

Summary
HDAC8 is a member of the family of Histone Deacetylases (HDAC) that catalyze the deacetylation of acetyl lysine residues within histone and non-histone proteins. The recent identification of novel non-histone HDAC8 substrates such as SMC3, ERRα and ARID1A indicates a complex functionality of this enzyme in cellular homeostasis. To discover additional HDAC8 substrates we developed a comprehensive, structure-based approach based on Rosetta FlexPepBind, a protocol that evaluates peptide-binding ability to a receptor from structural models of this interaction. Here we adapt this protocol to identify HDAC8 substrates using peptide sequences extracted from proteins with known acetylated sites. The many new in vitro HDAC8 peptide substrates identified in this study suggest that numerous cellular proteins are HDAC8 substrates, thus expanding our view of the acetylome and its regulation by HDAC8.
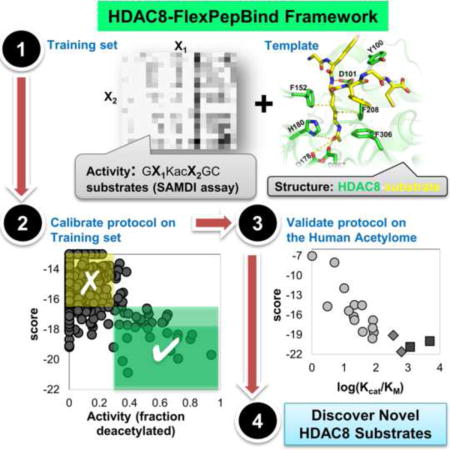
Graphical abstract
Introduction
Histone Deacetylases (HDACs) catalyze the hydrolysis of an acetyl group from Nε-acetylated lysines on many proteins. Through the process of deacetylation, HDACs alter the properties and activities of proteins and, in turn, affect cellular homeostasis. The best-characterized HDAC substrates are the N-terminal tails of histones where acetylation/deacetylation leads to an alteration in the chromatin structure and accessibility of the DNA to transcription factors and other proteins (Workman and Kingston, 1998). However, additional acetylated substrates have been reported that include transcription factors, chaperones, histone acetyltransferases (HATs) and histone deacetylases themselves (Glozak et al., 2005, Olson et al., 2014, Wolfson et al., 2013, Yang and Seto, 2008). Acetylome profiling in yeast has revealed overlap in regulation of diverse processes by the HDAC sirtuin and the HAT Gcn5, established by similar substrate profiles (Downey et al., 2015).
While non-histone substrates had been reported for other HDACs, no such substrates were known until recently for HDAC8 (Olson et al., 2014, Wolfson et al., 2013). HDAC8 has been invoked as a potential therapeutic target due to its association with a growing number of diseases (Chakrabarti et al., 2015). Because of this, determining the HDAC8 substrate set is a priority in the field. HDAC8 is proposed to regulate p53 levels (Yan et al., 2013), participate in skull morphogenesis (Haberland et al., 2009) and function as a key factor in smooth muscle contractility (Waltregny et al., 2005). Furthermore, various HDAC8 inhibitors induce apoptosis in Lymphoma cell lines (Balasubramanian et al., 2008), implying a role for HDAC8 in tumorigenesis in some tissues. Wilson et al. first proposed that HDAC8, together with Sirt-1 and p300, forms an acetylation switch that modulates the transcriptional activity of Estrogen-Related receptor (ERR α), and importantly, HDAC8 was found to catalyze deacetylation of the non-histone protein ERR α in vitro (Wilson et al., 2010). The best validated substrate of HDAC8 is SMC3 - a subunit of the cohesin complex that mediates sister chromatid cohesion. Mutations in SMC3, SMC1 and HDAC8 have been identified in patients with the Cornelia de Lange syndrome (CdLS), which is a genetic disease where patients suffer from a number of symptoms including mental retardation and facial deformity (Deardorff et al., 2012). Misregulation of cohesin complex proteins, including aberrant acetylation, is proposed to lead to this disease (Deardorff et al., 2012). Biochemical and structural characterization of HDAC8 mutants associated with CdLS spectrum disorders detected for all an effect on either protein stability or/and catalytic activity of HDAC8 (Decroos et al., 2015). A recent mass-spectrometry study has revealed an additional series of proteins that increased acetylation in cells when HDAC8 is specifically inhibited, suggesting that they are HDAC8 substrates (Olson et al., 2014). Reactivity of HDACs with libraries of acetylated peptides have identified amino acid preferences, but have not led to the identification of in vivo substrates (Riester et al., 2007, Gurard-Levin et al., 2009, Gurard-Levin et al., 2010).
HDAC8 is catalytically active as a monomer in vitro and has been proposed to function as a monomer or in small complexes in vivo (Yang and Seto, 2008), in contrast to other HDACs that function only within the context of high molecular weight, multi-protein complexes [reviewed in (Wolfson et al., 2013)]. From a structural biology perspective, HDAC8 is therefore the best choice among HDACs as a starting point for the study of deacetylation and its biological roles. The structure of catalytically inactive HDAC8 (Y306F) bound to a p53-derived diacetylated peptide substrate has defined the details of substrate binding by HDAC8 (Figure 1A), in particular the critical role of the highly conserved D101 at the rim of the active site, which establishes two specific hydrogen bonds with the substrate and whose mutation results in a complete loss of enzyme activity (Vannini et al., 2007, Dowling et al., 2008).
Figure 1.
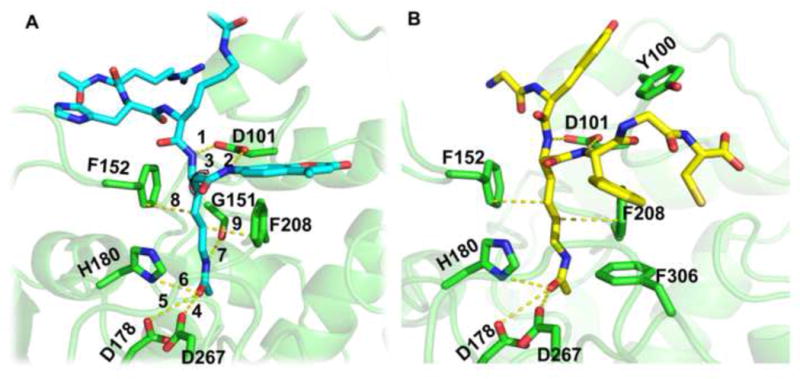
Structural features of the HDAC8-substrate interactions. (A) Key HDAC8 receptor - peptide substrate interactions imposed during optimization, including the tethering of the backbone of the acetylated lysine (i.e. the second acetylated lysine in the RHKacKac-coumarin substrate) to D101 and the acetyl group to the Zn2+ coordination site (see Text and Table S1). (B) Starting structure used in this study, generated by optimization of the most reactive substrate in our training set, Ac-GYKacFGC-NH2 (see Table S2). Structures are based on PDB id 2v5w (Vannini et al., 2007), and rendered using PyMOL (http://www.pymol.org).
Here we develop a structure-based computational high-throughput approach for the discovery of novel substrates of Zn2+-bound HDAC8 using Rosetta FlexPepBind (London et al., 2011). We used this in silico approach to identify HDAC8 substrate preferences and combined this method with experimental in vitro validation to further optimize the algorithm. Our study reveals that HDAC8 can bind and catalyze deacetylation of many acetylated peptides with sequences corresponding to cellular, non-histone proteins, thereby opening a new window to the functional role of HDAC8 in cells.
Results
For accurate and effective structure-based characterization of HDAC8 substrates, we first calibrated the Rosetta FlexPepBind algorithm to distinguish between HDAC8 substrates and non-substrates on a library of hexapeptides for which we measured HDAC8-catalyzed deacetylation rates. With the optimized protocol we then performed a large-scale screen of all reported known acetylated proteins to identify sites that can be deacetylated by HDAC8. Peptides corresponding to predicted targets of functional interest were shown experimentally to undergo deacetylation catalyzed by HDAC8.
