

Abstract
Most natural microbial systems have evolved to function in environments with temporal and spatial variations. A major limitation to understanding such complex systems is the lack of mathematical modeling frameworks that connect the genomes of individual species and temporal and spatial variations in the environment to system behavior. The goal of this review is to introduce the emerging field of spatiotemporal metabolic modeling based on genome-scale reconstructions of microbial metabolism. The extension of flux balance analysis to account for both temporal and spatial variations in the environment is termed Spatiotemporal Flux Balance Analysis (SFBA). Following a brief overview of flux balance analysis and its established dynamic extension, the SFBA problem is introduced and recent progress is described. Three case studies are reviewed to illustrate the current state-of-the-art and possible future research directions are outlined. The author posits that SFBA is the next frontier for microbial metabolic modeling and a rapid increase in methods development and system applications is anticipated.
Keywords: metabolic modeling, spatiotemporal models, genome-scale reconstructions, biofilms, microbial communities
Introduction
Metabolic flux balance analysis (FBA)
Whole cell metabolic modeling is highly challenging due to the large-scale and interconnected nature of microbial metabolic pathways. Rigorous modeling of metabolism requires descriptions of enzyme synthesis, kinetics and regulation at the individual reaction level. With the notable exception of primary metabolic pathways in model organisms [1–3], the development of such kinetic models is currently impractical due to lack of in vivo data on enzyme kinetics and regulation. As a result, a less detailed modeling approach commonly termed Flux Balance Analysis (FBA) based purely on metabolite mass conservation and reaction stoichiometry has emerged as the dominant methodology for describing whole cell metabolism [4, 5]. A stoichiometric model can be constructed by incorporating reactions derived from an annotated genome and augmented with additional “gap filling” reactions required to complete otherwise incomplete pathways [6–9]. Due to the key role of the annotated genome in reaction identification, the resulting model is usually called a Genome-scale Metabolic Reconstruction (GSM). GSMs are now available for dozens of microbial organisms [10–12], and new GSMs are published on a monthly basis.
Given the stoichiometric matrix and a specified set of available nutrient uptake rates, the goal is to solve the GSM for the unknown intracellular reaction rates (i.e. intracellular fluxes) and the metabolic byproduct secretion rates (i.e. secretion fluxes). Because the stoichiometric equations invariably contain more unknown fluxes than mass balance equations, the GSM has an infinite number of solutions and the flux distribution cannot be uniquely determined. To overcome this limitation, cell metabolism is assumed to be regulated to achieve some type of cellular objective [13, 14]. The most common objective is maximal growth, which requires the incorporation of a biomass stoichiometric equation in terms of biomass precursors (e.g. RNA, DNA, proteins, carbohydrates, etc.) [15]. The combination of the stoichiometric equations and the growth rate maximization objective produces a linear programming (LP) problem, which can be efficiently solved to generate predictions of the intracellular flux distribution, uptake and secretion (exchange) fluxes and the growth rate (Figure 1). FBA and other so-called constraint-based computational tools [16, 17] have been widely used to analyze the metabolism of both wild-type [18–22] and engineered [23–27] microbial strains.
Figure 1. Genome-scale flux balance analysis.

A GSM is used to formulate a LP to compute the unknown fluxes v from knowledge of the stoichiometric matrix A, biomass composition weights w, flux bounds vmin and vmax, and specified bounds on the nutrient uptake rates. This specific example shows glucose (g) and oxygen (o) as the growth limiting nutrients and ethanol (e) as the primary metabolic byproduct. From specified transport bounds on the two nutrient (vg, min, vo, min), the cellular growth rate (μ), the actual nutrient uptake rate (vg, vo), and the ethanol secretion rate (ve) are computed.
Dynamic flux balance analysis (DFBA)
FBA is based on assumptions that both intracellular metabolism and the extracellular environment are time invariant. While removal of the intracellular steady-state assumption requires the incorporation of enzyme kinetics, the steady-state assumption on the extracellular environment is more easily relaxed through an extension of FBA commonly termed Dynamic Flux Balance Analysis (DFBA) [28, 29]. A DFBA model is formulated by combining a GSM of intracellular metabolism with kinetic expressions for the uptake rates of growth limiting nutrients and dynamic mass balance equations for cellular biomass, limiting nutrients and secreted metabolic byproducts under the assumption that cells rapidly equilibrate to environmental changes (Figure 2). While difficult to verify, this assumption has been accepted for many systems including batch biochemical reactors where nutrient concentrations varying due to cellular consumption and possibly nutrient feeding [29]. A major advantage of DFBA is that the model outputs include biomass and extracellular metabolite concentrations rather than just the growth rate and exchange fluxes as in FBA [30]. Furthermore, the extracellular concentrations as well as intracellular fluxes are predicted with temporal resolution.
Figure 2. Genome-scale dynamic flux balance analysis.

