

Summary
CD4 T cells help immune responses, but knowledge of how memory CD4 T cells are regulated and how they regulate adaptive immune responses and induce immunopathology is limited. Using adoptive transfer of virus‐specific CD4 T cells, we show that naive CD4 T cells undergo substantial expansion following infection, but can induce lethal T helper type 1‐driven inflammation. In contrast, memory CD4 T cells exhibit a biased proliferation of T follicular helper cell subsets and were able to improve adaptive immune responses in the context of minimal tissue damage. Our analyses revealed that type I interferon regulates the expansion of primary CD4 T cells, but does not seem to play a critical role in regulating the expansion of secondary CD4 T cells. Strikingly, blockade of type I interferon abrogated lethal inflammation by primary CD4 T cells following viral infection, despite that this treatment increased the numbers of primary CD4 T‐cell responses. Altogether, these data demonstrate important aspects of how primary and secondary CD4 T cells are regulated in vivo, and how they contribute to immune protection and immunopathology. These findings are important for rational vaccine design and for improving adoptive T‐cell therapies against persistent antigens.
Keywords: CD4 T cell differentiation, CD4 T cell proliferation, gene regulation, inflammation, T cell
Introduction
CD4 T cells are necessary for the generation of protective immune responses following vaccination or infection. Following immunization, naive CD4 T cells proliferate and generate heterogeneous cell subsets that provide help to various arms of the immune response. These CD4 T cells can differentiate into T helper 1 (Th1), Th2, Th17, Th22, T follicular helper (Tfh), or T regulatory cells.1, 2, 3, 4 Two of these subsets (Th1 and Tfh cells) play critical roles in the control of viral infections and therefore have been a focus in vaccine research.2, 3, 5, 6, 7, 8, 9 Th1 cells express high levels of interferon‐γ (IFN‐γ), granzyme B, T‐bet and Ly6c, among other molecules, and are known to facilitate cell‐mediated immunity, whereas Tfh cells express ICOS and the follicle homing molecule CXCR5, which allows these cells to localize to germinal centres to help cognate B cells.2, 3 The process by which Tfh cells help B cells following immunization or acutely controlled infection is also dependent on the expression of various molecules, including interleukin‐21 and CD40L.
During chronic viral infection CD4 T cells also help adaptive immune responses. Absence of CD4 T cells at the time of a chronic viral challenge results in a protracted infection associated with severely impaired CD8 T‐cell and antibody responses.1, 5, 10, 11 Consistent with their role in sustaining cellular and humoral immune responses, adoptive transfer of virus‐specific CD4 T cells into chronically infected mice results in improvement of virus‐specific CD8 T‐cell and B‐cell responses, and reduction of viral loads.9 On the other hand, experimental ablation of CD4 T cells before chronic viral infection results in uncontrolled viral replication and severe immune exhaustion.10, 12 Nevertheless, experimental ablation of CD4 T cells can sometimes ameliorate host immunopathology during chronic viral infection, demonstrating that CD4 T cells mediate both beneficial and detrimental roles.13, 14, 15
Although CD4 T cells can promote lethal inflammatory responses in certain settings, these cells are typically one of the first lymphocyte subsets to undergo functional exhaustion following chronic viral infection. Interestingly, previous reports have suggested a role for type I interferon in regulating CD4 T‐cell function,7, 16, 17 but little is known about which specific CD4 T cells are regulated by type I interferon signalling.
Herein, we investigate how naive and memory CD4 T cells are regulated upon antigen challenge and we assess their specific contribution to the adaptive immune response and survival of the host following viral infection. We found that primary CD4 T cells derived from naive precursors expanded significantly more than secondary CD4 T cells derived from memory precursors following either acutely controlled infection or chronic infection. Importantly, primary and secondary CD4 T cells were regulated differently by type I interferon and induced different outcomes in the host. These data improve the current understanding of how memory CD4 T cells improve adaptive immunity, and provide a framework for the optimal design of vaccines and adoptive T‐cell therapies.
Materials and methods
Mice, infections and treatments
Approximately 6‐ to 8‐week‐old C57BL/6 mice from Jackson Laboratories (Bar Harbor, ME) were used as recipients in all experiments. SMARTA CD4 T‐cell receptor (TCR) transgenic mice (JAX Stock No. 030450) were originally developed by Drs Annette Oxenius, Rolf Zinkernagel and Hans Hengartner.18 For generating memory SMARTA CD4 T cells, naive CD45.1+ SMARTA cells were injected intravenously into CD45.2+ recipients, followed by lymphocytic choriomeningitis virus (LCMV) Armstrong immunization 1 day after. After 30 days, memory SMARTAs were MACS purified by negative selection using the mouse CD4 isolation kit (STEMCELL, Cambridge, MA, USA), and FACS sorted to ≥ 98% purity based on CD45.2 and CD8 exclusion, resulting in untouched memory SMARTA cells for adoptive transfers. The number of transferred CD4 T cells depended on the experimental question. Low numbers of co‐transferred CD4 T cells (105) allowed for longitudinal analyses of primary and secondary CD4 T‐cell responses in the same host, in the absence of excessive inflammation and mortality, whereas higher numbers of transferred CD4 T cells allowed for a comparative analysis of how primary or secondary CD4 T cells induced immune protection, immunopathology and mortality. Mice received acutely controlled antigen challenges or chronic antigen challenges 1 day after adoptive cell transfer. LCMV Cl‐13 challenges were performed intravenously at a dose of 2 × 106 plaque‐forming units (PFU). LCMV Armstrong challenges were performed intraperitoneally at a dose of 2 × 105 PFU. VV‐GP challenges were performed intraperitoneally at a dose of 2 × 105 PFU. LM‐GP61 challenges were performed intravenously at a dose of 2 × 105 CFU. LCMV stocks were prepared using BHK‐21 cells. IFNAR1 blocking antibodies (MAR1‐5A3) or isotype IgG control were administered at 1 mg per mouse on days −1, 0 and every 2 days after viral infection. Antibodies for in vivo treatments were purchased from BioXCell (West Lebanon, NH). These experiments were carried out in accordance with the recommendations of the Northwestern University Center for Comparative Medicine. All our animal experiments were performed following the guidelines of our approved animal protocol, set by the Northwestern University Institutional Animal Care and Use Committee (IACUC). All mice were treated and handed in accordance with the guidelines established by Northwestern University IACUC.
Flow cytometry
Intracellular cytokine staining was performed following the BD fixation and permeabilization protocol (Cytofix/Cytoperm, Perm Wash; BD Biosciences, Franklin Lakes, NJ) after 5‐hr peptide stimulation with GP61‐80 peptide (ANASPEC) in the presence of GolgiPlug and GolgiStop (BD Biosciences, San Jose, CA, USA). LCMV MHC class I tetramers were obtained from the NIH tetramer facility at Emory University. All antibodies were purchased from BD Biosciences. Samples were acquired using a Becton Dickinson LSRII and analysed using flowjo (FlowJo LLC, Ashland, Or, usa).
Histology
Mice were killed, and an incision was made in the abdomen and the back of the head, followed by immersion into Bouin's fixative (Polysciences, Inc., Warrington, PA). Haematoxylin & eosin stains were performed on the indicated tissues at day 7 following LCMV Cl‐13 challenge.
Microarrays and transcriptomics analysis
Microarrays were performed as described previously with three mice per group19, 20 and data were uplodaded (GSE number during process). Primary and memory SMARTA cells were MACS‐purified by negative selection (STEMCELL) and then FACS‐sorted to ≥ 98% purity on a FACS Aria (BD Biosciences) according to congenic marker expression (CD45.1+ for secondary, and CD45.1+ CD45.2+ for primary, CD4 T‐cell responses). Sorted cells were spun and resuspended in 1 ml of TRIzol (Life Sciences, Waltham, MA, USA), and stored at −80°. The next day, RNA was extracted with the RNAdvance Tissue Isolation kit (Agencourt, Beverly, MA, USA), and cDNA synthesis was performed using the Ovation Pico WTA v2 kit (NuGEN). cDNA was fragmented and biotinylated using the Encore Biotin Module 4200 (NuGEN), and hybridized to Mouse Genome 430 v2.0 chip (Affymetrix, Santa Clara, CA, USA) at the Microarray Core of Dana Farber Cancer Institute. Analysis of the genome array output data was conducted using the R statistical language and the limma statistical package from Bioconductor (www.bioconductor.org).21 First, arrays displaying unusually low median intensity, low variability, or low correlation relative to the bulk of the arrays were tagged as outliers and were discarded from the rest of the analysis. Quantile normalization, followed by a log2 transformation using the Bioconductor package limma, was applied to process microarrays. The limma package was used to fit a linear model to each probe and to perform a moderated Student's t‐test on various differences of interest. For data mining and functional analyses, genes that satisfied a P‐value < 0·05 were selected. Probes that did not map to annotated RefSeq genes and control probes were removed. When indicated, the expected proportions of false positives were estimated from the unadjusted P‐value. Enriched biological pathways were performed using gene set enrichment analyses.22 Gene interaction networks were generated using geneMANIA23, 24 and pathway module analyses was performed using cytoscape.25 The transcriptional data sets in this report are uploaded (ncbi.nlm.nih.gov) (GSE92474).
