

CIC (also known as Capicua) is a transcriptional repressor negatively regulated by RAS/MAPK signaling. Here, Simón-Carrasco et al. show that Cic inactivation in mice induces T-ALL by a mechanism involving derepression of its well-known target, Etv4. Cic inactivation renders T-ALL insensitive to MEK inhibitors in both mouse and human cell lines.
Keywords: CIC, Ras signaling, T-ALL, mouse models, Etv4
Abstract
CIC (also known as Capicua) is a transcriptional repressor negatively regulated by RAS/MAPK signaling. Whereas the functions of Cic have been well characterized in Drosophila, little is known about its role in mammals. CIC is inactivated in a variety of human tumors and has been implicated recently in the promotion of lung metastases. Here, we describe a mouse model in which we inactivated Cic by selectively disabling its DNA-binding activity, a mutation that causes derepression of its target genes. Germline Cic inactivation causes perinatal lethality due to lung differentiation defects. However, its systemic inactivation in adult mice induces T-cell acute lymphoblastic lymphoma (T-ALL), a tumor type known to carry CIC mutations, albeit with low incidence. Cic inactivation in mice induces T-ALL by a mechanism involving derepression of its well-known target, Etv4. Importantly, human T-ALL also relies on ETV4 expression for maintaining its oncogenic phenotype. Moreover, Cic inactivation renders T-ALL insensitive to MEK inhibitors in both mouse and human cell lines. Finally, we show that Ras-induced mouse T-ALL as well as human T-ALL carrying mutations in the RAS/MAPK pathway display a genetic signature indicative of Cic inactivation. These observations illustrate that CIC inactivation plays a key role in this human malignancy.
Ras proteins have been the subject of intense investigation due to their key role in mitogenic signaling (Malumbres and Barbacid 2003). Genetic studies have provided evidence that Ras signaling is essential for mammalian cell proliferation (Drosten et al. 2010). However, constitutive activation of individual members of the downstream mitogen-activated protein kinase (MAPK) pathway, including the Raf, Mek, and Erk kinases, can bypass the requirement for Ras proteins (Drosten et al. 2010). However, little is known regarding those events that take place beyond MAPK signaling, in part due to the complexity of the events triggered by this kinase cascade in which just the Erk proteins alone are known to phosphorylate >150 substrates (Roskoski 2012).
While searching for potential downstream effectors of the MAPK pathway, the Drosophila protein Capicua (Cic) (Jiménez et al. 2000) came to our attention. Cic proteins are evolutionarily conserved transcriptional repressors that directly bind DNA via their HMG-box with the assistance of a C-terminal motif (Forés et al. 2017). Both the Drosophila and mammalian Cic loci express short (Cic-S) and long (Cic-L) protein isoforms that are transcribed from two independent promoters (Lam et al. 2006; Jiménez et al. 2012; Forés et al. 2015). Moreover, Drosophila Cic proteins have a docking site that allows direct binding of Rolled, the Drosophila counterpart of the mammalian Erk kinases (Astigarraga et al. 2007). Cic repressor activity is down-regulated by receptor tyrosine kinase-mediated MAPK signaling (Jiménez et al. 2012). In Drosophila, the Cic locus has been implicated in a variety of developmental processes such as antero–posterior or dorso–ventral patterning of the embryo (Jiménez et al. 2000), specification of wing veins (Roch et al. 2002), and tissue growth in various contexts. Indeed, during cell proliferation of both larval tissues and adult intestinal stem cells, the mitogenic signals initiated by Ras signaling become dispensable when Cic is mutated (Tseng et al. 2007; Jin et al. 2015).
In contrast, the role of mammalian Cic has been less well characterized. However, it has been shown that MAPK signaling also regulates the repressor activity of human CIC proteins. Likewise, ERK and its downstream substrate, p90RSK, can phosphorylate human CIC, thereby interfering with its nuclear import and DNA-binding capabilities, respectively (Dissanayake et al. 2011). Furthermore, CIC has been implicated in several human pathologies. For instance, CIC is known to form nuclear complexes with Ataxin 1 (ATXN1) and Ataxin 1-like (ATXN1L, also known as BOAT [brother of Ataxin 1]) (Lam et al. 2006). ATXN1 is a glutamine-rich protein implicated in spinocerebellar ataxia type 1 (SCA1), and expansion of its polyglutamine tract, which is known to cause SCA1, alters the repressive properties of CIC (Crespo-Barreto et al. 2010). These results suggest that ATXN1 and ATXN1L act as Cic corepressors in mammals. Indeed, expression of polyglutamine-expanded ATXN1 causes deregulation of a subset of CIC target genes. Moreover, mice lacking both Atxn1 and Atxn1L show destabilization of Cic proteins accompanied by up-regulation of Cic target genes such as the Pea3 family members Etv4 or Etv5 (Lee et al. 2011). As a consequence, these mice display a series of developmental defects such as omphalocele and lung alveolarization deficiencies.
Mutations in CIC have been identified recently in several human cancers. The highest incidence of mutated CIC is found in oligodendrogliomas (53%–69%), where one allele is usually lost upon loss of heterozygosity (LOH) of chromosomes 1p and 19q, and the other is affected by missense or nonsense mutations (Bettegowda et al. 2011; Yip et al. 2012). Likewise, CIC mutations have also been identified, albeit at lower frequency, in several other malignancies, such as stomach adenocarcinomas (12.9%), endometrial carcinomas (6.9%), colorectal carcinomas (6.1%), or melanomas (5.2%) (Cerami et al. 2012; Gao et al. 2013). These mutations appear to be a late event during tumorigenesis and have been found to promote metastases (Okimoto et al. 2017). Most missense mutations in CIC cluster into codons encoding the HMG-box or the C1 motif, thus interfering with CIC's DNA-binding abilities and causing derepression of its target genes (Forés et al. 2017). Other mutations produce premature stop codons, altered splice sites, and frameshift insertions or deletions that are also thought to inactivate CIC's repressor function.
To better understand the role of Cic in mammals, we generated a genetically engineered mouse model expressing defective Cic proteins that cannot bind DNA and hence repress expression of its target genes. Our data show that Cic is an essential gene in mouse development and that systemic disruption of Cic DNA binding in adult mice causes T-cell acute lymphoblastic lymphoma (T-ALL). We also show that human T-ALL carrying mutations in the RAS/MAPK pathway displays a genetic signature indicative of CIC inactivation, thus suggesting that CIC inactivation plays a key role in this human malignancy.
Results
Generation of mice carrying inactive Cic alleles
To genetically analyze the role of Cic in mammals, we decided to generate a mouse strain carrying conditional loss-of-function (LOF) Cic alleles. To this end, we introduced loxP sequences flanking exons 2–6 by homologous recombination (Fig. 1A; Supplemental Fig. S1A). These exons encode the highly conserved HMG-box (Supplemental Fig. S1B), a domain required for DNA binding and hence repressor activity (Jiménez et al. 2012; Forés et al. 2017). Elimination of exons 2–6 maintains the Cic ORF, directing the synthesis of mutant Cic-L and Cic-S isoforms that lack 288 amino acids, a region that includes the HMG-box (Fig. 1B). In this study, the conditional allele is designated as Ciclox, whereas the recombined allele is designated as CicΔ2–6, and its gene products are designated as CicΔ2–6. To confirm that deletion of exons 2–6 eliminates Cic activity, we engineered a human CIC-S cDNA lacking those sequences corresponding to exons 2–6 fused to a GFP tag (Fig. 1C,D). The resulting GFP-CICΔ2–6 protein completely lacks the ability to bind to the promoters of well-known CIC target genes such as CCND1 (Cyclin D1) and the PEA3 family of transcription factors ETV1, ETV4, and ETV5, as demonstrated by chromatin immunoprecipitation (ChIP) assays using full-length GFP-CIC-S as a positive control (Fig. 1E).
Figure 1.
