A). In the case of protein synthesis, rates were chronically elevated in CBK hearts (genotype main effect; Figure 5A). Additional investigation of net [3H]-protein synthesis ex vivo confirmed increased rates in CBK hearts (compared to CON hearts) independent of insulin (i.e., genotype main effect; Figure 5B). Similarly, p-mTORSer2448 and p-S6Ser240/244 were both increased in ex vivo perfused CBK hearts (Figure 5B). Interestingly, biventricular weight (BVW) exhibited a trend (p=0.064) for a time-of-day-dependent pattern in CON mice (trough at ZT18), which is absent in CBK mice (Figure 5C). Collectively, these findings suggest that genetic disruption of the cardiomyocyte circadian clock leads to persistent elevation in the mTOR/S6/4EBP1 signaling axis, increased rates of protein synthesis, and increased heart weight.
Figure 5.
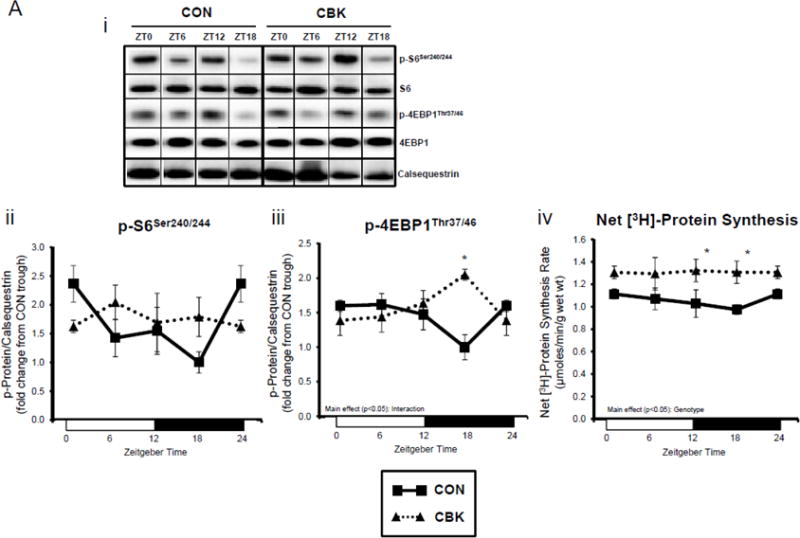
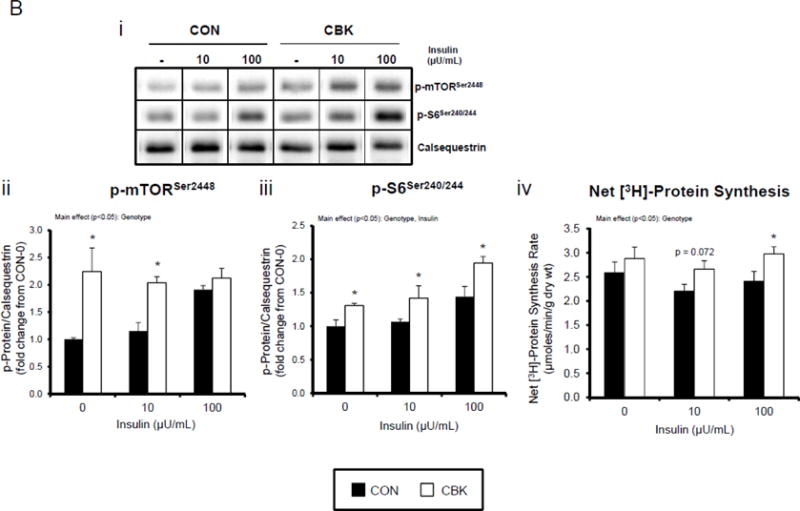
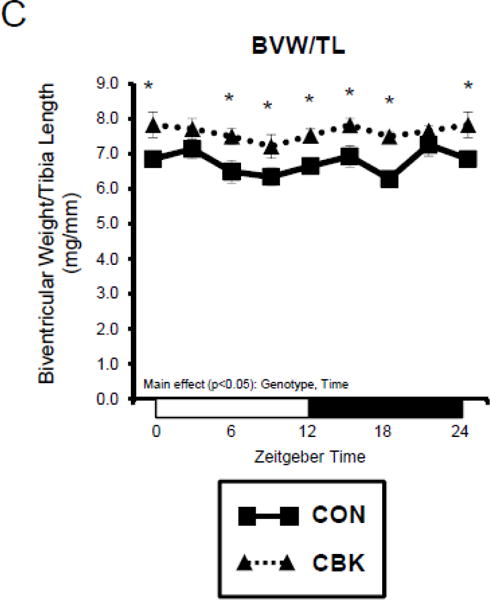
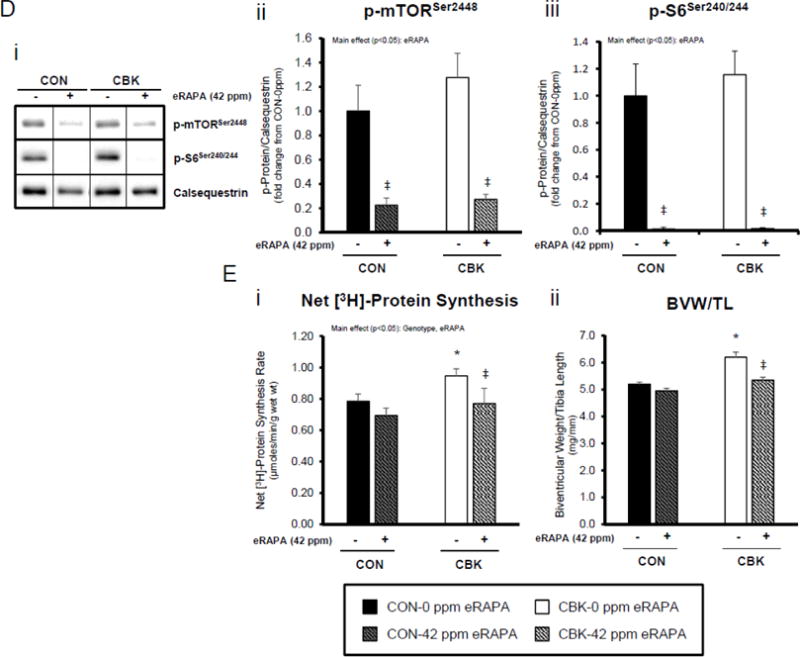
Impact of disruption of the cardiomyocyte circadian clock on protein synthesis in the heart. CBK and littermate control (CON) mouse hearts were collected at distinct times of day, followed by Western blot (A; 3–7 independent observations) and gravimetric (C; 6–12 independent observations) analysis. Rates of protein synthesis in CBK and CON hearts in vivo at distinct times of the day (A; 5 independent observations). Effects of challenging CBK and CON hearts with insulin (0, 10, or 100μU/mL; 30 minutes) ex vivo at ZT12 on mTOR and S6 phosphorylation, as well as protein synthesis (B; 5 independent observations). Impact of rapamycin (42 ppm in the diet; 10 days) on mTOR and S6 phosphorylation (D; 8–9 independent observations), as well as protein synthesis (Ei; 4–5 independent observations) and heart weight (Eii; 10–14 independent