

Abstract
Cancer treatments often require combinations of molecularly targeted agents to be effective. mTORi (rapamycin) and HDACi (MS-275/entinostat) inhibitors have been shown to be effective in limiting tumor growth, and here we define part of the cooperative action of this drug combination. More than 60 human cancer cell lines responded synergistically (CI<1) when treated with this drug combination compared to single agents. In addition, a breast cancer patient-derived xenograft, and a BCL-XL plasmacytoma mouse model both showed enhanced responses to the combination compared to single agents. Mice, bearing plasma cell tumors lived an average of 70 days longer on combination treatment compared to single agents. A set of 37 genes cooperatively affected (34 down-regulated; 3 up-regulated) by the combination responded pharmacodynamically in human myeloma cell lines, xenografts, and a P493 model, and were both enriched in tumors, and correlated with prognostic markers in myeloma patient datasets. Genes down-regulated by the combination were overexpressed in several untreated cancers (breast, lung, colon, sarcoma, head and neck, myeloma) compared to normal tissues. The MYC/E2F axis, identified by upstream regulator analyses and validated by immunoblots, was significantly inhibited by the drug combination in several myeloma cell lines. Furthermore, 88% of the 34 genes downregulated have MYC binding sites in their promoters, and the drug combination cooperatively reduced MYC half-life by 55% and increased degradation. Cells with MYC mutations were refractory to the combination. Thus, integrative approaches to understand drug synergy identified a clinically actionable strategy to inhibit MYC/E2F activity and tumor cell growth in vivo.
Keywords: mTOR, HDAC, rapamycin, entinostat, MYC, E2F, pharmacodynamic markers
Introduction
The complex interaction of multiple, often heterogeneous, genomic aberrations, microenvironment inputs, and non-linear signal transduction cascades promotes cancer progression and affects therapeutic response. To maximize clinical efficacy, combinations of targeted agents are often required to overcome compensatory cancer signaling networks and circumvent resistance mechanisms. Combinations of cytotoxic drugs, and more recently, molecularly targeted agents with cytotoxics, have become the mainstay of medical oncology(1). The ability of combinations to circumvent tumor resistance is well established and a spectrum of cooperative mechanisms between targeted agents is known, including enhanced response by vertically targeting the same pathway or by horizontally targeting parallel pathways(1,2). Identifying the mechanism(s) of molecular synergy for candidate combinations, while challenging, is critical for prioritization of combinations to evaluate clinically. In FDA-issued guidance for the development of combining unapproved drugs, a strong biological rationale for the combination and validated molecular response biomarkers are considered critically important(3). We have previously shown sensitivity and drug synergy for the combination of mTOR(mTORi) and histone deacetylase inhibitors(HDACi) in multiple myeloma(MM) cells isolated from patients as well as in genetically diverse sets of human MM, Burkitt’s lymphoma (BL), and mouse plasmacytoma cell lines(4). Preclinical findings have been reported for additional malignancies(5,6) and clinical activity has been found in an early phase trial of everolimus (mTORi) and panobinostat (HDACi) in patients with relapsed lymphoma(7). Panobinostat in combination with bortezomib and dexamethasone has been approved for use in treatment-refractory myeloma patients(8). Additionally, everolimus (9) and everolimus plus exemestane (10) has been approved for treatment in breast cancer. Entinostat (MS-275, an HDACi) was given breakthrough status in combination with exemesthane after a phase II trial demonstrated improvements in progression free survival and overall survival (11) and is currently being evaluated in a phase III trial in breast cancer. Here we have shown that mTORi/HDACi combinations are more effective than single agent therapy in a diverse array of cancers using cell lines, xenografts, patient-derived xenografts (PDX) and a genetically engineered mouse model. A biologically-integrated, network-based approach using myeloma cell lines and patient datasets was used to elucidate one of the mechanisms of action in combining mTOR and HDAC inhibitors.
Materials and Methods
Cell lines
The Developmental Therapeutics Program (DTP) screened the NCI-60 cell lines with single agent rapamycin, MS-275, or the combination as previously described(12). Human MM cell lines were obtained in 2014, cultured and authenticated as previously described(13). The tetracycline-repressible MYC P493-6 B cell line model, kindly provided by Chi-Van Dang (Ludwig Institute for Cancer Research) was treated as described previously(14) (summarized in the Supplemental Methods). For in vitro studies entinostat(MS-275) (15) and tamoxifen were purchased from Sigma-Aldrich and sirolimus(rapamycin) was provided by the Drug Synthesis and Chemistry Branch, DTP, Division of Cancer Treatment and Diagnosis, NCI, NIH). Panobinostat, bortezomib, and cycloheximide were purchased from LC Labs. Drugs were dissolved in dimethylsulfoxide (DMSO; Sigma) to 10mM and stored at −80°C.
In vivo experiments
Mice were tested using institutionally approved (LCBG-009, ACUC, NCI) animal protocols. In the L363 xenograft experiment, 5 × 106 cells were inoculated subcutaneously into each flank of athymic, NCr-nu/nu(Frederick, MD) mice, and allowed to grow for 11 days prior to daily treatment(oral gavage) for 5 days with rapamycin(2.5mg/kg) and entinostat(20mg/kg suspended in 20% hydroxypropyl B-cyclodextrin(Sigma)). BALB/c-Bcl-xL transgenic mice(16) were inoculated IP with 0.4ml pristane to induce tumors. After 8d, mice were randomized to four treatment groups, and dosed twice weekly with vehicle, 2.5mg/kg panobinostat, 2.5mg/kg rapamycin, or the combination(both drugs at 2.5mg/kg), all by IP injection. Details concerning preparations of tumor tissues, protein lysates, antibodies, immunoblots immunohistochemistry and Protein Simple protocols(17) are in the Supplemental Methods.
A patient derived breast cancer xenograft, MaCa4049, was treated with combinations of mTOR, tamoxifen, and HDAC inhibitors in NMRI nude mice (Experimental Pharmacology & Oncology Berlin, Germany; drug supplied by Syndax). Five groups of 8–10 female nude mice, each bearing subcutaneous tumors were given 1)vehicle(0.9%NaCl), 2)everolimus(2mg/kg), 3)everolimus(2mg/kg) and tamoxifen(10mg/kg), 4)entinostat(MS-275, 15mg/kg), and 5)everolimus(2mg/kg), tamoxifen(10mg/kg) and entinostat(15 mg/kg). Tumor diameter measurements were performed twice weekly between day 26(treatment start) and day 63(end of study). Tumor volumes were calculated by the formula: volume = (length × width2)/2, and analyzed for statistical significance among groups using two-way RM ANOVA(GraphPadPrism, Vers.6).
Microarray gene expression profiling
L363 cells were treated with either 1nM or 10nM rapamycin, 0.5uM MS-275 or the combination for 48 hours. Total RNA was extracted with TRIzol® (Invitrogen) from three separate experiments. Labeled aRNA prepared from 1 μg RNA (MessageAmpII aRNA Amplification kit; Ambion) was hybridized to Affymetrix (Santa Clara, CA, USA) HG-U133 Plus 2 array chips, processed on Workstation 450, and analyzed with Gene Chip Operating Software (Affymetrix)(GSE 97985). A detailed description of all bioinformatics procedures, including weighted gene coexpression network analysis (WGCNA)(18) and CHIP-Seq is provided in the Supplementary Methods.
Multiplexed digital gene expression profiling (Nanostring) and ChIP
100ng of total RNA (RNeasy® Mini Kit according to the manufacturer’s protocol(Qiagen)), was added to Nanostring reagents and processed using the nCounter® Analysis System (CCR DNA Sequencing/Digital Gene Expression Core Facility) according to manufacturer’s protocol(NanoString Technologies). nSolver Analysis Software was used for quality control checking, normalization, and initial analysis. In addition to normalization with “spiked in” positive and negative controls, counts were scaled to the geometric mean of the housekeeping genes G6PD, GUSB, OAZ1, SDHA, and SRP14. Unless otherwise indicated in the figure legend, all heat maps show the log2 fold change of a given sample to the expression measured in the vehicle treated control. MYC ChIP analyses were performed according to instructions from Pierce Agarose ChIP kit (26156, includes rabbit IgG control antibody) with the following MYC antibodies used in equal amounts: Santa Cruz 764 and 40 and Epitomics 1472-1. Triplicate qPCR reactions were performed with 100nM primers, 10ul 2xSYBR green PCR master mix (Applied Biosystems) and 2ul of DNA in a total of 20ul. Data were processed in GraphPad Prism7. Primer sequences flanking MYC binding sites in genes, validated in L363 cells, are shown in Supplemental Methods.
