

Abstract
Munc18-1/UNC-18 is believed to prime SNARE-mediated membrane fusion, yet the underlying mechanisms remain enigmatic. Here, we examine how potential gain-of-function mutations of Munc18-1/UNC-18 affect locomotory behavior and synaptic transmission, and how Munc18-1-mediated priming is related to Munc13-1/UNC-13 and Tomosyn/TOM-1, positive and negative SNARE regulators, respectively. We show that a Munc18-1(P335A)/UNC-18(P334A) mutation leads to significantly increased locomotory activity and acetylcholine release in Caenorhabditis elegans, as well as enhanced synaptic neurotransmission in cultured mammalian neurons. Importantly, similar to tom-1 null mutants, unc-18(P334A) mutants partially bypass the requirement of UNC-13. Moreover, unc-18(P334A) and tom-1 null mutations confer a strong synergy in suppressing the phenotypes of unc-13 mutants. Through biochemical experiments, we demonstrate that Munc18-1(P335A) exhibits enhanced activity in SNARE complex formation as well as in binding to the preformed SNARE complex, and partially bypasses the Munc13-1 requirement in liposome fusion assays. Our results indicate that Munc18-1/UNC-18 primes vesicle fusion downstream of Munc13-1/UNC-13 by templating SNARE complex assembly and acts antagonistically with Tomosyn/TOM-1.
SIGNIFICANCE STATEMENT At presynaptic sites, SNARE-mediated membrane fusion is tightly regulated by several key proteins including Munc18/UNC-18, Munc13/UNC-13, and Tomosyn/TOM-1. However, how these proteins interact with each other to achieve the precise regulation of neurotransmitter release remains largely unclear. Using Caenorhabditis elegans as an in vivo model, we found that a gain-of-function mutant of UNC-18 increases locomotory activity and synaptic acetylcholine release, that it partially bypasses the requirement of UNC-13 for release, and that this bypass is synergistically augmented by the lack of TOM-1. We also elucidated the biochemical basis for the gain-of-function caused by this mutation. Thus, our study provides novel mechanistic insights into how Munc18/UNC-18 primes synaptic vesicle release and how this protein interacts functionally with Munc13/UNC-13 and Tomosyn/TOM-1.
Keywords: C. elegans, exocytosis, membrane fusion, Munc18, SNARE, synapse
Introduction
At synapses, electrical activity in the presynaptic neuron triggers rapid release of neurotransmitters in a Ca2+-dependent manner. The neuronal SNARE complex, consisting of syntaxin-1, SNAP-25, and synaptobrevin/VAMP-2, plays a central role in mediating neurotransmitter release through membrane fusion (exocytosis; Söllner et al., 1993). This process is regulated by key proteins such as Munc18-1 (Hata et al., 1993; Verhage et al., 2000), Munc13 (Brose et al., 1995; Augustin et al., 1999), and Tomosyn (Fujita et al., 1998; McEwen et al., 2006). However, the precise regulatory mechanisms of exocytosis remain largely unclear.
Munc18-1, which belongs to the Sec1p/Munc18 (SM) family proteins, plays a critical role in synaptic exocytosis across species (Hosono et al., 1992; Harrison et al., 1994; Verhage et al., 2000; Voets et al., 2001; Weimer et al., 2003). Two main roles of Munc18-1 have been proposed to explain its essential function: chaperoning/trafficking of syntaxin-1 (Rowe et al., 1999; Arunachalam et al., 2008; McEwen and Kaplan, 2008) and activation of SNARE-mediated fusion (“priming”; Südhof and Rothman, 2009). While the former role is primarily mediated by an interaction between Munc18-1 and “closed” syntaxin-1 (L. Han et al., 2009; G. A. Han et al., 2011, 2014), the latter requires binding of Munc18-1 to the SNARE complex (Dulubova et al., 2007; Shen et al., 2007; Han et al., 2013). These two binding modes may not be mutually exclusive since SNARE complex assembly can start from the “closed” syntaxin-1–Munc18-1 complex, proceeding with the help of Munc13 in an N-ethylmaleimide sensitive factor (NSF)-soluble NSF attachment protein (SNAP)-resistant manner (Ma et al., 2013).
Several lines of evidence suggest that domain-3a of Munc18-1 plays a critical role in the transition between the two modes. Biochemical studies illustrated a direct interaction between the domain-3a and synaptobrevin, such that Munc18-1 can interact with the SNARE complex via this domain (Xu et al., 2010; Parisotto et al., 2014). In addition, the yeast SM protein Vps33 binds to the yeast homologues of synaptobrevin and syntaxin-1 at nearby sites involving domain-3a, suggesting that Vps33 and SM proteins generally act as templates to assemble SNARE complex (Baker et al., 2015). Indeed, Munc18-1 domain-3a mutants lose the ability to support exocytosis, despite retaining the ability to support syntaxin-1 chaperoning and docking of vesicles, indicating a postdocking or priming function for this domain (Han et al., 2013; Martin et al., 2013). Furthermore, a structural study revealed that a helical hairpin in domain-3a can adopt either bent or extended conformations, with the highly conserved Pro335 residue at the hinge point of these two conformations (Hu et al., 2011). Thus, these two conformations of domain-3a may allow Munc18-1 to transition from binding syntaxin-1 to the SNARE complex. This hypothesis has been supported by the finding that the P335A mutant, which was predicted to adopt the extended conformation, showed an increased activity in liposome fusion (Parisotto et al., 2014), as well as in dense-core vesicle secretion in both PC12 (Han et al., 2014) and chromaffin cells (Munch et al., 2016). This model and the notion that SM proteins function as a template for SNARE complex assembly have been further supported by previous studies with additional Munc18-1 domain-3a mutants (Sitarska et al., 2017).
However, many questions remain unanswered regarding the priming function of Munc18-1, including the effects of the P335A mutation. Specifically, (1) does the P335A mutation affect in vivo SNARE-mediated synaptic vesicle release and the behavior of animals? (2) How is the priming function of Munc18-1/UNC-18 related to two other key regulators of SNARE proteins, Munc13-1/UNC-13 and Tomosyn? (3) What is the biochemical basis of the gain-of-function effects of P335A on exocytosis? To address these, we examined the physiological function of mutant Munc18-1(P335A) and the corresponding mutant UNC-18(P334A) with respect to behavior and synaptic acetylcholine release using Caenorhabditis elegans. We further investigated genetic interactions among unc-18, unc-13, and tom-1 mutants. Finally, we examined the interactive properties of the mutant Munc18-1 protein with the SNAREs to explain the biochemical aspects underlying such gain-of-function effects.
Materials and Methods
Maintenance and generation of C. elegans strains.
All the strains used in the present study originated from the Bristol N2. They were maintained at 22°C on nematode growth medium (NGM) plates seeded with OP50, a strain of Escherichia coli. Nonintegrated transgenic lines were generated by injection of constructs into gonads. pMunc18-1 and punc-18 (kind gifts from Dr. Hitoshi Kitayama, Kyoto University, Kyoto, Japan) described in the study by Gengyo-Ando et al. (1996) were used as templates to generate the rescue constructs that express Munc18-1 and UNC-18 variants. These plasmids contained ∼3 kb of unc-18 promoter, ∼1.8 kb cDNA of Munc18-1 or UNC-18 variant, and ∼1 kb of 3′ poly(A) signal of the unc-18 gene (Gengyo-Ando et al., 1996). A mixture of each Munc18-1 or UNC-18 expression plasmid and a coinjection maker (Pmyo3::RFP::unc-54) was prepared such that the final concentration of each DNA was ∼50 ng/ul and injected into the gonads of young adult worms of the CB81 strain. Three to 4 d after the injection, the F1 generation was screened for red fluorescence, and only RFP-positive F1 worms were singled out. Integration of transgenes into the genome was achieved using a UV cross-linker (GS Gene-Linker, Bio-Rad). All new strains and transgenes are listed in Table 1.
Table 1.
C. elegans transgenes, alleles, and strains
| Transgene | DNA constructs | Genotype | Strain # |
|---|---|---|---|
| sksEx1 | Punc-18-Munc18-1 WT (cDNA) | unc-18 (e81) X | UHN1 |
| sksEx2 | Punc-18-Munc18-1 WT (cDNA) | unc-18 (e81) X | UHN2 |
| sksEx3 | Punc-18-Munc18-1 WT (cDNA) | unc-18 (e81) X | UHN3 |
| sksEx4 | Punc-18-Munc18-1 P335A (cDNA) | unc-18 (e81) X | UHN4 |
| sksEx5 | Punc-18-Munc18-1 P335A (cDNA) | unc-18 (e81) X | UHN5 |
| sksEx6 | Punc-18-Munc18-1 WT::GFP (cDNA) | unc-18 (e81) X | UHN6 |
| sksEx7 | Punc-18-Munc18-1 WT::GFP (cDNA) | unc-18 (e81) X | UHN7 |
| sksEx8 | Punc-18-Munc18-1 P335A::GFP (cDNA) | unc-18 (e81) X | UHN8 |
| sksEx9 | Punc-18-Munc18-1 P335A::GFP (cDNA) | unc-18 (e81) X | UHN9 |
| sksIs1 | Punc-18-Munc18-1 WT::GFP (sksEx6) | UHN10 | |
| sksIs2 | Punc-18-Munc18-1 P335A::GFP (sksEx8) | UHN11 | |
| unc-18(e81)X; Punc-18-Munc18-1 WT::GFP (sksIs1) | UHN12 | ||
| unc-18(e81)X; Punc-18-Munc18-1 P335A::GFP (sksIs2) | UHN13 | ||
| unc-18(md299)X; Punc-18-Munc18-1 WT::GFP (sksIs1) | UHN14 | ||
| unc-18(md299)X; Punc-18-Munc18-1 P335A::GFP (sksIs2) | UHN15 | ||
| sksIs3 | Punc-18-unc-18 WT (cDNA) | unc-18(md299)X; Punc-18-unc-18 WT | UHN16 |
| sksIs4 | Punc-18-unc-18 P334A (cDNA) | unc-18(md299)X; Punc-18-unc-18 P334A | UHN17 |
| unc-13(s69)I; Punc-18-Munc18-1 P335A::GFP (sksIs2) | UHN18 | ||
| unc-13(s69)I; Punc-18(md299)X; Punc-18-unc-18 WT (sksIs3) | UHN19 | ||
| unc-13(s69)I; Punc-18(md299)X; Punc-18-unc-18 P334A (sksIs4) | UHN20 | ||
| unc-18(sks1[P334A + loxP Pmyo-2-GFP; Prps-27-NeoR loxP])X | UHN21 | ||
| unc-18(sks2[P334A + loxP])X | UHN22 | ||
| tom-1(ok285)I; unc-18(sks2)X | UHN23 | ||
| tom-1(ok285)I; unc-13(s69)I | UHN24 | ||
| unc-13(s69)I; unc-18(sks2)X | UHN25 | ||
| tom-1(ok285)I; unc-13(s69)I; unc-18(sks2)X | UHN26 | ||
| unc-18(sks2)X; unc-49(e407)III | UHN27 |
Behavioral analysis.
The motility of C. elegans was evaluated by counting the number of thrashings per min in liquid medium. One-day-old adult worms free of OP50 bacteria were first transferred to a nonseeded plate containing 1 ml of M9 buffer. After worms were given a 2 min adjustment period in M9 buffer, they were video recorded for 1 min. A single trashing was defined as a bending at the midbody, with both head and tail pointing to the opposite direction of midbody movement.
For analysis of locomotion speed, 1-d-old adult C. elegans worms were transferred to fresh OP50 NGM plates and allowed to recover for 2 min. Their movement was recorded for 10 min using an OMAX A3580U camera and the OMAX ToupView program. At the beginning of each recording, a ruler was imaged so that calibration could be performed later during analysis. Video recordings were analyzed using the wrMTrck plug-in in ImageJ. Speed was calculated by dividing the distance traveled over the time tracked by the software.
Aldicarb assays.
The sensitivity of C. elegans to aldicarb was tested by examining 1-d-old adult worms on nonseeded NGM plates containing 1 mm of aldicarb. During the assays, animals were assessed for paralysis at multiple time points. A worm was considered paralyzed if the tail did not move after the head was tapped three times.
Western blot analysis of C. elegans worms expressing mammalian Munc18-1 proteins.
Protein extract was prepared as described previously by Weimer et al. (2003). Briefly, we plated ∼50 RFP (coinjected marker protein)–positive unc-18(e81) mutants expressing Munc18-1 proteins on 3.5 cm NGM plates and cultured them while manually removing RFP-negative offspring worms. As controls, we also cultured unc-18(e81). When the plates were full of worms with little OP50 left, worms were harvested and washed three times with a buffer containing 360 mm sucrose, 12 mm HEPES, and a protease inhibitor cocktail (1 μg/ml pepstatin A, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 0.1 mm PMSF). These worms were resuspended in ∼5 times volume of the buffer and frozen at −80°C until use. The defrosted worms were sonicated on ice 10 times with a 5 s burst. The resulting lysate was centrifuged for 15 min to pellet the cuticle, nuclei, and other debris. After centrifugation, the supernatant (final protein concentrations were ∼3–5 mg/ml) was transferred to a clean microcentrifuge tube with an equal volume of sample buffer (2X). Fifty micrograms of samples were subjected to SDS-PAGE electrophoresis followed by immunoblotting. The expression of transgenic Munc18-1 was detected by anti-Munc18-1 monoclonal antibody (1:1000) from BD Biosciences.
CRISPR-mediated genome editing.