observations). Data are represented as fold change from the trough of CON hearts (phosphoprotein data in A), as fold change from CON hearts challenged with saline (phosphoprotein data in B), as absolute values (protein synthesis and heart weight data in A, B, C, E), or as fold change from CON animals fed rapamycin-free diet (phosphoprotein data in D). Data for ZT0 is duplicated as ZT24 for visualization purposes only (A, C). All data are shown as mean ± SEM. *, p<0.05 for CBK versus CON hearts within a ZT (A), or CBK versus CON hearts within a ZT plus saline/insulin experimental group (A, B, C); ‡, p<0.05 for rapamycin versus control diet within a genotype (D, E).
Lastly, we tested the hypothesis that chronic activation of mTOR, and subsequent increased protein synthesis, plays a causal role in cardiac hypertrophy observed in CBK mice. CON and CBK mice were fed diets supplemented with encapsulated rapamycin at 42 ppm for 10 days. Consistent with data presented in Figures 4A and 5A, p-mTORSer2448 and p-S6Ser240/244 tended to be higher in CBK mice at ZT12, and were strongly inhibited by rapamycin in both CON and CBK mice (rapamycin main effects; Figure 5D). Myocardial net [3H]-protein synthesis was evaluated in vivo, 10 days after rapamycin supplementation was initiated. Importantly, rapamycin returned the rate of net [3H]-protein synthesis to control levels in CBK hearts (main effects rapamycin and genotype; Figure 5E). Furthermore, rapamycin reduced biventricular weight in CBK mice, with no effects in CON mice (genotype-rapamycin interaction; Figure 5E). Collectively, these findings suggest that chronic activation of mTOR following genetic disruption of the cardiomyocyte circadian clock contributes towards excessive cardiac growth, which can be restored to normal levels by rapamycin supplementation.
