

Abstract
Methyl-coenzyme M reductase (MCR), found in strictly anaerobic methanogenic and methanotrophic archaea, catalyzes the reversible production and consumption of the potent greenhouse gas methane. The α subunit of MCR (McrA) contains several unusual post-translational modifications, including a rare thioamidation of glycine. Based on the presumed function of homologous genes involved in the biosynthesis of thioviridamide, a thioamide-containing natural product, we hypothesized that the archaeal tfuA and ycaO genes would be responsible for post-translational installation of thioglycine into McrA. Mass spectrometric characterization of McrA from the methanogenic archaeon Methanosarcina acetivorans lacking tfuA and/or ycaO revealed the presence of glycine, rather than thioglycine, supporting this hypothesis. Phenotypic characterization of the ∆ycaO-tfuA mutant revealed a severe growth rate defect on substrates with low free energy yields and at elevated temperatures (39°C - 45°C). Our analyses support a role for thioglycine in stabilizing the protein secondary structure near the active site.
Research organism: Other
Introduction
Methyl-coenzyme M reductase (MCR) is a unique enzyme found exclusively in anaerobic archaea, where it catalyzes the reversible conversion of methyl-coenzyme M (CoM, 2-methylmercaptoethanesulfonate) and coenzyme B (CoB, 7-thioheptanoylthreoninephosphate) to methane and a CoB-CoM heterodisulfide (Ermler et al., 1997; Scheller et al., 2010):
This reaction, which has been proposed to proceed via an unprecedented methyl radical intermediate (Wongnate et al., 2016), plays a critical role in the global carbon cycle (Thauer et al., 2008). In the forward direction, MCR catalyzes the formation of methane in methane-producing archaea (methanogens). In the reverse direction, MCR catalyzes the consumption of methane in methanotrophic archaea (known as ANMEs, for anaerobic oxidation of methane). Together, these processes produce and consume gigatons of methane each year, helping to establish the steady-state atmospheric levels of an important greenhouse gas. MCR displays an α2β2γ2 protein domain architecture and contains two molecules of F430, a nickel porphinoid cofactor (Ermler et al., 1997; Zheng et al., 2016; Moore et al., 2017). The reduced Ni(I) form of F430 is essential for catalysis (Goubeaud et al., 1997), but is highly sensitive to oxidative inactivation, a feature that renders biochemical characterization of MCR especially challenging. As a result, many attributes of this important enzyme remain uncharacterized.
An unusual feature of MCR is the presence of several modified amino acids within the active site of the α-subunit. Among these are a group of methylated amino acids, including 3-methylhistidine (or N1-methylhistidine), S-methylcysteine, 5(S)-methylarginine, and 2(S)-methylglutamine, which are presumed to be installed post-translationally by S-adenosylmethionine-dependent methyltransferases (Selmer et al., 2000; Kahnt et al., 2007). 3-methylhistidine is found in all MCRs examined to date, whereas the presence of the other methylated amino acids is variable among methane-metabolizing archaea (Kahnt et al., 2007). A didehydroaspartate modification is also observed in some, but not all, methanogens (Wagner et al., 2016). Lastly, a highly unusual thioglycine modification, in which the peptide amide bond is converted to a thioamide, is present in all methanogens that have been analyzed to date (Figure 1—figure supplement 1) (Ermler et al., 1997; Kahnt et al., 2007).
The function(s) of the modified amino acids in MCR has/have not yet been experimentally addressed; however, several theories have been postulated. The 3-methylhistidine may serve to position the imidazole ring that coordinates CoB. This methylation also alters the pKa of histidine in a manner that would promote tighter CoB-binding (Grabarse et al., 2000). The variable occurrence of the other methylated amino acids suggests that they are unlikely to be directly involved in catalysis. Rather, it has been hypothesized that they tune enzyme activity by altering the hydrophobicity and flexibility of the active site pocket (Kahnt et al., 2007; Grabarse et al., 2000). A similar argument has been made for the didehydroaspartate residue (Wagner et al., 2016). In contrast, three distinct mechanistic hypotheses have implicated the thioglycine residue in catalysis. One proposal is that the thioglycine facilitates deprotonation of CoB by reducing the pKa of the sulfhydryl group (Grabarse et al., 2001). A second proposal suggests that thioglycine could serve as an intermediate electron carrier for oxidation of a proposed heterodisulfide anion radical intermediate (Horng et al., 2001). Lastly, cis-trans isomerization of the thioamide during catalysis could potentially play an important role in coupling the two active sites via a previously proposed two-stroke mechanism for the enzyme (Goenrich et al., 2005).
Thioamides are rare in biology. While cycasthioamide is plant-derived (Pan et al., 1997), most other naturally occurring thioamides are of bacterial origin. Among these are the ribosomally synthesized and post-translationally modified (RiPP) peptide natural products thioviridamide (Hayakawa et al., 2006) and methanobactin (Kim et al., 2004; Kenney and Rosenzweig, 2012; Kenney and Rosenzweig, 2013), as well as closthioamide, which appears to be an unusual non-ribosomal peptide (Lincke et al., 2010; Chiriac et al., 2015). The nucleotide derivatives thiouridine and thioguanine (Coyne et al., 2014), and two additional natural product antibiotics, thiopeptin and Sch 18640 (Puar et al., 1981; Hensens and Albers-Schönberg, 1983), also contain thioamide moieties. Although the mechanism of thioamide installation in peptides has yet to be established, the identification of the thioviridamide biosynthetic gene cluster provides clues to their origin (Izawa et al., 2013). Two of the proteins encoded by this gene cluster have plausible roles in thioamide synthesis. The first, TvaH, is a member of the YcaO superfamily, while the second, TvaI, is annotated as a ‘TfuA-like’ protein (Izawa et al., 2013). Biochemical analyses of YcaO-family proteins indicate that they catalyze the ATP-dependent cyclodehydration of Cys, Ser, and Thr residues to the corresponding thiazoline, oxazoline, and methyloxazoline (Figure 1). Many ‘azoline’ heterocycles undergo dehydrogenation to the corresponding azole, which are prominent moieties in various RiPP natural products classes, such as linear azol(in)e-containing peptides, thiopeptides, and cyanobactins (Dunbar et al., 2012; Dunbar et al., 2014; Arnison et al., 2013; Burkhart et al., 2017). The YcaO-dependent synthesis of peptidic azol(in)e heterocycles often requires a partner protein, which typically is the neighboring gene in the biosynthetic gene cluster (Burkhart et al., 2015; Dunbar et al., 2015; Wright et al., 2006). Based on enzymatic similarity, the TfuA protein encoded adjacent to the YcaO in the thioviridamide pathway may also enhance the thioamidation reaction, perhaps by recruiting a sulfurtransferase protein. Although not RiPPs, thiouridine and thioguanine biosynthesis requires the use of sulfurtransferases (Coyne et al., 2013; Palenchar et al., 2000).
Figure 1. Comparison of reactions catalyzed by YcaO proteins.
Top, Biochemically characterized YcaO proteins involved in the synthesis of azol(in)e-containing ribosomal natural products catalyze the ATP-dependent cyclodehydration of cysteine, serine, and threonine residues. Shown is the conversion of peptidic cysteine to thiazoline. This reaction proceeds via an O-phosphorylated hemiorthoamide intermediate. Bottom, An analogous reaction is believed to occur in the biosynthesis of the thioamide bond in thioviridamide. Rather than an adjacent cysteine acting as the nucleophile, an exogenous source of sulfide (S2-) is required for this reaction.
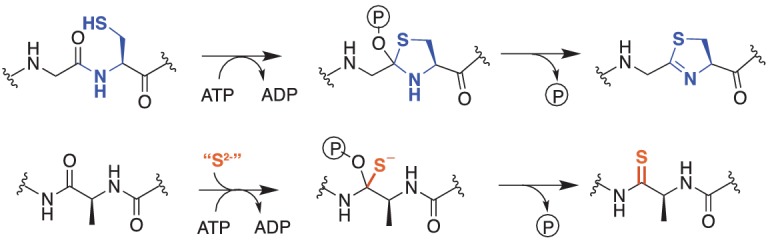
Figure 1—figure supplement 1. A view of the MCR active site using the crystal structure of M. barkeri (Protein DataBank entry 1E6Y).