FBA is performed with a GSM to predict the growth rate, nutrient uptake rates and byproduct secretion rates under the assumption that the intracellular dynamics are fast compared to the extracellular dynamics. The FBA fluxes serve as inputs to the ODEs for the extracellular environment, which are integrated to yield time resolved predictions of the biomass, nutrient and byproduct concentrations. These concentrations serve as inputs to the nutrient uptake kinetics, which are used to compute transport bounds on the nutrient uptake rates for the FBA problem. This specific example shows glucose (g) and oxygen (o) as the growth limiting nutrients and ethanol (e) as the primary metabolic byproduct where X, G, E and O are the biomass, glucose, ethanol and oxygen concentrations, respectively.
A DFBA model is comprised of ordinary differential equations (ODEs) describing the extracellular environment and a LP describing intracellular metabolism. As compared to FBA, a notable disadvantage of DFBA is the computational complexity of this hybrid ODE/LP system. Initial attempts to solve DFBA models were based on sequential strategies in which the LP was repeatedly solved according to a prespecified time step and the ODEs were integrated between time steps using the most current LP solution [28, 31]. Due to the unpredictable accuracy and stability of these sequential methods, more current solution techniques are typically based on simultaneous strategies in which the LP is embedded with the ODE solution [32, 33]. DFBA has emerged as a standard tool for the analysis [34–39] and optimization [32, 40–42] of individual species as well as microbial communities [43–45] in dynamic environments. While some DFBA models can be effectively solved by a straightforward combination of ODE and LP solvers, more advanced techniques may be required due to complications associated with the LP unpredictably becoming infeasible or producing alternative optima characterized by non-unique exchange fluxes. Sophisticated simulation methods that explicitly address and effectively overcome these complications are now available [46, 47].
Spatiotemporal Flux Balance Analysis (SFBA)
While DFBA accounts for the effects of extracellular dynamics on intracellular metabolism, the method is based on the assumption that the extracellular environment is well mixed and spatially homogeneous. Many engineered microbial systems such as biochemical reactors are carefully designed to achieve spatial homogeneity through liquid mixing [48]. However, most naturally occurring microbial systems exist in spatially heterogeneous environments that also exhibit time variations. The presence of spatial heterogeneity plays an essential role in the evolution and function of natural microbial species [49–52]. Perhaps the most common and important example of spatially heterogeneous environments is that established due to biofilm formation and development (Figure 3a) [53–56]. Concentration gradients in key nutrients due to limited diffusion establish unique metabolic niches within the biofilm that produce spatial variations in biomass density in the case of single species biofilms [57] and additionally spatial partitioning of species in the case of biofilm consortia [53]. The development of metabolic models that capture such spatial and temporal variations is important to analyze and manipulate complex microbial systems.
Figure 3. Spatial heterogeneities in microbial biofilms.
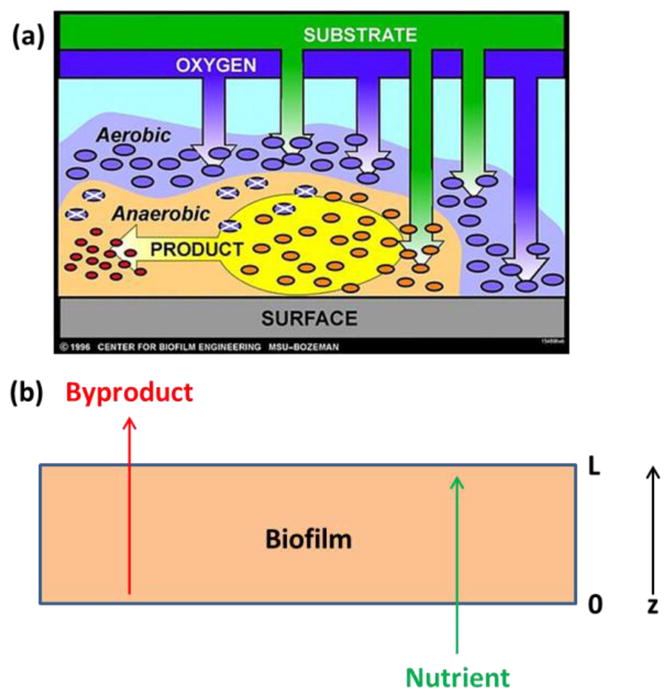
(a) Limited diffusion of a carbon containing nutrient (substrate) and oxygen can create metabolic niches with the biofilm that lead to aerobic and anaerobic regions with differential cell growth and product synthesis (image courtesy of the MSU Center for Biofilm Engineering). (b) A single species biofilm with thickness L in which a single growth limiting nutrient is available at the bottom of the biofilm and a single synthesized byproduct exits the top of the biofilm.
Model formulation
Mathematical models that account for both spatial and temporal variations are commonly termed spatiotemporal models [58, 59]. Therefore, we refer to the extension of DFBA to include spatial heterogeneity in the environment as Spatiotemporal Flux Balance Analysis (SFBA). The author believes that SFBA is the next frontier for microbial metabolic modeling and that the nascent activities to date will soon be followed by a rapid increase in methods development and system applications.