Results
mTORi/HDACi combination is synergistic for diverse cancer cell types
mTORi(rapamycin/sirolimus)/HDACi(entinostat/MS-275) combination activity and synergy was assessed across multiple tumor types in the NCI 60 cell line panel(Figures 1A,B, S1A–E). Broader single agent activity was observed with the mTORi(Figure S1A) than with the HDACi(Figure S1B). Upon combination, synergistic activity and sensitivity was observed across all but two cell lines(Fig. 1A,B) at several dose points(Figure S1C–E); NCI/ADR-RES and HCC-2998 cells remained insensitive. The NCI/ADR-RES cell line has high levels of the multidrug resistance 1(MDR1 P-glycoprotein) gene, and the HCC-2998 cell line carries mutations in at least 6 of the top 10 COSMIC cancer genes, including FBXW7(19).
Figure 1. Evaluation of mTORi/HDACi Synergy and gene co-expression network analysis defines cooperative drug response network.
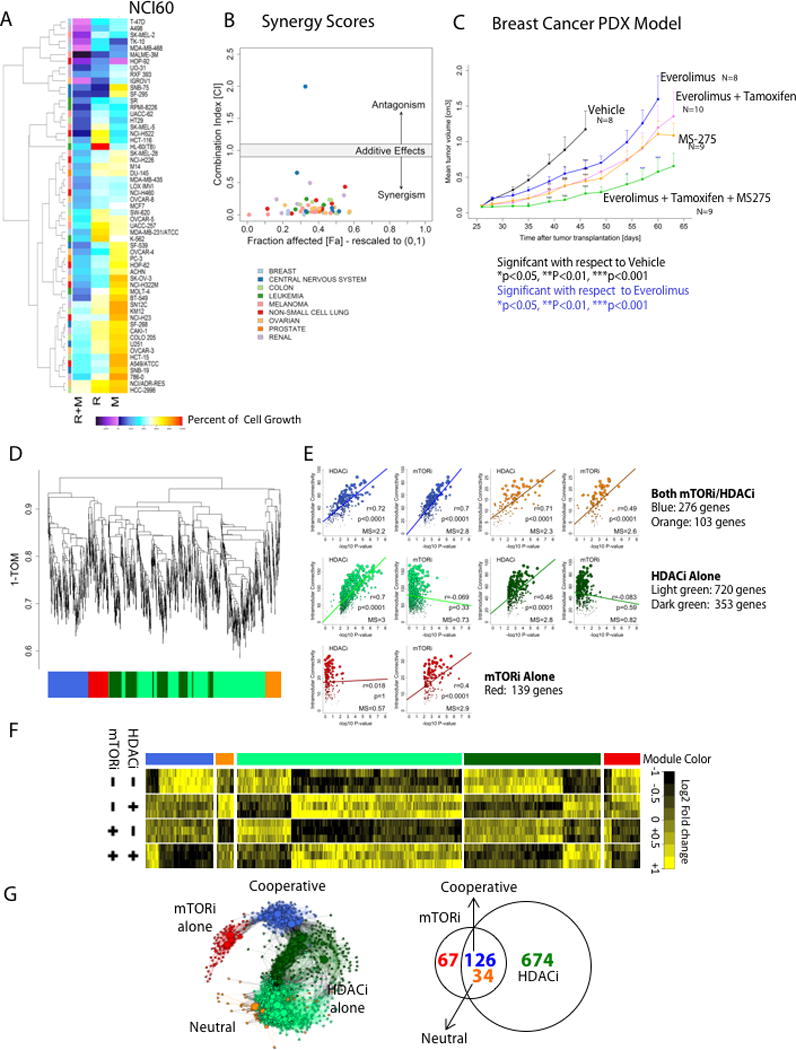
A) Percent Growth of cells relative to initial cell density in the NCI-60 panel after 48h of treatment with rapamycin (R)(10nM) and/or MS-275(M)(1uM)(percent growth from baseline is shown; see https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm for detailed methods and calculations; Figure S1 has additional doses), B) Distribution of synergy scores across the cell lines, based on the Combination Index calculation (CI<1 is synergistic) and the Fraction Affected (Fa), indicating proportion viable, C) In vivo activity of everolimus and/or MS-275 in combination with low-dose tamoxifen in the breast cancer patient derived xenograft (PDX) line MaCa4049. Black asterisks refer to comparisons of drug treatment time points significant relative to the vehicle; blue asterisks refer to comparisons with everolimus, D) Dendrogram from hierarchical clustering of genes into drug-response modules based on the topological overlap distance (1-TOM). The modules contain densely interconnected, highly co-expressed genes forming the dendrogram branches color coded underneath (See Figure S2 and Supplemental Methods for complete description of informatics procedures and parameters), E) Scatter plots show the relationship between drug treatment-based gene significance (negative log10 P-value from two-way ANOVA models) and intramodular connectivity for each drug-specific module identified. In the plots are shown Pearson’s correlation coefficients (r), correlation significance (Bonferroni corrected P-values), and significance scores (mean gene significance (MS)), which were used as criteria for mapping a network module to the treatment response (see Supplemental Methods and Figure S2D for further details), F) Gene co-expression profiles (mean centered by columns) for each the five drug-specific co-expression modules across individual treatments and the combination. The heatmaps of genes were generated with hierarchical clustering using (1 – uncentered correlation distance, and complete linkage). With respect to the combination: the cooperative (blue) module shows the concerted effect of both drugs; in the neutral (orange) module, individual drugs have opposing effects and therefore no net change in expression; HDACi contribution alone (light and dark green modules); mTORi contribution alone (red module), G) Network representation of the 901 most connected nodes (genes) from the drug-specific modules. Nodes are colored by module assignment (as in F), and sizes are proportional to within-module connectivity. A force-directed graph layout was generated with the Fruchterman-Reingold algorithm (igraph R package); topologically near nodes are close to each other and topologically distant nodes are farther apart. Venn diagram shows the number of genes with expression changes related to individual or combination treatment in the 901-gene drug response network.
Consistent with our observations of synergistic activity for combined mTOR/HDAC inhibition across multiple tumor types, we used a breast cancer PDX(MaCa4049) model to evaluate the effectiveness of single agent mTORi, HDACi and the combination with low-dose tamoxifen (an estrogen receptor antagonist frequently used in breast cancer treatment). The combination of everolimus and MS-275 with tamoxifen was superior to single agent treatment in limiting tumor growth (Fig. 1C).
mTORi/HDACi transcriptional network defined by WGCNA identifies genes cooperatively responding to the drug combination involved in cell cycle and DNA replication/repair
To assess the synergistic mechanism of action for this drug combination, we generated a transcriptional drug response network. Using a multiple myeloma line (L363) previously shown to have a synergistic response to combined mTORi/HDACi (4), we generated gene expression profiles(GEP) of cells treated with the individual drugs and the combination. Initially, we filtered out genes unlikely affected by any drug treatment and performed an assessment of additive and interaction effects by two-way ANOVA(20)(Table S1A; see Supplemental Methods). Weighted gene co-expression network analysis(18) of the dataset identified sets/modules of highly correlated genes associated with the single and combination drug effects. The gene co-expression network was constructed based on unsigned and power transformed (coefficient of eight) pairwise Pearson’s correlations across expression profiles of the control and treatment samples (Fig. S2A,B). Subsequently, unsupervised hierarchical clustering of the network topological overlap with the automated cluster separation algorithm (21) detected distinct modules of genes whose co-expression levels were shared(Figures 1D,E, S2C). Significant correlations between gene co-expression events in a module and the ANOVA mean gene significance identified modules associated with each single agent as well as the combination (Fig. 1E, S2D).
An overall drug response network of 901 genes were assigned to five distinct drug-related modules (color coded) were retained for further analysis after removing nodes with low module connectivity (Supplemental Methods, Figure 1D–G, genes assigned to each module are listed in Table S1B). Gene modules within the overall network result from a distinct perturbation, in this case, a particular drug or drug combination, thus illustrating the contribution of each drug to the overall molecular response of the combination(Figures 1F, S2B). Of the five modules, the HDACi alone elicited the largest gene expression response, and was solely responsible for two modules(light and dark green) totaling 674 genes. The mTORi module contained only 67 genes independently affected by rapamycin. Two modules contained genes affected by both drugs. In the neutral module(orange), there were antagonistic effects of the single agents leading to no net change in gene expression when the drugs were combined. The combination(blue) module contained 126 genes in which both individual drugs affected gene expression in the same direction, and when combined, resulted in an enhanced magnitude of response. The combination(blue) module was significantly enriched for genes involved in cell cycle and DNA replication/repair(Figs.2A, S2E;Table S1C). We focused on the 126 genes in the combination(blue) module to study the synergistic mechanism of combination treatment action.
