

Abstract
Introduction
T cell activation is a complex process that requires multiple cell signaling pathways, including a primary recognition signal and additional costimulatory signals. One of the best-characterized costimulatory pathways includes the Ig superfamily members CD28 and CTLA-4 and their ligands CD80 and CD86.
Areas Covered
This review discusses past, current and future biological therapies that have been utilized to block the CD28/CTLA-4 cosignaling pathway in the settings of autoimmunity and transplantation, as well the challenges facing successful implementation of these therapies.
Expert Opinion
The development of CD28 blockers Abatacept and Belatacept provided a more targeted therapy for transplant rejection and autoimmune disease relative to calcineurin inhibitors and anti-proliferatives, but overall efficacy may be limited due to their collateral effect of simultaneously blocking CTLA-4 coinhibitory signals. As such, current investigations into the potential of selective CD28 blockade to block the costimulatory potential of CD28 while exploiting the coinhibitory effects of CTLA-4 are promising. However, as selective CD28 blockade inhibits the activity of both effector and regulatory T cells, an important goal for the future is the design of therapies that will maximize the attenuation of effector responses while preserving the suppressive function of T regulatory cells.
1. Introduction
Limiting immune-mediated damage following transplantation and preventing autoimmune disease while maintaining protective immunity requires precise regulation of the immune system. In both instances, a major challenge is the activation of allo- or auto-reactive T lymphocytes, which are capable of directly mediating tissue damage during graft rejection and autoimmunity and also of providing T cell help for generation of allo- and auto-antibody. Simultaneously, the maintenance or enhancement of regulatory T cell number and functionality is also beneficial for the diminution of unwanted auto- and allo-immune responses. All of these processes are carefully controlled by the balance of costimulatory and coinhibitory signals T cells receive. Although T cell costimulation was first described to control initial priming of naive T cells, T cell costimulatory and coinhibitory pathways are now known to have much broader functions, controlling many aspects of naïve, effector, memory, and regulatory T cell activation and differentiation. Indeed, early work showed that TCR ligation alone induces T cell anergy or unresponsiveness and that the necessary costimulatory signal that prevents T cell unresponsiveness after TCR ligation was present on B cells and antigen presenting cells (1). The CD28/CTLA-4 pathway is the prototypic cosignaling pathway in T cells, with CTLA-4 coinhibition acting as the counter-signal to CD28 costimulation as they bind the same receptors (CD80 and CD86). Since CD28 costimulation is crucial for T-cell activation, immunomodulation via blockade of this pathway is a promising approach to prevent inappropriate T-cell activation in the setting of transplantation and also to potentially treat T cell mediated autoimmune diseases.
The results of studies using biologics to therapeutically target this pathway are outlined in the paragraphs below. This large body of work informs us that while critically important, the CD28/CTLA-4 cosignaling pathway is highly complex. Further detailed understanding of the kinetics, cellular distribution, binding partners, and intracellular signaling networks of cosignaling molecules in auto- and alloimmunity will aid in the rational development of novel immunomodulatory strategies to better target this pathway and improve outcomes following transplantation and autoimmunity.
2. Immunobiology of CD28/CTLA-4 signaling
CD28 is a costimulatory receptor that is constitutively expressed on the cell surface of naïve T cells and is required for optimal activation and function. Additionally, after an early, transient decrease, surface expression levels are increased about 2-fold following peptide stimulation both in vitro and in vivo (2–4). Given the critical balance between stimulation and inhibition that is necessary to prevent immune pathology, it has been shown that along with the upregulation of CD28 following activation is a concomitant increase in the ratio of CTLA-4 to CD28 (2, 4) (Figure 1A).
Figure 1.
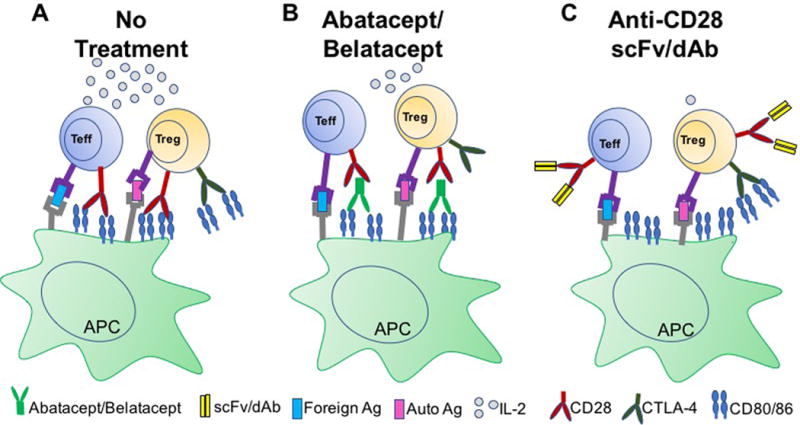
Modes of action of Abatacept/Belatacept and anti-CD28 domain antibodies. (A) During a normal immune response, both conventional T effector cells (Teff) and T regulatory cells (Treg) receive a primary activation signal via TCR engagement with peptide/MHC complexes presented on antigen presenting cells (APCs). To become fully activated, a second, co-stimulatory signal is also required, shown here as CD28 on the surface of T cells binding its ligand CD80/86. CTLA-4 is also capable of binding CD80/86, resulting in coinhibition of T cells. In addition, CTLA-4 expression on Tregs is important for their function. (B) Treatment with Abatacept/Belatacept blocks the shared ligands for CD28 and CTLA-4, thus blocking CTLA-4 mediated coinhibitory signals that serve to dampen effector T cell responses and promote Treg-mediated suppression. (C) Anti-CD28 domain antibodies selectively target CD28, while leaving CTLA-4 intact.
Unlike CD28, CTLA-4 is a negative regulator, and its expression is dependent on activation (5, 6) with resting murine T cells expressing little to no CTLA-4 on their surface (7). Readouts of intracellular CTLA-4 are important as CTLA-4 in resting cells is intracellularly localized to clathrin-associated complexes and is only relocated to the cell surface upon cell activation (2). CTLA-4 expression has been demonstrated at time points as early as one hour post-stimulation, with functional effects by 12 hours and peaking at 48h (2, 8). Interestingly, although surface expression of CTLA-4 only ever reaches ~1/50th that of CD28, its affinity for their common ligands, CD80 and CD86, is ~1000-fold greater (2, 9). Additionally, there is sustained expression of CTLA-4 with a significant proportion of T cells still expressing it at 6–10 days post-activation, a time at which CD28 is no longer elevated above constitutive levels (4).
Interestingly, upon the generation of CTLA-4 knockout (KO) mice it was observed that all animals developed a lymphoproliferative disease that caused death by three to four weeks of age (10). This was discovered to be due to the non-redundant role of CTLA-4 in inhibiting T-cell proliferation, cell cycle progression, and IL-2 production (11, 12). These data point to a critical role of CTLA-4 for controlling T-cell responses to foreign and self-antigens in an intrinsic manner. There is also evidence, however, for an extrinsic role for CTLA-4, in that ligation of CTLA-4 may also induce production of factors that inhibit the activation or proliferation of neighboring cells (13–15). The hypothesis was then raised that the high levels of CTLA-4 expressed on T regulatory cells (Tregs) may be involved in their suppressive function and that Tregs could therefore, be the CTLA-4-dependent population able to control CTLA-4 KO T cells in this chimeric system (16, 17). This hypothesis was confirmed to be true, in that control of CTLA-4 KO cells by WT cells required the presence of Foxp3+ Tregs in the wildtype population (18).
Alternatively, another interesting hypothesis has been proposed: could CTLA-4 function by actively engaging a positive event that could produce a negative outcome in terms of dampening the response to antigen (19)? For instance, TCR ligation has long been known to slow or reduce T-cell motility; an event termed the ‘stop signal’. Without the stop signal, T cells continue to move or remain tethered to the APC, but are unable to form an immunological synapse. Stable immunological-synapse formation is needed for the scanning of peptide–MHC complexes, their engagement by the TCR and the induction of signaling cascades (20–22). Evidence shows that CTLA-4 ligation reverses the stop signal, thereby interfering with the information of the immunological synapse. Therefore, T cells fail to make prolonged contact, despite remaining tethered, leading to an overall reduction in activation and cytokine production (23).
3. Biological Therapies to Target the CD28/CTLA-4 Pathway: The Benefits
Given the data described above, the blockade of the CD28 co-signaling pathway is an attractive strategy for preventing both allogeneic and autoreactive T cell responses.
3.1 CD28/CTLA-4 directed biological therapies in autoimmunity
A recombinant fusion protein, Abatacept, or CTLA4-Ig, is a drug that comprises the extracellular domain of human CTLA-4 fused with a fragment of the Fc portion of human IgG1. The use of a CTLA-4 fusion protein as a means to block the interactions of CD28 with CD80 and CD86 has been well documented in experimental autoimmunity (24, 25) (Figure 1B). The first study was conducted in patients with psoriasis, among whom 46% achieved a 50% or greater sustained improvement in clinical disease activity (26).
