

Abstract
Aging is associated with increased cardiac interstitial fibrosis and diastolic dysfunction. Our previous study has shown that mesenchymal fibroblasts in the C57BL/6J (B6J) aging mouse heart acquire an inflammatory phenotype and produce higher levels of chemokines. Monocyte chemoattractant protein-1 (MCP-1) secreted by these aged fibroblasts promotes leukocyte uptake into the heart. Some of the monocytes that migrate into the heart polarize into M2a macrophages/myeloid fibroblasts. The number of activated mesenchymal fibroblasts also increases with age and consequently both sources of fibroblasts contribute to fibrosis. Here we further investigate mechanisms by which inflammation influences activation of myeloid and mesenchymal fibroblasts and their collagen synthesis.
We examined cardiac fibrosis and heart function in three aged mouse strains; we compared C57BL/6J (B6J) with two other strains that have reduced inflammation via different mechanisms. Aged C57BL/6N (B6N) hearts are protected from oxidative stress and fibroblasts derived from them do not develop an inflammatory phenotype. Likewise these mice have preserved diastolic function. Aged MCP-1 null mice on the B6J background (MCP-1KO) are protected from elevated leukocyte infiltration; they develop moderate but reduced fibrosis and diastolic dysfunction. Based on these studies we further delineated the role of resident versus monocyte-derived M2a macrophages in myeloid-dependent fibrosis and found that the number of monocyte-derived M2a (but not resident) macrophages correlates with age-related fibrosis and diastolic dysfunction. In conclusion, we have found that ROS and inflammatory mediators are necessary for activation of fibroblasts of both developmental origins, and prevention of either led to better functional outcomes.
Keywords: heart, fibrosis, inflammation, aging, fibroblast
INTRODUCTION
Interstitial fibrosis is a pathologic process that increases the passive stiffness of the heart and contributes significantly to impairment of diastolic function [3, 39]. Diastolic dysfunction limits the maximum exertion of healthy elders [53] and predisposes them to heart failure with preserved ejection fraction (HFpEF). This is now the most common type of heart failure for those over 65 and it has proved very challenging to ameliorate. Efforts to treat HFpEF have consistently been unrewarding in comparison with heart failure with reduced systolic function (HFrEF). In some recent studies HFpEF now has a worse prognosis than HFrEF. Aging is one of the strongest risk factors for HFpEF, and aging is associated with interstitial fibrosis in the heart.
Aging myocardium is characterized by increased collagen content, reduced elasticity and increased hypertrophy [33, 46, 47]. Interstitial fibrosis is associated with dysregulation of resident mesenchymal fibroblasts in the aged myocardium [8]. Recently, inflammation has been implicated as a primary factor contributing to both fibrosis and HFpEF [20, 55]. In our previous reports we have postulated that fibrosis in aging C57BL/6J (B6J) hearts depends on fibroblasts of two different developmental origins: mesenchymal and myeloid [9, 49], and here we present compelling evidence that inflammatory mechanisms promote fibrosis involving both cell types.
Myeloid fibroblast
Monocyte chemoattractant protein-1 (MCP-1) has been identified as a factor critical for infiltration of blood leukocytes into the tissue of young mice in injury models such as intermittent ischemia/reperfusion cardiomyopathy (I/RC) [17] and angiotensin II (Ang II) infusion [22]. In both models, monocytes (that migrated in response to MCP-1) polarize into alternatively activated M2a macrophages expressing collagen (and effectively become myeloid fibroblasts) and inflammation directly correlates with the degree of fibrosis. MCP-1 null mice (MCP-1KO) subjected to Ang II infusion do not display increased leukocyte infiltration, do not have an increased number of myeloid fibroblasts and do not develop fibrosis [22], which demonstrates the critical role of MCP-1 in the development of myeloid-dependent fibrosis in an acute injury model. Our current data demonstrate the importance of MCP-1 in the development of myeloid-dependent fibrosis in the aged uninjured heart that is dependent on reactive oxygen species (ROS).
Mesenchymal fibroblast
We have extensively studied cardiac fibrosis in aged B6J mice [6] that manifest a similar age-related dysfunction as the human aging heart [18]. We have shown that mesenchymal fibroblasts with age progression become dysregulated predominantly because they develop a reduced TGF-β sensitivity due to decreased expression of TGF-β receptor I [10]. These fibroblasts become pro-inflammatory and secrete higher levels of cytokines such as MCP-1 and interleukin-6 (IL-6) among others [11] and also produce collagen type I a (Col1a) in response to a Ras-Erk pathway [8].