Definition of critical features for substrate-HDAC8 interactions
The first step in the development and validation of a structure-based protocol for the identification of new HDAC8 substrates requires the definition of a template for substrate-enzyme binding, and subsequent modeling of different potential substrate peptides into this template binding site. This simulated binding allows evaluation of the ability of HDAC8 to bind various peptide sequences in a catalysis-competent conformation. Critical features of the peptide-HDAC8 binding model were defined based on the crystal structure of the catalytically inactive Y306F HDAC8 mutant bound to a p53-derived peptide AcRHKacKac-(7-methoxycoumarin-4-yl) methyl] [the acetylated lysine interacting with the active site residues is highlighted in bold and italics; protein data bank (PDB) id 2v5w (Vannini et al., 2007); see Figure 1A and Table S1]. The nine constraints used in our model include tethering the peptide backbone of the acetylated lysine to the receptor via two hydrogen bonds to the side chain of HDAC8 residue D101, enforcing a cis dihedral angle conformation between the two adjacent backbone nitrogen atoms (constraints 1–3). The side chain of D101 has been shown to play a critical role for HDAC8 substrate binding and activity (Vannini et al., 2007, Dowling et al., 2008). In addition, we tether the acetyl group of the lysine to the position of the Zn2+ ion using Zn2+-binding residues D178, H180 and D267 (constraints 4–6; the Zn2+ ion is not included in the simulations). Finally, the backbone oxygen atom of G151 forms a hydrogen bond with the nitrogen atom of the acetyl lysine, and stacking interactions between the side-chain rings of F152 and F208 residues and the acetylated lysine side-chain were fixed (constraints 7–9). This set of constraints locks the acetyl lysine side chain within the binding pocket while allowing variable interactions between HDAC8 and the remainder of the peptide.
After constraining the structure, the protocol was optimized in two stages. Initially we used as the starting structure a template where the missing residues on the C-terminal side [replacing the (7-methoxycoumarin-4-yl) methyl moiety in the starting structure] were added in extended conformation. Subsequently we found that our predicted in silico results better matched experimental data when peptides were threaded onto an optimized structure of HDAC8 bound to the hexapeptide Ac-GYKacFGC-NH2, the substrate of Set_GX1KacX2GC with the highest activity (Figure 1B; see below and Methods). Below we report results from this optimized protocol, unless stated otherwise (corresponding results from the initial protocol are provided in the Supplementary Materials).
Calibration and validation of protocol on hexapeptide substrates
We measured HDAC8 deacetylation activity for a set of 361 hexapeptide substrates, Set_GX1KacX2GC (See DataSets in the Methods section). This set (Table S2) was used to calibrate our protocol for the reliable identification of HDAC8 substrates. The two main parameters evaluated during the protocol optimization were the sampling protocol (e.g. FlexPepDock refinement vs. minimization) and the scoring measure (e.g. peptide score vs. interface score) (see Methods for more details).
In the initial stage of calibration, we used a template with a peptide with an extended C-terminus (see above), minimized the threaded sequence, and ranked the different peptides using peptide score. This initial protocol was developed on half of Set_GX1KacX2GC and evaluated on the second half (Table S2). It was able to effectively eliminate a large fraction of non-binders (inactive peptides), and thus performed decently at binary distinction between strong substrates and non-substrates. Substrates were best distinguished using > 0.34 fraction deacetylation as threshold (p-values of 4.5e–10 and 1.3e–05 for Kolmogorov-Smirnov tests on the first and second halves, respectively; see Figure S1). Similar results were obtained for other ranking schemes used (data not shown).
However, using this protocol we also observed a high rate of false negatives, potentially because well-scoring conformations in the substrate-binding site were missed due to inadequate sampling. After evaluation of performance of this initial protocol on a set of potential HDAC8 targets (see below), we further improved our initial protocol by incorporating several changes (see Methods). In particular, a better starting structure was used as a template, by extensive optimization of a known substrate, GYKacFGC (see Figure 1B and Methods). In addition, we found that the interface score between enzyme and substrate (I_sc), rather than the peptide score (Pep_sc), provides the best discrimination and correlation to experimental values (see below).
Modeling the binding of the peptide substrates in Set_GX1KacX2GC using this optimized protocol clearly distinguishes substrates from non-substrates (p-value of 2.9e–14 for Kolmogorov-Smirnov - KS test, Figure 2A; see Figure S2 for corresponding results using other scoring measures). We note that the few false positive hexapeptide substrates with good scores (< −18.0; the stringent threshold defined below) that are nevertheless not deacetylated, include all a Tryptophan residue at position X1. Predicted values also correlate well with the experimentally measured HDAC8-catalyzed deacetylation (R=−0.33, p-value=5.1e–06). Application of our approach to a related set, Set_GRKacX2X3C [(Gurard-Levin et al., 2010); see Table S3 and Methods], demonstrates the robustness of our approach to the prediction of the influence of amino acids at specified positions to substrate strength (p-value of 1.7e–07 for KS test, and correlation coefficient of R=−0.6, p-value=6.2e–36; Figure 2B).
Figure 2.
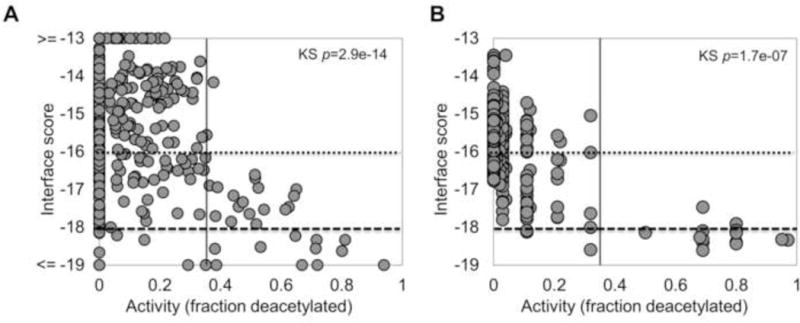
Performance of the optimized HDAC8 FlexPepBind protocol on two different sets of hexapeptides, Set_GX1KacX2GC (A), and Set_GRKacX2X3C (B) measured with a mass-spectrometry-based assay, SAMDI (see Methods). The binding score (y-axis) vs. substrate activity (x-axis) plots show good discrimination between substrates (vertical line; >0.34 fraction of deacetylation) and non-substrates using our measure of the ability of different substrates to bind to HDAC8 in a catalysis-competent conformation. The horizontal lines indicate the thresholds for predicted substrate definition derived from plot (A): a stringent cutoff of −18.0 detects 35% of the substrates (True Positives) at the expense of 4% selected non-substrates (False Positives), while the second cutoff of −16.5 identifies more substrates (91%), but also more wrong hits (19%).
Based on the results on these two datasets, we defined two different scoring thresholds: one stringent threshold (interface score Rosetta Energy Units, REU −18.0) to select high-confidence binders and another less strict threshold (interface score REU −16.5) which identifies most binders but at the expense of a larger fraction of false positives (See Figure 2 and Figure 3).
Figure 3.
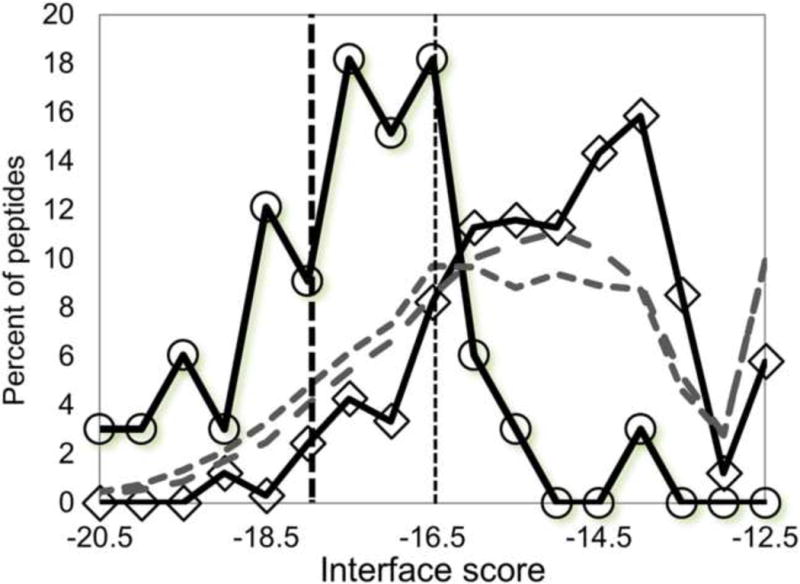
Histogram of binding scores for substrates (circles) and non-substrates (diamonds) of Set_GX1KacX2GC (at least 0.34 deacetylation, see Figure 2), as well as two large scale screens of peptides representing all known acetylated sites (1) PhosphoSitePlus release 6/2012: 3048 unique hexapeptides representing 3191 reported sites (short dashed grey line), and (2) PhosphoSitePlus release 11/2015: 17904 unique hexapeptides representing 20397 reported sites (long dashed grey line). The thresholds for predicted substrates defined in Figure 2 are indicated by vertical lines. Using the stringent threshold of −18.0, the large-scale screen identifies many additional potential substrates.