Statistical analysis
Survival plots were analysed using the Mantel–Cox test. All other data were analysed with the Mann–Whitney test using graphpad software (Prism, La Jolla, CA, USA).
Results
Secondary CD4 T cells are tightly regulated following chronic viral challenge
To evaluate how primary and secondary CD4 T cells are regulated following chronic viral infection, we performed adoptive transfers of splenic naive or memory virus‐specific CD4 T cells (Fig. 1a); 105 naive transgenic, congenically marked CD4 T cells specific for LCMV (CD45.1+ I‐Abgp66‐77 SMARTA cells) were adoptively transferred into naive CD45.2+ recipient mice. On the following day, recipient mice were immunized with LCMV Armstrong to generate memory CD4 T cells. After day 30 post‐immunization, ~ 50% of primary SMARTA cells exhibited a Th1 cell phenotype, whereas only ~ 35% of primary SMARTA cells exhibited a Tfh cell phenotype, using T‐bet and CXCR5 markers (ref. 2 and data not shown). We MACS‐purified and FACS‐purified primary SMARTA cells from spleen to > 98% and co‐transferred these with an equal number of naive SMARTA cells into naive recipient CD45.2+ mice. We corroborated that memory SMARTA cells were antigen‐experienced based on CD44 expression (Fig. 1b). One day after, the recipient mice were challenged with chronic LCMV Cl‐13 to assess CD4 T‐cell expansion.
Figure 1.
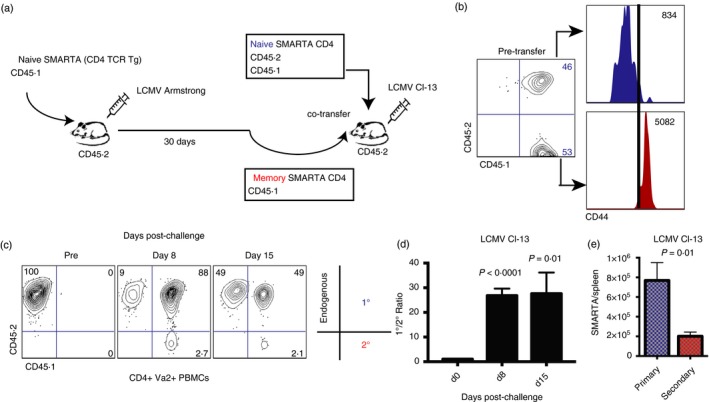
Memory CD4 T cells are tightly regulated following chronic viral challenge. (a) Experimental outline to assess the expansion of naive versus memory CD4 T cells following chronic viral challenge. (b) CD44 expression on naive and memory SMARTA CD4 T cells before injection to assess antigen experience. (c) Representative FACS plots showing the frequencies of primary and secondary CD4 T cells at various time‐points post infection (gated on total CD4 T cells). (d) Summary of the ratio between primary and secondary CD4 T‐cell responses. (e) Number of SMARTA cells in spleen at day 8 after lymphocytic choriomeningitis virus (LCMV) Cl‐13 infection. 105 naive and 105 memory SMARTA cells from spleen were co‐transferred (1 : 1 ratio) into congenically distinct recipient mice, followed by LCMV Cl‐13 challenge 1 day after CD4 T‐cell transfer. SMARTA cells were assessed in peripheral blood mononuclear cells (PBMCs) in all panels unless indicated otherwise. Data are from two experiments, n = 4 or n = 5 mice/group per experiment. Error bars indicate SEM. [Colour figure can be viewed at wileyonlinelibrary.com]
Strikingly, at day 8 post‐infection, there were 27‐fold greater numbers of primary SMARTA CD4 T cells (derived from naive precursors) relative to secondary SMARTA CD4 T cells (derived from memory precursors) (Fig. 1c,d, P < 0·0001). The pattern of increased primary CD4 T‐cell responses relative to secondary CD4 T‐cell responses was also evident in spleen. Primary CD4 T cells also exhibited higher immune activation than secondary CD4 T cells (see Supplementary material, Fig. S1). These results demonstrated that secondary CD4 T‐cell responses were more tightly regulated than primary CD4 T‐cell responses.
Secondary CD4 T cells also undergo tight regulation following acutely controlled challenges
The previous experiments demonstrated that memory CD4 T cells expand minimally following chronic viral challenge. This suggested a role for persistent antigen in curtailing the recall of memory CD4 T cells. To ascertain this, we interrogated whether secondary CD4 T‐cell responses were also tightly regulated after acutely controlled pathogen challenges. We performed adoptive co‐transfers of naive and memory CD4 T cells (similar to those in Fig. 1a), but challenging mice with acutely controlled antigens instead, which included viral (LCMV Armstrong and vaccinia‐GP) and bacterial (Listeria‐GP61) challenges that were all cleared within a week (Fig. 2a). Similar to our experiments with chronic LCMV Cl‐13 challenge, all acutely controlled challenges resulted in limited expansion of memory CD4 T cells (Fig. 2b–e). These findings demonstrate that secondary CD4 T‐cell responses are more tightly regulated than primary CD4 T‐cell responses by a mechanism that is not dependent on antigen persistence.
Figure 2.
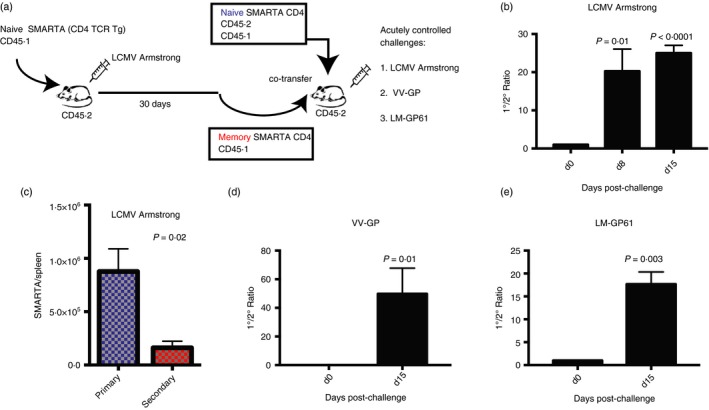
Memory CD4 T cells are also tightly regulated following acutely controlled challenges. (a) Experimental outline to assess the expansion of naive versus memory CD4 T cells following acutely controlled challenges. (b) Summary of the ratio between primary and secondary CD4 T‐cell responses following lymphocytic choriomeningitis virus (LCMV) Armstrong challenge. (c) Number of SMARTA cells in spleen at day 8 after LCMV Armstrong infection. (d) Summary of the ratio between primary and secondary CD4 T‐cell responses following vaccinia‐GP (VV‐GP) challenge. (e) Summary of the ratio between primary and secondary CD4 T‐cell responses following Listeria‐GP61 (LM‐GP61) challenge. 105 naive and 105 memory SMARTA cells from spleen were co‐transferred (1 : 1 ratio) into congenically distinct recipient mice, followed by various acutely controlled antigen challenges 1 day after CD4 T‐cell transfer. SMARTA cells were assessed in peripheral blood mononuclear cells (PBMCs) in all panels unless indicated otherwise. Data are from two experiments, n = 3 or n = 4 mice/group per experiment. Error bars indicate SEM. [Colour figure can be viewed at wileyonlinelibrary.com]
We then compared the early kinetics of primary and secondary virus‐specific CD4 T‐cell responses following acutely controlled infection and chronic infection. Our studies demonstrate that acutely controlled infection results in delayed priming of SMARTA cells within the hyperacute phase of infection (96 hr), but after this, acutely controlled infection resulted in greater CD4 T‐cell responses relative to chronic infection (see Supplementary material, Fig. S2). These data also demonstrate that secondary CD4 T cells derived from memory precursors are also tightly regulated during the hyperacute phase of viral infection.