From these promising results and understanding of the biology of the CD28/CTLA-4 pathway, proof of principle work was also undertaken in a mouse model of collagen induced arthritis to determine whether Abatacept might also be beneficial in this setting. It was found that administration of Abatacept at the time of immunization prevented disease. Interestingly, administration after disease onset also ameliorated disease. Similar effects were obtained when a combination of anti-CD80 and anti-CD86 antibodies were used to block the co-stimulation, indicating the need to block both pathways (27).
Abatacept has also been used in patients with Rheumatoid Arthritis (RA) with beneficial effects (28, 29), specifically in patients with methotrexate (MTX)- (29–32) and anti-TNF refractory disease (33, 34), as well as early RA patients who have yet to try other anti-rheumatic drug treatments (35). One year out from the start of Abatacept treatment, RA patients enrolled in the AIM (Abatacept in Inadequate responders to Methotrexate) trial experienced both clinical and X-ray improvements over time (32). In the ATTAIN (Abatacept Trial in Treatment of Anti-TNF INadequate responders) trial, ACR20, −50 and −70 responses increased up to 81.8%, 53.6% and 25.7% response respectively at three years out from the start of treatment (36).
Importantly, the positive results seen with Abatacept treatment in RA patients was stable over time, as those patients enrolled in the AGREE trial showed that 81% of patients who were in remission at year one were still in remission at year two (35). Additionally, their disease had not progressed, with 91.1% of year-1 non-progressors remaining non-progressors in year two. The AIM study went even further and followed patients for five years in order to evaluate the persistence of drug response over time. In this study, 60.3% of patients in remission at year one were still in remission at year five and almost 72% of patients who were X-ray non-progressors at year one were X-ray non-progressors at year five.
Another important autoimmune disease that could potentially benefit from costimulation blockade is systemic lupus erythematous (SLE). Although significant strides have been made in the management of lupus over the last several decades, mortality rates remain high with current treatment modalities, many of which have undesirable adverse effects that impact on patients’ health and quality of life (37). Early work in lupus-prone mice showed promise with treatment with a murine CTLA4-Ig fusion protein (muCTLA4Ig) blocking autoantibody production and prolonging life, even when treatment was delayed until the most advanced stage of clinical illness (24). More recently, it has been shown that Abatacept shows efficacy in mice and in a study of murine lupus nephritis using the drug in combination with an anti-CD154 molecule showed long-lasting inhibition of autoantibody production and a diminished occurrence of renal disease, even in animals with advanced nephritis (38, 39).
Although the translational animal models using Abatacept seemed promising, the human clinical trials were overall disappointing with a total of three Abatacept SLE trials all failing to achieve their primary outcomes (40–42). However, based on its mechanism of action, effects over the immune system in general, and the renal podocytes in particular, there is still a possibility that Abatacept may be useful for the treatment of lupus nephritis. Indeed, a very recent study suggests some efficacy of Abatacept in patients with refractory disease, particularly in cases of articular manifestations (43). These data support conducting new controlled trials of Abatacept in refractory SLE patients.
3.2 CD28/CTLA-4 directed biological therapies in transplantation
The use of Abatacept (CTLA4-Ig) as a therapeutic agent to inhibit the CD28 pathway was also established in the setting of transplantation, where early preclinical studies using CTLA4-Ig in murine and non-human primate models showed amelioration of GvHD (44). A first-in-disease trial of in vivo CD28:CD80/86-directed costimulation blockade with Abatacept tested the feasibility of adding in vivo T cell costimulation blockade for GvHD prevention (Clinical Trials.Org #NCT01012492). The decreased CD4+ T cell proliferation post-transplant and the encouragingly low rates of early, severe GvHD observed in this trial suggested that costimulation blockade may be an effective agent for GvHD prophylaxis and supported the conduct of a larger, randomized phase 2 study. Thus, a phase II multicenter, randomized, double-blind RCT of Abatacept combined with calcineurin inhibitor (CNI) and methotrexate versus placebo following unrelated donor HCT is currently underway (ClinicalTrials.gov NCT01743131).
Although preclinical studies demonstrated CTLA4-Ig mediated inhibition of T-cell–dependent antibody responses and prolongation of transplanted organ survival (45), it was found to be inadequate to maintain a hypo-responsive state to an allograft in nonhuman primate models (46). This lack of efficacy in the nonhuman primate model was hypothesized to be related to the lower avidity of Abatacept to CD86 compared with CD80. It has been demonstrated that CD80 and CD86 may differentially control the immune response because of the distinct properties of each molecule (47). Thus, the more rapid dissociation of Abatacept from CD86 than from CD80 may result in less effective inhibition of CD86-dependent responses than of CD80-dependent responses (48). Therefore, it was hypothesized that a compound that bound to CD86 with higher avidity than Abatacept would provide the inhibition of T-cell costimulation necessary to prevent allograft rejection. This led to the development of Belatacept (LEA29Y), which differs from Abatacept by two specific amino-acid substitutions, thus conferring greater binding avidity to CD80 and CD86, more potent inhibition of T-cell activation, and effective rejection prophylaxis in nonhuman primate models (46) (Figure 1B).
Following these promising preclinical nonhuman primate studies a phase II clinical trial was carried out with the primary objective to demonstrate the non-inferiority of Belatacept over cyclosporine with respect to the incidence of acute rejection at six months. The results of this study suggested that the two agents are similarly effective for the prevention of acute rejection in that patients treated with Belatacept regimens had rates of acute rejection similar to those among patients taking cyclosporine, satisfying pre-specified criteria for non-inferiority (49–53). Importantly, patients on Belatacept were able to avoid both the renal toxicity as well as the chronic non-renal effects associated with CNIs that can negatively impact patient and/or graft survival (53).
4. Biological Therapies to Target the CD28/CTLA-4 Pathway: The Challenges
Belatacept was approved by the U.S. Food and Drug Administration in 2011 (54) on the basis of 3-year data from two phase III studies: the Belatacept Evaluation of Nephroprotection and Efficacy as First-line Immunosuppression Trial (BENEFIT) and BENEFIT–Extended Criteria Donors (BENEFIT-EXT) (55–57). The results of these studies showed that Belatacept offers significantly improved long-term graft function and fewer toxicities compared to CNIs, with a 43% reduced risk of death or graft loss at 7 years post-transplantation (58). However, treatment with belatacept was also associated with a significantly higher incidence of acute rejection within one year of transplantation (59).
While the mechanisms underlying this observation are currently under investigation, the kinetics and severity of this phenomenon suggests that a CD28/CTLA-4 blockade resistant population of T cells mediates this rejection. Much work has gone into examining the mechanisms underlying this early costimulation blockade resistant rejection (CoBRR). They are likely two-fold: 1) the existence of a population of CD28-independent memory T cells that fail to be controlled by the drug and 2) a negative, collateral impact of the drug on immunosuppressive Treg populations.
4.1 The challenge of CD28-independent memory T cells
First, several mechanistic studies have identified a relationship between short-lived, alloreactive memory T cells that have differentiated beyond the requirements for CD28-B7 costimulation and graft rejection (60–62). These populations come to be through several different mechanisms, including encounters with pathogens, through the process of heterologous immunity (63, 64), prior sensitization through pregnancy, blood transfusion, or prior transplant (65), or through the process of homeostatic proliferation which allows for stable number and composition of peripheral T and B cells in the human body (66). Interestingly, the process of homeostatic proliferation has been shown to convert naïve T cells directly to effector memory cells in the absence of antigen (67, 68), and these cells have been shown to mediate rejection and are resistant to tolerance induction (69–71). Memory cells that are cross-reactive to transplant antigens exist at a frequency of 1:200,000 and memory cells that can recognize foreign MHC is 1–10% of the total T cell repertoire (72, 73).
The context and amount of stimulation also affect the role that memory cells play in costimulation blockade-resistant rejection (CoBRR). For instance, recipients possessing donor-reactive T cell memory responses that were generated under conditions of reduced antigen (Ag) exposure exhibited similar frequencies of Ag-specific T cells at day 30 post-infection, but, strikingly, failed to mediate rejection, demonstrating that the amount or duration of Ag exposure is a critical factor in determining the requirements of memory T cells for costimulation during the recall response after transplantation (74). Further, the context of stimulation also matters, in that memory generated by different pathogens leads to differential efficacy of a costimulation and integrin blockade based regimen (75). Intriguingly, the most sensitive memory T cell population to costimulation blockade was that composed primarily of central memory T cells which possessed greater recall potential, exhibited a less differentiated phenotype, and contained more multi-cytokine producing cells. These data suggest that the immune stimulation history of a given transplant patient may profoundly influence the relative barrier posed by heterologous immunity during transplantation (75).