Large-scale screens for new HDAC8 targets
Using our protocol we screened for substrates among all reported acetylated sites in the human proteome reported in the PhosphoSitePlus database (Hornbeck et al., 2004, Hornbeck et al., 2015) to identify novel HDAC8 substrates. We modeled the ability of hexapeptide fragments containing the known acetylated sites (XXKacXXX, X=all possible amino acids) to bind HDAC8 in a deacetylation-compatible conformation. Among the 3191 reported acetylated sites (PhosphoSitePlus release 6/2012; See Datasets in the Methods section), 418 passed the stringent threshold (interface score = −18.0) (Figure 3).
Experimental validation of de-novo predictions
In order to validate potential novel HDAC8 targets, and to test our prediction scheme, we compiled two sets of peptides at different stages of our project. The first, Novel_candidate_set1, consists of 26 peptide sequences selected after screening the PhosphoSitePlus database using our initial protocol (Table 1). Novel_candidate_set1 consists of 10 top-ranking peptides (Pep_sc ≤ −6; * in Table 1), 10 additional acetylated peptides that occur in the same proteins of the top-ranking hits, but are predicted not to undergo HDAC8-catalyzed deacetylation (# in Table 1), and finally 6 additional potential well-ranking substrates of further biological interest. Out of these 26 peptides, the reactivity of HDAC8 was experimentally characterized for 20 soluble acetylated peptides (Ac-XXKacXXX-COO−).
Table 1. Experimental validation of proposed novel substrates in Novel_candidate_set1.
Ac-XXKacXXX-COO− peptides were assessed [2.2 fold weaker than amidated C-termini used in Tables 2&3 below (Wolfson, 2014)]. Peptides are sorted and grouped according to enzymatic activity (kcat/KM): reactive (>1000), reasonable (>100), mild (>10), and poor.
| Peptide |
kcat/KM M−1s−1 |
Pep_sc(a) | Min. I_sc(b) |
Protein (Uniprot ID) | ||
|---|---|---|---|---|---|---|
| VS | K350 | GPF | 61.5 | −9.6 | −17.5 | Histone acetyltransferase KAT6A (Q92794)* |
| SF | K769 | SDQ | 12.3 | −7.7 | −16.6 | E3 ubiquitin-protein ligase TRIM33 (Q9UPN9)* |
|
| ||||||
| VS | K93 | GTL | 8.5 | −7.7 | −16.6 | Histone H1.5 (P16401)* |
| DI | K42 | YPL | 7.9 | −7.6 | −18.6 | Cytochrome b5 reductase B5R (P00387)* |
| DH | K604 | TLY | 7.5 | 174 | −16.5 | KAT6A (Q92794) # |
| SG | K256 | YDL | 6.6 | −7.2 | −17.1 | Alpha-enolase (P06733) |
| DS | K1583 | NAK | 5.2 | −4.4 | −17.1 | CREB Binding Protein CBP (Q92793) |
| KP | K209 | AAK | 4.1 | 145 | −15.4 | Histone H1.5 (P16401) # |
| PG | K117 | GVK | 3.7 | −7.6 | −14.4 | Transcription factor E2F1 (Q01094)* |
| FT | K252 | DHL | 3.2 | 965 | −16.1 | TRIM33 (Q9UPN9)# |
| KG | K953 | TAQ | 3.1 | 2.6 | −13.9 | TRIM33 (Q9UPN9) # |
| KK | K1588 | NNK | 1.7 | −5.2 | −15.6 | CBP (Q92793) |
| II | K593 | DGE | 1.5 | −7.5 | −16.8 | Apoptosis-inducing factor 1 AIFM1 (O95831)* |
| TQ | K755 | QEQ | 1.5 | −7.6 | −15.1 | Nucleoprotein TPR (P12270)* |
| GV | K168 | KVA | 1 | 624 | −15.1 | Histone H1.5 (P16401) # |
|
| ||||||
| DD | K169 | YTL | 0.67 | −7.6 | −16.3 | Signal peptidase complex subunit 2 SPC25 (Q15005)* |
| SG | K345 | KGQ | 0.66 | −4.5 | −16.0 | SWI/SNF complex subunit SMARCC1 (Q92922)* |
| SF | K109 | LNK | 0.61 | 16.2 | −17.1 | Histone H1.5 (P16401) # |
| NQ | K748 | LTA | < 0.6 | 974 | −14.3 | Nucleoprotein TPR (P12270) # |
| VG | K815 | SVS | < 0.6 | 1750 | −13.9 | KAT6A (Q92794) # |
|
| ||||||
| ES | K896 | FQQ | NA(c) | −9.6 | −19.4 | Retinoblastoma-associated protein (P06400)* |
| VS | K64 | AVE | NA | −5.1 | −16.6 | Alpha-enolase (P06733) |
| MG | K60 | GVS | NA | −5.2 | −13.2 | Alpha-enolase (P06733) |
| FS | K355 | VRT | NA | 1792 | −15.8 | Histone acetyltransferase KAT6A (Q92794) # |
| MI | K548 | HLE | NA | 186 | −17.1 | Retinoblastoma-associated protein (P06400) # |
| IV | K428 | EVE | NA | 1430 | −15.3 | Nucleoprotein TPR (P12270) # |
Predicted strong substrates (according to original protocol)
Predicted non-substrates in proteins for which we found predicted strong substrates (*)
Score of initial prediction scheme
Score of optimized protocol
NA-Not Assessed: peptide was not soluble and could not be assessed
Efficient enzymes in the cell typically show catalytic values of kcat/KM >104 M−1s−1 (Fersht, 1985). Since the reactivity of HDAC8 with peptide substrates is up to 1,000-fold lower than protein substrates (Wolfson, 2014), peptides with values of kcat/KM > 10 M−1s−1 may be cellular substrates. For discussion purposes we have grouped the peptides by their kcat/KM values: reactive (>1000), reasonable (>100), mild (>10), and poor. Our top-ranking substrate, Kac350 of Lysine Acetyl Transferase KAT6 (kcat/KM=61, M−1s−1) is deacetylated efficiently by HDAC8 and may be a cellular target. Overall however, the reactivity of HDAC8 with the peptides detected with our initial protocol is low and the overall correlation between the predicted score and experimental activity is weak (R=−0.39, p-value=0.083), due to many false positive predictions (Figure 4A).
Figure 4.
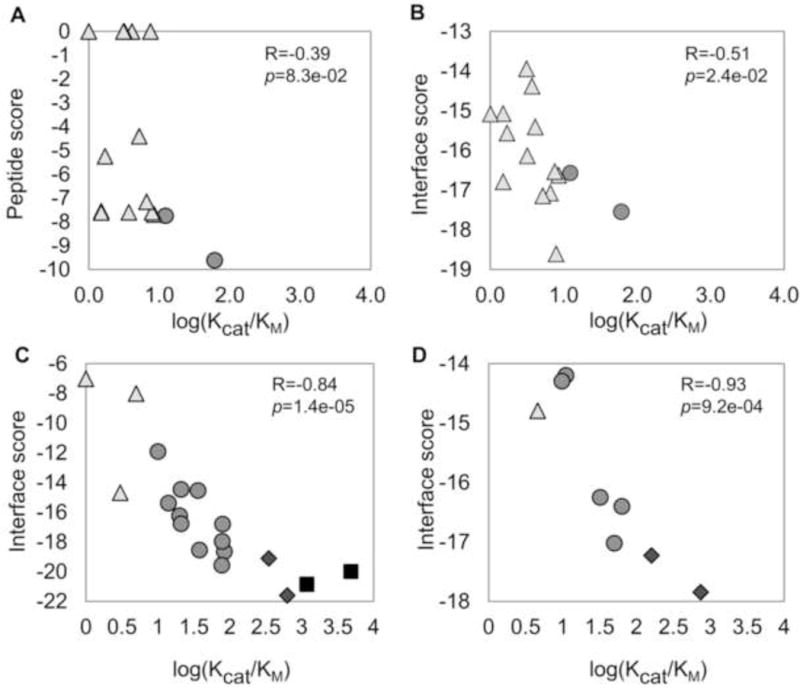
Correlation between predicted binding to HDAC8 and experimental deacetylation of protein substrates. (A) Novel_candidate_set1, using the initial protocol (R=−0.39). All following correlations were obtained with the optimized protocol: (B) Novel_candidate_set1 (R=−0.51), (C) Novel_candidate_set2 (R=−0.84) and (D) Proteometargets_set3 (R=−0.93). Substrate Strength: Reactive (
 ), reasonable (
), reasonable (
 ), mild (
), mild (
 ) and poor (△).
