

Abstract
Macrocyclic peptides are highly promising as inhibitors of protein–protein interactions. While many bond-forming reactions can be used to make cyclic peptides, most have limitations that make this chemical space challenging to access. Recently, a variety of cysteine alkylation reactions have been used in rational design and library approaches for cyclic peptide discovery and development. We and others have found that this chemistry is versatile and robust enough to produce a large variety of conformationally constrained cyclic peptides. In this chapter, we describe applications, methods, mechanistic insights, and troubleshooting for dithiol bis-alkylation reactions for the production of cyclic peptides. This method for efficient solution-phase macrocyclization is highly useful for the rapid production and screening of loop-based inhibitors of protein–protein interactions.
1. INTRODUCTION
In the last two decades, early-stage drug discovery has expanded to include targets outside the traditionally druggable classes of enzymes and cell surface receptors (Arkin, Tang, & Wells, 2014). Classically “undruggable” protein–protein interactions can make viable drug targets, but often have large interaction surfaces that are difficult for small molecules to bind with high affinity (Thompson, Dugan, Gestwicki, & Mapp, 2012). Peptides are an attractive option for targeting protein–protein interactions, as they are intermediate in size between small molecules and large biologics and offer many advantages over both (Kawamoto et al., 2012). Peptides are synthetically tractable, they can be optimized to high affinity and selectivity, and they often have good safety and tolerability profiles in animals and humans (Fosgerau & Hoffmann, 2014). These strengths have led to the FDA approval of several peptide-based therapeutics including linaclotide for gastrointestinal disorders, peginesatide for anemia, and carfilzomib for multiple myeloma (Kaspar & Reichert, 2013). While peptides hold many distinct advantages, they also have some inherent weaknesses including their short half-life and sensitivity to proteases (Fosgerau & Hoffmann, 2014). Also, short peptides are often poorly structured in aqueous solution, which can limit their affinity for their targets. One of the largest limitations of peptide drugs is poor membrane permeability, making delivery to intracellular targets difficult (Hill, Shepherd, Diness, & Fairlie, 2014). Chemical modification of peptides, such as isosteric backbone replacement and N-methylation, has substantial promise as a solution to many of these drawbacks (Avan, Hall, & Katritzky, 2014; Bockus et al., 2015; Brust et al., 2013; Chatterjee, Gilon, Hoffman, & Kessler, 2008; Yamagishi et al., 2011). Many modification strategies have been pursued in the effort to transform linear peptides into more natural product-like molecules with high potency, selectivity, cell penetration, and even oral bioavailability (Bock, Gavenonis, & Kritzer, 2013; Clark et al., 2010; Dahan, Tsume, Sun, Miller, & Amidon, 2011; Renukuntla, Vadlapudi, Patel, Boddu, & Mitra, 2013).
1.1 Stabilizing Alpha-Helical Structures
One solution to the inherent limitations of peptides is macrocyclization, which is typically applied as a structure-promoting conformational constraint. The effects of conformational constraints on peptide structure and function have been particularly well-studied in the field of alpha-helix stabilization (Henchey, Jochim, & Arora, 2008). A stable helical structure is difficult to achieve for a short peptide in aqueous solution, and according to the Zimm–Bragg model for helix–coil transition, the primary barrier is nucleation of an initial alpha-helical turn (Zimm & Bragg, 1959). Thus, a primary motivation for developing new peptide cyclization strategies has been stabilization of a nucleating alpha-helical turn, which can then propagate to stabilize an overall helical structure.
There are a host of cyclization strategies that have been developed for alpha-helix stabilization (Table 1). One of the most successful examples is hydrocarbon side-chain “stapling” using olefin metathesis (Miller et al., 1996; Walensky et al., 2004). Specific combinations of alpha-methyl groups and covalent cross-linking of (i, i+3), (i, i+4), or (i, i+7) residues with carbon chains of different lengths have been used to stabilize a variety of helical peptides (Bird et al., 2014; Kim, Kutchukian, & Verdine, 2010; Schafmeister et al., 2000). This method was used to stabilize an alpha-helix that mimics the hDM2-binding epitope of p53 (Chang et al., 2013), and a “stapled helix” of this type is currently in Phase I clinical trials for advanced hematologic and solid malignancies with wild-type p53 (Khoo, Hoe, Verma, & Lane, 2014). Another approach for alpha-helix stabilization using ring-closing olefin metathesis is the “hydrogen-bond surrogate” approach, which replaces the characteristic alpha-helix hydrogen bond between the N-terminal amino acid and the i+4 carbonyl with a carbon–carbon bond (Patgiri, Jochim, & Arora, 2008). Lactam bridges between lysine and aspartate residues located at (i, i+4) positions can also dramatically stabilize helical structures and promote potent protein binding (Shepherd et al., 2005). Copper-catalyzed Huisgen cycloaddition, or “click chemistry,” has also been used to join azide and alkyne side-chains at (i, i+4) positions (Kawamoto et al., 2012) to stabilize overall helical structure.
Table 1.
Some Common Reactions Used for Intramolecular Peptide Cross-Linking
| Cross-Linking Reaction | Reaction Conditionsa | Synthesis Mode | References |
|---|---|---|---|
|
|
10 mM Grubbs catalyst in 1,2-dichloroethane, 2 h | On resin | Miller, Blackwell, and Grubbs (1996) and Schafmeister, Po, and Verdine (2000) |
|
|
1.5 equiv. BOP, 2 equiv. DIPEA, DMF, 24 h | On resin | Shepherd, Hoang, Abbenante, and Fairlie (2005) |
|
|
Aqueous 6 M Gdm-HCl, pH 8.0, 2 h | In solution | Brunel and Dawson (2005) |
|
|
50:50 CH3CN/H2O with 5 mM NH4HCO3 pH 8.0, m-dibromoxylene, 15–120 min | In solution and on resin | Jo et al. (2012) and Timmerman, Beld, Puijk, and Meloen (2005) |
|
|
2:1 H2O/t-BuOH, 4.4 equiv. CuSO4·5H2O, 30–90 min | In solution and on resin | Kawamoto et al. (2012) and White and Yudin (2011) |
|
|
Aqueous 50 mM Tris, DMF, hexafluorobenzene, 4.5 h | In solution | Spokoyny et al. (2013) |
|
|
DMF, radical initiator, 365 nm UV, 1 h | In solution | Wang and Chou (2015) |
All reactions listed were run at room temperature for the times indicated.
BOP, (benzotriazol-1-yloxy) tris(dimethylamino)phosphonium hexafluorophosphate; DIPEA, N,N-diisopropylethylamine; DMF, N,N-dimethylformamide; Gdm-HCl, guanadinium hydrochloride.
Several chemistries are available that capitalize on the reactivity of cysteine residues. Thioether ligation was used to link a cysteine to a bromoacetylated ornithine, yielding an alternative to lactam bridge formation (Brunel & Dawson, 2005). A recent study also presented a thiol-based alternative to ring-closing metathesis, where peptides were stapled using thiolene chemistry between two cysteines and a diene linker (Wang & Chou, 2015). Cross-linking two or three unprotected cysteines can also be achieved using arylation reactions with perfluoroaryl groups (Spokoyny et al., 2013), or using alkylation reactions with bis-bromomethyl or tris-bromomethyl linkers ( Jo et al., 2012; Timmerman et al., 2005). The diverse chemistries for nucleating alpha-helical structure in short peptides have led to the successful development of many helical inhibitors of protein–protein interactions and hold promise for the development of many more therapeutic leads (Robertson & Jamieson, 2015).
In all cases of alpha-helix stabilization, the intramolecular cross-link promotes helix-nucleating hydrogen-bonding patterns, thus stabilizing overall alpha-helix structure. Another effective result is that these cyclic peptides are generally more stable to proteolytic degradation (Bird et al., 2014; Bock et al., 2013; Kawamoto et al., 2012; Shepherd et al., 2005). In some cases, cyclic peptides have increased cytosolic penetration compared to linear peptides (Bock et al., 2013; Chang et al., 2013; Muppidi, Wang, Li, Chen, & Lin, 2011; Qian et al., 2013). This sought-after property is essential for targeting intracellular protein–protein interactions.
