


Abstract
We have developed a straightforward fluorometric assay to measure primase-polymerase activity of human PrimPol (HsPrimPol). The sensitivity of this procedure uncovered a novel RNA-dependent DNA priming-polymerization activity (RdDP) of this enzyme. In an attempt to enhance HsPrimPol RdDP activity, we constructed a smart mutant library guided by prior sequence-function analysis, and tested this library in an adapted screening platform of our fluorometric assay. After screening less than 500 variants, we found a specific HsPrimPol mutant, Y89R, which displays 10-fold higher RdDP activity than the wild-type enzyme. The improvement of RdDP activity in the Y89R variant was due mainly to an increased in the stabilization of the preternary complex (protein:template:incoming nucleotide), a specific step preceding dimer formation. Finally, in support of the biotechnological potential of PrimPol as a DNA primer maker during reverse transcription, mutant Y89R HsPrimPol rendered up to 17-fold more DNA than with random hexamer primers.
INTRODUCTION
Human PrimPol (HsPrimPol) is a recently identified monomeric DNA-dependent DNA (and RNA) primase-polymerase encoded by the human CCDC111 gene, now renamed PRIMPOL (1). Unlike regular RNA primases, which exclusively behave as DNA-dependent RNA polymerases (DdRPs) in vivo (2), PrimPol preferentially incorporates dNTPs to initiate copying of a DNA template (DNA primase) and can additionally act as a distributive DNA polymerase during the elongation stage (1). In vivo analysis has shown that HsPrimPol has an important role in nuclear DNA maintenance, as it is required for repriming DNA synthesis under stress conditions such as UV irradiation or nucleotide depletion, and its downregulation provokes genome instability (3–5). Moreover, it has been observed that mouse cells lacking PrimPol display inefficient mitochondrial DNA (mtDNA) replication (1). The impact of HsPrimPol in DNA replication is thus a consequence of its potential to operate in translesion synthesis (TLS), tolerating different types of DNA damages such 8-oxo-7,8-dihydrodeoxyguanosine, abasic sites or pyrimidine dimers (1,6). PrimPol might exert its TLS activity in one of three possible ways: (i) by displaying a downstream-lesion repriming activity as occurs during the recovery of nuclear DNA replication forks (3), (ii) by avoiding unreadable lesions such as pyrimidine 6–4 pyrimidine photoproducts by primer realignment (6) or (iii) directly acting as a conventional TLS DNA polymerase, by incorporating either dNTPs or NTPs opposite lesions such as 8oxoG (6). Incorporation of NTPs opposite to either damaged or undamaged DNA by PrimPol, but also by replicative DNA polymerases, might entail the existence of copied DNA chains containing a ribonucleotide tract that, if it persists, might pose the problem of copying an RNA-containing template to PrimPol or other polymerases. However, an RNA-directed nucleotide polymerization activity has not yet been reported for HsPrimPol. Overall, the above mentioned activities exerted by HsPrimPol suggest that it is not a highly specialized polymerase, and support the notion that it might be considered the archetype of modern primases. These promiscuous properties, in combination with the presence of both primase and polymerase activities in the same catalytic active site, suggests that PrimPols are highly evolvable, and that their potential for biotechnological applications (7) could be enhanced.
To date, a large number of studies pursuing different biotechnological goals have documented in vitro evolution of DNA polymerases. Accordingly, DNA polymerases with expanded recognition of natural (8–11) or unnatural (12–19) substrates, increased processivity (20–22), altered fidelity (23–26), enhanced resistance to inhibitors (27,28) or thermostability (29) have been engineered by different methods. In most of these cases, researches have taken advantage of the thermostability or processivity traits of polymerases, allowing the use of exponential amplification or directed selection techniques to find desired polymerase variants (30). However, application of the above mentioned methods for engineering mesophilic polymerases, or those displaying low processivity, is challenging (16,31,32).
In this work, we have developed a PrimPol-specific fluorometric assay that has uncovered the RNA-dependent DNA primase-polymerase (RdDP) activity of purified HsPrimPol. This assay was adapted to directly sample crude supernatants extract of recombinant Escherichia coli cells overexpressing different HsPrimPol variants. The screening approach is based on real time quantification of de novo DNA synthesis catalyzed by HsPrimPol, detected as fluorescence emitted by the tight binding of a fluorescent dye to dsDNA. About 130 variants were generated by randomization of the conserved motif (WFYY) in HsPrimPol that contains tyrosine (Tyr) at position 89, a residue whose mutation to aspartate has been related to magna myopia (33–35), and their priming potential was investigated. Strikingly, the HsPrimPol variant Y89R showed ≥10-fold higher RNA-directed priming activity than the WT protein. A detailed analysis demonstrated that the increased activity in variant Y89R was related to an improved stabilization of the incoming nucleotide opposite an RNA template base. The Y89R mutant showed a high biotechnological potential when used to generate DNA primers to drive reverse transcription (RT) reactions. The physiological role of the RdDP activity inherent to WT HsPrimPol is discussed in the context of both nuclear and mitochondrial DNA replication.
MATERIALS AND METHODS
Reagents
Electro-competent E. coli BL21(DE3)-pRIL cells were prepared in-house according to standard protocols (36). DpnI Restriction enzyme, polynucleotide kinase (PNK), bovine serum albumin (BSA) and 100 mM stock solutions of dATP, dTTP, dGTP and dCTP were purchased from New England Biolabs. Phusion Hot Start II DNA polymerase, random decamer and hexamer primers, TRIzol® and Ni-NTA resin were obtained from ThermoFisher Scientific. Oligonucleotides were synthesized in standard desalted form or purified by PAGE by Sigma-Aldrich. Avian myeloblastosis virus Reverse Transcriptase (AMV-RT), RNAsin® (RNAse inhibitor) and Go-Taq® ‘G2 Flexi DNA’ polymerase were purchased from Promega.SYBR® Green I, Trizma base (Tris), NaCl, MnCl2, MgCl2, PMSF, imidazole, isopropyl-β-d-1-thiogalactopyranoside (IPTG), 1,4-dithiothreitol (DTT), poly(ethylene glycol) 4000 (PEG-4000) and β-mercaptoethanol were obtained from Sigma-Aldrich. Lysozyme, LB medium as a premixed powder, ampicillin (used at 100 μg/ml) and chloramphenicol (used at 17 μg/ml) were obtained from Applichem. Radiolabeled nucleotides [α-32P]dGTP and [γ-32P]ATP (250 μCi; 3000 Ci/mmol) were purchased from Perkin Elmer. Heparin-Sepharose resin and Sephadex® G50 were obtained from GE Healthcare.
Construction of PRIMPOL gene libraries
PRIMPOL/CCDC111 gene libraries were constructed by site directed mutagenesis (37) of the DNA vector pET16::HISCCDC111 (1) using the PCR primers 88/89-NBG/NDT-Fw and 88/89-NBG/NDT-rv (Supplementary Table S1), which contain two degenerated codons at encoded amino acids 88 and 89. PCR reactions contained 1× Phusion DNA polymerase buffer, 200 μM dNTPs, 250 nM of degenerated primers, 2.5 mM MgSO4, 50 ng of template DNA and 0.25 μl of Phusion DNA polymerase. Amplification conditions consisted of an initial denaturation step at 98°C (3 min), followed by 35 cycles of 98°C (1 min), 65.8 °C (1 min) and 72°C (4 min), with a final extension of 15 min at 72°C. PCR products were digested with DpnI at 37°C for 3 h to eliminate the template and then dialyzed on Millipore MF-membrane filters (0.05 μm) against distilled water for 20 min. Five microliters of the dialyzed plasmid were used to transform 50 μl of electro-competent BL-21 (DE3)-pRIL cells. The transformation mixture was spread onto LB agar plates containing ampicillin (100 μg/ml) and chloramphenicol (17 μg/ml). Codon degeneracy of each library was evaluated by scraping colonies from the corresponding plates (≥100 colonies) into 2 ml of LB medium as previously described (38). The plasmid pools were extracted by miniprep and sequenced to assess the quality of the degeneration.