Discussion
The purpose of this study was to determine the impact of cardiomyocyte circadian clock disruption on insulin-mediated processes in the heart. We provide evidence that Akt is persistently activated in the heart (baseline and/or insulin-mediated) in two models of cardiomyocyte circadian clock disruption (namely CBK and CCM). Our studies reveal a dissociation between insulin-mediated Akt activation and glucose utilization following circadian disruption, potentially due to depression of I-1. Conversely, Akt activation following circadian disruption was associated with persistent activation of mTOR, concomitant with autophagy attenuation and increased rates of protein synthesis. Consistent with chronic activation of the Akt/mTOR/S6 axis promoting cardiac growth in CBK mice, rapamycin administration normalized cardiac size within 10 days. Collectively, these observations suggest that disruption of the cardiomyocyte circadian clock differentially influences a number of insulin-mediated processes in the heart. We speculate that these findings may provide novel insights regarding the mechanisms by which circadian disruption adversely affects cardiac structure and metabolism.
Time-of-day-dependent rhythms have been described for a number of cardiometabolic parameters in both humans and animal models, ranging from circulating glucose and insulin levels, to tissue insulin sensitivity.(41–44) One example includes the early morning spike in blood glucose known as the “dawn phenomenon”, which is mediated in part by fluctuations in tissue insulin sensitivity.(45) Such daily perturbations in insulin sensitivity have classically been attributed to fluctuations in neurohumoral factors, such as cortisol and growth hormone.(44) However, recent studies have begun to highlight the importance of cell autonomous circadian clocks as modulators of insulin sensitivity. For example, genetic disruption of the clock reduces expression of TBC1D1, GLUT4, and Hexokinase, associated with impaired insulin-mediated glucose uptake.(46,47) In the liver, clock disruption decreases insulin signaling and results in impaired insulin and glucose tolerance.(48,49) Although the clock appears to regulate adipogenesis, ablation of the clock in adipose tissue does not appear to greatly affect insulin signaling or sensitivity.(50,51) However, the effects of circadian regulation of insulin sensitivity have not been previously investigated at the level of the heart.
We recently identified p85α, the regulatory subunit of the PI3K complex, as being directly regulated by the cardiomyocyte circadian clock, through binding of the CLOCK/BMAL1 heterodimer to E-box elements in the promoter of this gene.(25) Here, we confirm that cardiomyocyte clock disruption led to reduced mRNA and protein levels of p85α, as well as other key insulin signaling components (e.g., Akt) in both CBK and CCM hearts (Figures 1–2 and Supplemental Figures 1–2). Despite reduced expression of insulin signaling components, we observed evidence of increased basal and insulin-mediated signaling in clock disrupted hearts (i.e., increased p-AktSer473 and p-AktThr308; Figure 2 and Supplemental Figure 2). This observation is consistent with previous studies reporting that partial knockout or knockdown of p85α increases insulin-mediated signaling.(52,53) Importantly, no differences were seen in activation of upstream kinases p-IRS1 or p-PDPK1 (Figure 2 and Supplemental Figure 2), consistent with the concept that chronic activation of Akt following circadian disruption is most likely secondary to diminished p85α levels. Somewhat surprisingly, no significant time-of-day-dependent differences were observed in myocardial insulin sensitivity (although insulin-mediated Akt phosphorylation appears somewhat augmented at the beginning at the sleep phase [ZT0]; Figure 2, Supplemental Figure 2).