1.2 Stabilizing Nonhelical Structures
Work on helical peptides has demonstrated the major benefits of macrocyclization for promoting peptide structure and function, but applying cyclic constraints to nonhelical structures has not been as straightforward. While the results from screening large, unbiased libraries of cyclic peptides clearly indicate that this is a valuable chemical space for protein inhibitors (Gao, Amar, Pahwa, Fields, & Kodadek, 2015; Gartner et al., 2004; Passioura, Katoh, Goto, & Suga, 2014; Xiao et al., 2010), rational design of small, nonhelical cyclic peptides is still largely trial and error. The rational design approach is greatly assisted by starting with a peptide epitope derived from a known protein binding partner. Ideally, this epitope accounts for a majority of the binding energy of the interaction by comprising the most important “hot spot” residues (Clackson & Wells, 1995). These hot spots can be identified either through experimental mutagenesis or through computational mutagenesis methods (Kortemme & Baker, 2002; Kortemme, Kim, & Baker, 2004). Several groups have used computational techniques to comprehensively identify peptide epitopes from the Protein Data Bank, focusing on continuous segments or more specifically on helix, sheet, or loop structures (Gavenonis, Sheneman, Siegert, Eshelman, & Kritzer, 2014; Henchey et al., 2008; London, Raveh, Movshovitz-Attias, & Schueler-Furman, 2010; Watkins & Arora, 2014; Wuo, Mahon, & Arora, 2015). Translating these epitopes to effective inhibitory peptides requires replacing the entire protein tertiary structure with a synthetic linker that stabilizes the epitope’s highest-affinity 3D structure. For helices, sheets, and beta turns, generalizable approaches for structural stabilization have been developed that can be immediately applied to many epitopes (Yin, 2012). Other, less common 3D structures are loosely grouped as “loop” structures. These require a more general strategy since one cross-link chemistry, spacing and length cannot be used to stabilize the large variety of loop structures that mediate protein–protein interactions (Gavenonis et al., 2014).
To date, computational methods for the prediction of cyclic peptide structure have only begun to be rigorously designed and tested (Damas et al., 2013; Razavi, Wuest, & Voelz, 2014; Wakefield, Wuest, & Voelz, 2015; Yu & Lin, 2015). This makes it very challenging to predictively design specific cross-links to stabilize a desired loop structure. Currently, identifying the proper linker chemistry, length, and positioning can only be done in an iterative process (Berthelot, Gonçalves, Laïn, Estieu-Gionnet, & Déléris, 2006; Cudic, Wade, & Otvos, 2000; Hayouka et al., 2012; Kamens, Eisert, Corlin, Baleja, & Kritzer, 2014; Qvit et al., 2009). One way to accelerate, this process is to introduce diverse conformational constraints at a late stage of synthesis. In this chapter, we provide protocols for a robust, efficient method for late-stage conformational diversification of peptide epitopes using thiol bis-alkylation chemistry (originally reported by Timmerman et al., 2005). This allows for rapid preparation and screening of many conformations of a given loop using a panel of linkers, experimentally searching for the highest-affinity conformation.
2. USING THIOL ALKYLATION TO CONSTRAIN PEPTIDES
2.1 Libraries of Peptides That Are Constrained Through Alkylated Cysteines
Thiol bis-alkylation has been applied in several studies to constrain peptides and introduce structure. This chemistry has been particularly useful for introducing conformational constraints within large, randomized peptide libraries. In a landmark paper, Heinis and coworkers generated a trillion-member phage-display library of bicyclic peptides containing three reactive cysteines which were cyclized using tris-(bromomethyl)benzene (linker tmb, Fig. 1) (Heinis, Rutherford, Freund, & Winter, 2009). This library was applied in iterative selections for binding to the plasma protease kallikrein, ultimately producing a low-nanomolar inhibitor which was highly specific for kallikrein over other plasma proteases. Using the same strategy, Heinis and coworkers also discovered a submicromolar bicyclic ligand for the negative regulatory region of the Notch receptor (Urech-Varenne, Radtke, & Heinis, 2015). Thiol bis-alkylation has also been applied to mRNA display libraries (Schlippe, Hartman, Josephson, & Szostak, 2012). In one such selection, all the selected peptides were cyclic, even those containing a single cysteine; the mxy linker unexpectedly formed a small ring with the N-terminal methionine (White et al., 2015). The rapid cross-linking of cysteine and methionine using dibromoxylene linkers is an observation that we have confirmed in our lab (see later).
Fig. 1.
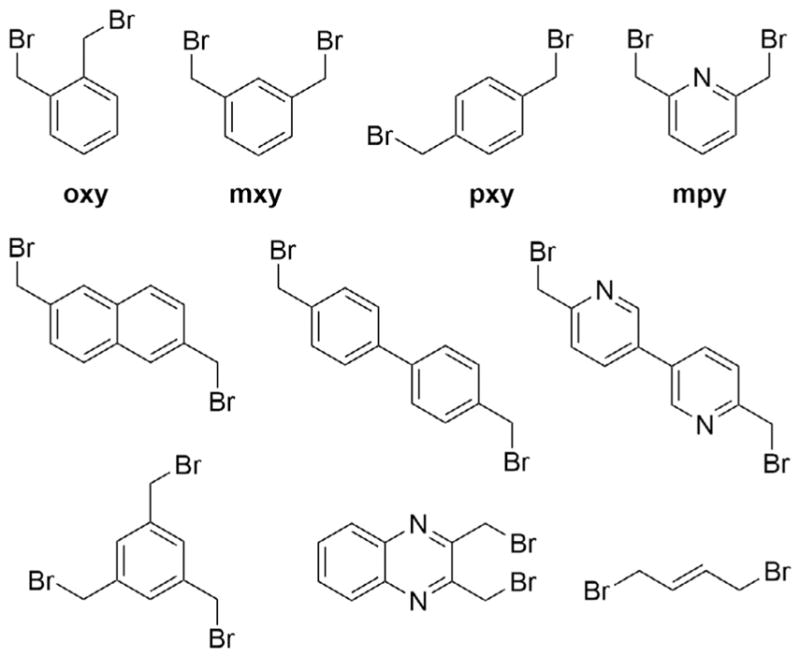
Common linkers used for thiol cross-linking. Top row: Dibromo-o-xylene (oxy), dibromo-m-xylene (mxy), dibromo-p-xylene (pxy), and 2,6-bis(bromomethyl) pyridine (mpy). Second row: 2,6-bis(bromomethyl)naphthalene, 4,4′-bis(bromomethyl)biphenyl and 6,6′-bis-bromomethyl-[3,3′]bipyridine. Bottom row: 1,3,5-tris(bromomethyl)benzene (tmb), 2,3-bis(bromomethyl)quinoxaline, and trans-1,4-dibromo-2-butene.
More recent work on thiol-alkylated libraries has pushed this chemistry even further. A novel method was reported by the Suga lab where they synthesized tricyclic peptides using flexizyme-assisted translation (Goto, Katoh, & Suga, 2011) and the tmb linker (Bashiruddin, Nagano, & Suga, 2015). With this technology, they replaced the N-terminal methionine with a chloroacetyl-containing amino acid, which spontaneously macrocyclizes by forming a thioether bond with a nonadjacent cysteine. The three remaining cysteines were then cyclized using the tmb linker to form tricyclic peptides (Bashiruddin et al., 2015).
2.2 Rational Design Using Cysteine Alkylation
As discussed earlier, cysteine alkylation has been used for the generation of libraries via phage or mRNA display, yielding cyclic, bicyclic, or tricyclic peptides with increased conformational rigidity. Alternatively, this chemistry has also been used to induce specific secondary structures in peptides. DeGrado, Greenbaum, and coworkers used thiol bis-alkylation to find the linker that would yield the cleanest cyclization reaction and induce alpha-helical structure in a peptide inhibitor of the protease calpain ( Jo et al., 2012). The cysteines were placed at (i, i+4) positions within a model peptide that had moderate helicity. A panel of 24 different linkers was screened, and the hits were compared using circular dichroism, from which they determined that cross-linking with dibromo-m-xylene (linker mxy, Fig. 1) showed the highest increase in helical structure. Then, they scanned the peptide for the best (i, i+4) staple location by moving the cysteines and then cross-linking with linker mxy ( Jo et al., 2012). Similarly, Muppidi et al. designed stapled helices containing cysteines in (i, i+7) positions, using the longer linkers 4,4′-bis-bromomethyl-biphenyl and 6,6′-bis-bromomethyl-[3,3′]bipyridine (Muppidi et al., 2011, 2014) (see Fig. 1). They showed that this cross-linking chemistry led to increased helicity and bioactivity, and even increased cell permeability for a series of BH3-peptide-derived ligands for MCL-1.
