

Abstract
Aberrant canonical NF-κB signaling is implicated in diseases from autoimmune disorders to cancer. A major therapeutic challenge is the need for selective inhibition of the canonical pathway without impacting the many non-canonical NF-κB functions. Here we show that a synthetic replacement of a loop that mediates formation of a critical regulator of canonical NF-κB signaling, the IKK complex, acts as a selective inhibitor with >10-fold increased potency over the peptide alone. Not only is this molecule, NBD2, a powerful tool for dissection of canonical NF-κB signaling in disease models and healthy tissues, the success of the synthetic loop replacement suggests that the general strategy could be useful for discovering modulators of the many protein-protein interactions mediated by such structures.
Keywords: protein-protein interactions, inhibitor, peptide loop, NEMO, canonical NF-κB signaling
Graphical Abstract
Here we show that a synthetic replacement of a loop that mediates formation of a critical regulator of canonical NF- κB signaling, the IKK complex, acts as a selective inhibitor with >10-fold increased potency over the peptide alone. Not only is this molecule, NBD2, a powerful tool for dissection of canonical NF-κB signaling in disease models and healthy tissues, the success of the synthetic loop replacement suggests that the general strategy could be broadly useful for discovering modulators of the many protein-protein interactions mediated by such structures.
Many regulatory protein-protein interactions (PPIs) operate only in certain cellular contexts, and synthetic modulators of such complexes have high intrinsic value due to their inherent context specificity.[1] Although numerous cellular signals converge on the master transcriptional activator NF-κB, for example, the PPIs formed in the so-called Inhibitor of κB Kinase (IKK) complex only control the canonical activation pathway of the activator, the path often dysregulated in inflammatory diseases and cancer (Figure 1a).[2,3] The only reported inhibitor of this complex is a peptide taken from two of the IKK components, and it selectively attenuates the canonical NF-κB pathway in vitro and in vivo, albeit with modest activity due to its short half life. [4–6] A recent elegant dissection of the IKK complex identified the linchpin as a short peptide loop stabilized by an intramolecular hydrogen bond.[7,8] Here, we show that this structural feature can be exploited by use of a Synthetic Loop Replacement (SLR) strategy to develop a synthetic inhibitor of the canonical NF-κB signaling pathway that is >10-fold more effective than the peptide itself. This structure, NBD2, directly engages the IKK component NEMO and blocks NF-κB-mediated transcription of both a reporter construct and endogenous genes.
Figure 1.

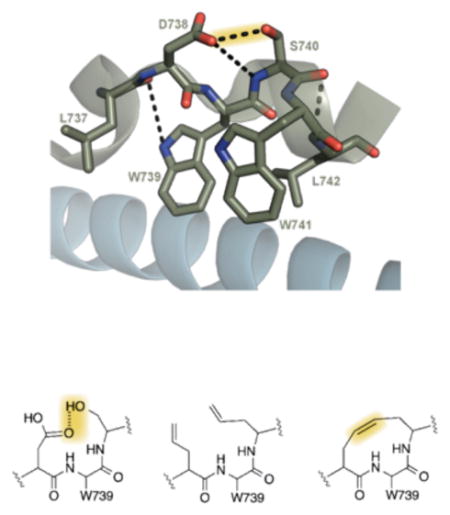
A) The signaling cascade of NF-κB can be defined by two pathways, the canonical and non-canonical pathway. The canonical pathway is tightly regulated through the assembly of the IKK complex, a heterotetramer of NEMO-IKKα-IKKβ. B) The critical assembly point of the IKK complex is mediated by 4-helix bundle where the hot-spot hydrophobic tryptophan residues of IKKα-IKKβ make critical interactions with the NEMO dimer. C) The tryptophan residues of IKKα-IKKβ are able to adopt the unique conformation needed to interact with the NEMO dimer through a hydrogen bond stabilized loop conformation between D738 and S740. D) Replacement of the critical hydrogen bond is possible through incorporation of allylglycine amino acids and subsequent metathesis, allowing us to develop a proteolytically stable inhibitor molecule of this critical PPI. PDB 3BRV
The IKK complex is a heterotetramer composed of the scaffold protein NEMO and the two kinase components IKKα and IKKβ.[9,10] NEMO is the unique component of this complex, while IKKα and IKKβ are required in crosstalk for noncanonical NF-κB signaling and other unrelated signaling pathways. [11,12]