A SFBA model is formulated by replacing the time-varying ODEs in DFBA with partial differential equations (PDEs) expressed in terms of time and some spatial coordinate(s) as independent variables [60]. The PDEs represent extracellular mass balance equations for biomass, metabolite and possibly other chemical species concentrations and account for the transport mechanisms that induce the spatial variations, which commonly include metabolite diffusion and liquid/gas phase convection. Boundary conditions must be imposed at the boundaries of the spatial domain to ensure that the PDEs are well posed. As in DFBA, the GSM and the extracellular mass balance equations are linked through nutrient uptake kinetics.
Some systems such as microbial biofilms involve mixed boundary conditions when nutrients enter and/or byproducts exit the biofilm at different locations [60, 61]. Furthermore, the spatial domain may shrink and/or grow with time due to cellular growth and/or death [62]. For the sake of illustration, consider a single species biofilm (Figure 3b) with a fixed thickness L in which a single growth limiting nutrient is available at the bottom of the biofilm (z = 0) and a single synthesized byproduct exits the top of the biofilm (z = L). The PDEs describing biofilm diffusional processes can be written as follows assuming that spatial variations occur only in the axial direction z of the biofilm,
| (1) |
Here X(z, t), S(z, t) and P(z, t) represent the biomass, substrate and byproduct concentrations at location z and time t. The growth rate μ, substrate uptake rate vS and byproduct synthesis rate vP are obtained by solution of the GSM. The substrate and byproduct diffuse through the biofilm with efficient diffusion coefficients DS and DP, respectively, while the biomass is assumed to be non-motile. The second column lists the boundary conditions at the bottom of the biofilm, which are based on the assumptions that substrate is available at a concentration S0 and the biomass and byproduct do not flux across this boundary. The boundary conditions at the top of the biofilm listed in the third column are based on the assumptions that the biomass and substrate do not flux across this boundary while the byproduct removal rate is sufficiently high that the boundary concentration is zero. Finally, the initial conditions in the fourth column are based on the assumption that the biofilm is spatially uniform at t = 0.
Model solution
A SFBA model is comprised of PDEs describing the extracellular environment and LPs describing intracellular metabolism [60]. Pure species systems have a single LP [63], while multispecies systems require solution of a LP for each species [64]. Because no computational methods exist to directly solve such hybrid PDE/LP models, the PDE transport behavior must be approximated in some manner to generate a solvable model. To date, three alternative methods have been proposed for implementing this approximation: (M1) table lookups of computed FBA solutions combined with integration of the PDEs on a coarse spatial grid [61, 63, 65]; (M2) real-time FBA solution combined with lattice-based descriptions of metabolite transport [62, 64]; and (M3) spatial discretization of the PDEs followed by time integration of the resulting ODE/LP system (Figure 4) [60, 66]. These methods differ according to whether the LP solutions are precomputed (M1) or generated in real-time (M2, M3) and whether the PDEs are discretized directly (M1, M3) or approximated indirectly (M2). While exhaustive studies of these methods have not yet been performed, the most appropriate method is very likely to be problem dependent.
Figure 4. Genome-scale spatiotemporal flux balance analysis [60].

A microbial system with temporal and spatial variations is described by a spatiotemporal model that accounts for relevant transport processes within the system. PDEs are written with respect to the species concentration (Xi), liquid-phase metabolite concentrations (Mj) and gas-phase metabolite concentrations (Pj) assuming that spatial variations are limited to a single direction z. FBA is performed with GSMs of the participating species to predict the growth rates, nutrient uptake rates and byproduct secretion rates. The PDEs are spatially discretized to yield a large-set of ODEs with embedded LPs that are solved with the MATLAB code DFBAlab [46] to generate time and spatially resolved predictions.
Representative studies
To illustrate the current state-of-the-art, three representative studies that represent the breadth of the spatiotemporal modeling methods described above are reviewed. In the first study [61], method M1 was used to study electricity generation in a microbial fuel cell where the anaerobic bacterium Geobacter sulfurreducens formed a biofilm on the anode. A metabolic model was developed to analyze the biofilm that oxidized acetate (the electron donor) to carbon dioxide with electron transfer to Fe(III) (the electron acceptor) mimicking the anode. Cellular growth was assumed to create an axial velocity that drove biofilm expansion and along with acetate diffusion created spatial heterogeneities across the biofilm. The steady-state assumption was invoked such that time was eliminated as an independent variable and the transport equations only involved spatial derivatives in the axial direction, thereby simplifying the spatiotemporal model to a time-invariant spatial model. The spatial model consisted of a published G. sulfurreducens GSM [67], Michaelis-Menten uptake kinetics for acetate and Nerst kinetics for Fe(III) reduction, and reaction-convection-diffusion type equations for the fractions of active, respiring and inert biomass, acetate concentration, current density, local potential and axial velocity.