) and poor (△).
We revisited the starting conformation as well as the scoring scheme in a new step of calibration (see above and Methods), considering both this new, quantitative experimental information from Novel_candidate_set1 as well as the previous sets Set_GX1KacX2GC and Set_GFKacX2X3C (see above). This improved the correlation for Novel_candidate_set1 (R=−0.51, p-value=0.023; Figure 4B).
In order to validate the new version of our prediction scheme, we performed a second screen of the PhosphoSitePlus database with the optimized protocol. For Novel_candidate_set2, we chose a total of 19 peptides with a range of different predicted binding strengths according to scoring measures I_sc and/or Pep_sc_noref, to identify the preferred scoring scheme (Table 2 & Supplementary Table S4). In this library, the peptides contained an acetylated N-terminus and an amino C-terminus (Ac-XXKacXXX-NH2) to increase peptide reactivity [a 2.2 fold increase in reactivity was observed for the KAT6A K350 substrate using peptide with an amino vs. carboxylated C-terminus, respectively (Wolfson, 2014)]. Experimental validation of this set identified the most efficient peptide substrate observed to date (corresponding to Zinc finger protein 318, peptide FGKacFSW at position 1275, kcat/KM=4826; assigned one of the top-scores of −20.0, well beyond the stringent threshold), as well as a substrate derived from EF1a1 (kcat/KM =1176), and two other substrates with kcat/KM ≥300. In addition, we observed an excellent correlation between prediction and experiment using the interface score measure (R=−0.84, p-value=1.3e–05, Figure 4C).
Table 2. Experimental validation of proposed novel substrates in Novel_candidate_set2.
Ac-XXKacXXX-NH2 peptides were assessed.
| Peptide |
kcat/KM M−1s−1 |
Min. I_sc |
Protein (Uniprot ID) | ||
|---|---|---|---|---|---|
| FG | K1275 | FSW | 4826 | −20 | Zinc finger protein 318 Znf 318 (Q5VUA4) |
| SF | K55 | YAW | 1176 | −20.8 | EF1 alpha 1 (P68104) |
|
| |||||
| LS | K747 | FLR | 630 | −21.6 | CAD protein (P27708) |
| LG | K1017 | FRR | 349 | −19.1 | La-related protein 1 LARP1(Q6PKG0) |
|
| |||||
| IS | K504 | YDR | 83.5 | −18.6 | Msh6 (DNA mismatch repair) (P52701) |
| RL | K713 | YSQ | 79.5 | −16.8 | SMC1A (Q14683) |
| TW | K78 | ANF | 77 | −19.6 | Interferon regulatory factor 2 IRF2 (P14316) |
| SL | K269 | EFY | 78 | −18 | Aldehyde dehydrogenase ALDHIII (P30838) |
| KI | K31 | RLR* | 38 | −18.5 | 60S ribosomal protein L7 (P18124) |
| KG | K878 | DAE | 36 | −14.5 | Nuclear receptor corepressor 2 N-CoR2 (Q9Y618) |
| SG | K149 | YYY | 21 | −16.8 | E3 SUMO-protein ligase CBX4 (O00257) |
| GG | K11 | AFG | 21 | −14.5 | Allograft inflammatory factor 1 AIF1 (P55008) |
| SG | K516 | YFA | 20 | −16.2 | AMP deaminase 2 AMPD2 (Q01433) |
| TF | K8 | GVD | 14 | −15.4 | Hematological and neurological expressed 1 protein HN1 (Q9UK76) |
| RK | K292 | GEP | 10 | −11.9 | Cellular tumor antigen p53 (P04637) |
|
| |||||
| SE | K1970 | PEK | 5 | −8 | DNA-dependent kinase, cat. subunit PKcs (P78527) |
| FV | K358 | AFA | 3 | −14.7 | Succ.-semialdehyde dehydrogenase ALDH5A1 (P51649) |
| GI | K371 | PFL | 0 | −7 | Actin-binding protein anillin (Q9NQW6) |
| SQ | K359 | EED | NA | −14.6 | SWI/SNF complex subunit SMARCC1 (Q92922) |
This acetylation site was reported in the PhosphoSitePlus release of 6/2012 but not in 11/2015. Legends as in Table 1.
In parallel to our efforts, the Holson group performed an unbiased, proteomic approach to identify substrates of HDAC8. Our predictions show excellent correlation with the reactivity of peptides corresponding to the identified protein substrates, termed here Proteometargets_set3 (Table 3 & Figure 4D; R=−0.93, p-value=2.8e–04). Among these substrates, the values for kcat/KM for corresponding acetylated peptides ranged from 4 – 740 M−1s−1 (Olson et al., 2014) (i.e., poor to reasonable), corresponding to interface scores of −14 to −18. This highlights the complementarity of the two approaches in the identification of HDAC8 substrates.
Table 3. Predicted values for substrates identified in Proteometargets_set3.
(Olson et al., 2014) Ac-XXKacXXX-NH2 peptides were assessed.
| Peptide(a) |
kcat/KM M−1s−1 |
Min. I_sc |
Protein (Uniprot ID) | ||
|---|---|---|---|---|---|
| KLIS | K1808 | FDKL | 740 ± 36 | −17.8 | ARID1A (chromatin remodeling) (O14497) |
| STPV | K292 | FISR | 160 ± 27 | −17.2 | CSRP2BP (HAT) (Q9H8E8) |
|
| |||||
| RVIGAK | K106 | DQY | 63 | −16.4 | SMC3 (mitosis) (Q9UQE7) |
| KRILH | K687 | LLQN | 50 ± 5.0 | −17.0 | NCOA3 (transcription) (Q9Y6Q9) |
| SKIQ | K3579 | QLDQ | 32 ± 5.7 | −16.2 | MLL2 (methyltransferase) (O14686) |
| KLGG | K1087 | QRAA | 11 ± 1.4 | −14.2 | RAI1 (transcription) (Q7Z5J4) |
|
| |||||
| KLSG | K167 | EING | 9.8 ± 1.9 | −14.3 | SRSF5 (splicing) (Q13243) |
| LGDG | K387 | MKS | 4.6 ± 0.10 | −14.8 | THRAP3 (splicing) (Q9Y2W1) |
| TEIG | K54 | TLAEK | 4.1 ± 0.30 | −14.0 | ZRANB2 (splicing) (O95218) |
The number of known acetylated sites in the human proteome reported in the PhosphoSitePlus database has increased over time; from 3191 sites (06/2012 release) in our initial screens, to 20397 sites in the most recent release (11/2015). This new dataset contains a similar proportion of potential novel targets that pass the stringent threshold (Figure 3), suggesting many additional biologically relevant substrates that await yet to be characterized (scores of all hexamer peptide stretches around known acetylated sites have been included for both datasets in the Appendix).
Discussion
We have described a structure-based protocol to identify peptides that are efficiently deacetylated by HDAC8. This approach is based on modeling hexameric peptides with a central acetylated lysine residue into the substrate-binding site of the HDAC8 structure. Using a combination of a computational screen of known acetylated proteins, and in vitro experimental validation of corresponding peptides, we detected novel HDAC8 substrates that were not identified by proteomic approaches, nor by simple, sequence-based screens (Tables 1–3; Figure 4). In the following we discuss the strongest of these proposed substrates. By integrating information about the location of the deacetylation site relative to globular domains in the protein, known adjacent additional modifications [from PhosphoSitePlus (Hornbeck et al., 2004, Hornbeck et al., 2015)], as well as reported cancer-related sites, and inspection of protein partners [from the Biogrid database (Chatr-Aryamontri et al., 2015), unless noted otherwise], we obtain a picture of potential functional regulatory roles of deacetylation in these proteins. This highlights in particular opportunities for the crosstalk of acetylation with other post-translational modifications, as well as the modulation of binding of target proteins to partners, including protein, DNA and RNA.