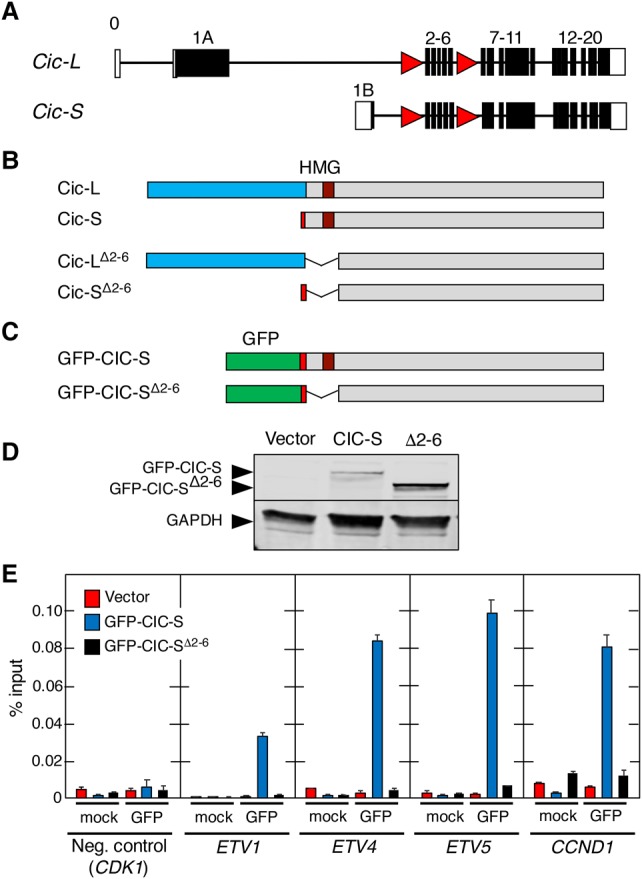
CicΔ2–6 proteins cannot bind to Cic target gene promoters. (A) Schematic representation of the targeted mouse Cic locus. Exons corresponding to Cic-L and Cic-S proteins are indicated. Coding exons are indicated by filled boxes, and noncoding exons are indicated by open boxes. LoxP sites are depicted as red triangles. (B) Representation of intact Cic-L and Cic-S proteins (top) and corresponding mutant products after Cre-mediated recombination (bottom). Exon 1A (unique to Cic-L) is indicated by blue boxes, and exon 1B (present only in Cic-S) is indicated by red boxes. (C) Schematic representation of human CIC-S and CIC-SΔ2–6 proteins fused to GFP. Sequences encoded by exon 1B (red boxes) and the GFP tag (green boxes) are shown. (D) Western blot analysis of GFP-CIC-S and GFP-CIC-SΔ2–6 (Δ2–6) proteins expressed in Flp-In T-REx 293 cells. Protein expression was detected using anti-GFP antibodies. As a control, cells were transfected with an empty vector (vector). GAPDH expression served as a loading control. (E) ChIP assay using GFP antibodies in Flp-In T-REx 293 cells stably expressing GFP-CIC-S (blue bars) or GFP-CIC-SΔ2–6 (black bars). Flp-In T-REx 293 cells stably transfected with an empty vector were used as a control (red bars). Specificity of GFP immunoprecipitations was validated using an unrelated antibody (mock). Association with the CIC-binding elements in the ETV1, ETV4, ETV5, and CCND1 promoters as well as binding to the CDK1 promoter lacking known CIC-binding sites (negative control) were analyzed by quantitative RT–PCR and normalized to the amount of input DNA. Data represent mean ± SD.
Cic activity is essential for late embryonic development
To assess the role of Cic in development, we generated germline Cic+/Δ2–6 mice by mating Cic+/lox animals (see the Supplemental Material) with EIIa-Cre transgenic mice that express the bacterial Cre recombinase in their zygotes (Lakso et al. 1996). Heterozygous Cic+/Δ2–6 animals were viable and did not show obvious defects for up to 1 yr of age. Homozygous CicΔ2–6/Δ2–6 embryos were present at Mendelian ratios at embryonic day 18.5 (E18.5). However, E18.5 CicΔ2–6/Δ2–6 embryos were significantly smaller than their wild-type or heterozygous counterparts and died immediately after birth (Fig. 2A). Most of these embryos (70%) presented an omphalocele, an abdominal wall closure defect (Fig. 2B). Omphaloceles occur naturally during mammalian development when the gut transiently herniates the umbilical space due to limited space in the peritoneal cavity (Doyonnas et al. 2001). This structure is usually resolved by E16 in mice, thus indicating that Cic function may be required for retraction of the gut from the umbilical cord. Interestingly, a similar phenotype was observed in embryos lacking the Cic corepressors Atxn1 and Atxn1L, an observation attributed to the decrease of Cic protein levels in Atxn1−/−;Atxn1L−/− mice (Lee et al. 2011). CicΔ2–6/Δ2–6 embryos did not display hydrocephali, another phenotype frequently detected in Atxn1−/−;Atxn1L−/− mice (Lee et al. 2011). In addition, we could not analyze whether lung alveolarization was affected, as shown previously in mice lacking the Cic-L isoform (Lee et al. 2011), since CicΔ2–6/Δ2–6 embryos died right after birth. However, expression of matrix metalloproteases known to be responsible for defective lung alveolarization was not affected at E18.5, although Cic target genes were markedly derepressed (Supplemental Fig. S2A,B).
Figure 2.

Cic repressor activity is essential for mouse development. (A) Body weight of Cic+/+, Cic+/Δ2–6, and CicΔ2–6/Δ2–6 embryos at E18.5. Data represent mean ± SD. Asterisks depict statistically significant differences. (*) P < 0.05; (***) P < 0.001, unpaired Student's t-test. (B) Representative images of Cic+/+ (left) and CicΔ2–6/Δ2–6 (middle) embryos at E18.5. (Right) Hematoxilin and eosin (H&E) staining of a whole CicΔ2–6/Δ2–6 embryo section at E18.5. Bar, 2 mm. Arrowheads indicate omphaloceles. (C) H&E, Ki67, TTF-1, and SP-C immunohistochemistry (IHC) as well as periodic acid Schiff (PAS) staining in lung sections of Cic+/+ and CicΔ2–6/Δ2–6 embryos at E18.5. Bars: H&E, Ki67, TTF-1, and SP-C, 50 µm; PAS stainings, 25 µm. Inlays in PAS stainings represent magnifications.
Further characterization of E18.5 CicΔ2–6/Δ2–6 embryos revealed a dramatic increase in proliferating (Ki67+) lung cells, suggesting a defect in terminal differentiation of the respiratory epithelium (Fig. 2C; Supplemental Fig. S2C). Immunohistochemistry (IHC) studies involving γH2AX and active Caspase 3 failed to reveal signs of DNA damage or increased apoptosis (Supplemental Fig. S2D). To determine whether terminal differentiation of the lung is affected in CicΔ2–6/Δ2–6 embryos, we analyzed lung differentiation markers by IHC. E18.5 CicΔ2–6/Δ2–6 embryos displayed persistent TTF-1 (also known as Nkx2-1) expression levels in a high proportion of epithelial cells, a phenotype reminiscent of lungs in the canalicular stage (Naltner et al. 2000; Moreno-Barriuso et al. 2006), which suggests delayed or altered alveolar maturation (Fig. 2C). Consistent with this defect, we detected reduced expression levels of SP-C, a marker of type II alveolar cells, and increased periodic acid Schiff (PAS) staining, thus demonstrating that the lungs of CicΔ2–6/Δ2–6 embryos cannot produce sufficient amounts of surfactant for postnatal life (Fig. 2C).
Cic inactivation in mouse embryonic fibroblasts (MEFs)
MEFs obtained from E13.5 CicΔ2–6/Δ2–6 embryos displayed the same proliferative properties as wild-type MEFs (Supplemental Fig. S3A). As expected, homozygous expression of CicΔ2–6 proteins resulted in derepression of Cic target genes, such as those encoding the Pea3 family of transcription factors (Supplemental Fig. S3B,C). Likewise, we did not detect significant differences in the activation of the MAPK or PI3K signaling pathways (Supplemental Fig. S3D). In addition, CicΔ2–6/Δ2–6 MEFs are as efficiently transformed as wild-type MEFs by H-RasG12V and E1A but not by E1A alone, thus suggesting that Cic inactivation does not phenocopy Ras activation in MEFs (Supplemental Fig. S3E,F). These results indicate that Cic proteins do not play an important role in normal cell proliferation, at least in MEFs.
Previous studies have demonstrated that ablation of Cic activity in the Drosophila eye imaginal disc as well as in intestinal stem cells sustains proliferation in the absence of Ras (Tseng et al. 2007; Jin et al. 2015). To test whether inactivation of Cic could sustain cell proliferation in MEFs devoid of Ras proteins, we generated H-Ras−/−;N-Ras−/−;K-Raslox/lox and H-Ras−/−;N-Ras−/−;K-Raslox/lox;Ciclox/lox MEFs. These cells were infected with adenoviral particles expressing the Cre recombinase in order to ablate K-Ras expression and generate “Rasless” cells. These Rasless cells failed to proliferate regardless of whether they retained Cic repressor activity (Supplemental Fig. S3G). These results indicate that Ras signaling must be mediated by additional effectors, at least in MEFs.