Fluorescent assay for HsPrimPol primase-polymerase activity
Primase-polymerase activity was determined as the increase in fluorescence emitted by the SYBR® green I after binding to dsDNA, which is obtained during the replication of an ssDNA template by HsPrimPol triggered by its DNA primase activity. To optimize fluorescence detection, purified HsPrimPol protein (1 μl from a ±1 μg/μl sample concentration; depending on the particular preparation) was added to 96-well black flat bottom reaction plates containing different volumes (50–200 μl total volume per well) of various reaction buffers [50 mM Tris–HCl (pH 7.5), 1 mM MnCl2 or 1–10 mM MgCl2, 20–100 μM dATP and dGTP, 0–400 mM NH4AcO, 0.5–2 μM GTCC oligonucleotide used as DNA priming template (Supplementary Table S1) and 1×, 2×, 4× or 8x SYBR® Green I]. Reaction plates were incubated in a plate reader (Fluostar Optima, BMG Labtech) at 30°C and the fluorescence intensity was quantified during 1 h using 485 nm (excitation) and 520 nm (emission) wavelengths. When using either a DNA template not containing a preferential 5′-dCdCdTdG-3′ priming site (1), Mg ions or no metal, or when the two required nucleotides (dA and dG) were not provided, the background fluorescence was indistinguishable from that of a negative control without HsPrimPol (Figure 1A). Consistent with results published elsewhere (39), high amounts of cyanine dye SYBR® Green I in the assay hampered HsPrimPol polymerization activity on a duplex template/primer. However, de novo synthesis in the presence of a ssDNA template was not impeded until reaching a 8×–10× dye concentration (results not shown), allowing real time fluorescent measurement at lower SYBR® Green I concentration during the whole reaction time period. Conditions producing the highest fluorescence graph slope were used for further activity analysis of mutant purified HsPrimPol protein and PRIMPOL mutant libraries.
Figure 1.

Fluorometric assay to detect RNA-dependent HsPrimPol DNA primase/polymerase activity. (A) Fluorescence kinetics as a measure of de novo DNA synthesis by purified HsPrimPol (450 nM), in combination with different components as indicated. An increase in fluorescence as a function of time occurred only when dATP and dGTP (100 μM each), the GTCC template (1 μM; see Supplementary Table S1), and MnCl2 (1 mM) were added. The use of the ATCC template (1 μM; Supplementary Table S1), or MgCl2 as metal donor, did not render a measurable fluorescence signal. (B) Fluorescence kinetics using the optimal conditions described in A, but comparing GTCC and GUCC templates (see Supplementary Table S1). A negative control assay using GTCC template in the absence of dNTPs is represented as a comparison. Note that the scale of arbitrary units in the y-axis is different with respect to A. (C) Representative electropherogram of a priming experiment carried out with purified HsPrimPol (450 nM) and 16 nM [γ-32P]ATP, 100 μM dGTP and either GTCC or GUCC as template. The position of the labeled ‘5′-AdG-3′ dimer is indicated. Detailed experimental conditions, as well as templates sequences are described in Materials and Methods.
PRIMPOL gene library platform: pilot experiments
Colonies grown under selection expressing the wild-type (WT) PRIMPOL/CCDC111 gene were picked and inoculated into a 96 deep well plate containing 1000 μl of LB medium supplemented with antibiotics, and sealed with a gas permeable seal. Cultures were grown overnight at 37°C with gentle agitation. After incubation, aliquots of different volumes (10–1000 μl) were transferred to new 96 deep-well plates containing different volumes of LB (0–990 μl) with corresponding antibiotics in the presence of different concentrations of IPTG (0, 0.25, 0.5 or 1 mM) and sealed as before. Cultures were grown for different time periods (2–16 h) at 30°C with vigorous agitation for protein overexpression. Plates were centrifuged at 3000 rpm at 4°C for 30 min to collect the cells. Cell pellets were subjected to two cycles of freezing-thawing and then resuspended in 100 μl of different lysis buffers containing 50 mM Tris–HCl (pH 7.0 to 8.0), 14 mg/ml lysozyme, increasing amounts of NH4AcO (0–400 mM) and increasing amounts of NaCl (0–1000 mM)]. An aliquot of each sample was analyzed by SDS-PAGE to assess HsPrimPol expression. Aliquots from the resuspended pellets (5–30 μl) were added to 96-well black flat-bottom reaction plate wells containing 50 μl of reaction buffer, and the samples assayed varying the afore mentioned parameters. The combination of conditions that provided the best assay performance was used for further screening of mutant libraries.
PRIMPOL gene libraries first activity screening
Individual colonies containing a PRIMPOL/CCDC111 gene variant were randomly picked and inoculated as before into 96 deep well plates containing 800 μl of LB medium supplemented with antibiotics. The first well of each plate was inoculated with a colony harbouring wild type pET16::HISCCDC111 plasmid as a control. Cultures were grown overnight at 37°C with gentle agitation. Subsequently, samples of 40 μl from this preculture were transferred to new 96 deep-well plates as before containing 360 μl of LB with the corresponding antibiotics and 1 mM IPTG. Cultures were grown for 5 h at 30°C with vigorous agitation for protein overexpression. Additionally, glycerol stock cultures were prepared from 150 of preculture and stored in sterile 96-well microtiter plates at –80°C. After protein overexpression, plates were centrifuged as before to harvest cells. Cell pellets were subjected to two cycles of freezing-thawing and then resuspended in 100 μl of lysis buffer [50 mM Tris–HCl (pH 7.5), 14 mg/ml lysozyme and 150 mM NaCl] and incubated for 45 min at 37°C with vigorous agitation. Plates were centrifuged again at 3000 rpm at 4°C for 30 min to pellet the cellular debris and then 700 μl of lysis buffer was carefully added to each well. From each sample, 15 μl was transferred to a 96-well black flat bottom reaction plate containing 35 μl of reaction buffer [50 mM Tris–HCl (pH 7.5), 1 mM MnCl2, 100 μM dATP and dGTP, 1 μM GUCC oligonucleotide (Supplementary Table S1) and 4× SYBR® Green I] per well. Plates were incubated in a plate reader at 30°C. Fluorescence intensity was quantified during 1 h as described above. Plasmid DNA from samples showing kinetic slopes greater than those of the control was extracted and sequenced for further analysis.
Expression and purification of HsPrimPol
pET16::HISCCDC111 vectors containing either WT or mutant PrimPol variants obtained after PRIMPOL gene library screening were transformed and plated as before, and a single colony was inoculated into 10 mL of LB supplemented with both antibiotics and grown overnight at 37°C. This preinoculum was added to 200 ml of LB supplemented with antibiotics and cultured at 37°C until an O.D. of 0.8–1 at 600 nm was reached, after which IPTG was added to 1 mM and the culture was incubated for further 3 h at 30°C for HsPrimPol overexpression. Cells were then pelleted by centrifugation at 4000 rpm for 20 min at 4°C, and the pellets frozen at –20°C until further use. Pellets were resuspended in 20 ml of lysis buffer (50 mM Tris–HCl pH 8.0, 1 M NaCl, 10% glycerol, 1 mM PMSF, 2 mM β-mercaptoethanol, 10 mM imidazole and 400 mM NH4AcO) and disrupted by sonication (5 cycles of 30 s). HsPrimPol was purified on a Ni-NTA agarose column as previously described (1). Protein was eluted with 2 ml of elution buffer (50 mM Tris–HCl pH 8.0, 50 mM NaCl, 10% glycerol and 400 mM imidazole). Eluted samples were loaded onto a Heparin–Sepharose column previously equilibrated with 50 mM Tris–HCl pH 8.0, 50 mM NaCl, 10% glycerol buffer. Samples were washed, and purified following the protocol described elsewhere. (1) Finally, 500 μl of purified protein were dialyzed against dialysis buffer (50 mM Tris–HCl pH 8, 50% glycerol, 500 mM NaCl and 1 mM DTT) and stored at –20°C for further biochemical characterization.
Primase assays on GTCC and GUCC oligonucleotide templates
Priming assays using HsPrimPol with GTCC and GUCC oligonucleotides were performed using radiolabeled [γ-32P]ATP (16 nM), 1 mM MnCl2, 1 μM template and 450 nM HsPrimPol. Reactions were carried out at 30°C for 30 min, and terminated with stop buffer [10 mM EDTA, 95% (v/v) formamide, 0.03% (w/v), xylene–cyanol]. Radioactive reaction products were separated in acrylamide gels as previously described (1) and analyzed using a Phosphorimager (BAS 1500, Fuji). The specific oligonucleotide used as template (Supplementary Table S1), as well as the concentrations of additional dGTP used in each assay, are indicated in the corresponding figure legends.