First, a two-step PCR was performed to generate a DNA product encoding single guide RNA (sgRNA) that would target a site within the unc-18 genomic region. Second, a repair vector was constructed that contained a dual-marker selection cassette (GFP and neomycin-resistant gene; Norris et al., 2015) flanked by left and right homology arms of unc-18. Upstream (left arm) and downstream (right arm) sequences of the CRISPR target site were amplified by PCR using pCFJ676, which contains the genomic sequence of the unc-18 gene (purchased from Addgene). The left arm PCR product was digested with EcoRI and XhoI, while the right arm PCR product was digested with NotI and SacII. Each of the digested products was ligated into pBluescript, followed by sequencing of the ligated products. To generate a knock-in (KI) mutant allele, site-directed mutagenesis was conducted on pBluescript containing the right arm; the protospacer adjacent motif site was also mutated so that the repair template would be refractory to the action of Cas9. The mutagenesis product was verified by sequencing. The dual-marker selection cassette digested with SpeI and NotI was ligated into pBluescript containing the left arm. In the meantime, the right arm harboring the mutation of interest was digested with NotI and SacII and then ligated into the vector containing the left arm and the dual-marker selection cassette. Third, multiple plasmids were injected into N2 animals: a vector encoding Cas9, the repair vector, the DNA product of sgRNA, and several injection markers (Pmyo-3-mCherry and Prab-3-mCherry). Fourth, screening for knock-in animals began by applying neomycin to the progeny of the injected animals. Then, animals that were neomycin resistant, GFP positive, and mCherry negative were selected as knock-in candidates and genotyped for verification.
Measurement of body length, area, and brood size.
After taking images of 1-d-old worms on bacterial lawn, they were analyzed using ImageJ. First, the backgrounds of images were subtracted, followed by adjustment of the threshold. This adjustment converted images to black and white binary ones, which were further skeletonized. Skeletonized worms were selected and measured for their perimeters, which were divided by 2 to calculate the lengths. For area measurement, worms were selected in binary images and analyzed for area.
To determine the brood size, L4 worms of each line were singled out. For 3 to 4 consecutive days, they were transferred to new plates, and the numbers of progeny they produced were counted.
Electrophysiological recordings from the neuromuscular junction in C. elegans.
The dissection of the C. elegans was described previously (Richmond and Jorgensen, 1999; Gao and Zhen, 2011). Briefly, 1- to 2-d-old hermaphrodite adults were glued to a Sylgard-coated cover glass covered with bath solution. Animals were immobilized on Sylgard 184 Silicone Elastomer (Dow Corning)–coated glass coverslips using tissue adhesive glue (Histoacryl Blue, Braun). The curved dorsal side of the animal was dissected using a sharpened tungsten or glass needle. After clearing the viscera by suction through a glass pipette, the cuticle flap was turned and gently glued down using WORMGLU (GluStitch) to expose the neuromuscular system. The muscle cells were patched using fire-polished 4–6 MΩ resistant borosilicate pipettes (World Precision Instruments). Membrane currents were recorded in the whole-cell configuration using a EPC-9 Patch-Clamp Amplifier (HEKA Elektronik). Data were digitized at 10 kHz and filtered at 2.6 kHz.
The recording solutions were as described in our previous studies (Richmond and Jorgensen, 1999). Specifically, the pipette solution contained the following (in mm): 115 K-gluconate, 25 KCl, 0.1 CaCl2, 5 MgCl2, 1 BAPTA, 10 HEPES, 5 Na2ATP, 0.5 Na2GTP, 0.5 cAMP, and 0.5 cGMP, pH 7.2 with KOH, ∼320 mOsm. The bath solution consisted of the following (in mm): 150 NaCl, 5 KCl, 5 CaCl2, 1 MgCl2, 10 glucose, 5 sucrose, and 15 HEPES, pH 7.3 with NaOH, ∼330 mOsm. Leak currents were not subtracted. All chemicals were from Sigma-Aldrich. Experiments were performed at room temperature (20−22°C). Subsequent analysis and graphing were performed using Excel (Microsoft), Igor Pro (Wavemetrics), and Clampfit (Molecular Devices). In this experiment, N refers to the number of recordings (one cell per animal). All data are presented as mean ± SEM.
Lipid and content mixing assays.
The assays were performed as described previously (Liu et al., 2016). Briefly, V liposomes with reconstituted rat synaptobrevin-2 (protein-to-lipid ratio, 1:500) contained 39% 1-Palmitoyl, 2-oleoyl phosphatidylcholine (POPC), 19% 1,2-Dioleoyl phosphatidylserine (DOPS), 19% 1-palmitoyl-2-oleoyl-sn-phosphatidylethanolamine (POPE), 20% cholesterol, 1.5% N-(7-Nitrobenz-2-Oxa-1,3-Diazol-4-yl)-1,2-Dihexadecanoyl-sn-Glycero-3-Phosphoethanolamine (NBD PE), and 1.5% Marine Blue Phosphatidylethanolamine (PE). T liposomes with reconstituted rat syntaxin-1A and rat SNAP-25A (protein-to-lipid ratio, 1:800) contained 38% POPC, 18% DOPS, 20% POPE, 20% cholesterol, 2% Phosphatidylinositol 4,5-bisphosphate (PIP2), and 2% Diacylglycerol (DAG). The liposomes were prepared in 25 mm HEPES, pH 7.4, 150 mm KCl, 0.5 mm Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and 10% glycerol (v/v). V liposomes (0.125 mm lipids) were mixed with T liposomes (0.25 mm lipids) in a total volume of 200 μl in the presence of 2.5 mm MgCl2, 2 mm ATP, 0.1 mm EGTA, 5 μm streptavidin, 0.8 μm Cricetulus griseus NSF V155M mutant, 2 μm Bos taurus αSNAP, 1 μm rat Munc18-1 [wild-type (WT) or P335A mutant], 1 μm rat synaptotagmin-1 C2AB (residues 131–421), and 1 μm excess SNAP-25 with or without 0.5 μm rat Munc13-1 C1C2BMUNC2C (residues 529–1735, with residues 1408–1452 replaced by EF). Before mixing, T liposomes were incubated with NSF, MgCl2, ATP, EGTA, streptavidin, NSF, αSNAP, and Munc18-1 at 37°C for 25 min. CaCl2 at a final concentration of 0.6 mm was added 300 s after the start of the reaction. A PTI spectrofluorometer was used to measure lipid mixing from dequenching of the fluorescence of Marina Blue–labeled lipids (excitation at 370 nm, emission at 465 nm) and content mixing from the development of FRET between PhycoE-Biotin trapped in the T liposomes and Cy5-streptavidin trapped in the V liposomes (PhycoE-biotin excitation at 565 nm, Cy5-streptavidin emission at 670 nm). All experiments were performed at 30°C. At the end of each reaction, 1% w/v octyl-beta-D-glucopyranoside (β-OG) was added to solubilize the liposomes, and all the lipid mixing data were normalized to the maximum fluorescence signal achieved after addition of β-OG. Control experiments without streptavidin were performed to measure the maximum Cy5 fluorescence attainable upon detergent addition.
Purification of recombinant proteins for binding experiments.
The expression plasmids used were described previously (Han et al., 2013). Several colonies of transformed bacteria were grown in a small volume of Lysogeny broth (LB)-ampicillin medium at 37°C overnight. The next day, these bacteria were further grown in a larger volume at 37°C for 3 h. Expression of recombinant proteins was induced by adding 125 μm isopropylthio-β-d-galactoside at 15°C overnight. The next day, this culture was centrifuged at 3000 rpm for 10 min at 4°C using a JA-14 rotor (Beckman). The pellet expressing GST–syntaxin-1A or strep-SNAP-25 was resuspended in PBS containing 1 mm PMSF and 1 mm EDTA. The pellet expressing His-tagged proteins was resuspended in a lysis buffer containing 20 mm Tris-HCl, 300 mm NaCl, 0.4% NP-40, 10 mm imidazole, 10% glycerol, 10 mm 2-mercaptoethanol, 1 mm PMSF, and 20 ng/ml DNase. Bacterial cell lysis was performed by applying a cell pressure of 1000 psi for 30 s three times using a French pressure cell press (Thermo Fisher Scientific). The lysate was then centrifuged at 15,000 rpm for 30 min at 4°C using a JA-21 rotor. The lysed product containing GST–syntaxin-1A was mixed with glutathione agarose beads (Pierce Biotechnology), the lysate containing strep-SNAP-25 with Strep-Tactin Sepharose (IBA Lifesciences), and the lysates containing His-tagged Munc18-1 proteins and His-tagged synaptobrevin with HisPur Ni-NTA resin (Thermo Fisher Scientific). The lysates were mixed with the beads on a rotator at 4°C for 2 h. Subsequent washing with GST or strep-tagged proteins was performed with PBS containing 1 mm PMSF and 1 mm EDTA. Washing for the His-tagged proteins was performed first using high-salt buffer (20 mm Tris-HCl, 500 mm NaCl, 20 mm imidazole, 10 mm 2-mercaptoethanol, 10% glycerol) followed by midsalt buffer (same contents as high-salt buffer but with 300 mm NaCl). To achieve tag-free syntaxin-1A, the GST moiety was removed from GST–syntaxin-1A recombinant proteins by thrombin cleavage for 2–4 h at room temperature. After centrifugation, the cleaved proteins were eluted in PBS containing PMSF. His-tagged recombinant Munc18 and synaptobrevin proteins immobilized on HisPur Ni-NTA resin were eluted in elution buffer (20 mm Tris-HCl, 150 mm NaCl, 250 mm imidazole, 10 mm 2-mercaptoethanol, 10% glycerol). SNAP-25 immobilized on Strep-Tactin Sepharose was also eluted in an elution buffer containing desthiobiotin.
Pulldown assays for analyzing SNARE assembly and interaction of SNARE complex with Munc18-1.
To examine SNARE assembly, GST–syntaxin-1A was incubated with His-tagged wild-type or mutant Munc18-1 in PBS containing EDTA, PMSF, and 0.1% Triton X-100 for 30 min. Then, eluted His-tagged synaptobrevin and strep-SNAP-25 were added and mixed with the incubated products for 1 h. After extensive washing with a PBS solution containing PMSF, EDTA, and 0.1% Triton X-100, sample buffer was added to the mixed products, which was followed by SDS-PAGE and Coomassie blue staining.
To examine the interaction of Munc18-1 with the preformed SNARE complex, cleaved recombinant syntaxin-1A (1–264), eluted His-tagged synaptobrevin (1–94), and SNAP-25 immobilized on Strep-Tactin Sepharose were incubated in a PBS solution containing PMSF and EDTA at room temperature overnight. On the next day, the SNARE complex immobilized on Strep-Tactin Sepharose was pelleted by centrifugation. After resuspending the pellet in a PBS solution containing PMSF, EDTA, and 0.1% Triton X-100, it was divided into three groups; one group was incubated with His-tagged wild-type Munc18-1, another with His-tagged mutant Munc18-1, and the other without any Munc18-1 proteins for 2–4 h at room temperature. The Munc18-1–SNARE complex immobilized on the Sepharose was washed multiple times with a PBS solution containing PMSF, EDTA, and 0.1% Triton X-100. After pelleting the Munc18-1–SNARE complex by centrifugation, sample buffer was added to the pellet, which was then run on SDS-PAGE gels and stained with Coomassie blue.
In vitro primary neuron preparation and electrophysiology using WT CD1 mice.
Embryonic cortical cultures were prepared from CD1 mice of both sexes at embryonic day 16 (E16). Cortices were digested with 0.025% trypsin/EDTA at 37°C for 15 min. Cell density was determined using an improved Neubauer hemocytometer, and 1.0 × 104 cells were plated on poly-d-lysine-coated glass coverslips (Sigma; 12 mm No. 1 German glass, Bellco, catalog #1943-10012). The cells were kept in 5% CO2 at 37°C in serum-free culture medium (Neurobasal medium supplemented with 1.8% B-27 (Invitrogen), 0.25% GlutaMAX, and 1% penicillin-streptomycin). Cells were infected on DIV 1 (4 h after plating) and exposed to the lentiviral vectors (either GFP, Munc18-1-wt-GFP, or Munc18-1-P335A-GFP) for 3 h, after which the medium was replaced. To assess infection efficiency, cells at DIV 16 were fixed with 4% paraformaldehyde for 15 min and mounted on slides with ProLong Gold antifade reagent (P36930, Thermo Fisher Scientific). Cells were imaged with a Zeiss LSM 700 confocal laser scanning microscope (Zeiss) using a 40× oil lens to examine GFP expression. To record spontaneous postsynaptic currents (PSCs), slides with nonfixed cells (DIV 16) were transferred to a submerged recording chamber at room temperature and perfused with bath (extracellular) solution, which contained the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 20 HEPES, and 10 glucose, pH 7.4 and osmolarity ∼300 mOsm (adjusted with NaOH and sucrose, respectively). To assess the presynaptic effects of Munc18-1 expression, all recordings were obtained from the corresponding GFP-negative neurons in each group. Cells were visualized on an Olympus CKX41 inverted fluorescence microscope. Pipette resistance was between 3 and 6 MΩ after filling the with pipette (intracellular) solution, which contained the following (in mm): 145 Cesium methanesulfonate (CsMSF), 1 MgCl2, 10 HEPES, 10 EGTA, and 5 MgATP, pH adjusted to 7.2 with CsOH (Yan et al., 2013). Patch-clamp gap-free recording was done in the whole-cell configuration using an Axopatch 700B (Molecular Devices) with the pClamp 9.2 software (Molecular Devices) at 2 kHz (digitized at 5 kHz). Analyses were done with the Mini Analysis Program (Synaptosoft) and Clampfit 9.2 (Molecular Devices).
Generation of Munc18-1 floxed mice.
Two targeted ES cell lines (D12, H11), which are derived from C57BL/6N mice, of Stxbp1tm1a(EUCOMM)Hmgu were obtained from the European Conditional Mouse Mutagenesis (EUCOMM) Program (Munich, Germany). D12 ES cells generated chimeric mice that exhibited germline transmission. Munc18-1 floxed mice were generated at the Toronto Center for Phamacogenetics using a standard protocol.