2.3 Thiol Bis-Alkylation Is Ideal for Constraining Epitopes into Diverse Conformations
We considered all the above cyclization chemistries when searching for methods for rapid translation of loop epitopes into useful protein–protein interaction inhibitors (Gavenonis et al., 2014). Thiol bis-alkylation has rapid kinetics and broad sequence tolerance, as evidenced by several discrete studies and by its many applications in cyclic peptide libraries ( Jo et al., 2012; Smeenk, Dailly, Hiemstra, van Maarseveen, & Timmerman, 2012; Timmerman et al., 2005; Todorova-Balvay, Stoilova, Gargova, & Vijayalakshmi, 2007). Also, studies that used diverse linkers to search for cross-links that specifically stabilize helical conformations indicated that this chemistry is optimal for late-stage conformational diversification ( Jo et al., 2012; Muppidi et al., 2011). This chapter describes adaptations and extensions of these prior studies toward general methods for the rapid production of cyclic peptides with diverse conformational constraints.
Late-stage conformational diversification is introduced by including two thiol-containing amino acids at positions known to be nonessential for target binding. Such peptides are readily cross-linked into macrocycles using a wide variety of different linkers. Fig. 1 lists commercially available or readily synthesized linkers that have been used to cross-link cysteine-containing peptides, which range from simple ortho, meta, and para dibromomethylxylenes (oxy, mxy, and pxy, respectively) to 2,3-bis (bromomethyl)quinoxaline ( Jo et al., 2012; Muppidi et al., 2011, 2014; Timmerman et al., 2005). While nearly all prior reports have used L-cysteine, one reported application used D-cysteine as the thiol-containing amino acid (Muppidi et al., 2011). We have found that many thiol-containing amino acids are compatible with this approach. To maximize the variety of conformational constraints, we have applied various combinations of L- and D-cysteine, L- and D-homocysteine, and L- and D-penicillamine, all without reduction in cross-linking efficiency. Dithiol-containing amino acids could also potentially be used (Chen et al., 2014), since the rigidity of the linkers should preclude bis-alkylation of both thiols on one side chain. A major advantage of using this chemistry is that it is orthogonal to natural amino acid functional groups including lysine and free N-termini. The exception is methionine, as discussed below. By strategically varying the thiol-containing amino acids, the relative positioning of the thiol-containing amino acids, and the linkers, it is possible to use this chemistry to prepare a large library of peptides with diverse 3D conformations from a single peptide epitope.
3. PROTOCOLS FOR PEPTIDE SYNTHESIS AND CROSS-LINKING
We use standard fluorenylmethoxycarbonyl (Fmoc) solid-phase peptide synthesis (SPPS) to synthesize the linear precursor peptides (Fields & Noble, 1990; Moss, 2005). For an amidated C-terminus, we use Rink Amide Resin (100–200 mesh) with a loading of 0.3–0.6 mmol/g (Han & Kim, 2004). In order to produce a panel of cross-linked peptides, we begin with the synthesis of one parent linear peptide at a scale of 50–100 μmol. Peptides can be synthesized by hand or using an automated synthesizer. After synthesizing the linear sequence, the N-terminus can be capped or left as a free amine, and the peptide is cleaved off the resin. The peptide is precipitated using cold ether to separate it from protecting groups and cleavage reagents, particularly scavengers such as ethanedithiol (EDT). The crude linear peptide can either be purified using reverse-phase high-performance liquid chromatography (RP-HPLC) or directly used in thiol bis-alkylation reactions. The linear peptide is divided into multiple reaction vessels and reacted with different linkers in a 50:50 mixture of acetonitrile (CH3CN) and water buffered at pH 8.0. The reaction is typically complete within 1 h at room temperature. After bis-alkylation, solvents can be concentrated by lyophilizing the reaction and resuspending in a smaller volume of CH3CN/H2O. The crude reaction is purified by RP-HPLC to obtain the final cyclic product. An abbreviated procedure is provided later, and a scheme is shown in Fig. 2.
Fig. 2.

Scheme for SPPS of linear peptides using Fmoc chemistry. DMF is N,N-dimethylformamide, DCM is dichloromethane, AA refers to the protected monomer form of the desired amino acid, HATU is 1-[bis(dimethylamino)methylene]-1H-1,-2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, HOAt is 1-hydroxy-7-azabenzotriazole, DIPEA is N,N-diisopropylethylamine, TFA is trifluoroacetic acid, EDT is ethanedithiol, TIPS is triisopropylsilane. Though the peptide in this example has cysteines at the N- and C-termini, it is possible to use many other thiol-containing amino acids and to place the thiol-containing amino acids at any positions within the peptide.
-
(1)
Swell resin in 5–10 mL of DMF for at least 30 min with shaking.
-
(2)
Deprotect the resin using 5–10 mL of 20% piperidine in DMF for 2× 7 min.
-
(3)
Wash the resin with 5–10 mL of DMF, 2× 30 s, DCM 2× 30 s, DMF 2× 30 s.
The presence of a free amine can be confirmed using a Kaiser test (Shelton & Jensen, 2013).
-
(4)
Dissolve 5 equiv. of the Fmoc-AA-OH, 5 equiv. of coupling reagent, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b] pyridinium 3-oxid hexafluorophosphate (HATU), and 5 equiv. of the coupling additive 1-hydroxy-7-azabenzotriazole (HOAt) in 5–10 mL of DMF or NMP. Add to resin and also add 13 equiv. of DIPEA. Shake at room temp for 30 min. The completion of the reaction can also be checked with a negative Kaiser test.
-
(5)
Wash the resin extensively as in step 3.
-
(6)
Repeat Fmoc deprotection and coupling steps 2–5 until the final amino acid has been coupled to the growing peptide chain.
-
(7)
After coupling the last amino acid, remove the last Fmoc group as described in step 2. Then, if an acetylated N-terminus is desired, cap the resin using 5–10 mL 10% acetic anhydride/10% 2,6-lutidine/80% DMF for 2× 10 min.
-
(8)
Wash the resin extensively using DMF and DCM, finishing with a methanol wash. Dry out the resin completely using vacuum or dry nitrogen or argon gas.
-
(9)
For global deprotection and cleavage, use 1–4 mL of a standard cleavage cocktail: 95% trifluoroacetic acid (TFA), 2.5% 1,2-ethanedithiol (EDT), 2.5% H2O, and 1% triisopropylsilane (TIPS). Allow to deprotect for 3–4 h depending on the amino acids in the sequence.
-
(10)
Chill 40 mL of diethyl ether on dry ice for 15 min.
-
(11)
Once the cleavage is complete, filter the cleavage solution to separate it from the resin and add dropwise to chilled ether. You should see your peptide crashing out in the ether and the solution should become opaque.
-
(12)
Centrifuge at 3500 rpm for 10 min. Decant the ether and wash the pellet three times with 40 mL of freshly chilled diethyl ether to ensure removal of cleavage cocktail components. Centrifuge again at 3500 rpm for 10 min.
-
(13)
Decant the ether and dry the pellet under dry argon or nitrogen gas. A precipitated pellet of crude peptide is shown in Fig. 3.
Fig. 3.

Scheme for thiol bis-alkylation of crude linear peptides. As described in Fig. 2, the identities of the thiol-containing amino acids and their relative positions within the peptide can be varied with only minor effects on overall yield.
At this point, the linear peptide can be purified by reverse-phase HPLC. However, in most cases, the crude peptide is relatively pure and can be immediately bis-alkylated in solution. We highly recommend skipping HPLC purification of most linear peptides prior to bis-alkylation, unless major byproducts are present. We have had good results by performing the bis-alkylation as follows (Fig. 3):
-
(14)
Dissolve the ether-precipitated pellet (or purified linear peptide) in 50:50 CH3CN/H2O.
-
(15)
If the linear peptide has a Tyr or Trp, estimate the concentration by UV–vis spectrophotometry using UV absorbance at 280 nm. If the concentration cannot be estimated by UV, we have more roughly estimated the maximum amount of peptide present by assuming a 100% yield of the overall solid-phase synthesis. This works well because one would rather have the peptide more dilute than expected for the bis-alkylation reaction than more concentrated.
-
(16)
Prepare a 1 mM solution of peptide in 50:50 solution of CH3CN and H2O buffered with 20 mM ammonium bicarbonate, pH 8.0.