Another memory population that has sparked recent interest regarding the mechanism behind CoBRR is the population of cells that is CD4+ CD57+. Espinosa and colleagues (76) studied patients that received Belatacept or conventional CNI-based immunosuppression and identified a population of CD57+ PD1− CD4+ T cells that were present prior to transplantation and which correlated with CoBRR. This is contrary to data that recognizes CD57 as a marker of senescence on CD8 T cells, and intriguingly these authors uncovered a non-senescent, cytolytic phenotype that was associated with CD57 on CD4 T cells. These cells expressed high levels of adhesion molecules which have been implicated in experimental CoBRR, expressed a transcriptional phenotype broadly defining allograft rejection and were shown to be present in rejecting human kidney allografts, strongly implicating CD57+ CD4+ T cells in clinical CoBRR and potentially identifying patients who are at greater risk for acute rejection on this treatment regimen (76).
Finally, several studies have shown that the propensity of memory T cells to mediate CoBRR may depend on their CD28 status. At birth, nearly all human T cells express CD28, however as we age the population of CD28null cells continues to grow, with over 50–60% of CD8 T cells being CD28null in individuals over 80 years old (77). Similar trends have been observed in CD4 T cells, though the effect is less dramatic. It has been shown that proliferation of CD4+CD28− T cells reactivated with renal tubular epithelial cells (RTECs) are resistant to tacrolimus and everolimus, which suggests a potential CD28− T cell mediated mechanism for organ rejection under standard immunosuppression therapy (78).
It is also possible that Abatacept and Belatacept treatment contributes to the expansion of CD28null cells paradoxically by inhibiting CTLA-4 coinhibitory signals. Interestingly, human CD4 and CD8 populations of CD28null cells rapidly express CTLA-4 on their surface and this expression is sustained compared to naïve T cells, likely due to the significant intracellular reservoirs of CTLA-4 found in memory T cell populations (79). The discrepancy in sustained expression may relate to the ‘need’ for CTLA-4. For instance, naïve cells, which neither have to be long-lived, nor function as regulatory cells, may only be needed early after activation to control the response, whereas memory cells do have to be long-lived, and CTLA-4 has been linked to protection from apoptosis. This suggests that the sustained expression after activation of CTLA-4 may be important to memory cell survival (80). Indeed, it has been shown that crosslinking CTLA-4 on CD28null cells leads to a decrease in activation induced cell death (AICD) through decreased caspase activity (81). These results provide premise for the hypothesis that CD28null cells could be associated with CoBRR not by virtue of their CD28null status, but by their reliance on CTLA-4 coinhibitory signaling.
Importantly, a more recent study from our lab in which we conducted a retrospective immunophenotypic analysis of adult renal transplant recipients who experienced acute rejection on Belatacept treatment as compared to those that did not, uncovered that the pre-transplant frequency of CD28+ cells among CD4+ TEM was very significantly increased in patients who went on to reject versus those that did not (Cortes-Cerisuelo et al, in press). There was also a statistically significant increase in the pre-transplant frequency of CD28+ cells among CD4+ TEMRA cells in these patients, which suggests that patients possessing higher frequency of CD28null CD4+ TEM prior to transplant were actually protected from acute rejection following treatment with a Belatacept-based immunosuppressive regimen. Mechanistically, CD28null CD4+ TEM were found to contain significantly fewer IL-2 producers and expression of the 2B4 coinhibitory molecule was significantly increased on CD28null CD4+ TEM isolated from stable Belatacept-treated patients versus patients who had rejected. These data raise the possibility that pre-transplant frequencies of CD28+ CD4+ TEM could be used as a biomarker to predict risk of rejection following treatment with Belatacept (Cortes-Cerisuelo, et al. In press).
It is also possible that Th17 cells could be at least partially responsible for CoBRR. Given that Abatacept showed mixed results in the treatment of the Th17-mediated diseases Muscular Sclerosis (MS) and Inflammatory Bowel Disease (IBD) (82) and the early severe rejection observed in renal transplant recipients, Th17 cells might be uniquely resistant to CD28/CTLA-4 blockade. Indeed, it has recently been found that an elevated level of Th17 memory cells is associated with acute rejection with Belatacept treatment (83). In addition, Candida albicans infection is able to polarize T cell differentiation toward a Th17 phenotype and also enhances expression of CTLA-4 on these cells. Immunization of mice with this pathogen conferred resistance to costimulation blockade following transplantation, whereas infection with Mycobacterium tuberculosis, which polarizes the response toward Th1 cells, did not confer such resistance (84). These data suggest that Th17 cells might be particularly sensitive to regulation by CTLA-4, and therefore may be a key player in the mechanism surrounding CoBRR.
4.2 The challenge of preserving Foxp3+ Treg functionality
A second major mechanism underlying CoBRR may be related to the impact of these drugs on the function of Foxp3+ Tregs. It is critical to note that because Belatacept blocks the shared ligands CD80 and CD86, both CD28 and CTLA-4 signals are impaired. Importantly, CTLA-4 has been implicated in mediating the functionality of CD4+CD25+ regulatory cells (15, 16); both mouse and human CD4+ CD25+ Tregs constitutively express it (79). Indeed, CTLA-4 has been shown to be required for Treg function by the use of CTLA-4 conditional KO (cKO) mice which lack CTLA-4 specifically in Foxp3-expressing Tregs. These mice succumb to a fatal lymphoproliferative disease similar to that seen in the total CTLA-4 KO animals, albeit with slower kinetics, demonstrating an effector T-cell-intrinsic role of CTLA-4 in maintaining T-cell homeostasis and tolerance (85). Interestingly, in humans it was found that despite a loss of Treg function following CTLA-4 blockade, several labs observed that CTLA-4-deficient Tregs could suppress effector responses in vitro and in autoimmunity models in vivo by inhibiting Teff cells through immunoregulatory pathways such as TGF-β or IL-10 (86, 87). This is distinct from other mechanisms often used by wildtype Tregs in these systems and led investigators to conclude that Tregs developing in a CTLA-4-deficient environment may be able to overcome the need for CTLA-4 through compensatory mechanisms of suppression.
While the mechanisms behind early acute rejection episodes experienced by patients on Belatacept continue to be examined, the pool of patients currently being treated on this regimen continue to experience reduced rates of renal toxicity and a better quality of life.
4.3 The challenge of protective immunity
It is also important to note that Belatacept-treated patients experience higher rates of Epstein-Barr virus (EBV)-associated post-transplant lymphoproliferative disorder (PTLD). Although the mechanisms underlying this clinical observation remain to be demonstrated experimentally, they are likely related to impaired immune surveillance of EBV-infected B cells by antigen-specific CD8+ T cells and EBV-specific antibody, as a result of reduced CD4+ T cell help in the setting of CD28 blockade. It is also interesting to speculate that impaired Treg functionality in the setting of CD28 blockade could contribute to unrestrained EBV-driven B cell proliferation (88).
Currently, continuous administration of Belatacept is required to maintain graft survival, as evidenced by the fact that cessation of treatment led to rejection in studies of NHP renal transplantation (46). Interestingly, continuous administration of RA patients with Abatacept for several years showed beneficial changes in their immunologic profiles; specifically, a decrease in the frequency of CD28null cells and an increase in the frequency of CD28+ cells was observed, suggesting that long-term administration of a CD28 blocker mollifies the inflammatory milieu in these patients (89). However, as with any continuous immunosuppression, belatacept-treated patients are at increased risk of infection. Thus, it has been speculated that pulsing doses of belatacept could afford an “immunosuppression holiday” and potentially allow for improved maintenance of protective immunity while still maintaining graft acceptance. This hypothesis, however, remains to be investigated.
5. Biological Therapies to Target the CD28/CTLA-4 Pathway: The Opportunities
As noted above, Abatacept and Belatacept block the shared ligands for CD28 and CTLA-4, thus blocking CTLA-4 mediated coinhibitory signals that could serve to dampen effector T cell responses and promote Treg-mediated suppression (Figure 1B). Thus, a reagent that could selectively target CD28, while leaving CTLA-4 intact, would be expected to better control alloreactive T cell responses. To this end, non-crosslinking anti-CD28 blocking Fc-devoid or Fc-silent antibodies were developed over the last decade (90–93) (Figure 1C). These reagents were found to be roughly 5 times more potent than Belatacept against CD86-driven T cell proliferation (92), and most studies concluded that the increased efficacy is primarily a result of preserved CTLA-4 coinhibitory signaling, as demonstrated by the fact that anti-CD28 dAb lose much of their potency in the presence of anti-CTLA-4 mAbs (90, 91). However, selective CD28 blockade would in theory also inhibit potential CD28-ICOS-L costimulatory interactions and spare PD-L1-CD80 coinhibitory interactions (94, 95), although the functional consequences of this has not been well-established.