Patch-clamp recordings from cultured hippocampal neurons from Munc18-1 floxed mice.
Hippocampi were dissected out from postnatal day 0 (P0)–P1 Munc18-1 floxed mice of both sexes. Neurons were prepared and cultured as described previously (Kavalali et al., 1999) with a little modification. Briefly, neurons were dissociated by treatment with trypsin (10 mg/ml) and DNase (0.25 mg/ml) for 10 min at 37°C, were triturated with a P200 pipette tip attached onto a P1000 pipette tip, and were then plated onto 12 mm coverslips coated with Matrigel (BD Biosciences) plus poly-d-lysine. The typical plating density was two hippocampi per six coverslips. Culture media consisted of minimal essential media, 5 mg/ml glucose, 0.1 mg/ml transferrin, 0.5 mm GlutaMAX, 5% iron supplemented calf serum (Hyclone), 2% B-27 supplement, 1 μm cytosine arabinoside, 50 units/ml of penicillin, and 50 μg/ml of streptomycin. Cultures were maintained at 37°C in a humidified incubator gassed with 95% air and 5% CO2. Cells were infected with lentiviral particles that coexpressed GFP, WT Munc18-1-GFP or P335A Munc18-1-GFP and Cre recombinase at DIV 3.
A coverslip was placed on a recording chamber with an extracellular solution that contained the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 20 HEPES, and 10 glucose, pH 7.4, with osmolarity ∼300 mOsm. Recording electrodes were made with thin-wall borosilicate glass tubes (1.5 mm diameter; World Precision Instruments) using a two-stage puller (Model PC-10; Narishige). Electrodes for whole-cell patch-clamp recordings were filled with a solution that contained 160 mm potassium gluconate, 2 mm HEPES, and 0.1 mm EGTA (pH 7.28; 292 mOsm; pipette resistance, ∼5 MΩ). Recording was performed at room temperature under visualization of neurons with an upright microscope (Axioscope, Zeiss) equipped with a 40× water-immersion objective. Signals were recorded via a dual-channel amplifier (700A, Molecular Devices). Data acquisition, storage, and analysis were done using PClamp software (version 9, Molecular Devices). These signals were recorded in frequencies of 0–5 kHz and digitized at 50 kHz (Digidata 1320, Molecular Devices). Only neurons that had resting membrane potentials more negative than −50 mV with series resistance of ≤20 MΩ after whole-cell configuration were used for analysis. Spontaneous excitatory postsynaptic currents were recorded by holding voltage at −70 mV at least 10 min.
For data analysis, recording traces were first 60 Hz cycle noise filtered by Clampfit (version 9, Molecular Devices) and then were processed in MATLAB 2016b (MathWorks). Custom functions were written to automatically count excitatory postsynaptic currents that were more than a noise level (∼5 pA). The occurrence of events was expressed as a cumulative probability as well as number of occurrences per 10 s.
Statistical analysis.
For two-group experiments, independent t tests were conducted, in which a level of difference was considered significant if p was <0.05. For comparison of multiple groups, one-way ANOVA followed by Tukey's range test was conducted, with a significance level of 0.05. All statistical tests were conducted using OriginPro 9.0.
Results
Conformational change of a hairpin in domain-3a of Munc18-1 confers phenotypes consistent with increased synaptic activity in vivo
The P335A mutation was predicted to favor an extended conformation of the domain-3a loop of Munc18-1 over the bent conformation favored by WT Munc18-1 (Hu et al., 2011). Previous studies have demonstrated that introduction of this mutation causes increased SNARE-mediated fusion in liposome fusion assays and in dense core vesicle exocytosis from both PC12 cells (Han et al., 2014) and chromaffin cells (Munch et al., 2016). To examine how the mutation affects synaptic function in vivo, we compared the ability of Munc-18–1(P335A) and WT Munc18-1 to rescue the phenotype of C. elegans unc-18(e81) mutants (Brenner, 1974; Hosono et al., 1992; Fig. 1). The e81 allele contains a premature stop codon mutation and thus is likely null (Sassa et al., 1999; Weimer et al., 2003). We used rescue constructs similar to those used by Gengyo-Ando et al. (1996), who demonstrated that Munc18-1, but not Munc18-3, can partially rescue paralysis of unc-18(e81).
Figure 1.
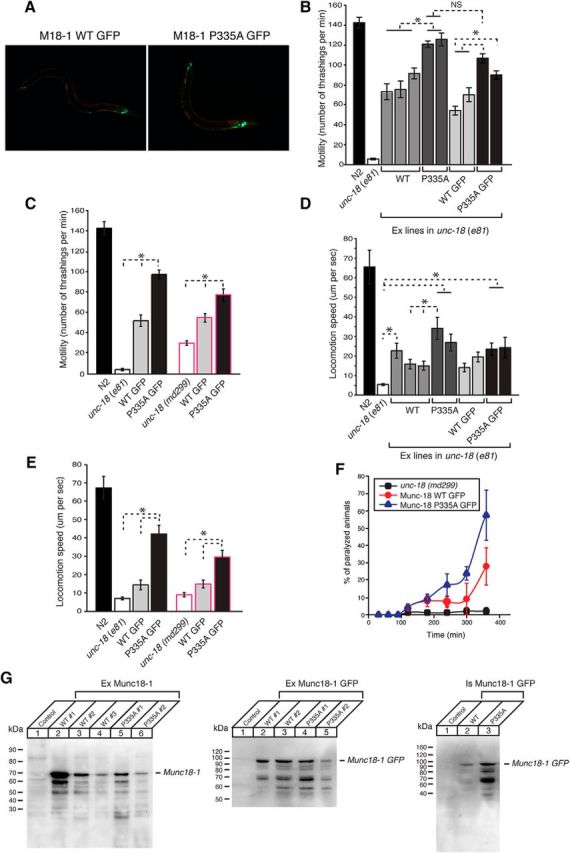
Munc18-1(Pro335A) enhances the protein's ability to rescue the defects associated with unc-18 severe loss-of-function mutants. A, GFP-tagged wild-type (left) and mutant (right) Munc18-1 proteins are expressed under the control of the unc-18 promoter. mCherry (red) is expressed in body wall muscles only. B, The motility of C. elegans is measured by counting the number of thrashings per min in liquid. unc-18 mutants expressing multiple copies of mutant Munc18-1 exhibit higher thrashing rates than those expressing multiple copies of wild-type Munc18-1, regardless of whether or not Munc18-1 is GFP tagged. n = 10–25. In one-way ANOVA statistical tests, F(4,53) = 12.7 and p = 2.50E-7 for comparison between non-GFP constructs, F(3,50) = 23.2 and p = 1.5E-9 for comparison between GFP-tagged constructs, and F(3,48) = 17.0 and p = 1.2E-7 for comparison between non-GFP mutant Munc18-1 and GFP-tagged mutant Munc18-1. C, Stable expression of mutant Munc18-1 in two independent unc-18 mutant backgrounds leads to higher motility of C. elegans than that of wild-type Munc18-1. n = 12–16. In one-way ANOVA statistical tests, F(2,46) = 209.5 and p = 0 for the e81 background, and F(2,47) = 37.2 and p = 2.1E-10 for the md299 background. D, unc-18 mutants expressing multiple copies of mutant Munc18-1 move at higher speeds on a bacterial lawn than those expressing wild-type Munc18-1. n = 14–17. In a one-way ANOVA statistical test, F(9,144) = 4.9 and p = 1.0E-5. E, Stable expression of mutant Munc18-1 in two independent unc-18 mutant backgrounds causes C. elegans to move at higher speeds than that of wild-type Munc18-1. n = 15–18. In one-way ANOVA statistical tests, F(2,45) = 34.6 and p = 7.9E-10 for the e81 background, and F(2,43) = 17.5 and p = 2.7E-6 for the md299 background. F, Aldicarb assays were conducted to assess the sensitivity of rescued unc-18 mutants to 1 mm aldicarb. Stable expression of mutant Munc18-1 causes unc-18 mutants to be more sensitive to aldicarb than that of wild-type Munc18-1. n = 7. *p < 0.05. Tukey's test was performed for mean comparisons in ANOVA. Error bars indicate SEM. G, Fifty micrograms of indicated samples were subjected to SDS-PAGE followed by immunoblotting. The expression of transgenic Munc18-1 was detected by anti-Munc18-1 monoclonal antibody (1:1000) followed by enhanced chemiluminescence detection.
To accommodate for the phenotypic variations arising from the extrachromosomal array expression of Munc18-1, we generated multiple transgenic lines expressing each rescue construct and examined the motility by performing thrashing assays. The expression of Munc18-1 proteins was driven by the promoter of unc-18 gene (Punc-18). We found that WT Munc18-1 rescued the motility defect of unc-18(e81) to ∼50% of N2 wild-type level, while Munc18-1(P335A) rescued the motility of unc-81(e81) significantly better than WT Munc18-1 (Fig. 1B). Using GFP-tagged Munc18-1 proteins (Munc18-1::GFP), we confirmed the neuronal expression of both the WT and the P335A mutant in transgenic strains (Fig. 1A). Expression of the GFP-tagged mutant Munc18-1 also rescued the motility defect better than GFP-tagged WT Munc18-1 (Fig. 1B), in both extrachromosomal and integrated arrays (Fig. 1A,C). We observed similar rescuing ability of mutant Munc18-1 expressed in C. elegans harboring another loss-of-function allele of unc-18, md299, which contains deletions of the promoter and 5′ half of the gene (Weimer et al., 2003; Fig. 1C).
To further evaluate the effect of this mutation, we measured the locomotion speed of C. elegans in the presence of food. Similar to the results from the thrashing assays, the locomotion speed of unc-18(e81) was partially rescued by expression of multiple copies of WT Munc18-1, but mutant Munc18-1 exhibited a more robust rescue (Fig. 1D). Locomotion speed rescued by expression of GFP-tagged mutant Munc18-1 was significantly higher than that by the WT integrated arrays in both unc-18 mutant alleles (e81, md299; Fig. 1E). Overall, our results indicate that the Munc18-1(P335A) mutant exhibits enhanced function compared to the WT protein in rescuing locomotion of unc-18 mutants.
C. elegans locomotion is driven by the activity of cholinergic motor neurons. We investigated whether the behavioral enhancement of unc-18 null animals expressing mutant Munc18-1 can be explained by increased synaptic transmission. To evaluate synaptic acetylcholine release, we examined the sensitivity of C. elegans to aldicarb, an inhibitor of acetylcholine esterase, a key enzyme that clears acetylcholine from the synaptic cleft (Materials and Methods). Aldicarb treatment in C. elegans leads to accumulation of acetylcholine in the neuromuscular junction, persistent muscle contraction, paralysis, and lethality (Miller et al., 1996; Nonet et al., 1998; Mahoney et al., 2006). unc-18 loss-of-function mutants are resistant to aldicarb (Miller et al., 1996), consistent with impaired release of acetylcholine from motor neurons (Weimer et al., 2003). We found that GFP-tagged mutant Munc18-1 integrants exhibited higher sensitivity to aldicarb (1 mm) than GFP-tagged WT Munc18-1 integrants (Fig. 1F). This indicates that cholinergic synaptic transmission was enhanced in animals expressing mutant Munc18-1 compared to those expressing WT Munc18-1. Collectively, these results reveal that changing the conformation of a helical hairpin in domain-3a of Munc18-1 augments the ability of this protein to mediate synaptic acetylcholine release, thereby enhancing locomotory behavior.
To examine the level of WT and mutant Munc18-1 transgenically expressed in unc-18 mutant C. elegans, we performed Western blot analysis. We found that either WT or mutant Munc18-1 was expressed at various levels in different rescued lines, but, overall, a comparable expression between WT and P335A mutant proteins (Fig. 1G). This indicates that the increased motility and aldicarb sensitivity by the mutant over WT are not simply due to differences in their expression level.
Effect of the P335A hinge point mutation is conserved in the C. elegans orthologue of Munc18-1, UNC-18
The region surrounding Pro335 in domain-3a of Munc18-1 is highly conserved among homologues in various species including C. elegans (P334 in UNC-18). To determine whether the conformational change in domain-3a of UNC-18 also results in a similar effect, we tested the ability of the P334A mutant of UNC-18 to rescue the phenotype of unc-18(md299) in comparison with WT UNC-18. Here, we measured the motility and locomotion speed of unc-18(md299) carrying an integrated WT or mutant UNC-18 array. The reduction of both parameters seen in unc-18(md299) was fully rescued by expression of WT UNC-18 under the control of the Punc-18 promoter (Fig. 2A,B). Furthermore, the expression of mutant UNC-18 induced a modest, but significant, enhancement of thrashing compared to WT UNC-18 (Fig. 2A), though we did not observe a difference in locomotion speed (Fig. 2B).
Figure 2.

Mutation of Pro334 confers a gain-of-function effect on UNC-18. A, Mutation of Pro334 enhances the ability of UNC-18 to restore the motility of unc-18 mutants. n = 18–19. In a two-sample t test, t(35) = −2.09, p = 0.044. B, Locomotion speed measured in the presence of food is fully restored by expression of wild-type or mutant UNC-18 in unc-18 mutants. In a two-sample t test, t(29) = 0.08, p = 0.94. n = 15–19. C, unc-18 null mutants rescued by expression of mutant UNC-18 are more responsive to aldicarb than N2 animals and those rescued by expression of wild-type UNC-18. tom-1 (ok285) indicates C. elegans lacking the gene encoding TOM-1, the ortholog of Tomosyn. n = 8. *p < 0.05. Error bars indicate SEM. NS, Statistically nonsignificant.