Elongation assays using a template–primer DNA complex
Five picomoles Sp1C primer (Supplementary Table S1) was 5′[γ-32P]-labeled using [γ-32P]ATP and PNK as described elsewhere (1) and purified by G50 Sephadex chromatography. This labelled primer was hybridized to 10 pmol of a 28-mer ssDNA or ssRNA oligonucleotide template (T13T or T13U, respectively; Supplementary Table S1). Polymerase assays were carried out in reaction buffer [50 mM Tris–HCl pH 7.5, 75 mM NaCl, 2.5% (w/v) glycerol, 1 mM DTT, 0.1 mg/ml BSA, 1 mM MnCl2], different dNTPs mixes (100 μM each), 2.5 mM template/primer complex and 400 nM of PrimPol. Assays were performed at 30°C for 20 min. Reactions were stopped as before, and elongation products were separated by electrophoresis at 30 W, for 120 min, using 8 M urea-containing 20% polyacrylamide gels in TBE buffer.
Determination of the kinetic parameters (KM and kcat) for PrimPol-catalyzed nucleotide incorporation
The Sp1C primer was labeled and hybridized with the T13T template as described above. Kinetic parameters were determined in a standard polymerization assay buffer as described above using 200 nM template/primer complex and 40 nM PrimPol. After 10 min incubation at 37°C, reaction was started by adding different concentrations of dATP (1, 1.5, 2, 4, 5, 7.5, 10, 12.5, 15, 20, 25 and 50 μM). Samples were allowed to react for 4 min (linear polymerization), after which they were and then quenched with stop buffer (see above). Reactions were solved and the relative dATP incorporation with respect to non-elongated primer was quantified using a Phosphorimager (BAS 1500, Fuji). Data were plotted (percentage of elongated primer as a function of dATP concentration) and fitted by nonlinear regression using the SigmaPlot 11.0 software (Systal Software). Kinetic parameters values and their corresponding standard deviations were deducted from the obtained graph equation.
Gel mobility-shift assay for enzyme:ss-template binary complex
A binary complex between PrimPol and ssDNA (GTCC or GUCC templates, end-labeled and purified as described earlier) was determined by electrophoretic shift assay (EMSA). Briefly, the DNA-binding mixture included 2 nM [γ-32P]-labeled oligonucleotide template, 50 mM Tris–HCl pH 7.5, 25 mM NaCl, 2.5% (w/v) glycerol, 1 mM DTT, 2.5% (w/v) PEG-4000, 0.1 mg/ml BSA and increasing concentrations (0–100 nM) of either WT or Y89R HsPrimPol. The mixtures (20 μl final volume) were incubated for 15 min at 30°C and directly loaded onto a non-denaturing 6% polyacrylamide–acrylamide gel and subjected to electrophoresis at 150 V and 4°C for 120 min, in Tris-glycine pH 8.3 buffer. After electrophoresis the gel was dried for 1 h at 80°C in a vacuum gel dryer. The radioactivity corresponding to the enzyme:ss-template complex relative to the free template was quantified by phosphorimaging from three independent replicates, adwas used as an estimate of the enzyme:template affinity. Binary complex formation was quantified as the percentage of retarded template with the standard deviation of the means.
Gel mobility-shift assay for enzyme:ss-template:dNTP preternary complex
Preternary complex characterization was carried out using either 1 μM GTCC or GUCC templates (Supplementary Table S1) with 16 nM [α-32P]dGTP, 500 nM PrimPol (WT or Y89R variant), 50 mM Tris–HCl pH 7.5, 25 mM NaCl, 2.5% (w/v) glycerol, 1.25% (w/v) PEG-4000, 1 mM MnCl2, 1 mM DTT and 0.1 mg/ml BSA. Reactions (20 μl final volume) were incubated for 30 min at 30°C and directly loaded onto a non-denaturing 6% polyacrylamide–acrylamide gel. Electrophoresis and gel drying were carried out as described before. The radioactive band corresponding to the enzyme:ss-template:[α-32P]dGTP preternary complex from five independent replicates was quantified (in arbitrary units) by phosphorimaging, subtracting background values from an control assay containing all aforementioned reagents except the template molecule. The relative efficiency (WT versus Y89R mutant PrimPol) of preternary complex formation was calculated as an average ratio quantified as the percentage of retarded template, with the standard deviation of the means.
PrimPol-primed reverse transcription (PP-RT) PCR coupling
This method relies on three consecutive reaction steps. First, a HsPrimPol-mediated priming reaction (PP) was carried out in 10 μl containing reaction buffer [50 mM Tris–HCl pH 7.5, 50 mM NaCl, 5% (w/v) glycerol, 1 mM DTT, 0.1 mg/ml BSA, 1 mM MnCl2], 100 nM dNTPs, six units of RNAsin® and different concentrations of purified RNA from hepatitis C virus (HCV) (see Figure 6), kindly provided by Prof. Esteban Domingo (CBMSO, Madrid). Reactions were started ading of either 500 nM Y89R or WT HsPrimPol, followed by incubation at 37°C for 2 min and rapid cooling at 4°C. Next, one half of the PP reaction mixture (5 μl) was used as a template for an RT reaction containing 1× AMV-RT reaction buffer, 1 mM dNTPs and 1.5 units of AMV-RT (20 μl final volume); samples were incubated at 42°C for 45 min and then heated at 90°C for 5 min for protein inactivation. Finally, a 5 μl sample of this PP-RT reaction was added to 45 μl of a PCR reaction mixture containing 1× Go taq® polymerase buffer, 2.5 mM MgCl2, 200 μM dNTPs, 0.75 Units of Go taq® polymerase and 200 nM of specific NS5A-F2 and NS5R3 primers (Supplementary Table S1). PCR: 98°C (3 min), 30 cycles of 98°C (30 s), 51°C (30 s) and 72°C (1 min) and 10 min at 72°C. The PP-RT/PCR products were visualized in agarose gels stained with ethidium bromide. As a positive control, an RT reaction was performed using different amounts of HCV RNA (see Figure 6) with RNAsin® (6 units), NS5R3 reverse (150 nM) or random decamer (50 μM) primers (RP-RT; see Results section), 1× AMV-RT reaction buffer, 1 mM dNTPs and 1.5 units of AMV-RT. Samples were incubated at 37°C for 45 min and then heated at 90°C 5 min for protein inactivation. As a negative control, a similar reaction in the absence of the NS5R3 reverse primer was performed. After RP-RT, both reactions were subjected to PCR as described above.
Figure 6.

RT-PCR of RNA primed by HsPrimPol. (A) Scheme of the RT-PCR reaction carried out with HsPrimPol. Red lines are RNA, blue lines are ssDNA, black lines are dsDNA. a: binding of HsPrimPol to the RNA, b: synthesis of DNA primers. c & d: synthesis of ssDNA by AMV-RT. e: PCR reaction with specific primers and synthesis of the dsDNA amplicon. (B) RT-PCR amplification of NS5A HCV gene using WT HsPrimPol or Y89R variant to produce primers during the first reaction step. The amount of RNA template is indicated above. Positive RT-PCR control (C+) using the corresponding reverse primer during the RT process, and negative RT-PCR controls in the presence. (C-1) or absence (C-2) of RNA, are indicated. Molecular size markers and the corresponding sizes (base pairs) are indicated on the left. Size of the expected amplicon is 1051 nt. (C) RT-PCR amplifications of NS3 HCV gene using HsPrimPol or Y89R HsPrimPol to produce primers during the first reaction step. Amount of RNA templates is indicated. Positive RT-PCR control (C+) using random decamers to prime the RT, and negative controls (C-1 and ‘C-2, as in B), are indicated. Size of the expected amplicon is 937 nt. (D) Y89R mutant renders higher RT-qPCR efficiency. The efficiency of DNA synthesis in RT-qPCR reactions primed with either WT (white), or Y89R HsPrimPol (gray), for six different housekeeping genes (see Materials and Methods), is calculated from the Cq values and compared to RT-qPCR primed with random hexamers normalized to 1 (‘RH’; black). For the indicated genes, the ratio of DNA produced in the RT-qPCR primed with Y89R variant respect to that primed with WT HsPrimPol or RH is shown. Averages for each set of ratios, with the standard errors, are shown.