The G. sulfurreducens GSM was solved off-line at different values of the acetate uptake and Fe(III) reduction rates to generate a 20×20 lookup table of LP solutions. The boundary value ODEs representing the reaction-diffusion-convection equations were solved using a continuation method [68] with the LP lookup table queried as needed to resolve intracellular metabolism at different points in the biofilm. Unfortunately, details concerning the continuation solution method and the spatial grid used are not reported. The spatial metabolic model was solved for varying extracellular conditions (i.e. the presence/absence of NH4) and maintenance energy demands to investigate their effects on biofilm thickness and electrical current production. Simulations combined with validation experiments provided key predictions into biofilm and fuel cell behavior including that: (1) limited acetate diffusion through the biofilm induced low local acetate concentrations that restricted biomass formation and current generation; and (2) respiring cells that did not grow but produce current located in acetate limited regions had a substantial impact on fuel cell performance.
In the second study [64], a spatiotemporal modeling framework called Computation of Microbial Ecosystems in Time and Space (COMETS) was developed to investigate the emergent behavior of two- and three-member synthetic communities. A two-dimensional lattice was defined, with each box within the lattice representing a distinct spatial location. GSMs of the participating species were used to solve an independent DFBA problem for each box assuming that each species had the potential to consume available carbon sources according to the same uptake kinetics. A sequential solution strategy was used where the species LPs were solved to generate local growth rates and fluxes, and then the extracellular ODEs were integrated over a fixed time step with these constant LP solutions to generate local biomass and metabolite concentrations. Before resolving the LPs to start the next iteration, two-dimensional diffusion equations were solved over the same time step to allow species cell mass and metabolites to diffuse between boxes.
COMETS was used to investigate engineered metabolite cross-feeding in a two-species synthetic community consisting of Salmonella enterica and Escherichia coli. The spatiotemporal model was able to reproduce experiments showing the two species robustly co-existed and converged to a population fraction of 79% E. coli regardless of the initial fractions. Similar results were obtained for a considerably more complex three-species synthetic community consisting of S. enterica, E. coli and Methylobacterium extorquens. The two-species system was used to investigate the impact of spatial structure. The model correctly predicted that community growth would diminish as the two species were inoculated further apart from each other due to diffusional limitations of the cross-fed metabolites. Moreover, the model reproduced experiments showing community growth would be enhanced by placement of a second colony of engineered S. enterica between the S. enterica and E. coli populations but that placement of a wild-type S. enterica colony which did not require the cross-fed metabolite from E. coli would reduce community growth.
In the third study [66], method M3 was used to predict synthesis gas (CO, H2) conversion to ethanol by the anaerobic bacterium Clostridium ljungdahlii in a vertical bubble column reactor. Synthesis gas and liquid media feed streams were introduced into the bottom of the column and flowed up the column with different velocities, producing large spatial gradients due to cellular growth and gas depletion. The metabolic byproducts ethanol and acetate were recovered in the liquid phase stream exiting the top of the column. The spatiotemporal metabolic model consisted of a published C. ljungdahlii GSM [69], Michaelis-Menten uptake kinetics for CO and H2, and reaction-convection type PDEs for C. ljungdahlii biomass, liquid phase CO, H2, ethanol and acetate, and gas phase CO and H2. The PDEs were discretized with 100 spatial node points, and lexicographic optimization [47] with 6 LPs at each node point was used to ensure unique exchange fluxes. The large discretized model consisting of 900 ODEs in time and 600 LPs was efficiently solved within MATLAB (MathWorks, Natick, Massachusetts, USA) using the DFBAlab tool (Figure 4) [46].
Dynamic simulations performed for a wide range of column operating conditions and nutrient uptake parameters generated predictions about column behavior and bottlenecks to ethanol production consistent with available experiments including: (1) typical CO rich syngas will produce substantial acetate due to H2 depletion in the upper part of the column, suggesting that H2 augmentation of the syngas feed may be beneficial; (2) efficient gas-liquid mass transfer is critical to achieve high ethanol production and high conversions, demonstrating the need for continued development of advanced bubble column designs that achieve very high gas-liquid mass transfer rates; and (3) enhanced H2 uptake rates substantially increase the ethanol titer and the ethanol/acetate ratio, suggesting that C. ljungdahlii engineering efforts should focus on increasing H2 uptake rates.
Conclusions
Spatially heterogeneous environments are the rule rather than the exception for naturally occurring microbial systems. Our continued ability to fundamentally understand and rationally manipulate microbial systems will depend on the development of predictive modeling frameworks that connect the genetic capabilities of individual species and the spatiotemporal characteristics of growth environments to system function. Over the past five years, spatiotemporal metabolic modeling techniques that leverage the increasing availability of genome-scale reconstructions have been developed and evaluated. Despite their limitations with regard to intracellular organization, dynamics and regulation, these Spatiotemporal Flux Balance Analysis (SFBA) methods hold great promise for analysis of natural microbial systems as well as the design of engineered systems that exploit the evolved capabilities of microbes to optimally function in highly dynamic and spatially heterogeneous environments [70, 71]. Possible research directions for the nascent SFBA field are myriad and include: (1) continued identification and study of microbial systems such as the human microbiome [72, 73] and lignocellulosic degrading communities [74, 75] that would benefit from spatiotemporal analysis; (2) incorporation of higher fidelity intracellular models that extend beyond just reaction stoichiometry [76, 77]; (3) development of alternative methods for formulating SFBA models that are more amenable to efficient numerical solution; (4) development and testing of general purpose software such as DFBAlab [46] for the solution of the large-scale differential equation and linear program systems that result from SFBA models; and (5) experimental testing of SFBA model predictions through the collection of omics data with both temporal and spatial resolution [78–80]. Based on the overarching importance of the problem and the initial successes reported this review, SFBA can be expected to become the next major frontier for microbial metabolic modeling.