5.1 Preserving CTLA-4 mediated coinhibitory signals via selective CD28 blockade
Several versions of selective CD28 blockers have been generated: first as single-chain Fv Ab fragments from a high-affinity anti-human CD28 Ab which were then fused to human α1-antitrypsin (sc28AT) to increase the half-life in circulation (96). More recently, this same group described a humanized PEGylated anti-CD28 Fab Ab fragment (FR104) (97). Importantly, the VH and VL domains are joined with a flexible polypeptide linker in these single-chain Fvs thus preventing dissociation. Additionally, antibody Fab and scFv fragments, comprising both VH and VL domains, retain the specific, monovalent, antigen-binding affinity of the parent IgG, while showing improved pharmacokinetics for tissue penetration (98).
Another selective CD28 blocker, dAb-00a, was selected for binding to a recombinant biotinylated monomeric human CD28 fragment by phage display from a large synthetic Vκ dAb library (99). This library is based on a fully human germline scaffold with targeted diversification of a subset of hypervariable loop residues known to be involved in Ag interaction. dAb-00a was a stable monomeric Vκ dAb that expressed well in the supernatant of Escherichia coli cultures and had a KD of 1.6 × 10−7 M and EC50 of 1.3 μM in a cell-based luciferase reporter gene assay. The affinity of dAb-00a was improved in two steps. First, a library was created in which dAb-00a was diversified using random mutagenesis. Phage selection of this library led to isolation of dAb-00b that retained good biophysical properties of the parental dAb-00a molecule and had a 130-fold increase in potency in an IL-2 reporter luciferase assay (EC50 10 nM) and KD of 8.4 × 10−9 M. In the second step, a library was created in which diversification was targeted toward CDR residues of dAb-00b using oligo-directed mutagenesis. This strategy led to isolation and identification of dAb-001 and dAb-002 (92). These molecules were then pegylated to increase serum half-life.
In vitro anti-CD28 dAbs inhibit T cell activation by preventing CD28 engagement with CD80 and CD86 while preserving the ability of CTLA-4 to bind to these same ligands. This effect was demonstrated by the ability of the dAbs to potently inhibit T cell proliferation and cytokine production in the context of a DC-MLR while not affecting the ability of CTLA-4–bearing cells to promote downregulation of CD86 (92). Importantly, as expected, these effects were reversed in the presence of antagonistic anti-CTLA-4 mAbs (90, 91). As discussed earlier, CTLA-4 expression on conventional T cells inhibits T cell activation by competing with CD28 for binding to CD80 and CD86 on APCs and is a suppressive mechanism in T cell homeostasis (48). In contrast, CTLA-4 expression on Tregs is important for the suppression of autoreactive T cells and the prevention of in vivo autoimmunity (85). Indeed, use of CD28 antagonists is associated with Treg accumulation in the graft, where they most likely modulate pathogenic T cells and promote prolonged allograft survival (91).
In a head to head assessment of FR104 and Belatacept in kidney allotransplantation in baboons, four of five recipients receiving Belatacept developed severe, steroid–resistant acute cellular rejection, whereas FR104-treated animals did not. This was not due to higher regulatory T cell frequencies in FR104-treated animals or in differences in Th17 cytokines, but may have been due, in part to higher levels of IL-21, the main cytokine secreted by CD4 T follicular helper (Tfh) cells, in belatacept-treated animals. Indeed, in vitro, FR104 controlled the proliferative response of human preexisting Tfh cells more efficiently than Belatacept. In mice, selective CD28 blockade also controlled Tfh memory cell responses to KLH stimulation more efficiently than CD80/86 blockade. This study reveals that selective CD28 blockade and Belatacept exert differential effects on the mechanisms of renal allograft rejection, particularly at the level of Tfh cell stimulation (100). Indeed, several recent studies have highlighted the critical role of CTLA-4 coinhibition in limiting CXCR5+ T follicular helper (Tfh) cell responses, thus impairing the development of high affinity antibodies (101–103). Loss of CTLA-4 on effector Tfh cells led to a spontaneous Tfh cell differentiation and exaggerated GC B cell responses in vivo (102, 103). Furthermore, even short-term blockade of CTLA-4 resulted in a significant increase in Tfh cell differentiation and GC development (103). Thus, it is possible that next generation selective CD28 blockers that leave CTLA-4 coinhibition intact will more effectively inhibit de novo donor-specific antibody and prevent antibody-mediated rejection during transplantation.
A recent report by Poirier et al. demonstrates that a monovalent anti-human CD28 antagonist (CD28-specific single-chain Fv Ab fragment linked to α1-antitrypsin, sc28AT) synergizes with the in vitro suppressive activity of Tregs, whereas anti–CTLA-4 Ab blocks the suppression (91). Furthermore, treatment of nonhuman primates with sc28AT plus tacrolimus, during and following kidney or heart allograft transplantation, results in the prevention of acute rejection, attenuation of chronic rejection, and the influx of functional Tregs into the graft (91). Similarly, treatment of mice receiving cardiac allografts with a monovalent mouse anti-CD28 scFv (α28scFv) combined with either anti-CD154 or cyclosporine treatment significantly increased the proportion of intragraft Tregs compared with recipients that received either treatment alone. There was also a positive correlation between the number of intragraft Tregs and prolonged cardiac allograft survival in mice (93).
Furthermore, one study identified a critical role for the co-inhibitory SLAM family member 2B4 (CD244) in attenuating primary antigen-specific CD8+ T cell responses in the presence of immune modulation with selective CD28 blockade. This study found a specific up-regulation of 2B4 on antigen-specific CD8+ T cells in animals in which CD28 signaling was blocked. However, 2B4 up-regulation was not observed in animals treated with Abatacept or CD28 blockade in the presence of anti–CTLA-4 mAb. This upregulation was functionally significant, as the inhibitory impact of CD28 blockade was diminished when antigen-specific CD8+ T cells were deficient in 2B4 (90).
5.2 The challenge of Treg survival
Despite the preservation of signaling through their critical functional modulator CTLA-4, Tregs are likely still negatively impacted by selective CD28 blockade. This is due to the finding that CD28 is also required for Treg development and survival, where it has been found that mice deficient in CD28 or its ligands, have a dramatically reduced number of natural Tregs and develop accelerated autoimmunity (17). It was known that blockade of CD28 signaling by Abatacept lead to a rapid decrease of Tregs, both in the thymus and in the periphery, which in turn lead to a break in self-tolerance or transplantation-tolerance in models in which Tregs play a major role in maintaining these states (17, 86, 104). To get at the mechanism behind this observation, Turka’s group generated CD28-conditional knockout mice that targeted CD28 specifically on Foxp3+ Tregs. It was found that although these mice had normal numbers of Tregs, there was both a cell-intrinsic proliferation and survival defect, which manifested only under competitive conditions, and a functional impairment in vivo, which was accompanied by decreased expression of the molecules CTLA-4, PD-1, and CCR6, leading to the development of a systemic autoimmunity characterized by prominent skin inflammation (105). More recent work from the same group dug deeper and showed that while these conditional CD28-deficient Tregs are able to regulate inflammation normally when injected directly into the skin, they fail to home properly to inflamed skin (106).
One potential strategy to overcome the challenge of diminished Treg survival in the setting of CD28 blockade is through the use of CD154-directed biological therapeutics. Blockade of CD40–CD154 interactions during T cell priming has been shown to be a highly effective means of inducing long-term survival of allografts and transplantation tolerance in both murine and nonhuman primate models (107, 108), and many groups have shown that this is due to enhanced regulatory T cell responses in animals treated with CD154 blockade (109–112). With this in mind, our lab conducted a study to determine whether iTreg generation would be preserved when Abatacept was given in combination with a potentially clinically translatable anti-CD154 dAb relative to an Fc-intact anti-CD154 antibody. This study demonstrated that blockade of the CD40–CD154 pathway is sufficient to generate high frequencies of antigen-specific CD4+ iTregs even in the presence of Abatacept (113). These data are potentially clinically important in that they provide evidence that combinatorial use of these costimulation blockers may be beneficial in attenuating the effector T cell responses and promoting immune regulation.
In sum, the dynamic interplay of co-stimulatory and co-inhibitory signals received during T cell priming orchestrates antigen-specific T cell responses that ultimately result in either tolerance or immunity, the selective CD28 blockade through the use of domain antibodies holds promise as a clinically translatable strategy for the mitigation of unwanted immune responses in transplantation.
6. Conclusion
Costimulation and coinhibition are fundamental mechanisms evolved to control immune activation. Continuous integration of these two processes provides precise, but complex regulation. From the growing understanding of this multiplicity of immune function, CD28 and CTLA-4 have emerged as critically important regulatory molecules. Cosignaling is central to allo- and auto-reactive T cell responses and thus remains a viable target for inhibiting graft rejection and autoaggressive T cells. Indeed, as both pathways have been shown to affect both alloreactive and regulatory T cells, an important goal for the future development of blockade of each of these pathways clinically is how to utilize them in a manner that optimizes the attenuation of effector cells while preserving the suppressive function of T regulatory cells. In summary, these signals are vital for optimal immune homeostasis, protective immunity and tolerance, therefore making T cell cosignaling molecules an attractive target during transplantation and autoimmunity.