PP-RT quantitative PCR coupling
In a first reaction, total RNA from mouse spleen (purified by TRIzol®) was used as template for a PP reaction in a final volume of 20 μl. The reaction mixture contained 500 nM WT or Y89R HsPrimPol variant, 50 mM Tris–HCl pH 7.5, 1 mM DTT, 0.1 mg/ml BSA, 25 mM NaCl, 2.5% (w/v) glycerol, 0.1 mg/ml BSA, 1 mM MnCl2, 6 units of RNAsin®, 10 μl dNTPs and 400 ng of RNA; reactions were incubated at 37°C for 5 min and then rapidly cooled on ice. Five independent PP reactions performed with each HsPrimPol variant were pooled into a single tube, and further distributed in five independent RT reactions carried out in a 20 μl volume, which contained 1× AMV-RT reaction buffer, 1 mM dNTPs, 1.5 units of AMV-RT and 5 μl (100 ng RNA from the original PP reaction) from the corresponding WT or Y89R HsPrimPol pooled tube. Samples were incubated at 37°C for 60 min and then heated at 90°C for 5 min for protein denaturation. Finally, the five reactions were pooled into a single tube and stored at –20°C for further use. As a positive control, five independent RT reactions were performed with 100 ng total RNA from mouse spleen purified as above, 6 units of RNAsin®, and 50 μM random hexamer primers (RP-RT). As a negative control, five independent RT reactions were performed as described above but in the absence of random hexamers. Positive and negative reaction tubes were respectively pooled and stored at –20°C for further use. cDNA amplification was validated by a standard PCR protocol as described in the previous section, using as template 5 μl from either WT HsPrimPol, Y89R variant, or positive or negative experimental control pools, and as primers either TFAM-Fw and TFAM-rv, or Polγ-Fw and Polγ-rv pairs (that specifically amplify a cDNA fragment from the mitochondrial transcription factor A or polymerase γ genes, respectively; Supplementary Table S1). Amplification using both primers pairs was observed in all samples apart from the negative control, indicating no DNA contamination in the original RNA samples. An additional control to assess the specificity of the RNA-dependent priming reaction by HsPrimPol was carried out. Briefly, 5 μl from a PP reaction performed with WT HsPrimPol, but in the absence of RNA, was used to drive an RT reaction as described above. Then, 5 μl of the RT reaction mixture was subjected to PCR as described in the previous section using the TFAM-Fw and TFAM-rv as primer pair (Supplementary Table S1). No amplification was observed here (not shown), demonstrating total template-dependence of HsPrimPol during the PP-RT reactions.
Five independent sets of reactions, using two different PrimPol and RNA batches, encompassing pooled samples from either WT HsPrimPol, Y89R variant or positive experimental control, obtained as described above, were then subjected to quantitative PCR (qPCR) to estimate the relative RT yield by amplification of the following six different medium-high expression housekeeping genes: glyceraldehyde 3-phosphate dehydrogenase, 18S rRNA, β-actin, peptidylprolyl isomerase A, hypoxanthine-guanine phosphoribosyltransferase 1 and acidic ribosomal phosphoprotein (GDPH, 18S, ACTB, PPIA, HPRT1 and ARBP, respectively). RNA (2 μl, 10 ng) was mixed with 6 μl of a reaction buffer containing the corresponding pair of specific primers (5 nM) and 5 μl of SsoFast™ EvaGreen® Supermix (Bio-Rad). Reactions were carried out in a CFX 384 qPCR apparatus (Bio-Rad) using the following program: 95°C (5 s), 40 cycles (95°C, 5 s; 60°C, 5 s), 60°C (5 s) and 95°C 5 s. Fluorescence acquisition was performed at the end of each elongation step during the entire amplification period. Amplification specificity was evaluated in an additional denaturation step (from 60 to 95°C) of the amplified product. Only one melting peak was observed in all the cases. An additional negative control in the absence of RNA was performed and no amplification was observed. All qPCR experiments were carried out and assessed by the Genomics and Massive Sequencing Service (at CBMSO, Madrid, Spain). Two samples were considered different if the DNA amount of their corresponding quantification cycles (Cq) differed by at least a half-cycle. All results analyzed are averages of three independent technical replicates.
RESULTS
A fluorometric assay to measure HsPrimPol activity
To enhance and facilitate in vitro detection of the HsPrimPol primase-polymerase activity, we have established a new method derived from previous studies carried out by the group of Prof. Andreas Marx, that is centered on an enhanced fluorescence emission by the chromophore SYBR® Green I upon interaction with dsDNA (23,31). At the start of the assay, SYBR® Green emits a low background fluorescence due to the presence of the unreplicated ssDNA; however, as the reaction proceeds, HsPrimPol primase-polymerase activity will produce dsDNA, optimal for dye intercalation, thus increasing the fluorescence of SYBR® green I brightness (40,41), which can be recorded in real-time using a fluorimeter. Validation of this method using purified HsPrimPol protein confirmed that PrimPol activity is evident when using specific DNA template sequences containing a favored priming site (i.e. containing the preferential 5′-dCdCdTdG-3′ priming site flanked by two homopolymeric thymine tract referred to as ‘GTCC’ template). In agreement with previous results (1), fluorescence was detectable when using manganese as metal and dNTPs as incoming nucleotides (Figure 1A). Importantly, the ability of HsPrimPol to perform de novo DNA synthesis directed by a ssDNA template was not inhibited by the large concentrations of SYBR® Green I, unlike the inhibitory effect reported for other enzymes using similar dyes (39), thus increasing the sensitivity of the screening method. Such an improvement in sensitivity allowed us to perform primase-polymerase experiments using a DNA template containing an ‘RNA-version’ of the preferential priming site (5′-rCrCrUrG-3′; ‘GUCC’ template). As shown in Figure 1B, purified HsPrimPol displayed a residual but measurable activity. We also ran priming experiments in the presence of [γ-32P]ATP and using denaturing PAGE to confirm the results of our fluorescent assay (Figure 1C; see Materials and Methods). Thus, a first conclusion is that HsPrimPol exhibits an RdDP activity.
Randomization and screening of HsPrimPol to enhance priming activity on RNA
In order to boost the RdDP activity, we subjected the human PRIMPOL gene to random site-directed mutagenesis, and selection of improved variants by a selective pressure screening, a powerful strategy successfully implemented for the catalytic improvement of several DNA and RNA polymerases (30,42,43). Recently, we have determined the critical role of 87WFYY90 motif in HsPrimPol for both priming and polymerization reactions, contributing to the stabilization of the 3′ incoming nucleotide (Calvo et al., manuscript in preparation). With the aim of increasing the priming potential of HsPrimPol when facing an ‘unfavorable RNA template, we followed a semi-rational randomization approach of the less conserved residues in the 87WFYY90 motif of HsPrimPol (i.e. Phe88 and Tyr89), since our previous results indicated that altering the invariant Trp87 or Tyr90 caused a strong loss of HsPrimPol activities with a critical defect in binding the incoming 3′ nucleotide (Calvo et al., manuscript in preparation). Interestingly, a mutation of Tyr89 to aspartate has been related to magna myopia (33–35).
To minimize the oversampling and facilitate the full library coverage (44), we performed site-directed mutagenesis of positions Phe88 and Tyr89, using a pair of primers with degenerated codons for these specific amino acid positions, thus rendering a reduced amino acid repertoire defined by NBC (N: adenine/cytosine/guanine/thymine, B: cytosine/guanine/thymine, C: cytosine, which encodes for A, R, C, G, I, L, F, P, S, T, V) for position Phe88, and NDT (D: adenine/guanine/thymine, T: thymine, which encodes for R, N, D, C, G, H, I, L, F, S, Y, V) for position Tyr89 (Supplementary Table S1). This randomization design only required the screening of 430 colonies to ensure a theoretical non-biased 95% library coverage (44,45). Mutants with improved RdDP activity were screened using a 96-well format version of the SYBR® Green I fluorescence assay, but testing crude cell extracts supernatants overexpressing HsPrimPol. After pilot experiments demonstrated the suitability of the system to support screenings of crude extracts samples (Supplementary Figure S1), 480 transformants were tested for an enhanced DNA primase-polymerase on the GUCC template, and 18 PrimPol variants showing increased activities were shortlisted (Supplementary Figure S1; Supplementary Table S2). These selected variants were overexpressed in E. coli BL21(DE3)-pRIL cells, purified and further evaluated to confirm the RdDP improvement. Primase experiments comparing GTCC and GUCC templates confirmed an enhanced RdDP activity for 9 variants (Supplementary Table S2). The variant showing the greatest increase in RdDP (RNA as template) was the Y89R mutant (Figure 2). The similar activity observed in lanes 13 versus lane 5, and lanes 14 versus lane 6, corresponding to a 10-fold difference in dGTP provided, we can estimate that the Y89R mutant displays at least 10-fold higher activity than WT HsPrimPol.