Cic inactivation in the brain is not sufficient to initiate oligodendroglioma development
Human oligodendrogliomas often display losses of chromosomes 1p and 19q. Recently, the CIC locus, located in 19q, has been found to be mutated in these tumors following a mutational pattern indicative of LOF, thus suggesting that CIC may act as a tumor suppressor (Bettegowda et al. 2011; Yip et al. 2012). To determine whether inactivation of CIC is sufficient to initiate oligodendroglioma development, we crossed Ciclox/lox mice with a strain expressing the Cre recombinase under the control of the human glial fibrillary acidic protein promoter (hGFAP-Cre) (Zhuo et al. 2001). This strain expresses Cre in the central nervous system from E13.5 onward, resulting in efficient recombination in cells derived from GFAP-positive progenitors, such as neurons, astrocytes, and oligodendrocytes (Casper and McCarthy 2006). Indeed, crossing the hGFAP-Cre mice with the Rosa26LSLlacZ reporter strain (Mao et al. 1999) revealed efficient recombination in most if not all cells of the brain (Supplemental Fig. S4A). hGFAP-Cre-mediated recombination of Ciclox alleles did not result in tumor formation even at 1 yr of age. Indeed, we could not detect any significant alterations in the brain at the histopathological level despite efficient recombination of Ciclox alleles followed by significant derepression of Cic target genes such as Etv4 and Etv5 (Supplemental Fig. S4B,C). These observations indicate that Cic inactivation in the brain does not initiate oligodendroglioma formation, thus suggesting the need for other cooperating events in this tumor type.
Cic inactivation in adult mice causes T-cell lymphoblastic lymphoma
CIC mutations have also been identified in a variety of other human tumors (Cerami et al. 2012; Gao et al. 2013). Hence, we decided to systemically inactivate Cic in adult mice. To this end, we crossed Ciclox/lox animals with mice carrying a transgene encoding a ubiquitously expressed CreERT2 recombinase (hUBC-CreERT2) (Ruzankina et al. 2007). Continuous exposure of these compound mice to a tamoxifen (Tmx) diet from 4 wk of age resulted in efficient recombination of Ciclox alleles after 3 mo of treatment (Supplemental Fig. S5A,B). At this time, mice expressing the inactive CicΔ2–6 proteins throughout most of their tissues were normal and did not show obvious phenotypes at the histopathological level. However, when we subjected Ciclox/lox;hUBC-CreERT2+/T mice for longer times to the Tmx diet, most of them (92%) died before 1 yr of age, with a median survival of 36 wk (Fig. 3A). Characterization of these mice at a humane end point revealed dramatically enlarged thymuses (Fig. 3B). No other tissue displayed detectable alterations. Further analysis revealed the presence of T-cell lymphoblastic lymphomas (T-ALL) as indicated by CD3 IHC staining on the cell surface and moderate expression of the terminal deoxynucleotidyl transferase (TDT) (Fig. 3C). Moreover, these tumors displayed a prominent population of CD4/CD8 double-positive cells, although the levels of single CD4+ and CD8+ cells were variable, as suggested by immunophenotyping of three different tumors, indicating that the tumors harbored features of immature T cells from cortical stages (Fig. 3D; Belver and Ferrando 2016). In most cases, tumor cells also spread to other organs, including the lungs, liver, and spleen, and could be transplanted into immunodeficient mice (Supplemental Fig. S6A,B). Consistent with these observations, the tumors harbored monoclonal or oligoclonal T-cell receptor (TCR) rearrangements, indicating clonal expansion of tumor cells (Fig. 3E).
Figure 3.
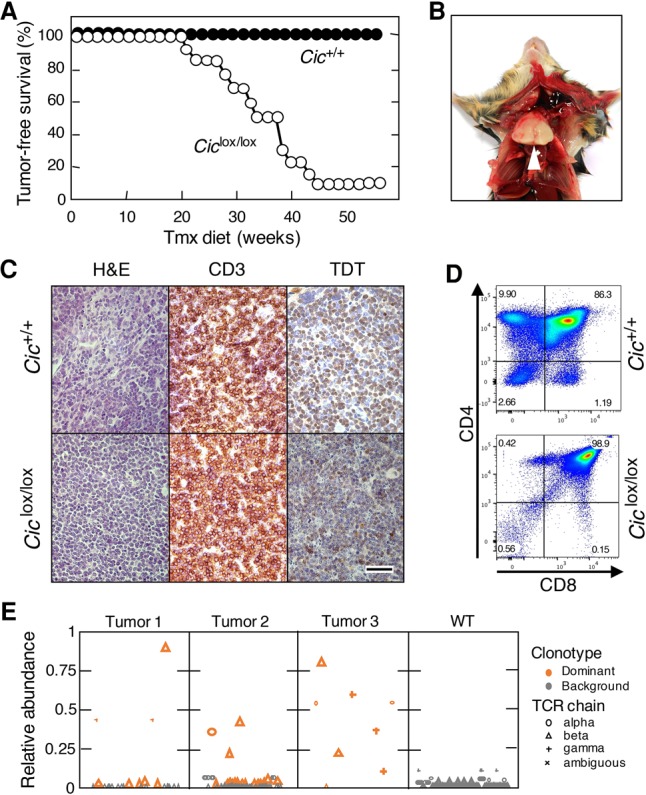
Cic inactivation in adult mice causes T-ALL. (A) Tumor-free survival of Cic+/+;hUBC-CreERT2+/T (black dots; n = 13) or Ciclox/lox;hUBC-CreERT2+/T (white dots; n = 11) mice subjected to a continuous Tmx diet at 4 wk of age. (B) Representative image of the thoracic cavity of a Ciclox/lox;hUBC-CreERT2+/T mouse treated for 8 mo with a Tmx diet and sacrificed at a humane end point. (C) H&E as well as CD3 or TDT IHC staining of thymus sections obtained from Cic+/+;hUBC-CreERT2+/T mice at 6 mo of age or Ciclox/lox;hUBC-CreERT2+/T mice at a humane end point. Bar, 50 µm. (D) Representative flow cytometry analyses of CD4+ and CD8+ cells in thymuses obtained from Cic+/+;hUBC-CreERT2+/T mice at 6 mo of age or Ciclox/lox;hUBC-CreERT2+/T mice at a humane end point. (E) Abundance of TCR clonotypes in three independent T-ALL tumors obtained from Tmx-treated Ciclox/lox;hUBC-CreERT2+/T mice (tumors 1–3) or a representative thymus from a Cic+/+;hUBC-CreERT2+/T mouse (wild type). The relative abundance of each clonotype is shown, calculated as the abundance of each clonotype relative to the total abundance of all clonotypes of the same TCR chain in a sample. Clonotypes are plotted in lexicographical order. The read abundance of each clonotype is represented by the size of the symbol (large symbols, 1:1,000,000; small symbols, 1:10,000,000). Clonotypes determined to be dominant are shown in orange. (Circles) α TCR chains; (triangles) β TCR chains; (cross) γ TCR chains; (X) ambiguous TCR chains.
Similar transcriptional profiles in T-ALL induced by Cic inactivation and Ras oncogenes
To understand the molecular bases of T-ALL development induced by ablation of Cic activity, we performed transcriptional profiling analysis of these tumors by RNA sequencing (RNA-seq). As illustrated in Supplemental Figure S7A and Supplemental Table S1, we detected significant overexpression of 55 genes and down-regulation of 181 loci (log2 fold change ≥ +3 or log2 fold change ≤ −3; adjusted P-value < 0.05). As expected, the well-known Cic target genes Etv4 and Etv5 were among the 30 most up-regulated genes in these tumors. Next, we interrogated whether the transcriptional profiles of these tumors resemble those induced by Ras oncogenes (Kindler et al. 2008; Sabnis et al. 2009), including those present in KHRasV12 mice, a strain that expresses an oncogenic H-RasG12V protein from the K-Ras locus (Drosten et al. 2017). Analysis by RNA-seq followed by principal component analysis (PCA) of gene expression profiles of H-RasG12V and Cic inactivation-induced T-ALLs revealed significant overlaps (Fig. 4A,B). In contrast, similar tumors obtained from p53−/− mice (Jacks et al. 1994) were clearly distinct. These results suggest that Ras oncoproteins and inactivation of Cic induced T-ALL by similar mechanisms. Notably, Cic target gene expression also depends on Ras signaling in normal thymuses, thus suggesting that the Cic repressor activity is regulated by Ras signaling in normal thymuses (Supplemental Fig. S7B).