Insulin signaling in the heart potently regulates numerous cellular processes, including substrate metabolism. Insulin promotes trafficking of glucose transporters (GLUTs) to the plasma membrane in an AS160-dependent manner, thus facilitating glucose uptake.(54) Myocardial glucose utilization exhibits marked time-of-day-dependent oscillations, which are mediated by the cardiomyocyte circadian clock; disruption of the cardiomyocyte clock depresses glucose uptake, glycolysis, glycogen synthesis, and glucose oxidation.(24) Following confirmation of diminished basal glucose utilization in CBK hearts, the current study reports attenuated insulin-mediated glucose disposal (Figure 3). The apparent dissociation between Akt activation and glucose utilization may be due, in part, to impaired AS160 phosphorylation/activation in CBK hearts. Similar observations have been reported in the heart with type 2 diabetes, wherein myocardial glucose uptake is attenuated despite chronic activation of Akt.(55) Furthermore, genetic models of constitutive Akt activation (e.g., myrAkt/caAkt) similarly display reduced myocardial insulin-mediated glucose utilization.(55–57) Interestingly, AS160 total protein levels were also decreased in CBK hearts (Figure 3). Dyar et al found that TBC1D1, an AS160 paralog more highly expressed in skeletal muscle, was regulated in a circadian fashion in the gastrocnemius, and was significantly reduced in muscle of Bmal1 null mice, concomitant with reduced insulin-mediated glucose uptake.(46) It is noteworthy that reduced GLUT1, GLUT4, and/or HKII (Supplemental Figure 5) in CBK mice could also contribute towards decreased glucose utilization; again, similar findings have been reported in skeletal muscle of Bmal1 null mice.(46) In addition to glucose catabolism, insulin promotes the channeling of glucose into glycogen, in part through Akt-dependent GSK3β phosphorylation/inactivation. Circadian rhythms in tissue glycogen content have been observed in several peripheral tissues including liver, skeletal muscle and heart, which often correlate with fasting/feeding cycles (consistent with a role of insulin).(58–60) p-GSK3βSer9 displayed a biphasic pattern in CON hearts, which was absent in CBK hearts (Figure 3). Somewhat surprisingly, p-GSK3βSer9 was chronically lower in CBK hearts (despite increased Akt activation). Consistent with lower p-GSK3βSer9, glycogen synthesis was lower in CBK hearts, independent of insulin (Figure 3). Collectively, these studies highlight depression of insulin-mediated myocardial glucose utilization following disruption of the cardiomyocyte circadian clock.
Given the apparent dissociation between the phosphorylation status of Akt and its downstream targets AS160 and GSK3β, we decided to interrogate PP1, which is responsible for dephosphorylating AS160 and GSK3β. Although prior microarray studies suggest that PP1 is not transcriptionally altered in CCM and CBK hearts, an established negative regulator of PP1 was identified (namely Inhibitor-1). Previous studies have shown that the activation status of I-1 is subject to time-of-day-dependent regulation in the heart.(61) Consistent with those, we found that I-1 expression is reduced at the mRNA and protein level in both CBK (Figure 3) and CCM (data not shown) hearts; a reduction in I-1 would be predicted to promote PP1 activity, thus dephosphorylating AS160 and GSK3β, as observed following clock disruption. Interestingly, peak AS160 phosphorylation in control hearts occurred at ZT12 in the current study, which corresponds to the timing of peak I-1 activation.(61) Future studies are required to establish a causative role of PP1 in the dissociation of AKT from its downstream targets AS160 and GSK3β following circadian disruption in the heart.
Another important downstream target of Akt is mTOR, which modulates essential cardiac processes, including autophagy and protein synthesis. Consistent with Akt activation, p-mTORSer2448 was increased in CBK hearts (Figure 4). mTOR affects autophagy through a number of mechanisms, including direct phosphorylation (and inactivation) of ULK1.(62,63) Interrogation of time-of-day-dependent oscillations in p- ULK1Ser757 revealed a biphasic pattern in control hearts, which troughs at both ZT6 and ZT18 (in a manner similar to mTOR). Conversely, LC3 lipidation exhibited an antiphase pattern in CON hearts. Similar to p-mTORSer2448, p-ULK1Ser757 was persistently elevated in CBK hearts. Interestingly, p62 was elevated in CBK hearts independent of feeding status, while autophagosomes accumulated in fasted CBK hearts (relative to CON hearts). These data are consistent with impairment of autophagy in CBK hearts, particularly the later stages, resulting in insufficient clearance of autophagosomes. Future studies are required to interrogate this relationship further.
Consistent with its anabolic nature, insulin is important in regulating heart size.(64) This is in large part mTOR-dependent; activation of mTOR leads to increased translational activity, protein synthesis and hypertrophic growth through downstream mediators S6/4EBP1. In agreement with chronic activation of PI3K/Akt/mTOR signaling, we have previously shown that clock disruption (in CBK and CCM mice) leads to a pro-hypertrophic phenotype.(25,29,34) Here we report that CON hearts exhibit a trend for time-of-day-dependent regulation of S6/4EBP1 signaling, protein synthesis, and heart weight, with a trough in the middle of the active period (ZT18), which is absent in CBK hearts (Figure 5). Similar to diurnal variations in heart weight observed in the current study, Sinturel et al recently reported daily fluctuations in hepatic ribosomal function and translation, resulting in oscillations in liver weight.(65) Importantly, elevated myocardial protein synthesis, as well as heart weight, observed in CBK hearts is normalized within 10 days by pharmacological inhibition of mTOR with rapamycin (Figure 5). Previous findings suggest that mTOR inhibition can extend lifespan in germline Bmal1−/− mice.(66) Whether normalization of mTOR activity improves cardiac function in either germline or cardiomyocyte-specific Bmal1 knockout mice requires further elucidation.