Figure 2. Identification of core cooperative response signature by integration of disease datasets.
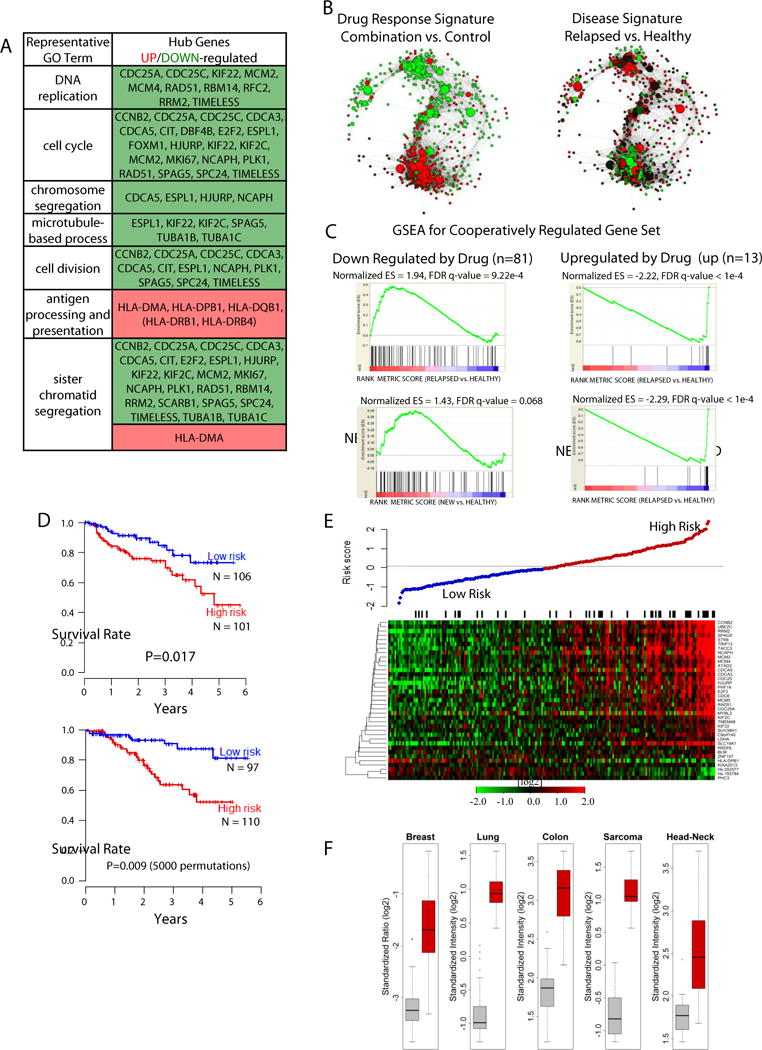
A) Genes responsible for functional enrichment in the representative gene ontology (GO) term for the cooperative combination (blue) module (see Table S1C, Fig. S2E for REVIGO and DAVID functional annotation). B) The expression pattern representing the disease signature, assessed by comparing relapsed MM patients with healthy controls was the opposite of the drug response signature, assessed by treating L363 cells with the drug combination. Node color reflects the direction of gene expression (t-statistic): red identifies genes overexpressed (patients) or up-regulated (drug treated cell line) and green is under-expressed (patients) or down-regulated (drug treated cell line). Note: Of the 901 top connected genes (Fig. 1G, 594 were available for gene expression analysis in dataset GSE6477). C) Gene set enrichment analysis (GSEA) of the combination cooperative (blue) module up- and down-regulated genes; 94 of the 126 genes were enriched in the myeloma dataset. One-way ANOVA contrast t-statistics were used to rank the genes according to their correlation with either the cancer phenotype (red bar) or the healthy donor phenotype (blue bar)(from GSE6477). In this dataset, four comparisons to healthy donor were possible: relapsed MM, newly diagnosed MM, smoldering (asymptomatic) myeloma (SMM), and the pre-malignant condition monoclonal gammopathy of undetermined significance (MGUS). Significant enrichment for blue module genes down-regulated by the combination was only seen in the GSEA results for the new and relapsed myeloma comparisons. All other modules are shown in Figure S3 and detailed in Table S1D. D) Multivariate survival risk predictor built from cooperative module genes using myeloma dataset GSE4587 (37-gene classifier) using the principal components method of Bair and Tibshirani(28). For development of the classifier, 414 MM patients were randomly split into two independent datasets of equal size. Kaplan-Meier overall survival curves are shown for predicted risk groups using 50th percentile cutoff. Single-split test set stratified into low risk (N=97) and high risk (N=110) groups. Asymptotic p-value computed for the log-rank test. Leave-one-out cross-validated Kaplan-Meier curves between low risk (N=106) and high risk (N=101) patients (cross-validated “training set”). Permutation P-value computed by the log-rank test. E) Median centered gene expression for 37 genes of the cooperative module found to be predictive of survival. Samples ordered by increasing risk score computed by the survival classifier and plotted above the heatmap. Black bars indicate death during follow-up time period. F) Signature enrichment was found in five additional tumor types. Box plots comparing cancer (red) versus normal (grey) expression of the down-regulated genes from the cooperative 37-gene signature in five different tumor types (datasets from Oncomine). The box plots shown indicate the mean expression value of the thirty-three down-regulated genes in the 37-gene signature that are also significantly over-expressed in cancer (Oncomine). The Oncomine datasets, the enrichment statistics, and the percent rank in the over-expressed list were: “TCGA Breast” (26 genes, OR=72.6, q-value=4e-25; top 5%), “Hou Lung” (27 genes, OR=88, q-value=9e-27; top 5%), “Sabates-Bellver Colon” (26 genes, OR=33.9, q-value=6e-18; top 10%), “Detwiller Sarcoma” (20 genes, OR=43.6, q-value=1e-17; top 5%), “Ye Head-Neck” (17 genes, OR=20.5, q-value=5e-12; top 5%).
Gene Set Enrichment Analyses (GSEA) identify a disease associated cooperative response signature induced by the drug combination
Integration of our drug response network of 126 genes cooperatively affected by the combination with disease-based transcriptional networks (22–28) from large, well-annotated MM patient cohorts resulted in narrowing the set of genes that are both cooperatively regulated by the mTORi/HDACi combination and relevant to the malignant state to 94. We defined gene sets differentially expressed in CD138+ plasma cells from healthy (volunteer donors) versus myeloma patients and found that patterns were largely inverse—most genes down-regulated by the drug combination were over-expressed in the disease state and vice versa(Figure 2B). In four of the five drug response modules, GSEA found significant enrichment of genes down-regulated by the combination among genes over-expressed in patients, and similarly, genes up-regulated by treatment were enriched in the set of genes under-expressed in patients(Figure 2C, S3A,B; Table S1D,E). The genes from the neutral(orange; no net change in expression) module were not enriched among gene sets either over- or under-expressed in myeloma patients (Figure S3, Table S1D).
Having found 94 of the genes from the combination(blue) module enriched in the patient dataset, we explored relationships between the expression of any of these genes and patient survival. Using the algorithm of Bair and Tibshirani(29), we were able to develop a multivariate prognostic classifier with 37 of the 126 genes significantly associated with overall survival(Figures 2D, Tables S1F,G, Supplemental Methods). Kaplan-Meier curves for predicted low-risk and high-risk groups (Figure 2D) show strong separation both in the split-set validation (p = 0.017) and leave-one-out cross-validation (permutation p = 0.009). The direction of drug combination-induced gene expression change which we observed experimentally would predict improved prognosis in the patient cohort retrospectively evaluated in this analysis(Figure 2E). Hypothetically, the patients identified as highest risk by this classifier may benefit most from combination treatment, but prospective clinical testing would be required to demonstrate this.