The biosynthesis of the thioglycine in MCR was proposed to occur by a mechanism similar to that used to produce thioamide-containing natural products (Kahnt et al., 2007), a prediction made six years prior to the discovery of the thioviridamide biosynthetic gene cluster (Izawa et al., 2013). Given their putative role in thioamidation reactions, it is notable that YcaO homologs were found to be universally present in an early analysis of methanogen genomes, resulting in their designation as ‘methanogenesis marker protein 1’ (Basu et al., 2011). Genes encoding TfuA homologs are also ubiquitous in methanogens, usually encoded in the same locus as ycaO, similar to their arrangement in the thioviridamide biosynthetic gene cluster. Therefore, both biochemical and bioinformatic evidence are consistent with these genes being involved in thioglycine formation. In this report, we use the genetically tractable methanogen Methanosarcina acetivorans to show that installation of the thioamide bond at Gly465 in the α-subunit of MCR requires the ycaO-tfuA locus (Figure 1—figure supplement 1). As MCR is essential for growth and survival (Rother et al., 2005), the viability of ycaO-tfuA mutants precludes the hypothesis that the thioamide residue is essential for MCR activity. Instead, our phenotypic analyses support a role for thioglycine in maintaining a proper structural conformation of residues near the active site, such that absence of this modification in MCR might be particularly detrimental on growth substrates that have low free energy yields as well as under additionally destabilizing conditions such as elevated temperatures.
Results
Phylogenetic analyses of TfuA and YcaO in methanogenic and methanotrophic archaea
To examine the possibility that YcaO and TfuA are involved in McrA thioamidation, we reassessed the distribution, diversity, and phylogeny of genes encoding these proteins in sequenced microbial genomes, which today comprise a much larger dataset than when ‘methanogenesis marker protein 1’ was first proposed (Basu et al., 2011). Significantly, all complete methanogen and ANME genome sequences encode a YcaO homolog, with a few strains encoding two copies (Figure 2). The YcaO sequences form a distinct, well-supported clade that also includes homologs from ammonia-oxidizing archaea (Figure 2—figure supplement 1). Excluding the second YcaO in strains that encode two copies, the YcaO tree is congruent with the phylogeny of methanogens reconstructed using housekeeping or mcr genes (Figure 2). Thus, it is likely that YcaO was acquired early in the evolution of methanogens and maintained largely through vertical inheritance, as expected for a trait that coevolved with MCR (Borrel et al., 2013; Vanwonterghem et al., 2016).
Figure 2. A maximum-likelihood phylogenetic tree of YcaO homologs in archaea.
Taxa in green, blue, and orange depict methanogens and anaerobic methane-oxidizing archaea (ANMEs) with sequenced genomes. Taxa shown in green contain a single copy of ycaO and tfuA, those in blue contain two copies of ycaO and one copy of tfuA, while those in orange contain one copy of ycaO, but do not encode tfuA. Taxa shown in pink are other archaea that also encode ycaO and tfuA homologs. The gene neighborhoods of selected taxa are also depicted, showing the common co-localization with genes annotated as having a role in sulfur metabolism or methanogenesis. The node labels indicate support values calculated using the Shiomdaira-Hasegawa test using 1000 resamples. Support values less than 0.5 have not been shown. The outgroup displayed derives from a variety of bacterial YcaO proteins.

Figure 2—figure supplement 1. (Panel A) Unrooted maximum-likelihood phylogeny of 1,000 YcaO sequences retrieved from the NCBI non-redundant protein sequence database using the corresponding sequence from Methanosarcina acetivorans as a search query.

Figure 2—figure supplement 2. Unrooted maximum-likelihood phylogeny of 1000 TfuA sequences retrieved from the NCBI non-redundant protein sequence database using the corresponding sequence from Methanosarcina acetivorans as a search query.

TfuA homologs are encoded in the overwhelming majority of MCR-encoding taxa, although a few species lack this gene. The latter include Methanopyrus kandleri and Methanocaldococcus janaschii, two species previously shown to contain the thioglycine modification (Kahnt et al., 2007) (Figure 2). Thus, TfuA cannot be obligatorily required for thioglycine installation. The archaeal TfuA proteins also fall within a single clade that is largely congruent with the evolutionary history of archaea (Figure 2—figure supplements 1–2); however, unlike YcaO, the methanogen-containing clade includes numerous bacterial homologs (Figure 2—figure supplement 2).
The genomic context of tfuA and ycaO in methanogens supports a shared or related function, perhaps involving sulfur incorporation and/or methanogenesis (Figure 2). When both are present, the two genes comprise a single locus in which the stop codon of the upstream ycaO gene overlaps with the start codon of the downstream tfuA gene, suggesting that they are co-transcribed. In several instances, additional genes involved in sulfur metabolism such as thiF, sufS, as well as moaD, moaE and moeB (involved in molybdopterin biosynthesis) (Zhang et al., 2010; Park et al., 2003), are found in the genomic vicinity. Occasionally, genes encoding enzymes involved in methanogenesis, including the Methanomassiliicoccus MCR operon, are locally encoded (Figure 2).
TfuA and YcaO are not essential in Methanosarcina acetivorans
To test their role in thioglycine installation, we generated a mutant lacking the ycaO-tfuA locus in the genetically tractable methanogen Methanosarcina acetivorans. Based on the hypothesis that thioglycine may be imperative for MCR activity, and the knowledge that the MCR-encoding operon (mcrBCDAG) is essential (Guss et al., 2008), we first examined the viability of ΔycaO-tfuA mutants using a recently developed Cas9-based assay for gene essentiality (Nayak and Metcalf, 2017). This assay compares the number of transformants obtained using a Cas9 gene-editing plasmid with and without a repair template that creates a gene deletion. Essential genes give similar, low numbers of transformants with or without the repair template; whereas non-essential genes give ca. 103-fold higher numbers with the repair template. Our data demonstrate that deletion of the ycaO-tfuA locus has no impact on viability (Figure 3). Several independent mutants were clonally purified and verified by PCR prior to phenotypic characterization (Figure 3—figure supplement 1).
Figure 3. Panel A, Experimental strategy for deletion of the ycaO-tfuA locus in M. acetivorans using a Cas9-mediated genome editing technique.
Co-expression of the Cas9 protein along with a single guide RNA (sgRNA) with a 20 bp target sequence flanked by a 3’ NGG protospacer adjacent motif on the chromosome (PAM; in red) generates double-stranded breaks at the ycaO-tfuA locus. Repair of this break via homologous recombination with an appropriate repair template (HR Template) generates an in-frame deletion on the host chromosome. Panel B, A Cas9-based assay for gene essentiality. Mean transformation efficiencies of plasmids with (dark gray) or without (light gray) an appropriate repair template are shown. The known essential gene mcrA is included as a positive control. The error bars represent one standard deviation of three independent transformations.

Figure 3—figure supplement 1. A PCR-based screen to genotype the ycaO-tfuA locus in Methanosarcina acetivorans.

Figure 3—figure supplement 2. The experimental strategy for generating an in-frame deletion of either the tfuA locus (Panel A) or the ycaO locus (Panel B) in M. acetivorans using a Cas9-mediated genome editing technique.

The ∆ycaO-tfuA mutant lacks the McrA Gly465 thioamide
To test whether YcaO and TfuA are involved in the post-translational modification of Gly465 in McrA, we isolated the McrA protein from cell extracts of the isogenic parent (hereafter referred to as wild-type or WT), the ∆ycaO-tfuA mutant, as well as from ∆tfuA and ∆ycaO individual mutants (Figure 3—figure supplement 2). After performing SDS-PAGE, the appropriate Coomassie-stained bands were excised and then subjected to in-gel trypsin digestion. The resulting peptides were then analyzed by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF-MS). The mass spectrum from the WT strain showed a peak at m/z 3432 Da (Figure 4) corresponding to the peptide (461LGFFGFDLQDQCGATNVLSYQGDEGLPDELR491) with Gly465 being thioamidated (Kahnt et al., 2007; Grabarse et al., 2000). The identity of this peptide was confirmed by high-resolution electrospray ionization tandem MS analysis (HR-ESI-MS/MS, [M + 3H]3+ expt. m/z 1144.8608 Da; calc. m/z 1144.8546 Da; 5.4 ppm error; Figure 4—figure supplement 1A). This peptide also contains the recently reported didehydroaspartate modification at Asp470 (Wagner et al., 2016) and S-methylation at Cys472 (Selmer et al., 2000). Consistent with the involvement of TfuA-associated YcaO proteins in thioamide formation, we noted the absence of the m/z 3432 Da species in the mass spectrum of similarly treated ∆ycaO-tfuA, ∆tfuA, and ∆ycaO samples. Instead, a predominant m/z 3416 Da species appeared, which was 16 Da lighter, consistent with replacement of sulfur by oxygen (Figure 4). HR-ESI-MS/MS analysis of the corresponding peptide from the ∆ycaO-tfuA mutant confirmed the identity of this peptide as being McrA Leu461-Arg491 ([M + 3H]3+ expt. m/z 1139.5316 Da; calc. m/z 1139.5290 Da; 2.3 ppm error; Figure 4—figure supplement 1B). During HR-ESI-MS/MS analysis of the WT, thioamide-containing peptide, we did not observe fragmentation between Gly465 and Phe466. In contrast, fragmentation was seen at this location in the ∆ycaO-tfuA strain (b5 ion, Figure 4—figure supplement 1B). The lack of fragmentation in the WT peptide was anticipated based on the greater double bond character of C-N bonds in thioamides (Wiberg and Rablen, 1995).