Figure 4.

Similar transcriptional profiles in T-ALL driven by Cic inactivation or Ras hyperactivation. (A) PCA of gene expression profiles obtained from wild-type thymuses (black dots) and T-ALLs developed in Tmx-treated Ciclox/lox;hUBC-CreERT2+/T (red dots), KHRasV12;hUBC-CreERT2+/T (green dots), or p53−/− mice (blue dots) at a humane end point. (B) Heat map displaying differentially expressed genes (log2 fold change ≥ +3 or log2 fold change ≤ −3; adjusted P-value < 0.05; false discovery rate [FDR] 0.05) as estimated by RNA-seq from samples shown in A. Relative expression (log2 fold change) is scaled in color (as indicated) from dark blue (−3) to dark red (+3). Gene symbols are listed in Supplemental Table S1.
A common event in T-ALL development is hyperactivation of the Notch signaling pathway (Belver and Ferrando 2016). Indeed, all H-RasG12V or Cic inactivation-driven tumors show activation of the Notch pathway based on IHC staining of cleaved Notch1 and its target, Hes1 (Supplemental Fig. S8A,B; Drosten et al. 2017). Moreover, we detected a significant enrichment of a transcriptional signature indicative of Notch pathway activation (Supplemental Fig. 8C; Liberzon et al. 2015). Previous data have shown that T-ALL driven by K-RasG12D carries frequent mutations in Notch1 that were not tumorigenic on their own but cooperated with the activated Ras oncogene to induce T-ALL (Chiang et al. 2008). RNA-seq analysis of five tumors induced upon Cic inactivation revealed only a rare miscoding mutation (A1639T) in one of the tumors, thus suggesting that Notch pathway activation occurred mostly via other mechanisms.
Growth of Cic-induced T-ALL is independent of MAPK activation
Next, we interrogated whether T-ALL tumors induced by inactivation of Cic proteins require an active MAPK signaling pathway. These tumors, unlike those induced by H-RasG12V expression, did not display elevated levels of Erk phosphorylation (Fig. 5A). Next, we treated tumor cells derived from both CicΔ2–6/Δ2–6- and H-RasG12V-driven tumors with GSK1120212 (trametinib), a selective MEK inhibitor that effectively blocks MAPK signaling driven by Ras oncogenes (Wright and McCormack 2013). As illustrated in Figure 5, A and B, trametinib effectively blocked MAPK signaling as well as proliferation of H-RasG12V-driven tumor cells. In contrast, this MEK inhibitor had no effect on proliferation of tumor cells driven by Cic inactivation. These results demonstrate that Cic inactivation occurs downstream from Ras signaling in T-ALL. Furthermore, they also predict that human tumors carrying inactivating mutations in the CIC locus are likely to be resistant to inhibitors of the MAPK pathway (see below).
Figure 5.

Murine T-ALL driven by Cic inactivation is resistant to MEK inhibition and requires Etv4. (A) Western blot analysis of p-Erk1/2 and total Erk1/2 in T-ALL cells obtained from tumors developed in Tmx-treated KHRasV12;hUBC-CreERT2+/T (left) or Ciclox/lox;hUBC-CreERT2+/T (right) mice treated with the indicated concentrations of trametinib for 24 h. GAPDH expression served as a loading control. (B) Growth curve of T-ALL cells obtained from tumors developed in Tmx-treated KHRasV12;hUBC-CreERT2+/T (left) or Ciclox/lox;hUBC-CreERT2+/T (right) mice treated with the indicated concentrations of trametinib for the indicated time. (C) Tumor-free survival of Ciclox/lox;Etv4+/NLZ;hUBC-CreERT2+/T (open dots; n = 11) or Ciclox/lox;Etv4NLZ/NLZ;hUBC-CreERT2+/T (closed dots; n = 5) mice subjected to a continuous Tmx diet starting at 4 wk of age. (D) Percentage of thymocyte populations in 1-mo-old wild-type (open bars; n = 3) or Etv4NLZ/NLZ (closed bars; n = 3) mice. CD4/CD8-double-negative, CD4/CD8-double-positive, CD4-single-positive, and CD8-single-positive populations are shown. Data represent mean ± SD. (E) Percentage of CD4/CD8 double-negative (DN) thymocyte subpopulations in 1-mo-old wild-type (open bars; n = 3) or Etv4NLZ/NLZ (closed bars; n = 3) mice. DN1 (CD44+/CD25−), DN2 (CD44+/CD25+), DN3 (CD44−/CD25+), and DN4 (CD44−/CD25−) subpopulations are shown. Data represent mean ± SD. (F) Relative expression levels of Etv1, Etv4, and Etv5 mRNAs in wild-type thymuses (open bars; n = 2); T-ALLs developed in Tmx-treated Ciclox/lox;hUBC-CreERT2+/T mice (CicΔ2–6/Δ2–6; red bars; n = 2); or the only tumor that developed in Tmx-treated Ciclox/lox;Etv4NLZ/NLZ;hUBC-CreERT2+/T mice (CicΔ2–6/Δ2–6;Etv4NLZ/NLZ; closed bars). β-Actin expression levels were used for normalization. Data represent mean ± SD.
Tumor induction by Cic inactivation is mediated by the Etv4 transcription factor
As indicated above, some of the main targets of Cic are the genes encoding members of the Pea3 family of transcription factors: Etv1, Etv4, and Etv5 (Kawamura-Saito et al. 2006; Dissanayake et al. 2011). Hence, we examined whether any of these proteins might be responsible for mediating tumor induction upon inactivation of Cic. Since Etv4 was the gene most up-regulated in cells lacking active Cic repression, we introduced Etv4-deficient alleles (Etv4NLZ/NLZ) (Livet et al. 2002) into Ciclox/lox;UBC-CreERT2+/T mice. As illustrated in Figure 5C, reduced expression of Etv4 in Ciclox/lox;Etv4+/NLZ;UBC-CreERT2+/T animals had no effect on tumor development, since all of the mice developed T-ALL with a median survival similar to that of animals carrying wild-type Etv4 alleles. However, complete ablation of Etv4 dramatically prevented the induction of T-ALL upon inactivation of Cic. Indeed, four of five Ciclox/lox;Etv4NLZ/NLZ;UBC-CreERT2+/T mice had no signs of thymic disease by 1 yr of age, a time when all Ciclox/lox;Etv4+/+;UBC-CreERT2+/T animals had already succumbed to the disease. Only one out of these five mice displayed limited tumor formation by 1 yr of age, indicating that tumor growth was dramatically delayed. These results strongly indicate that Etv4 is a key mediator of the tumorigenic consequences of Cic inactivation. Notably, absence of Etv4 did not per se affect normal thymocyte development (Fig. 5D,E). Interestingly, CicΔ2–6/Δ2–6;Etv4NLZ/NLZ tumor cells displayed increased levels of Etv5 and, to a lesser extent, Etv1 expression, indicating that in the absence of Etv4, Etv5 is the primary member of the Pea3 family to respond to Cic inactivation (Fig. 5F). However, this increased expression is not sufficient to induce T-ALL, at least in most mice. Whether concomitant ablation of Etv4 and Etv5 expression will completely block T-ALL development or whether overexpression of Etv4 alone is sufficient to cause T-ALL remains to be determined.
Identification of a gene signature indicative of CIC inactivation in mouse and human T-ALL
Human T-ALL has been classified based on transcriptional profiling. Work by Ferrando et al. (2002) identified gene signatures that correspond to specific stages of T-cell development. We compared these human T-ALL signatures with expression data obtained from T-ALL tumors induced by either H-RasG12V expression or Cic inactivation. As illustrated in Supplemental Figure S8D, tumors induced by Cic inactivation were highly enriched for the TLX1+ signature (also known as HOX11+), thus suggesting that these tumors share features of human T-ALL arrested at the early cortical stage (Belver and Ferrando 2016). In agreement with the PCA data, H-RasG12V-driven tumors also displayed enrichment of this signature but to a slightly lesser degree. These observations suggest that tumors driven by Ras oncogenes should display a transcriptional signature indicative of Cic inactivation. To this end, we first established a Cic LOF gene signature based on genes that carried evolutionarily conserved Cic-binding sites and became derepressed in tumors induced by Cic inactivation. Specifically, we selected a 32-gene signature (Cic LOF signature CIC_LOF_4) based on genes that were up-regulated at least a log2 fold change of 1.5 and harbored at least one Cic-binding sequence (CBS) (Kawamura-Saito et al. 2006) with a conservation score of >0.875 in their promoters (see the Supplemental Material; Supplemental Fig. S9A; Supplemental Table S2). Next, we performed unsupervised clustering of the independent T-ALL tumor samples using this Cic LOF signature. As illustrated in Figure 6A, Cic LOF- and H-RasG12V-induced tumors formed a common cluster. Although they did not cluster with the aforementioned tumors, p53-null tumors showed a limited enrichment of the Cic LOF signature (Supplemental Fig. S9B). These observations indicate that Cic target genes are derepressed in H-RasG12V-driven tumors, suggesting that hyperactive Ras signaling causes inactivation of Cic to promote T-ALL.