In MM, a number of expression-based classifiers have been reported that relate to patient prognosis(26–28). In one reported classifier, six disease subtypes were validated as related to known MM driver lesions(e.g. c-MAF- or MMSET-activating translocations), while one additional subtype was most related to relapse after high-dose therapy and autologous stem cell transplant (28). In co-classifying patients by these disease subtypes with our cooperative response signature, we saw deviations from expected frequencies (chi-square) in the PR (proliferation): and CD2 (CCND1/CCND3 subgroup2) groups (χ2=81, df=6, p<0.001, Fig. S4A). In the PR group, predicted high-risk patients by the 37 gene signature were over-represented and predicted low-risk patients were under-represented), while the opposite was true in the CD2 group (low risk patients were over-represented, and high-risk patients were under-represented). For the remaining 5 groups, there were no deviations from expected by chance alone. In addition, another frequently used classifier is the expression-based proliferation index (based on the expression of 11 genes associated with proliferation: TOP2A, BIRC5, CCNB2, NEK2, ANAPC7, STK6, BUB1, CDC2, C10Orf3, ASPM and CDCA1 (28). It is noteworthy that our 37 gene signature only contains two (CCNB2, STK6) of these genes. When the 7 subgroups were stratified using the PI classifier, deviations from random were observed in subgroups CD2, MS and PR of the high PI patients and CD2 subgroup of the low PI patients. Interestingly, for the CD2 and MS subgroups, a large proportion of the patients falling within the high PI group are classified as low risk by the 37 gene classifier and would not be “predicted” to respond to the drug combination.
Using the Oncomine Molecular Concepts Analysis (30) to query patient datasets for a diversity of tumor types, we asked whether the 37-gene signature was also enriched in any other tumor types. Significant overlap (Fisher’s test p-value < 10−3, odds ratio greater than 2) of the down-regulated genes with the top 5 or 10% over-expressed genes in malignant versus corresponding normal tissues was seen for a diverse array of tumor types (Figure 2F).
Pharmacodynamic behavior of cooperative signature
The pharmacodynamic behavior of the 37 genes comprising the disease-relevant cooperative response to combined mTORi/HDACi across cell lines and tumor types was used to identify and validate upstream regulators of these genes. A linear correlation(R2 = 0.89) of the magnitude of expression effect with drug dose was observed in two separate microarray experiments in the L363 cell line (Figure S4C,D).
A digital gene expression codeset (Nanostring) for our signature was designed as a more tractable means to evaluate the pharmacodynamic behavior of the 37 genes in a variety of settings. Our initial microarray gene expression studies were confirmed with the Nanostring codeset (R2=0.89)(Figures 3A and S4E); 33 genes were downregulated and 4 genes were upregulated by the mTORi/HDACi(R+M) combination. Using a xenograft model of L363 in nude mice, 4 tumors treated in vivo with the combination for 5 days showed similar gene expression responses as cell lines treated in vitro (compare all tumors Fig. 3B(T1–T4) with the cell line in Fig. 3A, column R+M).
Figure 3. Pharmacodynamic evaluation of cooperative signature.
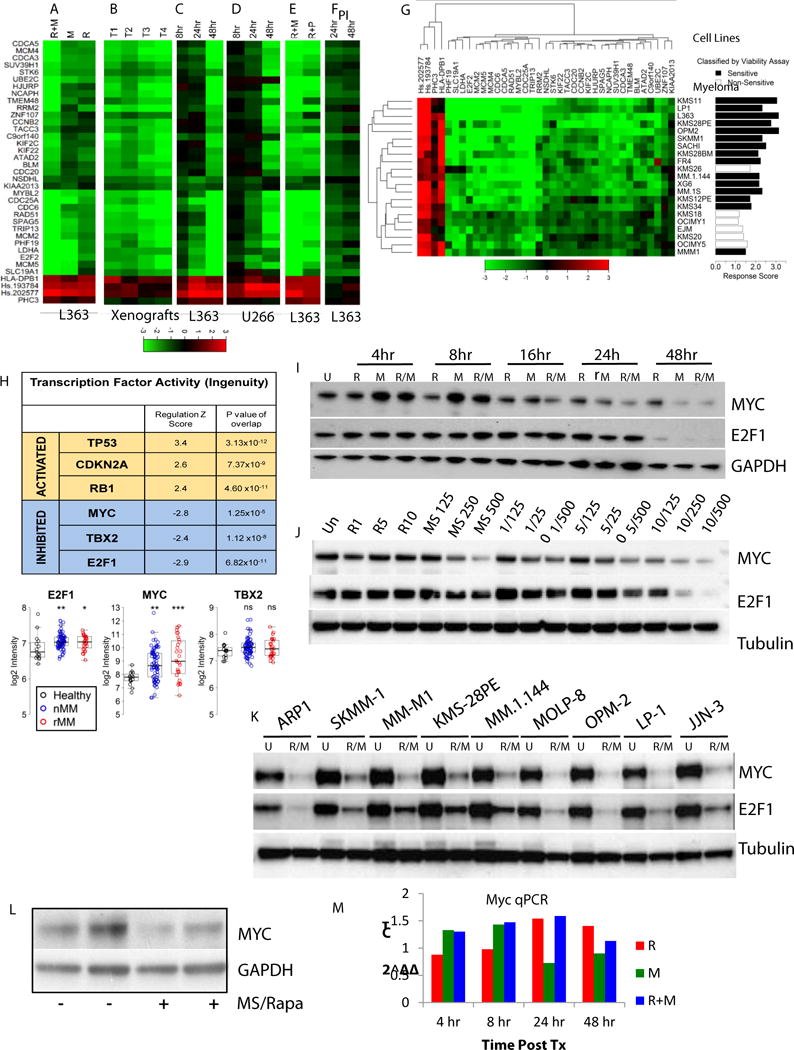
Heatmaps of log2 fold change expression in the cooperative signature compared to respective untreated controls measured by Nanostring: A) L363 cell line treated for 48h with 10nM rapamycin(R), 500nM MS-275(M), and the combination(R+M), B) gene expression comparison of L363 xenografts (tumors T1-4) from vehicle or combination(rapamycin 2.5mg/kg and MS-275 20mg/kg) treated mice. Palpable tumors were treated for 5 days. C) L363 and D) U266 MM cells were treated for 8, 24, and 48h with R+M, E) comparison of combination of 10nM rapamycin with either 500nM MS-275 (R+M) or 2.5nM panobinostat (R+P) at 48h, F) L363 treated with 2.5nM of the proteasome inhibitor (PI) bortezomib for 24 and 48h, G) Heatmap and hierarchical clustering (Euclidean distance) of the log2 expression changes due to 48h treatment with R+M in 21 human MM cell lines. Cell lines with decreases in viability greater than 50% were considered sensitive; transcriptional response scores for a cell line were calculated as a weighted average of log2 fold changes from the 37 genes; the response scores are significantly higher in the cell lines identified as sensitive to the combination treatment (p-value=0.0002). H) Six potential transcriptional “master regulators” of the cooperative signature, identified by Ingenuity Upstream Regulator Analysis, were predicted from the cooperative (37-gene) signature expression pattern. Box plots of log2 intensity of E2F1, MYC, and TBX2 in healthy volunteer plasma cells versus CD138+ B cells from newly diagnosed (nMM) or refractory MM (rMM) patients in dataset GSE6477. p-values (*<0.05, **<0.005, ***<0.0005) are pairwise comparisons by ANOVA (patients vs. healthy donors): MYC and E2F1 expression is significantly different between healthy volunteers and new or relapsed myeloma patients. I–J) Western blots for MYC and E2F1 in L363 cells across a I) treatment (R:10nM, M:500nM, or R/M) time-course, and J) dose course. K) Western blots of MYC and E2F1 after 48 hours R+M treatment in a panel of MM cell lines sensitive (>50% decrease in viability) to R+M). L) MYC protein levels from L363 xenografts treated with vehicle(−) or the combination(+) for 5 days. M) qPCR for MYC transcripts from L363 cells over treatment (Tx) time course (10nM rapamycin, 500nM MS-275, and the combination)(see also Figure S5B for E2F1 and MYC expression levels measured with Nanostring in the same panel of lines from Figure 3G).
Time course experiments found a small subset of genes responding as early as 8 hours post-combination treatment, however the largest changes occurred between 24–48 hours of treatment in two separate cell lines (Figure 3C(L363), D (U266)). This temporal relationship of gene expression suggests the possibility that these genes are controlled by an upstream regulator affected by the combination, rather than necessarily being direct targets.