The strongest HDAC8 peptidic substrate identified so far: ZNF318, K1275 [kcat/KM=4826]
Zinc Finger Protein 318 acts as a co-repressor of the Androgen Receptor and is involved in spermatogenesis (Tao et al., 2006). The FGKac1275FSW sequence is located right after two Zn Fingers of this otherwise largely unstructured protein, and is therefore expected to be accessible to bind to HDAC8 in vivo within the full protein context. The fact that an adjacent mutation, G1274R, has been found in breast cancer (Sjoblom et al., 2006), and that an adjacent position, S1277, undergoes phosphorylation may indicate that this region functions as a regulatory hotspot. The connection between phosphorylation and acetylation is reinforced by the observation that both HDAC8 and ZNF318 have been reported to bind to PPP1CC phosphatase (Gao et al., 2009, Esteves et al., 2013). Previously identified protein partners of ZNF318 include Sirt7 (Tsai et al., 2012), and HDAC2 as well as the Androgen Receptor (AR), with whom ZNF318 associates to prevent its transforming activity (Tao et al., 2006). To summarize, deacetylation of ZNF318 by HDAC8 could very well play an important role in the cross-talk between phosphorylation, acetylation, and the resulting modulation of cellular homeostasis.
Cross-regulation of translation and transcription: EF1a1 and LARP1
One of the most efficient peptide substrates detected in this study, SFKac55YAW [kcat/KM=1176], resides in a conserved, accessible loop of the GTP binding domain of Elongation Factor 1 alpha 1 (EF1α1). The same position has also been shown to undergo methylation (Dever et al., 1989), indicating a possible role for HDAC8 in the cross-regulation between acetylation and methylation. EF1α1 promotes GTP-dependent binding of aminoacyl-tRNA to the A-site of ribosomes during protein biosynthesis, but acts also as T helper 1 (Th1) - specific transcription factor of interferon gamma. This important functional role of EF1α1 in both translation as well as transcription (Vera et al., 2014) may provide evidence for HDAC8 regulation of functions related to gene regulation beyond chromatin conformation.
The La-related protein 1 (LARP1) belongs to the LARP family involved in transcriptional and translational regulation. LARP1 binds to 5′TOP pyrimidine-rich UTR mRNA sequences, as well as to PolyAdenylate Binding Protein (PABP) via its unique C-terminal DM15 HEAT repeats. The specific role of LARP1 in translational regulation depends on the context: both phosphorylation and dimerization have been suggested to modulate LARP1 activity (Stavraka and Blagden, 2015). We suggest that acetylation (and deacetylation) might be an additional player: the acetylated lysine substrate, LSK1017FRR [kcat/KM=349], is located adjacent to the suggested RNA binding site in the recently solved structure of these HEAT repeats (Lahr et al., 2015), and its modification might thus affect binding to RNA, or to PABP.
Cross-regulation of KATs and HDACs: KAT6A and CAD
For lysine acetyltransferase KAT6A (also called MYST3 and MOZ), we identified two potential HDAC8 lysine substrates: K350 and K604 (kcat/KM=61.5 and 7.5, respectively, for charged peptide substrates). VSKac350GPF is located in an unstructured region right after two Zinc finger domains (similar to the ZNF318 substrate described above). Not much is known about the potential impact of deacetylation (and acetylation) at this position. However, evaluation of further targets in the same protein reveals a second potential deacetylation site in the KAT6A sequence, DHKac604TLY. This highly conserved lysine has previously been shown in closely related MYST proteins to undergo autoacetylation that is critical for acetyltransferase activity (Yuan et al., 2012). Thus, KAT6A and HDAC8 could participate in a double feedback loop that is potentially responsible for regulating the acetylation level of proteins inside the cell and the nucleus; HDAC8 would not only deacetylate the substrate (e.g. histone tails), but also shut down re-acetylation catalyzed by KAT6A, by inactivating the latter through deacetylation of K604. Indeed, a genome-wide synthetic lethality screen of HDACs and other proteins revealed that a knockdown of HDAC8 can partly be rescued by MYST3 and other related acetyl transferases (Lin et al., 2012). A similar example of cross-regulation between HATs and HDACs has been observed for the HAT hMOF and the corresponding SIRT1 deacetylase (Lu et al., 2011).
The CAD enzyme (Carbamoyl-phosphate synthase - Aspartate carbamoyltransferase – Dihydroorotase) catalyzes several enzymatic steps of the pyrimidine synthesis pathway. The identified peptide substrate in CAD including K747 [kcat/KM=630] is located in a linker region that connects two enzymatic domains, suggesting that this site may be accessible to HDAC8 in the context of the full protein. Interestingly, CAD has been reported to interact with several HDACs, including HDACs 5, 6, 11, and Sirt7, as well as with the KAT Nuclear receptor coactivator 2, CoA-2, and p53, a suggested HDAC8 target. These binding partners highlight a probable cross-regulation achieved by both interactions and cross-modification of KATs and HDACs, as also described for KAT6A above.
Functional impact of deacetylation on DNA mismatch repair: Msh6
According to the structure of the Mutsα (Msh2-Msh6)-DNA complex [PDB id 2o8b (Warren et al., 2007)], the substrate lysine of HDAC8 in Msh6 identified in this study, K504 [kcat/KM=83.5], is located adjacent to the DNA, suggesting significant functional consequences of the addition of a positive charge upon its deacetylation by HDAC8, perhaps enhancement of DNA binding of Muts α, and consequently increase in mismatch repair. Similar modulation of DNA binding affinity upon acetylation has been reported for the C-terminal domain of p53, where acetylation of residues 372, 373, 381 and 382 resulted in significant decrease of DNA binding affinity (Friedler et al., 2005). Our protocol predicts three among these residues to be strong substrates for HDAC8 (residues 372, 381 and 382, with score values of −18.3, −18.3, and 18.0, respectively). And indeed, it was recently shown that HDAC8 inhibition specifically targets Inv(16) acute myeloid leukemic stem cells by restoring p53 acetylation (Qi et al., 2015).
Structure-based and proteomic approaches are complementary and identify distinct substrates
Application of HDAC8 FlexPepBind to a set of targets identified by the Holson group (Olson et al., 2014) (Figure 4C) showed good correlation between the predicted score and the substrate efficiency measured in vitro (R=0.84). Importantly however, our approach also identified proteins where the corresponding peptide substrates have considerably higher in vitro catalytic activities for deacetylation that were not detected by the proteomic study, including the most efficient substrate ever identified (Table 2). In addition, while one of the top putative substrates, ERR1α (Wilson et al., 2010), was not detected by the proteomic study, it ranks well according to HDAC8-FlexPepBind (I_sc=−18.5). Both approaches thus provide distinct information about the HDAC8 deacetylome and should optimally be used in combination.
We note that not all substrates identified by the proteomic assay showed enzymatic activity in a subsequent in vitro enzymatic assay of corresponding peptide substrates (Olson et al., 2014). In particular more than half (5/9) of the substrates contain a glycine residue preceding the acetylated lysine, i.e. GKac, but only minor in vitro enzymatic activity was measured for this subset, indicating additional possible biases of this approach [Table 3 and (Olson et al., 2014)]. This method identifies changes in acetylation levels upon addition of an HDAC8 inhibitor; however, the change could be due to a downstream effect rather than demonstrating deacetylation catalyzed directly by HDAC8. Alternatively, the reactivity of these proteins with HDAC8 in vivo may be mediated by additional cofactors, including other binding protein partners.
The benefit of a structure-based approach compared to simple sequence-based screens
Sequence based computational approaches have been widely used to predict novel substrates for different enzymes (Maurer-Stroh and Eisenhaber, 2005, Rawlings et al., 2014, Dinkel et al., 2014). Even though those methods are computationally less expensive and overall perform very well, structure based approaches can provide a detailed understanding at the atomic level and consequently a more complete picture of the substrate space (London et al., 2011). A retrospective analysis of the targets evaluated in this study (Tables 1–3) reveals that all but one of the seven most reactive substrates (kcat/KM values >100) would have been detected by screening for the sequence KacF/Y, i.e. for an acetylated site followed by phenylalanine or tyrosine. The additional substrate contains a SKacG combination that shows reasonable deacetylation according to Set_GX1KacX2GC (0.35, see Table S2 and Methods), and could therefore have also been identified using sequence information only. However, these sequence-based screens identify a large number of sites, many of which are likely false positive substrate predictions. Furthermore, among the 21 substrates identified by our screen and the proteomic assay with kcat/KM values in the range of 10.0–100.0 (Tables 1–3), six contain tyrosine, but none phenylalanine, at the position following the acetylated lysine, including a peptide from the best-validated in vivo substrate, SMC3. Moreover, only two contain a preferred X1KacX2 combination. Still, most of these acetylated peptides are well-scored by our scheme, 5 and 13 peptides passing the stringent and lenient threshold of −18.0 and −16.5, respectively.