Figure 4. MALDI-TOF MS analysis of McrA.
Panel A, Spectrum obtained from trypsinolysis of WT MCR, which contains the thioglycine-modified L461-R491 peptide (m/z 3432 Da). A small amount of the unmodified L461-R491 peptide (m/z 3416 Da) is also observed, which probably arises via non-enzymatic hydrolysis of the thioamide bond as previously reported (Kahnt et al., 2007). Panels B, C, D, Spectrum obtained from trypsinolysis of McrA from the ∆ycaO-tfuA, ∆tfuA, ∆ycaO strains, respectively, containing only the unmodified L461-R491 peptide (m/z 3416 Da). Panels E, F, G. Spectrum obtained from trypsinolysis of MCR from the ∆ycaO-tfuA mutant complemented with PmcrB(tetO4)-ycaO-tfuA, ∆tfuA mutant complemented with PmcrB(tetO4)-tfuA, ∆ycaO mutant complemented with PmcrB(tetO4)-ycaO, respectively. Tetracycline was added to a final concentration of 100 µg/mL to the growth medium to induce expression from the PmcrB(tetO4) promoter. All three spectra contain the thioglycine-modified Leu461-Arg491 peptide (m/z 3432 Da). The m/z 3375 Da species present in all samples corresponds to McrA residues D392-R421.

Figure 4—figure supplement 1. (Panel A) High-resolutions electrospray ionization tandem mass spectrometry (HR-ESI MS/MS) analysis of a wild-type tryptic peptide from Methanosarcina acetivorans McrA (Leu461-Arg491, m/z 3432 Da).

Figure 4—figure supplement 2. HR-ESI-MS/MS of the tryptic peptide from Methanosarcina acetivorans McrA (Asp392-Arg421, m/z 3375 Da) present in the wild-type and ∆ycaO-tfuA, ∆tfuA, ∆ycaO deletion strains.

Figure 4—figure supplement 3. (Panel A) Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis of the wild-type (WT, top) and ∆ycaO-tfuA deletion strain (bottom) spectra identify the McrA tryptic peptide, His271-Arg284 (m/z 1496 Da), containing the known N-methylhistidine modification.

To confirm that mutations in ycaO and/or tfuA were responsible for loss of the thioglycine modification, we transformed the ∆ycaO-tfuA mutant with a self-replicating plasmid containing the ycaO-tfuA locus fused to the tetracycline-inducible promoter PmcrB(tetO4) (Guss et al., 2008). Upon tryptic digestion of MCR from cultures supplemented with tetracycline, a peak at m/z 3432 Da reappeared (Figure 4). Similar results were obtained when complementation experiments were performed with the ycaO or tfuA genes in the corresponding single mutants (Figure 4).
The m/z 3375 Da species present in all samples corresponds to an unrelated tryptic peptide, McrA Asp392-Arg421 (Figure 4—figure supplement 2). We also observed a peak at m/z 1496 in the WT and mutant samples corresponding to a N-methylhistidine-containing tryptic peptide (His271-Arg284, Figure 4—figure supplement 3A). Thus, thioglycine formation is not a prerequisite for this modification. Additionally, a peak at m/z 1561 was observed, corresponding to McrA Phe408-Arg421, shows that Gln420 remains unmodified in M. acetivorans, as previously been observed for Methanosarcina barkeri (Figure 4—figure supplement 3B) (Kahnt et al., 2007).
Growth defects associated with loss of the McrA thioglycine modification
To understand potential phenotypic consequences of losing the thioglycine modification, we quantified the doubling time and growth yield by monitoring changes in optical density during growth of M. acetivorans on a variety of substrates (Table 1). While no significant differences were observed during growth on methanol or trimethylamine (TMA) at 36°C, the ∆ycaO-tfuA mutant had substantially longer generation times and lower cell yields on both dimethylsulfide (DMS) and acetate (Table 1). Interestingly, the growth phenotype on methanol medium was strongly temperature-dependent, with no observed differences at 29°C and 36°C, but severe defects observed for the ∆ycaO-tfuA mutant at 39°C and 42°C. Furthermore, unlike WT, the mutant was incapable of growth at 45°C (Figure 5).
Table 1. Growth phenotypes of M. acetivorans WT and the ∆ycaO-tfuA mutant on different substrates at 36°C.
| Substrate* | Strain | Doubling time (h)† | p-value |
Maximum OD600† | p-value |
|---|---|---|---|---|---|
| Methanol | WT | 6.94 ± 0.41 | 0.87 | 5.35 ± 0.31 | 0.26 |
| ∆ycaO-tfuA | 6.89 ± 0.15 | 5.06 ± 0.39 | |||
| TMA | WT | 10.92 ± 0.54 | 0.84 | 9.57 ± 0.67 | 0.31 |
| ∆ycaO-tfuA | 10.98 ± 0.43 | 8.91 ± 0.73 | |||
| Acetate | WT | 54.23 ± 4.03 | 0.0002 | 0.44 ± 0.01 | 0.0005 |
| ∆ycaO-tfuA | 124.33 ± 12.52 | 0.35 ± 0.02 | |||
| DMS | WT | 31.27 ± 1.28 | <0.0001 | 0.60 ± 0.01 | <0.0001 |
| ∆ycaO-tfuA | 58.28 ± 1.45 | 0.44 ± 0.01 |
*TMA, trimethylamine; DMS, dimethylsulfide.
†The error represents the 95% confidence interval of the mean for three biological replicates and the p-values were calculated using two-tailed unpaired t-tests.
Figure 5. Growth rate of WT (blue) and the ∆ycaO-tfuA mutant (green) in HS medium with 125 mM methanol at the indicated temperatures.
A statistically significant difference (p<0.01) using a two-tailed unpaired t-test is indicated with an *. NG indicates that no growth was detected after one month of incubation.

Figure 5—figure supplement 1. (Panel A) Nucleotide and amino acid sequence of the N-terminal TAP (tandem affinity purification) tag inserted immediately upstream of the start codon of mcrG (locus tag, MA4547) using the Cas9-mediated genome editing technique.

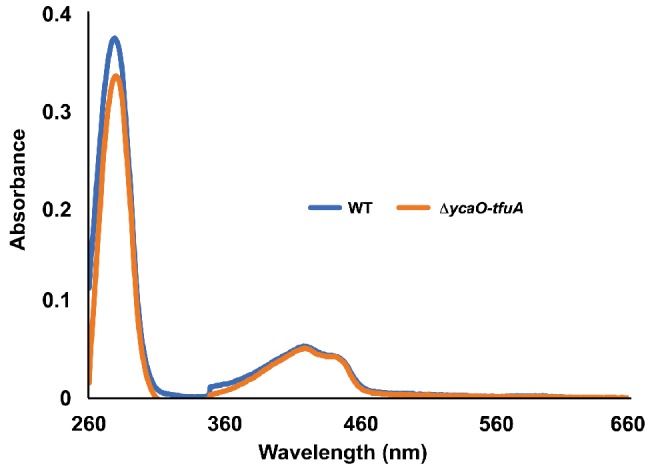
Figure 5—figure supplement 2. UV-Vis absorbance spectra of tagged-MCR purified from the WT strain (blue) and the ∆ycaO-tfuA mutant (orange).