Figure 6.

A gene signature indicative of Cic LOF is enriched in mouse and human T-ALL driven by inactivation of Cic or activation of the Ras/MAPK pathway. (A) Heat map generated by unsupervised clustering of differentially expressed genes present in a 32-gene Cic LOF signature (CIC_LOF_4), as estimated by RNA-seq from wild-type thymuses and T-ALLs developed in Tmx-treated Ciclox/lox;hUBC-CreERT2+/T as well as KHRasV12;hUBC-CreERT2+/T mice or p53−/− animals at a humane end point. Gene symbols are indicated, and relative expression (log2 fold change) is scaled in color (as indicated) from dark blue (−3) to dark red (+3). (B) Indication of point mutations and gene fusions present in a published data set of human T-ALL samples (from Atak et al. 2013). Samples carrying protein-altering mutations and fusions that are predicted to activate the RAS/MAPK/CIC axis are indicated in dark blue, and those harboring mutations affecting other pathways are indicated in light blue. The mutations affecting specific genes in each sample are indicated in red (only for the RAS/MAPK/CIC group). NUP214-ABL1 and SSBP2-FER represent gene fusions identified in selected samples. Mutations in NOTCH1 are indicated in green. The SUPT1 cell line harbors a gene fusion that causes overexpression of NOTCH1 (light green). (C) Enrichment plot showing significant enrichment (FDR < 25%) of a 143-gene signature indicative of CIC LOF (CIC_LOF_3; left) or a TLX1+ (HOX11+; right) gene signature in human T-ALL samples predicted to have an active RAS/MAPK/CIC axis (from B). (NES) Normalized enrichment score.
We next wanted to interrogate whether the Cic LOF gene signature derived from our murine T-ALL expression data is conserved in human T-ALL samples harboring activation of the RAS/MAPK/CIC axis. To this end, we took advantage of a published T-ALL data set that includes gene expression and mutation data from 31 T-ALL patients and 18 T-ALL cell lines (Atak et al. 2013). We selected those samples that carried either a mutation in CIC or any other alteration that predicts CIC inactivation via activation of the MAPK pathway, such as mutations in NRAS, KRAS, NF1, or PTK2B. In addition, we included samples carrying the NUP214-ABL1 or SSBP2-FER fusions, since their activated tyrosine kinases are known to induce MAPK activation. Following this strategy, we assembled 16 out of the 49 samples into a group that was expected to harbor inactive CIC via one of the mechanisms described above (Fig. 6B). Since our Cic LOF signature derived from mouse tumors was composed of only 32 genes, we did not find enrichment of this restricted signature in the RAS/MAPK/CIC sample group. However, when we used a more relaxed signature composed of 143 genes that were significantly up-regulated in Cic LOF-driven T-ALL and contained at least one CBS (CIC_LOF_3) (Supplemental Table S3; Supplemental Fig. S9A), we found this signature to be significantly enriched in the RAS/MAPK/CIC sample group (Fig. 6C). In addition, we found the TLX1+ signature to be enriched in this group of 16 samples, providing further evidence for the similarity between human and mouse tumors (Fig. 6C). Taken together, these data indicate that human T-ALL samples driven by the RAS/MAPK/CIC axis and mouse tumors driven by H-RasG12V expression or Cic inactivation share a gene signature characteristic of CIC inactivation.
CIC inactivation in human T-ALL causes resistance to MAPK inhibition
Sequencing analysis of primary T-ALL clinical specimens revealed the presence of CIC mutations in 10% (n = 7 of 69) of cases analyzed (Supplemental Fig. S10A–C). Other studies also identified CIC mutations, albeit at a lower frequency (Supplemental Fig. S10D; Atak et al. 2013; Kataoka et al. 2015). These mutations are predicted to cause resistance to MAPK inhibition, similar to our observation in murine T-ALL cells. To test this hypothesis, we first determined the sensitivity of a variety of human T-ALL cell lines to trametinib. Notably, exposure to trametinib had no effect in CCRFCEM and JURKAT cells (Supplemental Fig. S11A,B). In contrast, MOLT4 and HSB2 cells were sensitive to this MEK inhibitor. To interrogate whether CIC activity determines sensitivity to MAPK pathway inhibition, we focused on MOLT4 cells (known to carry mutated N-Ras) and disabled CIC via Crispr/Cas9-mediated gene editing using two different single-guide RNAs (sgRNAs). Indeed, as illustrated in Figure 7A–C, MOLT4 cells lacking CIC became resistant to trametinib treatment, indicating that CIC inactivation confers resistance to MAPK inhibition in human T-ALL.
Figure 7.
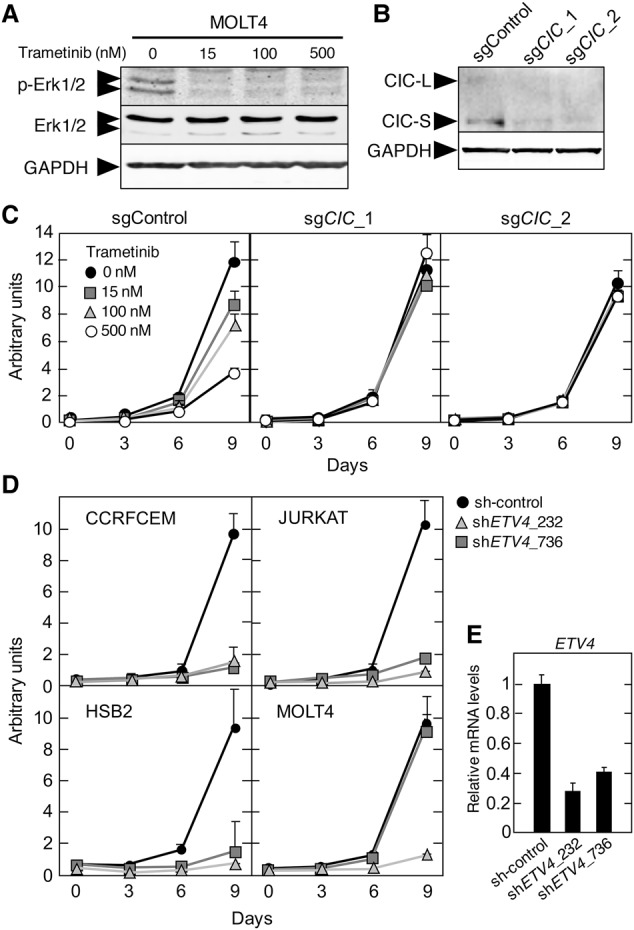
Resistance to MEK inhibition and requirement for ETV4 expression in human T-ALL cell lines. (A) Western blot analysis of p-ERK1/2 and total ERK1/2 expression in MOLT4 cells treated with the indicated concentrations of trametinib for 24 h. GAPDH expression served as a loading control. (B) Western blot analysis of CIC expression levels in MOLT4 cells 2 wk after infection with lentiviruses expressing Cas9 and the indicated sgRNAs. GAPDH expression served as a loading control. (C) Proliferation of MOLT4 cells 2 wk after infection with lentiviruses expressing Cas9 and the indicated sgRNAs and treated with the indicated concentrations of trametinib. (D) Proliferation of CCRFCEM, JURKAT, HSB2, and MOLT4 cells infected with lentiviruses expressing two independent shRNAs targeting ETV4 or a nontargeting control. (E) Relative ETV4 mRNA expression levels in 293T cells infected with lentiviruses expressing a control shRNA or two different shRNAs against ETV4.