Figure 2.

RNA-dependent DNA primase activity shown by WT and Y89R variant. DNA primase activity of purified WT HsPrimPol (left panel) and Y89R variant (right panel) was determined using the GTCC or GUCC templates and the indicated concentration (1, 10 or 100 μM) of dGTP. The position of the labeled 5′-AdA-3′ and 5′-AdGdG-3′ products are indicated. Vertical lines separate non-contiguous lanes.
At this point, we deemed that the increased RdDP activity displayed by the Y89R variant may be related to three non-exclusive concepts: (i) increased template-directed affinity for incoming nucleotides after primer formation, thus rendering a much higher amount of elongated product after several catalytic cycles; (ii) increased RNA template binding activity; (iii) enhanced de novo DNA synthesis opposite RNA, as a consequence of a more stable binding of the 3′ nucleotide preceding dimer formation (2).
RNA-directed DNA polymerization by HsPrimPol
To assess any increased affinity for the incoming nucleotide during elongation, we measured the incorporation of dNTPs directed by DNA or RNA in polymerization reactions using heteropolymeric template/primer complexes (Figure 3A). Upon addition of different mixes of dNTPs (Figure 3B), WT HsPrimPol and mutant Y89R showed similar patterns of incorporation in the presence of both templates, with preferential accumulation of elongated products whose size corresponds to both DNA and RNA template-directed incorporation. Reverse transcription, that is, DNA polymerization activity using RNA as a template, was observed for both enzymes, but was less efficient than polymerization using a DNA template. At the dNTP concentration used in this experiment, performed in multiple turnover conditions, the Y89R variant synthesized a slightly larger amount of products than the WT HsPrimPol, either using DNA or RNA as template.
Figure 3.

RNA-dependent DNA polymerase activity shown by WT HsPrimPol and Y89R variant. (A) Names and sequences of oligonucleotides used as template/primer hybrids to measure primer-dependent dNTP incorporation by HsPrimPol; RNA sequences are indicated in italics; ‘*’ indicates the radiolabeled oligonucleotide used as primer. (B) Polymerization reactions by WT or Y89R variant HsPrimPol was evaluated using the indicated combinations of dNTPs (100 μM each) and either DNA (T13T/Spc1) or RNA (T13U/Spc1) templates. As shown by electrophoretic analysis, primers (15-mer) elongated in 2, 4, 6 or 13 residues represent the expected products in the presence of either dATP, dATP+dGTP, dATP+dGTP+dTTP, or the four dNTPs, respectively.
To quantify any general improvement in binding the incoming nucleotide due to the Y89R substitution, stationary-state kinetics analysis was performed with WT and mutant proteins, using a DNA template/primer complex. In agreement with earlier results (Table 1), the Y89R substitution scarcely affected the catalytic efficiency of PrimPol (Y89R/WT catalytic efficiency ratio was 1.4), mainly due to a modest decrease of the KM (3.6 and 2.5 μM for WT and Y89R variant, respectively). We were not able to obtain any elongated product when RNA was used as template, either with WT or mutant protein, in the limiting conditions required for steady-state kinetic analysis. Thus, these results suggest that the Y89R mutation modestly improves the affinity for dNTPs during primer extension, at least when copying a DNA template.
Table 1. Kinetic constants for dATP incorporation by WT and Y89R PrimPola.
| PrimPol | K M (μM) | k cat (min−1) | k cat/ K M (μM−1 min−1) |
|---|---|---|---|
| WT | 3.59±0.41 | 0.23±0.01 | 0.07±0.01 |
| Y89R | 2.49±0.52 | 0.22±0.01 | 0.09±0.02 |
aExperiments were carried out as described in Materials and Methods. R2 values of the nonlinear regression analysis from data obtained in kinetic assays using the WT HsPrimPol and Y89R were 0.985 and 0.964, respectively.
PrimPol variant Y89R can form a preternary complex on RNA
To evaluate the competence of Y89R for template binding, we performed EMSA assays with radiolabeled GTCC and GUCC ssDNA templates and WT or Y89R PrimPols (Figure 4). Both proteins produced a similar retarded band (Figure 4), representing an enzyme:ssDNA binary complex, but the WT enzyme was more efficient at low protein concentration (10 nM) as compared with the Y89R protein (43.7 ± 3.5% and 30.5 ± 5.8% of retarded product using WT or Y89R, respectively); however, at the highest protein concentration tested (40 nM) both proteins retard most of the ssDNA molecules containing the GTCC sequence (94.0 ± 3.7% and 86.7 ± 11.2% of retarded product using WT or Y89R, respectively). In agreement with the RdDP activity described earlier, both enzymes also bound the ssDNA molecules containing the ribo-tetramer GUCC less efficiently than the deoxyribo-tetramer GTCC, but maintained their relative binding enzyme:template affinity (18.0 ± 0.4.6% and 8.3 ± 1.0% of retarded product using either WT or Y89R at 10 nM, respectively). These small differences in the formation of the binary complex observed by EMSA cannot explain the enhanced priming activity on RNA observed in Figure 2 for Y89R. Indeed, at the higher protein concentration used in the primase assay (450 nM), a similar efficiency in binding the GUCC template can be inferred for both WT and mutant Y89R in view of the EMSA results.
Figure 4.
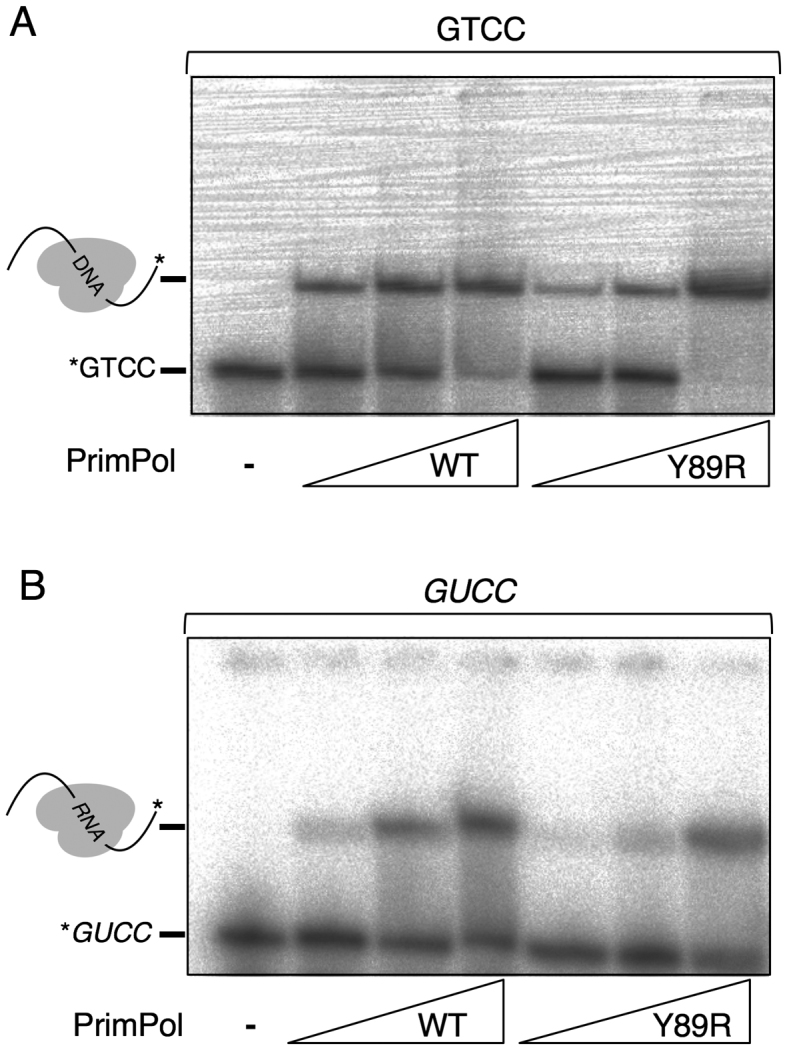
WT PrimPol and Y89R can form binary complexes with DNA and RNA. Radiolabeled GTCC (A) or GUCC (B) oligonucleotides were incubated with increasing amounts (10, 20 and 40 nM) of either WT or Y89R variant HsPrimPol, and binary complexes were analyzed by EMSA.