Note: At this step, check the pH of the solution before you add the linker, especially if you are performing the alkylation on a crude peptide. It is possible for TFA to carry over from ether precipitation or RP-HPLC, which could potentially lower the pH of the reaction.
-
(17)
Dissolve 1.5 equiv. of linker in 1–2 mL of CH3CN and add to the peptide. The reaction is typically completed in under 1 h. You can monitor the formation of the product using mass spectrometry. For instance, adding the oxy, mxy, or pxy linker will result in a cyclic peptide product that is 102 Daltons higher in mass than the linear peptide, as observed in Fig. 5. We have observed that, for most peptides, the appearance of the product peak by MALDI-TOF coincides with the disappearance of the starting material, and that the mass spectrometry peak for the starting material frequently becomes tiny or unobservable after 1 h.
-
(18)
The reaction can be stopped by lowering the pH with HCl or TFA and can be immediately purified. We often freeze the reaction, lyophilize, and redissolve in a smaller volume prior to purification.
-
(19)
Once the cyclic peptide is purified, it can be stored as lyophilized powder at −20°C or directly used in assays.
Note: While dimethyl sulfoxide (DMSO) is a common solvent for concentrated stocks of purified peptides, DMSO can also accelerate the oxidation of sulfides. If you store dithiol bis-alkylated peptides in DMSO, you should monitor their purities and masses over time to ensure sulfur oxidation has not occurred. In a minority of cases, we have observed sulfide oxidation and loss of bioactivity of bis-alkylated peptides that had been stored in DMSO for longer than 4 weeks.
Fig. 5.

Bis-alkylation of peptide 2 using the mxy linker, forming cyclic peptide 2-mxy. MALDI mass spectrometry of peptide 2 and the reaction mixture after 60 min are shown at the right.
4. APPLICATIONS, TIPS, AND TROUBLESHOOTING
4.1 Applying Dithiol Bis-Alkylation for Constraining Loop Epitopes
We have found thiol bis-alkylation to be an efficient and robust method to add conformational constraints to peptide epitopes. In one application, we designed peptide 1, which contains a loop epitope derived from the Eps15–stonin2 interaction (Gavenonis et al., 2014; Rumpf et al., 2008). Peptide 1 was synthesized as described earlier, and crude peptide was divided into fractions that were separately cyclized using oxy, mxy, and pxy linkers. Each reaction was then lyophilized and purified by preparative RP-HPLC. Fig. 4 shows the preparative-scale chromatograms along with the analytical reinjections of the major peaks. For each reaction, one pass on the HPLC produced cyclic peptides with purity well over 95%. These peptides can be used directly in biochemical assays and/or cell-based assays to screen for the desired activity.
Fig. 4.

Crude peptide 1 was redissolved from an ether-precipitated pellet, split into three fractions, and each was bis-alkylated as described in Fig. 3. This produced three peptides with identical epitopes but three different conformational constraints. Shown at the right are representative preparative-scale HPLC chromatograms, with peaks corresponding to the product cyclic peptides highlighted in gray. Preparative-scale HPLC was performed using a Agilent ZORBAX SB-C8 Vydac Maxisorb column with a flow rate of 20 mL/min and a gradient of 5% solvent B to 100% solvent B over 30 min (solvent A is H2O with 0.2% TFA, solvent B is CH3CN with 0.2% TFA). Insets in these chromatograms show analytical HPLC chromatograms of the highlighted fractions, demonstrating greater than 95% purity after one pass. Analytical-scale HPLC was performed using an Agilent ZORBAX Eclipse Plus C18 Vydac Maxisorb column with a flow rate of 1 mL/min and a gradient of 5% solvent B to 100% solvent B over 30 min.
4.2 Monitoring Progression of Thiol Bis-Alkylation
Model peptide 2, whose sequence was adapted from the autophagy regulator Beclin 1 (Shoji-Kawata et al., 2013), was bis-alkylated using the mxy linker (Fig. 5). The linear peptide was purified and lyophilized, then brought up in a 1:1 mixture of acetonitrile and water (water was buffered to pH 8.0 to a final concentration of 20 mM ammonium bicarbonate). Three equivalents of the linker were dissolved in 500 μL of acetonitrile and added to the reaction, which was then placed on a shaker. At different time points over the course of 1 h, 100 μL were removed from the reaction mixture and quenched with 50 μL of 0.1 M HCl to stop the reaction. Samples at 0, 5, 15, 30, and 60 min were then analyzed by analytical RP-HPLC to monitor the conversion of linear to cyclic peptide. Fig. 6 shows how a peptide bis-alkylation reaction can be monitored for progression and efficiency. The excess linker can be seen eluting at 20 min. The corresponding MALDI traces show that the product (expected mass 1364.61 Da, observed mass m/z 1364.81) has a molecular weight of 102 Da larger than the linear peptide (expected mass 1262.47 Da, observed m/z 1262.42 Da) (Fig. 5).
Fig. 6.
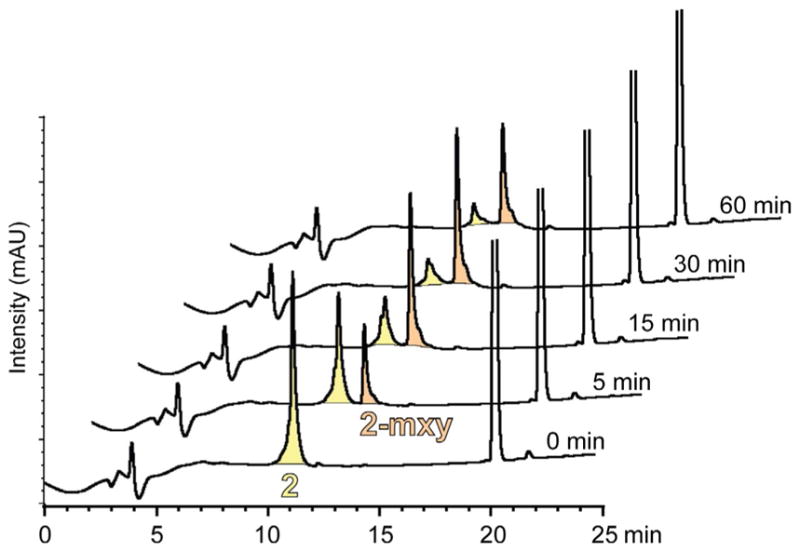
Monitoring reaction progress by HPLC. Aliquots from the reaction shown in Fig. 5 were removed and quenched at 5, 15, 30, and 60 min and analyzed by analytical HPLC. This stacked plot of HPLC chromatograms shows progression of the reaction from peptide 2 (peaks highlighted in yellow (light gray in the print version)) to cyclic peptide 2-mxy (peaks highlighted in orange (gray in the print version)).
4.3 Reaction Scope and Versatility
We have obtained excellent yields with a wide variety of dithiol peptides and linkers. Based on this experience, we provide some general observations about the scope and versatility of this macrocyclization reaction.
In most peptides, varying the positions of the thiol-containing amino acids does not affect the efficiency of the reaction. In general, we have observed that relative positions from (i, i+3) to (i, i+10) can be readily cyclized using dithiol bis-alkylation. For example, we prepared model peptides 3, 4, and 5 (see Table 2) and observed no difference in cyclization efficiencies among these (i, i+4), (i, i+5), and (i, i+6) macrocyclization reactions. For each of these peptides, the desired bis-alkylated peptide (calculated m/z 1326.56, observed m/z 1327.32 Da) was the major product after only 2 min (Fig. 7A). Minor formation of xylene cross-linked dimer with one linker (calculated m/z 2550.99, observed m/z 2549.60 Da) and two linkers (calculated m/z 2653.12, observed m/z 2654.97 Da) was also observed by mass spectrometry at different time points.
Various linkers can be used, though some require additional optimization. In our hands, bis-alkylation of dithiol-containing peptides has been consistently most efficient with the xylene-based linkers and 2,6-bis(bromomethyl) pyridine (linkers oxy, mxy, pxy, and mpy, Fig. 1). Biphenyl and napthyl linkers can also be used, but obtaining high yields of macrocyclized product with these linkers sometimes requires case-by-case optimization to find the most favorable reaction solvents, stoichiometries, times, and conditions. Under similar reaction conditions, we have also used allyl bromide and benzyl bromide to generate bis-alkylated analogs with no macrocyclic constraint. In our hands, linkers lacking benzylic bromides such as 1,4-dibromo-2,3-butanedione, 1,2-dibromoethane, and 1,4-dibromobutane have seldom yielded cleanly cyclized peptides.