Structural and biochemical studies have demonstrated that a six amino acid sequence (737LDWSWL742, also known as the NEMO binding domain or NBD peptide) present in both IKKα and IKKβ is critical for interaction with NEMO and subsequent activation of canonical NF-κB signaling (Figure 1b).[7,13,14] This short peptide forms a loop stabilized by a hydrogen bond bridge between D738 and S740,[8] allowing the hot spot hydrophobic residues W739 and W741 to interact with NEMO over a surface area of ~1800 Å2 (Figure 1c–d).[7] When conjugated to a cell-penetrating peptide, this six amino acid sequence selectively blocks canonical NF-κB activation (IC50 ~100 μM),[13,15–17]. We hypothesized that replacement of the hydrogen bond bridge in the NBD peptide with a non-labile connection would both stabilize this atypical secondary structure and render it less susceptible to degradation, thus producing an effective inhibitor of the NEMO-IKKα/β interaction (Figure 1e).[18–25]
Molecular dynamics (MD) simulations were used to test the initial hypothesis. As illustrated in Figure 2, simulations of the native NBD sequence produced lowest energy conformers that overlap with the reported crystal structure in the docked configuration at the position of W739 (indicated by the blue dot in Figure 2, panel A).[8] When D738 and S740 are substituted with allylglycine residues, the lowest free energy conformer is observed to have backbone dihedral angles at W739 that are dissimilar to those in the native sequence, suggesting these modifications increase the local flexibility of the modified NBD peptide (Figure 2, panel B).[7,14] However, MD simulations of a peptide in which a C-C bond replaces the loop hydrogen bond reveal that the key dihedral angles are analogous to those in the wild-type NBD peptide (Figure 2, panel C), demonstrating that the constrained version of the NBD sequence can adopt the key dihedral angles needed to interact with NEMO.
Figure 2.

Ramachandran plots generated from molecular dynamics (MD) experiments performed with the indicated peptides. Highlighted in blue, are the observed dihedral angles of hot-spot residue W739 in the NEMO-IKKα/β complex crystal structure (PDB: 3BRV). The lowest free energy conformers observed in MD simulations are indicated in dark red for A) wild-type peptide B) the peptide incorporating the allylglycine residues unmetathesized (denoted by X) and C) the peptide with allyglycine residues metathesized (denoted by interconnected X residues).
A series of NEMO-binding domain (NBD)-derived peptides were designed based upon the above results (Figure 3a). Each of the structures contains a cell penetrating peptide (CPP) previously demonstrated to be important for the function of the native peptide.[13,26] Synthesis of the peptides was accomplished by standard methods and for peptides NBD2 and NBD3, ring-closing metathesis was performed using Hoveyda-Grubbs generation II catalyst. All of the modified peptides (NBD1, 2 and 3) have circular dichroism spectra analogous to that of the wild-type sequence (Supplementary Figure 1).[27] The modification does confer significant proteolytic stability to the sequence, however. A time course experiment with NBDWT, NBD1, and NBD2 in the presence of chymotrypsin type VII (Sigma), a protease that cleaves the C-terminus of F, Y, L, and W, revealed that NBDWT and NBD1 were completely degraded after 15 minutes. Conversely, no proteolytic degradation occurred over 4h for NBD2 under identical conditions, highlighting the protection afforded to the key hot spot residues (L737, W739, W741, and L742) by the hydrocarbon bridge modification. (Supplemental Figure 2).[7] Significantly, the modification does not alter the ability of the hydrophobic hot spot residues to engage NEMO in the context of cellular lysates. As shown in Figure 3b, photoactive variants of NBDWT (bNBDWT) and NBD2 (bNBD2) capture NEMO in a pulldown experiment in HEK293T cell lysates.
Figure 3.

A) Peptides synthesized for characterization and cellular experiments. The model used to synthesize the peptides is a Cell Penetrating Peptide (CPP), a diglycine linker (GG), and the inhibitory peptide (NBD peptide). B) NEMO capture experiment performed with a biotinylated, photoreactive version of the NBDWT peptide and NBD2 peptide. Pull-down assays were performed with HeLa cellular extracts. C) Effects of the NBD peptides against an NF-κB luciferase reporter. NBD1 displayed significant toxicity and was thus excluded from further study.[28] The error is SDOM, determined from three independent experiments.