One of the limitations of studying CD4 T‐cell responses is that they can disappear following immunization.26 Class II tetramers can effectively identify LCMV‐specific CD4 T cells during the early, but not the late stages of an LCMV Armstrong infection, suggesting a time‐dependent mechanism by which memory CD4 T cells reduce TCR recognition (data not shown). To assess potential TCR down‐regulation by virus‐specific CD4 T cells, we interrogated whether memory SMARTA cells down‐regulate their TCR Va2 chain during memory differentiation. Intriguingly, our data demonstrate that TCR Va2 progressively declines after infection (see Supplementary material, Fig. S3).
Biased T helper cell subset expansion of primary and secondary CD4 T cells
We then interrogated T helper subset differentiation of adoptively transferred naive and memory CD4 T cells following viral challenge. Secondary CD4 T cells derived from memory precursors exhibited a greater expansion in Tfh cell subsets, whereas primary CD4 T cells derived from naive precursors exhibited greater expansion in Th1 cell subsets (average CXCR5+ was 69% for secondary, and only 44% for primary CD4 T cells; P = 0·01) (Fig. 3a,b). Primary CD4 T cells expressed higher levels of granzyme B (Fig. 3c) and IFN‐γ (average IFN‐γ + was 66% for primary, and only 18% for secondary CD4 T cells; P < 0·0001) (Fig. 3d,e), but interleukin‐2 expression was not significantly different between secondary and primary CD4 T cells (P = 0·1). A similar pattern of phenotypic differentiation was observed when we co‐transferred higher numbers (5 × 106) of naive and memory CD4 T cells (see Supplementary material, Fig. S4), suggesting that the number of transferred cells did not substantially affect the relative distribution of CD4 T‐cell helper subsets. Altogether, these phenotypic differences suggested that primary and secondary CD4 T cells may play different roles during viral infection.
Figure 3.
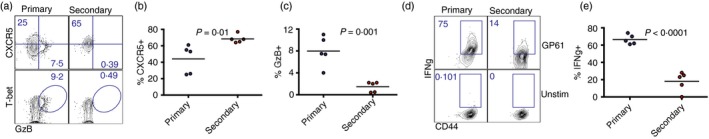
Memory CD4 T cells exhibit biased expansion of follicular helper T (Tfh) cell subsets. (a) Representative FACS plots showing the expression of Tfh and T helper type 1 (Th1) markers on primary and secondary SMARTA CD4 T cells. (b) Summary of CXCR5 expression. (c) Summary of granzyme B expression. (d) Representative FACS plots showing the expression of interferon‐γ (IFN‐γ) on primary and secondary SMARTA CD4 T cells. (e) Summary of IFN‐γ expression. 105 naive and 105 memory SMARTA cells from spleen were co‐transferred (1 : 1 ratio) into different recipient mice, followed by lymphocytic choriomeningitis virus (LCMV) Cl‐13 challenge 1 day after (similar to Fig. 1). Data are from spleen at day 7 following LCMV Cl‐13 challenge. Data shown are from one experiment. Experiment was repeated with similar results, n = 5 mice/group per experiment. [Colour figure can be viewed at wileyonlinelibrary.com]
Primary CD4 T cells are substantially more inflammatory than secondary CD4 T cells
As mentioned earlier, primary CD4 T cells derived from naive precursors exhibited a biased expansion of Th1 cell subsets, whereas secondary CD4 T cells derived from memory precursors showed a biased expansion of Tfh cell subsets. Since uncontrolled Th1 polarization can cause immunopathological disease,15 we reasoned that primary, but not secondary, CD4 T cells could cause mortality when present in high numbers. We transferred a high number (5 × 106 cells) of naive or memory SMARTAs into naive recipient mice, and challenged these with LCMV Cl‐13 the next day to compare host survival and immunopathology.
Mice that received naive SMARTA cells exhibited only a 20% survival and exaggerated weight loss by day 15 post‐challenge, whereas mice that received memory SMARTA cells showed 100% survival and only 7% weight loss (P = 0·0003) (Fig. 4a). Mortality was also observed in mice that received a high number of naive SMARTA cells followed by challenge with acutely controlled LCMV Armstrong or vaccinia‐GP (data not shown), suggesting that antigen persistence was not necessary to induce lethal immunopathology.
Figure 4.
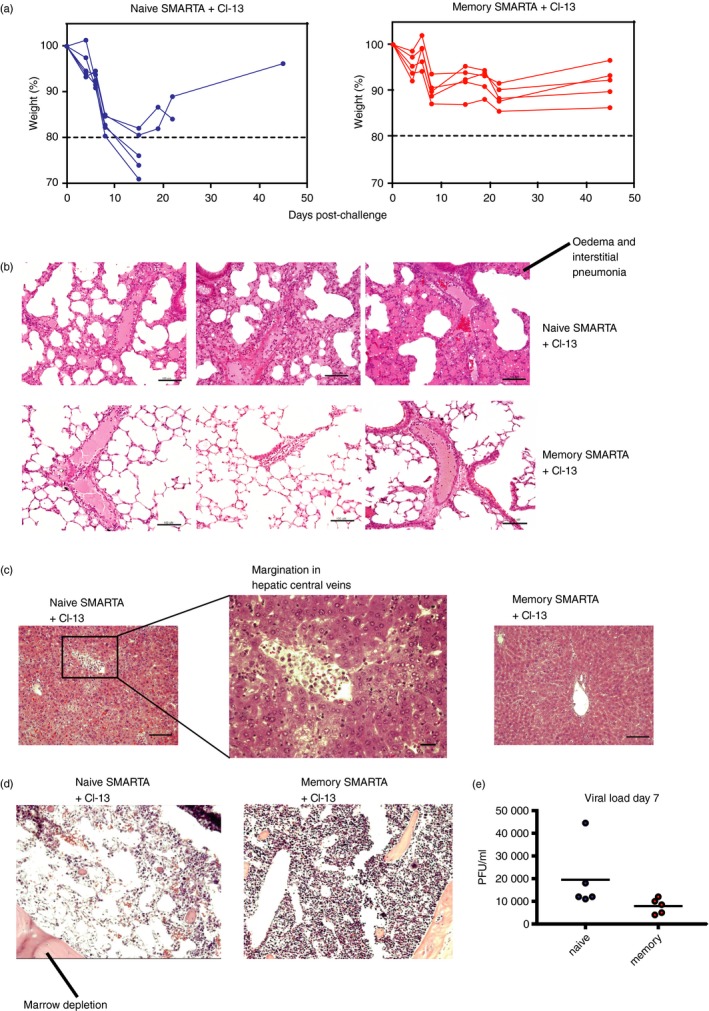
Memory CD4 T cells are significantly less inflammatory than naive CD4 T cells. (a) Survival plots. (b) Haematoxylin & eosin (H&E) stain of lung from different mice. (c) H&E stain of liver. (d) H&E stain of bone marrow. (e) Viral loads in sera at day 7 post‐challenge. 5 × 106 naive or 5 × 106 memory SMARTA cells from spleen were transferred into congenically distinct recipient mice, followed by lymphocytic choriomeningitis virus (LCMV) Cl‐13 challenge 1 day after. Histopathology data are from day 7 following LCMV Cl‐13 challenge. These experiments represent single SMARTA transfers (naive versus memory) to specifically assess the effect of transferring a high number of each cell population in host survival and immunopathology. Note that lower numbers of adoptively transferred CD4 T cells (105) do not result in mortality, and so, only high cell numbers enable us to detect differences in immunopathology. Scale bars represent 100 μm. Data shown are from one experiment. Experiment was repeated with similar results, n = 3 to n = 5 mice/group per experiment. [Colour figure can be viewed at wileyonlinelibrary.com]
Histological analyses 7 days after LCMV Cl‐13 challenge revealed excessive pulmonary oedema (Fig. 4b), abnormal monocyte margination at the hepatic endothelium (Fig. 4c), and bone marrow depletion (Fig. 4d) in the mice that received a high dose of naive, but not memory, CD4 T cells. There was a pattern of decreased viral loads in mice that received memory SMARTA CD4 T cells relative to mice that received naive SMARTA cells, but this was not statistically significant (P = 0·1) (Fig. 4e). Altogether, these findings demonstrate that primary CD4 T cells can be lethal and substantially more inflammatory than secondary CD4 T cells.