Thioglycine does not influence the global stability of MCR
Given that the C-N bond rotation barrier is higher for thioamides relative to amide (Wiberg and Rablen, 1995), we tested if the presence of thioglycine promotes the thermal stability of MCR. We evaluated thermal stability by assaying the melting temperature of MCR purified from WT and the ∆ycaO-tfuA mutant. A tandem affinity purification (TAP) tag consisting of a 3 × FLAG tag and a Twin-Strep tag was introduced immediately upstream of the start codon of mcrG (Figure 5—figure supplement 1A). The proteins were affinity-purified using a Strep-tactin resin under aerobic conditions (Figure 5—figure supplement 1B). Using the SYPRO Orange-based Thermofluor assay (Huynh and Partch, 2015), the melting temperature of WT MCR was 67.3 ± 0.3°C (mean ± 95% confidence interval of three technical replicates) whereas the melting temperature for the MCR variant lacking thioglycine was 69.1 ± 0.2°C. Additionally, quantification of the protein and F430 concentrations by Bradford assay and absorption at 280/430 nm demonstrated that the F430 content was not significantly affected by the presence of thioglycine (Figure 5—figure supplement 2).
Discussion
The loss of the thioglycine modification in the ∆ycaO, ∆tfuA, and ΔycaO-tfuA mutants shows that both of these genes are required for the thioamidation of McrA. Although this could be an indirect requirement, we believe it is more likely that the YcaO/TfuA proteins directly catalyze modification of McrA. This conclusion is mechanistically compatible with biochemical analyses of YcaO homologs. YcaO enzymes that carry out the ATP-dependent cyclodehydration of beta-nucleophile-containing amino acids have been extensively investigated (Burkhart et al., 2017). Such cyclodehydratases coordinate the nucleophilic attack of the Cys, Ser, and Thr side chain on the preceding amide carbonyl carbon in a fashion reminiscent of intein splicing (Perler et al., 1997) (Figure 1). The enzyme then O-phosphorylates the resulting oxyanion and subsequently N-deprotonates the hemiorthoamide, yielding an azoline heterocycle. An analogous reaction can be drawn for the YcaO-dependent formation of peptidic thioamides. The only difference is that an exogenous equivalent of sulfide is required for the thioamidation reaction, rather than an adjacent beta-nucleophile-containing amino acid for azoline formation (Figure 1).
Most YcaO cyclodehydratases require a partner protein for catalytic activity. The earliest investigated YcaO partner proteins are homologs of the ThiF/MoeB family, which are related to E1 ubiquitin-activating enzymes (Dunbar et al., 2014; Schulman et al., 2009). These YcaO partner proteins, as well as the more recently characterized ‘ocin-ThiF’ variety (Dunbar et al., 2015), contain a ~ 90 residue domain referred to as the RiPP precursor peptide Recognition Element (RRE), which facilitates substrate recognition by interacting with the leader peptide. Considering these traits of azoline-forming YcaOs, it is possible that thioamide-forming YcaOs require the TfuA partner to facilitate binding to the peptidic substrate. Alternatively, TfuA may recruit and deliver sulfide equivalents by a direct or indirect mechanism. In this regard, it is noteworthy that the ycaO-tfuA locus can be found adjacent to genes involved in sulfur and molybdoterin metabolism (Figure 2). Many of these genes encode proteins with rhodanese-like homology domains, which are well-established sulfurtransferases. These enzymes typically carry sulfur in the form of a cysteine persulfide, a non-toxic but reactive equivalent of H2S (Palenchar et al., 2000; Matthies et al., 2005). Akin to rare cases of azoline-forming, partner-independent YcaOs, certain methanogens lack a bioinformatically identifiable TfuA (e.g. Methanopyrus kandleri and Methanocaldococcus sp., Figure 2). Whether these YcaOs act independently or use an as yet-unidentified partner protein remains to be seen. Clearly, further in vitro experimentation will be required to delineate the precise role of TfuA in the thioamidation reaction.
The viability of the ΔycaO-tfuA mutant raises significant questions as to the role of thioglycine in the native MCR enzyme, especially considering its universal presence in all MCRs examined to date. We considered three hypotheses to explain this result. First, we examined the possibility that thioglycine modification is involved in enhancing the reaction rate (Kahnt et al., 2007; Horng et al., 2001). Although it has not been explicitly determined, MCR is thought to catalyze the rate-limiting step of methanogenesis (Scheller et al., 2010; Wongnate et al., 2016). Therefore, the absence of thioglycine might lead to a corresponding decrease in the growth rate, with more pronounced defects on substrates that lead to the fastest growth. However, we observed the opposite: the most pronounced defects were observed with growth substrates that support the slowest WT growth rates (Table 1). Next, we considered the possibility that the thioglycine influences substrate affinity. C1 units enter methanogenesis at the level of N5-methyl-tetrahydrosarcinapterin (CH3-H4SPt) during growth on acetate (Galagan et al., 2002; Deppenmeier et al., 1999), but at the level of methyl-CoM during growth on DMS, methanol, and TMA (Figure 6) (Bose et al., 2008; Fu and Metcalf, 2015). Significantly, these entry points are separated by an energy-dependent step that is coupled to production or consumption of the trans-membrane Na+ gradient. As a result, intracellular levels of methyl-CoM, CoM, and CoB could possibly be significantly different depending on the entry point into the methanogenic pathway. Because we observed growth defects on substrates that enter at both points (i.e. DMS and acetate), we suspect that the growth deficiency phenotype is unlikely to be related to changes in substrate affinity. A third explanation we considered was that thioglycine increases the stability of MCR. In this model, unstable MCR protein would need to be replaced more often, creating a significant metabolic burden for the mutant. Consistent with our results, this additional burden would be exacerbated on lower energy substrates like DMS and acetate, especially given that MCR comprises ~10% of the total protein (Rospert et al., 1990). Further, one would expect that a protein stability phenotype would be exaggerated at higher temperatures, which we observed during growth on methanol (Figure 5). Thus, multiple lines of evidence support the idea that the growth-associated phenotypes stemming from the deletion of TfuA and YcaO are caused by decreased MCR stability. However, the melting temperature of MCR from the ∆ycao-tfuA mutant was modestly higher than that purified from the WT. As the thioglycine is buried deep within the active site of MCR (Ermler et al., 1997), these data are consistent with the notion that thioamidation does not affect the global stability of the protein. Instead, it suggests that the thioglycine modification impacts the local stability in the vicinity of the buried active site of MCR (Figure 1—figure supplement 1), which in turn might have a detrimental effect under catalytic turnover conditions.
Figure 6. An overview of methanogenic metabolism in M. acetivorans.

Methylotrophic substrates such as methanol (CH3OH) or dimethylsulfide (DMS, CH3-S-CH3) enter the methanogenic pathway via S-methylation of coenzyme M (CoM) and subsequently disproportionate to methane (CH4) and carbon dioxide (CO2; metabolic flux is shown as blue arrows). Notably, the first step in oxidation of CH3-CoM to CO2 is the energy-requiring transfer of the methyl moiety to generate methyl-tetrahydrosarcinapterin (CH3-H4SPt). In contrast, acetic acid (CH3COOH) enters the pathway at the CH3-H4SPt level, followed by reduction to CH4 (green arrows). Thus, the second step of the pathway is exergonic.