Acknowledgments
The author wishes to acknowledge assistance from Jin Chen in compiling the references and financial support from the National Institutes of Health (1U01EB019416-01) and the National Science Foundation (CBET 1511346).
Abbreviations
- COMETS
computation of microbial ecosystems in time and space
- DFBA
dynamic flux balance analysis
- DFBAlab
dynamic flux balance analysis laboratory
- FBA
flux balance analysis
- GSM
genome-scale metabolic reconstruction
- LP
linear program
- ODE
ordinary differential equation
- PDE
partial differential equation
- SFBA
spatiotemporal flux balance analysis
References
- 1.Chakrabarti A, Miskovic L, Soh KC, Hatzimanikatis V. Towards kinetic modeling of genome-scale metabolic networks without sacrificing stoichiometric, thermodynamic and physiological constraints. Biotechnol J. 2013;8:1043–1057. doi: 10.1002/biot.201300091. [DOI] [PubMed] [Google Scholar]
- 2.Khodayari A, Zomorrodi AR, Liao JC, Maranas CD. A kinetic model of Escherichia coli core metabolism satisfying multiple sets of mutant flux data. Metab Eng. 2014;25:50–62. doi: 10.1016/j.ymben.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Zielinski D, Palsson B. Kinetic Modeling of Metabolic Networks. In: Wittmann C, Lee SY, editors. Systems Metabolic Engineering. Springer Netherlands; 2012. pp. 25–55. [Google Scholar]
- 4.Price ND, Papin JA, Schilling CH, Palsson BO. Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 2003;21:162–169. doi: 10.1016/S0167-7799(03)00030-1. [DOI] [PubMed] [Google Scholar]
- 5.Santos F, Boele J, Teusink B. A Practical Guide to Genome-Scale Metabolic Models and Their Analysis. Method Enzymol. 2011;500:509–532. doi: 10.1016/B978-0-12-385118-5.00024-4. [DOI] [PubMed] [Google Scholar]
- 6.Feng XY, Xu Y, Chen YX, Tang YJJ. MicrobesFlux: a web platform for drafting metabolic models from the KEGG database. BMC Syst Biol. 2012:6. doi: 10.1186/1752-0509-6-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28:977-U922. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 8.Latendresse M, Krummenacker M, Trupp M, Karp PD. Construction and completion of flux balance models from pathway databases. Bioinformatics. 2012;28:388–396. doi: 10.1093/bioinformatics/btr681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5:93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henry CS, Overbeek R, Xia FF, Best AA, Glass E, Gilbert J, Larsen P, Edwards R, Disz T, Meyer F, Vonstein V, DeJongh M, Bartels D, Desai N, D’Souza M, Devoid S, Keegan KP, Olson R, Wilke A, Wilkening J, Stevens RL. Connecting genotype to phenotype in the era of high-throughput sequencing. Bba-Gen Subjects. 2011;1810:967–977. doi: 10.1016/j.bbagen.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Milne CB, Kim PJ, Eddy JA, Price ND. Accomplishments in genome-scale in silico modeling for industrial and medical biotechnology. Biotechnol J. 2009;4:1653–1670. doi: 10.1002/biot.200900234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu C, Liu LL, Zhang Z, Jin DF, Qiu JP, Chen M. Genome-scale metabolic model in guiding metabolic engineering of microbial improvement. Appl Microbiol Biotechnol. 2013;97:519–539. doi: 10.1007/s00253-012-4543-9. [DOI] [PubMed] [Google Scholar]
- 13.Burgard AP, Maranas CD. Optimization-based framework for inferring and testing hypothesized metabolic objective functions. Biotechnol Bioeng. 2003;82:670–677. doi: 10.1002/bit.10617. [DOI] [PubMed] [Google Scholar]
- 14.Schuetz R, Kuepfer L, Sauer U. Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol Syst Biol. 2007:3. doi: 10.1038/msb4100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senger RS. Biofuel production improvement with genome-scale models: The role of cell composition. Biotechnol J. 2010;5:671–685. doi: 10.1002/biot.201000007. [DOI] [PubMed] [Google Scholar]
- 16.Papin JA, Stelling J, Price ND, Klamt S, Schuster S, Palsson BO. Comparison of network-based pathway analysis methods. Trends Biotechnol. 2004;22:400–405. doi: 10.1016/j.tibtech.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 17.Schellenberger J, Que R, Fleming RMT, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J, Hyduke DR, Palsson BO. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balagurunathan B, Jonnalagadda S, Tan L, Srinivasan R. Reconstruction and analysis of a genome-scale metabolic model for Scheffersomyces stipitis. Microb Cell Fact. 2012:11. doi: 10.1186/1475-2859-11-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrgard MJ, Swainston N, Dobson P, Dunn WB, Arga KY, Arvas M, Bluthgen N, Borger S, Costenoble R, Heinemann M, Hucka M, Le Novere N, Li P, Liebermeister W, Mo ML, Oliveira AP, Petranovic D, Pettifer S, Simeonidis E, Smallbone K, Spasic I, Weichart D, Brent R, Broomhead DS, Westerhoff HV, Kirdar B, Penttila M, Klipp E, Palsson BO, Sauer U, Oliver SG, Mendes P, Nielsen J, Kell DB. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat Biotechnol. 2008;26:1155–1160. doi: 10.1038/nbt1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loira N, Dulermo T, Nicaud JM, Sherman DJ. A genome-scale metabolic model of the lipid-accumulating yeast Yarrowia lipolytica. BMC Syst Biol. 2012:6. doi: 10.1186/1752-0509-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pitkanen E, Jouhten P, Hou J, Syed MF, Blomberg P, Kludas J, Oja M, Holm L, Penttila M, Rousu J, Arvas M. Comparative genome-scale reconstruction of gapless metabolic networks for present and ancestral species. PLoS Comput Biol. 2014:10. doi: 10.1371/journal.pcbi.1003465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Senger RS, Papoutsakis ET. Genome-scale model for Clostridium acetobutylicum: Part I. Metabolic network resolution and analysis. Biotechnol Bioeng. 2008;101:1036–1052. doi: 10.1002/bit.22010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgard AP, Pharkya P, Maranas CD. OptKnock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng. 2003;84:647–657. doi: 10.1002/bit.10803. [DOI] [PubMed] [Google Scholar]
- 24.Fong SS, Marciniak JY, Palsson BO. Description and interpretation of adaptive evolution of Escherichia coli K-12 MG1655 by using a genome-scale in silico metabolic model. J Bacteriol. 2003;185:6400–6408. doi: 10.1128/JB.185.21.6400-6408.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JW, Kim TY, Jang YS, Choi S, Lee SY. Systems metabolic engineering for chemicals and materials. Trends Biotechnol. 2011;29:370–378. doi: 10.1016/j.tibtech.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Pharkya P, Maranas CD. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab Eng. 2006;8:1–13. doi: 10.1016/j.ymben.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Segre D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci U S A. 2002;99:15112–15117. doi: 10.1073/pnas.232349399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahadevan R, Edwards JS, Doyle FJ. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys J. 2002;83:1331–1340. doi: 10.1016/S0006-3495(02)73903-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hjersted JL, Henson MA. Optimization of fed-batch Saccharomyces cerevisiae fermentation using dynamic flux balance models. Biotechnol Prog. 2006;22:1239–1248. doi: 10.1021/bp060059v. [DOI] [PubMed] [Google Scholar]
- 30.Hjersted JL, Henson MA. Steady-state and dynamic flux balance analysis of ethanol production by Saccharomyces cerevisiae. IET Syst Biol. 2009;3:167–179. doi: 10.1049/iet-syb.2008.0103. [DOI] [PubMed] [Google Scholar]
- 31.Varma A, Palsson BO. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia-Coli W3110. Appl Environ Microbiol. 1994;60:3724–3731. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hjersted JL, Henson MA, Mahadevan R. Genome-scale analysis of Saccharomyces cerevisiae metabolism and ethanol production in fed-batch culture. Biotechnol Bioeng. 2007;97:1190–1204. doi: 10.1002/bit.21332. [DOI] [PubMed] [Google Scholar]
- 33.Henson MA, Hanly TJ. Dynamic flux balance analysis for synthetic microbial communities. IET Syst Biol. 2014;8:214–229. doi: 10.1049/iet-syb.2013.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hohenschuh W, Hector R, Murthy GS. A dynamic flux balance model and bottleneck identification of glucose, xylose, xylulose co-fermentation in Saccharomyces cerevisiae. Bioresour Technol. 2015;188:153–160. doi: 10.1016/j.biortech.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 35.Jouhten P, Wiebe M, Penttila M. Dynamic flux balance analysis of the metabolism of Saccharomyces cerevisiae during the shift from fully respirative or respirofermentative metabolic states to anaerobiosis. FEBS J. 2012;279:3338–3354. doi: 10.1111/j.1742-4658.2012.08649.x. [DOI] [PubMed] [Google Scholar]
- 36.Meadows AL, Karnik R, Lam H, Forestell S, Snedecor B. Application of dynamic flux balance analysis to an industrial Escherichia coli fermentation. Metab Eng. 2010;12:150–160. doi: 10.1016/j.ymben.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Oddone GM, Mills DA, Block DE. A dynamic, genome-scale flux model of Lactococcus lactis to increase specific recombinant protein expression. Metab Eng. 2009;11:367–381. doi: 10.1016/j.ymben.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 38.Waldherr S, Oyarzun DA, Bockmayr A. Dynamic optimization of metabolic networks coupled with gene expression. J Theor Biol. 2015;365:469–485. doi: 10.1016/j.jtbi.2014.10.035. [DOI] [PubMed] [Google Scholar]