7. Expert Opinion
Both proof-of-concept studies in experimental models and clinical trials in patients with autoimmunity and following transplantation have provided empirical evidence that therapeutic targeting of the CD28/CTLA-4 pathway is a powerful means to control unwanted adaptive immune responses. Early generation reagents Abatacept and belatacept are limited, however, by the fact that they disrupt CTLA-4 signaling, and thus block an important physiologic mechanism that controls adaptive immunity. While the critical role of CTLA-4 in the function of Foxp3+ Treg is relatively well understood, the unique reliance of Th17 cells on CTLA-4 signaling is less well elucidated. Does CTLA-4 signaling participate in cell fate decisions during Th1 vs. Th17 differentiation? Or does CTLA-4 signaling in established Th17 populations differ from that of Th1 such that CTLA-4 blockade has a greater impact on Th17 cells? Future research should be directed at determining the gene expression and signaling changes that render Th17 cells particularly sensitive to CTLA-4 coinhibitory signals. This line of inquiry is particularly important in the setting of autoimmune diseases such as multiple sclerosis and inflammatory bowel disease, which have a strong Th17 component.
Next generation blockers that selectively block CD28 while preserving CTLA-4 coinhibitory signaling represent a big leap forward in the field, and clinical trials in both GVHD and several autoimmune diseases are so far promising. The great challenge that still presents itself, however, is the reliance of Foxp3+ Treg, a population critical for optimal control of allo- and autoimmunity, on CD28 signals for survival. Indeed, due to their higher affinity selective CD28 blockers may have an even more severe effect on Foxp3+ Treg populations than do the CTLA-4 Ig fusion proteins. Thus, the next great challenge and opportunity for the field is devising a strategy to block CD28 signals specifically on naïve and effector T cells, while preserving the CD28 signals critical for Treg survival. This could potentially be accomplished through the development of bi-specific antibodies, although selection of appropriate co-targets could prove difficult as many are shared between effector and regulatory T cells. Alternatively, one could imagine it might be possible to specifically agonize CD28 signals selectively in Foxp3+ Treg in order to “artificially” send survival signals in animals or patients in which CD28 is blocked. At the moment, these strategies appear a long way off, but may represent what is coming on the horizon in order to maximally capitalize on intrinsic physiologic mechanisms through which adaptive immune responses are controlled.
Further, it is also apparent that there are subsets of memory T cells that have diminished requirements for CD28 costimulation, and that may be the drivers of graft rejection or recurrent autoimmunity in the setting of CD28 blockade. Additional mechanistic insight into the pathways utilized by these populations for survival, proliferation, and/or effector function will yield novel therapeutic targets on which to intervene. For example, targeting cytokine signaling pathways preferentially used by memory T cells such IL-15 (by blocking CD122) has shown promise as a potential therapeutic target to inhibit memory T cell-mediated rejection (Dr. Andrew B. Adams, Emory University, personal communication). Furthermore, agonizing memory T cell-specific coinhibitory pathways such as 2B4 and FcgRIIb may also hold promise for limiting allo- and autoaggressive CD28-independent T cell responses (90) (and unpublished data). In sum, developing novel therapeutics to control memory T cell responses while preserving and promoting effective regulation will result in more precise control of allo- and autoimmunity and result in better outcomes for patients.
Highlights.
CD28 costimulation blockade is advantageous for inhibiting unwanted T cell responses during autoimmunity and transplantation because the targets are confined to the immune system, limiting off-target toxicities associated with calcineurin inhibitors and anti-proliferatives
Blocking CD28 using conventional CTLA-4 Ig fusion proteins (abatacept, belatacept) that bind to the shared receptors CD80/CD86 has the collateral effect of also inhibiting CTLA-4 coinhibitory signaling
Due to both relative CD28 independence and the loss of regulation via CTLA-4 coinhibitory signals, memory T cells, Th17 cells, and impaired Treg function may play a role in breakthrough T cell responses in patients treated with CTLA-4Ig fusion proteins
Novel CD28-specific domain antibodies have been developed in order to selectively block CD28 signals while leaving CTLA-4 coinhibitory signaling intact
Inhibition of CD28 signals on Foxp3+ Treg populations may still pose a challenge for the use of selective CD28 blocking reagents in transplantation and autoimmunity
References
- 1.Mueller DL, Jenkins MK, Schwartz RH. An accessory cell-derived costimulatory signal acts independently of protein kinase C activation to allow T cell proliferation and prevent the induction of unresponsiveness. J Immunol. 1989;142(8):2617–28. Epub 1989/04/15. [PubMed] [Google Scholar]
- 2.Linsley PS, Greene JL, Tan P, et al. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med. 1992;176(6):1595–604. doi: 10.1084/jem.176.6.1595. Epub 1992/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linsley PS, Bradshaw J, Urnes M, et al. CD28 engagement by B7/BB-1 induces transient down-regulation of CD28 synthesis and prolonged unresponsiveness to CD28 signaling. J Immunol. 1993;150(8 Pt 1):3161–9. Epub 1993/04/15. [PubMed] [Google Scholar]
- 4.Metzler B, Burkhart C, Wraith DC. Phenotypic analysis of CTLA-4 and CD28 expression during transient peptide-induced T cell activation in vivo. Int Immunol. 1999;11(5):667–75. doi: 10.1093/intimm/11.5.667. Epub 1999/05/18. [DOI] [PubMed] [Google Scholar]
- 5.Freeman GJ, Lombard DB, Gimmi CD, et al. CTLA-4 and CD28 mRNA are coexpressed in most T cells after activation. Expression of CTLA-4 and CD28 mRNA does not correlate with the pattern of lymphokine production. J Immunol. 1992;149(12):3795–801. Epub 1992/12/15. [PubMed] [Google Scholar]
- 6.Guinan EC, Gribben JG, Boussiotis VA, et al. Pivotal role of the B7:CD28 pathway in transplantation tolerance and tumor immunity. Blood. 1994;84(10):3261–82. Epub 1994/11/15. [PubMed] [Google Scholar]
- 7.Metz DP, Farber DL, Taylor T, et al. Differential role of CTLA-4 in regulation of resting memory versus naive CD4 T cell activation. J Immunol. 1998;161(11):5855–61. Epub 1998/12/02. [PubMed] [Google Scholar]
- 8.Lindsten T, Lee KP, Harris ES, et al. Characterization of CTLA-4 structure and expression on human T cells. J Immunol. 1993;151(7):3489–99. Epub 1993/10/01. [PubMed] [Google Scholar]
- 9.Linsley PS, Brady W, Urnes M, et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–9. doi: 10.1084/jem.174.3.561. Epub 1991/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–7. doi: 10.1016/1074-7613(95)90125-6. Epub 1995/11/01. [DOI] [PubMed] [Google Scholar]
- 11.Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405–13. doi: 10.1016/1074-7613(94)90071-x. Epub 1994/08/01. [DOI] [PubMed] [Google Scholar]
- 12.Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. 1996;183(6):2541–50. doi: 10.1084/jem.183.6.2541. Epub 1996/06/01. **The first paper to show that CTLA-4 acts as a negative regulator of T cell activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bachmann MF, Kohler G, Ecabert B, et al. Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol. 1999;163(3):1128–31. Epub 1999/07/22. [PubMed] [Google Scholar]
- 14.Chen W, Jin W, Wahl SM. Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor beta (TGF-beta) production by murine CD4(+) T cells. J Exp Med. 1998;188(10):1849–57. doi: 10.1084/jem.188.10.1849. Epub 1998/11/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192(2):295–302. doi: 10.1084/jem.192.2.295. Epub 2000/07/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192(2):303–10. doi: 10.1084/jem.192.2.303. Epub 2000/07/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salomon B, Lenschow DJ, Rhee L, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12(4):431–40. doi: 10.1016/s1074-7613(00)80195-8. Epub 2000/05/05. **This paper provided critical evidence that CD28 costimulation is essential for the maintenance of regulatory T cells. [DOI] [PubMed] [Google Scholar]