Combinations of rapamycin with panobinostat(P), an HDACi structurally distinct from entinostat, elicited a similar signature response, with some genes being less affected (Figure 3E). Treatment with a dose of the proteasome inhibitor bortezomib eliciting a similar viability effect on L363 cells did not have an affect similar to that seen with mTORi/HDACi combinations on the expression of most of these genes(cf. Figure 3F with 3E, column R+M)). A large panel of MM cell lines was screened, and the overall magnitude of expression change for the gene signature was related to the relative sensitivity of the cell line to the drug combination (Figure 3G). These analyses also suggested that some of the genes (eg. NSDHL, KIAA2013, ZNF107 and PHC3) were not consistently cooperative across cell lines in the combination response. A similar sensitivity-expression response relationship was seen from screening a subset of the NCI-60 lines (Figure S4F), with two lung cancer (H522, H460) and one renal (TK10) cell line treated with the combination showing the greatest degree of similarity with our myeloma lines.
Master regulators of mTORi/HDACi cooperation
As the cooperative signature consists of a biologically-enriched set of genes responding to each individual drug, and synergistically to combination treatment, we tested whether there were any known transcriptional regulators likely to yield this gene expression pattern. Ingenuity upstream regulator analysis was used to identify transcriptional regulators that could explain the gene expression changes observed in the cooperative signature in response to combination treatment. Six transcriptional regulators were found to have both a highly significant p-value for overlap between the individual regulator dataset and the cooperative signature, as well as activation Z-scores of >2 or <−2(Figure 3H). A negative Z–score implies that inhibition of the regulator would account for the gene expression pattern queried, whereas a positive Z-score suggests an activated state for that regulator. All three “activated” transcriptional regulators (CDKN2A, RB1, TP53) are known tumor suppressors, and we have previously implicated these regulators in responses to the combination(4).
We were particularly interested in the contribution of the predicted “inhibited” transcriptional regulators, MYC, TBX2, and E2F1 in driving the responses of the 37 genes. Both E2F1 and MYC were significantly overexpressed in CD138+ cells from MM patients compared to healthy donor CD138+ cells in dataset GSE6477, while TBX2 was not(Figure 3H). At the protein level, both individual drugs, and to a greater extent the combination, diminished MYC and E2F1 expression(Figure 3I). Expression of both proteins was also decreased in a dose-dependent manner(Figure 3J), but MYC protein levels decreased in response to the combination sooner after treatment than did E2F1(Figure 3I). In a panel of nine MM cell lines sensitive to the combination, expression of both proteins was decreased in the combination treated cells versus control(Figure 3K).
MYC and E2F1 often coordinately regulate gene expression, with ChIP-PET(pair-end ditag sequencing) studies finding E2F1 binding motifs enriched within MYC binding clusters(31). Further, E2F1 collaborates with MYC to regulate a large number of genes; E2F1 itself is transcriptionally up-regulated in response to MYC activation(31). These reports taken together with our observation of MYC protein expression diminishing much sooner after drug treatment than E2F1 expression, led us to focus on the role of MYC in driving the gene expression response to the mTORi/HDACi combination. Analysis of a MYC ChIP-Seq dataset from the MM cell line MM.1s(14), confirmed the findings of our Ingenuity analysis. MYC binds to the promoters of ~88% of the down-regulated genes in the cooperative signature (p< 10−5)(Figure 4A,B, S5A). These findings were validated with conventional ChIP-qPCR (Fig. 4C) in L363 cells for 7 of 7 target genes queried. L363 xenografts treated with the combination in vivo exhibited decreases in MYC protein levels 5d post-treatment (Fig. 3L).
Figure 4. Evaluation of the role of MYC in mTORi/HDACi combination response and cooperative signature expression.
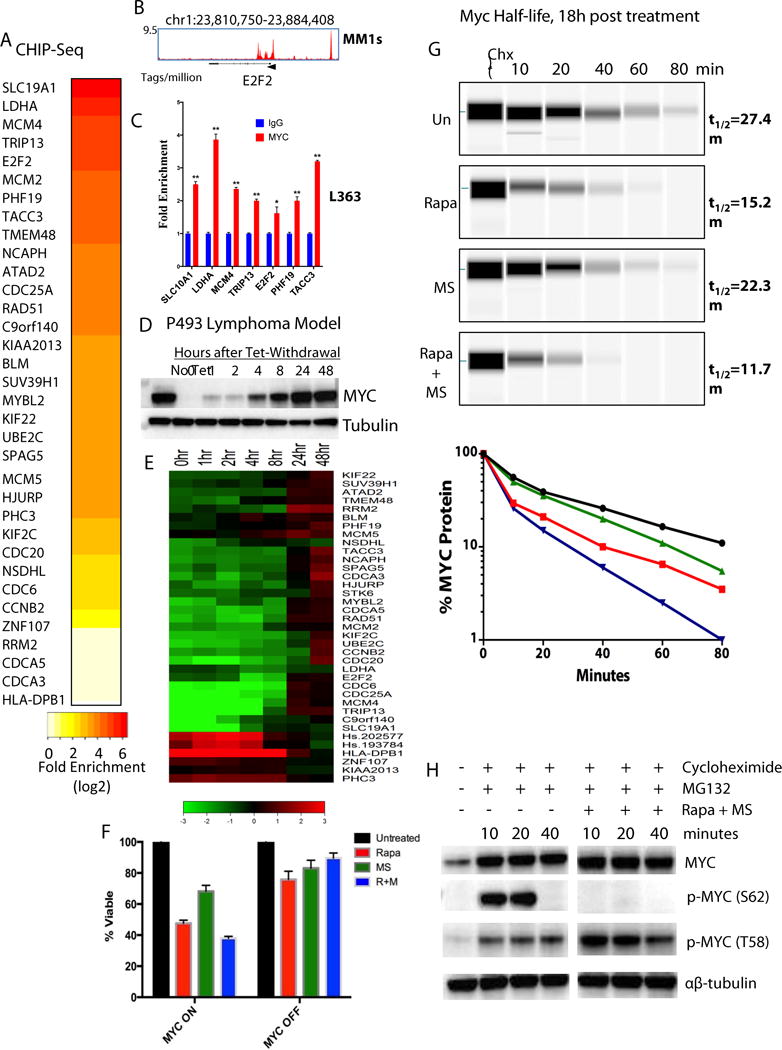
A) Heatmap of the fold enrichment of cMyc ChIP-seq (ChIP signal versus input signal) in the promoter region for each gene in MM1s cells (see Figure S5A for other gene regions). B) Representative genomic view of MYC binding on E2f2 in MM1s cells, C) ChIP-qPCR analysis of seven genomic proximal promoters of human MYC target genes in L363 (mean ± SE, *p<0.05, **p<0.001). D) Western blot for MYC in P493-6 tet-repressible cell line. Cells treated with 0.1mg/ml doxycycline for 72 hours for full repression of exogenous MYC expression (timepoint 0), before washing and changing media to tet (dox)-free. E) Cooperative signature heatmap of log2 fold change versus P493-6 (1-48hrs) after removal of tet treatment. F) Cell viability relative to untreated control for MYC-ON and -OFF P493 cells after 48 hours of single agent or combination treatment (n = 2 experiments with 3 technical replicates in each). The interaction of combination treatment and MYC effect was significant by ANOVA (p<0.0001) (See also Figure S6). G) Combined mTOR and HDAC inhibition affects MYC protein stability. MYC stability following cycloheximide treatment. Representative western blot pseudo-images (Simple Western automated capillary immunoassay) for MYC in L363 cells after 18 hours of single agent rapamycin (10nM) or MS-275 (500nM) or combination treatment. Cells were treated with cycloheximide to block protein translation to determine drug effects on MYC half-life. This experiment was repeated 3×. Graph showing percent MYC protein relative to no cycloheximide/untreated control over an 80 minute time course (time 0 is 5 minutes after addition of cycloheximide); MYC protein half-life is estimated from the slope of lines (see Fig. S7 for equation and half-life estimations immediately and 4 hours after initiating mTORi/HDACi treatment), H) Western blots of MYC phospho-threonine 58 residue (known to destabilize the protein) and MYC phospho-serine 62 residue (protein stabilizing). Cells were pre-treated with cycloheximide to inhibit translation and MG-132 to prevent immediate degradation of the phosphorylated MYC species.