- 39.Willemsen AM, Hendrickx DM, Hoefsloot HCJ, Hendriks MMWB, Wahl SA, Teusink B, Smilde AK, van Kampen AHC. MetDFBA: incorporating time-resolved metabolomics measurements into dynamic flux balance analysis. Mol BioSyst. 2015;11:137–145. doi: 10.1039/c4mb00510d. [DOI] [PubMed] [Google Scholar]
- 40.Birch EW, Udell M, Covert MW. Incorporation of flexible objectives and time-linked simulation with flux balance analysis. J Theor Biol. 2014;345:12–21. doi: 10.1016/j.jtbi.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gadkar KG, Doyle FJ, Edwards JS, Mahadevan R. Estimating optimal profiles of genetic alterations using constraint-based models. Biotechnol Bioeng. 2005;89:243–251. doi: 10.1002/bit.20349. [DOI] [PubMed] [Google Scholar]
- 42.Zhuang K, Yang L, Cluett WR, Mahadevan R. Dynamic strain scanning optimization: an efficient strain design strategy for balanced yield, titer, and productivity. DySScO strategy for strain design. BMC Biotechnol. 2013:13. doi: 10.1186/1472-6750-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanly TJ, Henson MA. Dynamic Model-based analysis of furfural and HMF detoxification by pure and mixed batch cultures of S. cerevisiae and S. stipitis. Biotechnol Bioeng. 2014;111:272–284. doi: 10.1002/bit.25101. [DOI] [PubMed] [Google Scholar]
- 44.Pardelha F, Albuquerque MGE, Reis MAM, Oliveira R, Dias JML. Dynamic metabolic modelling of volatile fatty acids conversion to polyhydroxyalkanoates by a mixed microbial culture. New Biotechnol. 2014;31:335–344. doi: 10.1016/j.nbt.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 45.Zomorrodi AR, Islam MM, Maranas CD. d-OptCom: Dynamic multi-level and multi-objective metabolic modeling of microbial communities. Acs Synth Biol. 2014;3:247–257. doi: 10.1021/sb4001307. [DOI] [PubMed] [Google Scholar]
- 46.Gomez JA, Hoffner K, Barton PI. DFBAlab: a fast and reliable MATLAB code for dynamic flux balance analysis. BMC Bioinformatics. 2014:15. doi: 10.1186/s12859-014-0409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffner K, Harwood SM, Barton PI. A reliable simulator for dynamic flux balance analysis. Biotechnol Bioeng. 2013;110:792–802. doi: 10.1002/bit.24748. [DOI] [PubMed] [Google Scholar]
- 48.Singh H, Hutmacher DW. Bioreactor studies and computational Fluid dynamics. Bioreactor Systems for Tissue Engineering. 2009;112:231–249. doi: 10.1007/978-3-540-69357-4_10. [DOI] [PubMed] [Google Scholar]
- 49.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10:538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 51.Hol FJ, Galajda P, Woolthuis RG, Dekker C, Keymer JE. The idiosyncrasy of spatial structure in bacterial competition. BMC Res Notes. 2015;8:245. doi: 10.1186/s13104-015-1169-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kovacs AT. Impact of spatial distribution on the development of mutualism in microbes. Front Microbiol. 2014;5:649. doi: 10.3389/fmicb.2014.00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burmolle M, Ren DW, Bjarnsholt T, Sorensen SJ. Interactions in multispecies biofilms: do they actually matter? Trends Microbiol. 2014;22:84–91. doi: 10.1016/j.tim.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 54.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: From the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 55.Mazumdar V, Amar S, Segre D. Metabolic proximity in the order of colonization of a microbial community. PLoS One. 2013:8. doi: 10.1371/journal.pone.0077617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tolker-Nielsen T, Molin S. Spatial organization of microbial biofilm communities. Microb Ecol. 2000;40:75–84. doi: 10.1007/s002480000057. [DOI] [PubMed] [Google Scholar]
- 57.Stewart PS. A review of experimental measurements of effective diffusive permeabilities and effective diffusion coefficients in biofilms. Biotechnol Bioeng. 1998;59:261–272. doi: 10.1002/(sici)1097-0290(19980805)59:3<261::aid-bit1>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 58.Grimson MJ, Barker GC. Continuum model for the spatiotemporal growth of bacterial colonies. Phys Rev E. 1994;49:1680–1684. doi: 10.1103/physreve.49.1680. [DOI] [PubMed] [Google Scholar]
- 59.Monds RD, O’Toole GA. The developmental model of microbial biofilms: ten years of a paradigm up for review. Trends Microbiol. 2009;17:73–87. doi: 10.1016/j.tim.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 60.Chen J, Gomez JA, Höffner K, Phalak P, Barton PI, Henson MA. Spatiotemporal modeling of microbial metabolism. Biotechnol Bioeng. 2015 doi: 10.1186/s12918-016-0259-2. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jayasinghe N, Franks A, Nevin KP, Mahadevan R. Metabolic modeling of spatial heterogeneity of biofilms in microbial fuel cells reveals substrate limitations in electrical current generation. Biotechnol J. 2014;9:1350–1361. doi: 10.1002/biot.201400068. [DOI] [PubMed] [Google Scholar]