Importantly, mutant UNC-18(P334A) exhibited dramatically higher sensitivity to aldicarb compared to the WT UNC-18 integrants or N2 animals, indicating an increase in synaptic acetylcholine release (Fig. 2C). Such a strikingly high aldicarb sensitivity was previously reported for tom-1(ok285) null animals (Dybbs et al., 2005). Tomosyn/TOM-1 is a key negative regulator of SNARE-mediated fusion that competes with synaptobrevin for binding to the other SNAREs (Malkin et al., 1990; Hatsuzawa et al., 2003; Gracheva et al., 2006; McEwen et al., 2006). The mutant UNC-18-rescued line exhibited even higher sensitivity to aldicarb than tom-1(ok285) (Fig. 2C). These results indicate that acetylcholine release is significantly facilitated by expression of UNC-18(P334A) in unc-18(md299) compared to WT, and the degree of facilitation is comparable to or even greater than in the tom-1 null condition.
Overexpression of WT or mutant UNC-18(P334A) in the wild-type background shows opposite effects on motility and synaptic release
Since we introduced multiple copies of WT or mutant UNC-18 in unc-18(md299), either protein was likely to be expressed at higher than endogenous level. UNC-18 and its Drosophila orthologue, ROP, were shown to inhibit synaptic release when they were strongly overexpressed far beyond their endogenous levels in wild-type C. elegans and Drosophila, respectively (Schulze et al., 1994; Wu et al., 1998; Weimer et al., 2003). We questioned how expression of WT and mutant UNC-18 in the presence of endogenous UNC-18 would affect the behavior of animals and synaptic release, and hence examined the resulting phenotype.
We found dramatic differences between the effects of WT and mutant UNC-18 in the presence of endogenous UNC-18. Specifically, overexpression of WT UNC-18 strongly decreased motility and aldicarb sensitivity (Fig. 3). It is quite surprising that expression of the identical WT UNC-18 integrant causes such a dramatic difference in motility depending on the genetic background [unc-18 null(md299) background, Fig. 2A, vs N2 WT background, Fig. 3A]. In contrast, overexpression of mutant UNC-18 increased motility (Fig. 3A) and strongly enhanced sensitivity to aldicarb (Fig. 3B). These results indicate that when the expression level of WT UNC-18 is higher than that of the endogenous UNC-18, synaptic release is strongly inhibited, as previously reported (Weimer et al., 2003). Furthermore, the greater rescuing ability of mutant UNC-18 in unc-18(md299) (Fig. 2) cannot simply be explained by its higher expression than multiple copies of the transgene encoding WT UNC-18. Rather, mutant UNC-18 exhibits a positive effect on motility and synaptic release without showing overexpression-induced inhibition of synaptic release. Hence, we conclude that the P334A mutation causes strikingly different phenotypes from WT in synaptic release and locomotion.
Figure 3.

Overexpression of WT UNC-18 suppresses locomotion and acetylcholine release, whereas the P334A mutant accelerates locomotion and release in wild-type (N2) animals. A, Overexpression of wild-type UNC-18 severely impairs the motility of C. elegans compared to N2, whereas overexpression of mutant UNC-18 enhances the motility. n = 28–32. In a one-way ANOVA statistical test, F(2,86) = 1835.8 and p = 0. Tukey's test was performed for mean comparisons. B, Overexpression of wild-type UNC-18 decreases the sensitivity to aldicarb compared to N2, while overexpression of mutant UNC-18 increases aldicarb sensitivity. n = 7. *p < 0.05. Error bars indicate SEM.
Overexpressed mutant UNC-18 partially bypasses the requirement of UNC-13
Similar to Munc18-1/UNC-18, Munc-13/UNC-13 is believed to be involved in synaptic vesicle priming. Munc13-1 performs its function in priming at least in part by opening “closed” syntaxin-1/UNC-64 (Richmond et al., 2001; Yang et al., 2015). Since mutant UNC-18(P334A) shows strong enhancement of synaptic release, presumably by increasing vesicle priming, we asked whether it could bypass the requirement for UNC-13. The s69 allele of unc-13 harbors a 5 bp deletion, which causes a frame shift in the common domain of short and long isoforms of UNC-13, and is thus considered to be null (Richmond et al., 1999). As demonstrated in a previous study (Sassa et al., 1999), unc-13(s69);unc-18(md299) double mutants exhibit lethality. Either WT or mutant UNC-18 rescued the lethality (Fig. 4). We compared the motility of these animals to that of unc-13(s69). The motility of double mutants expressing WT UNC-18 was comparable to that of unc-13(s69) (Fig. 4A). However, the double mutants expressing UNC-18(P334A) markedly restored motility, with an increase in thrashing by 15 times compared to unc-13(s69). This result indicates that the phenotype of unc-13(s69) was partially suppressed by the P334A mutation in unc-18.
Figure 4.

The behavioral defects associated with unc-13 mutant animals are partially rescued by the expression of the P334A mutant UNC-18. A, Expression of mutant UNC-18 in the unc-13 and unc-18 double loss-of-function mutant background partially restores motility measured by thrashing assays. In a one-way ANOVA statistical test, mutant UNC-18 was excluded: F(2,69) = 8.9, p = 3.74E-4. n = 19–28. B, Expression of mutant UNC-18 in the unc-13 and unc-18 double mutant background partially restores locomotion speed measured in the presence of food. n = 15–16. In a one-way ANOVA statistical test, F(2,43) = 6.3 and p = 0.004. *p < 0.05. Tukey's test was performed for means comparisons in ANOVA. Error bars indicate SEM.
We further asked whether expression of mutant Munc18-1(P335A) would also lead to suppression of the behavioral defects associated with unc-13(s69). We were unable to obtain viable unc-13 and unc-18 double mutants that expressed WT Munc18-1, but those expressing mutant Munc18-1 were viable. More importantly, these viable animals displayed an even slightly higher motility than unc-13(s69) mutants, suggesting that only Munc18-1(P335A) could partially bypass the requirement for UNC-13, albeit less significantly than the C. elegans UNC-18(P334A). Locomotion speed of unc-13 and unc-18 double mutants expressing Munc18-1(P335A) was rescued to a comparable level to that of unc-13(s69) mutants (Fig. 4B). The double mutants expressing C. elegans UNC-18(P334A) exhibited twice the locomotion speed of unc-13 single mutants. These results indicate that the mutant form of UNC-18, which presumably harbors the constitutively extended conformation of the helical hairpin in domain-3a, partially bypasses the requirement of UNC-13 in vivo.
C. elegans harboring the unc-18(P334A) KI allele exhibits enhanced synaptic release
During the course of our study, the CRISPR/Cas9-mediated gene replacement method became feasible in C. elegans (Levine, 1997; Dickinson et al., 2013; Friedland et al., 2013; Norris et al., 2015). To characterize mutant UNC-18(P334A) at the endogenous level, we generated C. elegans harboring the P334A KI allele using CRISPR/Cas9 technology (Fig. 5A; see Materials and Methods). Owing to the large size of the repair template which contained two selection markers, GFP and neomycin resistant gene, we generated a novel unc-18 null (KO) allele (sks1), in which the insertion of the repair template fully disrupted the unc-18 gene (Fig. 5B; see Materials and Methods). C. elegans harboring this putative null allele exhibited almost no thrashing, which is a more severe locomotion phenotype than what is observed in animals carrying the canonical severe loss-of-function allele e81 or md299 (Fig. 5B). Following the expression of Cre recombinase and removal of selection markers, the motility of C. elegans was fully restored. Using PCR for genotyping, we verified that the animals displaying restored motility harbored the KI allele (Fig. 5C).
Figure 5.
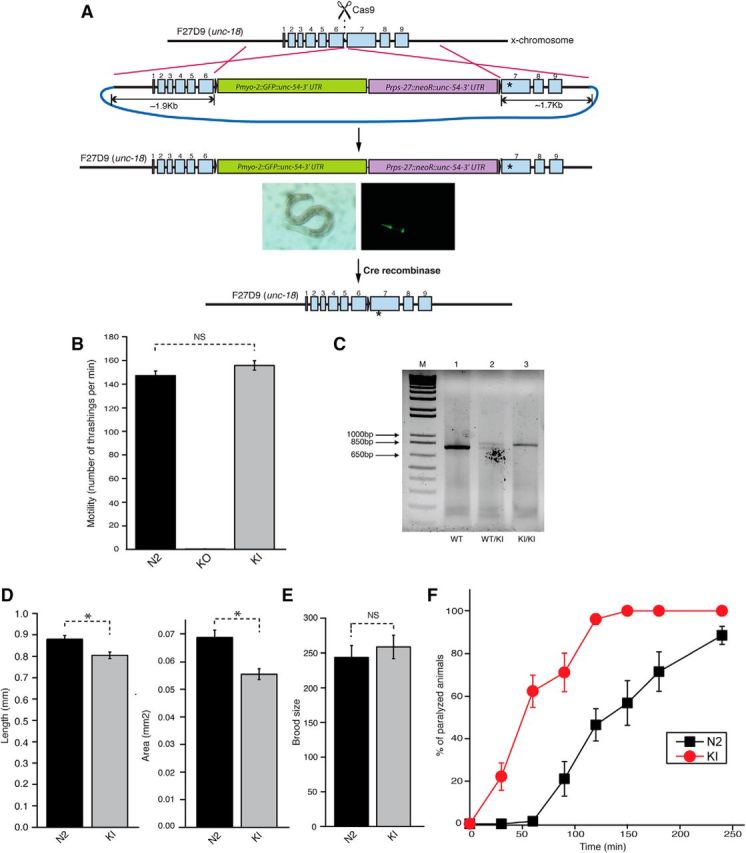
C. elegans harboring the knock-in unc-18 allele (KI, P334A) exhibits enhanced acetylcholine release. A, A schematic diagram represents how CRISPR-mediated homologous recombination replaces the wild-type allele of unc-18 with the P334A allele. One of the selection markers is GFP expressed by a pharynx-specific promoter. B, In thrashing assays, unc-18(sks1) mutant animals referred to as KO, harboring the selection markers in the unc-18 locus, exhibit completely eliminated motility. Upon expression of Cre recombinase, the selection markers are excised, generating the KI allele (sks2) containing a mutation that changes Pro334 to Ala334. Thrashing of the unc-18 KI animals is comparable to that in N2 animals (t(42) = −1.53, p = 0.13). n = 21–23. C, PCR-based genotyping of unc-18(P334A) KI allele in comparison with WT allele. Note that the KI allele is longer than the WT allele by ∼60 bp due to the inclusion of 34 bp of a loxP site as well as restriction enzyme sites to generate the left and right arms for homologous recombination. D, Morphology of C. elegans harboring the KI allele. Comparisons of body length (left) and area (right) were made between N2 (n = 26) and the KI (n = 17) C. elegans worms. The KI animals exhibited reduced body size as determined by measuring their body length and area. Two-sample t tests were performed: t(41) = 3.05, p = 0.004 for length and t(41) = 2.51, p = 0.016 for area. E, Comparison of brood size is made between N2 (n = 5) and the KI (n = 5) C. elegans worms. The brood size of these animals is comparable to that of N2 animals. A two-sample t test was performed: t(8) = −0.63, p = 0.55. F, Aldicarb assays were conducted to indirectly compare synaptic acetylcholine release between N2 and KI animals. Compared to N2 animals, the KI animals exhibited hypersensitivity to aldicarb. n = 4. *p < 0.05. NS, Statistically nonsignificant. Error bars indicate SEM.
We analyzed the phenotype of the P334A KI animals, unc-18(sks2). We observed a trend of increased thrashing activity in unc-18(sks2) compared to the N2 wild-type animals, although this increase did not reach statistical significance (Fig. 5B). Hence, the slight but statistically significant increase in the motility of unc-18 null mutants expressing multiple copies of UNC-18(P334A) may reflect the effect of overexpression (Fig. 3A). Nevertheless, unc-18(sks2) mutants were strongly hypersensitive to aldicarb when compared to the WT N2 animals, indicating that synaptic release was facilitated in the KI animals (Fig. 5F). Thus, transgenic overexpression of P334A in unc-18(md299) animals or KI of unc-18(P334A) caused behavioral enhancement correlated with increased acetylcholine release in C. elegans.
We also observed that the unc-18(sks2) mutants manifested a reduction in body size that was measured as their body length and area (Fig. 5D), yet their brood size was still comparable to that of N2 C. elegans (Fig. 5E). Notably, we made an observation that tom-1 null animals, which are reported to exhibit enhancement of neurotransmitter release (Gracheva et al., 2006; McEwen et al., 2006), also displayed reduced body length, as reported previously (Lee et al., 2011). In C. elegans, mutations in components of BBSome (bbs-1, bbs-2, bbs-5, bbs-7, bbs-8, bbs-9) consistently result in hypersecretion from dense-core vesicles and reduction of the body size (Lee et al., 2011). Thus, it is possible that the decrease in body size seen in the KI mutant is caused by hypersecretion from dense-core vesicles in addition to that from synaptic vesicles.
To obtain direct evidence for the increased release from presynaptic neurons, we performed electrophysiological recordings from the neuromuscular junctions of unc-18(sks2) KI animals (Fig. 6). We found that the frequency of miniature postsynaptic currents (mPSCs) was significantly increased in unc-18(sks2) compared to WT animals (Fig. 6A). Body wall muscles receive synaptic inputs not only from excitatory (acetylcholine) motor neurons but also from inhibitory (GABA) motor neurons (Richmond and Jorgensen, 1999). To exclude the inhibitory effects of GABA-mediated synaptic transmission, we also generated unc-18(sks2) in a GABAA receptor mutant background, unc-49(e407) (Richmond and Jorgensen, 1999). We found that the frequency of spontaneous acetylcholine release in uns-49(e407);unc-18(sks2) double mutants was significantly higher than in unc-49(e407) (Fig. 6B, left). The amplitude of mPSCs in unc-49(e407), unc-18(sks2), and double mutants, however, was comparable to that of WT animals (Fig. 6B, right). Thus, we conclude that the KI mutation in unc-18 significantly increases synaptic release.