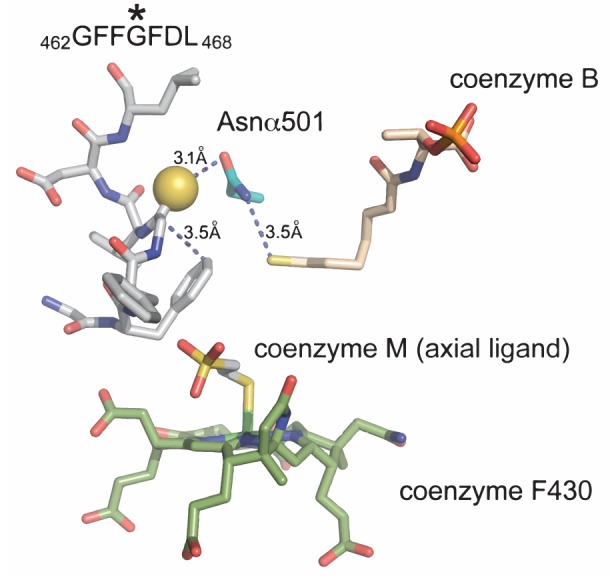
The chemical properties of amides relative to thioamides are consistent with our hypothesis. Although amides tend to be planar, their rotational barriers are lower than for the corresponding thioamide and thus they are more conformationally flexible. This is especially true for glycine. Considering the negligible electronegativity difference between sulfur and carbon, the thioamide carbonyl is not polarized like an amide carbonyl (Wiberg and Rablen, 1995). Further, sulfur has a larger van der Waals radius than oxygen resulting in a thioamide bond length that is ~40% longer than the amide bond (1.71 Å versus 1.23 Å) (Petersson et al., 2014), which presents additional steric hindrances to backbone flexibility. Finally, the pKa of thioamides is lower than amides, making thioglycine a stronger hydrogen bond donor than glycine (Lee et al., 2002), which again could reduce conformational flexibility. Taken together, it is chemically reasonable to state that the increased flexibility of the unmodified glycine in the ΔycaO-tfuA mutant would render the contorted conformation of the peptide backbone in the Gly462-Leu469 region of McrA considerably less stable (Figure 1—figure supplement 1). Indeed, this region adopts a rather strange configuration, with the side chain of Phe463 bending back towards the thioamide of Gly465. The molecular alignment and distance (3.5 Å) suggests a potential π-cation interaction between these residues(Figure 1—figure supplement 1) The thioamide sulfur is also optimally distanced (3.6 Å) and geometrically positioned to form an H-bond with the side chain of Asn501 and the backbone N-H of Leu469. There also appears to be a hydrophobic interaction between the thioamide and the beta carbon of Leu469, which would be energetically unfavorable with a more polarized amide. These observations suggest that the local stability in the vicinity of the active site imparted by the thioamide is required for protein stability and increased half-life of the MCR protein. A conclusive test of this hypothesis will require comprehensive structural and biochemical characterization of the active MCR variant lacking the thioglycine modification, which is beyond the scope of this work.
Finally, the temperature-sensitive phenotype of the ΔycaO-tfuA mutant has potential implications regarding the evolution and ecology of methanogenic archaea. Based on this result, it seems reasonable to speculate that the thioglycine modification would be indispensable for thermophilic methanogens. It is often posited that the ancestor of modern methanogens was a thermophilic organism (Gribaldo and Brochier-Armanet, 2006; Forterre, 2015; Weiss et al., 2016; López-García et al., 2015). If so, one would expect the thioglycine modification to be present in most methanogenic lineages, being stochastically lost due to genetic drift only in lineages that grow at low temperatures where the modification is not required. In contrast, if methanogenesis evolved in a cooler environment, one might expect the distribution of the modification to be restricted to thermophilic lineages. Thus, the universal presence of the thioglycine modification supports the thermophilic ancestry of methanogenesis. Indeed, the ycaO-tfuA locus is conserved even in Methanococcoides burtonii, a psychrophilic methanogen isolated from Ace Lake in Antarctica, where the ambient temperature is always below 2°C (Franzmann et al., 1992). It will be interesting to see whether this modification is maintained by other methanogenic and methanotrophic archaea growing in low temperature environments.
Materials and methods
Bioinformatics analyses
The 1000 closest homologs were extracted from the NCBI non-redundant protein database using the YcaO amino acid sequence (MA0165) or the TfuA amino acid sequence (MA0164) as queries in BLAST-P searches. The amino acid sequences of these proteins were aligned using the MUSCLE plug-in (Edgar, 2004) with default parameters in Geneious version R9 (Kearse et al., 2012). Approximate maximum-likelihood trees were generated using FastTree version 2.1.3 SSE3 using the Jones-Taylor-Thornton (JTT) model +CAT approximation with 20 rate categories. Branch support was calculated using the Shimodaira-Hasegawa (SH) test with 1000 resamples. Trees were displayed using Fig Tree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
Strains, media, and growth conditions
All M. acetivorans strains were grown in single-cell morphology (Sowers et al., 1993) in bicarbonate-buffered high salt (HS) liquid medium containing 125 mM methanol, 50 mM trimethylamine hydrochloride (TMA), 40 mM sodium acetate, or 20 mM dimethylsulfide (DMS). Cultures were grown in sealed tubes with N2/CO2 (80/20) at 8–10 psi in the headspace. Most substrates were added to the medium prior to autoclaving. DMS was added from an anaerobic stock solution maintained at 4°C immediately prior to inoculation. Growth rate measurements were conducted with three independent biological replicate cultures acclimated to the energy substrate or temperature as indicated. A 1:10 dilution of a late-exponential phase culture was used as the inoculum for growth rate measurement. Plating on HS medium containing 50 mM TMA solidified with 1.7% agar was conducted in an anaerobic glove chamber (Coy Laboratory Products, Grass Lake, MI) as described previously (Metcalf et al., 1996). Solid media plates were incubated in an intra-chamber anaerobic incubator maintained at 37°C with N2/CO2/H2S (79.9/20/0.1) in the headspace as described previously (Metcalf et al., 1998). Puromycin (CalBiochem, San Diego, CA) was added to a final concentration of 2 μg/mL from a sterile, anaerobic stock solution to select for transformants containing the pac (puromycin transacetylase) cassette. The purine analog 8-aza-2,6-diaminopurine (8ADP) (R. I. Chemicals, San Diego, CA) was added to a final concentration of 20 µg/mL from a sterile, anaerobic stock solution to select against the hpt (phosphoribosyltransferase) cassette encoded on pC2A-based plasmids. Anaerobic, sterile stocks of tetracycline hydrochloride in deionized water were prepared fresh shortly before use and added to a final concentration of 100 µg/mL to the growth medium of strains containing ycaO and/or tfuA fused to PmcrB(tetO4). E. coli strains were grown in LB broth at 37°C with standard antibiotic concentrations. WM4489, a DH10B derivative engineered to control copy-number of oriV-based plasmids (Kim et al., 2012), was used as the host strain for all plasmids generated in this study (Supplementary file 1). Plasmid copy number was increased dramatically by supplementing the growth medium with sterile rhamnose to a final concentration of 10 mM.
Plasmids
All plasmids used in this study are listed in Supplementary file 1. Plasmids for Cas9-mediated genome editing were designed as described previously (Nayak and Metcalf, 2017). For designing the complementation plasmids, the ycaO and/or tfuA coding sequence(s) were amplified and fused to the PmcrB(tetO4) promoter in pJK029A (Guss et al., 2008) linearized with NdeI and HindIII by the Gibson assembly method as described previously in Nayak and Metcalf, 2017. Standard techniques were used for the isolation and manipulation of plasmid DNA. WM4489 was transformed by electroporation at 1.8 kV using an E. coli Gene Pulser (Bio-Rad, Hercules, CA). All pDN201-derived plasmids were verified by Sanger sequencing at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign and all pAMG40 cointegrates were verified by restriction endonuclease analysis.
In silico design of sgRNAs for gene-editing
All target sequences used for Cas9-mediated genome editing in this study are listed in Supplementary file 2. Target sequences were chosen using the CRISPR site finder tool in Geneious version R9 (61). The M. acetivorans chromosome and the plasmid pC2A were used to score off-target binding sites.
Transformation of M. acetivorans
All M. acetivorans strains used in this study are listed in Supplementary file 3. Liposome-mediated transformation was used for M. acetivorans as described previously (Metcalf et al., 1997) using 10 mL of late-exponential phase culture of M. acetivorans and 2 µg of plasmid DNA for each transformation.
In-gel tryptic digest of McrA
Mid-exponential phase cultures of M. acetivorans grown in 10 mL HS medium containing 50 mM TMA were harvested by centrifugation (3000 × g) for 15 min at 4°C (DuPont, Wilmington, DE). The cell pellet was resuspended in 1 mL lysis buffer (50 mM NH4HCO3, pH = 8.0) and harvested by centrifugation (17,500 × g) for 30 min at 4°C (DuPont, Wilmington, DE). An equal volume of the supernatant was mixed with 2 × Laemmli sample buffer (Bio-Rad, Hercules, CA) containing 5% β-mercaptoethanol, incubated in boiling water for 10 min, loaded on a 4–20% gradient Mini-Protean TGX denaturing SDS-PAGE gel (Bio-Rad, Hercules, CA) and run at 70 V until the dye-front reached the bottom of the gel. The gel was stained using the Gel Code Blue stain reagent (Thermo Fisher Scientific, Waltham, MA) as per the manufacturer’s instructions. Bands corresponding to McrA (ca. 60 kDa) were excised and cut into ca. 1 × 1 mm cubes. The gel slices from a single lane were destained with 50% acetonitrile, the corresponding protein was reduced with 10 mM dithiothreitol (DTT), and digested with 1.5 µg sequencing-grade trypsin (Promega, Madison, WI) at 37°C for 16–20 hr in the presence of 5% (v/v) n-propanol. The digested peptides were extracted and dried as described previously (Shevchenko et al., 2006).