Other thiol-containing amino acids undergo bis-alkylation with similar efficiency. Our lab has used L- and D-cysteine, L- and D-homocysteine, and L- and D-penicillamine in various permutations at the N-terminal and C-terminal cyclization positions. We have seen no decrease in the efficiency of cyclization even with the more hindered penicillamines, though we have observed a slight decrease in solubility which can be addressed by reducing peptide concentration in the bis-alkylation reaction. We have also used cysteamine 2-chlorotrityl resin to introduce an achiral, thiol-containing moiety to the C-terminus in place of one cysteine. This cysteine-to-cysteamine substitution at the C-terminus produces peptides that cyclize robustly with a variety of linkers.
Table 2.
Primary Sequences of Model Peptides
| Peptide | Sequence | Calculated m/z | Observed m/z |
|---|---|---|---|
| 1 | CPWRATNPFC | 1235.45 | 1234.91 |
| 2 | pa-VCNATCHIWH | 1262.47 | 1262.42 |
| 3 | VCNATCHIWH | 1224.43 | 1224.57 |
| 4 | VCNATHCIWH | 1224.43 | 1223.26 |
| 5 | VCNATIHCWH | 1224.43 | 1223.26 |
| 6 | VCNATSHIWH | 1208.36 | 1208.54 |
| 7 | VCNATMHIWH | 1252.48 | 1252.59 |
| 8 | VMNATMHIWH | 1280.53 | 1280.73 |
All peptides were prepared with C-terminal amides and N-terminal acetyl groups, except for peptide 2 which was prepared with an N-terminal pentynyl group (denoted pa-) as shown in Fig. 5. Sulfur-containing amino acids capable of participating in macrocyclization reactions are shown in bold. Observed m/z are reported as major peaks from MALDI mass spectra.
Fig. 7.

Peptides listed in Table 2 were incubated at 1 mM with 1.5 mM mxy linker and 200 μM TCEP in 50% CH3CN, 50% H2O with 20 mM NH4HCO3, pH 8.0. At time points between 2 and 60 min, aliquots of the reactions were removed, quenched with HCl, and analyzed qualitatively by MALDI mass spectrometry. Data are shown for selected time points for (A) peptide 3, (B) peptide 6, (C) peptide 7, and (D) peptide 8.
4.4 Investigating the Bis-Alkylation Mechanism
The thiol bis-alkylation reaction is a series of two nucleophilic substitutions. In each, a nucleophilic thiolate attacks the benzylic bromide, and the bromide ion is the leaving group. The robustness of the bis-alkylation reaction with benzylic and allylic bromides compared to alkyl bromides suggests the possibility of some SN1 character in either or both substitutions. In order to investigate the role of the nucleophiles, we carried out test reactions using peptides 3, 6, 7, and 8 (see Table 2 for sequences). These peptides are identical except that peptide 3 contains two cysteines at (i, i+4) positions, peptide 6 substitutes one cysteine with serine, peptide 7 substitutes one cysteine with methionine, and peptide 8 substitutes both cysteines with methionine. Each peptide was incubated at 1 mM with 1.5 mM mxy linker for 1 h in the presence of 200 μM tris(2-carboxyethyl)phosphine (TCEP), and results at different time points were monitored by MALDI mass spectrometry. Based on prior observations and the results of this limited study (Fig. 7), we conclude the following:
The full extent of the reaction usually occurs within the first 30 min, with no significant change typically observed after 60 min. We note that more time may be necessary for peptides with unusually hindered structures, and for bis-alkylation conducted on-resin ( Jo et al., 2012).
A peptide containing one cysteine will mostly form cross-linked dimer. Under these conditions, peptide 6 reacts sluggishly, and primarily forms a xylene-cross-linked dimer. At 2 min, we observe the xylene-cross-linked dimer (calculated m/z 2518.86, observed m/z 2519.92) as well as a peak which corresponds to mono-alkylated product where the linker still has one bromine (calculated m/z 1391.41, observed m/z 1391.73) (Fig. 7B). We note that this mono-alkylated intermediate is detected for peptide 6, but not for any peptides that bis-alkylate. This indicates that the second substitution step occurs very rapidly for those peptides that can readily bis-alkylate. By t=30 min, a new peak at 1561.69 Da is observed, which corresponds to mono-alkylated peptide lacking bromine plus the mass of TCEP (calculated m/z 1562.70). Noncovalent, charge-paired TCEP adducts can be observed by mass spectrometry for some peptides. The loss of bromine could be due to an attack by the serine hydroxyl, histidine imidazole, or alkylated cysteine thioether on the second benzylic bromide. The resulting sulfonium or imidazolium ion would be stabilized by forming a salt with TCEP. Overall, the slow reaction and many difficult-to-characterize products are typically observed only for reactions where rapid intramolecular bis-alkylation is not possible.
A peptide-containing one cysteine and one methionine will bis-alkylate. Fig. 7C shows the results over time for bis-alkylation of peptide 7 under the same conditions. At 30 and 60 min, the major products are bis-alkylated product (calculated m/z 1355.62, observed m/z 1355.37 Da) and cross-linked dimer (calculated m/z 2607.09, observed m/z 2608.67). The peak at 1606.90 Da is a noncovalent TCEP adduct of mono-alkylated peptide (calculated m/z 1606.82).
A peptide with no cysteines is not altered under these conditions. Peptide 8, which has methionines in place of both cysteines, does not undergo any reaction under these conditions (Fig. 7D). Notably, starting material is relatively unchanged and little dimeric product is observed. This shows that functional groups such as the histidine imidazole and methionine sulfide are not nucleophilic enough to react under these conditions, at least in the absence of any thiol groups.
The above observations provide some clues as to the overall mechanism. Clearly, a first step must be nucleophilic substitution of a thiolate on the benzyl bromide, but the direct product of this step can only be observed transiently and for peptides that do not rapidly undergo a second alkylation. The second, intramolecular substitution is accelerated due to the high effective concentration of the second bromide in the vicinity of the second nucleophile. Electronic effects at the benzylic bromides may also play an important role. Specifically, the presence of the sulfide directly above or below the benzene ring likely makes the bromomethyl benzyl sulfide even more electrophilic and may even promote a more SN1-like mechanism for the second bis-alkylation (see Fig. 8). This explains why peptide 7, with cysteine and methionine in (i, i+4) positions, bis-alkylated so rapidly even though peptide 8, with two methionines, did not. This would also explain why the oxy, mxy, pxy, and mpy linkers are so reactive toward bis-alkylation, whereas the other linkers require more optimization. These electronic effects would also explain the high reactivity of the mono-alkylated peptide to side reactions when a second strong nucleophile is not present in the peptide.
Fig. 8.

Nucleophilic attacks for closing peptide macrocycles. (A) Lactam formation is widely used in macrocyclization reactions, but requires attack of a primary amine on an activated ester. (B) If the second substitution is SN2-like, the ring closure step for thiol bis-alkylation will involve attack by a thiolate on the benzylic bromide. (C) If the second substitution is SN1-like, the ring closure step for thiol bis-alkylation will involve attack by a thiolate on a benzylic carbocation. Note that the nucleophiles shown in (B) and (C) could also be sulfides (as in the case of methionine).
Thiol bis-alkylation appears to form intramolecular cross-links with minimal dependence on the sequence or structure of the intervening peptide. This is generally not the case with other macrocyclization approaches (Table 1). We surmise that this major advantage is due to the high reactivity of the bromomethyl benzyl sulfide, as well as an increased tolerance in the angle of the approach of the nucleophile in the macrocyclization step. To illustrate this point, Fig. 8 contrasts lactam formation (where a carboxylic-acid-derived ester is attacked by a primary amine) with a thiolate attacking a benzylic bromide (SN2-like mechanism) and a thiolate attacking a carbocation (SN1-like mechanism). While more in-depth investigation is needed, this model illustrates why this chemistry is so robust and versatile for the formation of peptide macrocycles with varied 3D structures.
4.5 Optimizing and Troubleshooting Bis-Alkylation Reactions
Though this cross-linking strategy is very robust and can be easily used for any peptide containing reactive thiols, optimal conditions may vary for different peptides. Here we briefly discuss different conditions with respect to troubleshooting reactions with low yield, or optimizing reactions to maximize total yield.