Memory CD4 T cells improve humoral and cytotoxic responses
As shown previously, secondary CD4 T cells derived from memory precursors are enriched in Tfh cell subsets. Therefore, we hypothesized that secondary CD4 T cells could be better poised to help humoral responses relative to primary CD4 T cells. To test this, we transferred 106 naive or 106 memory SMARTA cells into naive recipient mice and infected these with LCMV Cl‐13 1 day after. Consistent with our hypothesis, we noticed a slight, but not statistically significant (P = 0·06) increase in the numbers of germinal centre B cells in mice that received memory CD4 T cells relative to those that received naive CD4 T cells (Fig. 5a). This increase in germinal centre B‐cell responses in mice that received memory CD4 T cells was associated with 14·6‐fold higher antibody levels (P = 0·02) (Fig. 5b), and sixfold greater viral control (P = 0·05) (Fig. 5c) relative to mice that received naive CD4 T cells at day 14. Although virus‐specific CD8 T cells expanded similarly during the early phase of the chronic infection, mice that received memory SMARTAs exhibited improved maintenance of virus‐specific CD8 T cells at late time‐points (Fig. 5d,e). Taken together, our data suggest that memory CD4 T‐cell responses exhibit limited anamnestic expansion, but are nonetheless better poised to help adaptive immune responses compared with naive CD4 T‐cell responses.
Figure 5.
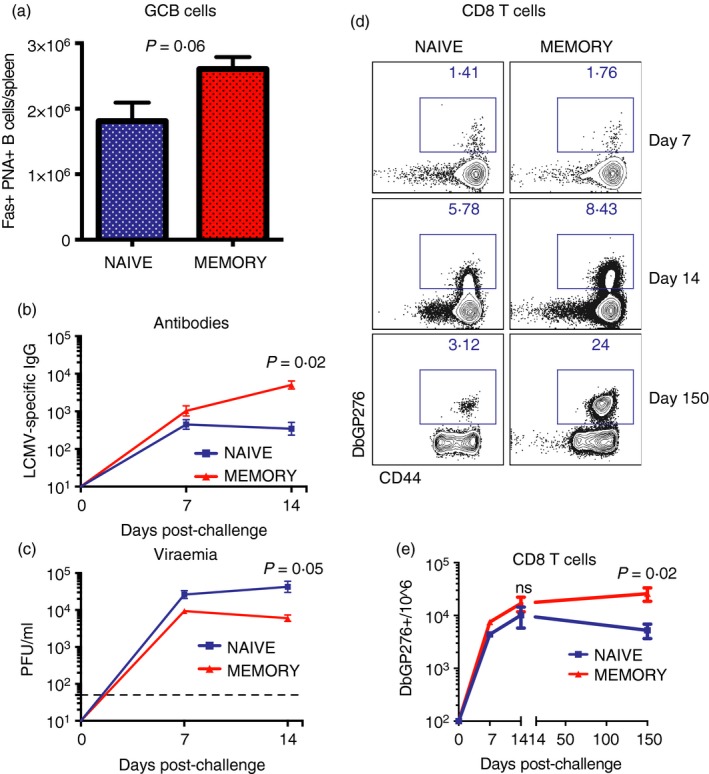
Memory CD4 T cells are more effective at helping B‐cell responses and sustaining CD8 T‐cell responses compared with naive CD4 T cells. (a) Summary of germinal centre (GC) B‐cell responses. (b) Summary of antibody responses. (c) Summary of viral control. (d) Representative FACS plots showing the frequencies of lymphocytic choriomeningitis virus (LCMV) ‐specific CD8 T‐cell responses at various time‐points. (e) Summary of LCMV‐specific CD8 T‐cell responses. 106 naive or 106 memory SMARTA cells from spleen were transferred into different recipient mice, followed by LCMV Cl‐13 challenge 1 day after. (a) Spleen at day 14; (b, c) sera and (d, e) are from peripheral blood mononuclear cells (PBMCs). These experiments represent single SMARTA transfers (naive versus memory) to specifically assess their contribution to the immune response. Data are from two experiments, n = 4 or n = 5 mice/group per experiment. Error bars indicate SEM. [Colour figure can be viewed at wileyonlinelibrary.com]
Type I interferon signalling regulates primary CD4 T cells and determines host survival
The data from above showed that memory CD4 T cells can provide help to adaptive immune responses despite their limited recall expansion. To elucidate the transcriptional signature of ‘memory CD4 T‐cell help’, we compared gene expression of primary and secondary SMARTA cells at day 7 post‐challenge. The experiment setup was similar to that of Fig. 1, but MACS‐purifying CD4 T cells and FACS‐sorting primary and secondary SMARTA cells (based on CD45.1/CD45.2 expression) from spleen at day 7 after LCMV Cl‐13 challenge for gene expression analyses. This resulted in untouched splenic CD4 T cells for transcriptional analyses. Primary and secondary CD4 T cells showed distinct transcriptional profiles (Fig. 6a,b), including various genes involved in cell division pathways (Fig. 6c), consistent with the low recall expansion of memory CD4 T cells. In addition, primary CD4 T cells were enriched in various metabolic pathways, such as oxophos, glycolysis and mitochondrial import, suggestive of high metabolic activity in the rapidly proliferating primary CD4 T‐cell response (Fig. 6d). Moreover, inhibitory and co‐stimulatory receptor gene analyses showed that primary CD4 T cells expressed higher levels of Cd160, Ctla4, Lag3, Pdcd1, Cd28, whereas secondary CD4 T cells exhibited greater expression of Cd86 and Cd244 (Fig. 6e,f).
Figure 6.
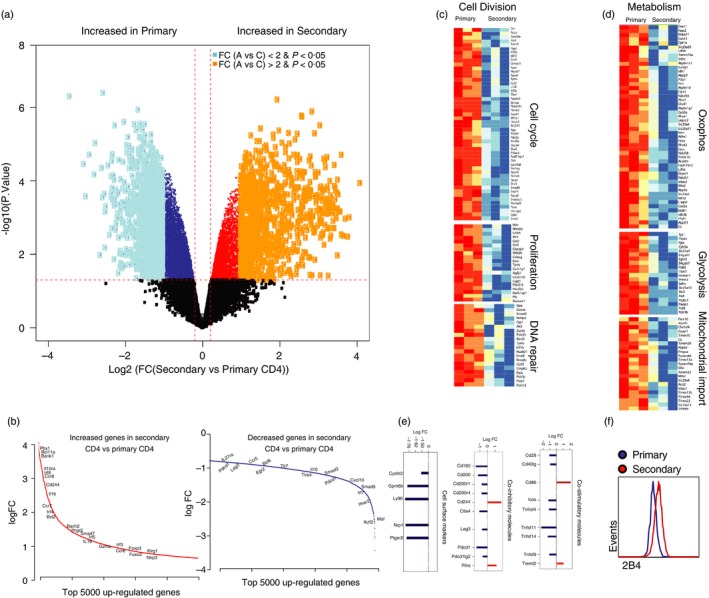
Transcriptional signature of primary and secondary CD4 T‐cell responses. (a) Volcano plot showing the overall gene expression in secondary and primary CD4 T cells. Genes were coloured according to their statistical significance in secondary compared with primary CD4. The x‐axis represents the log 2‐fold change (FC) expression in secondary versus primary and y‐axis represents the –log 10 P‐value. The horizontal red line represents the P‐value cut‐off of 0·05. Dark dots represent not significant genes (P < 0·05). Yellow dots represent significant genes with FC > 2 and light blue dots genes with FC < 2. Dark red (increased) and dark blue (decreased) represent the remaining significant genes (P < 0·05). (b) Illustration of selected significant leading genes ranked by their fold change from the highest to the lowest fold change. x‐axis represents the top 5000 increased (red) or decreased (blue) in secondary versus primary CD4 cells. y‐axis represents the log 2‐fold change expression of these genes. (c, d) Heat maps illustrating the scaled expression of the top leading genes of the top pathways that are significantly enriched by gene set enrichment analyses (FDR 5%) in secondary versus primary CD4 T cells. (e) Regulation of co‐stimulatory and co‐inhibitory receptor gene by memory and naive CD4 T cells following viral infection. (f) Representative FACS plots showing 2B4 expression. Bar plots representative of the log 2‐fold change expression in secondary versus primary CD4 cells for cell surface markers; co‐inhibitory and co‐stimulatory molecules. Genes that are decreased in secondary versus primary CD4 are depicted in blue, and genes that are increased in secondary versus primary CD4 are depicted in red. Experimental set‐up was similar to that in Fig. 1, FACS‐sorting primary and secondary SMARTA cells from spleen at day 7 for gene expression analyses. [Colour figure can be viewed at wileyonlinelibrary.com]
Many cellular pathways were differentially enriched by gene set enrichment analysis scoring (Fig. 7a), and transcription factor expression analyses revealed that primary CD4 T cells were enriched in the transcriptional regulators Ikzf2 (Helios), Prdm15 and Prdm16 (Fig. 7b,c). Moreover, primary CD4 T cells were enriched in the Th1 transcription factor T‐bet, whereas secondary CD4 T cells were enriched in Eomes (Fig. 7c). Primary CD4 T cells also showed enrichment in TCR signalling, mammalian target of rapamycin signalling, and type I interferon signalling (Fig. 7d).