It is important to note that tissue specific effects of circadian disruption on insulin-mediated processes appear to exist. Here we show that Bmal1 is a negative regulator of the Akt/mTOR pathway, such that cardiomyocyte specific deletion of Bmal1 leads to activation both in vivo and ex vivo (thus promoting protein synthesis and hypertrophy). Similarly, Khapre et al have shown increased activation of mTOR signaling in various tissues isolated from germline Bmal1 null mice (including the heart), which persisted in vitro.(66) However, Zhang et al. reported that germline Bmal1 loss led to reduced activation of Akt/mTOR in the liver.(48) Lipton et al also described a role for Bmal1 as part of the translational machinery in the cytosol of the liver, and found that hepatic protein synthesis was decreased in germline Bmal1 null mice.(67) Similarly, Sinturel et al reported circadian regulation of hepatic translation, through alterations in ribosomal function.(65) Taken together, it is likely that distinct tissues respond differentially to circadian clock disruption.
In summary, the current study provides evidence suggesting that genetic disruption of the cardiomyocyte circadian clock leads to hyperactivation of the PI3K/Akt/mTOR signaling axis, which likely contributes towards attenuation of autophagy and augmentation of protein synthesis (Figure 6). In contrast, myocardial glucose utilization is impaired following clock disruption, likely secondary to diminished AS160 and GSK3β phosphorylation (perhaps due to decreased I-1 levels) and/or decreased GLUT1/GLUT4/HKII expression (Figure 6). The phenotype observed following circadian disruption (increased Akt/mTOR activity, concomitant with diminished glucose utilization and autophagy, as well as increased cardiac hypertrophy) parallels many of the characteristic cardiac changes that develop in type 2 diabetes. Given the association between circadian disruption, insulin resistance and increased risk for type 2 diabetes and its cardiovascular complications, the present study sheds novel insights linking circadian disruption, cardiac dysfunction, and insulin resistance.
Figure 6.
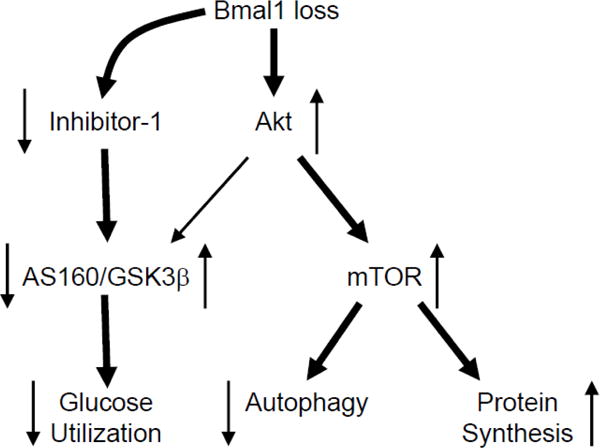
Hypothetical model. Bmal1 loss leads to chronic activation of Akt, under both basal states, as well as in response to insulin. Chronic Akt activation is associated with mTOR activation, and concomitant attenuation of autophagy and augmentation of protein synthesis. Bmal1 loss, also leads to decreased I-1 levels, which is anticipated to activate PP1; PP1 decreases phosphorylation of AS160 and GSK3β, leading to a concomitant decrease and increase in activity, respectively. Consistent with decreased AS160 and GSK3β phosphorylation, Bmal1 loss leads to diminished glucose utilization. Directionality of arrows indicate activity status of protein/process. Thickness of line signifies magnitude of relationship.
Supplementary Material
Highlights.