ETV4 expression is required for human T-ALL cell lines
Finally, our data indicate that Etv4 expression is required for T-ALL development induced by Cic inactivation in mice. To analyze whether ETV4 is also required for growth of human T-ALL cells, we inhibited ETV4 expression in CCRFCEM, JURKAT, MOLT4, and HSB2 cells with two independent shRNAs. As indicated in Figure 7, D and E, ETV4 expression is required for efficient proliferation of these cell lines independently of the mutational status of CIC or RAS. These observations suggest that ETV4 plays a relevant role in murine and human T-ALL. In summary, these data indicate that the same genes and mechanisms operate to induce T-ALL in mice and humans.
Discussion
Cic is a transcriptional repressor negatively regulated by MAPK signaling that is known to play essential roles during development of Drosophila embryos (Jiménez et al. 2000, 2012; Astigarraga et al. 2007). In contrast, the role of Cic in mammalian development has not been thoroughly studied. Zoghbi and coworkers (Fryer et al. 2011; Lee et al. 2011) used a gene trap approach to generate a strain that expresses only the Cic-S isoform (Cic-L−/−). Most Cic-L−/− mice died before weaning, and those that survived postnatal development were of smaller size and presented lung alveolarization defects, a phenotype similar to that of mice lacking the Cic corepressor Atxn1L (Lee et al. 2011).
In this study, we completely ablated Cic repressor activity by targeting exons 2–6 of the Cic locus, a domain encoding the HMG-box required for DNA binding and repressor activity of both Cic isoforms, Cic-L and Cic-S. As expected, the truncated CicΔ2–6 proteins did not bind to the promoters of their target genes. When Cic activity was inactivated in the germline, none of the CicΔ2–6/Δ2–6 mice survived after birth despite being present at Mendelian ratios at E18.5. We detected dramatic defects in lung maturation, thus suggesting that these immature lungs were primarily responsible for the perinatal lethality in CicΔ2–6/Δ2–6 mice. However, we could not assess the presence of lung alveolarization defects such as those observed in surviving Cic-L−/− and Atxn1L−/− animals because lung alveolarization occurs at later stages of postnatal life.
The majority of E18.5 CicΔ2–6/Δ2–6 embryos (70%) also presented omphaloceles, a phenotype observed in Atxn1−/−;Atxn1L−/− mice (Lee et al. 2011), thus suggesting that the Cic/Atxn1 or Cic/Atxn1L complex is essential for the regulation of abdominal wall closure. However, our CicΔ2–6/Δ2–6 embryos did not recapitulate all of the defects present in Atxn1−/−;Atxn1L−/− mice. For instance, we did not observe hydrocephali, suggesting that Atxn1/Atxn1L proteins may possess Cic-independent functions. Alternatively, the different phenotypes may be explained by low residual levels of Cic proteins in Atxn1−/−;Atxn1L−/− mice (Lee et al. 2011).
Previous studies have shown that Cic inactivation in the eye imaginal disc and adult intestinal stem cells in Drosophila rescues the proliferation defects caused by ablation of the single Ras gene (Tseng et al. 2007; Jin et al. 2015). This indicates that Cic is a key mediator that connects MAPK signaling with the transcriptional program required to process mitogenic signals. However, ablation of Cic activity in cells lacking the three Ras isoforms failed to restore their proliferative properties, indicating that Ras mitogenic signals must be mediated by additional components of the transcriptional machinery, at least in these cells.
CIC is recurrently mutated in human oligodendrogliomas. These tumors display mutations in one CIC allele, whereas the other is lost as part of larger 19q chromosomal deletions, suggesting that CIC functions as a tumor suppressor (Bettegowda et al. 2011; Yip et al. 2012). However, selective inactivation of Cic proteins in the mouse brain was not sufficient to drive oligodendroglioma formation. As illustrated here, selective expression of inactive Cic proteins in most brain cells, including neuronal and glial precursors derived from hGFAP-Cre-expressing progenitors, failed to induce any detectable alterations in 1-yr-old mice. In human oligodendrogliomas, CIC is always comutated with IDH1 or IDH2 (Yip et al. 2012) or, less frequently, FUBP1, thus raising the possibility that tumor formation requires cooperation of these cancer genes with CIC (Bettegowda et al. 2011). Interestingly, Idh1 mutant mice also fail to develop brain tumors (Sasaki et al. 2012). Finally, it should also be noted that CIC mutations are not always maintained in recurrent oligodendrogliomas, arguing that these mutations may be subclonal secondary events that do not necessarily provide a growth advantage for tumor cells (Aihara et al. 2017).
Conditional inactivation of Cic proteins in adult mice resulted in the development of T-ALL after 6–9 mo with almost complete penetrance (>90%), thus demonstrating that Cic can act as a tumor suppressor in mice. These tumors were significantly enriched in the TLX1+ signature, thus suggesting that Cic inactivation causes thymocyte arrest at the early cortical stage (Belver and Ferrando 2016). Interestingly, Cic is likely a target of the Tlx1 repressor, a fact that raises the possibility that Tlx1-mediated repression of Cic plays a role in T-ALL development induced by this transcriptional repressor (De Keersmaecker et al. 2010; Della Gatta et al. 2012). However, the functional relevance of this pathway for T-ALL development remains to be determined.
T-ALL development via activation of Ras oncogenes has also been described in mouse tumor models (Kindler et al. 2008; Sabnis et al. 2009). More recently, we observed T-ALL in mice that express the H-RasG12V oncoprotein driven from the K-Ras promoter (Drosten et al. 2017). These tumors are indistinguishable from those induced upon Cic inactivation. Indeed, the gene expression profiles of tumors induced by H-RasG12V expression or Cic inactivation are highly similar. Hence, we propose that a significant proportion of gene expression changes in Ras-driven T-ALL occurs via inactivation of Cic. Likewise, a gene signature indicative of Cic inactivation was highly enriched in Ras-driven T-ALL. More importantly, this signature is also significantly enriched in human T-ALL samples harboring mutations that are predicted to cause aberrant signaling through the RAS/MAPK/CIC axis. These observations indicate that CIC acts downstream from the RAS/MAPK pathway in T-ALL. Although RAS mutations are not frequent in these cancers (∼5%), it has been suggested that up to 50% of all human T-ALL cases have aberrant RAS signaling (von Lintig et al. 2000). In addition, RAS/MAPK-activating mutations are much more prevalent in relapsed T-ALL (Oshima et al. 2016), thus suggesting that CIC inactivation through hyperactive Ras signaling may play a relevant role in a significant number of T-ALL cases.
However, these T-ALL tumors may present different responses to pharmacological inhibition of the MAPK pathway. For instance, tumors that developed as a consequence of K-RasG12D activation in mice are sensitive to Mek inhibition (Dail et al. 2010). Likewise, as demonstrated here, tumor cells derived from lymphomas driven by H-RasG12V expression could be efficiently inhibited upon exposure to the MEK inhibitor trametinib. However, T-ALLs that arose upon Cic inactivation were completely resistant to this inhibitor, indicating that mutations in this locus activate mitogenic signaling downstream from the MAPK cascade. Indeed, CIC inactivation in human T-ALL cell lines also caused resistance to MEK inhibition. Hence, CIC mutations may develop as a potential mechanism of resistance to MAPK inhibitors. Of note, mutations in CIC have been identified in human T-ALL, albeit at a low frequency (Atak et al. 2013; Kataoka et al. 2015; this study). Consistent with our observations, CIC has been identified recently in a screen to detect novel mechanisms that cause resistance to trametinib in other tumor models (Wang et al. 2017). Similarly, CIC has also been identified in a screen for genes whose absence promotes resistance to EGFR inhibition (Liao et al. 2017). Finally, Bivona and colleagues (Okimoto et al. 2017) have shown that CIC inactivation is associated with advanced tumor stages and metastasis formation in lung and gastric tumors, thus suggesting that these tumors may be intrinsically resistant to MEK or EGFR inhibition.
The emerging role of CIC mutations in human cancers makes it necessary to understand the mechanisms that mediate its repressor activity. As indicated above, the best-known CIC targets are the members of the PEA3 family of transcription factors: ETV1, ETV4, and ETV5. However, the functional consequences of inhibiting CIC repression in human tumors are unknown. Here, we provide genetic evidence that in T-ALL induced by inactivation of Cic proteins, the main Cic effector is the transcription factor Etv4. Indeed, ablation of Etv4 expression in Cic inactivation-induced T-ALL completely prevented the development of this disease in four out of five mice and significantly delayed tumor formation in the remaining animal. Notably, thymocyte development was not altered in Etv4-deficient mice, indicating that Etv4 (and possibly other Pea3 family members such as Etv5) is selectively required for tumor formation. Whether Etv4 is also required for Ras-driven T-ALL remains to be determined. However, similar observations were made upon depletion of ETV4 in human T-ALL cell lines, although growth inhibition appears to be independent of the mutational status of CIC or RAS. Thus, a better understanding of the molecular events triggered by CIC inactivation should provide more effective therapeutic approaches to treat those human tumors driven by either RAS hyperactivation or CIC mutations.