Before catalysis, some specialized polymerases and primases form an enzyme:template complex that recruits and stabilizes and incoming nucleotide at the elongation site, thus forming the so-called preternary complex, as an intermediate step preceding primer binding (46). For HsPrimPol, this preternary complex can be assayed by a modified EMSA in the presence of [α-32P]dGTP, able to detect the preternary complex as a labeled retarded band, and an ori sequence (3′[dGdTdC]5′) at the ssDNA template in which the dC promotes the template-directed stabilization of dGTP (see Figure 5). When the template contains an RNA equivalent of the ori sequence (3′[rGrUrC]5′), WT PrimPol did not stabilize the preternary complex (Figure 5, right panel). To explore the possibility that the Y89R mutation promotes an improvement in the formation of the preternary complex on both DNA and RNA templates, we performed EMSA in the presence of [α-32P]dGTP and either GTCC or GUCC templates, using similar experimental conditions to those in Figure 2 for the primase assay. Under these conditions, the Y89R variant showed a slight improvement in the preternary complex formation when using the GTCC template (1.4-fold over the WT; Figure 5, left panel). Strikingly, the Y89R variant was able to form a measurable preternary complex when using the GUCC template (Figure 5, right panel). These results support the notion that the increased RdDP activity of Y89R is the consequence of an enhanced capacity to form a preternary complex on ssRNA.
Figure 5.

Y89R can form a preternary complex on ssRNA. EMSA analysis and mobility of the preternary complex. WT and Y89R proteins (450 nM) were incubated with either GTCC or GUCC templates (1 μM) and labeled dGTP to form a preternary complex. Cartoons represent preternary complex formation between HsPrimPol (gray), a DNA template (GTCC) or an RNA template (GUCC), and dGTP, which occupies the 3′ elongation site (white circle) in HsPrimPol.
Y89R PrimPol as a DNA primer maker in RT reactions
We next explored whether the gain in priming activity of Y89R could be demonstrated in heteropolymeric templates exclusively made of RNA. To initially test this hypothesis, we performed additional primase assays using a dNTP mixture containing [α-32P]dGTP and with both synthetic and natural heteropolymeric ssRNAs as templates. Results confirmed the higher activity of the Y89R variant when compared with WT HsPrimPol (Supplementary Figure S2). These results encouraged us to use the Y89R variant as a RNA-directed primase to synthesize DNA primers that can be directly used by a reverse transcriptase in a reverse transcription (RT) reaction. To explore this possibility, we designed an experiment in which HsPrimPol, AMV-RT and Taq polymerase, were sequentially used (see Figure 6A). In this set-up, in which no synthetic DNA primers are provided during the RT step, the final amount of specific dsDNA obtained by RT-PCR will depend upon the DNA primase reaction performed by PrimPol on RNA. To this end, priming reactions either with WT or Y89R HsPrimPols were carried out with different concentrations of HCV RNA (previously shown to be a valid template; Supplementary Figure S2). Next, one half of the reaction volume was used as substrate in a two-step RT-PCR reaction for NS5A HCV gene amplification (see Material and Methods for additional details). The reaction primed by Y89R PrimPol rendered a ≥10-fold larger amount of dsDNA product after the final PCR step than those reactions primed by WT HsPrimPol (Figure 6B), establishing the biotechnological potential of HsPrimPol intrinsic RdDP activity. In a follow-up experiment, we compared the efficiency of RT-PCR amplification of the NS3 HCV gene using HsPrimPol versus random decamers to prime RT. DNA amplification yield obtained with WT HsPrimPol was similar to that obtained when using the random decamer primers whereas mutant Y89R yielded a much greater DNA amplification (Figure 6C).
To further explore the potential biotechnological value of our findings RT-quantitative PCR (RT-qPCR) was used to assess the superiority of the Y89R variant to generate primers on RNA. Total RNA (400 ng) from mouse spleen was used as a template for priming by either WT or Y89R PrimPol, subsequently,100 ng from this reaction was used as a substrate for RT, and only 10 ng from the latter reaction was used to amplify six different housekeeping genes by qPCR. The average (6 genes) enhancement of RT-PCR using Y89R versus WT HsPrimPol was 4-fold (Figure 6D). RT-qPCR control experiments using random hexamer primers added during the RT step were also performed in parallel to detect the same set of genes. As shown in Figure 6D, the DNA amplification yield was higher when using the Y89R variant (on average, 8-fold higher than using random hexamers).
Collectively, these results show the great biotechnological potential of HsPrimPol, and particularly mutant Y89R, to prime reverse transcription of heterogeneous or even unknown RNA sequences.
DISCUSSION
HsPrimPol is a single polypeptide displaying DNA-dependent DNA and RNA primase activities, and also behaves as primer-dependent DNA and RNA polymerase with strand-displacement and TLS capabilities (1,3,6). Here, we have developed a direct fluorimetric assay to measure de novo DNA synthesis by HsPrimPol. The standard assay exploits the HsPrimPol hallmark reaction, DNA-dependent DNA priming, using as template a ssDNA containing an HsPrimPol preferred priming site, and is monitored by the increase of SYBR® green I fluorescence emission by its incorporation into dsDNA. Using a variation of this assay we demonstrate an inherent RdDP activity of HsPrimPol in vitro. The physiological role of such an activity in the nucleus and mitochondria, however, remains unknown, although it could facilitate the bypass, either by TLS or repriming, of template ribonucleotides sporadically present in mitochondrial and nuclear DNA (47,48) thus facilitating replication fork progression. In addition to these sporadic ribonucleotides, long RNA tracts of transcriptional origin called RITOLS (Ribonucleotides are Incorporated ThroughOut the Lagging Strand) (49–51) are specifically hybridized to the H-strand of mitochondrial DNA. It has been proposed that this RNA patch can serve as a back template to bypass unreadable damage in the leading strand. The reverse transcription ability of HsPrimPol described here, would be well suited for this task.
We used crude extracts from recombinant E. coli to identify HsPrimPol variants with increased RdDP activity in a 96-well plate screening platform. A small library of <500 transformants (132 theoretical variants) was designed on a semi-rational approach basis where randomization occurs at defined amino acid positions using ‘smart’ libraries (52) (i.e. containing reduced amino acid alphabet degenerations) instead of using completely random methods (53). Selection of mutable positions at motif 87WFYY90 of HsPrimPol was based on recent findings indicating the critical involvement of Trp87 and Tyr90 in the initial steps of priming and elongation (Calvo et al., manuscript in preparation). As a consequence, we elected to randomize only amino acids Phe88 and Tyr89, which show greater variability among eukaryotic and archaeal PrimPols than the most invariant, flanking positions of the motif. A main conclusion of our study is that introducing a positively charged amino acid (Arg) instead of Tyr89 selectively improves the capacity to generate DNA primers on RNA template sequences. This enhanced RdDP activity in mutant Y89R is due to an increased stabilization of the preternary complex (PrimPol:template:nucleotide) that precedes dimer formation, whereas the mutation only has a minor effect on template binding and nucleotide incorporation further during polymerization.
During the initial screening, 18 variants were selected with an increased RdDP activity (Supplementary Table S2) and purified, and 9 of them were confirmed to have an enhanced RNA-dependent primase activity using the assay described in Figure 2. All the variants showed polar (Asn, Ser and Cys) or basic (Arg and His) changes in Tyr89, either alone, or in combination with almost any possible substitutions (except Pro or Cys) at position Phe88. Our study suggests that the change at Tyr89 is the main responsible for the RNA-directed activity gain; however, a second change at Phe88 can modulate this enhancement. Thus, double mutant F88S-Y89C showed increased RdDP activity compared with WT HsPrimPol, while single mutant Y89C, or double mutant F88P-Y89R did not (data not shown). Collectively, these results emphasize the high degree of functional plasticity shown by this protein, represented by the number of different structural solutions to achieve the same goal. The three-dimensional structure of HsPrimPol catalytic core complexed with a template–primer and an incoming nucleotide has been recently solved (54), and suggests that Tyr89 could be indirectly involved in the template-directed stabilization of the 3′- incoming nucleotide, in agreement with our data.