The role of ROS in fibrosis
The increased expression of cytokines and collagen seen in aged B6J hearts has been attributed to elevated levels of ROS. We found that mesenchymal fibroblasts derived from old hearts display both increased levels of superoxide and elevated cytokine secretion [9]. The role of ROS in the pathology of the aging heart has been widely hypothesized and studied [13, 43]. We noted that another mouse strain, C57BL/6N (B6N), when compared with B6J, has less cardiac fibrosis and related cardiac dysfunction during aging when studied in our laboratory under identical conditions. Genetic comparisons of B6J and B6N strains show differences in coding sequences manifested as SNPs and a partial deletion of the nicotinamide nucleotide transhydrogenase (Nnt) gene [26, 42]. Nnt catalyzes the reversible reduction of nicotinamide adenine dinucleotide phosphate (NADP+) by reduced nicotinamide adenine dinucleotide (NADH) into reduced nicotinamide adenine dinucleotide phosphate (NADPH) and converts NADH into nicotinamide adenine dinucleotide (NAD+) [25]. NADPH is required to reduce glutathione and thereby decrease ROS levels [1]. A lack of functional Nnt is seen in B6J mice (as a result of a spontaneous mutation) and reduced levels of Nnt in humans with heart failure [41] may predispose them to cardiac fibrosis. However it is unclear if aged B6N mice (with functional Nnt) can develop inflammation and fibrosis.
Here, we analyze fibrosis in three aging heart models, in which MCP-1KO and B6N hearts display diminished inflammation compared to B6J, which have a pro-inflammatory phenotype. We demonstrate that fibroblasts from two developmental origins (myeloid and mesenchymal) engage in collagen production. We provide evidence that elevated ROS levels correlate with the pro-inflammatory phenotype that drives infiltration of leukocytes into the heart. Within the infiltrating leukocyte pool, monocytes give rise to M2a alternatively polarized macrophages/myeloid fibroblasts that participate in myeloid-dependent fibrosis.
The inflammatory mesenchymal fibroblasts in aged B6J hearts are also a source of augmented collagen synthesis that is dependent on ROS and/or inflammatory dysregulation. The cellular/histological findings are consistent with observed levels of passive stiffness (functional analysis), wherein B6J ventricles were stiffer than the ones from MCP-1KO or B6N hearts. Overall, our data suggest that inflammation can dictate the severity of interstitial fibrosis and can affect mesenchymal fibroblast activation and cardiac dysfunction.
METHODS
Animals
Young (3 month-old) C57BL6/J (#000664) male mice were purchased from The Jackson Laboratory (Bar Harbor, ME), 20–30 month-old male C57BL/6J mice were obtained from the National Institute of Aging, and 8 month-old MCP-1KO males (#004434, B6.129S4-CCL2tm1Rol/J) were purchased from The Jackson Laboratory and aged until they were 21–24 months old. MCP-1KO mice were backcrossed to the B6J background for more than 11 generations. 8 month-old C57BL/6N males (#027) were purchased from Charles River (Shrewsbury, MA) and aged to 21–24 months of age. All animals were treated in accordance with the NIH Guide for the Care and Use of Laboratory Animals [DHHS publication (NIH) 85-23, revised 1996], and approved by the Baylor College of Medicine Institutional Animal Care and Use Committee.
Human frozen heart section immunofluorescence staining
Frozen sections of human hearts were purchased from Zyagen (San Diego, CA). For immunofluorescence analysis CD44-PE (#103007, BioLegend, San Diego, CA), CD45-PE (#IM2078U, Beckman Coulter, Brea, CA), collagen type I a (#600-401-103, Rockland Immunochemicals, Gilbertsville, PA), the secondary affinity-purified F(ab′)2 anti-rabbit antibody conjugated to fluorescein (#711-096-152, Jackson ImmunoResearch, West Grove, PA) or phycoerythrin (#711-116-152, Jackson ImmunoResearch) and appropriate isotype controls were used.
IF mouse
Paraffin-embedded zinc-Tris fixed mouse heart sections were stained with the following antibodies: CD45-PE (#103106, BioLegend), collagen type 1 a (#600-401-103, Rockland Immunochemicals), CD44 (#25340, Abcam), procollagen type 1a (# sc25973, Santa Cruz Bioechnology, Santa Cruz, CA), followed by the secondary affinity-purified F(ab′)2 anti-rabbit antibody conjugated to FITC (#705-096-147, Jackson ImmunoResearch) or conjugated to DyLight 549 (#712-506-153, Jackson ImmunoResearch).
Cell isolation
Hearts were cut into 1 mm3 pieces, digested with Liberase TH (Roche Diagnostics, Indianapolis, IN) and incubated in a 37°C shaking water bath with regular trituration by pipet to obtain a single cell suspension (all non-myocytes). Afterwards, cells were centrifuged at 250xg for 5 min. The cell pellet was washed and then suspended in cell growth medium [5].