MS analysis of tryptic peptides
MALDI-TOF-MS analysis was performed using a Bruker UltrafleXtreme MALDI TOF-TOF mass spectrometer (Bruker Daltonics, Billerica, MA) in reflector positive mode at the University of Illinois School of Chemical Sciences Mass Spectrometry Laboratory. The samples were desalted using C-18 zip-tips using aqueous acetonitrile and sinapic acid in 70% acetonitrile as the matrix. Data analysis was carried out using the Bruker FlexAnalysis software. For HR-ESI MS/MS, samples were dissolved in 35% aq. acetonitrile and 0.1% formic acid. Samples were directly infused using an Advion TriVersa Nanomate 100 into a ThermoFisher Scientific Orbitrap Fusion ESI-MS. The instrument was calibrated weekly, following the manufacturer’s instructions, and tuned daily with Pierce LTQ Velos ESI Positive Ion Calibration Solution (Thermo Fisher Scientific, Waltham, MA). The MS was operated using the following parameters: resolution, 100,000; isolation width (MS/MS), 1 m/z; normalized collision energy (MS/MS), 35; activation q value (MS/MS), 0.4; activation time (MS/MS), 30 ms. Data analysis was conducted using the Qualbrowser application of Xcalibur software (Thermo Fisher Scientific, Waltham, MA). HPLC-grade reagents were used to prepare samples for mass spectrometric analyses.
MCR purification
MCR was purified from 250 mL of mid-exponential phase culture grown in HS +50 mM TMA at 36°C. Protein purification was performed under aerobic conditions and cells were harvested by centrifugation (3000 × g) for 15 min at 4°C. The cell pellet was lysed in 5 mL of Wash buffer (50 mM NaH2PO4, 300 mM NaCl, pH = 8.0) and the cell lysate was clarified by centrifugation (17,500 × g) for 30 min at 4°C. The supernatant fraction was loaded on a column containing 2 mL Strep-tactin XT Superflow resin (50% suspension; IBA Lifesciences, Goettingen, Germany) equilibrated with 4 mL of the Wash buffer. The column was washed twice with 4 mL of Wash buffer and the purified protein was eluted in four fractions with 0.5 mL Elution buffer (50 mM NaH2PO4, 300 mM NaCl, 50 mM biotin, pH = 8.0) per fraction. The protein concentration in each fraction was estimated using the Coomassie Plus (Bradford) assay kit (Pierce Biotechnology, Thermo-Scientific, Rockford, IL, USA) with BSA (bovine serum albumin) as the standard per the manufacturer’s instructions. The highest protein concentration (ca. 600 μg/mL) was obtained in fraction 3 therefore this fraction was used to conduct the Thermofluor assay. To visualize the purified protein, 10 µL of the crude cell extract as well as fraction 3 were mixed with an equal volume of 2 × Laemmli sample buffer (Bio-Rad, Hercules, CA) containing 5% β-mercaptoethanol, incubated in boiling water for 10 min, loaded on a 4–20% gradient Mini-Protean TGX denaturing SDS-PAGE gel (Bio-Rad, Hercules, CA) and run at 70 V until the dye-front reached the bottom of the gel. The gel was stained using the Gel Code Blue stain reagent (Thermo Fisher Scientific, Waltham, MA) as per the manufacturer’s instructions. All three bands corresponding to the MCR subunits were observed and verified by LC-MS analyses of the tryptic peptides from fraction 3 of the purified protein sample at the Protein Sciences facility at the Roy J. Carver Biotechnology Center at the University of Illinois, Urbana-Champaign, USA. A fourth band, identified as a hypothetical protein encoded by MA3997, was also detected in each sample (Figure 5—figure supplement 1).
Thermofluor assay
A 5000 × concentrate of SYPRO Orange protein gel stain in DMSO (Thermo-Fisher Scientific, Waltham, MA, USA) was diluted with the Elution buffer to generate a 200 × stock solution. 5 µL of the 200 × stock solution of SYPRO Orange protein gel stain was added to 45 µL of purified protein with a concentration of ca. 600 µg/mL in 96-well optically clear PCR plates. A melt curve was performed on a Mastercycler ep realplex machine (Eppendorf, Hamburg, Germany) using the following protocol: 25°C for 30 s, ramp to 99°C over 30 min, return to 25°C for 30 s. SYPRO Orange fluorescence was detected using the VIC emission channel and the temperature corresponding to the inflection point of the first derivative (dI/dT) was determined to be the melting temperature (Tm). Appropriate no-dye controls and no-protein controls were examined and each sample was run in triplicate. The Thermofluor assay was conducted within 12 hr of protein purification.
UV-Vis spectrum of MCR
The spectrum was obtained on a Cary 4000 UV-Vis spectrophotometer (Varian, Palo Alto, CA, USA) using 200 µL of protein sample (1.5 µM each) in a quartz supracil cuvette (Hellma Analytics, Mullheim, Germany). The scanning range was 200–800 nm and the Elution buffer was used as a blank.
Acknowledgements
We thank Graham A Hudson (DAM lab) for technical assistance with the HR and tandem MS data acquisition and Dr. Peter Yau at the Protein Sciences facility at the Roy J Carver Biotechnology Center at the University of Illinois for identifying the protein that co-purified with MCR.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Douglas A Mitchell, Email: douglasm@illinois.edu.
William W Metcalf, Email: metcalf@illinois.edu.
Dianne K Newman, Howard Hughes Medical Institute, California Institute of Technology, United States.
Funding Information
This paper was supported by the following grants:
National Institute of General Medical Sciences GM097142 to Douglas A Mitchell.
Life Sciences Research Foundation Simons Foundation to Dipti D Nayak.
U.S. Department of Energy DE-FG02-02ER15296 to William W Metcalf.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Data curation, Formal analysis, Supervision, Funding acquisition, Investigation, Methodology, Writing—original draft, Project administration, Writing—review and editing.
Data curation, Formal analysis, Investigation, Writing—original draft, Writing—review and editing.
Conceptualization, Supervision, Funding acquisition, Writing—original draft, Project administration, Writing—review and editing.
Conceptualization, Data curation, Formal analysis, Supervision, Funding acquisition, Investigation, Methodology, Writing—original draft, Project administration, Writing—review and editing.