Materials and methods
Mice
The hUBC-CreERT2 (Ruzankina et al. 2007), EIIa-Cre (Lakso et al. 1996), p53−/− (Jacks et al. 1994), hGFAP-Cre (Zhuo et al. 2001), Etv4NLZ/NLZ (Livet et al. 2002), H-Ras−/−;N-Ras−/−;K-Raslox/lox (Drosten et al. 2010), and KHRasV12 (Drosten et al. 2017) strains have been described previously. Activation of the inducible CreERT2 recombinase was achieved by feeding the mice with a Tmx-containing diet (Harlan Laboratories). All mice were maintained in a mixed 129/Sv-C57BL/6 background. For tumor cell transplantation assays, single-cell suspensions (106 cells per 200 µL of PBS) were injected into the tail veins of immunodeficient mice. All animal experiments were approved by the Ethical Committees of the Spanish National Cancer Research Centre (CNIO), the Carlos III Health Institute, and the Autonomous Community of Madrid and were performed in accordance with the guidelines stated in the International Guiding Principles for Biomedical Research Involving Animals, developed by the Council for International Organizations of Medical Sciences (CIOMS).
Cell lines
MEFs were extracted from E13.5 embryos and cultured in DMEM supplemented with 10% FBS. T-ALL cell lines established from tumor-bearing mice and human T-ALL cell lines were propagated in RPMI1640 supplemented with 10% FBS. For growth inhibition assays, murine cells were seeded at a density of 2 × 105 cells per milliliter with various concentrations of trametinib. Human cell lines were seeded in 96-well plates at a density of 10,000 cells per well, and proliferation was assessed using the (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Roche).
Plasmids
The GFP-tagged human CIC-S construct in pcDNA5/FRT/TO (Dissanayake et al. 2011) was a kind gift of Dr. Carol MacKintosh (University of Dundee, UK). The deletion of exons 2–6 (spanning residues 24–311) was introduced using the QuikChange site-directed mutagenesis kit (Agilent) following the manufacturer's guidelines. All plasmids were stably introduced into Flp-In T-REx 293 cells (Invitrogen) following instructions from the manufacturer. Human CIC targeting sgRNAs (#1, CCCCTCCGTGC AGCCGAGCG; #2, GCCTCGCTCGGCTGCACGGA) or a nontargeting control sgRNA (CTCGTGAACAAGATCCGAC) were cloned into lentiCRISPRv2 (Addgene 52961). Sigma MISSION shRNA lentiviral vectors were used to knock down ETV4 expression in human cell lines (#232, TRCN0000013937; and #736, TRCN0000013934). A nontargeting shRNA vector (SHC002) was used as control.
Histopathology and IHC
For histological analyses, tissues were fixed in 10% buffered formalin and embedded in paraffin. Hematoxilin and eosin (H&E) staining and IHC analyses were performed on 3-µm paraffin sections. For IHC, the following antibodies were used: CD3 (Pharmingen, 553057), cleaved Notch1 (Abcam, ab8925), Hes1 (CNIO), TDT (DAKO, A3524), Ki67 (Master Diagnostica, 0003110QD), γH2AX (Millipore, 05-636), and active Caspase 3 (Cell Signaling, 9661). PAS staining was performed by incubation for 5 min with periodic acid followed by 20 min of incubation with Schiff reagent and hematoxylin counterstaining.
Western blot analysis
Western blot analysis of protein extracts obtained from tissues, cell lines, or MEFs was performed as described (Drosten et al. 2010). To specifically probe for Cic expression levels, protein extracts were prepared in TST buffer as described (Lam et al. 2006). Primary antibodies used included polyclonal Cic antisera for the mouse proteins (Lam et al. 2006; Kim et al. 2015), human CIC (Abcam, ab123822), pErk1/2 (Cell Signaling Technology, 9101), pAkt (Cell Signaling Technology, 9271), Akt (Cell Signaling Technology, 9272), Erk1 (Santa Cruz Biotechnology, C16), and GAPDH (Sigma-Aldrich, G8795).
Flow cytometry analysis
Thymocytes were obtained by mechanical dissociation of the thymus in RPMI1640. Before analysis, cells were preincubated with purified anti-mouse CD16/32 antibodies (1:200; BD Pharmingen) for 15 min on ice to block nonspecific Fc receptor-mediated binding. Aliquots of 5 × 106 cells were stained for 20 min at room temperature with the following monoclonal antibodies: PECy7 α-Gr1 (1:200), BUV737 α-CD19 (1:400), APCCy7 α-Ter119 (1:200), PE α-Nk1.1/α-DX5 (1:200; to exclude non T cells), FITC α-CD4 (1:800), BUV395 α-CD8 (1:400), APC α-CD44 (1:200), and PerCPCy5.5 α-CD25 (1:200). Samples were processed on a LSRFortessa flow cytometer (BD Pharmingen) and analyzed using FlowJo (Tree Star).
Statistical analysis
Data are mean ± SD for Figures 1E, 5D–F, and 7C–E. Comparison between different genotypes in Figure 2B was performed using an unpaired Student's t-test. P-values of <0.05 were considered to be statistically significant (P < 0.05 [*] and P < 0.001 [***]).
Supplementary Material
Acknowledgments
We are grateful to Carol MacKintosh (University of Dundee, UK) for the pcDNA5/FRT/TO-GFP-CIC plasmid, and Huda Zoghbi (Baylor College of Medicine, Houston, TX) and Yoontae Lee (University of Pohang, Korea) for Cic antisera. We thank Scott Brown and Robert Holt (University of Vancouver, Canada) for their help with TCR abundance calculations. We also thank Carmen G. Lechuga, Marta San Roman, Raquel Villar, Beatriz Jiménez, and Nuria Cabrera for excellent technical assistance. We value the support of Sagrario Ortega (Transgenic Mice Core Unit, CNIO) for help in generating the Cic mutant mice, Orlando Dominguez (Genomics Core Unit, CNIO) for the RNA-seq analysis, and the Histopathology Core Unit. This work was supported by grants from the Fundació La Marató de TV3 (20131730/1) to G.J. and M.B., and the European Research Council (ERC-AG/ 250297-RAS AHEAD), the EU-Framework Programme (HEALTH-F2-2010-259770/LUNGTARGET and HEALTH-2010-260791/EUROCANPLATFORM), the Spanish Ministry of Economy and Competitiveness (SAF2014-59864-R), the Autonomous Community of Madrid (S2011/BDM-2470/ONCOCYCLE), and the Asociación Española contra el Cáncer (AECC) (GC16173694BARB) to M.B. M.B. is the recipient of an Endowed Chair from the AXA Research Fund. L.S.-C. was supported by a fellowship from the Programa de Formación de Personal Investigator (FPI) of the Spanish Ministry of Economy and Competitiveness. M.D. and M.B. conceived and designed the study. L.S.-C., O.G., G.J., M.D., and M.B. developed the methodology. L.S.-C., O.G., M.S., and M.D. acquired the data. L.S.-C., O.G., M.S., H.K.C.J., G.J., M.D., and M.B. analyzed and interpreted the data. L.S-C., O.G., G.J., M.D., and M.B. wrote, reviewed, and/or revised the manuscript. G.J. provided material support. A.G. analyzed the T-ALL sequencing. M.D. and M.B. supervised the study.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.300244.117.