- 62.Cole JA, Kohler L, Hedhli J, Luthey-Schulten Z. Spatially-resolved metabolic cooperativity within dense bacterial colonies. BMC Syst Biol. 2015:9. doi: 10.1186/s12918-015-0155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang YL, Scheibe TD, Mahadevan R, Garg S, Long PE, Lovley DR. Direct coupling of a genome-scale microbial in silico model and a groundwater reactive transport model. J Contam Hydrol. 2011;122:96–103. doi: 10.1016/j.jconhyd.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 64.Harcombe WR, Riehl WJ, Dukovski I, Granger BR, Betts A, Lang AH, Bonilla G, Kar A, Leiby N, Mehta P, Marx CJ, Segre D. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014;7:1104–1115. doi: 10.1016/j.celrep.2014.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scheibe TD, Mahadevan R, Fang YL, Garg S, Long PE, Lovley DR. Coupling a genome-scale metabolic model with a reactive transport model to describe in situ uranium bioremediation. Microb Biotechnol. 2009;2:274–286. doi: 10.1111/j.1751-7915.2009.00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J, Gomez J, Hoffner K, Barton P, Henson M. Metabolic modeling of synthesis gas fermentation in bubble column reactors. Biotechnol Biofuels. 2015;8:89. doi: 10.1186/s13068-015-0272-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mahadevan R, Bond DR, Butler JE, Esteve-Nunez A, Coppi MV, Palsson BO, Schilling CH, Lovley DR. Characterization of metabolism in the Fe(III)-reducing organism Geobacter sulfurreducens by constraint-based modeling. Appl Environ Microbiol. 2006;72:1558–1568. doi: 10.1128/AEM.72.2.1558-1568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ascher UM, Mattheij RMM, Russell RD. Numerical Solution of Boundary Value Problems for Ordinary Differential Equations. Society for Industrial and Applied Mathematics; Philadelphia, PA: 1994. [Google Scholar]
- 69.Nagarajan H, Sahin M, Nogales J, Latif H, Lovley DR, Ebrahim A, Zengler K. Characterizing acetogenic metabolism using a genome-scale metabolic reconstruction of Clostridium ljungdahlii. Microb Cell Fact. 2013:12. doi: 10.1186/1475-2859-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brenner K, You LC, Arnold FH. Engineering microbial consortia: a new frontier in synthetic biology. Trends Biotechnol. 2008;26:483–489. doi: 10.1016/j.tibtech.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 71.Wintermute EH, Silver PA. Dynamics in the mixed microbial concourse. Genes Dev. 2010;24:2603–2614. doi: 10.1101/gad.1985210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Greenblum S, Chiu HC, Levy R, Carr R, Borenstein E. Towards a predictive systems-level model of the human microbiome: progress, challenges, and opportunities. Curr Opin Biotechnol. 2013;24:810–820. doi: 10.1016/j.copbio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heinken A, Sahoo S, Fleming RM, Thiele I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes. 2013;4:28–40. doi: 10.4161/gmic.22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gowen CM, Fong SS. Exploring biodiversity for cellulosic biofuel production. Chem Biodiversity. 2010;7:1086–1097. doi: 10.1002/cbdv.200900314. [DOI] [PubMed] [Google Scholar]
- 75.Shong J, Diaz MRJ, Collins CH. Towards synthetic microbial consortia for bioprocessing. Curr Opin Biotechnol. 2012;23:798–802. doi: 10.1016/j.copbio.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 76.Blazier AS, Papin JA. Integration of expression data in genome-scale metabolic network reconstructions. Front Physiol. 2012:3. doi: 10.3389/fphys.2012.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Macklin DN, Ruggero NA, Covert MW. The future of whole-cell modeling. Curr Opin Biotechnol. 2014;28:111–115. doi: 10.1016/j.copbio.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Crosetto N, Bienko M, van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nat Rev Genet. 2015;16:57–66. doi: 10.1038/nrg3832. [DOI] [PubMed] [Google Scholar]
- 79.Gatto L, Breckels LM, Burger T, Nightingale DJH, Groen AJ, Campbell C, Nikolovski N, Mulvey CM, Christoforou A, Ferro M, Lilley KS. A foundation for reliable spatial proteomics data analysis. Mol Cell Proteomics. 2014;13:1937–1952. doi: 10.1074/mcp.M113.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oetjen J, Veselkov K, Watrous J, McKenzie JS, Becker M, Hauberg-Lotte L, Kobarg JH, Strittmatter N, Mroz AK, Hoffmann F, Trede D, Palmer A, Schiffler S, Steinhorst K, Aichler M, Goldin R, Guntinas-Lichius O, von Eggeling F, Thiele H, Maedler K, Walch A, Maass P, Dorrestein PC, Takats Z, Alexandrov T. Benchmark datasets for 3D MALDI- and DESI-imaging mass spectrometry. Gigascience. 2015;4:20. doi: 10.1186/s13742-015-0059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]