Selection of Y89R as a PrimPol variant with enhanced RdDP offered us the opportunity to explore its biotechnological potential to produce DNA primers for a RT reaction. Thus, the expression of 6 standard housekeeping genes was evaluated by RT of total RNA from mouse spleen, in a reaction primed either with WT or Y89R PrimPol, or by providing random hexameric primers, followed by qPCR analysis. The process primed by Y89R rendered, on average, a 4-fold greater amount of qPCR-amplified DNA than that using WT PrimPol for all genes tested, and yielded up to 17-fold more DNA than the reactions primed with random hexamers (one of the most common methods for RT priming). Along this line, PrimPol from Thermus thermophilus (TthPrimPol) has been recently characterized (7), and its capacity for generating DNA primers has been successfully exploited in a new DNA amplification method named TruPrime™. Unlike its human orthologue, a putative RdDP activity has not yet been described for TthPrimPol. The screening method presented here might be directly applied to TthPrimPol in order to improve its putative RNA-dependent priming activity. Indeed, the use of a thermostable PrimPol in combination with a thermostable reverse transcriptase might result in a striking benefit in the field of whole transcriptome amplification (55–57) or RNA quasispecies deep sequencing analyses (58).
Concluding remarks
We have developed a simple fluorometric assay to specifically evaluate the primase-polymerase activity of HsPrimPol, allowing the affordable screening of hundreds of HsPrimPol variants engineered for the improved use of RNA as template. This study has identified Y89R as a mutation that improves the stabilization of the preternary complex, as an intermediate preceding primer synthesis. The improved activity of mutant Y89R on RNA templates allowed its use to prime RT reactions, when combined with a reverse transcriptase. Thus, the present study represents a proof-of-principle of the potential biotechnological value of HsPrimPol as a DNA primase, and demonstrates the power of direct evolution not just to improve or alter its enzymatic properties, but to unravel its intimate structure-function details. The high specificity and selectivity of the assay to detect HsPrimPol primase has a high potential to evolve and enhance HsPrimPol recognition and the use of natural and chemically-modified nucleotides as substrates. Furthermore, slight modifications to this method might expand the application of the platform to check HsPrimPol activity in the presence of antibodies, inhibitors, aptamers and drugs, and can likely extrapolated to other primases and polymerases (such as de novo viral RNA-dependent RNA polymerases). These aforementioned assays may be germane to develop biotechnological tools, anticancer therapies and high-throughput analyses of de novo RdRP and DdRP libraries (59).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank to Dr Sara García-Gómez (The Crick Institute, London) for preliminary data on the HsPrimPol RdDP activity, Prof. Esteban Domingo (CBMSO, Madrid) for providing the HCV RNA and to the Genomics and Massive Sequencing Service (CBMSO, Madrid) for its technical guidance on qPCR. We also thank thanks also to Dr Armando Arias (DTU National Veterinary Institute) for critical review of the manuscript.
Footnotes
Present address: Rubén Agudo, Departamento de Ciencias Farmacéuticas y de la Salud, Facultad de Farmacia, Universidad San Pablo CEU, Alcorcón, Madrid, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie [654615 to R.A]; Spanish Ministry of Economy and Competitiveness [BFU2015-65880-P to L.B]. P.A.C. is recipient of a JAE-predoctoral fellowship from the Spanish Ministry of Economy and Competitiveness. Funding for open access charge: Spanish Ministry of Economy and Competitiveness [grant number BFU2015-65880-P to L.B].
Conflict of interest statement. None declared.
REFERENCES
- 1. Garcia-Gomez S., Reyes A., Martinez-Jimenez M.I., Chocron E.S., Mouron S., Terrados G., Powell C., Salido E., Mendez J., Holt I.J. et al. . PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell. 2013; 52:541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frick D.N., Richardson C.C.. DNA primases. Annu. Rev. Biochem. 2001; 70:39–80. [DOI] [PubMed] [Google Scholar]
- 3. Mouron S., Rodriguez-Acebes S., Martinez-Jimenez M.I., Garcia-Gomez S., Chocron S., Blanco L., Mendez J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013; 20:1383–1389. [DOI] [PubMed] [Google Scholar]
- 4. Keen B.A., Jozwiakowski S.K., Bailey L.J., Bianchi J., Doherty A.J.. Molecular dissection of the domain architecture and catalytic activities of human PrimPol. Nucleic Acids Res. 2014; 42:5830–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wan L., Lou J., Xia Y., Su B., Liu T., Cui J., Sun Y., Lou H., Huang J.. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013; 14:1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martinez-Jimenez M.I., Garcia-Gomez S., Bebenek K., Sastre-Moreno G., Calvo P.A., Diaz-Talavera A., Kunkel T.A., Blanco L.. Alternative solutions and new scenarios for translesion DNA synthesis by human PrimPol. DNA Repair (Amst.). 2015; 29:127–138. [DOI] [PubMed] [Google Scholar]
- 7. Picher A.J., Budeus B., Wafzig O., Kruger C., Garcia-Gomez S., Martinez-Jimenez M.I., Diaz-Talavera A., Weber D., Blanco L., Schneider A. TruePrime is a novel method for whole-genome amplification from single cells based on TthPrimPol. Nat. Commun. 2016; 7:13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cozens C., Pinheiro V.B., Vaisman A., Woodgate R., Holliger P.. A short adaptive path from DNA to RNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:8067–8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ellefson J.W., Gollihar J., Shroff R., Shivram H., Iyer V.R., Ellington A.D.. Synthetic evolutionary origin of a proofreading reverse transcriptase. Science. 2016; 352:1590–1593. [DOI] [PubMed] [Google Scholar]
- 10. Xia G., Chen L., Sera T., Fa M., Schultz P.G., Romesberg F.E.. Directed evolution of novel polymerase activities: mutation of a DNA polymerase into an efficient RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:6597–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Patel P.H., Loeb L.A.. Multiple amino acid substitutions allow DNA polymerases to synthesize RNA. J. Biol. Chem. 2000; 275:40266–40272. [DOI] [PubMed] [Google Scholar]
- 12. Aschenbrenner J., Marx A.. Direct and site-specific quantification of RNA 2′-O-methylation by PCR with an engineered DNA polymerase. Nucleic Acids Res. 2016; 44:3495–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen F., Gaucher E.A., Leal N.A., Hutter D., Havemann S.A., Govindarajan S., Ortlund E.A., Benner S.A.. Reconstructed evolutionary adaptive paths give polymerases accepting reversible terminators for sequencing and SNP detection. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:1948–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hansen C.J., Wu L., Fox J.D., Arezi B., Hogrefe H.H.. Engineered split in Pfu DNA polymerase fingers domain improves incorporation of nucleotide gamma-phosphate derivative. Nucleic Acids Res. 2011; 39:1801–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen T., Hongdilokkul N., Liu Z., Adhikary R., Tsuen S.S., Romesberg F.E.. Evolution of thermophilic DNA polymerases for the recognition and amplification of C2′-modified DNA. Nat. Chem. 2016; 8:556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pinheiro V.B., Taylor A.I., Cozens C., Abramov M., Renders M., Zhang S., Chaput J.C., Wengel J., Peak-Chew S.Y., McLaughlin S.H. et al. . Synthetic genetic polymers capable of heredity and evolution. Science. 2012; 336:341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loakes D., Gallego J., Pinheiro V.B., Kool E.T., Holliger P.. Evolving a polymerase for hydrophobic base analogues. J. Am. Chem. Soc. 2009; 131:14827–14837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Staiger N., Marx A.. A DNA polymerase with increased reactivity for ribonucleotides and C5-modified deoxyribonucleotides. Chembiochem. 2010; 11:1963–1966. [DOI] [PubMed] [Google Scholar]