Figure 6.
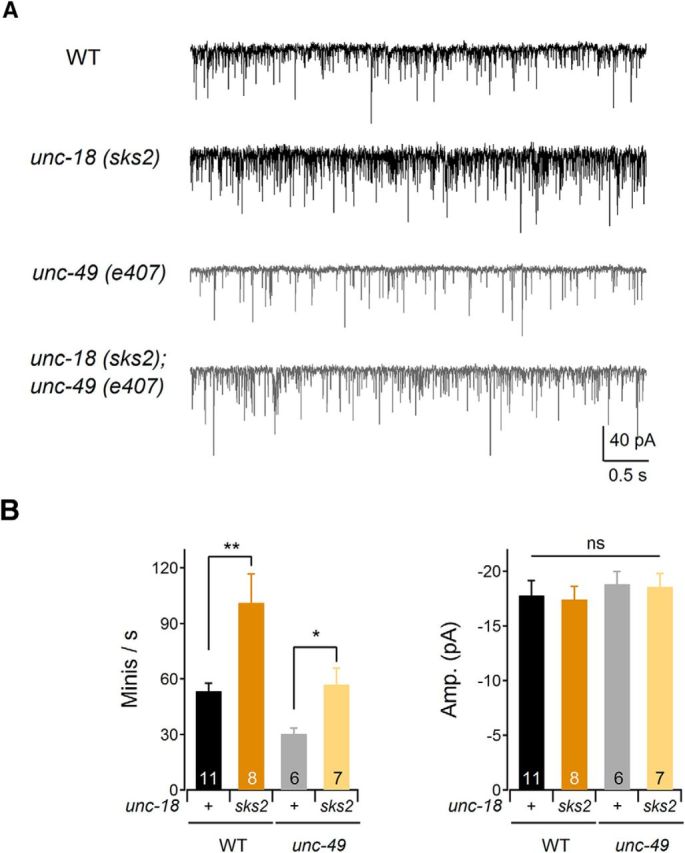
Frequency of mPSCs is increased in unc-18(sks2) mutants. A, Representative traces of spontaneous mPSCs in the WT, unc-18(sks2), unc-49(e407), and unc-18(sks2);unc-49(e407) mutant animals. Muscles were holding at −60 mV. B, Left, The frequency of mPSCs was increased in unc-18(sks2) mutant animals compared to WT animals. Two-sample t tests were performed: t(17) = −3.38, **p < 0.01; t(11) = −2.67, *p < 0.05. Right, The amplitude of mPSCs in unc-49(e407), unc-18(sks2), and double mutants was comparable to that of WT animals. In a one-way ANOVA statistical test, F(3,28) = 0.27 and p = 0.85. ns, Statistically nonsignificant. Error bars indicate SEM.
unc-18(P334A) KI and tom-1 null mutants show strong synergy in suppressing the unc-13 null mutant phenotype
While the majority of mutations identified in synaptic genes cause reduced synaptic activity, the unc-18(P334A) KI augmented synaptic vesicle release and the motility of C. elegans (Fig. 5B,F, Fig. 6). Thus, we exploited such rare mutation in unc-18 to investigate the potential genetic interactions of this gene and others, such as tom-1. The locomotory behavior of the KI animals was assessed in comparison to that of tom-1 null (ok285) animals. The KI animals exhibited higher motility than tom-1(ok285) animals (Fig. 7A). Nonetheless, they were comparable to the tom-1 null mutants when subjected to aldicarb (Fig. 7B). We then asked how both locomotion behavior and aldicarb sensitivity would be altered by the double mutation of unc-18(sks2) and tom-1(ok285). Interestingly, these double mutants displayed higher motility than wild-type N2 or tom-1 single mutants (Fig. 7A). Not only did they exhibit higher sensitivity to aldicarb than N2 wild-type animals, but they were also more sensitive than either tom-1 or unc-18 single mutants (Fig. 7B). These results show that the combination of the unc-18(P334A) mutation and the null mutation in tom-1 causes greater enhancement of locomotion behavior and synaptic transmission than the single mutations.
Figure 7.
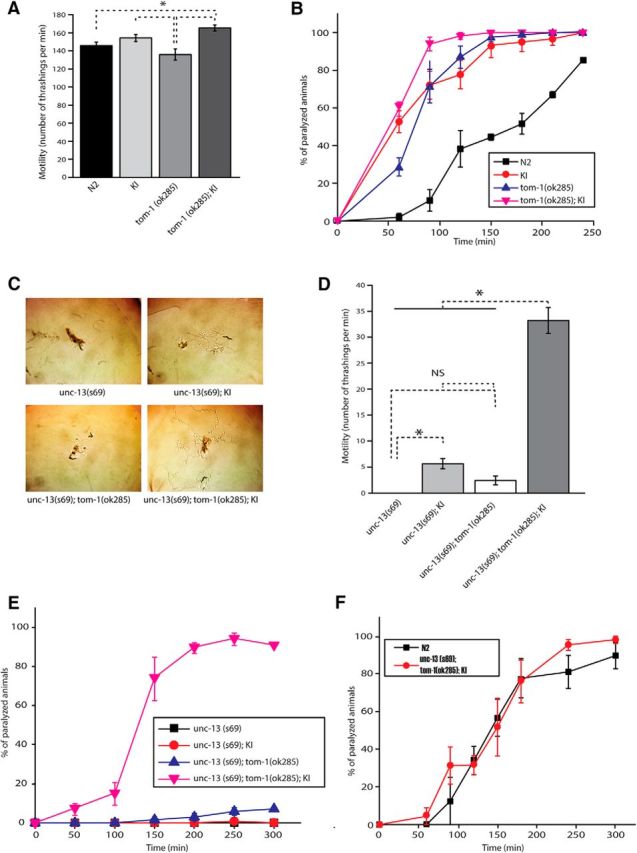
unc-18 KI and tom-1 null mutants show strong synergy in rescuing unc-13 mutant defects. A, unc-18 KI and tom-1 null double mutant animals swim at higher thrashing rates than N2 and tom-1 null mutant animals. n = 17–23. In a one-way ANOVA statistical test, F(3,76) = 7.8 and p = 1.3E-4. B, unc-18 KI and tom-1 null double mutant animals are more responsive to aldicarb than N2 animals, unc-18 KI animals, and tom-1 null mutants. n = 4. C, Traces of unc-13 single, double (unc-13 with unc-18 KI or tom-1), and triple mutant (tom-1, unc-13, and unc-18 KI) C. elegans. These animals were placed on fresh plates seeded with OP50 and recorded for 1.5 h. The images shown were taken at the end of recording. Crawling of unc-13 mutants is partially restored by unc-18 KI or the combination of unc-18 KI and tom-1(ok285). D, Thrashing assays of the same set of mutants as in C reveal that the motility of unc-13 mutants is partially restored by unc-18 KI or tom-1;unc-18 KI. n = 12–18. In a one-way ANOVA statistical test, F(3,57) = 128.8 and p = 0. Tukey's test was performed for mean comparisons in ANOVA. E, Aldicarb assays were conducted to assess synaptic acetylcholine release among the same set of mutants C. n = 6. F, N2 WT and the triple mutants (unc-13;unc-18 KI;tom-1) showed comparable sensitivity to aldicarb. n = 4. An asterisk indicates statistical significance at p < 0.05. Error bars indicate SEM.
To further investigate the effect of unc-18(P334A) with other synaptic genes, we generated unc-13(s69) harboring the unc-18 KI, tom-1(ok285), or both mutant alleles. We already found that the unc-18(md299) and unc-13(s69) double mutant animals expressing multiple copies of mutant UNC-18 showed drastic improvement in their locomotion behavior compared to unc-13(s69) single mutants (Fig. 4). Consistent with this result, unc-13(s69) harboring the unc-18(P334A) KI allele exhibited increased locomotion on bacterial lawn and enhanced motility in liquid medium compared to the unc-13 single mutants (Fig. 7C,D). The tom-1 null mutation conferred partially restored motility and locomotion (Fig. 7C,D) and increased aldicarb sensitivity (Fig. 6E) in the unc-13(s69) single mutant background. In contrast, unc-13(s69) harboring the unc-18(P334A) KI allele still showed strong resistance to aldicarb (Fig. 7E), indicating that depression of the synaptic transmission arising from UNC-13 deficiency was not rescued by expression of UNC-18(P334A) at the endogenous level.
Surprisingly, unc-13(s69); tom-1(ok285); unc-18(sks2) triple mutants displayed the most striking rescue of locomotion and motility (Fig. 7C,D). The degree of rescue was greater than the additive effect of tom-1(ok285) and unc-18(sks2). More importantly, these triple mutants were as sensitive to aldicarb as N2 wild-type animals (Fig. 7F). This result suggests that acetylcholine release was almost completely restored in the triple mutants, although the locomotion behavioral defects did not seem to be fully rescued (Fig. 7C,D). UNC-13 has two isoforms (short and long) and controls the timing of release via these two isoforms. It has been suggested that the long form (UNC-13L) is critical for fast transmitter release, while the short form (UNC-13S) is important for slow release (Hu et al., 2013; Zhou et al., 2013). Since TOM-1/Tomosyn selectively inhibits slow release mediated by UNC-13S (Hu et al., 2013), it is likely that our triple mutants exhibit primarily slow release, accounting for a lack of full rescue of movement.
Munc18-1(P335A) enhances liposome fusion, partially bypassing the requirement of Munc13-1. Using in vivo systems, it is difficult to directly address whether the enhanced function of UNC-18(P334A) in synaptic transmission, including its ability to bypass the requirement for UNC-13, is due to the direct action of the mutant protein on SNARE proteins or the indirect effects of interactions between the mutant and other proteins. We therefore tested whether the effect of Munc18/UNC-18 (P335A/P334A) on vesicle release and the behavior of C. elegans can be reproduced in vitro using liposome fusion assays with defined protein factors and Munc18-1 (WT or P335A). Note that enhancing effects of mutant Munc18(P335A) had already been observed in previous reconstitution experiments, but fusion in these experiments did not require Munc13 (Parisotto et al., 2014). Our recent reconstitution studies showed that fusion requires both Munc18-1 and Munc13-1 when NSF and αSNAP are included, because NSF-αSNAP disassembles syntaxin-1–SNAP-25 complexes, and Munc18-1 together with Munc13-1 mediate SNARE complex assembly in an NSF-αSNAP-resistant manner (Ma et al., 2013; Liu et al., 2016). Using this new assay, we can directly test the enhancement of lipid and content mixing by Munc18-1(P335A) in the presence and absence of Munc13-1.
We first performed both lipid and content mixing assays where we included the three SNAREs, NSF, α-SNAP, Munc18-1, a fragment spanning the synaptotagmin-1 C2 domains (C2AB), and a fragment spanning the conserved C-terminal region of Munc13-1 (referred to as the C1C2BMUNC2C fragment; Fig. 8A,B). Interestingly, WT Munc18-1 exhibited very little content mixing before adding Ca2+, whereas the P335A mutant induced efficient content mixing. Once Ca2+ was added, both WT and mutant Munc18-1 induced rapid and strong content mixing. These results show that the P335A mutant helps to efficiently overcome an energy barrier that strongly hinders fusion in the absence of Ca2+, and that is relieved by Ca2+ binding to the Munc13-1 C2B domain in this system, as observed for the Munc18-1 D326K mutant described previously (Sitarska et al., 2017). A dramatic difference between WT and the P335A mutant was observed when we did analogous experiments without the Munc13-1 fragment (Fig. 8C,D). While there was very little content mixing without Munc13-1 C1C2BMUNC2C in the case of the WT Munc18-1, there was considerable content mixing with the P335A mutant (Fig. 8C–E). Note that fusion was only observed upon Ca2+ addition in these experiments because, unlike the Munc13-1 C1C2BMUNC2C fragment, the synaptotagmin-1 requires Ca2+ for stimulation of SNARE complex formation and fusion (Liu et al., 2016). These results suggest that our in vitro fusion assays can reproduce, at least to some extent, the gain-of-function phenotype of the P335A mutant, including partial bypassing of the Munc13-1 requirement. Since our fusion assays are composed of the three SNAREs, NSF, α-SNAP, Munc18-1, and synaptotagmin-1 C2AB, the effects of the P335A mutation are likely to arise from interactions of Munc18-1 with some of these proteins, most notably the SNAREs.
Figure 8.