- 18.Friedline RH, Brown DS, Nguyen H, et al. CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. J Exp Med. 2009;206(2):421–34. doi: 10.1084/jem.20081811. Epub 2009/02/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudd CE. The reverse stop-signal model for CTLA4 function. Nat Rev Immunol. 2008;8:153–60. doi: 10.1038/nri2253. England. [DOI] [PubMed] [Google Scholar]
- 20.Dustin ML, Bromley SK, Kan Z, et al. Antigen receptor engagement delivers a stop signal to migrating T lymphocytes. Proc Natl Acad Sci U S A. 1997;94(8):3909–13. doi: 10.1073/pnas.94.8.3909. Epub 1997/04/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bousso P, Robey E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol. 2003;4(6):579–85. doi: 10.1038/ni928. Epub 2003/05/06. [DOI] [PubMed] [Google Scholar]
- 22.Hugues S, Fetler L, Bonifaz L, et al. Distinct T cell dynamics in lymph nodes during the induction of tolerance and immunity. Nat Immunol. 2004;5(12):1235–42. doi: 10.1038/ni1134. Epub 2004/11/02. [DOI] [PubMed] [Google Scholar]
- 23.Schneider H, Valk E, da Rocha Dias S, et al. CTLA-4 up-regulation of lymphocyte function-associated antigen 1 adhesion and clustering as an alternate basis for coreceptor function. Proc Natl Acad Sci U S A. 2005;102(36):12861–6. doi: 10.1073/pnas.0505802102. Epub 2005/08/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science (New York, NY) 1994;265(5176):1225–7. doi: 10.1126/science.7520604. Epub 1994/08/26. *The first use of CTLA4-IG (Abatacept) to treat autoimmune disease. [DOI] [PubMed] [Google Scholar]
- 25.Reynolds J, Tam FW, Chandraker A, et al. CD28-B7 blockade prevents the development of experimental autoimmune glomerulonephritis. J Clin Invest. 2000;105(5):643–51. doi: 10.1172/jci6710. Epub 2000/03/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abrams JR, Lebwohl MG, Guzzo CA, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest. 1999;103(9):1243–52. doi: 10.1172/jci5857. Epub 1999/05/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Webb LM, Walmsley MJ, Feldmann M. Prevention and amelioration of collagen-induced arthritis by blockade of the CD28 co-stimulatory pathway: requirement for both B7-1 and B7-2. European journal of immunology. 1996;26(10):2320–8. doi: 10.1002/eji.1830261008. Epub 1996/10/01. [DOI] [PubMed] [Google Scholar]
- 28.Moreland LW, Alten R, Van den Bosch F, et al. Costimulatory blockade in patients with rheumatoid arthritis: a pilot, dose-finding, double-blind, placebo-controlled clinical trial evaluating CTLA-4Ig and LEA29Y eighty-five days after the first infusion. Arthritis and rheumatism. 2002;46(6):1470–9. doi: 10.1002/art.10294. Epub 2002/07/13. [DOI] [PubMed] [Google Scholar]
- 29.Kremer JM, Westhovens R, Leon M, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. The New England journal of medicine. 2003;349(20):1907–15. doi: 10.1056/NEJMoa035075. Epub 2003/11/14. [DOI] [PubMed] [Google Scholar]
- 30.Schiff M, Keiserman M, Codding C, et al. Efficacy and safety of abatacept or infliximab vs placebo in ATTEST: a phase III, multi-centre, randomised, double-blind, placebo-controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Annals of the rheumatic diseases. 2008;67(8):1096–103. doi: 10.1136/ard.2007.080002. Epub 2007/12/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinblatt M, Combe B, Covucci A, et al. Safety of the selective costimulation modulator abatacept in rheumatoid arthritis patients receiving background biologic and nonbiologic disease-modifying antirheumatic drugs: A one-year randomized, placebo-controlled study. Arthritis and rheumatism. 2006;54(9):2807–16. doi: 10.1002/art.22070. Epub 2006/09/02. [DOI] [PubMed] [Google Scholar]
- 32.Kremer JM, Genant HK, Moreland LW, et al. Effects of abatacept in patients with methotrexate-resistant active rheumatoid arthritis: a randomized trial. Annals of internal medicine. 2006;144(12):865–76. doi: 10.7326/0003-4819-144-12-200606200-00003. Epub 2006/06/21. [DOI] [PubMed] [Google Scholar]
- 33.Schiff M, Pritchard C, Huffstutter JE, et al. The 6-month safety and efficacy of abatacept in patients with rheumatoid arthritis who underwent a washout after anti-tumour necrosis factor therapy or were directly switched to abatacept: the ARRIVE trial. Annals of the rheumatic diseases. 2009;68(11):1708–14. doi: 10.1136/ard.2008.099218. Epub 2008/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Genovese MC, Becker JC, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. The New England journal of medicine. 2005;353(11):1114–23. doi: 10.1056/NEJMoa050524. Epub 2005/09/16. [DOI] [PubMed] [Google Scholar]
- 35.Westhovens R, Kremer JM, Moreland LW, et al. Safety and efficacy of the selective costimulation modulator abatacept in patients with rheumatoid arthritis receiving background methotrexate: a 5-year extended phase IIB study. The Journal of rheumatology. 2009;36(4):736–42. doi: 10.3899/jrheum.080813. Epub 2009/03/11. [DOI] [PubMed] [Google Scholar]
- 36.Genant HK, Peterfy CG, Westhovens R, et al. Abatacept inhibits progression of structural damage in rheumatoid arthritis: results from the long-term extension of the AIM trial. Annals of the rheumatic diseases. 2008;67(8):1084–9. doi: 10.1136/ard.2007.085084. Epub 2007/12/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aringer M, Burkhardt H, Burmester GR, et al. Current state of evidence on ‘off-label’ therapeutic options for systemic lupus erythematosus, including biological immunosuppressive agents, in Germany, Austria and Switzerland–a consensus report. Lupus. 2012;21(4):386–401. doi: 10.1177/0961203311426569. Epub 2011/11/11. [DOI] [PubMed] [Google Scholar]
- 38.Daikh DI, Finck BK, Linsley PS, et al. Long-term inhibition of murine lupus by brief simultaneous blockade of the B7/CD28 and CD40/gp39 costimulation pathways. J Immunol. 1997;159(7):3104–8. Epub 1997/10/08. [PubMed] [Google Scholar]
- 39.Daikh DI, Wofsy D. Cutting edge: reversal of murine lupus nephritis with CTLA4Ig and cyclophosphamide. J Immunol. 2001;166(5):2913–6. doi: 10.4049/jimmunol.166.5.2913. Epub 2001/02/24. [DOI] [PubMed] [Google Scholar]
- 40.Furie R, Nicholls K, Cheng TT, et al. Efficacy and safety of abatacept in lupus nephritis: a twelve-month, randomized, double-blind study. Arthritis & rheumatology (Hoboken, NJ) 2014;66(2):379–89. doi: 10.1002/art.38260. Epub 2014/02/08. [DOI] [PubMed] [Google Scholar]
- 41.Merrill JT, Burgos-Vargas R, Westhovens R, et al. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis and rheumatism. 2010;62(10):3077–87. doi: 10.1002/art.27601. Epub 2010/06/10. [DOI] [PubMed] [Google Scholar]
- 42.Treatment of lupus nephritis with abatacept: the Abatacept and Cyclophosphamide Combination Efficacy and Safety Study. Arthritis & rheumatology (Hoboken, NJ) 2014;66(11):3096–104. doi: 10.1002/art.38790. Epub 2014/11/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danion F, Rosine N, Belkhir R, et al. Efficacy of abatacept in systemic lupus erythematosus: a retrospective analysis of 11 patients with refractory disease. Lupus. 2016;25(13):1440–7. doi: 10.1177/0961203316640911. Epub 2016/03/26. [DOI] [PubMed] [Google Scholar]
- 44.Blazar BR, Taylor PA, Linsley PS, et al. In vivo blockade of CD28/CTLA4: B7/BB1 interaction with CTLA4-Ig reduces lethal murine graft-versus-host disease across the major histocompatibility complex barrier in mice. Blood. 1994;83(12):3815–25. Epub 1994/06/15. [PubMed] [Google Scholar]
- 45.Kirk AD, Harlan DM, Armstrong NN, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci U S A. 1997;94(16):8789–94. doi: 10.1073/pnas.94.16.8789. Epub 1997/08/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46**.Larsen CP, Pearson TC, Adams AB, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5(3):443–53. doi: 10.1111/j.1600-6143.2005.00749.x. Epub 2005/02/15. This publication describes the development of the second generation CD28 blockade agent Belatacept. [DOI] [PubMed] [Google Scholar]
- 47.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annual review of immunology. 1996;14:233–58. doi: 10.1146/annurev.immunol.14.1.233. Epub 1996/01/01. [DOI] [PubMed] [Google Scholar]
- 48.Linsley PS, Greene JL, Brady W, et al. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1(9):793–801. doi: 10.1016/s1074-7613(94)80021-9. Epub 1994/12/01. [DOI] [PubMed] [Google Scholar]