4.5.1 Concentration
We and others (Dewkar, Carneiro, & Hartman, 2009; Timmerman et al., 2005) have shown that the peptide concentration for this reaction is optimal between 0.01 and 1 mM, but can also be achieved at higher concentrations. One of the byproducts encountered if the peptide is too concentrated is the cross-linked dimer. Trace amounts of this byproduct can often be observed even in highly successful reactions—see, for instance, the tiny peak at m/z=2520 in the mass spectrum of the reaction that produced peptide 2-mxy (Fig. 5). This is rarely a major product at concentrations 1 mM or below, and it can easily be avoided with a more dilute reaction mixture.
4.5.2 Relative Amount of Linker
While initial reports of cysteine bis-alkylation used a marginal excess (1.05 equiv.) of linker (Timmerman et al., 2005), we have found that using 1.5-fold excess to threefold excess generally results in clean reactions and rapid reaction times. At higher concentrations, using a threefold excess can increase the amount of cross-linked dimer. We have never observed higher yields or faster reactions above threefold excess of linker.
4.5.3 Solvent Selection
The optimal solvent for the reaction is very much dependent on the solubility of the peptide and the linker. A 1:1 mixture of buffered water and acetonitrile has worked well in our hands for most peptides and most linkers, particularly the oxy, mxy, pxy, and mpy linkers. Increasing the amount of acetonitrile can be useful for more hydrophobic peptide sequences and larger, more hydrophobic linkers, but it slows the overall reaction. The reaction can also be performed using a 1:1 mixture of buffered water and DMF, which would help with solubility (Hacker, Almohaini, Anbazhagan, Ma, & Hartman, 2015). Since reactions are more easily concentrated if they are run in acetonitrile/water, we have not extensively explored the use of DMF as a cosolvent.
4.5.4 Presence of Reducing Agents
One might assume that a major competing reaction would be disulfide formation, either intermolecular to form a disulfide-linked dimer or intramolecular to form a disulfide-bridged macrocycle. Using degassed, deionized water and paying careful attention to pH (see later), we have rarely observed disulfide formation outcompeting bis-alkylation at the optimized conditions described earlier. In rare cases where bis-alkylation reactions are slow and are incubated overnight, we have observed some disulfide formation. One preventative measure is to use TCEP, a nonnucleophilic, nonsulfur-containing reducing agent which is not reactive with the linker. If disulfide formation has been observed, 100–200 μM TCEP can be added to the peptide prior to cyclization. As observed in Fig. 7, TCEP adducts (+250 Da) can sometimes be observed by mass spectrometry following the reaction, but these are not covalent adducts and typically disappear after acidification and HPLC purification. We do not recommend using TCEP routinely, as the thiol bis-alkylation is typically much faster than oxidation. Rather, we recommend using degassed aqueous buffers and, in general, storing all thiol-containing and sulfide-containing peptides as lyophilized powders rather than DMSO stocks. Overall, we have had excellent success using freshly prepared peptides in bis-alkylation reactions without reducing agents, compared to peptides that have been stored for prolonged periods of time.
4.5.5 pH Dependence
In very few cases, we have observed no product formation with a dithiol-containing peptide. In these cases, the major problem has typically been pH. For instance, TFA can be carried over with the peptides following ether precipitation, altering the resulting pH of the reaction solution. We check reaction pH immediately prior to adding the linker, usually by spotting a small drop onto pH paper, in order to verify the pH is still around 8.0. Overall, we have found that maintaining the pH above 7.6 is critical for clean, fast bis-alkylation reactions. At lower pH, reactions are sluggish or do not progress, typically yielding only starting material. In systematic testing, we have not observed significant improvements in yields or reaction times at pH 8.5, 9, or 9.5.
5. FUTURE DIRECTIONS
This chapter describes a methodology that can be used to produce a large synthetic library of loops as inhibitors for protein–protein interactions. By varying the thiol-containing amino acids, their relative positions, and the linkers, it is possible to lock any individual epitope into a large variety of conformations and rapidly screen them for a desired function. Though these macrocycles make great chemical biology tools for studying specific protein–protein interactions, they are not considered “drug-like” molecules. The cross-link affords structural stability, which generally increases metabolic stability and cell penetration compared to linear peptides (Bock et al., 2013). However, an anticipated problem for thiol bis-alkylated peptides is that benzyl sulfides may be readily oxidized in vivo. Still, these molecules could be regarded as a starting point for drug development, and medicinal chemistry could rapidly identify a suitable isosteric replacement for the thioethers, if necessary. Thus, for chemical biology and drug development, this strategy represents an efficient, rapid, and robust method for the discovery of macrocyclic inhibitors of protein–protein interactions.
References
- Arkin MR, Tang Y, Wells JA. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chemistry and Biology. 2014;21(9):1102–1114. doi: 10.1016/j.chembiol.2014.09.001. http://doi.org/10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avan I, Hall CD, Katritzky AR. Peptidomimetics via modifications of amino acids and peptide bonds. Chemical Society Reviews. 2014;43(10):3575–3594. doi: 10.1039/c3cs60384a. http://doi.org/10.1039/c3cs60384a. [DOI] [PubMed] [Google Scholar]
- Bashiruddin NK, Nagano M, Suga H. Bioorganic Chemistry Synthesis of fused tricyclic peptides using a reprogrammed translation system and chemical modification. Bioorganic Chemistry. 2015;61:45–50. doi: 10.1016/j.bioorg.2015.06.002. http://doi.org/10.1016/j.bioorg.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Berthelot T, Gonçalves M, Laïn G, Estieu-Gionnet K, Déléris G. New strategy towards the efficient solid phase synthesis of cyclopeptides. Tetrahedron. 2006;62(6):1124–1130. http://doi.org/10.1016/j.tet.2005.10.080. [Google Scholar]
- Bird GH, Irimia A, Ofek G, Kwong PD, Wilson IA, … Walensky LD. Stapled HIV-1 peptides recapitulate antigenic structures and engage broadly neutralizing antibodies. Nature Structural & Molecular Biology. 2014;21(12):1058–1067. doi: 10.1038/nsmb.2922. http://doi.org/10.1038/nsmb.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock JE, Gavenonis J, Kritzer JA. Getting in shape: Controlling peptide bio-activity and bioavailability using conformational constraints. ACS Chemical Biology. 2013;8(3):488–499. doi: 10.1021/cb300515u. http://doi.org/10.1021/cb300515u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockus AT, Schwochert JA, Pye CR, Townsend CE, Sok V, Bednarek MA, Lokey RS. Going out on a limb: Delineating the effects of beta-branching, N-methylation, and side chain size on the passive permeability, solubility, and flexibility of sanguinamide a analogues. Journal of Medicinal Chemistry. 2015;58(18):7409–7418. doi: 10.1021/acs.jmedchem.5b00919. http://doi.org/10.1021/acs.jmedchem.5b00919. [DOI] [PubMed] [Google Scholar]
- Brunel FM, Dawson PE. Synthesis of constrained helical peptides by thioether ligation: Application to analogs of gp41. Chemical Communications (Cambridge, England) 2005:2552–2554. doi: 10.1039/b419015g. http://doi.org/10.1039/b419015g. [DOI] [PubMed]
- Brust A, Wang CIA, Daly NL, Kennerly J, Sadeghi M, Christie MJ, … Alewood PF. Vicinal disulfide constrained cyclic peptidomimetics: A turn mimetic scaffold targeting the norepinephrine transporter. Angewandte Chemie. 2013;125(46):12242–12245. doi: 10.1002/anie.201304660. http://doi.org/10.1002/ange.201304660. [DOI] [PubMed] [Google Scholar]
- Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, … Sawyer TK. Stapled α-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(36):E3445–E3454. doi: 10.1073/pnas.1303002110. http://doi.org/10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee J, Gilon C, Hoffman A, Kessler H. N-methylation of peptides: A new perspective in medicinal chemistry. Accounts of Chemical Research. 2008;41(10):1331–1342. doi: 10.1021/ar8000603. http://doi.org/10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- Chen S, Gopalakrishnan R, Schaer T, Marger F, Hovius R, Bertrand D, … Heinis C. Dithiol amino acids can structurally shape and enhance the ligand-binding properties of polypeptides. Nature Chemistry. 2014;6(11):1009–1016. doi: 10.1038/nchem.2043. http://doi.org/10.1038/nchem.2043. [DOI] [PubMed] [Google Scholar]
- Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267(5196):383–386. doi: 10.1126/science.7529940. http://doi.org/10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angewandte Chemie International Edition. 2010;49(37):6545–6548. doi: 10.1002/anie.201000620. http://doi.org/10.1002/anie.201000620. [DOI] [PubMed] [Google Scholar]
- Cudic M, Wade JD, Otvos L., Jr Convenient synthesis of a head-to-tail cyclic peptide containing an expanded ring. Tetrahedron Letters. 2000;41(23):4527–4531. http://doi.org/10.1016/S0040-4039(00)00629-8. [Google Scholar]
- Dahan A, Tsume Y, Sun J, Miller JM, Amidon GL. Amino acids, peptides and proteins in organic chemistry. Vol. 4. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2011. Oral bioavailability of peptide and peptidomimetic drugs; pp. 277–292. http://doi.org/10.1002/9783527631827.ch8. [Google Scholar]
- Damas JM, Filipe LCS, Campos SRR, Lousa D, Victor BL, Baptista AM, Soares CM. Predicting the thermodynamics and kinetics of helix formation in a cyclic peptide model. Journal of Chemical Theory and Computation. 2013;9(11):5148–5157. doi: 10.1021/ct400529k. http://doi.org/10.1021/ct400529k. [DOI] [PubMed] [Google Scholar]
- Dewkar GK, Carneiro PB, Hartman MCT. Synthesis of novel peptide linkers: Simultaneous cyclization and labeling. Organic Letters. 2009;11(20):4708–4711. doi: 10.1021/ol901662c. http://doi.org/10.1021/ol901662c. [DOI] [PubMed] [Google Scholar]
- Fields G, Noble R. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. International Journal of Peptide and Protein Research. 1990;35(3):161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. http://doi.org/10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- Fosgerau K, Hoffmann T. Peptide therapeutics: Current status and future directions. Drug Discovery Today. 2014;20(1):122–128. doi: 10.1016/j.drudis.2014.10.003. http://doi.org/10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Gao Y, Amar S, Pahwa S, Fields G, Kodadek T. Rapid lead discovery through iterative screening of one bead one compound libraries. ACS Combinatorial Science. 2015;17:49–59. doi: 10.1021/co500154e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. DNA-templated organic synthesis and selection of a library of macrocycles. Science (New York, NY) 2004;305(September):1601–1605. doi: 10.1126/science.1102629. http://doi.org/10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavenonis J, Sheneman BA, Siegert TR, Eshelman MR, Kritzer JA. Comprehensive analysis of loops at protein-protein interfaces for macrocycle design. Nature Chemical Biology. 2014;10(9):1–8. doi: 10.1038/nchembio.1580. http://doi.org/10.1038/nchembio.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Katoh T, Suga H. Flexizymes for genetic code reprogramming. Nature Protocols. 2011;6(6):779–790. doi: 10.1038/nprot.2011.331. http://doi.org/10.1038/nprot.2011.331. [DOI] [PubMed] [Google Scholar]
- Hacker DE, Almohaini M, Anbazhagan A, Ma Z, Hartman MCT. Methods in molecular biology. Vol. 1248. New York: Springer; 2015. Peptide and peptide library cyclization via bromomethylbenzene derivatives; p. 278. http://doi.org/10.1007/978-1-4939-2020-4. [DOI] [PubMed] [Google Scholar]
- Han SY, Kim YA. Recent development of peptide coupling reagents in organic synthesis. Tetrahedron. 2004;60(11):2447–2467. http://doi.org/10.1016/j.tet.2004.01.020. [Google Scholar]
- Hayouka Z, Levin A, Hurevich M, Shalev DE, Loyter A, Gilon C, Friedler A. A comparative study of backbone versus side chain peptide cyclization: Application for HIV-1 integrase inhibitors. Bioorganic and Medicinal Chemistry. 2012;20(10):3317–3322. doi: 10.1016/j.bmc.2012.03.039. http://doi.org/10.1016/j.bmc.2012.03.039. [DOI] [PubMed] [Google Scholar]
- Heinis C, Rutherford T, Freund S, Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nature Chemical Biology. 2009;5(7):502–507. doi: 10.1038/nchembio.184. http://doi.org/10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- Henchey LK, Jochim AL, Arora PS. Contemporary strategies for the stabilization of peptides in the α-helical conformation. Current Opinion in Chemical Biology. 2008;12(6):692–697. doi: 10.1016/j.cbpa.2008.08.019. http://doi.org/10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill TA, Shepherd NE, Diness F, Fairlie DP. Constraining cyclic peptides to mimic protein structure motifs. Angewandte Chemie International Edition. 2014;53(48):13020–13041. doi: 10.1002/anie.201401058. http://doi.org/10.1002/anie.201401058. [DOI] [PubMed] [Google Scholar]
- Jo H, Meinhardt N, Wu Y, Kulkarni S, Hu X, Low KE, … Greenbaum DC. Development of α-helical calpain probes by mimicking a natural protein-protein interaction. Journal of the American Chemical Society. 2012;134(42):17704–17713. doi: 10.1021/ja307599z. http://doi.org/10.1021/ja307599z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamens AJ, Eisert RJ, Corlin T, Baleja JD, Kritzer JA. Structured cyclic peptides that bind the EH domain of EHD1. Biochemistry. 2014;53:4758–4760. doi: 10.1021/bi500744q. http://doi.org/10.1021/bi500744q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discovery Today. 2013;18(17–18):807–817. doi: 10.1016/j.drudis.2013.05.011. http://doi.org/10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Kawamoto SA, Coleska A, Ran X, Yi H, Yang CY, Wang S. Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. Journal of Medicinal Chemistry. 2012;55(3):1137–1146. doi: 10.1021/jm201125d. http://doi.org/10.1021/jm201125d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo KH, Hoe KK, Verma CS, Lane DP. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nature Reviews Drug Discovery. 2014;13(3):217–236. doi: 10.1038/nrd4236. http://doi.org/10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- Kim YW, Kutchukian PS, Verdine GL. Introduction of all-hydrocarbon i, i+3 staples into alpha-helices via ring-closing olefin metathesis. Organic Letters. 2010;12(13):3046–3049. doi: 10.1021/ol1010449. http://doi.org/10.1021/ol1010449. [DOI] [PubMed] [Google Scholar]
- Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(22):14116–14121. doi: 10.1073/pnas.202485799. http://doi.org/10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme T, Kim DE, Baker D. Computational alanine scanning of protein-protein interfaces. Science’s STKE: Signal Transduction Knowledge Environment. 2004;2004(219):l2. doi: 10.1126/stke.2192004pl2. http://doi.org/10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- London N, Raveh B, Movshovitz-Attias D, Schueler-Furman O. Can self-inhibitory peptides be derived from the interfaces of globular protein-protein interactions? Proteins: Structure, Function, and Bioinformatics. 2010;78(15):3140–3149. doi: 10.1002/prot.22785. http://doi.org/10.1002/prot.22785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Blackwell H, Grubbs R. Application of ring-closing metathesis to the synthesis of rigidified amino acids and peptides. Journal of the American Chemical Society. 1996;49(50):9606–9614. Retrieved from, http://pubs.acs.org/doi/abs/10.1021/ja961626l. [Google Scholar]
- Moss J. Guide for resin and linker selection in solid-phase peptide synthesis. Current Protocols in Protein Science. 2005;Chapter 18:18.7.1–18.7.19. doi: 10.1002/0471140864.ps1807s40. http://doi.org/10.1002/0471140864.ps1807s40. [DOI] [PubMed] [Google Scholar]