Consistent with the stability and target engagement data obtained in vitro, NBD2 is an effective inhibitor of canonical NF-κB signaling in cells. In a luciferase reporter assay, the canonical NF-κB stimulator interleukin-1 β (IL-1β)[2] potently upregulates the NF-κB-driven reporter (13-fold). Consistent with previous reports, NBDWT displays dose-dependent inhibition in this system with approximately 50% inhibition at 100 μM (Figure 3c).[13,26] In contrast, the negative control peptide (NBDMut) in which critical tryptophan residues W739 and W741 are mutated to alanine has no activity, highlighting the importance of these tryptophan hot spot residues. [7,8,14] Notably, NBD2 is approximately an order of magnitude more potent than NBDWT. Analogous to the wild type system, the W739A/W741A double mutant (NBD3) is inactive.[28] The activity of NBD2 extends to canonical NF-κB-regulated endogenous genes. MIP-3α is an NF-κB regulated gene implicated in inflammatory bowel disease,[29,30] whereas IL-8 has been identified as a critical factor required for oncogenic metastasis. [31,32]. In Figure 4, the inhibitory effects of the modified peptides on MIP-3α and IL-8 gene expression are outlined. Consistent with the reporter gene results, a 10-fold increase in potency for NBD2 compared to the unmodified NBDWT peptide was observed. Furthermore, the alanine mutant versions of the peptides (NBDMut and NBD3) displayed significantly attenuated inhibition. Consistent with selectivity for the canonical pathway, no impact on the noncanonical gene target Cyclin D1 was observed (Supplementary Figure 4).[33–38]
Figure 4.

Effects of NBD peptides against NF-κB regulated gene expression of A) MIP3α and B) IL-8. The error is SDOM, determined from three technical replicates. Three biological replicates were performed (see SI for additional data)
In summary, we implemented a Synthetic Loop Replacement (SLR) strategy to develop an inhibitor against the NEMO-IKKα/β interaction. Utilizing this strategy, we demonstrated that installation of a hydrocarbon bridge can be used to stabilize the IKK loop structure, a feature necessary for interaction with NEMO. MD simulations demonstrate that the modified NBD2 peptide adopts dihedral angles and conformations similar to the docked configuration observed in the reported crystal structure. Furthermore, we demonstrated the enhanced proteolytic stability afforded by this synthetic loop replacement modification. Collectively, these characteristics resulted in a synthetic inhibitor that was 10-fold more potent in both an NF-κB driven luciferase reporter assay and against NF-κB endogenous genes. Future structural studies of NBD2 and derivatives alone and in complex with NEMO will be key to dissecting precise role that the hydrocarbon tether plays in the protein-protein interaction, although this will be challenging given the recalcitrant nature of NEMO.[7] A unique aspect of the synthetic loop replacement is that the hydrocarbon bridge is installed in place of a polar interaction, a hydrogen bond, rendering it potentially general.[39–43] A recent study by Gavenonis et al. found 1,407 PPIs mediated by hot spot containing loop structures, analogous to the NEMO-IKK interaction.[44] A focused analysis of the identified loops presented by Gavenonis et al demonstrates that there are several loops bearing an i to i+2 hydrogen bond stabilizing feature similar to D738 and S740 in IKKα/β bound to NEMO. These identified loops should be ideal candidates for the SLR strategy presented herein (Figure 5) with potential inhibitor applications for use ranging from autoimmune diseases, anticoagulants, antivenom, and metabolic therapies. A recent study of characterized PPI interfaces found that α-helices and β-strands make up only 50% of the secondary structure observed at PPI interfaces while the other 50% of characterized PPIs are mediated by non-canonical loop regions, a class of PPIs not readily amenable to previously developed strategies.[45] This finding highlights both the opportunity and the necessity of implementing new chemical strategies such as SLR to target PPIs mediated by non-canonical loop regions.[44]
Figure 5.