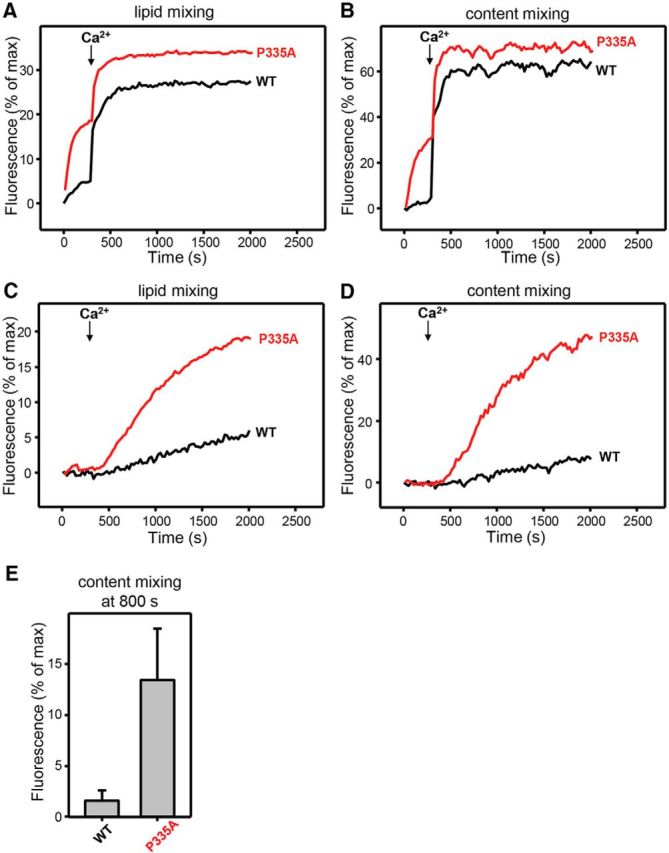
P335A mutant of Munc18-1 can substantially bypass the requirement of Munc13-1 in lipid and content mixing assays. A–D, Lipid mixing (A, C) between V and T liposomes was measured from the fluorescence dequenching of Marina Blue–labeled lipids, and content mixing (B, D) was monitored from the development of FRET between PhycoE-Biotin trapped in the T liposomes and Cy5-streptavidin trapped in the V liposomes. In A and B, the assays were performed in the presence of NSF-αSNAP, Munc13-1 C1C2BMUNC2C, synaptotagmin-1 C2AB fragment, and WT or mutant Munc18-1s as indicated. In C and D, the assays were performed in the presence of NSF-αSNAP, synaptotagmin-1 C2AB fragment, and WT or mutant Munc18-1s as indicated. Experiments were started in the presence of 100 μm EGTA and 5 μm streptavidin, and Ca2+ (600 μm) was added after 300 s. E, Quantification of content mixing in the experiments of D. Bars represent averages of the normalized fluorescence observed after 800 s (500 s after Ca2+ addition) in experiments performed at least in triplicate. Error bars represent SDs.
The P335A mutation in Munc18-1 facilitates SNARE complex formation and enhances binding of Munc18-1 to the preformed SNARE complex
We explored the mechanisms of how the Munc18-1(P335A)/UNC-18(P334A) mutants manifest the gain-of-function phenotypes that include enhanced synaptic release and motility, the lack of suppression of transmitter release when overexpressed, and the partial bypassing of the Munc13-1/UNC-13 requirement in vitro and in vivo. While the bent conformation of the helical hairpin in domain-3 of Munc18-1 is compatible with binding to syntaxin-1, the extended conformation has been predicted to be inconsistent with binding to closed syntaxin-1 (Hu et al., 2011). The structural consequences of the P335A mutation are unclear, but our finding suggests that it facilitates SNARE complex assembly starting from the binary syntaxin-1–Munc18-1 complex. To test this idea, we examined the assembly of the SNARE complex in the presence of Munc18-1. GST-fused syntaxin-1 was first incubated with His-tagged WT or mutant Munc18-1 to form the binary complex, followed by the addition of SNAP-25 and VAMP2 (Fig. 9A). The incubated proteins were run on SDS-PAGE gels. The interaction of syntaxin-1 with WT Munc18-1 was so strong that the formation of the SNARE complex did not occur. In contrast, the P335A Munc18-1 clearly led to SNARE complex formation. As reported previously, these SNARE complexes were SDS resistant, and their thermal stability was disrupted upon boiling. Note also that the binding of syntaxin-1 to the mutant Munc18-1 was not as strong as that to WT Munc18-1 (Fig. 9A), likely because the mutation of the hinge point disturbed the bent conformation of Munc18-1. Moreover, these results show that the P335A mutation in Munc18-1 promotes the formation of the SNARE complex, as predicted by the template model. These results also explain why overexpressed WT UNC-18 inhibits neurotransmission, whereas overexpressed UNC-18(P334A) does not (Fig. 3).
Figure 9.

P335A Munc18-1 increases SNARE assembly and its binding to the SNARE complex. A, WT or mutant Munc18-1 is initially incubated with syntaxin-1 only, and these two proteins are further incubated with SNAP-25 and synaptobrevin-2. Mutant Munc18-1 but not WT Munc18-1 allows syntaxin-1 to form the SNARE complex with synaptobrevin-2 and SNAP-25. Coomassie staining reveals that the SNARE complex formed in the presence of mutant Munc18-1 is SDS resistant but destabilized upon boiling. n = 4. B, To examine the interaction between the SNARE complex and Munc18-1, formation of the SNARE complex is first induced by incubating SNAP-25, syntaxin-1, and synaptobrevin-2. These SNARE proteins are further incubated with wild-type or mutant Munc18-1. Coomassie staining shows that the binding of mutant Munc18-1 to the SNARE complex seems to be greater than that of the wild type. C, The interaction between the SNARE complex and Munc18-1 variants is quantified by measuring the intensity of Munc18-1 bands in the Coomassie blue staining. The intensity values were first normalized to those of SNAP-25 bands (left) or syntaxin-1A bands (right) and then to the background staining (no Munc18-1 condition) such that the value for the no Munc18-1 condition was set to 1. A paired t test was performed. For normalization to SNAP-25, t(3) = 8.32, p = 0.004, n = 4; for normalization to syntaxin-1, t(3) = 4.86, p = 0.017, n = 4. *p < 0.05; **p < 0.01. Error bars indicate SEM. D, Nonspecific binding of WT and mutant Munc18-1 to beads onto which strep-SNAP-25 was immobilized.
Since we observed that a loss-of-function mutation (KE/5I) in domain-3a of Munc18-1 significantly reduces the ability of Munc18-1 to bind the preformed SNARE complex (Han et al., 2013), we speculated that the P335A mutation may enhance the binding, which could also provide an explanation for increased function caused by the mutation. To test this possibility, we examined the direct interaction between the SNARE complex and WT or mutant Munc18-1 (Fig. 9B). The formation of the SNARE complex was induced by incubating the three SNARE proteins: syntaxin-1, strep-SNAP-25, and His-tagged synaptobrevin-2. We then added WT or mutant Munc18-1 to allow binding to the SNARE complex. Next, the mixture was run on SDS-PAGE gels to examine the binding of Munc18-1 to the SNARE complex. We found that Munc18-1(P335A) was able to bind to the SNARE complex more effectively than WT Munc18-1 (Fig. 9B). When normalized with the amount of immobilized strep-SNAP-25 or syntaxin-1A within the preformed SNARE complex, the amount of mutant Munc18-1 bound to the preassembled complex was significantly higher than that in WTs (Fig. 9C). Collectively, these results suggest that promoting the extended conformation of the helical hairpin in Munc18-1/UNC-18 increases both SNARE complex assembly and the affinity for the SNARE complex. To test whether mutant Munc18-1 nonspecifically binds to agarose beads more than the WT, we examined the binding of this mutant versus WT to the agarose beads onto which only strep-SNAP-25 is immobilized. We found that both mutant and WT proteins displayed very little binding to the beads and that there was no difference in nonspecific binding between the two proteins (Fig. 9D). This result indicates that the P335A mutant exhibits increased binding specific to the preassembled SNARE complex.
Munc18-1(P335A) exhibits dominant-positive effects on the frequency of miniature EPSCs in mammalian neurons
The enhanced ability of Munc18-1(P335A) to form and to bind the SNARE complex seems to explain the positive phenomenon in which the P334A mutant of UNC-18 can facilitate synaptic transmission even in the presence of endogenous UNC-18 (Fig. 3). We asked whether Munc18-1(P335A) can also increase synaptic transmission of mammalian neurons that express endogenous WT Munc18-1 in a dominant-positive manner. The precise electrophysiology available in mammalian neurons allows us to test whether the mutant increases the frequency and/or kinetics of SNARE-mediated fusion events. Therefore, we infected E16 cortical neurons with lentiviruses expressing GFP, WT Munc18-1-GFP, or Munc18-1(P335A)-GFP and subsequently examined the effects of expressed proteins on synaptic transmission.
To determine the presynaptic effects of Munc18-1 expression, only GFP-negative neurons were patched. Infection of neurons with lentivirus yielded an efficiency of ∼50% across all three groups (example images shown in Fig. 10A). Thus, GFP-negative cells were readily accessible for patch clamp. In the absence of stimulation, there is spontaneous fusion of synaptic vesicles (Fig. 10B), which enables the measurement of the quantal size and kinetics of these single fusion events (PSCs). As summarized in Figure 10C, there was a significant difference among the three groups (one-way ANOVA, F(2,33) = 19.69, p = 2.36 * 10−6, n = 12/group); the frequency of spontaneous events was significantly higher in the P335A Munc18-1-expressing groups compared to the control or WT Munc18-1 groups (54.36 ± 1.64 events/10 s vs, respectively, 44.39 ± 1.43 and 41.96 ± 1.35 events/10 s; p < 0.01 and p < 0.001, respectively). However, there was no significant difference across the three groups for PSC size (27.16 ± 1.31 pA, 30.04 ± 2.06 pA, and 25.66 ± 1.09 pA for control, WT Munc18, and P335A Munc18 groups, respectively; ANOVA, F(2,33) = 2.09, p = 0.11; n = 12/group) or decay kinetics (8.77 ± 0.14 ms, 8.29 ± 0.25 ms, and 8.38 ± 0.13 ms for control, WT Munc18, and P335A Munc18 groups, respectively; ANOVA, F(2,33) = 1.96, p = 0.16; n = 12/group). Our results indicate that the P335A mutant of Munc18-1 can stimulate the frequency of the SNARE-mediated fusion events in the presence of endogenous Munc18-1, but it does not seem to accelerate the kinetics of fusion events.
Figure 10.
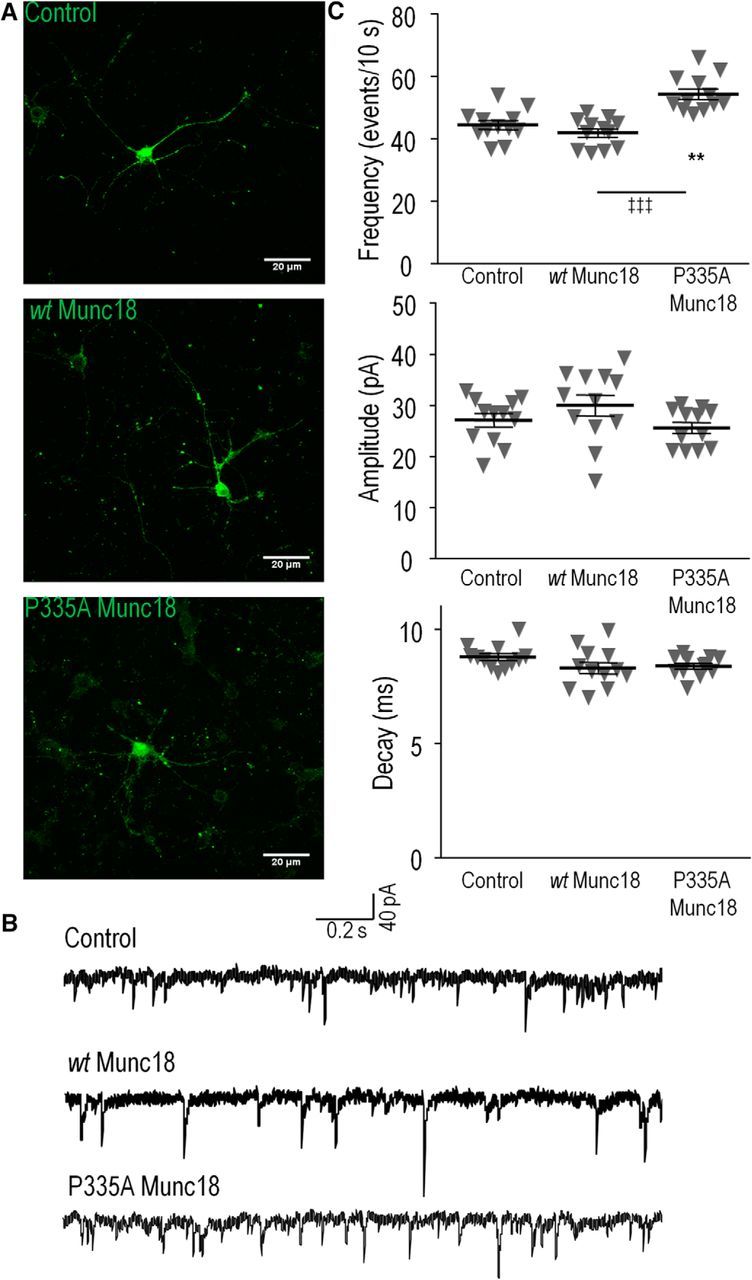
Munc18-1(P335A) increases spontaneous PSC frequency in the presence of endogenous Munc18-1. GFP alone (control), wild-type Munc18-1 + GFP (wt Munc18), or P335A Munc18-1 + GFP (P335A GFP) was introduced using lentivirus into primary mouse cortical neurons at DIV 1. A, Representative images of DIV 16 neurons expressing GFP (control), Munc18-1-wt-GFP (wt Munc18), or Munc18-1-P335A-GFP (P335A Munc18). B, Example traces of spontaneous PSCs in the absence of stimulation. C, Summary charts comparing the PSC frequency, amplitude, decay kinetics between the three groups. **p < 0.01 (GFP vs P335A); ‡‡‡p < 0.001 (WT vs P335A; one-way ANOVA with Tukey's HSD post hoc test; n = 12/group). Error bars indicate SEM.