Figure 7.
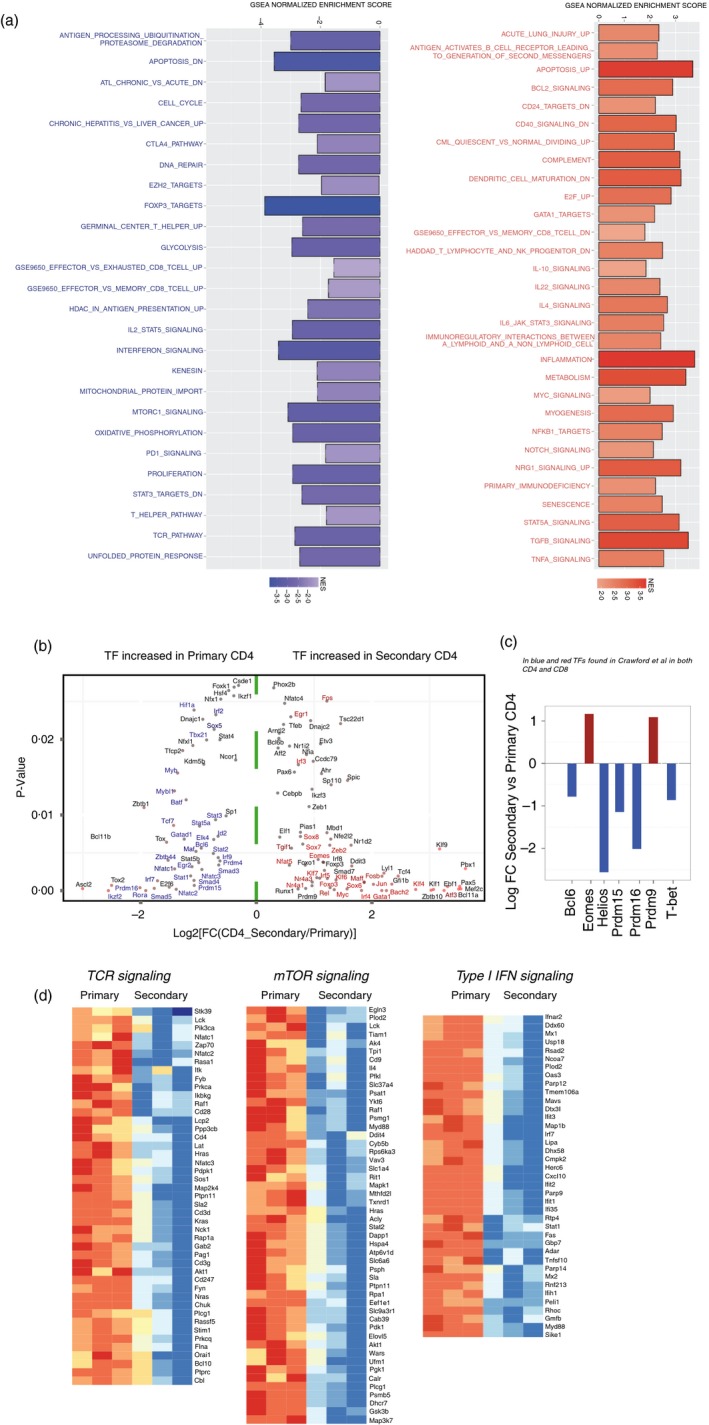
Regulation of transcription factor genes and type I interferon‐induced genes by primary and secondary CD4 T cells following viral infection. (a) Gene set enrichment analyses normalized enrichment score showing various cellular pathways that were differentially enriched in primary versus secondary CD4 T cells. Pathways increased in secondary CD4 T cells are shown in red (right), and pathways increased in primary CD4 T cells are shown in blue (left). (b) 2D plot showing the transcription factors (TF) that are significantly (P < 0·05) increased (red) or decreased (blue) in secondary versus primary CD4 T cells. x‐axis represents the log 2‐fold change and y‐axis the P‐value. (c) Bar plot representation of the log 2‐fold change in secondary versus primary CD4 T cells of TFs found to be expressed in both CD4 and CD8 T cells in Crawford et al.48 (d) Heat map representing the log 2‐fold change gene expression for the top downstream targets of T‐cell receptor (TCR) signalling, mammalian target of rapamycin (mTOR) signalling, and type I interferon signalling increased or decreased in secondary versus primary CD4 T cells. Experiment set‐up was similar to that in Fig. 1, FACS‐sorting primary and secondary SMARTA cells from spleen at day 7 for gene expression analyses. [Colour figure can be viewed at wileyonlinelibrary.com]
To ascertain the role of type I interferon signalling on CD4 T‐cell expansion and immunopathology, we transferred high numbers (5 × 106) of naive or memory SMARTA cells into naive recipient mice, and infected the mice 1 day after with LCMV Cl‐13. Mice were treated with anti‐IFNAR1 blocking antibody or isotype control as previously described.16 Interestingly, mice that received naive SMARTA cells and chronic viral challenge exhibited a more striking expansion of primary CD4 T cells following IFNAR1 blockade (3·6‐fold, P = 0·006) (Fig. 8a), but such treatment did not increase the expansion of secondary CD4 T cells (Fig. 8b). Importantly, mice that received naive SMARTA cells and IFNAR1 blocking antibodies did not succumb despite the increased primary CD4 T‐cell response (Fig. 8c,d), demonstrating a role for type I interferon in regulating immunopathology by primary CD4 T‐cell responses independently of CD4 T‐cell expansion. Such abrogation of mortality following IFNAR1 blockade was associated with significantly reduced levels of granzyme B on primary virus‐specific CD4 T cells, suggesting that interferon type I signalling positively regulated cytotoxicity by CD4 T cells (Fig. 9a). Future studies will ascertain whether granzyme B expression alone by primary CD4 T cells is sufficient to induce immunopathology, which may arguably depend on cytotoxic CD4 T‐cell function. Furthermore, interferon type I blockade in mice that received naive SMARTA cells resulted in inverted T helper subset differentiation with greater numbers of Tfh cells and lower numbers of Th1 cells relative to control treated animals (Fig. 9a,c). Consistent with this increase in Tfh cell differentiation, antibody responses were substantially improved following interferon type I blockade relative to control mice (Fig. 9d,f), consistent with recent reports that show a negative role for interferon in regulating antibody responses.27, 28 Altogether, our data show that the expansion, function and regulation of memory CD4 T cells are substantially different from those of naive CD4 T cells following antigen challenge, and that type I interferon plays a critical role in regulating T helper differentiation and the balance between immunopathology and immune protection.
Figure 8.
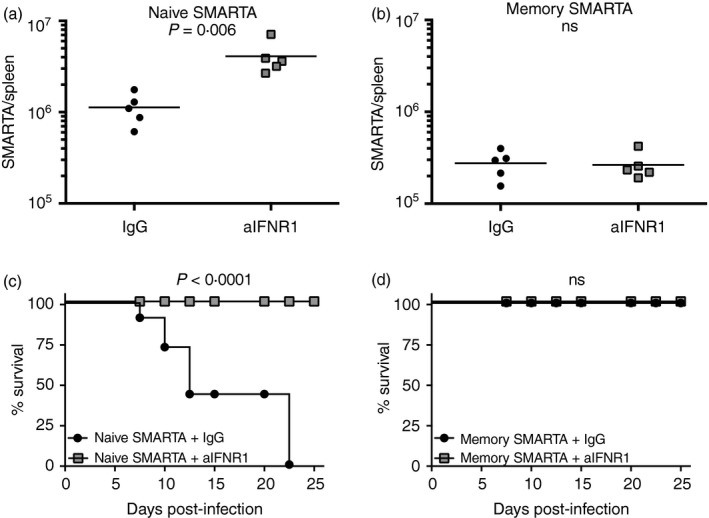
Type I interferon regulates the expansion of naive CD4 T cells and is critical for controlling lethal inflammation by primary CD4 T cells. (a) Summary of primary CD4 T‐cell responses following type I interferon blockade. (b) Summary of secondary CD4 T‐cell responses following type I interferon blockade. (c) Survival following type I interferon blockade in mice that received naive SMARTA cells. (d) Survival following type I interferon blockade in mice that received memory SMARTA cells. (a, b) Day 6 post‐infection (gated on SMARTA cells). Experiment layout was similar to that of Fig. 4, transferring 5 × 106 naive or 5 × 106 memory SMARTA cells into congenically distinct recipient mice, followed by lymphocytic choriomeningitis virus (LCMV Cl‐13 challenge 1 day later. These experiments represent single SMARTA transfers (naive versus memory) to specifically assess the effect of interferon type I blockade in host survival. Data shown are from one experiment. Experiment was repeated with similar results, n = 5 mice/group per experiment.