- 19. Huber C., von Watzdorf J., Marx A.. 5-methylcytosine-sensitive variants of Thermococcus kodakaraensis DNA polymerase. Nucleic Acids Res. 2016; 44:9881–9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davidson J.F., Fox R., Harris D.D., Lyons-Abbott S., Loeb L.A.. Insertion of the T3 DNA polymerase thioredoxin binding domain enhances the processivity and fidelity of Taq DNA polymerase. Nucleic Acids Res. 2003; 31:4702–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y., Prosen D.E., Mei L., Sullivan J.C., Finney M., Vander Horn P.B.. A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Res. 2004; 32:1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Williams J.G., Steffens D.L., Anderson J.P., Urlacher T.M., Lamb D.T., Grone D.L., Egelhoff J.C.. An artificial processivity clamp made with streptavidin facilitates oriented attachment of polymerase-DNA complexes to surfaces. Nucleic Acids Res. 2008; 36:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Summerer D., Rudinger N.Z., Detmer I., Marx A.. Enhanced fidelity in mismatch extension by DNA polymerase through directed combinatorial enzyme design. Angew. Chem. Int. Ed. Engl. 2005; 44:4712–4715. [DOI] [PubMed] [Google Scholar]
- 24. Strerath M., Gloeckner C., Liu D., Schnur A., Marx A.. Directed DNA polymerase evolution: effects of mutations in motif C on the mismatch-extension selectivity of thermus aquaticus DNA polymerase. Chembiochem. 2007; 8:395–401. [DOI] [PubMed] [Google Scholar]
- 25. Patel P.H., Kawate H., Adman E., Ashbach M., Loeb L.A.. A single highly mutable catalytic site amino acid is critical for DNA polymerase fidelity. J. Biol. Chem. 2001; 276:5044–5051. [DOI] [PubMed] [Google Scholar]
- 26. Ghadessy F.J., Ramsay N., Boudsocq F., Loakes D., Brown A., Iwai S., Vaisman A., Woodgate R., Holliger P.. Generic expansion of the substrate spectrum of a DNA polymerase by directed evolution. Nat. Biotechnol. 2004; 22:755–759. [DOI] [PubMed] [Google Scholar]
- 27. Kermekchiev M.B., Kirilova L.I., Vail E.E., Barnes W.M.. Mutants of Taq DNA polymerase resistant to PCR inhibitors allow DNA amplification from whole blood and crude soil samples. Nucleic Acids Res. 2009; 37:e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baar C., d’Abbadie M., Vaisman A., Arana M.E., Hofreiter M., Woodgate R., Kunkel T.A., Holliger P.. Molecular breeding of polymerases for resistance to environmental inhibitors. Nucleic Acids Res. 2011; 39:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghadessy F.J., Ong J.L., Holliger P.. Directed evolution of polymerase function by compartmentalized self-replication. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen T., Romesberg F.E.. Directed polymerase evolution. FEBS Lett. 2014; 588:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gieseking S., Bergen K., Di Pasquale F., Diederichs K., Welte W., Marx A.. Human DNA polymerase beta mutations allowing efficient abasic site bypass. J. Biol. Chem. 2011; 286:4011–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Povilaitis T., Alzbutas G., Sukackaite R., Siurkus J., Skirgaila R.. In vitro evolution of phi29 DNA polymerase using isothermal compartmentalized self replication technique. Protein Eng. Des. Sel. 2016; 29:617–628. [DOI] [PubMed] [Google Scholar]
- 33. Keen B.A., Bailey L.J., Jozwiakowski S.K., Doherty A.J.. Human PrimPol mutation associated with high myopia has a DNA replication defect. Nucleic Acids Res. 2014; 42:12102–12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao F., Wu J., Xue A., Su Y., Wang X., Lu X., Zhou Z., Qu J., Zhou X.. Exome sequencing reveals CCDC111 mutation associated with high myopia. Hum. Genet. 2013; 132:913–921. [DOI] [PubMed] [Google Scholar]
- 35. Li J., Zhang Q.. PRIMPOL mutation: functional study does not always reveal the truth. Invest. Ophthalmol. Vis. Sci. 2015; 56:1181–1182. [DOI] [PubMed] [Google Scholar]
- 36. Sambrook J., Green M.R.. Molecular Cloning. A Laboratory Manual. 2012; 4th edn, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 37. Hogrefe H.H., Cline J., Youngblood G.L., Allen R.M.. Creating randomized amino acid libraries with the QuikChange Multi Site-Directed Mutagenesis Kit. Biotechniques. 2002; 33:1158–1160. [DOI] [PubMed] [Google Scholar]
- 38. Agudo R., Roiban G.D., Reetz M.T.. Achieving regio- and enantioselectivity of P450-catalyzed oxidative CH activation of small functionalized molecules by structure-guided directed evolution. Chembiochem. 2012; 13:1465–1473. [DOI] [PubMed] [Google Scholar]
- 39. Driscoll M.D., Rentergent J., Hay S.. A quantitative fluorescence-based steady-state assay of DNA polymerase. FEBS J. 2014; 281:2042–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneeberger C., Speiser P., Kury F., Zeillinger R.. Quantitative detection of reverse transcriptase-PCR products by means of a novel and sensitive DNA stain. PCR Methods Appl. 1995; 4:234–238. [DOI] [PubMed] [Google Scholar]
- 41. Dragan A.I., Pavlovic R., McGivney J.B., Casas-Finet J.R., Bishop E.S., Strouse R.J., Schenerman M.A., Geddes C.D.. SYBR Green I: fluorescence properties and interaction with DNA. J. Fluoresc. 2012; 22:1189–1199. [DOI] [PubMed] [Google Scholar]
- 42. Henry A.A., Romesberg F.E.. The evolution of DNA polymerases with novel activities. Curr. Opin. Biotechnol. 2005; 16:370–377. [DOI] [PubMed] [Google Scholar]
- 43. Kranaster R., Marx A.. Engineered DNA polymerases in biotechnology. Chembiochem. 2010; 11:2077–2084. [DOI] [PubMed] [Google Scholar]
- 44. Patrick W.M., Firth A.E., Blackburn J.M.. User-friendly algorithms for estimating completeness and diversity in randomized protein-encoding libraries. Protein Eng. 2003; 16:451–457. [DOI] [PubMed] [Google Scholar]
- 45. Patrick W.M., Firth A.E.. Strategies and computational tools for improving randomized protein libraries. Biomol. Eng. 2005; 22:105–112. [DOI] [PubMed] [Google Scholar]
- 46. Brissett N.C., Martin M.J., Pitcher R.S., Bianchi J., Juarez R., Green A.J., Fox G.C., Blanco L., Doherty A.J.. Structure of a preternary complex involving a prokaryotic NHEJ DNA polymerase. Mol. Cell. 2011; 41:221–231. [DOI] [PubMed] [Google Scholar]
- 47. Grossman L.I., Watson R., Vinograd J.. The presence of ribonucleotides in mature closed-circular mitochondrial DNA. Proc. Natl. Acad. Sci. U.S.A. 1973; 70:3339–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Williams J.S., Kunkel T.A.. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst.). 2014; 19:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reyes A., Kazak L., Wood S.R., Yasukawa T., Jacobs H.T., Holt I.J.. Mitochondrial DNA replication proceeds via a ‘bootlace’ mechanism involving the incorporation of processed transcripts. Nucleic Acids Res. 2013; 41:5837–5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yasukawa T., Reyes A., Cluett T.J., Yang M.Y., Bowmaker M., Jacobs H.T., Holt I.J.. Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J. 2006; 25:5358–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kolesar J.E., Wang C.Y., Taguchi Y.V., Chou S.H., Kaufman B.A.. Two-dimensional intact mitochondrial DNA agarose electrophoresis reveals the structural complexity of the mammalian mitochondrial genome. Nucleic Acids Res. 2013; 41:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reetz M.T., Kahakeaw D., Lohmer R.. Addressing the numbers problem in directed evolution. Chembiochem. 2008; 9:1797–1804. [DOI] [PubMed] [Google Scholar]
- 53. Gillam E.M.J., Copp J.N., Ackerley D.F.. Directed Evolution Library Creation: Methods and Protocols. 2014; 2nd edn, NY: Humana Press. [Google Scholar]
- 54. Rechkoblit O., Gupta Y.K., Malik R., Rajashankar K.R., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K.. Structure and mechanism of human PrimPol, a DNA polymerase with primase activity. Sci. Adv. 2016; 2:e1601317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jiang Z., Zhou X., Li R., Michal J.J., Zhang S., Dodson M.V., Zhang Z., Harland R.M.. Whole transcriptome analysis with sequencing: methods, challenges and potential solutions. Cell Mol. Life Sci. 2015; 72:3425–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Z., Gerstein M., Snyder M.. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009; 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Greenbaum D., Colangelo C., Williams K., Gerstein M.. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003; 4:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lopez-Bueno A., Rastrojo A., Peiro R., Arenas M., Alcami A.. Ecological connectivity shapes quasispecies structure of RNA viruses in an Antarctic lake. Mol. Ecol. 2015; 24:4812–4825. [DOI] [PubMed] [Google Scholar]
- 59. Eltahla A.A., Lackovic K., Marquis C., Eden J.S., White P.A.. A fluorescence-based high-throughput screen to identify small compound inhibitors of the genotype 3a hepatitis C virus RNA polymerase. J. Biomol. Screen. 2013; 18:1027–1034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.