In addition, we also compared the ability of mutant Munc18-1 versus WT to support neurotransmitter release from neurons in which the endogenous Munc18-1 level is reduced. Munc18-1 KO neurons are known to suffer from severe neurodegeneration starting at E12 in mice (Verhage et al., 2000). To circumvent this problem, we generated Munc18-1 floxed mice using ES cells from EUCOMM (see Materials and Methods). We infected the cultured neurons isolated from Munc18-1 floxed mice with the lentivirus coexpressing GFP or WT Munc18-1-GFP or P335A Munc18-1-GFP with Cre recombinase at DIV 3. GFP expression in the infected neurons escalated daily, and expression of Cre recombinase plus GFP started to deteriorate the health of the neurons at DIV 15. By DIV 25, we found no surviving GFP positive neurons, in accordance with the neurodegenerative Munc18-1 KO phenotype. Therefore, we performed whole-cell patch-clamp recordings from the infected neurons at DIV 13–15. Spontaneous EPSCs were recorded by holding the membrane potential at −70 mV, and only neurons with resting membrane potential more negative than −50 mV and series resistance of ≤20 MΩ for >10 min were included for analysis. We found that reexpression of Munc18-1 was able to rescue diminished synaptic transmission, as the neurons with Munc18-1-WT-GFP or P335A-GFP exhibited higher frequency of spontaneous EPSCs than those with GFP only (Fig. 11). However, consistent with the results from our C. elegans studies (Fig. 6), mutant Munc18-1 supported enhanced synaptic transmission compared to WT; the number of events of spontaneous EPSCs was significantly higher in neurons expressing Munc18-1 P335A than in WTs (62.7 ± 6.7 vs 44.4 ± 5.5, p < 0.05; n = 17 and 16, respectively). Thus, we conclude that the P335A mutant of Munc18-1 can increase synaptic release regardless of the presence or absence of endogenous Munc18-1 protein.
Figure 11.
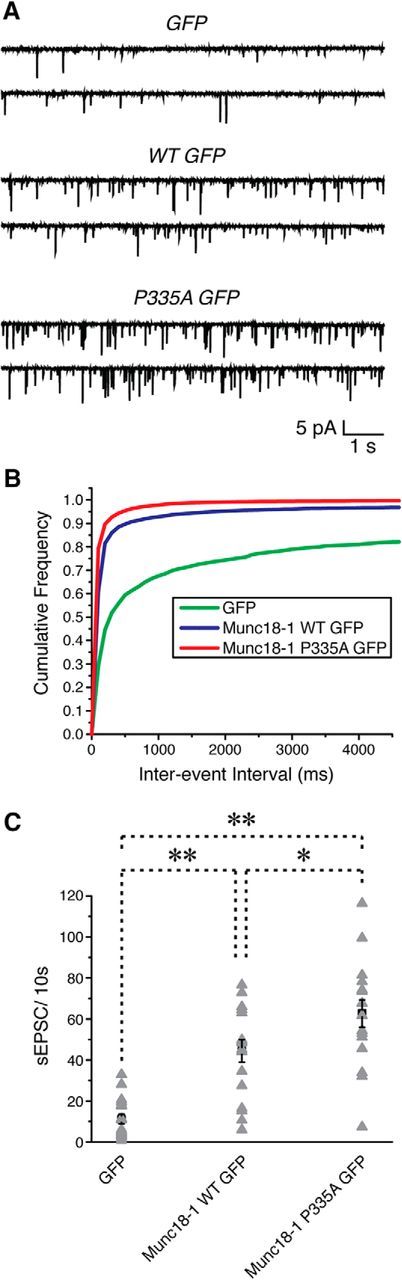
Expression of Munc18-1 P335A enhances synaptic transmission in mouse hippocampal neurons deficient of endogenous Munc18-1. Hippocampal neurons isolated from homozygous Munc18-1 floxed mice (P0–P1) were infected with the lentivirus that coexpressed GFP, WT Munc18-1-GFP, or P335A Munc18-1 GFP with Cre recombinase at DIV 3. Spontaneous EPSCs were recorded from the cultured neurons at DIV 13–15 by holding membrane potential at −70 mV. Only neurons with series resistance of ≤20 MΩ for >10 min and resting membrane potentials more negative than −50 mV were included for analysis. Spontaneous EPSCs with an amplitude of <5 pA (noise) were omitted. A, Example traces of spontaneous PSCs in the absence of stimulation. B, Cumulative probability plot of spontaneous EPSC frequency distribution from GFP, WT Munc18-1 GFP, and P335A Munc18-1 GFP. C, Average number of spontaneous EPSC events recorded per 10 s from an individual cell (gray triangle). Black squares represent means of spontaneous EPSCs in each group. Error bars indicate SEM. *p < 0.05; **p < 0.01 (one-way ANOVA, F(2,47) = 25.99, p = 0, with Tukey's HSD post hoc test; n = 16–17/group).
Discussion
Using a combination of approaches, the in vivo C. elegans system, in vitro liposome fusion assays, and synaptic transmission in primary mammalian neurons, we show that a specific missense mutation in the hinge point of a helical hairpin in domain-3a of Munc18-1/UNC-18 confers phenotypes consistent with increased synaptic exocytosis. These phenotypes include (1) enhanced synaptic release and the motility of animals in the absence (Figs. 1, 2, 5, 6, 11) and presence (Figs. 3, 10) of WT endogenous UNC-18 and (2) the ability to partially bypass the requirement of Munc13-1/UNC-13 in membrane fusion in vitro (Fig. 8) and in vivo (Fig. 4), particularly in the absence of Tomosyn/TOM-1 (Fig. 7).
Our results offer crucial insights into the functional interplay between Munc18-1/UNC-18 and Munc13-1/UNC-13, and at the same time illustrate vehemently the complexity of the regulation of neurotransmitter release. Munc18-1 is a central component of the release machinery, having both inhibitory and active functions. The binary interaction of Munc18-1 with syntaxin-1 stabilizes both proteins (Verhage et al., 2000; Arunachalam et al., 2008; L. Han et al., 2009; G. A. Han et al., 2010, 2011), serving a chaperone-like role. At the same time, this interaction imposes an energy barrier for progression toward synaptic vesicle fusion, because the closed conformation of syntaxin-1 is incompatible with the formation of SNARE complex (Dulubova, 1999; Misura et al., 2000). The mechanism underlying the active role(s) of Munc18-1 was highly enigmatic, but previous data strongly suggested that Munc18-1 orchestrates the SNARE complex assembly in an NSF-SNAP-resistant manner together with Munc13-1 (Ma et al., 2013). Furthermore, the crystal structures of the yeast SM protein Vps33 bound to the Vam3 or Nyv1 SNARE motifs (Baker et al., 2015) yielded compelling evidence that SM proteins in general provide a template to assemble the SNARE complex. On the other hand, structural studies indicated that Munc18-1 can adopt at least two conformations, one where the loop of domain-3a is furled and binds to syntaxin-1 (Misura et al., 2000), and another where the loop is unfurled and one of the helices in domain-3a is extended (Hu et al., 2011), as observed in the structure of Vps33 bound to the synaptobrevin homolog Nyv1. These findings imply that Munc18-1 needs to adopt an unfurled conformation to template the SNARE complex assembly, which was supported by recent experiments with a Munc18-1 mutation that favors the unfurled conformation (Sitarska et al., 2017).
The structural basis for the effects of the P335A mutation is unclear, although this mutation was designed to extend one of the helices of domain-3a of Munc18-1 (Hu et al., 2011; Parisotto et al., 2014). The structural studies of Vps33 show that, even in the unfurled conformation, there is a kink in the extended helix with a proline in the corner (Baker et al., 2015). Regardless of the structural consequences of the P335A mutation, it is clear that the mutation increases the activity of Munc18-1 in synaptic function, likely because it decreases the affinity for closed syntaxin-1 (Han et al., 2014) and/or because it facilitates the transition from the binary Munc18-1-closed syntaxin-1 complex to the SNARE complex (Fig. 9A). Since the P335A mutation also increases binding to the assembled SNARE complex (Fig. 9B), it is also plausible that the increased activity arises in part because it stabilizes the Munc18-1-SNARE macromolecular assembly and this assembly is critical for fusion.
The observation that a closed syntaxin-1 conformation binds to Munc18-1 with a furled loop conformation indicates that this binary complex imposes at least two energy barriers for SNARE complex formation even if it constitutes a critical starting point for the pathway that leads to fusion. Genetic evidence that an open form of syntaxin-1 (Dulubova et al. 1999) exhibited partial rescue of the unc-13 mutant phenotype (Richmond et al., 2001), together with biophysical and mutational data (Ma et al., 2011; Yang et al., 2015), has established that Munc13-1 plays a key role in opening syntaxin-1. Our results here demonstrate that the unc-13 mutant phenotype can also be partially rescued by the P334A mutation in UNC-18, which may enhance its activity in templating SNARE complex assembly and binding to the SNARE complex. Deletion of Tomosyn/TOM-1, a negative regulator of SNARE complex assembly, also partially rescues the unc-13 mutant's phenotype (McEwen et al., 2006). The rescuing effect of tom-1 null and unc-18(P334) mutants exhibit a strong synergy, although the underlying mechanism is still unclear. Importantly, such synergy was not observed between unc-64/syntaxin-1 open mutant and tom-1 null mutants in the rescue of defects in unc-13 null animals (McEwen et al., 2006). Tomosyn is known to inhibit release because it contains a domain homologous to the synaptobrevin SNARE motif that can form a tight four-helix bundle with syntaxin-1 and SNAP-25, thus preventing SNARE complex assembly (Malkin et al., 1990; Hatsuzawa et al., 2003). It is plausible that SNARE complex formation normally occurs through the Munc18–Munc13–dependent pathway and that Tomosyn inhibits release through this same pathway. In this case, it is unlikely that mutant UNC-18/Munc18-1 acts merely by decreasing its binding to the closed syntaxin-1 conformation, as such decrease would be expected to facilitate formation of both the SNARE complex and the Tomosyn–syntaxin-1–SNAP-25 complex. Therefore, the synergy with the Tomosyn deletion could arise if the P334A mutation specifically increases the binding of UNC-18/Munc18-1 to synaptobrevin and/or to the SNARE complex, but not to Tomosyn and/or the Tomosyn–syntaxin-1–SNAP-25 complex. Alternatively, formation of the Tomosyn–syntaxin-1–SNAP-25 complex may follow a parallel pathway that does not involve Munc18-1 and Munc13 but competes with the Munc18-1–Munc13–dependent pathway.
We favor the latter model to explain our results. This model is more consistent with the role of Munc18-1 in templating the SNARE complex assembly (Baker et al., 2015; Sitarska et al., 2017). At the plasma membrane, syntaxin-1 is assumed to switch between its SNAP-25-bound state and its Munc18-1-bound state, the latter of which is more prevalent due to the higher affinity of syntaxin-1 for Munc18-1 (Ma et al., 2013). When Munc18-1 forms a binary complex with syntaxin-1, it proceeds to template SNARE complex formation in a Munc13-dependent and NSF-SNAP-resistant manner, and likely remains bound to the SNARE complex. For Munc18-1 to selectively act as a template for SNARE complex assembly but not for formation of the syntaxin-1–SNAP-25–Tomosyn complex, it needs to bind to residues of synaptobrevin that are not conserved in Tomosyn. In this model, Tomosyn binds to the syntaxin-1–SNAP-25 binary complex irreversibly via its synaptobrevin homologous SNARE motif through an Munc18-1- and Munc13-independent manner. In this scenario, the P334A mutation would tilt the balance between the syntaxin–SNAP-25 pathway and the syntaxin–Munc18-1 pathway in favor of the latter because of the increased ability to template SNARE complex assembly, thus explaining the synergy with the Tomosyn deletion.
Much attention has been paid (naturally) to the mechanism of membrane fusion, but, from the point of view of neuroscience, understanding how fusion does not occur prematurely is equally important: regulation of the probability of release though a diversity of mechanisms in different presynaptic plasticity processes shapes the properties of neural networks and underlies multiple forms of information processing in the brain (Nicoll and Schmitz, 2005; Regehr, 2012). Munc13s are master regulators of release because of their crucial role in orchestrating SNARE complex assembly and because of their multiple domains; thus, a calmodulin-binding motif, the C1 domain and the C2B domain respond to factors that mediate some types of presynaptic plasticity, such as Ca2+ and DAG, and the C2A domain binds to RIMs and Rab3s, which in turn control other forms of short- and long-term presynaptic plasticity (Rizo and Südhof, 2012). The picture that emerges from our findings and those of previous studies is that all of these forms of regulation of synaptic vesicle fusion, which are crucial for brain function and survival, are made possible by the existence of energy barriers that hinder fusion, such as those imposed by the furled loop conformation of Munc18-1, by the closed conformation of syntaxin-1, or by Tomosyn/TOM-1. This notion adds to evidence suggesting that other proteins such as synaptotagmin-1 and complexins also play inhibitory and active roles in release (Rizo and Xu, 2015). Although a few years ago there were hotly debated arguments as to whether a protein such as complexin played an active or an inhibitory role in release, it seems that having both active and inhibitory roles is the rule rather than the exception for the components of the neurotransmitter release machinery, as in fact should be expected for proteins that control such an exquisitely regulated biological process.
Footnotes
This work was supported by Natural Sciences and Engineering Research Council of Canada Grants RGPIN-2015-06438 (S.S.), 2014-06471 (Z.P.F.), and RGPIN-2016-04574 (H.S.S.); Heart and Stroke Foundation of Ontario Grant 0171 (S.S.), Canadian Institute of Health Research Grants MOP-93665 and 130573 (S.S.) and MOP-93619 and 123250 (M.Z.); NIH Research Project Award R35 NS097333 (J.R.), which continues research performed previously under NIH Grants NS037200 and NS049044 (J.R.); and National Natural Science Foundation of China Grant 31671052 and Junior Thousand Talents Program of China and Wuhan Morning Light Plan of Youth Science and Technology Grant 2017050304010295 (S.G.). We thank Dr. Hitoshi Kitayama (Kyoto University, Kyoto, Japan) for providing us with punc-18 and pMunc18-1 plasmids. The CB81 strain with a genotype of unc-18(e81) and RM299 with unc-18(md299) were provided by the Caenorhabditis Genetics Center at the University of Minnesota, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). The VC223 strain with a genotype of tom-1(ok285) was provided by the C. elegans Reverse Genetics Core Facility at the University of British Columbia, which is part of the international C. elegans Gene Knockout Consortium. We thank Dr. Natali L. Chanaday in the Ege T. Kavalali's lab (UT Southwestern, Dallas) for a protocol and advice for culturing hippocampal neurons from postnatal mice. We thank all the members of the Zhen lab for their invaluable contributions to the C. elegans work in this study.