Figure 9.
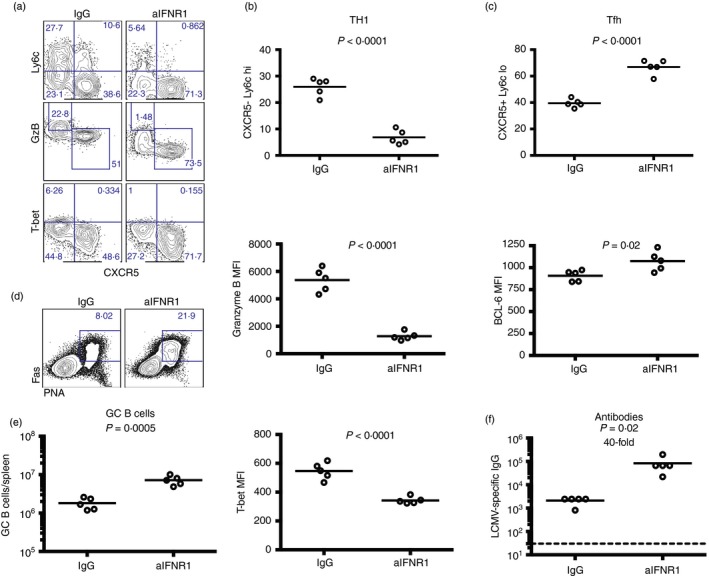
Blockade of interferon type I signalling results in biased follicular T helper (Tfh) cell differentiation. (a) Representative FACS plots showing T helper subset differentiation following type I interferon blockade (gated on CD45·1+ SMARTA cells). (b) Percentage of SMARTA cells that are T helper type 1 (Th1) and their expression of granzyme B and T‐bet. (c) Percentage of SMARTA cells that are Tfh cells and their expression of Bcl‐6. (d) Representative FACS plots showing germinal centre (GC) B cells (gated from IgM− and IgD− B220+ CD3− live lymphocytes). (e) Numbers of GC B cells. (f) lymphocytic choriomeningitis virus (LCMV) ‐specific IgG responses by ELISA. 5 × 106 naive SMARTA from spleen cells were transferred into congenically distinct recipient mice, followed by LCMV Cl‐13 challenge 1 day later, similar to Fig. 8. All panels represent data from spleen, except for (f), which is from sera. Data are from day 6 post‐infection. Data shown are from one experiment. Experiment was repeated with similar results, n = 5 mice/group per experiment. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
CD4 T cells help adaptive immune responses following acutely controlled infection or vaccination. In addition, CD4 T‐cell responses are crucial for sustaining adaptive immune responses during persistent viral infection, and so, a major goal in the field is to understand the biology and differentiation of CD4 T cells. We focused on two main CD4 T‐cell subsets that play predominant roles in the control of viral infections. These include Th1 cells, characterized by their high expression of T‐bet, granzyme B, Ly6c and IFN‐γ and Tfh cells, characterized by the expression of CXCR5, which selectively localize these cells to B‐cell follicles.8 Cognate Tfh cells provide help to B cells in part via CD40L and interleukin‐21, which provide signals to help generate neutralizing antibodies. Importantly, immune protection elicited by most licensed vaccines is thought to be dependent on antibodies, and therefore, knowledge of the pathways that skew Tfh cell differentiation is important for rational vaccine design.
Following acutely controlled infection, primary CD4 T cells are highly functional, but during chronic viral infections and cancers, they undergo functional exhaustion and deletion, which limits their ability to sustain CD8 T‐cell function and antibody responses. Consistent with this, adoptive transfer of LCMV‐specific CD4 T cells during a chronic LCMV infection results in rescue of CD8 T‐cell and antibody responses, highlighting the helper roles of CD4 T cells in maintaining immune responses to persistent antigens.9 Similarly, adoptive transfer of antigen‐specific T cells after chimeric antigen receptor engineering has been demonstrated to be a promising therapy against chronic infections and cancers, but it is currently unknown whether the level of ‘antigen experience’ of adoptively transferred CD4 T cells can impact inflammation and the clearance of the persistent antigen differently. To address this simple question, we compared the effect of transferring naive and memory CD4 T cells into mice followed by a chronic viral challenge. Intriguingly, co‐transfer of equal numbers of naive and memory CD4 T cells followed by chronic LCMV Cl‐13 challenge resulted in preferential expansion of primary CD4 T cells derived from naive precursors. Similarly, we observed a more limited expansion of secondary CD4 T cells following acutely controlled viral (LCMV Armstrong and vaccinia) and bacterial (Listeria) challenges. It is important to mention that Listeria is a potent Th1‐inducer.29 As shown in our study, primary CD4 T cells are Th1‐biased, but the increased primary CD4 T‐cell expansion in Listeria‐challenged mice may also be partially due to the infection itself. Altogether, we showed that primary CD4 T cells derived from naive precursors proliferate extensively following acute or chronic challenges, whereas secondary CD4 T cells derived from memory precursors undergo a more limited expansion. This is a counterintuitive observation, given that memory immune responses are thought to exhibit greater expansion compared with naive immune responses following antigen encounter.
A previous paper from the Ahmed laboratory showed that memory CD8 T cells expand more than naive CD8 T cells in response to acutely controlled viral challenges,30 which was strikingly different from what we report for CD4 T cells, demonstrating a critical difference between CD8 and CD4 T cells. The aforementioned paper, however, showed that memory CD8 T cells disappear during a chronic infection, which was similar to what we observe for memory CD4 T cells. Deletion of memory CD8 T cells following chronic viral challenge was shown to be dependent on the inhibitory receptor CD244, which was shown to be highly expressed on secondary, but not primary CD8 T cells.30 Similarly, we also noted that this was one of the most up‐regulated genes on memory CD4 T cells relative to primary CD4 T cells.
Overall, our findings demonstrate that secondary CD4 T cells derived from memory precursors are tightly regulated irrespective of the persistence of their cognate viral antigen. Memory CD4 T‐cell deletion is a process that may occur during breakthrough infections, and may be critical for preventing excessive tissue damage by vaccine‐induced CD4 T‐cell responses. Our results are consistent with previous reports that demonstrated limited expansion of secondary CD4 T cells following peptide or LCMV Armstrong immunization,31, 32 but we now show that secondary CD4 T cells not only expand poorly in response to rapidly controlled viral and bacterial antigens, but also chronic viral antigens that provide a more protracted inflammation and antigen stimulation. Importantly, our data demonstrate how primary and secondary CD4 T‐cell responses can impact immune protection versus immunopathology. Taken together, memory T‐cell differentiation is not a fixed process, and memory T cells can become exhausted or deleted as a way to prevent excessive tissue damage following viral infection.
The secondary CD4 T‐cell response derived from memory precursors exhibited a biased Tfh phenotype. A previous study demonstrated that memory Th1 and Tfh cells exhibit lineage commitment, since after antigen rechallenge, each memory subset tends to preserve its Th1 or Tfh profile.2 Therefore, our observation that secondary CD4 T cells were enriched in Tfh cell responses is probably explained by a preferential expansion of ‘committed Tfh cell subsets’ and not de‐differentiation of Th1 subsets into Tfh cell subsets. Moreover, we assessed the in vivo effect of transferring naive or memory CD4 T cells following viral infection. Strikingly, a high dose of naive SMARTA cells followed by LCMV Cl‐13 challenge induced exaggerated adhesion of inflammatory cells at the endothelium, and resulted in lethal pulmonary oedema and bone marrow depletion. Our data are consistent with previous data showing that high doses of naive SMARTA cells could mediate immunopathology during uncontrolled LCMV infection by a mechanism that is partially dependent on tumour necrosis factor and involves persistent viral antigen.33, 34, 35 However, we also observed immunopathology in terms of hunched posture and lethargy when mice received high doses of naive SMARTA CD4 T cells followed by various acutely controlled pathogen challenges, including LCMV Armstrong or VV‐GP (data not shown). This suggests that antigen persistence is not always required to induce CD4 T‐cell immunopathology by primary CD4 T cells, since high precursor frequencies of CD4 T cells can similarly induce immune‐mediated damage. In contrast, transfer of a high number of memory SMARTA cells followed by viral challenge improved antibody and cytotoxic responses and resulted in 100% survival with minimal weight loss. Consistent with our findings, a previous paper demonstrated that memory CD4 T cells can improve neutralizing antibody responses,36 but we now demonstrate an undescribed aspect of memory CD4 T cells, namely the ability to help adaptive immune responses in the context of minimal collateral damage (help without harm). This important feature renders memory CD4 T‐cell responses better able to protect the host, especially since uncontrolled CD4 T‐cell responses can be highly inflammatory and detrimental to the host. It is important to mention that the outcomes of our experiments were dependent on the number of CD4 T cells transferred: very low numbers of memory CD4 T cells were not sufficient to improve antibody and CD8 T‐cell responses, and low numbers of naive CD4 T cells were not sufficient to kill the mice. Therefore, the dose of CD4 T cells used in each experiment depended on the question asked, for example, to ascertain the ability of CD4 T cells to help adaptive immunity versus their potential to induce immunopathological disease we adoptively transferred low or high CD4 T‐cell numbers, respectively.