Interestingly, while the combination inhibited both E2F1 mRNA and protein (Fig. S5B), discordance was seen between MYC mRNA(Figure 3M, S5B) and protein(Figure 3I–K) levels after drug treatment. For example, while the MYC protein successively decreased in the L363 line from 18 to 48 hours post-combination treatment, mRNA expression was slightly increased relative to control as measured by both qPCR and Nanostring(Figures 3M, S5B). This drug combination effect on MYC protein, but not RNA, explains why MYC itself was not identified as part of the cooperative response signature in the original microarray.
Role of MYC inhibition in the cooperative drug response
Since mTORi/HDACi treatment led to decreased MYC protein expression at time points as early as 16h post-treatment, we sought to experimentally validate the role of MYC in the synergistic drug response. MYC activation, found in 67% of MM cases, has been attributed to malignant progression in MM as well as numerous other tumor types(14,32) and MYC rearrangements have been found in ~50% of primary MM samples(33). To investigate the relationship between MYC expression and the expression of our cooperative response signature, we used the P493-6 EBV-transformed tonsillar B cell line—a model for studying MYC activation in later-stage B cells(31). This cell line contains a tetracycline (Tet)-repressible MYC transgene, in which MYC expression is low in the presence of Tet and restored to a level nearly 30× higher 24 hours after Tet removal(14,31,34). MYC protein levels are completely inhibited in the P493-6 cells after 72 hours of Tet treatment(0hr time point, Figure 4D), and gradually increase after removal of Tet from the media to full restoration by 24 hours(Figure 4D protein; S6A mRNA). Total RNA collected from cells in the same experiment was used to measure expression of the cooperative signature by Nanostring. The responsiveness of these genes to MYC expression is shown as a heat map of the log2 fold change in gene expression compared to P493-6 cells without Tet(Figure 4E). The gene expression changes seen in cells treated with the mTORi/HDACi combination(Figure 3A–G) are largely recapitulated by MYC repression in the P493 model(0hr)(Figure 4E).
Importantly, MYC was found to be critical to the biological effects of the combination. In the P493 model, when MYC expression was “on”, the combination synergistically decreased viability by ~60%, while less than 20% viability was observed in MYC “off” cells(Figure 4F). The interaction of MYC status and combination drug (treatment) effect in these experiments was significant by two-way ANOVA (p<0.0001)(Figure S6B,C). These results point to a critical role for MYC in response to mTORi/HDACi treatment, and link the expression of the cooperative signature to MYC modulation.
mTORi/HDACi decreases MYC protein stability
We went on to investigate the mechanism of mTORi/HDACi action on MYC protein. The broad transcriptional capabilities and potent oncogenic properties of MYC require highly regulated control of expression for normal cellular proliferation, homeostasis, and differentiation(14,34). In normal mammalian cells, numerous mechanisms for regulating MYC expression exists ranging from regulation of transcription, mRNA stability, and translation, to rapid proteolysis orchestrated by a complex convergence of signaling pathways, post-translational modifications, and multiple ubiquitin ligases leading to proteasomal degradation in generally less than 20–30 minutes(35,36).
Since MYC mRNA and protein levels were discordant after mTORi/HDACi treatment(compare Figure 3M with 3I), we evaluated the activity of the combination on MYC protein. Mechanisms of MYC translational control have previously been linked to mTORi sensitivity(37). MYC protein stability following single agent and combination treatment was assessed within 10 min(Figure S7A), 4h(Figure S7B) and 18h(Figure 4G, S7C) after inhibition of protein synthesis with cycloheximide. MYC stability decreased after overnight treatment with both drugs alone and to a greater extent with the combination(Figure 4G). Large effects of mTORi on MYC stability were seen immediately after rapamycin treatment(Fig. S7A), whereas the greatest decreases in MYC stability induced by HDACi treatment were seen after 18h (Fig. 4G). Both the RAS-ERK and PI3K pathways have been shown to regulate MYC stability through modulation of a series of phosphorylation and dephosphorylation events involving residues 58 and 62, leading to Fbox-mediated ubiquitinylation and subsequent degradation(38).
Increased phosphorylation of the threonine 58 residue, known to destabilize the MYC protein(36,39), was observed after combination treatment in L363 cells, while phosphorylation levels of the stabilizing serine 62 residue were already absent just 10 minutes after treatment(Figure 4H). In contrast, combination treatment of the Burkitt Lymphoma RAJI cell line, carrying alanine at residue 58, does not undergo phosphorylation(Fig. 5A), total MYC protein levels remain high (Fig.5A), and the RAJI cells remain 80% viable(Fig. 5B), showing a limited response to the drug combination. Mutations in exon 2 of Myc affecting T58, S62 and surrounding residues have been documented in as many as 29% of AIDS associated lymphomas and 26% of Burkitt’s lymphomas (40). Mining sequence data in TCGA and CCLE via cbioportal (http://www.cbioportal.org), COSMIC (http://cancer.sanger.ac.uk/cosmic) and at TGEN (http://www.keatslab.org), found more than 70 tumor samples harboring mutations in MYC between residues 57 and 62 (Supplementary Table 1H). Hematopoietic tumors comprised roughly two-thirds of the samples, with variants also existing in sarcomas, melanomas, head and neck, esophageal, and lung cancers.
Figure 5. Combination response of a B cell line (RAJI) carrying a MYC phosphorylation mutant, and long-term in vivo efficacy of mTORi/HDACi in a MYC-driven plasma cell model.
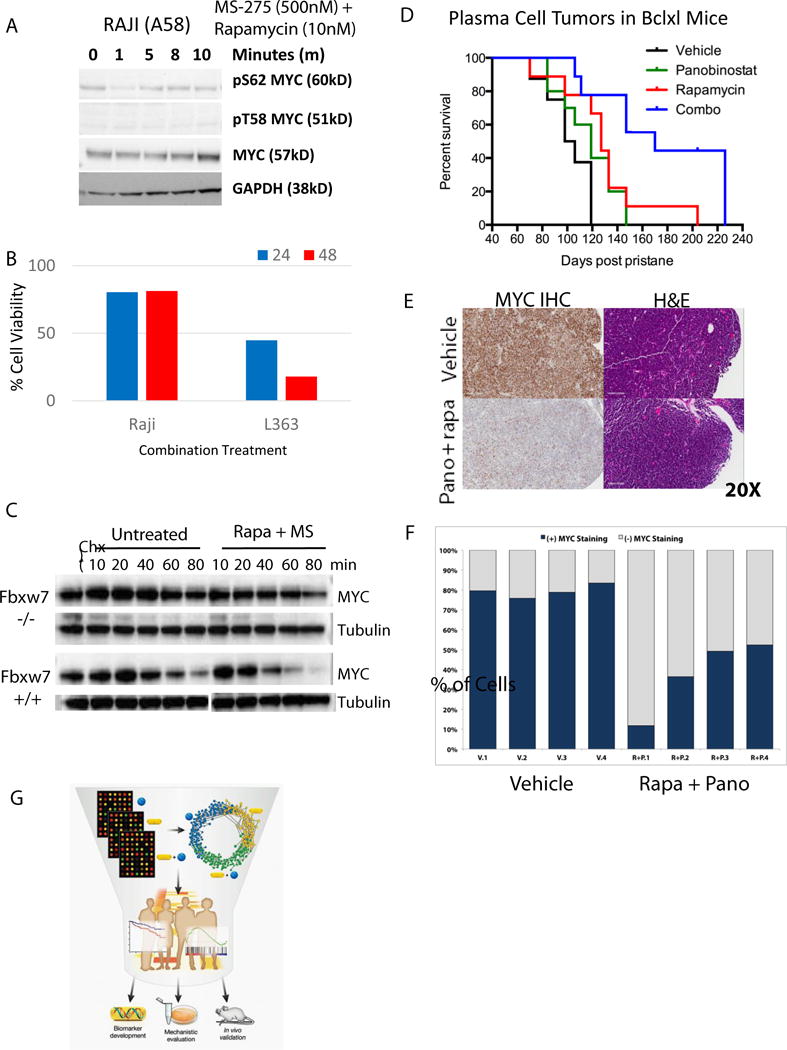
A) Expression of MYC and phospho-MYC in Burkitt lymphoma RAJI cells (which carry an Alanine instead of Threonine at position 58) treated over time (0–10 min) with the combination, B) Growth of RAJI cells known to carry a MYC T58 mutant (Alanine at position 58) vs L363 cells (T58 site intact) treated with the combination, C) Western blot for MYC in cycloheximide experiment with the HCT-116 isogenic FBXW7 (−/−) and (+/+) lines. D) Kaplan-Meier survival curve of Bcl-Xl transgenic mice with pristane-induced plasmacytomas treated twice weekly with vehicle (n=8; median survival 102 days), 2.5mg/kg rapamycin (n=9; median survival 127 days), 2.5mg/kg panobinostat (n=10; median survival 119 days), or the combination (n=9; median survival 170 days); survival of mice treated with Rapamycin (p<0.01) and the combination (p<0.001) was significantly different from control mice by the Log Rank Mantel-Cox test. There was a ~6 week increase in median survival between the combination and rapamycin only arms (p<0.01). E) Representative images of H&E and MYC immunohistochemistry of tumors collected after 5 days of treatment with vehicle or panobinostat/rapamycin combination. F) Pixel quantification by Aperio of MYC IHC intensity (4 tumors from each group). G) Schematic illustrating the integrated approach, utilizing GEP, WGCNA, GSEA and multivariate risk predictors to uncover a cooperative gene signature responding to the mTORi/HDACi combination treatment that led to in vivo validation and mechanistic understanding of the combination (see Fig. S2F).