Additional files
References
- Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013;30:108–160. doi: 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu MK, Selengut JD, Haft DH. ProPhylo: partial phylogenetic profiling to guide protein family construction and assignment of biological process. BMC Bioinformatics. 2011;12:434. doi: 10.1186/1471-2105-12-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrel G, O'Toole PW, Harris HM, Peyret P, Brugère JF, Gribaldo S, Borrel G. Phylogenomic data support a seventh order of Methylotrophic methanogens and provide insights into the evolution of Methanogenesis. Genome Biology and Evolution. 2013;5:1769–1780. doi: 10.1093/gbe/evt128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose A, Pritchett MA, Metcalf WW. Genetic analysis of the methanol- and methylamine-specific methyltransferase 2 genes of Methanosarcina acetivorans C2A. Journal of Bacteriology. 2008;190:4017–4026. doi: 10.1128/JB.00117-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nature Chemical Biology. 2015;11:564–570. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart BJ, Schwalen CJ, Mann G, Naismith JH, Mitchell DA. YcaO-Dependent Posttranslational Amide Activation: Biosynthesis, Structure, and Function. Chemical Reviews. 2017;117:5389–5456. doi: 10.1021/acs.chemrev.6b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriac AI, Kloss F, Krämer J, Vuong C, Hertweck C, Sahl HG. Mode of action of closthioamide: the first member of the polythioamide class of bacterial DNA gyrase inhibitors. Journal of Antimicrobial Chemotherapy. 2015;70:2576–2588. doi: 10.1093/jac/dkv161. [DOI] [PubMed] [Google Scholar]
- Coyne S, Chizzali C, Khalil MN, Litomska A, Richter K, Beerhues L, Hertweck C. Biosynthesis of the antimetabolite 6-thioguanine in Erwinia amylovora plays a key role in fire blight pathogenesis. Angewandte Chemie International Edition. 2013;52:10564–10568. doi: 10.1002/anie.201305595. [DOI] [PubMed] [Google Scholar]
- Coyne S, Litomska A, Chizzali C, Khalil MN, Richter K, Beerhues L, Hertweck C. Control of plant defense mechanisms and fire blight pathogenesis through the regulation of 6-thioguanine biosynthesis in Erwinia amylovora. ChemBioChem. 2014;15:373–376. doi: 10.1002/cbic.201300684. [DOI] [PubMed] [Google Scholar]
- Deppenmeier U, Lienard T, Gottschalk G. Novel reactions involved in energy conservation by methanogenic archaea. FEBS Letters. 1999;457:291–297. doi: 10.1016/S0014-5793(99)01026-1. [DOI] [PubMed] [Google Scholar]
- Dunbar KL, Melby JO, Mitchell DA. YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations. Nature Chemical Biology. 2012;8:569–575. doi: 10.1038/nchembio.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar KL, Chekan JR, Cox CL, Burkhart BJ, Nair SK, Mitchell DA. Discovery of a new ATP-binding motif involved in peptidic azoline biosynthesis. Nature Chemical Biology. 2014;10:823–829. doi: 10.1038/nchembio.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar KL, Tietz JI, Cox CL, Burkhart BJ, Mitchell DA. Identification of an Auxiliary Leader Peptide-Binding Protein Required for Azoline Formation in Ribosomal Natural Products. Journal of the American Chemical Society. 2015;137:7672–7677. doi: 10.1021/jacs.5b04682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science. 1997;278:1457–1462. doi: 10.1126/science.278.5342.1457. [DOI] [PubMed] [Google Scholar]
- Forterre P. The universal tree of life: an update. Frontiers in Microbiology. 2015;6:1–18. doi: 10.3389/fmicb.2015.00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmann PD, Springer N, Ludwig W, Conway De Macario E, Rohde M. A Methanogenic Archaeon from Ace Lake, Antarctica: Methanococcoides burtonii sp. nov. Systematic and Applied Microbiology. 1992;15:573–581. doi: 10.1016/S0723-2020(11)80117-7. [DOI] [Google Scholar]
- Fu H, Metcalf WW. Genetic basis for metabolism of methylated sulfur compounds in Methanosarcina species. Journal of Bacteriology. 2015;197:1515–1524. doi: 10.1128/JB.02605-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Nusbaum C, Roy A, Endrizzi MG, Macdonald P, FitzHugh W, Calvo S, Engels R, Smirnov S, Atnoor D, Brown A, Allen N, Naylor J, Stange-Thomann N, DeArellano K, Johnson R, Linton L, McEwan P, McKernan K, Talamas J, Tirrell A, Ye W, Zimmer A, Barber RD, Cann I, Graham DE, Grahame DA, Guss AM, Hedderich R, Ingram-Smith C, Kuettner HC, Krzycki JA, Leigh JA, Li W, Liu J, Mukhopadhyay B, Reeve JN, Smith K, Springer TA, Umayam LA, White O, White RH, Conway de Macario E, Ferry JG, Jarrell KF, Jing H, Macario AJ, Paulsen I, Pritchett M, Sowers KR, Swanson RV, Zinder SH, Lander E, Metcalf WW, Birren B. The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Research. 2002;12:532–542. doi: 10.1101/gr.223902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goenrich M, Duin EC, Mahlert F, Thauer RK. Temperature dependence of methyl-coenzyme M reductase activity and of the formation of the methyl-coenzyme M reductase red2 state induced by coenzyme B. JBIC Journal of Biological Inorganic Chemistry. 2005;10:333–342. doi: 10.1007/s00775-005-0636-6. [DOI] [PubMed] [Google Scholar]
- Goubeaud M, Schreiner G, Thauer RK. Purified methyl-coenzyme-M reductase is activated when the enzyme-bound coenzyme F430 is reduced to the nickel(I) oxidation state by titanium(III) citrate. European Journal of Biochemistry. 1997;243:110–114. doi: 10.1111/j.1432-1033.1997.00110.x. [DOI] [PubMed] [Google Scholar]
- Grabarse W, Mahlert F, Shima S, Thauer RK, Ermler U. Comparison of three methyl-coenzyme M reductases from phylogenetically distant organisms: unusual amino acid modification, conservation and adaptation. Journal of Molecular Biology. 2000;303:329–344. doi: 10.1006/jmbi.2000.4136. [DOI] [PubMed] [Google Scholar]
- Grabarse W, Mahlert F, Duin EC, Goubeaud M, Shima S, Thauer RK, Lamzin V, Ermler U. On the mechanism of biological methane formation: structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. Journal of Molecular Biology. 2001;309:315–330. doi: 10.1006/jmbi.2001.4647. [DOI] [PubMed] [Google Scholar]
- Gribaldo S, Brochier-Armanet C. The origin and evolution of Archaea: a state of the art. Philosophical Transactions of the Royal Society B: Biological Sciences. 2006;361:1007–1022. doi: 10.1098/rstb.2006.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guss AM, Rother M, Zhang JK, Kulkarni G, Metcalf WW. New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea. 2008;2:193–203. doi: 10.1155/2008/534081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Sasaki K, Nagai K, Shin-ya K, Furihata K. Structure of thioviridamide, a novel apoptosis inducer from Streptomyces olivoviridis. The Journal of Antibiotics. 2006;59:6–10. doi: 10.1038/ja.2006.2. [DOI] [PubMed] [Google Scholar]
- Hensens OD, Albers-Schönberg G. Total structure of the highly modified peptide antibiotic components of thiopeptin. The Journal of Antibiotics. 1983;36:814–831. doi: 10.7164/antibiotics.36.814. [DOI] [PubMed] [Google Scholar]
- Horng YC, Becker DF, Ragsdale SW. Mechanistic studies of methane biogenesis by methyl-coenzyme M reductase: evidence that coenzyme B participates in cleaving the C-S bond of methyl-coenzyme M. Biochemistry. 2001;40:12875–12885. doi: 10.1021/bi011196y. [DOI] [PubMed] [Google Scholar]
- Huynh K, Partch CL. Analysis of protein stability and ligand interactions by thermal shift assay. Current Protocols in Protein Science. 2015;79:28.9.1–28.928. doi: 10.1002/0471140864.ps2809s79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa M, Kawasaki T, Hayakawa Y. Cloning and heterologous expression of the thioviridamide biosynthesis gene cluster from Streptomyces olivoviridis. Applied and Environmental Microbiology. 2013;79:7110–7113. doi: 10.1128/AEM.01978-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahnt J, Buchenau B, Mahlert F, Krüger M, Shima S, Thauer RK. Post-translational modifications in the active site region of methyl-coenzyme M reductase from methanogenic and methanotrophic archaea. FEBS Journal. 2007;274:4913–4921. doi: 10.1111/j.1742-4658.2007.06016.x. [DOI] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney GE, Rosenzweig AC. Chemistry and biology of the copper chelator methanobactin. ACS Chemical Biology. 2012;7:260–268. doi: 10.1021/cb2003913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney GE, Rosenzweig AC. Genome mining for methanobactins. BMC Biology. 2013;11:17. doi: 10.1186/1741-7007-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Graham DW, DiSpirito AA, Alterman MA, Galeva N, Larive CK, Asunskis D, Sherwood PM. Methanobactin, a copper-acquisition compound from methane-oxidizing bacteria. Science. 2004;305:1612–1615. doi: 10.1126/science.1098322. [DOI] [PubMed] [Google Scholar]