Currently, a major limitation of adoptive T‐cell therapies in cancer patients is overt inflammation following T‐cell transfer, which can sometimes threaten the patient's survival,19, 37 warranting a deeper understanding of how T‐cell responses should be harnessed. Our data demonstrate that adoptive transfer of a high number of naive, but not memory, virus‐specific CD4 T cells results in a lethal inflammation following viral challenge. At first glance, this may appear to contradict an earlier paper from our laboratory, in which we demonstrated that memory CD4 T cells elicited by various CD4 T‐cell vaccines can trigger a lethal immunopathology following chronic LCMV Cl‐13 challenge.15 The detrimental effect of memory CD4 T cells reported in this previous paper could be explained by the fact that memory CD4 T cells induced by CD4 T‐cell vaccines are widely distributed in many tissues (their numbers surpass 107), exceeding the number of CD4 T cells that can be engrafted following adoptive CD4 T‐cell transfers assuming a donor cell engraftment of 10%. Therefore, secondary CD4 T cells pose a lower risk of immunopathology relative to primary CD4 T cells, but the former could nonetheless kill the host if they are present at excessively high numbers without CD8 T cells and antibodies to help control the infection. This could only be achieved by selective CD4 T‐cell vaccination. However, single induction of CD4 T‐cell responses (in the complete absence of CD8 T‐cell responses) is difficult to achieve in nature, as many CD4 T‐cell epitopes contain shorter overlapping CD8 T‐cell epitopes,15, 20, 38, 39, 40, 41, 42 ensuring that both cytotoxic CD8 T cells and helper CD4 T cells are generated following antigen challenge.
Is there a reason why primary CD4 T‐cell responses are more enriched in Th1 subsets, whereas secondary CD4 T‐cell responses are more enriched in Tfh subsets? It is reasonable to hypothesize that, since a primary viral challenge in a naive host results in vigorous intracellular virus replication in the absence of antibodies or CD8 T cells, immune control would depend more on potent Th1 responses to help eliminate infected cells. Following resolution of this primary infection, however, cytotoxic and humoral responses are generated, providing multiple lines of defence that may confer protection upon re‐infection. Nevertheless, rechallenge with a higher dose of virus or a mutated virus could result in breakthrough infection, and a biased expansion of Tfh cells during secondary challenge would help to improve further antibody responses to achieve sterilizing protection, while minimizing Th1‐driven inflammation after each rechallenge. The preferential expansion of Tfh cells in secondary CD4 T‐cell responses may also explain the efficacy of booster immunizations at inducing potent antibody responses. In addition, biased expansion of Tfh cells over Th1 cells is also observed during chronic viral infections, such as those with LCMV, simian immunodeficiency virus and human immunodeficiency virus (HIV).6, 43, 44 In the case of HIV or LCMV Cl‐13, generation of neutralizing antibodies occurs only after months or years of uncontrolled viral replication, which coincides with the gradual accumulation of Tfh cells and deletion of Th1 cells.45, 46, 47 Altogether, our data and that of others using multiple experimental systems suggest that continuous TCR signalling preferentially expands Tfh cell responses, which appears to be an evolutionarily conserved feature of the virus‐specific CD4 T‐cell response.
To understand the molecular pathways that regulate primary and secondary CD4 T‐cell responses, we performed transcriptional profiling. Of note, Cd244, which encodes inhibitory receptor 2B4, was enriched in secondary CD4 T‐cell responses relative to primary CD4 T‐cell responses. A previous study demonstrated that high 2B4 expression on secondary CD8 T cells is also associated with deletion of these cells following chronic viral challenge,30 suggesting a mechanistic overlap between memory CD8 T‐cell deletion and memory CD4 T‐cell exhaustion. In addition, secondary CD4 T cells derived from memory precursors expressed higher levels of Eomes, which is a transcription factor that drives CD4 T‐cell exhaustion, whereas primary CD4 T cells derived from naive precursors expressed higher T‐bet levels, which is a transcription factor expressed in functional CD4 T cells.48 These results suggest that secondary CD4 T cells undergo an accelerated terminal differentiation toward an exhausted state. Importantly, this natural exhaustion process was necessary for the survival of the host. It is important to mention that our transcriptional and adoptive transfer analyses were performed with ‘bulk’ primary versus secondary CD4 T cells, and future experiments of purified Th1/Tfh cell subsets may reveal additional differences between primary and secondary CD4 T‐cell responses.
Furthermore, TCR signalling, mammalian target of rapamycin signalling, and type I interferon signalling pathways were differentially enriched in primary and secondary CD4 T cells. This last result suggested a distinct role of interferons in regulating primary versus secondary responses. Consistent with our gene expression profiling, blockade of type I interferon signalling resulted in improved expansion of primary CD4 T cells, but not secondary CD4 T cells. A previous study showed that blockade of type I interferon improves CD4 T‐cell responses during a chronic LCMV infection,7, 16, 49 but until now it was unknown whether this pathway selectively regulates primary (de novo) or secondary CD4 T‐cell responses. Strikingly, interferon type I blockade in chronically infected mice polarized CD4 T‐cell responses almost exclusively toward Tfh cell subsets, which resulted in significantly improved antibody responses. A recent study demonstrated that interferon signalling suppresses B‐cell responses by regulating CD8 T cells, which could recognize and kill B cells.28 Other papers have suggested that interferon can regulate Th1 versus Tfh cell differentiation.49, 50 However, our study is novel,because we now show that interferon regulates not only Th1/Tfh cell differentiation, but also controls the interplay between host inflammation and survival during chronic viral infection. Our data also demonstrate a previously unrecognized feature of memory CD4 T cells that distinguishes them from naive CD4 T cells: their ability to bypass immune regulation by type I interferons during their expansion. However, we do not currently understand whether type I interferon signalling is acting in a CD4 T‐cell intrinsic manner, and this will be the focus of future experiments.
The extent to which our findings may generalize to humans has not been rigorously determined, but previous data with experimental HIV vaccines and various clinically approved vaccines show that sequential boosting immunization increases neutralizing antibody responses. This suggests that recall of memory CD4 T cells can improve the antibody response by a mechanism dependent on preferential Tfh cell anamnestic expansion. Taken together, our data demonstrate critical aspects of memory CD4 T‐cell responses that render them better poised to help adaptive immunity. These findings may be important for optimizing adoptive T‐cell transfer therapies for the control of cancers or chronic infections, and may also provide valuable insights for rational vaccine design.
Disclosures
The authors declare no financial or commercial conflicts of interests.
Supporting information
Figure S1. Memory CD4 T cells exhibit lower antigen‐driven activation compared with naive CD4 T cells following lymphocytic choriomeningitis virus (LCMV) challenge.
Figure S2. Chronic viral challenge induces accelerated priming of virus‐specific CD4; T‐cell responses compared with acutely controlled viral challenge.
Figure S3. Progressive loss of the T‐cell receptor by lymphocytic choriomeningitis‐specific CD4 T cells.
Figure S4. High number of co‐transferred naive and memory CD4 T cells also result in similar follicular helper t (Tfh) cell differentiation of secondary CD4 T‐cell responses.
Acknowledgements
This work was supported by NIH grants (AI007245, AI07387, 1K22AI118421 to PPM); Chicago Third Coast CFAR grants (P30 AI117943) to PPM; the Bill and Melinda Gates Foundation (OPP1033091 to DHB), and the Ragon Institute of MGH, MIT, and Harvard. The authors thank Drs Rafi Ahmed, David Brooks and all members of the Penaloza laboratory for important discussions.