Cardiomyocyte clock disruption differentially influences insulin signaling proteins
Cardiomyocyte clock disruption attenuates glucose utilization
Cardiomyocyte clock disruption attenuates autophagy
Cardiomyocyte clock disruption augments protein synthesis
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute (HL106199, HL074259, and HL123574 to MEY; HL122975 to JCC and MEY; HL118067 to NSR), the National Institute of Diabetes and Digestive and Kidney Diseases (DK107441 to SJF), the Veterans Association (Merit Award to SJF), and an UAB AMC21 reload multi-investigator grant (NSR, ARW, VDU, JCC, JZ, MEY). GRM was supported by the National Institutes of Health (T32HD071866) and American Heart Association (16POST27010009). We would like to thank Maximiliano Grenett, Sabrina Moon, Luisa Szimmtenings, and Lan He for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
References
- 1.Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lejay A, Fang F, John R, et al. Ischemia reperfusion injury, ischemic conditioning and diabetes mellitus. J Mol Cell Cardiol. 2016;91:11–22. doi: 10.1016/j.yjmcc.2015.12.020. [DOI] [PubMed] [Google Scholar]
- 3.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–95. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abel E, Litwin S, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abel ED. Myocardial insulin resistance and cardiac complications of diabetes. Current drug targets Immune, endocrine and metabolic disorders. 2005;5:219–26. doi: 10.2174/1568008054064869. [DOI] [PubMed] [Google Scholar]
- 6.Riehle C, Abel ED. Insulin Signaling and Heart Failure. Circ Res. 2016;118:1151–69. doi: 10.1161/CIRCRESAHA.116.306206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–75. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edery I. Circadian rhythms in a nutshell. Physiol Genomics. 2000;3:59–74. doi: 10.1152/physiolgenomics.2000.3.2.59. [DOI] [PubMed] [Google Scholar]
- 9.Gekakis N, Staknis D, Nguyen H, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 10.Hogenesch J, Gu Y, Jain S, Bradfield C. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Aad Sci USA. 1998;95:5474–5479. doi: 10.1073/pnas.95.10.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woon PY, Kaisaki PJ, Braganca J, et al. Aryl hydrocarbon receptor nuclear translocator-like (BMAL1) is associated with susceptibility to hypertension and type 2 diabetes. Proc Natl Acad Sci U S A. 2007;104:14412–7. doi: 10.1073/pnas.0703247104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knutsson A, Akerstedt T, Jonsson B, Orth-Gomer K. Increased risk of ischaemic heart disease in shift workers. Lancet. 1986;12:89–92. doi: 10.1016/s0140-6736(86)91619-3. [DOI] [PubMed] [Google Scholar]
- 13.Scott EM, Carter AM, Grant PJ. Association between polymorphisms in the Clock gene, obesity and the metabolic syndrome in man. Int J Obes (Lond) 2008;32:658–62. doi: 10.1038/sj.ijo.0803778. [DOI] [PubMed] [Google Scholar]
- 14.Scott EM, Carter AM, Grant PJ. Diabetes and cardiovascular disease: related disorders created by disturbances in the endogenous clock. Journal of the Indian Medical Association. 2008;106:736–8, 740. [PubMed] [Google Scholar]
- 15.Fagard RH, Thijs L, Staessen JA, Clement DL, De Buyzere ML, De Bacquer DA. Night-day blood pressure ratio and dipping pattern as predictors of death and cardiovascular events in hypertension. J Hum Hypertens. 2009;23:645–53. doi: 10.1038/jhh.2009.9. [DOI] [PubMed] [Google Scholar]
- 16.Ohkubo T, Hozawa A, Yamaguchi J, et al. Prognostic significance of the nocturnal decline in blood pressure in individuals with and without high 24-h blood pressure: the Ohasama study. J Hypertens. 2002;20:2183–9. doi: 10.1097/00004872-200211000-00017. [DOI] [PubMed] [Google Scholar]
- 17.Curtis AM, Cheng Y, Kapoor S, Reilly D, Price TS, Fitzgerald GA. Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc Natl Acad Sci U S A. 2007;104:3450–5. doi: 10.1073/pnas.0611680104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcheva B, Ramsey KM, Buhr ED, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–31. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turek FW, Joshu C, Kohsaka A, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–5. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA. Development of dilated cardiomyopathy in Bmal1-deficient mice. Am J Physiol Heart Circ Physiol. 2012;303:H475–85. doi: 10.1152/ajpheart.00238.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eckel-Mahan KL, Patel VR, de Mateo S, et al. Reprogramming of the circadian clock by nutritional challenge. Cell. 2013;155:1464–78. doi: 10.1016/j.cell.2013.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohsaka A, Laposky AD, Ramsey KM, et al. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 2007;6:414–21. doi: 10.1016/j.cmet.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Young ME, Wilson CR, Razeghi P, Guthrie PH, Taegtmeyer H. Alterations of the circadian clock in the heart by streptozotocin-induced diabetes. J Mol Cell Cardiol. 2002;34:223–31. doi: 10.1006/jmcc.2001.1504. [DOI] [PubMed] [Google Scholar]