- 49.Kode R, Fa K, Chowdhury S, et al. Basiliximab plus low-dose cyclosporin vs. OKT3 for induction immunosuppression following renal transplantation. Clinical transplantation. 2003;17(4):369–76. doi: 10.1034/j.1399-0012.2003.00061.x. Epub 2003/07/19. [DOI] [PubMed] [Google Scholar]
- 50.Lawen JG, Davies EA, Mourad G, et al. Randomized double-blind study of immunoprophylaxis with basiliximab, a chimeric anti-interleukin-2 receptor monoclonal antibody, in combination with mycophenolate mofetil-containing triple therapy in renal transplantation. Transplantation. 2003;75(1):37–43. doi: 10.1097/01.tp.0000040864.73222.67. Epub 2003/01/25. [DOI] [PubMed] [Google Scholar]
- 51.Lebranchu Y, Bridoux F, Buchler M, et al. Immunoprophylaxis with basiliximab compared with antithymocyte globulin in renal transplant patients receiving MMF-containing triple therapy. Am J Transplant. 2002;2(1):48–56. doi: 10.1034/j.1600-6143.2002.020109.x. Epub 2002/07/04. [DOI] [PubMed] [Google Scholar]
- 52.Vincenti F, Ramos E, Brattstrom C, et al. Multicenter trial exploring calcineurin inhibitors avoidance in renal transplantation. Transplantation. 2001;71(9):1282–7. doi: 10.1097/00007890-200105150-00017. Epub 2001/06/09. [DOI] [PubMed] [Google Scholar]
- 53.Vincenti F, Larsen C, Durrbach A, et al. Costimulation blockade with belatacept in renal transplantation. The New England journal of medicine. 2005;353(8):770–81. doi: 10.1056/NEJMoa050085. Epub 2005/08/27. [DOI] [PubMed] [Google Scholar]
- 54.Archdeacon P, Dixon C, Belen O, et al. Summary of the US FDA approval of belatacept. Am J Transplant. 2012;12(3):554–62. doi: 10.1111/j.1600-6143.2011.03976.x. Epub 2012/02/18. [DOI] [PubMed] [Google Scholar]
- 55.Durrbach A, Pestana JM, Pearson T, et al. A phase III study of belatacept versus cyclosporine in kidney transplants from extended criteria donors (BENEFIT-EXT study) Am J Transplant. 2010;10(3):547–57. doi: 10.1111/j.1600-6143.2010.03016.x. Epub 2010/04/27. [DOI] [PubMed] [Google Scholar]
- 56.Rostaing L, Vincenti F, Grinyo J, et al. Long-term belatacept exposure maintains efficacy and safety at 5 years: results from the long-term extension of the BENEFIT study. Am J Transplant. 2013;13(11):2875–83. doi: 10.1111/ajt.12460. Epub 2013/09/21. [DOI] [PubMed] [Google Scholar]
- 57.Pestana JO, Grinyo JM, Vanrenterghem Y, et al. Three-year outcomes from BENEFIT-EXT: a phase III study of belatacept versus cyclosporine in recipients of extended criteria donor kidneys. Am J Transplant. 2012;12(3):630–9. doi: 10.1111/j.1600-6143.2011.03914.x. Epub 2012/02/04. [DOI] [PubMed] [Google Scholar]
- 58.Vincenti F, Rostaing L, Grinyo J, et al. Belatacept and Long-Term Outcomes in Kidney Transplantation. 2016 doi: 10.1056/NEJMoa1506027. http://dxdoiorg/101056/NEJMoa1506027. doi: NJ201601283740408. [DOI] [PubMed]
- 59.Vincenti F, Blancho G, Durrbach A, et al. Five-year safety and efficacy of belatacept in renal transplantation. Journal of the American Society of Nephrology : JASN. 2010;21(9):1587–96. doi: 10.1681/asn.2009111109. Epub 2010/07/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ford ML, Koehn BH, Wagener ME, et al. Antigen-specific precursor frequency impacts T cell proliferation, differentiation, and requirement for costimulation. J Exp Med. 2007;204:299–309. doi: 10.1084/jem.20062319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ford ML, Larsen CP. Overcoming the memory barrier in tolerance induction: molecular mimicry and functional heterogeneity among pathogen-specific T-cell populations. Curr Opin Organ Transplant. 2010;15(4):405–10. doi: 10.1097/MOT.0b013e32833b7916. Epub 2010/07/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ford ML, Wagener ME, Hanna SS, et al. A critical precursor frequency of donor-reactive CD4+ T cell help is required for CD8+ T cell-mediated CD28/CD154-independent rejection. J Immunol. 2008;180(11):7203–11. doi: 10.4049/jimmunol.180.11.7203. Epub 2008/05/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pantenburg B, Heinzel F, Das L, et al. T cells primed by Leishmania major infection cross-react with alloantigens and alter the course of allograft rejection. J Immunol. 2002;169(7):3686–93. doi: 10.4049/jimmunol.169.7.3686. Epub 2002/09/24. [DOI] [PubMed] [Google Scholar]
- 64.Adams AB, Williams MA, Jones TR, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111(12):1887–95. doi: 10.1172/jci17477. Epub 2003/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deacock SJ, Lechler RI. Positive correlation of T cell sensitization with frequencies of alloreactive T helper cells in chronic renal failure patients. Transplantation. 1992;54(2):338–43. doi: 10.1097/00007890-199208000-00026. Epub 1992/08/01. [DOI] [PubMed] [Google Scholar]
- 66.Sprent J, Surh CD, Tough D. Fate of T and B cells transferred to SCID mice. Research in immunology. 1994;145(5):328–31. doi: 10.1016/s0923-2494(94)80194-0. Epub 1994/06/01. [DOI] [PubMed] [Google Scholar]
- 67.Tchao NK, Turka LA. Lymphodepletion and homeostatic proliferation: implications for transplantation. Am J Transplant. 2012;12(5):1079–90. doi: 10.1111/j.1600-6143.2012.04008.x. Epub 2012/03/17. [DOI] [PubMed] [Google Scholar]
- 68.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192(4):557–64. doi: 10.1084/jem.192.4.557. Epub 2000/08/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moxham VF, Karegli J, Phillips RE, et al. Homeostatic proliferation of lymphocytes results in augmented memory-like function and accelerated allograft rejection. J Immunol. 2008;180(6):3910–8. doi: 10.4049/jimmunol.180.6.3910. Epub 2008/03/07. [DOI] [PubMed] [Google Scholar]
- 70.Wu Z, Bensinger SJ, Zhang J, et al. Homeostatic proliferation is a barrier to transplantation tolerance. Nature medicine. 2004;10(1):87–92. doi: 10.1038/nm965. Epub 2003/12/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iida S, Suzuki T, Tanabe K, et al. Transient lymphopenia breaks costimulatory blockade-based peripheral tolerance and initiates cardiac allograft rejection. Am J Transplant. 2013;13(9):2268–79. doi: 10.1111/ajt.12342. Epub 2013/07/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blattman JN, Antia R, Sourdive DJ, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195(5):657–64. doi: 10.1084/jem.20001021. Epub 2002/03/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suchin EJ, Langmuir PB, Palmer E, et al. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166(2):973–81. doi: 10.4049/jimmunol.166.2.973. Epub 2001/01/06. [DOI] [PubMed] [Google Scholar]
- 74.Floyd TL, Koehn BH, Kitchens WH, et al. Limiting the amount and duration of antigen exposure during priming increases memory T cell requirement for costimulation during recall. J Immunol. 2011;186(4):2033–41. doi: 10.4049/jimmunol.1003015. Epub 2011/01/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Badell IR, Kitchens WH, Wagener ME, et al. Pathogen Stimulation History Impacts Donor-Specific CD8(+) T Cell Susceptibility to Costimulation/Integrin Blockade-Based Therapy. Am J Transplant. 2015;15(12):3081–94. doi: 10.1111/ajt.13399. Epub 2015/08/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Espinosa J, Herr F, Tharp G, et al. CD57(+) CD4 T Cells Underlie Belatacept-Resistant Allograft Rejection. Am J Transplant. 2016;16(4):1102–12. doi: 10.1111/ajt.13613. Epub 2015/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fagnoni FF, Vescovini R, Mazzola M, et al. Expansion of cytotoxic CD8+ CD28− T cells in healthy ageing people, including centenarians. Immunology. 1996;88(4):501–7. doi: 10.1046/j.1365-2567.1996.d01-689.x. Epub 1996/08/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Demmers MW, Baan CC, Janssen M, et al. Substantial proliferation of human renal tubular epithelial cell-reactive CD4+CD28null memory T cells, which is resistant to tacrolimus and everolimus. Transplantation. 2014;97(1):47–55. doi: 10.1097/01.TP.0000435697.31148.b2. Epub 2013/10/26. [DOI] [PubMed] [Google Scholar]
- 79.Jago CB, Yates J, Camara NO, et al. Differential expression of CTLA-4 among T cell subsets. Clin Exp Immunol. 2004;136(3):463–71. doi: 10.1111/j.1365-2249.2004.02478.x. Epub 2004/05/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.da Rocha Dias S, Rudd CE. CTLA-4 blockade of antigen-induced cell death. Blood. 2001;97(4):1134–7. doi: 10.1182/blood.v97.4.1134. Epub 2001/02/13. [DOI] [PubMed] [Google Scholar]