References
- 1. Penaloza‐MacMaster P, Kamphorst AO, Wieland A, Araki K, Iyer SS, West EE et al Interplay between regulatory T cells and PD‐1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med 2014; 211:1905–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS et al Distinct memory CD4+ T cells with commitment to T follicular helper‐ and T helper 1‐cell lineages are generated after acute viral infection. Immunity 2013; 38:805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hale JS, Ahmed R. Memory T follicular helper CD4 T cells. Front Immunol 2015; 6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4+ T cells in immunity to viruses. Nat Rev Immunol 2012; 12:136–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elsaesser H, Sauer K, Brooks DG. IL‐21 is required to control chronic viral infection. Science 2009; 324:1569–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med 2011; 208:987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M et al Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013; 340:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 2011; 29:621–63. [DOI] [PubMed] [Google Scholar]
- 9. Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, Barber DL et al Antigen‐specific CD4 T‐cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci USA 2011; 108:21182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T‐cell responses during chronic viral infection. J Virol 1994; 68:8056–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yi JS, Du M, Zajac AJ. A vital role for interleukin‐21 in the control of a chronic viral infection. Science 2009; 324:1572–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ortiz AM, Klatt NR, Li B, Yi Y, Tabb B, Hao XP et al Depletion of CD4+ T cells abrogates post‐peak decline of viremia in SIV‐infected rhesus macaques. J Clin Invest 2011; 121:4433–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature 2012; 481:394–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stamm A, Valentine L, Potts R, Premenko‐Lanier M. An intermediate dose of LCMV clone 13 causes prolonged morbidity that is maintained by CD4+ T cells. Virology 2012; 425:122–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Penaloza‐MacMaster P, Barber DL, Wherry EJ, Provine NM, Teigler JE, Parenteau L et al Vaccine‐elicited CD4 T cells induce immunopathology after chronic LCMV infection. Science 2015; 347:278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G et al Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013; 340:202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Snell LM, Osokine I, Yamada DH, De la Fuente JR, Elsaesser HJ, Brooks DG. Overcoming CD4 Th1 cell fate restrictions to sustain antiviral CD8 T cells and control persistent virus infection. Cell Rep 2016; 16:3286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oxenius A, Bachmann MF, Zinkernagel RM, Hengartner H. Virus‐specific MHC‐class II‐restricted TCR‐transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur J Immunol 1998; 28:390–400. [DOI] [PubMed] [Google Scholar]
- 19. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K et al Efficacy and toxicity management of 19‐28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dao T, Korontsvit T, Zakhaleva V, Haro K, Packin J, Scheinberg DA. Identification of a human cyclin D1‐derived peptide that induces human cytotoxic CD4 T cells. PLoS ONE 2009; 4:e6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S et al Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 2005; 102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mostafavi S, Morris Q. Fast integration of heterogeneous data sources for predicting gene function with limited annotation. Bioinformatics 2010; 26:1759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mostafavi S, Ray D, Warde‐Farley D, Grouios C, Morris Q. GeneMANIA: a real‐time multiple association network integration algorithm for predicting gene function. Genome Biol 2008; 9(Suppl 1):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network‐based method for gene‐set enrichment visualization and interpretation. PLoS ONE 2010; 5:e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams MA, Ravkov EV, Bevan MJ. Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity 2008; 28:533–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fallet B, Narr K, Ertuna YI, Remy M, Sommerstein R, Cornille K et al Interferon‐driven deletion of antiviral B cells at the onset of chronic infection. Sci Immunol 2016; 1:pii: eaah6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moseman EA, Wu T, de la Torre JC, Schwartzberg PL, McGavern DB. Type I interferon suppresses virus‐specific B cell responses by modulating CD8+ T cell differentiation. Sci Immunol 2016; 1:eaah3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL‐12 produced by Listeria‐induced macrophages. Science 1993; 260:547–9. [DOI] [PubMed] [Google Scholar]
- 30. West EE, Youngblood B, Tan WG, Jin HT, Araki K, Alexe G et al Tight regulation of memory CD8+ T cells limits their effectiveness during sustained high viral load. Immunity 2011; 35:285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. MacLeod MK, McKee A, Crawford F, White J, Kappler J, Marrack P. CD4 memory T cells divide poorly in response to antigen because of their cytokine profile. Proc Natl Acad Sci USA 2008; 105:14521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Merica R, Khoruts A, Pape KA, Reinhardt RL, Jenkins MK. Antigen‐experienced CD4 T cells display a reduced capacity for clonal expansion in vivo that is imposed by factors present in the immune host. J Immunol 2000; 164:4551–7. [DOI] [PubMed] [Google Scholar]
- 33. Oxenius A, Zinkernagel RM, Hengartner H. Comparison of activation versus induction of unresponsiveness of virus‐specific CD4+ and CD8+ T cells upon acute versus persistent viral infection. Immunity 1998; 9:449–57. [DOI] [PubMed] [Google Scholar]
- 34. Hunziker L, Recher M, Ciurea A, Martinic MM, Odermatt B, Hengartner H et al Antagonistic variant virus prevents wild‐type virus‐induced lethal immunopathology. J Exp Med 2002; 196:1039–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ciurea A, Hunziker L, Martinic MM, Oxenius A, Hengartner H, Zinkernagel RM. CD4+ T‐cell‐epitope escape mutant virus selected in vivo . Nat Med 2001; 7:795–800. [DOI] [PubMed] [Google Scholar]
- 36. Khanolkar A, Williams MA, Harty JT. Antigen experience shapes phenotype and function of memory Th1 cells. PLoS ONE 2013; 8:e65234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T‐cell therapy. Mol Ther Oncolytics 2016; 3:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dow C, Oseroff C, Peters B, Nance‐Sotelo C, Sidney J, Buchmeier M et al Lymphocytic choriomeningitis virus infection yields overlapping CD4+ and CD8+ T‐cell responses. J Virol 2008; 82:11734–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. May RJ, Dao T, Pinilla‐Ibarz J, Korontsvit T, Zakhaleva V, Zhang RH et al Peptide epitopes from the Wilms' tumor 1 oncoprotein stimulate CD4+ and CD8+ T cells that recognize and kill human malignant mesothelioma tumor cells. Clin Cancer Res 2007; 13:4547–55. [DOI] [PubMed] [Google Scholar]
- 40. Ou D, Mitchell LA, Decarie D, Gillam S, Tingle AJ. Characterization of an overlapping CD8+ and CD4+ T‐cell epitope on rubella capsid protein. Virology 1997; 235:286–92. [DOI] [PubMed] [Google Scholar]
- 41. Abrams SI, Stanziale SF, Lunin SD, Zaremba S, Schlom J. Identification of overlapping epitopes in mutant ras oncogene peptides that activate CD4+ and CD8+ T cell responses. Eur J Immunol 1996; 26:435–43. [DOI] [PubMed] [Google Scholar]
- 42. Homann D, Lewicki H, Brooks D, Eberlein J, Mallet‐Designe V, Teyton L et al Mapping and restriction of a dominant viral CD4+ T cell core epitope by both MHC class I and MHC class II. Virology 2007; 363:113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC et al CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest 2012; 122:3281–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lindqvist M, van Lunzen J, Soghoian DZ, Kuhl BD, Ranasinghe S, Kranias G et al Expansion of HIV‐specific T follicular helper cells in chronic HIV infection. J Clin Invest 2012; 122:3271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Battegay M, Moskophidis D, Waldner H, Brundler MA, Fung‐Leung WP, Mak TW et al Impairment and delay of neutralizing antiviral antibody responses by virus‐specific cytotoxic T cells. J Immunol 1993; 151:5408–15. [PubMed] [Google Scholar]
- 46. Sather DN, Carbonetti S, Malherbe DC, Pissani F, Stuart AB, Hessell AJ et al Emergence of broadly neutralizing antibodies and viral coevolution in two subjects during the early stages of infection with human immunodeficiency virus type 1. J Virol 2014; 88:12968–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eschli B, Zellweger RM, Wepf A, Lang KS, Quirin K, Weber J et al Early antibodies specific for the neutralizing epitope on the receptor binding subunit of the lymphocytic choriomeningitis virus glycoprotein fail to neutralize the virus. J Virol 2007; 81:11650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE et al Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 2014; 40:289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ et al Type I interferon suppresses de novo virus‐specific CD4 Th1 immunity during an established persistent viral infection. Proc Natl Acad Sci USA 2014; 111:7409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity 2014; 40:367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Memory CD4 T cells exhibit lower antigen‐driven activation compared with naive CD4 T cells following lymphocytic choriomeningitis virus (LCMV) challenge.
Figure S2. Chronic viral challenge induces accelerated priming of virus‐specific CD4; T‐cell responses compared with acutely controlled viral challenge.
Figure S3. Progressive loss of the T‐cell receptor by lymphocytic choriomeningitis‐specific CD4 T cells.
Figure S4. High number of co‐transferred naive and memory CD4 T cells also result in similar follicular helper t (Tfh) cell differentiation of secondary CD4 T‐cell responses.