- Muppidi A, Wang Z, Li X, Chen J, Lin Q. Achieving cell penetration with distance-matching cysteine cross-linkers: A facile route to cell-permeable peptide dual inhibitors of Mdm2/Mdmx. Chemical Communications (Cambridge, England) 2011;47(33):9396–9398. doi: 10.1039/c1cc13320a. http://doi.org/10.1039/c1cc13320a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muppidi A, Zhang H, Curreli F, Li N, Debnath AK, Lin Q. Design of antiviral stapled peptides containing a biphenyl cross-linker. Bioorganic & Medicinal Chemistry Letters. 2014;24(7):1748–1751. doi: 10.1016/j.bmcl.2014.02.038. http://doi.org/10.1016/j.bmcl.2014.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passioura T, Katoh T, Goto Y, Suga H. Selection-based discovery of druglike macrocyclic peptides. Annual Review of Biochemistry. 2014;83:727–752. doi: 10.1146/annurev-biochem-060713-035456. http://doi.org/10.1146/annurev-biochem-060713-035456. [DOI] [PubMed] [Google Scholar]
- Patgiri A, Jochim AL, Arora PS. A hydrogen bond surrogate approach for stabilization of short peptide sequences in α-helical conformation. Accounts of Chemical Research. 2008;41(10):1289–1300. doi: 10.1021/ar700264k. http://doi.org/10.1021/ar700264k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Liu T, Liu YY, Briesewitz R, Barrios AM, Jhiang SM, Pei D. Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chemical Biology. 2013;8(2):423–431. doi: 10.1021/cb3005275. http://doi.org/10.1021/cb3005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvit N, Hatzubai A, Shalev DE, Friedler A, Ben-Neriah Y, Gilon C. Design and synthesis of backbone cyclic phosphorylated peptides: The IkB model. Biopolymers. 2009;91(2):157–168. doi: 10.1002/bip.21098. http://doi.org/10.1002/bip.21098. [DOI] [PubMed] [Google Scholar]
- Razavi AM, Wuest WM, Voelz VA. Computational screening and selection of cyclic peptide hairpin mimetics by molecular simulation and kinetic network models. Journal of Chemical Information and Modeling. 2014;54(5):1425–1432. doi: 10.1021/ci500102y. http://doi.org/10.1021/ci500102y. [DOI] [PubMed] [Google Scholar]
- Renukuntla J, Vadlapudi AD, Patel A, Boddu SHS, Mitra AK. Approaches for enhancing oral bioavailability of peptides and proteins. International Journal of Pharmaceutics. 2013;447(1–2):75–93. doi: 10.1016/j.ijpharm.2013.02.030. http://doi.org/10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson NS, Jamieson AG. Regulation of protein-protein interactions using stapled peptides. Reports in Organic Chemistry. 2015;5:65–74. http://doi.org/10.2147/ROC.S68161. [Google Scholar]
- Rumpf J, Simon B, Jung N, Maritzen T, Haucke V, Sattler M, Groemping Y. Structure of the Eps15-stonin2 complex provides a molecular explanation for EH-domain ligand specificity. The EMBO Journal. 2008;27(3):558–569. doi: 10.1038/sj.emboj.7601980. http://doi.org/10.1038/sj.emboj.7601980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. Journal of the American Chemical Society. 2000;122(6):5891–5892. [Google Scholar]
- Schlippe YV, Hartman MC, Josephson K, Szostak JW. In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. Journal of the American Chemical Society. 2012;134(25):10469–10477. doi: 10.1021/ja301017y. http://doi.org/10.1021/ja301017y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton PT, Jensen KJ. Linkers, resins, and general procedures for solid-phase peptide synthesis. Methods in Molecular Biology. 2013;1047:23–41. doi: 10.1007/978-1-62703-544-6_2. [DOI] [PubMed] [Google Scholar]
- Shepherd NE, Hoang HN, Abbenante G, Fairlie DP. Single turn peptide alpha helices with exceptional stability in water. Journal of the American Chemical Society. 2005;127(9):2974–2983. doi: 10.1021/ja0456003. http://doi.org/10.1021/ja0456003. [DOI] [PubMed] [Google Scholar]
- Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, … Levine B. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. doi: 10.1038/nature11866. http://doi.org/10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk LEJ, Dailly N, Hiemstra H, van Maarseveen JH, Timmerman P. Synthesis of water-soluble scaffolds for peptide cyclization, labeling, and ligation. Organic Letters. 2012;14(5):1194–1197. doi: 10.1021/ol203259a. http://doi.org/10.1021/ol203259a. [DOI] [PubMed] [Google Scholar]
- Spokoyny A, Zou Y, Ling J, Yu H, Lin YS, Pentelute B. A perfluoroaryl-cysteine SNAr chemistry approach to unprotected peptide stapling. Journal of the American Chemical Society. 2013;135:5946–5949. doi: 10.1021/ja400119t. Retrieved from, http://pubs.acs.org/doi/abs/10.1021/ja400119t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AD, Dugan A, Gestwicki JE, Mapp AK. Fine-tuning multiprotein complexes using small molecules. ACS Chemical Biology. 2012;7(8):1311–1320. doi: 10.1021/cb300255p. http://doi.org/10.1021/cb300255p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman P, Beld J, Puijk WC, Meloen RH. Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem: A European Journal of Chemical Biology. 2005;6(5):821–824. doi: 10.1002/cbic.200400374. http://doi.org/10.1002/cbic.200400374. [DOI] [PubMed] [Google Scholar]
- Todorova-Balvay D, Stoilova I, Gargova S, Vijayalakshmi MA. An efficient two step purification and molecular characterization of beta-galactosidases from Aspergillus oryzae. Journal of Molecular Recognition: JMR. 2007;19(4):299–304. doi: 10.1002/jmr.788. http://doi.org/10.1002/jmr. [DOI] [PubMed] [Google Scholar]
- Urech-Varenne C, Radtke F, Heinis C. Phage selection of bicyclic peptide ligands of the Notch1 receptor. ChemMedChem. 2015;10(10):1754–1761. doi: 10.1002/cmdc.201500261. http://doi.org/10.1002/cmdc.201500261. [DOI] [PubMed] [Google Scholar]
- Wakefield AE, Wuest WM, Voelz VA. Molecular simulation of conformational pre-organization in cyclic RGD peptides. Journal of Chemical Information and Modeling. 2015;55(4):806–813. doi: 10.1021/ci500768u. http://doi.org/10.1021/ci500768u. [DOI] [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, … Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science (New York, NY) 2004;305(2002):1466–1470. doi: 10.1126/science.1099191. http://doi.org/10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chou DH-C. A thiolene coupling approach to native peptide stapling and macrocyclization. Angewandte Chemie International Edition. 2015;54(37):10931–10934. doi: 10.1002/anie.201503975. http://doi.org/10.1002/anie.201503975. [DOI] [PubMed] [Google Scholar]
- Watkins AM, Arora PS. Anatomy of β-strands at protein–protein interfaces. ACS Chemical Biology. 2014;9:1747–1754. doi: 10.1021/cb500241y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White ER, Sun L, Ma Z, Beckta JM, Danzig BA, Hacker DE, … Hartman MCT. Peptide library approach to uncover phosphomimetic inhibitors of the BRCA1 C-terminal domain. ACS Chemical Biology. 2015;10:1198–1208. doi: 10.1021/cb500757u. http://doi.org/10.1021/cb500757u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CJ, Yudin AK. Contemporary strategies for peptide macrocyclization. Nature Chemistry. 3(7):509–524. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- Wuo MG, Mahon AB, Arora PS. An effective strategy for stabilizing minimal coiled coil mimetics. Journal of the American Chemical Society. 2015;137(36):11618–11621. doi: 10.1021/jacs.5b05525. http://doi.org/10.1021/jacs.5b05525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Wang Y, Lau EY, Luo J, Yao N, Shi C, … Lam KS. The use of one-bead one-compound combinatorial library technology to discover high-affinity alphavbeta3 integrin and cancer targeting arginine-glycine-aspartic acid ligands with a built-in handle. Molecular Cancer Therapeutics. 2010;9(10):2714–2723. doi: 10.1158/1535-7163.MCT-10-0308. http://doi.org/10.1158/1535-7163.MCT-10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi Y, Shoji I, Miyagawa S, Kawakami T, Katoh T, Goto Y, Suga H. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chemistry & Biology. 2011;18(12):1562–1570. doi: 10.1016/j.chembiol.2011.09.013. http://doi.org/10.1016/j.chembiol.2011.09.013. [DOI] [PubMed] [Google Scholar]
- Yin H. Constrained peptides as miniature protein structures. ISRN Biochemistry. 2012;2012:1–15. doi: 10.5402/2012/692190. http://doi.org/10.5402/2012/692190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lin YS. Toward structure prediction of cyclic peptides. Physical Chemistry Chemical Physics. 2015;17(6):4210–4219. doi: 10.1039/c4cp04580g. http://doi.org/10.1039/C4CP04580G. [DOI] [PubMed] [Google Scholar]
- Zimm BH, Bragg JK. Theory of the phase transition between helix and random coil in polypeptide chains. The Journal of Chemical Physics. 1959;31(2):526–535. http://doi.org/10.1063/1.1730390. [Google Scholar]