While neither the proteomic, the sequence-based, nor our structure-based approach specifically selects strong targets, the present protocol is an excellent tool for HDAC8 target identification. We have detected the most reactive HDAC8 peptide targets to date, as well as a range of milder targets of potential biological significance. Thus, our approach complements both the proteomic as well as sequence-based approaches, thereby providing an increasingly comprehensive picture of the HDAC8 deacetylome.
From peptide substrate to the full protein
Our protocol performs impressively well at discriminating non-binding peptides from those that can bind HDAC8 and consequently undergo deacetylation. However, is there a direct relation between in vitro deacetylation of the short peptide, as measured in this present study, and the in vivo deacetylation of that site within the context of the full protein in the cell? Several additional features might need to be taken into consideration, including spatial accessibility of the target site to HDAC8 (Sirota et al., 2015). The acetylation/deacetylation site may reside within either an unstructured or structured region, and function as a signal or modulate protein activity. Disordered regions will be highly favored as they can easily adopt an HDAC8-compatible conformation (e.g. K1275 in ZNF318 discussed above). For sites located in and substantially buried in ordered domains, (e.g. K604 in KAT6A discussed above), the structural context might very well affect HDAC8 activity. Furthermore, the local structure propensity of the peptide, in particular its tendency to adapt, e.g., an ordered alpha helix, might reduce HDAC8 activity, as it will be more difficult to adopt a catalysis-compatible conformation (e.g. to form backbone mediated hydrogen bonds with D101; see Figure 1A). This might be the reason why the best substrate identified in this study (FGKac1275FSW [kcat/KM=4826], predicted to lie in a disordered region in ZNF 318) is considerable more active than the following two substrates (Ef1 alpha 1 SFKac55YAW [kcat/KM=1176] & CAD protein LSKac747FLR [kcat/KM=630], both predicted to adapt an ordered, helical conformation) even though the latter two have much better scores (−20.0 vs. −20.8 & −21.6) (Table 2 and Figure 4C; See Supplementary Table S5 for secondary structure propensities of the different peptide substrates in this study). Further parameters such as the local substrate protein concentration, as well as other cofactors may modulate the effective reactivity with HDAC8. Finally, other HDAC isozymes may also recognize a given acetylated site. All these factors will also influence experimental approaches, and new, more sensitive methods for the measure of protein interactions, in particular transient interactions, within the cell, need still to be developed [e.g. (Roux et al., 2013)].
Chromatin modifiers are increasingly recognized as major regulatory factors. In addition to the blooming research on the regulatory role of different chromatin states, and the consequent considerable functional effects of these modifiers, modification of additional substrates, beyond histones, have connected these enzymes to a wide range of regulatory roles (Choudhary et al., 2009, Joshi et al., 2013). One could speculate that an initial, non-specific action of chromatin modifiers, such as HDAC8, on a nearby protein present at high effective local concentration, could have resulted in benefits in selection, or been maintained due to its neutral effect. With time this modification could have evolved into a specific modification with a specific regulatory role. In this study we have demonstrated, using HDAC8 as an example, how a structural approach can efficiently identify additional targets and therefore boost insight into the biological function of an enzyme.
Methods
We adapted the Rosetta FlexPepBind protocol (London et al., 2011, London et al., 2012) to predict the substrate specificity of HDAC8. Using a template peptide-receptor complex structure we modeled each of the investigated peptide sequences, and distinguished binders from non-binders based on these structural models. Different parameters, such as the template structure used as starting structure for optimization, the amount of sampling, and the scoring function were calibrated. Conformational sampling was biased towards relevant conformations by implementation of constraints that reproduce conserved structural features identified from known structures of the interaction.
The underlying assumption of this approach is that the ability of a lysine-acetylated peptide to bind to the HDAC8 catalytic site in an appropriate conformation contributes significantly to the rate of deacetylation. Our goal was to obtain maximal energy separation between peptides that are HDAC8 substrates (i.e. binders) and those that are not (i.e. cannot bind), and a good correlation between predicted relative binding affinities and corresponding experimental values for catalytic efficiency.
Modeling the structures of peptide-protein complexes
The first step involves the generation of an approximate starting structure of the peptide-receptor complex. In the second step, this structure is optimized, followed by scoring in the third step. This results in a ranked list of peptides according to their predicted ability to bind to HDAC8. Details and runline commands used in the different modeling steps are included in the Supplementary Text.
Step 1: Preparation of a starting model of a peptide-HDAC8 complex
In a first step a coarse model of the complex was generated, based on a template crystal structure of a catalytically dead Y306F HDAC8 mutant in complex with a p53-derived peptide AcRHKacKac (7-methoxycoumarin-4-yl) methyl [PDB id 2v5w, (Vannini et al., 2007)]. Initially, we calibrated our protocol with a structure in which missing peptide residues were added in an extended conformation (we used peptide models with charged termini throughout this study). Later, this structure was extensively optimized for a known strong substrate, GYKacFGC in our training set (see below), using Rosetta FlexPepDock refinement [as described previously (Raveh et al., 2010); 1000 models were generated, and the top-scoring model, according to interface score was selected, see below]. The resulting structure was used as template for modeling the binding of different peptide sequences to HDAC8.
Step 2: Optimization of the peptide-HDAC8 complex structure for different peptide sequences
Each peptide sequence of interest was threaded onto the above template [using Rosetta fixed-backbone design (Kuhlman et al., 2003)], and optimized using FlexPepDock (London et al., 2011)].
Step 3: Ranking of the peptide substrates based on the optimized peptide-receptor complex structures
In the last step, different lysine-acetylated peptide-HDAC8 complexes were ranked to determine the relative binding ability of different peptides to HDAC8. In order to focus on the peptide-receptor interface, different subsets of the total score were investigated as possible ranking measures:
Peptide score (Pep_sc) - The sum of the internal energy of the peptide and the interactions across the interface. A similar term, Peptide score noref (Pep_sc_noref) does not contain the reference energy term, Eref.
Interface score (I_sc)- includes the sum of interactions across the interface.
Reweighted score - sum of peptide score, interface score and total score.
We started with the default scoring function in Rosetta (score12). Subsequently, we modified this energy function to better account for electrostatic effects, as in our study on prediction of BCL binding specificity [for details, see (London et al., 2012)].
Parameters optimized for HDAC8 FlexPepBind
Biasing conformations with constraints
HDAC8 catalyzes deacetylation of a wide range of different substrates, which all must bind to the catalytic site. Maintaining the structural features that are important for binding is critical for structure-based substrate specificity prediction. Inspection of the structure of HDAC8 bound to a p53-derived peptide [PDB id 2v5w, (Vannini et al., 2007)], identified potential critical interactions that we implemented as constraints (see Figure 1 and Table S1).
Definition of axes for rigid body movements
The rigid body orientation between the HDAC8 receptor and the peptide substrate is defined by two anchors that connect the receptor to the peptide. In order to optimize rigid body sampling to relevant conformations, we explicitly defined the Cα atom of the acetylated lysine as the peptide anchor.
Datasets
Set_GX1KacX2GC: 361 peptides
The ability of HDAC8 to deacetylate 361 six amino acid peptides with the sequence Ac-GX1KacX2GC-NH2 (where X1, X2 can be any amino acid, except cysteine; see Table S2 in the Supplementary Material) was tested using a mass spectrometry technique termed SAMDI (self-assembled monolayers for matrix assisted laser desorption/ionization time-of-flight mass spectrometry) (Gurard-Levin et al., 2010). The arrays were prepared by immobilizing the peptides to a self-assembled monolayer of alkanethiolates on gold. For each peptide, the fraction of deacetylation catalyzed by recombinant HDAC8 was measured after reacting the peptide substrates immobilized on a chip for 30 minutes at 30°C. In these experiments care was taken to maintain a specific 1:1 Zn2+ to HDAC8 ratio using methods specified in Gantt et al. (Gantt et al., 2006), to minimize inhibition by excess metal ion. The data for the Set_GRKacX2X3C: 361 peptides was taken directly from Gurard-Levin et al. (Gurard-Levin et al., 2010) (see Table S3).