- 24.Bray M, Shaw C, Moore M, et al. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function; metabolism; and gene expression. Am J Physiol Heart Circ Physiol. 2008;294:H1036–H1047. doi: 10.1152/ajpheart.01291.2007. [DOI] [PubMed] [Google Scholar]
- 25.Young ME, Brewer RA, Peliciari-Garcia RA, et al. Cardiomyocyte-specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms. 2014;29:257–76. doi: 10.1177/0748730414543141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroder EA, Lefta M, Zhang X, et al. The cardiomyocyte molecular clock, regulation of Scn5a, and arrhythmia susceptibility. Am J Physiol Cell Physiol. 2013;304:C954–65. doi: 10.1152/ajpcell.00383.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durgan D, Trexler N, Egbejimi O, et al. The circadian clock within the cardiomyocyte is essential for responsiveness of the heart to fatty acids. J Biol Chem. 2006;281:24254–24269. doi: 10.1074/jbc.M601704200. [DOI] [PubMed] [Google Scholar]
- 28.Durgan DJ, Pulinilkunnil T, Villegas-Montoya C, et al. Short communication: ischemia/reperfusion tolerance is time-of-day-dependent: mediation by the cardiomyocyte circadian clock. Circ Res. 2010;106:546–50. doi: 10.1161/CIRCRESAHA.109.209346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durgan DJ, Tsai JY, Grenett MH, et al. Evidence suggesting that the cardiomyocyte circadian clock modulates responsiveness of the heart to hypertrophic stimuli in mice. Chronobiol Int. 2011;28:187–203. doi: 10.3109/07420528.2010.550406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taegtmeyer H, Young ME, Lopaschuk GD, et al. Assessing Cardiac Metabolism: A Scientific Statement From the American Heart Association. Circ Res. 2016;118:1659–701. doi: 10.1161/RES.0000000000000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodwin GW, Ahmad F, Doenst T, Taegtmeyer H. Energy provision from glycogen, glucose, and fatty acids on adrenergic stimulation of isolated working rat hearts. Am J Physiol. 1998;274:H1239–47. doi: 10.1152/ajpheart.1998.274.4.H1239. [DOI] [PubMed] [Google Scholar]
- 32.Challiss RA, Lozeman FJ, Leighton B, Newsholme EA. Effects of the beta-adrenoceptor agonist isoprenaline on insulin-sensitivity in soleus muscle of the rat. Biochem J. 1986;233:377–81. doi: 10.1042/bj2330377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodwin GW, Arteaga JR, Taegtmeyer H. Glycogen turnover in the isolated working rat heart. J Biol Chem. 1995;270:9234–40. doi: 10.1074/jbc.270.16.9234. [DOI] [PubMed] [Google Scholar]
- 34.He L, Hamm JA, Reddy A, et al. Biotinylation: a novel posttranslational modification linking cell autonomous circadian clocks with metabolism. Am J Physiol Heart Circ Physiol. 2016;310:H1520–32. doi: 10.1152/ajpheart.00959.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKee EE, Cheung JY, Rannels DE, Morgan HE. Measurement of the rate of protein synthesis and compartmentation of heart phenylalanine. J Biol Chem. 1978;253:1030–40. [PubMed] [Google Scholar]
- 36.Gibson UE, Heid CA, Williams PM. A novel method for real time quantitative RT-PCR. Genome Res. 1996;6:995–1001. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- 37.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–94. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 38.Wagner BK, Arany Z. High-throughput real-time PCR for detection of gene-expression levels. Methods in molecular biology. 2009;486:167–75. doi: 10.1007/978-1-60327-545-3_12. [DOI] [PubMed] [Google Scholar]
- 39.Durgan DJ, Pat BM, Laczy B, et al. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem. 2011;286:44606–19. doi: 10.1074/jbc.M111.278903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins HE, He L, Zou L, et al. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am J Physiol Heart Circ Physiol. 2014;306:H1231–9. doi: 10.1152/ajpheart.00075.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goel N, Stunkard AJ, Rogers NL, et al. Circadian rhythm profiles in women with night eating syndrome. J Biol Rhythms. 2009;24:85–94. doi: 10.1177/0748730408328914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalsbeek A, la Fleur S, Fliers E. Circadian control of glucose metabolism. Mol Metab. 2014;3:372–83. doi: 10.1016/j.molmet.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.la Fleur SE, Kalsbeek A, Wortel J, Fekkes ML, Buijs RM. A daily rhythm in glucose tolerance: a role for the suprachiasmatic nucleus. Diabetes. 2001;50:1237–43. doi: 10.2337/diabetes.50.6.1237. [DOI] [PubMed] [Google Scholar]