- Kim SY, Ju KS, Metcalf WW, Evans BS, Kuzuyama T, van der Donk WA. Different biosynthetic pathways to fosfomycin in Pseudomonas syringae and Streptomyces species. Antimicrobial Agents and Chemotherapy. 2012;56:4175–4183. doi: 10.1128/AAC.06478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-J, Choi Y-S, Lee K-B, Park J, Yoon C-J. Hydrogen bonding abilities of thioamide. The Journal of Physical Chemistry A. 2002;106:7010–7017. doi: 10.1021/jp025516e. [DOI] [Google Scholar]
- Lincke T, Behnken S, Ishida K, Roth M, Hertweck C. Closthioamide: an unprecedented polythioamide antibiotic from the strictly anaerobic bacterium Clostridium cellulolyticum. Angewandte Chemie. 2010;49:2055–2057. doi: 10.1002/ange.200906114. [DOI] [PubMed] [Google Scholar]
- López-García P, Zivanovic Y, Deschamps P, Moreira D. Bacterial gene import and mesophilic adaptation in archaea. Nature Reviews Microbiology. 2015;13:447–456. doi: 10.1038/nrmicro3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthies A, Nimtz M, Leimkühler S. Molybdenum cofactor biosynthesis in humans: identification of a persulfide group in the rhodanese-like domain of MOCS3 by mass spectrometry. Biochemistry. 2005;44:7912–7920. doi: 10.1021/bi0503448. [DOI] [PubMed] [Google Scholar]
- Metcalf WW, Zhang JK, Shi X, Wolfe RS. Molecular, genetic, and biochemical characterization of the serC gene of Methanosarcina barkeri Fusaro. Journal of Bacteriology. 1996;178:5797–5802. doi: 10.1128/jb.178.19.5797-5802.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf WW, Zhang JK, Apolinario E, Sowers KR, Wolfe RS. A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. PNAS. 1997;94:2626–2631. doi: 10.1073/pnas.94.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf WW, Zhang JK, Wolfe RS. An anaerobic, intrachamber incubator for growth of Methanosarcina spp. on methanol-containing solid media. Applied and Environmental Microbiology. 1998;64:768–770. doi: 10.1128/aem.64.2.768-770.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SJ, Sowa ST, Schuchardt C, Deery E, Lawrence AD, Ramos JV, Billig S, Birkemeyer C, Chivers PT, Howard MJ, Rigby SE, Layer G, Warren MJ. Elucidation of the biosynthesis of the methane catalyst coenzyme F430. Nature. 2017;543:78–82. doi: 10.1038/nature21427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak DD, Metcalf WW. Cas9-mediated genome editing in the methanogenic archaeon Methanosarcina acetivorans. PNAS. 2017;114:2976–2981. doi: 10.1073/pnas.1618596114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palenchar PM, Buck CJ, Cheng H, Larson TJ, Mueller EG. Evidence that ThiI, an enzyme shared between thiamin and 4-thiouridine biosynthesis, may be a sulfurtransferase that proceeds through a persulfide intermediate. Journal of Biological Chemistry. 2000;275:8283–8286. doi: 10.1074/jbc.275.12.8283. [DOI] [PubMed] [Google Scholar]
- Pan M, Mabry TJ, Beale JM, Mamiya BM. Nonprotein amino acids from Cycas revoluta. Phytochemistry. 1997;45:517–519. doi: 10.1016/S0031-9422(96)00866-7. [DOI] [PubMed] [Google Scholar]
- Park JH, Dorrestein PC, Zhai H, Kinsland C, McLafferty FW, Begley TP. Biosynthesis of the thiazole moiety of thiamin pyrophosphate (vitamin B1) Biochemistry. 2003;42:12430–12438. doi: 10.1021/bi034902z. [DOI] [PubMed] [Google Scholar]
- Perler FB, Xu MQ, Paulus H. Protein splicing and autoproteolysis mechanisms. Current Opinion in Chemical Biology. 1997;1:292–299. doi: 10.1016/S1367-5931(97)80065-8. [DOI] [PubMed] [Google Scholar]
- Petersson EJ, Goldberg JM, Wissner RF. On the use of thioamides as fluorescence quenching probes for tracking protein folding and stability. Phys. Chem. Chem. Phys. 2014;16:6827–6837. doi: 10.1039/C3CP55525A. [DOI] [PubMed] [Google Scholar]
- Puar MS, Ganguly AK, Afonso A, Brambilla R, Mangiaracina P, Sarre O, MacFarlane RD. Sch 18640. A new thiostrepton-type antibiotic. Journal of the American Chemical Society. 1981;103:5231–5233. doi: 10.1021/ja00407a047. [DOI] [Google Scholar]
- Rospert S, Linder D, Ellermann J, Thauer RK. Two genetically distinct methyl-coenzyme M reductases in Methanobacterium thermoautotrophicum strain Marburg and delta H. European Journal of Biochemistry. 1990;194:871–877. doi: 10.1111/j.1432-1033.1990.tb19481.x. [DOI] [PubMed] [Google Scholar]
- Rother M, Boccazzi P, Bose A, Pritchett MA, Metcalf WW. Methanol-dependent gene expression demonstrates that methyl-coenzyme M reductase is essential in Methanosarcina acetivorans C2A and allows isolation of mutants with defects in regulation of the methanol utilization pathway. Journal of Bacteriology. 2005;187:5552–5559. doi: 10.1128/JB.187.16.5552-5559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller S, Goenrich M, Boecher R, Thauer RK, Jaun B. The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature. 2010;465:606–608. doi: 10.1038/nature09015. [DOI] [PubMed] [Google Scholar]
- Schulman BA, Harper JW, Wade Harper J. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nature Reviews Molecular Cell Biology. 2009;10:319–331. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmer T, Kahnt J, Goubeaud M, Shima S, Grabarse W, Ermler U, Thauer RK. The biosynthesis of methylated amino acids in the active site region of methyl-coenzyme M reductase. Journal of Biological Chemistry. 2000;275:3755–3760. doi: 10.1074/jbc.275.6.3755. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protocols. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Sowers KR, Boone JE, Gunsalus RP. Disaggregation of Methanosarcina spp. and Growth as Single Cells at Elevated Osmolarity. Applied and Environmental Microbiology. 1993;59:3832–3839. doi: 10.1128/aem.59.11.3832-3839.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nature Reviews Microbiology. 2008;6:579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- Vanwonterghem I, Evans PN, Parks DH, Jensen PD, Woodcroft BJ, Hugenholtz P, Tyson GW. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nature Microbiology. 2016;1:16170. doi: 10.1038/nmicrobiol.2016.170. [DOI] [PubMed] [Google Scholar]
- Wagner T, Kahnt J, Ermler U, Shima S. Didehydroaspartate modification in methyl-coenzyme M reductase catalyzing methane formation. Angewandte Chemie International Edition. 2016;55:10630–10633. doi: 10.1002/anie.201603882. [DOI] [PubMed] [Google Scholar]
- Weiss MC, Sousa FL, Mrnjavac N, Neukirchen S, Roettger M, Nelson-Sathi S, Martin WF. The physiology and habitat of the last universal common ancestor. Nature Microbiology. 2016;1:16116. doi: 10.1038/nmicrobiol.2016.116. [DOI] [PubMed] [Google Scholar]
- Wiberg KB, Rablen PR. Why does thioformamide have a larger rotational barrier than formamide? Journal of the American Chemical Society. 1995;117:2201–2209. doi: 10.1021/ja00113a009. [DOI] [Google Scholar]
- Wongnate T, Sliwa D, Ginovska B, Smith D, Wolf MW, Lehnert N, Raugei S, Ragsdale SW. The radical mechanism of biological methane synthesis by methyl-coenzyme M reductase. Science. 2016;352:953–958. doi: 10.1126/science.aaf0616. [DOI] [PubMed] [Google Scholar]
- Wright CM, Christman GD, Snellinger AM, Johnston MV, Mueller EG. Direct evidence for enzyme persulfide and disulfide intermediates during 4-thiouridine biosynthesis. Chemical Communications. 2006;29:3104–3106. doi: 10.1039/b604040c. [DOI] [PubMed] [Google Scholar]
- Zhang W, Urban A, Mihara H, Leimkühler S, Kurihara T, Esaki N. IscS functions as a primary sulfur-donating enzyme by interacting specifically with MoeB and MoaD in the biosynthesis of molybdopterin in Escherichia coli. Journal of Biological Chemistry. 2010;285:2302–2308. doi: 10.1074/jbc.M109.082172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, Ngo PD, Owens VL, Yang XP, Mansoorabadi SO. The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science. 2016;354:339–342. doi: 10.1126/science.aag2947. [DOI] [PubMed] [Google Scholar]