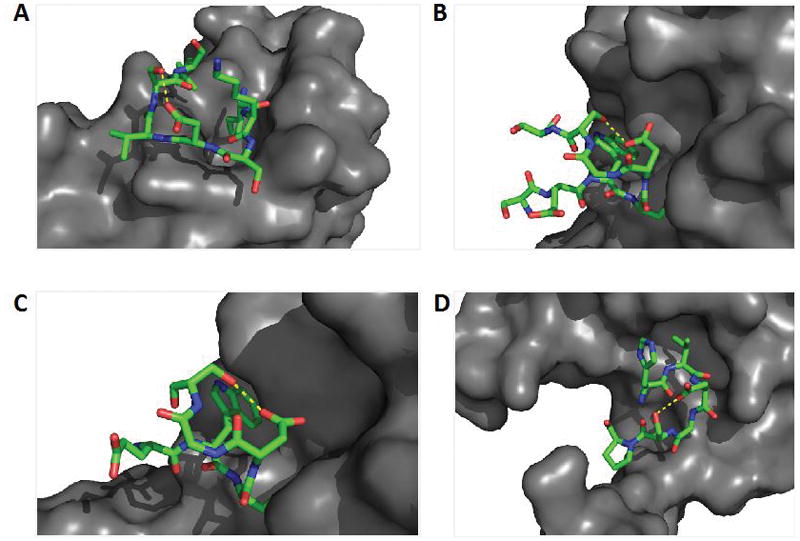
A subset of highlighted PPIs mediated by loop regions containing reported hot spot residues. This subset of highlighted loop regions are characterized by an i to i+2 hydrogen bond (yellow dashed line) between aspartic acid and serine residues that would be ideal candidates amenable to the described LBR strategy for inhibitor development. These potential LBR inhibitors could be used to a) inhibit dimerization of outer surface protein C (OspC) from Lyme disease spirochete; PDB 1F1M b) inhibit dimerization of von Willebrand Factor and Botrocetin, a snake venom protein involved in platelet aggregation; PDB 1FVU c) inhibit interaction of blood coagulation factors IX/X-binding protein (IX/X-bp), an anticoagulent isolated from habu snake venom; PDB 1IXX and d) inhibit assembly of acetoacetate decarboxylase, a major contributor to diabetic ketoacidosis; PDB 3BGT
Supplementary Material
Acknowledgments
A.K.M. gratefully acknowledges NIH CA140667 for support of this work. Support from the NIH through GM37554 is acknowledged by C.L.B.
Footnotes
Supporting information for this article is given via a link at the end of the document:
References
- 1.Garner AL, Janda KD. Curr Top Med Chem. 2011;11:258–280. doi: 10.2174/156802611794072614. [DOI] [PubMed] [Google Scholar]
- 2.Hoesel B, Schmid JA. Mol Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albert J, Baldwin S. Journal of Clinical Investigation. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villoutreix BO, Kuenemann MA, Poyet JL, Bruzzoni-Giovanelli H, Labbé C, Lagorce D, Sperandio O, Miteva MA. Mol Inform. 2014;33:414–437. doi: 10.1002/minf.201400040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makley LN, Gestwicki JE. Chem Biol Drug Des. 2013;81:22–32. doi: 10.1111/cbdd.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verdine G. Mol Cancer Ther. 2007 [Google Scholar]
- 7.Golden MS, Cote SM, Sayeg M, Zerbe BS, Villar EA, Beglov D, Sazinsky SL, Georgiadis RM, Vajda S, Kozakov D, et al. J Am Chem Soc. 2013;135:6242–6256. doi: 10.1021/ja400914z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rushe M, Silvian L, Bixler S, Chen LL, Cheung A, Bowes S, Cuervo H, Berkowitz S, Zheng T, Guckian K, et al. Structure. 2008;16:798–808. doi: 10.1016/j.str.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 9.Gasparian AV, Yao YJ, Kowalczyk D, Lyakh LA, Karseladze A, Slaga TJ, Budunova IV. Journal of Cell Science. 2002;115:141–151. doi: 10.1242/jcs.115.1.141. [DOI] [PubMed] [Google Scholar]
- 10.Zandi E, Rothwarf DM, Delhase M, Hayakawa M. Cell. 1997 doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 11.Makris C, Godfrey VL, Krähn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M. Mol Cell. 2000;5:969–979. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 12.Oeckinghaus A, Hayden MS, Ghosh S. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 13.May MJ, D’Acquisto F, Madge LA, Glöckner J, Pober JS, Ghosh S. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 14.May MJ, Marienfeld RB, Ghosh S. J Biol Chem. 2002;277:45992–46000. doi: 10.1074/jbc.M206494200. [DOI] [PubMed] [Google Scholar]
- 15.Strickland I, Ghosh S. Ann Rheum Dis. 2006;65(Suppl 3):iii75–82. doi: 10.1136/ard.2006.058438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delfín DA, Xu Y, Peterson JM, Guttridge DC, Rafael-Fortney JA, Janssen PM. J Transl Med. 2011;9:68. doi: 10.1186/1479-5876-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rehman KK, Bertera S, Bottino R, Balamurugan AN, Mai JC, Mi Z, Trucco M, Robbins PD. J Biol Chem. 2003;278:9862–9868. doi: 10.1074/jbc.M207700200. [DOI] [PubMed] [Google Scholar]
- 18.Inspired by success reported by others in the stabilization of canonical secondary structures such as α-helices and β-strands, references 19–25
- 19.Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. J Am Chem Soc. 2010;132:941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Cheng PN, Liu C, Zhao M, Eisenberg D, Nowick JS. Nat Chem. 2012;4:927–933. doi: 10.1038/nchem.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapman RN, Arora PS. Org Lett. 2006;8:5825–5828. doi: 10.1021/ol062443z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapman RN, Dimartino G, Arora PS. J Am Chem Soc. 2004;126:12252–12253. doi: 10.1021/ja0466659. [DOI] [PubMed] [Google Scholar]
- 24.Kutchukian PS, Yang JS, Verdine GL, Shakhnovich EI. J Am Chem Soc. 2009;131:4622–4627. doi: 10.1021/ja805037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schafmeister CE, Po J, Verdine GL. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]
- 26.Khaja K, Robbins P. Pharmaceuticals. 2010;3:110–124. doi: 10.3390/ph3010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenfield NJ. Nature Protocols. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The construct NBD1, which contains the unmetathesized allylglycine residues, also exhibited dose-dependent inhibition of NF-κB driven luciferase activity. However, the NBD1 construct also exhibited dose-dependent inhibition of the constitutively expressed β-Gal reporter signal consistent with off-target activities and thus, was excluded from further study.