- 44.Gamble KL, Berry R, Frank SJ, Young ME. Circadian clock control of endocrine factors. Nature reviews Endocrinology. 2014;10:466–75. doi: 10.1038/nrendo.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolli GB, De Feo P, De Cosmo S, et al. Demonstration of a dawn phenomenon in normal human volunteers. Diabetes. 1984;33:1150–3. doi: 10.2337/diab.33.12.1150. [DOI] [PubMed] [Google Scholar]
- 46.Dyar KA, Ciciliot S, Wright LE, et al. Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Molecular metabolism. 2014;3:29–41. doi: 10.1016/j.molmet.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harfmann BD, Schroder EA, Kachman MT, Hodge BA, Zhang X, Esser KA. Muscle-specific loss of Bmal1 leads to disrupted tissue glucose metabolism and systemic glucose homeostasis. Skelet Muscle. 2016;6:12. doi: 10.1186/s13395-016-0082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang D, Tong X, Arthurs B, et al. Liver clock protein BMAL1 promotes de novo lipogenesis through insulin-mTORC2-AKT signaling. J Biol Chem. 2014;289:25925–35. doi: 10.1074/jbc.M114.567628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou B, Zhang Y, Zhang F, et al. CLOCK/BMAL1 regulates circadian change of mouse hepatic insulin sensitivity by SIRT1. Hepatology. 2014;59:2196–206. doi: 10.1002/hep.26992. [DOI] [PubMed] [Google Scholar]
- 50.Shimba S, Ishii N, Ohta Y, et al. Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc Natl Acad Sci U S A. 2005;102:12071–6. doi: 10.1073/pnas.0502383102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paschos GK, Ibrahim S, Song WL, et al. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nature medicine. 2012;18:1768–77. doi: 10.1038/nm.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mauvais-Jarvis F, Ueki K, Fruman DA, et al. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest. 2002;109:141–9. doi: 10.1172/JCI13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terauchi Y, Tsuji Y, Satoh S, et al. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3-kinase. Nat Genet. 1999;21:230–5. doi: 10.1038/6023. [DOI] [PubMed] [Google Scholar]
- 54.Sano H, Kane S, Sano E, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 55.Zhu Y, Pereira RO, O’Neill BT, et al. Cardiac PI3K-Akt impairs insulin-stimulated glucose uptake independent of mTORC1 and GLUT4 translocation. Mol Endocrinol. 2013;27:172–84. doi: 10.1210/me.2012-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsui T, Nagoshi T, Hong EG, et al. Effects of chronic Akt activation on glucose uptake in the heart. Am J Physiol Endocrinol Metab. 2006;290:E789–97. doi: 10.1152/ajpendo.00564.2004. [DOI] [PubMed] [Google Scholar]
- 57.Wende AR, O’Neill BT, Bugger H, et al. Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes. Mol Cell Biol. 2015;35:831–46. doi: 10.1128/MCB.01109-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Conlee RK, Hickson RC, Winder WW, Hagberg JM, Holloszy JO. Regulation of glycogen resynthesis in muscles of rats following exercise. Am J Physiol. 1978;235:R145–50. doi: 10.1152/ajpregu.1978.235.3.R145. [DOI] [PubMed] [Google Scholar]
- 59.Ishikawa K, Shimazu T. Circadian rhythm of liver glycogen metabolism in rats: effects of hypothalamic lesions. Am J Physiol. 1980;238:E21–5. doi: 10.1152/ajpendo.1980.238.1.E21. [DOI] [PubMed] [Google Scholar]
- 60.Leighton B, Kowalchuk JM, Challiss RA, Newsholme EA. Circadian rhythm in sensitivity of glucose metabolism to insulin in rat soleus muscle. Am J Physiol. 1988;255:E41–5. doi: 10.1152/ajpendo.1988.255.1.E41. [DOI] [PubMed] [Google Scholar]
- 61.Sachan N, Dey A, Rotter D, et al. Sustained hemodynamic stress disrupts normal circadian rhythms in calcineurin-dependent signaling and protein phosphorylation in the heart. Circ Res. 2011;108:437–45. doi: 10.1161/CIRCRESAHA.110.235309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:643–4. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Belke DD, Betuing S, Tuttle MJ, et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. The Journal of clinical investigation. 2002;109:629–39. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sinturel F, Gerber A, Mauvoisin D, et al. Diurnal Oscillations in Liver Mass and Cell Size Accompany Ribosome Assembly Cycles. Cell. 2017;169:651–663.e14. doi: 10.1016/j.cell.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khapre RV, Kondratova AA, Patel S, et al. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging. 2014;6:48–57. doi: 10.18632/aging.100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lipton JO, Yuan ED, Boyle LM, et al. The Circadian Protein BMAL1 Regulates Translation in Response to S6K1-Mediated Phosphorylation. Cell. 2015;161:1138–51. doi: 10.1016/j.cell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.