Validation sets
We applied our calibrated protocol to a large comprehensive set of reported acetylation sites, which was downloaded from the PhosphoSitePlus database (3191 acetylated sites in the human proteome reported in the 06/2012 release), to extract a small set of peptides for validation. This was done twice: Novel_candidate_set1 (Table 1): The first validation set was used to evaluate the initial protocol and contained 26 peptides. Novel_candidate_set2 (Table 2): The second validation set was used to evaluate the optimized, final protocol (see Table S4 for details about the criteria for selection of each of the targets), and contained 19 peptides. Deacetylation of acetylated peptides catalyzed by recombinant Zn-HDAC8 was assayed using a coupled assay that measures the production of acetate, as described previously (Wolfson et al., 2014). More details of this experimental method are provided in the Supplementary Material.
Our final validation, Proteometargets_set3 (Table 3): consisted of HDAC8 targets independently identified in a large-scale proteomic assay led by the Holson group (Olson et al., 2014).
The PhosphoSitePlus database has accumulated many additional acetylation sites over the years. We therefore repeated our simulations on the latest available version (11/201511/20397 reported acetylated sites, identified either in a low-throughput assay, or in more than one independent high-throughput assays). Scores for all sites are included in the Appendix.
Statistical tests
We used two measures to evaluate the performance of HDAC8 FlexPepBind substrate prediction: (1) Binary distinction between substrates and non-substrates (applied to the SAMDI experiments), and (2) Linear correlation between predicted relative binding affinity and experimentally determined substrate activity. An optimal protocol should be able to reliably identify (at least the strongest) peptide substrates, and preferably to rank peptide substrates according to their degree of deacetylation by HDAC8.
Binary distinction between substrates and non-substrates
For binary distinction, we determined a deacetylation level, beyond/below which a peptide is defined as a substrate/non-substrate, respectively (see Results). The non-parametric Kolmogorov-Smirnov test was applied to determine whether substrates and non-substrates are derived from two distinct populations (i.e. they can be distinguished).
Prediction of relative substrate activity
In order to assess the ability of FlexPepBind to rank substrate peptides according to their degree of deacetylation by HDAC8, we calculated correlations and the associated statistical significance, using the non-parametric Pearson correlation test.
Supplementary Material
Acknowledgments
This work was supported, in whole or in part, by the Israel Science Foundation, founded by the Israel Academy of Science and Humanities (grant number 319/11 to O.S-F.), and by the European Research Council under the ERC Grant Agreement (No. 310873 to O.S-F). We thank the National Institutes of Health (NIH) (GM40602 to C.A.F.) for support.
Footnotes
Author Contributions
Concept and plan: N.A, L.Z., & O.S-F.; Performance: N.A. & L.Z.; Experimental validation: CG.J. & NA.W.; Manuscript: N.A, NA.W., CA.F. & O.S-F.
References
- BALASUBRAMANIAN S, RAMOS J, LUO W, SIRISAWAD M, VERNER E, BUGGY JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–34. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- CHAKRABARTI A, OEHME I, WITT O, OLIVEIRA G, SIPPL W, ROMIER C, PIERCE RJ, JUNG M. HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol Sci. 2015;36:481–92. doi: 10.1016/j.tips.2015.04.013. [DOI] [PubMed] [Google Scholar]
- CHATR-ARYAMONTRI A, BREITKREUTZ BJ, OUGHTRED R, BOUCHER L, HEINICKE S, CHEN D, STARK C, BREITKREUTZ A, KOLAS N, O’DONNELL L, REGULY T, NIXON J, RAMAGE L, WINTER A, SELLAM A, CHANG C, HIRSCHMAN J, THEESFELD C, RUST J, LIVSTONE MS, DOLINSKI K, TYERS M. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015;43:D470–8. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOUDHARY C, KUMAR C, GNAD F, NIELSEN ML, REHMAN M, WALTHER TC, OLSEN JV, MANN M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- DEARDORFF MA, BANDO M, NAKATO R, WATRIN E, ITOH T, MINAMINO M, SAITOH K, KOMATA M, KATOU Y, CLARK D, COLE KE, DE BAERE E, DECROOS C, DI DONATO N, ERNST S, FRANCEY LJ, GYFTODIMOU Y, HIRASHIMA K, HULLINGS M, ISHIKAWA Y, JAULIN C, KAUR M, KIYONO T, LOMBARDI PM, MAGNAGHI-JAULIN L, MORTIER GR, NOZAKI N, PETERSEN MB, SEIMIYA H, SIU VM, SUZUKI Y, TAKAGAKI K, WILDE JJ, WILLEMS PJ, PRIGENT C, GILLESSEN-KAESBACH G, CHRISTIANSON DW, KAISER FJ, JACKSON LG, HIROTA T, KRANTZ ID, SHIRAHIGE K. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–7. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DECROOS C, CHRISTIANSON NH, GULLETT LE, BOWMAN CM, CHRISTIANSON KE, DEARDORFF MA, CHRISTIANSON DW. Biochemical and Structural Characterization of HDAC8 Mutants Associated with Cornelia de Lange Syndrome Spectrum Disorders. Biochemistry. 2015;54:6501–13. doi: 10.1021/acs.biochem.5b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEVER TE, COSTELLO CE, OWENS CL, ROSENBERRY TL, MERRICK WC. Location of seven post-translational modifications in rabbit elongation factor 1 alpha including dimethyllysine, trimethyllysine, and glycerylphosphorylethanolamine. J Biol Chem. 1989;264:20518–25. [PubMed] [Google Scholar]
- DINKEL H, VAN ROEY K, MICHAEL S, DAVEY NE, WEATHERITT RJ, BORN D, SPECK T, KRUGER D, GREBNEV G, KUBAN M, STRUMILLO M, UYAR B, BUDD A, ALTENBERG B, SEILER M, CHEMES LB, GLAVINA J, SANCHEZ IE, DIELLA F, GIBSON TJ. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 2014;42:D259–66. doi: 10.1093/nar/gkt1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOWLING DP, GANTT SL, GATTIS SG, FIERKE CA, CHRISTIANSON DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–63. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOWNEY M, JOHNSON JR, DAVEY NE, NEWTON BW, JOHNSON TL, GALAANG S, SELLER CA, KROGAN N, TOCZYSKI DP. Acetylome profiling reveals overlap in the regulation of diverse processes by sirtuins, gcn5, and esa1. Mol Cell Proteomics. 2015;14:162–76. doi: 10.1074/mcp.M114.043141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESTEVES SL, KORRODI-GREGORIO L, COTRIM CZ, VAN KLEEFF PJ, DOMINGUES SC, DA CRUZ E SILVA OA, FARDILHA M, DA CRUZ E SILVA EF. Protein phosphatase 1gamma isoforms linked interactions in the brain. J Mol Neurosci. 2013;50:179–97. doi: 10.1007/s12031-012-9902-6. [DOI] [PubMed] [Google Scholar]
- FERSHT A. Enzyme structure and mechanism 1985 [Google Scholar]
- FRIEDLER A, VEPRINTSEV DB, FREUND SM, VON GLOS KI, FERSHT AR. Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure. 2005;13:629–36. doi: 10.1016/j.str.2005.01.020. [DOI] [PubMed] [Google Scholar]