The authors declare no competing financial interests.
References
- Arunachalam L, Han L, Tassew NG, He Y, Wang L, Xie L, Fujita Y, Kwan E, Davletov B, Monnier PP, Gaisano HY, Sugita S (2008) Munc18-1 is critical for plasma membrane localization of syntaxin1 but not of SNAP-25 in PC12 cells. Mol Biol Cell 19:722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Südhof TC, Brose N (1999) Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400:457–461. 10.1038/22768 [DOI] [PubMed] [Google Scholar]
- Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM (2015) A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science 349:1111–1114. 10.1126/science.aac7906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Hofmann K, Hata Y, Südhof TC (1995) Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J Biol Chem 270:25273–25280. 10.1074/jbc.270.42.25273 [DOI] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, Goldstein B (2013) Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods 10:1028–1034. 10.1038/nmeth.2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Südhof TC, Rizo J (1999) A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J 18:4372–4382. 10.1093/emboj/18.16.4372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Khvotchev M, Liu S, Huryeva I, Südhof TC, Rizo J (2007) Munc18-1 binds directly to the neuronal SNARE complex. Proc Natl Acad Sci U S A 104:2697–2702. 10.1073/pnas.0611318104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dybbs M, Ngai J, Kaplan JM (2005) Using microarrays to facilitate positional cloning: identification of tomosyn as an inhibitor of neurosecretion. PLoS Genet 1:6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA (2013) Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods 10:741–743. 10.1038/nmeth.2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Shirataki H, Sakisaka T, Asakura T, Ohya T, Kotani H, Yokoyama S, Nishioka H, Matsuura Y, Mizoguchi A, Scheller RH, Takai Y (1998) Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron 20:905–915. 10.1016/S0896-6273(00)80472-9 [DOI] [PubMed] [Google Scholar]
- Gao S, Zhen M (2011) Action potentials drive body wall muscle contractions in Caenorhabditis elegans. Proc Natl Acad Sci U S A 108:2557–2562. 10.1073/pnas.1012346108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gengyo-Ando K, Kitayama H, Mukaida M, Ikawa Y (1996) A murine neural-specific homolog corrects cholinergic defects in Caenorhabditis elegans unc-18 mutants. J Neurosci 16:6695–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva EO, Burdina AO, Holgado AM, Berthelot-Grosjean M, Ackley BD, Hadwiger G, Nonet ML, Weimer RM, Richmond JE (2006) Tomosyn inhibits synaptic vesicle priming in Caenorhabditis elegans. PLoS Biol 4:e261. 10.1371/journal.pbio.0040261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han GA, Malintan NT, Collins BM, Meunier FA, Sugita S (2010) Munc18-1 as a key regulator of neurosecretion. J Neurochem 115:1–10. [DOI] [PubMed] [Google Scholar]
- Han GA, Malintan NT, Saw NM, Li L, Han L, Meunier FA, Collins BM, Sugita S (2011) Munc18-1 domain-1 controls vesicle docking and secretion by interacting with syntaxin-1 and chaperoning it to the plasma membrane. Mol Biol Cell 22:4134–4149. 10.1091/mbc.E11-02-0135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han GA, Bin NR, Kang SY, Han L, Sugita S (2013) Domain 3a of Munc18-1 plays a crucial role at the priming stage of exocytosis. J Cell Sci 126:2361–2371. 10.1242/jcs.126862 [DOI] [PubMed] [Google Scholar]
- Han GA, Park S, Bin NR, Jung CH, Kim B, Chandrasegaram P, Matsuda M, Riadi I, Han L, Sugita S (2014) A pivotal role for Pro335 in balancing the dual functions of Munc18-1 domain-3a in regulated exocytosis. J Biol Chem 289:33617–33628. 10.1074/jbc.M114.584805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Jiang T, Han GA, Malintan NT, Xie L, Wang L, Tse FW, Gaisano HY, Collins BM, Meunier FA, Sugita S (2009) Rescue of Munc18-1 and -2 double knockdown reveals the essential functions of interaction between Munc18 and closed syntaxin in PC12 cells. Mol Biol Cell 20:4962–4975. 10.1091/mbc.E09-08-0712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SD, Broadie K, van de Goor J, Rubin GM (1994) Mutations in the Drosophila Rop gene suggest a function in general secretion and synaptic transmission. Neuron 13:555–566. 10.1016/0896-6273(94)90025-6 [DOI] [PubMed] [Google Scholar]
- Hata Y, Slaughter CA, Südhof TC (1993) Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature 366:347–351. 10.1038/366347a0 [DOI] [PubMed] [Google Scholar]
- Hatsuzawa K, Lang T, Fasshauer D, Bruns D, Jahn R (2003) The R-SNARE motif of tomosyn forms SNARE core complexes with syntaxin 1 and SNAP-25 and down-regulates exocytosis. J Biol Chem 278:31159–31166. 10.1074/jbc.M305500200 [DOI] [PubMed] [Google Scholar]
- Hosono R, Hekimi S, Kamiya Y, Sassa T, Murakami S, Nishiwaki K, Miwa J, Taketo A, Kodaira KI (1992) The unc-18 gene encodes a novel protein affecting the kinetics of acetylcholine metabolism in the nematode Caenorhabditis elegans. J Neurochem 58:1517–1525. 10.1111/j.1471-4159.1992.tb11373.x [DOI] [PubMed] [Google Scholar]
- Hu SH, Christie MP, Saez NJ, Latham CF, Jarrott R, Lua LH, Collins BM, Martin JL (2011) Possible roles for Munc18-1 domain 3a and Syntaxin1 N-peptide and C-terminal anchor in SNARE complex formation. Proc Natl Acad Sci U S A 108:1040–1045. 10.1073/pnas.0914906108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Tong XJ, Kaplan JM (2013) UNC-13L, UNC-13S, and Tomosyn form a protein code for fast and slow neurotransmitter release in Caenorhabditis elegans. Elife 2:e00967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Klingauf J, Tsien RW (1999) Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci U S A 96:12893–12900. 10.1073/pnas.96.22.12893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Liu J, Wong D, Srinivasan S, Ashrafi K (2011) Hyperactive neuroendocrine secretion causes size, feeding, and metabolic defects of C. elegans Bardet-Biedl syndrome mutants. PLoS Biol 9:e1001219. 10.1371/journal.pbio.1001219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ. (1997) p53, the cellular gatekeeper for growth and division. Cell 88:323–331. 10.1016/S0092-8674(00)81871-1 [DOI] [PubMed] [Google Scholar]
- Liu X, Seven AB, Camacho M, Esser V, Xu J, Trimbuch T, Quade B, Su L, Ma C, Rosenmund C, Rizo J (2016) Functional synergy between the Munc13 C-terminal C1 and C2 domains. Elife 5:e13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Li W, Xu Y, Rizo J (2011) Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat Struct Mol Biol 18:542–549. 10.1038/nsmb.2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Su L, Seven AB, Xu Y, Rizo J (2013) Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339:421–425. 10.1126/science.1230473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney TR, Luo S, Nonet ML (2006) Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat Protoc 1:1772–1777. 10.1038/nprot.2006.281 [DOI] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233–1238. 10.1126/science.1978757 [DOI] [PubMed] [Google Scholar]
- Martin S, Tomatis VM, Papadopulos A, Christie MP, Malintan NT, Gormal RS, Sugita S, Martin JL, Collins BM, Meunier FA (2013) The Munc18-1 domain 3a loop is essential for neuroexocytosis but not for syntaxin-1A transport to the plasma membrane. J Cell Sci 126:2353–2360. 10.1242/jcs.126813 [DOI] [PubMed] [Google Scholar]
- McEwen JM, Kaplan JM (2008) UNC-18 promotes both the anterograde trafficking and synaptic function of syntaxin. Mol Biol Cell 19:3836–3846. 10.1091/mbc.E08-02-0160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen JM, Madison JM, Dybbs M, Kaplan JM (2006) Antagonistic regulation of synaptic vesicle priming by Tomosyn and UNC-13. Neuron 51:303–315. 10.1016/j.neuron.2006.06.025 [DOI] [PubMed] [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB (1996) A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci U S A 93:12593–12598. 10.1073/pnas.93.22.12593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura K, Scheller R, Weis W (2000) Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 404:355–362. 10.2210/pdb1dn1/pdb [DOI] [PubMed] [Google Scholar]
- Munch AS, Kedar GH, van Weering JR, Vazquez-Sanchez S, He E, André T, Braun T, Söllner TH, Verhage M, Sørensen JB (2016) Extension of helix 12 in Munc18-1 Induces vesicle priming. J Neurosci 36:6881–6891. 10.1523/JNEUROSCI.0007-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D (2005) Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci 6:863–876. 10.1038/nrn1786 [DOI] [PubMed] [Google Scholar]
- Nonet ML, Saifee O, Zhao H, Rand JB, Wei L (1998) Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci 18:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris AD, Kim HM, Colaiácovo MP, Calarco JA (2015) Efficient genome editing in Caenorhabditis elegans with a toolkit of dual-marker selection cassettes. Genetics 201:449–458. 10.1534/genetics.115.180679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisotto D, Pfau M, Scheutzow A, Wild K, Mayer MP, Malsam J, Sinning I, Söllner TH (2014) An extended helical conformation in domain 3a of the SM protein Munc18-1 provides a template for SNARE (soluble N-ethylmaleimide sensitive factor attachment protein receptor) complex assembly. J Biol Chem 289:9639–9650. 10.1074/jbc.M113.514273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG. (2012) Short-term presynaptic plasticity. Cold Spring Harb Perspect Biol 4:a005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM (1999) One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci 2:791–797. 10.1038/12160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM (1999) UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci 2:959–964. 10.1038/14755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Weimer RM, Jorgensen EM (2001) An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 412:338–341. 10.1038/35085583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Südhof TC (2012) The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices–guilty as charged? Annu Rev Cell Dev Biol 28:279–308. 10.1146/annurev-cellbio-101011-155818 [DOI] [PubMed] [Google Scholar]
- Rizo J, Xu J (2015) The synaptic vesicle release machinery. Annu Rev Biophys 44:339–367. 10.1146/annurev-biophys-060414-034057 [DOI] [PubMed] [Google Scholar]
- Rowe J, Corradi N, Malosio ML, Taverna E, Halban P, Meldolesi J, Rosa P (1999) Blockade of membrane transport and disassembly of the Golgi complex by expression of syntaxin 1A in neurosecretion-incompetent cells: prevention by rbSEC1. J Cell Sci 112:1865–1877. [DOI] [PubMed] [Google Scholar]
- Sassa T, Harada S, Ogawa H, Rand JB, Maruyama IN, Hosono R (1999) Regulation of the UNC-18-Caenorhabditis elegans syntaxin complex by UNC-13. J Neurosci 19:4772–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze KL, Littleton JT, Salzberg A, Halachmi N, Stern M, Lev Z, Bellen HJ (1994) ROP, a Drosophila homolog of yeast Sec1 and vertebrate n-Sec1/Munc-18 proteins, is a negative regulator of neurotransmitter release in vivo. Neuron 13:1099–1108. 10.1016/0896-6273(94)90048-5 [DOI] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ (2007) Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128:183–195. 10.1016/j.cell.2006.12.016 [DOI] [PubMed] [Google Scholar]
- Sitarska E, Xu J, Park S, Liu X, Quade B, Stepien K, Sugita K, Brautigam CA, Sugita S, Rizo J (2017) Autoinhibition of Munc18-1 modulates synaptobrevin binding and helps to enable Munc13-dependent regulation of membrane fusion. Elife 6:e24278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362:318–324. 10.1038/362318a0 [DOI] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323:474–477. 10.1126/science.1161748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287:864–869. 10.1126/science.287.5454.864 [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Südhof TC, Neher E, Verhage M (2001) Munc18-1 promotes large dense-core vesicle docking. Neuron 31:581–591. 10.1016/S0896-6273(01)00391-9 [DOI] [PubMed] [Google Scholar]
- Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, Jorgensen EM (2003) Defects in synaptic vesicle docking in unc-18 mutants. Nature Neuroscience 6:1023–1030. 10.1038/nn1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MN, Littleton JT, Bhat MA, Prokop A, Bellen HJ (1998) ROP, the Drosophila Sec1 homolog, interacts with syntaxin and regulates neurotransmitter release in a dosage-dependent manner. EMBO J 17:127–139. 10.1093/emboj/17.1.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Su L, Rizo J (2010) Binding of Munc18-1 to synaptobrevin and to the SNARE four-helix bundle. Biochemistry 49:1568–1576. 10.1021/bi9021878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Yamasaki M, Straub C, Watanabe M, Tomita S (2013) Homeostatic control of synaptic transmission by distinct glutamate receptors. Neuron 78:687–699. 10.1016/j.neuron.2013.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wang S, Sheng Y, Zhang M, Zou W, Wu L, Kang L, Rizo J, Zhang R, Xu T, Ma C (2015) Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic-vesicle priming. Nat Struct Mol Biol 22:547–554. 10.1038/nsmb.3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Stawicki TM, Goncharov A, Jin Y (2013) Position of UNC-13 in the active zone regulates synaptic vesicle release probability and release kinetics. Elife 2:e01180. [DOI] [PMC free article] [PubMed] [Google Scholar]