- 81.Hoff H, Knieke K, Cabail Z, et al. Surface CD152 (CTLA-4) expression and signaling dictates longevity of CD28null T cells. J Immunol. 2009;182(9):5342–51. doi: 10.4049/jimmunol.0801624. Epub 2009/04/22. [DOI] [PubMed] [Google Scholar]
- 82**.Linsley PS, Nadler SG. The clinical utility of inhibiting CD28-mediated costimulation. Immunol Rev. 2009;229(1):307–21. doi: 10.1111/j.1600-065X.2009.00780.x. Epub 2009/05/12. This review summarizes the use of CD28 costimulation blockade therapy. [DOI] [PubMed] [Google Scholar]
- 83.Krummey SM, Cheeseman JA, Conger JA, et al. High CTLA-4 expression on Th17 cells results in increased sensitivity to CTLA-4 coinhibition and resistance to belatacept. Am J Transplant. 2014;14(3):607–14. doi: 10.1111/ajt.12600. Epub 2014/04/15. *This is the first publication to describe a role for Th17 cells in costimulation blockage resistant rejection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krummey SM, Floyd TL, Liu D, et al. Candida-elicited murine Th17 cells express high Ctla-4 compared with Th1 cells and are resistant to costimulation blockade. J Immunol. 2014;192(5):2495–504. doi: 10.4049/jimmunol.1301332. Epub 2014/02/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85**.Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 Control over Foxp3+ Regulatory T Cell function. 2008 doi: 10.1126/science.1160062. Describes the requirement for CTLA-4 expression for T regulatory cell function. [DOI] [PubMed] [Google Scholar]
- 86.Tang Q, Henriksen KJ, Boden EK, et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol. 2003;171(7):3348–52. doi: 10.4049/jimmunol.171.7.3348. Epub 2003/09/23. [DOI] [PubMed] [Google Scholar]
- 87.Zwar TD, Read S, van Driel IR, et al. CD4+CD25+ regulatory T cells inhibit the antigen-dependent expansion of self-reactive T cells in vivo. J Immunol. 2006;176(3):1609–17. doi: 10.4049/jimmunol.176.3.1609. Epub 2006/01/21. [DOI] [PubMed] [Google Scholar]
- 88.Zhao D-M, Thornton AM, DiPaolo RJ, et al. Activated CD4+CD25+ T cells selectively kill B lymphocytes. 2006 doi: 10.1182/blood-2005-11-4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Imberti L, Scarsi M, Zanotti C, et al. Reduced T-cell repertoire restrictions in abatacept-treated rheumatoid arthritis patients. Journal of translational medicine. 2015;13:12. doi: 10.1186/s12967-014-0363-2. Epub 2015/01/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu D, Krummey SM, Badell IR, et al. 2B4 (CD244) induced by selective CD28 blockade functionally regulates allograft-specific CD8+ T cell responses. J Exp Med. 2014;211:297–311. doi: 10.1084/jem.20130902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91*.Poirier N, Azimzadeh AM, Zhang T, et al. Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci Transl Med. 2010;2(17):17ra0. doi: 10.1126/scitranslmed.3000116. Epub 2010/04/08. This paper describes the critical role of CTLA-4 in the promotion of regulatory T cells in the setting of solid organ transplantation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Suchard SJ, Davis PM, Kansal S, et al. A monovalent anti-human CD28 domain antibody antagonist: preclinical efficacy and safety. J Immunol. 2013;191(9):4599–610. doi: 10.4049/jimmunol.1300470. Epub 2013/10/02. [DOI] [PubMed] [Google Scholar]
- 93.Zhang T, Fresnay S, Welty E, et al. Selective CD28 blockade attenuates acute and chronic rejection of murine cardiac allografts in a CTLA-4-dependent manner. Am J Transplant. 2011;11(8):1599–609. doi: 10.1111/j.1600-6143.2011.03624.x. Epub 2011/07/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Butte MJ, Keir ME, Phamduy TB, et al. PD-L1 interacts specifically with B7-1 to inhibit T cell proliferation. Immunity. 2007;27(1):111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yao S, Zhu Y, Zhu G, et al. B7-H2 is a costimulatory ligand for CD28 in human. Immunity. 2011;34(5):729–40. doi: 10.1016/j.immuni.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vanhove B, Laflamme G, Coulon F, et al. Selective blockade of CD28 and not CTLA-4 with a single-chain Fv-alpha1-antitrypsin fusion antibody. Blood. 2003;102(2):564–70. doi: 10.1182/blood-2002-08-2480. Epub 2003/03/22. [DOI] [PubMed] [Google Scholar]
- 97.Poirier N, Mary C, Dilek N, et al. Preclinical efficacy and immunological safety of FR104, an antagonist anti-CD28 monovalent Fab’ antibody. Am J Transplant. 2012;12(10):2630–40. doi: 10.1111/j.1600-6143.2012.04164.x. Epub 2012/07/05. [DOI] [PubMed] [Google Scholar]
- 98.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nature Biotechnology. 2005;23(9):1126–36. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 99.Ignatovich O, Jespers L, Tomlinson IM, et al. Creation of the large and highly functional synthetic repertoire of human VH and Vkappa domain antibodies. Methods in molecular biology (Clifton, NJ) 2012;911:39–63. doi: 10.1007/978-1-61779-968-6_4. Epub 2012/08/14. [DOI] [PubMed] [Google Scholar]
- 100.Ville S, Poirier N, Branchereau J, et al. Anti-CD28 Antibody and Belatacept Exert Differential Effects on Mechanisms of Renal Allograft Rejection. 2016 doi: 10.1681/ASN.2015070774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wing JB, Ise W, Kurosaki T, et al. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity. 2014;41(6):1013–25. doi: 10.1016/j.immuni.2014.12.006. Epub 2014/12/20. [DOI] [PubMed] [Google Scholar]
- 102.Sage PT, Paterson AM, Lovitch SB, et al. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. 2014;41(6):1026–39. doi: 10.1016/j.immuni.2014.12.005. Epub 2014/12/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang CJ, Heuts F, Ovcinnikovs V, et al. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc Natl Acad Sci U S A. 2015;112(2):524–9. doi: 10.1073/pnas.1414576112. Epub 2014/12/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Riella LV, Liu T, Yang J, et al. Deleterious effect of CTLA4-Ig on a Treg-dependent transplant model. Am J Transplant. 2012;12(4):846–55. doi: 10.1111/j.1600-6143.2011.03929.x. Epub 2012/02/04. [DOI] [PubMed] [Google Scholar]
- 105**.Zhang R, Huynh A, Whitcher G, et al. An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest. 2013;123(2):580–93. doi: 10.1172/jci65013. Epub 2013/01/03. This work describes the cell-intrinsic role of CD28 in the regulatory of T regulatory cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang R, Borges CM, Fan MY, et al. Requirement for CD28 in Effector Regulatory T Cell Differentiation, CCR6 Induction, and Skin Homing. J Immunol. 2015;195(9):4154–61. doi: 10.4049/jimmunol.1500945. Epub 2015/09/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kirk AD, Burkly LC, Batty DS, et al. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nature medicine. 1999;5(6):686–93. doi: 10.1038/9536. Epub 1999/06/17. [DOI] [PubMed] [Google Scholar]
- 108.Larsen CP, Elwood ET, Alexander DZ, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381(6581):434–8. doi: 10.1038/381434a0. Epub 1996/05/30. [DOI] [PubMed] [Google Scholar]
- 109.Ferrer IR, Wagener ME, Song M, et al. Antigen-specific induced Foxp3+ regulatory T cells are generated following CD40/CD154 blockade. Proc Natl Acad Sci U S A. 2011;108(51):20701–6. doi: 10.1073/pnas.1105500108. Epub 2011/12/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Taylor PA, Friedman TM, Korngold R, et al. Tolerance induction of alloreactive T cells via ex vivo blockade of the CD40:CD40L costimulatory pathway results in the generation of a potent immune regulatory cell. Blood. 2002;99(12):4601–9. doi: 10.1182/blood.v99.12.4601. Epub 2002/05/31. [DOI] [PubMed] [Google Scholar]
- 111.Dodd-o JM, Lendermon EA, Miller HL, et al. CD154 Blockade Abrogates Allospecific Responses and Enhances CD4+ Regulatory T Cells in Mouse Orthotopic Lung Transplant. Am J Transplant. 2011;11(9) doi: 10.1111/j.1600-6143.2011.03623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nakayama Y, Brinkman CC, Bromberg JS. Murine Fibroblastic Reticular Cells From Lymph Node Interact With CD4+ T Cells Through CD40-CD40L. Transplantation. 2015;99(8):1561–7. doi: 10.1097/tp.0000000000000710. Epub 2015/04/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113*.Pinelli D, Wagener M, Liu D, et al. An Anti-CD154 Domain Antibody Prolongs Graft Survival and Induces FoxP3+ iTreg in the Absence and Presence of CTLA-4 Ig. Am J Transplant. 2013;13(11):3021–30. doi: 10.1111/ajt.12417. This paper describes the use of an anti-CD154 domain antibody to induce iTreg in the setting of transplantation. [DOI] [PMC free article] [PubMed] [Google Scholar]