- GANTT SL, GATTIS SG, FIERKE CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–8. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- GAO J, SIDDOWAY B, HUANG Q, XIA H. Inactivation of CREB mediated gene transcription by HDAC8 bound protein phosphatase. Biochem Biophys Res Commun. 2009;379:1–5. doi: 10.1016/j.bbrc.2008.11.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLOZAK MA, SENGUPTA N, ZHANG X, SETO E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- GURARD-LEVIN ZA, KILIAN KA, KIM J, BAHR K, MRKSICH M. Peptide arrays identify isoform-selective substrates for profiling endogenous lysine deacetylase activity. ACS Chem Biol. 2010;5:863–73. doi: 10.1021/cb100088g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GURARD-LEVIN ZA, KIM J, MRKSICH M. Combining mass spectrometry and peptide arrays to profile the specificities of histone deacetylases. Chembiochem. 2009;10:2159–61. doi: 10.1002/cbic.200900417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HABERLAND M, MOKALLED MH, MONTGOMERY RL, OLSON EN. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009;23:1625–30. doi: 10.1101/gad.1809209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HORNBECK PV, CHABRA I, KORNHAUSER JM, SKRZYPEK E, ZHANG B. PhosphoSite: A bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–61. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- HORNBECK PV, ZHANG B, MURRAY B, KORNHAUSER JM, LATHAM V, SKRZYPEK E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–20. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOSHI P, GRECO TM, GUISE AJ, LUO Y, YU F, NESVIZHSKII AI, CRISTEA IM. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUHLMAN B, DANTAS G, IRETON GC, VARANI G, STODDARD BL, BAKER D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–8. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- LAHR RM, MACK SM, HEROUX A, BLAGDEN SP, BOUSQUET-ANTONELLI C, DERAGON JM, BERMAN AJ. The La-related protein 1-specific domain repurposes HEAT-like repeats to directly bind a 5′TOP sequence. Nucleic Acids Res. 2015;43:8077–88. doi: 10.1093/nar/gkv748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN YY, KIIHL S, SUHAIL Y, LIU SY, CHOU YH, KUANG Z, LU JY, KHOR CN, LIN CL, BADER JS, IRIZARRY R, BOEKE JD. Functional dissection of lysine deacetylases reveals that HDAC1 and p300 regulate AMPK. Nature. 2012;482:251–5. doi: 10.1038/nature10804. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- LONDON N, GULLA S, KEATING AE, SCHUELER-FURMAN O. In silico and in vitro elucidation of BH3 binding specificity toward Bcl-2. Biochemistry. 2012;51:5841–50. doi: 10.1021/bi3003567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LONDON N, LAMPHEAR CL, HOUGLAND JL, FIERKE CA, SCHUELER-FURMAN O. Identification of a novel class of farnesylation targets by structure-based modeling of binding specificity. PLoS Comput Biol. 2011;7:e1002170. doi: 10.1371/journal.pcbi.1002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LU L, LI L, LV X, WU XS, LIU DP, LIANG CC. Modulations of hMOF autoacetylation by SIRT1 regulate hMOF recruitment and activities on the chromatin. Cell research. 2011;21:1182–95. [Google Scholar]
- MAURER-STROH S, EISENHABER F. Refinement and prediction of protein prenylation motifs. Genome Biol. 2005;6:R55. doi: 10.1186/gb-2005-6-6-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLSON DE, UDESHI ND, WOLFSON NA, PITCAIRN CA, SULLIVAN ED, JAFFE JD, SVINKINA T, NATOLI T, LU X, PAULK J, MCCARREN P, WAGNER FF, BARKER D, HOWE E, LAZZARO F, GALE JP, ZHANG YL, SUBRAMANIAN A, FIERKE CA, CARR SA, HOLSON EB. An Unbiased Approach To Identify Endogenous Substrates of “Histone” Deacetylase 8. ACS Chem Biol. 2014;3:749–753. doi: 10.1021/cb500492r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QI J, SINGH S, HUA WK, CAI Q, CHAO SW, LI L, LIU H, HO Y, MCDONALD T, LIN A, MARCUCCI G, BHATIA R, HUANG WJ, CHANG CI, KUO YH. HDAC8 Inhibition Specifically Targets Inv(16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation. Cell Stem Cell. 2015;17:597–610. doi: 10.1016/j.stem.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAVEH B, LONDON N, SCHUELER-FURMAN O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins. 2010;78:2029–40. doi: 10.1002/prot.22716. [DOI] [PubMed] [Google Scholar]
- RAWLINGS ND, WALLER M, BARRETT AJ, BATEMAN A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014;42:D503–9. doi: 10.1093/nar/gkt953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIESTER D, HILDMANN C, GRUNEWALD S, BECKERS T, SCHWIENHORST A. Factors affecting the substrate specificity of histone deacetylases. Biochem Biophys Res Commun. 2007;357:439–45. doi: 10.1016/j.bbrc.2007.03.158. [DOI] [PubMed] [Google Scholar]
- ROUX KJ, KIM DI, BURKE B. BioID: a screen for protein-protein interactions. Curr Protoc Protein Sci. 2013;74 doi: 10.1002/0471140864.ps1923s74. Unit 19 23. [DOI] [PubMed] [Google Scholar]
- SIROTA FL, MAURER-STROH S, EISENHABER B, EISENHABER F. Single-residue posttranslational modification sites at the N-terminus, C-terminus or in-between: To be or not to be exposed for enzyme access. Proteomics. 2015;15:2525–46. doi: 10.1002/pmic.201400633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SJOBLOM T, JONES S, WOOD LD, PARSONS DW, LIN J, BARBER TD, MANDELKER D, LEARY RJ, PTAK J, SILLIMAN N, SZABO S, BUCKHAULTS P, FARRELL C, MEEH P, MARKOWITZ SD, WILLIS J, DAWSON D, WILLSON JK, GAZDAR AF, HARTIGAN J, WU L, LIU C, PARMIGIANI G, PARK BH, BACHMAN KE, PAPADOPOULOS N, VOGELSTEIN B, KINZLER KW, VELCULESCU VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- STAVRAKA C, BLAGDEN S. The La-Related Proteins, a Family with Connections to Cancer. Biomolecules. 2015;5:2701–22. doi: 10.3390/biom5042701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAO RH, KAWATE H, WU Y, OHNAKA K, ISHIZUKA M, INOUE A, HAGIWARA H, TAKAYANAGI R. Testicular zinc finger protein recruits histone deacetylase 2 and suppresses the transactivation function and intranuclear foci formation of agonist-bound androgen receptor competitively with TIF2. Mol Cell Endocrinol. 2006;247:150–65. doi: 10.1016/j.mce.2005.12.052. [DOI] [PubMed] [Google Scholar]
- TSAI YC, GRECO TM, BOONMEE A, MITEVA Y, CRISTEA IM. Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol Cell Proteomics. 2012;11:M111. doi: 10.1074/mcp.M111.015156. 015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANNINI A, VOLPARI C, GALLINARI P, JONES P, MATTU M, CARFI A, DE FRANCESCO R, STEINKUHLER C, DI MARCO S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–84. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERA M, PANI B, GRIFFITHS LA, MUCHARDT C, ABBOTT CM, SINGER RH, NUDLER E. The translation elongation factor eEF1A1 couples transcription to translation during heat shock response. Elife. 2014;3:e03164. doi: 10.7554/eLife.03164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALTREGNY D, GLENISSON W, TRAN SL, NORTH BJ, VERDIN E, COLIGE A, CASTRONOVO V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19:966–8. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- WARREN JJ, POHLHAUS TJ, CHANGELA A, IYER RR, MODRICH PL, BEESE LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. 2007;26:579–92. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- WILSON BJ, TREMBLAY AM, DEBLOIS G, SYLVAIN-DROLET G, GIGUERE V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol Endocrinol. 2010;24:1349–58. doi: 10.1210/me.2009-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLFSON NA. Determining HDAC8 substrate specificity. University of Michigan; 2014. PHD. [Google Scholar]
- WOLFSON NA, PITCAIRN CA, FIERKE CA. HDAC8 substrates: Histones and beyond. Biopolymers. 2013;99:112–26. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLFSON NA, PITCAIRN CA, SULLIVAN ED, JOSEPH CG, FIERKE CA. An enzyme-coupled assay measuring acetate production for profiling histone deacetylase specificity. Anal Biochem. 2014;456:61–9. doi: 10.1016/j.ab.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WORKMAN JL, KINGSTON RE. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu Rev Biochem. 1998;67:545–79. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- YAN W, LIU S, XU E, ZHANG J, ZHANG Y, CHEN X, CHEN X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene. 2013;32:599–609. doi: 10.1038/onc.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG XJ, SETO E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–18. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YUAN H, ROSSETTO D, MELLERT H, DANG W, SRINIVASAN M, JOHNSON J, HODAWADEKAR S, DING EC, SPEICHER K, ABSHIRU N, PERRY R, WU J, YANG C, ZHENG YG, SPEICHER DW, THIBAULT P, VERREAULT A, JOHNSON FB, BERGER SL, STERNGLANZ R, MCMAHON SB, COTE J, MARMORSTEIN R. MYST protein acetyltransferase activity requires active site lysine autoacetylation. The EMBO journal. 2012;31:58–70. doi: 10.1038/emboj.2011.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.