- 29.Kwon JH, Keates S, Simeonidis S, Grall F, Libermann TA, Keates AC. J Biol Chem. 2003;278:875–884. doi: 10.1074/jbc.M208241200. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama T, Fujisawa R, Yamada H, Horikawa T, Kawasaki H, Hieshima K, Izawa D, Fujiie S, Tezuka T, Yoshie O. Int Immunol. 2001;13:95–103. doi: 10.1093/intimm/13.1.95. [DOI] [PubMed] [Google Scholar]
- 31.Kunsch C, Rosen CA. Molecular and Cellular Biology. 1993;13:6137–6146. doi: 10.1128/mcb.13.10.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee S, Kim YJ, Kwon S, Lee Y, Choi SY, Park J. BMB rep. 2009 [Google Scholar]
- 33.Perkins ND. Nat Rev Mol Cell Biol. 2007 doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 34.Wang F, Shi Y, Yadav S, Wang H. Toxicology. 2010 [Google Scholar]
- 35.Albanese C, Wu K, D’Amico M, Jarrett C, Joyce D, Hughes J, Hulit J, Sakamaki T, Fu M, Ben-Ze’ev A, et al. Mol Biol Cell. 2003;14:585–599. doi: 10.1091/mbc.02-06-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Westerheide SD, Mayo MW, Anest V, Hanson JL, Baldwin AS. Molecular and Cellular Biology. 2001;21:8428–8436. doi: 10.1128/MCB.21.24.8428-8436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park KJ, Krishnan V, O’Malley BW, Yamamoto Y, Gaynor RB. Mol Cell. 2005;18:71–82. doi: 10.1016/j.molcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Warren MA, Shoemaker SF, Ip MM. Endocrinology. 2007;148:268–278. doi: 10.1210/en.2006-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.For previous work replacing nonpolar interactions with hydrocarbon tethers see references 40–42
- 40.Glas A, Bier D, Hahne G, Rademacher C, Ottmann C, Grossmann TN. Angew Chem Int Ed Engl. 2014;53:2489–2493. doi: 10.1002/anie.201310082. [DOI] [PubMed] [Google Scholar]
- 41.Shin Youngsook, Winans Katharine A, Backes Bradley J, Kent Stephen BH, Ellman A Jonathan A, Bertozzi Carolyn R. J Am Chem Soc. 1999;121:11864–11689. [Google Scholar]
- 42.Reichwein JF, Versluis C, Liskamp RM. J Org Chem. 2000;65:6187–6195. doi: 10.1021/jo000759t. [DOI] [PubMed] [Google Scholar]
- 43.Miles SM, Leatherbarrow RJ, Marsden SP, Coates WJ. Org Biomol Chem. 2004;2:281–283. doi: 10.1039/b312908j. [DOI] [PubMed] [Google Scholar]
- 44.Gavenonis J, Sheneman BA, Siegert TR, Eshelman MR, Kritzer JA. Nat Chem Biol. 2014;10:716–722. doi: 10.1038/nchembio.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guharoy M, Chakrabarti P. Bioinformatics. 2007;23:1909–1918. doi: 10.1093/bioinformatics/btm274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.