We have previously shown that this drug combination (but neither drug alone) can decrease ERK activation(4) in MM cell lines. Since, ERK can phosphorylate MYC on residue serine 62, which helps to stabilize MYC protein levels, decreased ERK activation is likely to contribute to diminished MYC stability.
Proteasomal degradation of MYC is often mediated by the Skp1-Cul1-F box ubiquitin ligase complex, including FBXW7(36). FBXW7 sequencing was reported for a number of MM cell lines, including L363, in the Cancer Cell Line Encyclopedia (41) and no mutations were found; interestingly, the HCC-2998 colorectal line which was relatively resistant to the mTORi/HDACi combination(Fig. 1A) does harbor an FBXW7 mutation. Using isogenic FBXW7 (+/+) and (−/−) HCT-116 cell lines, we found that MYC destabilization by mTORi/HDACi treatment was attenuated in the FBXW7(−/−) line (Figure 5C).
While the individual drugs clearly have some effect on MYC expression and stability, the combination caused an earlier and more sustained decrease in total MYC protein levels than either single agent alone(Figures 3I, 4E). This concerted action on MYC protein levels may well account for a large fraction of the synergistic gene expression effects attributed to the cooperative module, as MYC binding was evident by ChIP-Seq in a majority of the genes in the module(Figure 4A, S5A). The synergistic down regulation of Aurora-A (AURKA/STK6) and Polo-like kinase1 (PLK1), within the combination (blue) module(Table 1B; Figure 2A) may also further potentiate decreased MYC stability(42,43). No other genes/proteins demonstrated to directly modulate MYC stability were found in our transcriptional drug response network (901 genes, Table S1B).
Long-term in vivo efficacy of mTORi/HDACi in a mouse model of plasma cell neoplasia
We examined the in vivo efficacy of combining mTORi rapamycin with the HDACi panobinostat in an immune competent model of plasma cell tumors, with features similar to both human myeloma and Burkitt’s lymphoma(16). Additional plasma cell tumor models have been shown to have similar phenotypic, gene expression, and drug sensitivity patterns as human myeloma(44,45). In this Bcl-xl transgenic model, intraperitoneal administration of pristane induces development of plasma cell tumors harboring Myc translocations in nearly all mice within 4–6 weeks(16). While single agent treatment with rapamycin had a modest, but significant, survival improvement (Log Rank Mantel-Cox test p<0.01), there was no significant survival difference between groups treated with vehicle vs. panobinostat alone(Fig. 5D). Twice weekly treatment with the combination of panobinostat and rapamycin significantly improved overall survival (p<0.001) of these mice compared to control (median survival 24.3 weeks in the combination group versus 14.5 weeks in vehicle)(Figure 5D). There was also a significant increase in overall survival between the combination and rapamycin treatment groups (6 week increase in median survival in the combination group, p<0.01). The combination treatment was well tolerated. To test the combination effect on MYC expression in vivo, Bcl-Xl tumors were transplanted into syngeneic pristane-primed mice and allowed to grow for two weeks, at which time daily treatment with vehicle or the panobinostat/rapamycin combination was begun. After five days of treatment, animals were sacrificed and tumors collected. By immunohistochemistry, MYC protein expression was substantially diminished in the combination treated tumors (Figure 5E,F).
Discussion
We used a cross-disciplinary systems pharmacology approach (illustrated in Figure 5G, S2F) to evaluate the synergistic activity of combining mTOR and HDAC inhibitors at the disease, organismal, cellular, and molecular levels. Our network-based modeling (46) successively integrated three network-based filters, 1) transcriptional co-expression, 2) disease versus normal differential expression, and 3) upstream regulator analyses based on over-representation in knowledge-based transcriptional networks to identify and then validate the MYC/E2F axis as a master regulator controlling genes whose expression were also prognostically associated with survival in patient datasets.
The pleiotropic functions of oncogenic MYC have been implicated in the pathogenesis of many tumor types, with 70% of all human cancers estimated to have some degree of MYC deregulation(14,34). Extensive evidence exists validating MYC as a therapeutic target, yet, as with many transcription factors, the development of small molecule MYC inhibitors has been challenging. MYC transcriptional repression through BET bromodomain (47) or G-quadraplex inhibition (48,49) shows potential with the BET BRD compounds further along the path to clinical development.
The MYC-dependent p493 lymphoma model clearly demonstrated that the drug combination was most effective when MYC protein levels were high. Data from the myeloma cells highlighted that it was MYC protein levels, and not MYC transcripts that were reduced by the drug combination; as such, we could not have implicated MYC directly from gene expression profile data. The findings from the cell models led to the identification of decreased MYC stability as a major contributor to the cooperative inhibitory activity of the mTORi/HDACi combination. Although we focused our efforts on how decreased MYC protein levels ultimately led to decreases in many of the genes in our gene signature, presumably through decreased binding of their promoters, it is worth noting that the increase in Class II HLA could also be an indirect result of decreasing MYC levels (50). As such, the drug combination may also help to increase immune recognition by increasing antigen presentation.
Our results here offer an alternative strategy to inhibit MYC activity through the combined use of drug classes that have separately already entered clinical practice as single agents, and together are the subject of several on-going clinical trials. Broad mTORi/HDACi activity and synergy were observed previously in multiple myeloma (cells and xenografts) and Burkitt’s lymphoma cell lines (4), and currently within the NCI60 screen, the breast cancer PDX model, the P493 lymphoma model, and the improved survival in a MYC-driven plasmacytoma model. The differential expression of the cooperative signature in several solid tumor types (breast, lung, colon sarcoma and head and neck), together with our preclinical reports (4) and those of others for mTORi/HDACi activity in lymphomas (6,7,51), further supports exploring this drug combination in conjunction with its associated gene signature. Given the propensity for Myc exon 2 mutations in several hematopoietic tumors, similar to those seen in the RAJI cell line, evaluating the Myc mutational profile may also be warranted.
Supplementary Material
Acknowledgments
Insights from Doug Lowy, Martin Eilers, David Levens, Peter Blumberg, Kent Hunter Jonathan Ashwell, Pat Steeg, Gregoire Altan-Bonnet and anonymous reviewers contributed to the experimental design, preparation and editing of the manuscript. We thank Pat Steeg and Susan Holbeck for their assistance with access to and implementation of the NCI60 screen. We are grateful to Steven Shema, Kathleen Hartman, Patricia Johnson, Maudeline Etienne, and Alex Papageorge for advice, technical assistance and support. This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
Grant Support
John Simmons was the recipient of a Multiple Myeloma Research Foundation fellowship.