References
- Aihara K, Mukasa A, Nagae G, Nomura M, Yamamoto S, Ueda H, Tatsuno K, Shibahara J, Takahashi M, Momose T, et al. 2017. Genetic and epigenetic stability of oligodendrogliomas at recurrence. Acta Neuropathol Commun 5: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astigarraga S, Grossman R, Díaz-Delfín J, Caelles C, Paroush Z, Jimenéz G. 2007. A MAPK docking site is critical for downregulation of Capicua by Torso and EGFR RTK signaling. EMBO J 26: 668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atak ZK, Gianfelici V, Hulselmans G, De Keersmaecker K, Devasia AG, Geerdens E, Mentens N, Chiaretti S, Durinck K, Uyttebroeck A, et al. 2013. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet 9: 1003997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belver L, Ferrando AA. 2016. The genetics and mechanisms of T cell acute lymphoblastic leukemia. Nat Rev Cancer 16: 494–507. [DOI] [PubMed] [Google Scholar]
- Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G, et al. 2011. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 333: 1453–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper KB, McCarthy KD. 2006. GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol Cell Neurosci 31: 676–684. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. 2012. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MY, Xu L, Shestova O, Histen G, L'Heureux S, Romany C, Childs ME, Gimotty PA, Aster JC, Pear WS. 2008. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest 118: 3181–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo-Barreto J, Fryer JD, Shaw CA, Orr HT, Zoghbi HY. 2010. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet 6: e1001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dail M, Li Q, McDaniel A, Wong J, Akagi K, Huang B, Kang HC, Kogan SC, Shokat K, Wolff L, et al. 2010. Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc Natl Acad Sci 107: 5106–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker K, Real PJ, Della Gatta G, Palomero T, Sulis ML, Tosello V, Van Vlierberghe P, Barnes K, Castillo M, Sole X, et al. 2010. The TLX1 oncogene drives aneuploidy in T cell transformation. Nat Med 16: 1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Gatta G, Palomero T, Perez-Garcia A, Ambesi-Impiombato A, Bansal M, Carpenter ZW, De Keersmaecker K, Sole X, Xu L, Paietta E, et al. 2012. Reverse engineering of TLX oncogenic transcriptional networks identifies RUNX1 as a tumor suppressor in T-ALL. Nat Med 18: 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissanayake K, Toth R, Blakey J, Olsson O, Campbell DG, Prescott AR, MacKintosh C. 2011. ERK/p90(RSK)/14-3-3 signaling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicua. Biochem J 433: 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyonnas R, Kershaw DB, Duhme C, Merkens H, Chelliah S, Graf T, McGagny KM. 2001. Anuria, omphalocele, and perinatal lethality in mice lacking the Cd34-related protein podocalyxin. J Exp Med 194: 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, Barbacid M. 2010. Genetic analysis of Ras signaling pathways in cell proliferation, migration and survival. EMBO J 29: 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten M, Simón-Carrasco L, Hernández-Porras I, Lechuga CG, Blasco MT, Jacob HK, Fabbiano S, Potenza N, Bustelo XR, Guerra C, et al. 2017. H-Ras and K-Ras oncoproteins induce different tumor spectra when driven by the same regulatory sequences. Cancer Res 77: 707–718. [DOI] [PubMed] [Google Scholar]
- Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland DG, et al. 2002. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 1: 75–87. [DOI] [PubMed] [Google Scholar]
- Forés M, Ajuria L, Samper N, Astigarraga S, Nieva C, Grossman R, González-Crespo S, Paroush Z, Jiménez G. 2015. Origins of context-dependent gene repression by Capicua. PLoS Genet 11: e1004902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forés M, Simón-Carrasco L, Ajuria L, Samper N, González-Crespo S, Drosten M, Barbacid M, Jiménez G. 2017. A new mode of DNA binding distinguishes Capicua from other HMG-box factors and explains its recurrent mutation patterns in cancer. PLoS Genet 13: e1006622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Yu P, Kang H, Mandel-Brehm C, Carter AN, Crespo-Barreto J, Gao Y, Flora A, Shaw C, Orr HT, et al. 2011. Exercise and genetic rescue of SCA1 vie the transcriptional repressor Capicua. Science 334: 690–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. 1994. Tumor spectrum analysis in p53-mutant mice. Curr Biol 4: 1–7. [DOI] [PubMed] [Google Scholar]
- Jiménez G, Guichet A, Ephrussi A, Casanova J. 2000. Relief of gene repression by torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev 14: 224–231. [PMC free article] [PubMed] [Google Scholar]
- Jiménez G, Shvartsman SY, Paroush Z. 2012. The Capicua repressor—a general sensor of RTK signaling in development and disease. J Cell Sci 125: 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Ha N, Forés M, Xiang J, Glässer C, Maldera J, Jiménez G, Edgar BA. 2015. EGFR/Ras signaling controls Drosophila intestinal stem cell proliferation via Capicua-regulated genes. PLoS Genet 11: e1005634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga JI, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G, et al. 2015. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 47: 1304–1315. [DOI] [PubMed] [Google Scholar]
- Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H, Gotoh T, Motoi T, Fukayama M, Aburatani H, et al. 2006. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 15: 2125–2137. [DOI] [PubMed] [Google Scholar]
- Kim E, Park S, Choi N, Lee J, Yoe J, Kim S, Jung HY, Kim KT, Kang H, Fryer JD, et al. 2015. Deficiency of Capicua disrupts bile acid homeostasis. Sci Rep 5: 8272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE, Fröhling S, Lee BH, Gilliland DG. 2008. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to γ-secretase inhibitors. Blood 112: 3373–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. 1996. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci 93: 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun JD, Duvick LA, Orr HT, Botas J, et al. 2006. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127: 1335–1347. [DOI] [PubMed] [Google Scholar]
- Lee Y, Fryer JD, Kang H, Crespo-Barreto J, Bowman AB, Gao Y, Kahle JJ, Hong JS, Kheradmand F, Orr HT, et al. 2011. ATXN1 protein family and CIC regulate extracellular matrix remodeling and lung alveolarization. Dev Cell 21: 746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S, Davoli T, Leng Y, Li MZ, Xu Q, Elledge SJ. 2017. A genetic interaction analysis identifies cancer drivers that modify EGFR dependency. Genes Dev 31: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. 2015. The molecular signature database hallmark gene set collection. Cell Syst 1: 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livet J, Sigrist M, Stroebel S, De Paolo V, Price SR, Henderson CE, Jessell TM, Arber S. 2002. ETS gene Pea3 controls the central position and terminal arborization of specific motor neuron pools. Neuron 35: 877–892. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. 2003. RAS oncogenes: the first 30 years. Nat Rev Cancer 3: 459–465. [DOI] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Orkin SH. 1999. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci 96: 5037–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Barriuso N, López-Malpartida AV, de Pablo F, Pichel JG. 2006. Alterations in alveolar epithelium differentiation and vasculogenesis in lungs of LIF/IGF-I double deficient embryos. Dev Dyn 235: 2040–2050. [DOI] [PubMed] [Google Scholar]
- Naltner A, Wert S, Whitsett JA, Yan C. 2000. Temporal/spatial expression of nuclear receptor coactivators in the mouse lung. Am J Physiol Lung Cell Mol Physiol 279: L1066–L1074. [DOI] [PubMed] [Google Scholar]
- Okimoto RA, Breitenbuecher F, Olivas VR, Wu W, Gini B, Hofree M, Asthana S, Hrustanovic G, Flanagan J, Tulpule A, et al. 2017. Inactivation of Capicua drives cancer metastases. Nat Genet 49: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima K, Khiaganian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, Sanchez-Martin M, Carpenter Z, Penson A, Perez-Garcia A, et al. 2016. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci 113: 11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch F, Jiménez G, Casanova J. 2002. EGFR signaling inhibits Capicua-dependent repression during specification of Drosophila wing veins. Development 129: 993–1002. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr. 2012. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 66: 105–143. [DOI] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. 2007. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1: 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabnis AJ, Cheung LS, Dail M, Kang HC, Santaguida M, Hermiston ML, Passagué E, Shannon K, Braun BS. 2009. Oncogenic Kras initiates leukemia in hematopoietic stem cells. PLoS Biol 7: e1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J, et al. 2012. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev 26: 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng AS, Tapon N, Kanda H, Cigizoglu S, Edelmann L, Pellock B, White K, Hariharan IK. 2007. Capicua regulates cell proliferation downstream of the receptor tyrosine kinase/ras signaling pathway. Curr Biol 17: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lintig FC, Huvar I, Law P, Diccianni MB, Yu AL, Boss GR. 2000. Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin Cancer Res 6: 1804–1810. [PubMed] [Google Scholar]
- Wang B, Krall EB, Aguirre AJ, Kim M, Widlund HG, Doshi MB, Sicinska E, Sulahian R, Goodale A, Cowley GS, et al. 2017. ATXN1L, CIC, and ETS transcription factors modulate sensitivity to MAPK pathway inhibition. Cell Rep 18: 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CJ, McCormack PL. 2013. Trametinib: first global approval. Drugs 73: 1245–1254. [DOI] [PubMed] [Google Scholar]
- Yip S, Butterfield YS, Morozova O, Chittaranjan S, Blough MD, An J, Birol I, Chesnelong C, Chiu R, Chuah E, et al. 2012. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol 226: 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. 2001. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31: 85–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.