Footnotes
Authors’ Contributions
Conception and design: JK Simmons, A Michalowski, B Mock
Acquisition of data: JK Simmons, J Patel, B Gamache, K Zhang, W DuBois, J Gary, S Gaikwad, S Zhang, N Watson, D Connors, E Leon, J-Q Chen, P Ordentlich
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A Michalowski, J Simmons, J Huang, K Zhang, M Lee, B Mock
Writing, review, and/or revision of the manuscript: JK Simmons, A Michalowski, B Mock, M Kuehl, A Zingone, O Landgren, P Ordentlich
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Dancey JE, Chen HX. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat Rev Drug Discov. 2006;5(8):649–59. doi: 10.1038/nrd2089. doi nrd2089 [pii] 10.1038/nrd2089. [DOI] [PubMed] [Google Scholar]
- 2.Jia J, Zhu F, Ma X, Cao Z, Li Y, Chen YZ. Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov. 2009;8(2):111–28. doi: 10.1038/nrd2683. [DOI] [PubMed] [Google Scholar]
- 3.LoRusso PM, Canetta R, Wagner JA, Balogh EP, Nass SJ, Boerner SA, et al. Accelerating cancer therapy development: the importance of combination strategies and collaboration. Summary of an Institute of Medicine workshop. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(22):6101–9. doi: 10.1158/1078-0432.CCR-12-2455. [DOI] [PubMed] [Google Scholar]
- 4.Simmons JK, Patel J, Michalowski A, Zhang S, Wei BR, Sullivan P, et al. TORC1 and class I HDAC inhibitors synergize to suppress mature B cell neoplasms. Molecular oncology. 2014;8(2):261–72. doi: 10.1016/j.molonc.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beagle BR, Nguyen DM, Mallya S, Tang SS, Lu M, Zeng Z, et al. mTOR kinase inhibitors synergize with histone deacetylase inhibitors to kill B-cell acute lymphoblastic leukemia cells. Oncotarget. 2015;6(4):2088–100. doi: 10.18632/oncotarget.2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta M, Ansell SM, Novak AJ, Kumar S, Kaufmann SH, Witzig TE. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood. 2009;114(14):2926–35. doi: 10.1182/blood-2009-05-220889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oki Y, Buglio D, Fanale M, Fayad L, Copeland A, Romaguera J, et al. Phase I Study of Panobinostat plus Everolimus in Patients with Relapsed or Refractory Lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(24):6882–90. doi: 10.1158/1078-0432.CCR-13-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laubach JP, Moreau P, San-Miguel JF, Richardson PG. Panobinostat for the Treatment of Multiple Myeloma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(21):4767–73. doi: 10.1158/1078-0432.CCR-15-0530. [DOI] [PubMed] [Google Scholar]
- 9.Royce ME, Osman D. Everolimus in the Treatment of Metastatic Breast Cancer. Breast Cancer (Auckl) 2015;9:73–9. doi: 10.4137/BCBCR.S29268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hortobagyi GN. Everolimus plus exemestane for the treatment of advanced breast cancer: a review of subanalyses from BOLERO-2. Neoplasia. 2015;17(3):279–88. doi: 10.1016/j.neo.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31(17):2128–35. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Molecular cancer therapeutics. 2010;9(5):1451–60. doi: 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabrea A, Martelli ML, Qi Y, Roschke A, Barlogie B, Shaughnessy JD, Jr, et al. Secondary genomic rearrangements involving immunoglobulin or MYC loci show similar prevalences in hyperdiploid and nonhyperdiploid myeloma tumors. Genes, chromosomes & cancer. 2008;47(7):573–90. doi: 10.1002/gcc.20563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151(1):56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics. 2013;7:47–60. doi: 10.2147/BTT.S29965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovalchuk AL, duBois W, Mushinski E, McNeil NE, Hirt C, Qi CF, et al. AID-deficient Bcl-xL transgenic mice develop delayed atypical plasma cell tumors with unusual Ig/Myc chromosomal rearrangements. The Journal of experimental medicine. 2007;204(12):2989–3001. doi: 10.1084/jem.20070882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen JQ, Heldman MR, Herrmann MA, Kedei N, Woo W, Blumberg PM, et al. Absolute quantitation of endogenous proteins with precision and accuracy using a capillary Western system. Analytical biochemistry. 2013;442(1):97–103. doi: 10.1016/j.ab.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- 19.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73(14):4372–82. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slinker BK. The statistics of synergism. J Mol Cell Cardiol. 1998;30(4):723–31. doi: 10.1006/jmcc.1998.0655. doi S0022-2828(98)90655-1 [pii] 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- 21.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008;24(5):719–20. doi: 10.1093/bioinformatics/btm563. doi btm563[pii]10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 22.Broyl A, Hose D, Lokhorst H, de Knegt Y, Peeters J, Jauch A, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood. 2010;116(14):2543–53. doi: 10.1182/blood-2009-12-261032. [DOI] [PubMed] [Google Scholar]
- 23.Carrasco DR, Tonon G, Huang Y, Zhang Y, Sinha R, Feng B, et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;9(4):313–25. doi: 10.1016/j.ccr.2006.03.019. doi S1535-6108(06)00089-4 [pii]10.1016/j.ccr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 24.Chng WJ, Kumar S, Vanwier S, Ahmann G, Price-Troska T, Henderson K, et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007;67(7):2982–9. doi: 10.1158/0008-5472.CAN-06-4046. doi 67/7/2982 [pii]10.1158/0008-5472.CAN-06-4046. [DOI] [PubMed] [Google Scholar]
- 25.Davies FE, Dring AM, Li C, Rawstron AC, Shammas MA, O’Connor SM, et al. Insights into the multistep transformation of MGUS to myeloma using microarray expression analysis. Blood. 2003;102(13):4504–11. doi: 10.1182/blood-2003-01-0016. [DOI] [PubMed] [Google Scholar]
- 26.Decaux O, Lode L, Magrangeas F, Charbonnel C, Gouraud W, Jezequel P, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myelome. J Clin Oncol. 2008;26(29):4798–805. doi: 10.1200/JCO.2007.13.8545JCO.2007.13.8545. [pii] [DOI] [PubMed] [Google Scholar]
- 27.Shaughnessy JD, Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–84. doi: 10.1182/blood-2006-07-038430. doi blood-2006-07-038430 [pii]10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 28.Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–8. doi: 10.1182/blood-2005-11-013458. doi blood-2005-11-013458 [pi10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bair E, Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2(4):E108. doi: 10.1371/journal.pbio.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, et al. Oncomine 30: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9(2):166–80. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A. 2006;103(47):17834–9. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, Troska-Price T, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011;25(6):1026–35. doi: 10.1038/leu.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. 2014 doi: 10.1038/leu.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151(1):68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hann SR. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Seminars in cancer biology. 2006;16(4):288–302. doi: 10.1016/j.semcancer.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor Myc. Advances in cancer research. 2011;110:77–106. doi: 10.1016/B978-0-12-386469-7.00004-9. [DOI] [PubMed] [Google Scholar]
- 37.Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, et al. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. The Journal of biological chemistry. 2004;279(4):2737–46. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 38.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes & development. 2000;14(19):2501–14. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrell AS, Sears RC. MYC degradation. Cold Spring Harbor perspectives in medicine. 2014;4(3) doi: 10.1101/cshperspect.a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.AR W, LZ P. Myc: The Beauty and the Beast. Genes and Cancer. 2010;1(6):532–41. doi: 10.1177/1947601910378024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell. 2013;24(1):75–89. doi: 10.1016/j.ccr.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan J, Li Z, Lee PL, Guan P, Aau MY, Lee ST, et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer discovery. 2013;3(10):1156–71. doi: 10.1158/2159-8290.CD-12-0595. [DOI] [PubMed] [Google Scholar]
- 44.Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, et al. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood. 2012;120(2):376–85. doi: 10.1182/blood-2012-02-412783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linden M, Kirchhof N, Carlson C, Van Ness B. Targeted overexpression of Bcl-XL in B-lymphoid cells results in lymphoproliferative disease and plasma cell malignancies. Blood. 2004;103(7):2779–86. doi: 10.1182/blood-2003-10-3399. [DOI] [PubMed] [Google Scholar]
- 46.Harrold JM, Ramanathan M, Mager DE. Network-based approaches in drug discovery and early development. Clinical pharmacology and therapeutics. 2013;94(6):651–8. doi: 10.1038/clpt.2013.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov. 2011;10(4):261–75. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Felsenstein KM, Saunders LB, Simmons JK, Leon E, Calabrese DR, Zhang S, et al. Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression. ACS Chem Biol. 2016;11(1):139–48. doi: 10.1021/acschembio.5b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.God JM, Cameron C, Figueroa J, Amria S, Hossain A, Kempkes B, et al. Elevation of c-MYC disrupts HLA class II-mediated immune recognition of human B cell tumors. J Immunol. 2015;194(4):1434–45. doi: 10.4049/jimmunol.1402382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lemoine M, Derenzini E, Buglio D, Medeiros LJ, Davis RE, Zhang J, et al. The pan-deacetylase inhibitor panobinostat induces cell death and synergizes with everolimus in Hodgkin lymphoma cell lines. Blood. 2012;119(17):4017–25. doi: 10.1182/blood-2011-01-331421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.