Tissue culture
Cells were cultured in growth medium comprised of DMEM/F12 (1:1) (ThermoFisher Scientific, Waltham, MA) supplemented with 10% FBS (Atlanta Biologicals, Flowery Branch, GA) and 1% antibiotic-antimycotic (ThermoFisher Scientific). Quiescence was initiated 24 h before each experiment by switching the medium to low glucose DMEM without FBS.
Monocyte migration assay
Spleens were mechanically dissociated and the tissue digested with Liberase DL and DNase (Roche Diagnostics) [12]. Red cells were lysed using 1X RBC Lysis Buffer Solution (eBioscience, San Diego, CA). Monocytes were enriched by negative selection using the Easy Sep Mouse Monocyte Enrichment Kit (Stem Cell Technologies, Cambridge, MA). Monocytes (1×106) were placed within 3 μm pore cell culture inserts (Corning, Corning, NY). Wells below the inserts were supplemented with or without 10 ng/ml MCP-1 (R&D Systems, Minneapolis, MN). Cells that migrated through the pores were stained using crystal violet and counted.
Flow cytometry
Cells were incubated with antibodies to CD45-PECy5 (#103110, BioLegend) and CD44-PE (#ab25224, Abcam, Cambridge, MA) or the appropriate isotype controls. Calcein (ThermoFisher Scientific) was added to samples to determine cell viability and we proceeded with analysis of external antigens. Cells that were analyzed for internal antigens were fixed in 2% paraformaldehyde with 0.1% saponin for 20 minutes at 4°C, washed and then incubated with antibodies to collagen type 1 a (#600-401-103, Rockland Immunochemicals) or CD301-PE (#145704, BioLegend) or IgG controls. The secondary anti-rabbit antibody conjugated to fluorescein (#711-096-152, Jackson ImmunoResearch) was used for labeling intracellular collagen and cells were analyzed on a Cell Lab Quanta SC flow cytometer (Beckman Coulter). Quanta Analysis and FloJo (Ashland, OR) software were used for analysis.
Superoxide anion assay
Mesenchymal fibroblasts derived from young and aged hearts were cultured in growth medium (as specified above). 24 h before assay cells were serum-starved. 5 μM MitoSOX reagent (ThermoFisher Scientific) in Hank’s balanced salt solution with calcium and magnesium was applied to cells for 10 min. Next, cells were washed, nuclei were stained with Dapi and then cells were immediately analyzed by fluorescence microscopy.
Protein array
106 cells were synchronized via FBS withdrawal, and then after 24 h medium was changed again to low glucose DMEM (ThermoFisher Scientific) (without FBS) and subsequently medium was collected 24 h later and supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (ThermoFisher Scientific). Next, the medium was concentrated using centrifugal concentrators with a 5 kDA molecular weight cut off (Sartorius, Goettingen, Germany). 100 μg of protein was loaded onto mouse cytokine antibody array membranes (RayBiotech, Norcross, GA). Membranes were processed according to the manufacturer’s instructions, images were obtained using the Odyssey imaging system (Li-cor Biosciences, Lincoln, NE), and densitometry was analyzed by ImageJ software (NIH, Bethesda, MA). Data are expressed as integrated pixel density normalized to positive controls and with the background subtracted.
Doppler and two-dimensional echocardiography
Anatomic and functional parameters were obtained by B-mode and M-mode echocardiography (Vevo 770; VisualSonics, Toronto, Canada) as described previously with the mice under 1% Isoflurane gas in oxygen on a heated ECG board. Transmitral and aortic flow velocity data was recorded using a 10 MHz pulsed probe and Doppler Ultrasound Model DSPW (Indus Instruments, Houston, TX) as we described [6, 21].
Parametrized Diastolic Filling Formalism
Values for a spring model of diastolic filling were obtained as previously described by Kovacs [27]. The Mitral flow velocity envelope was used to generate velocity versus time data. We required the velocity to decrease to 50% of peak value before the start of the A wave. The values were input to MATLAB and fitted to a curve for a damped simple harmonic oscillator described by the following formula:
The fit for the E-wave was accepted when the r2 was ≥ 0.99 and values for c, k and x0 were obtained, (h is a time offset). Five beats were used for each animal.
Statistical analysis
Results are presented as mean ± SEM. Comparison between 2 groups was made by an unpaired Student’s t-test with Welch correction. For multiple group comparison ANOVA with Tukey-Kramer Multiple Comparisons test was applied. Non-parametric Anova (Kruskal-Wallis Test) was applied when Barlett’s test suggested that the differences among standard deviations were significant. Differences were considered statistically significant if p<0.05.
RESULTS
1. Leukocyte recruitment and macrophage polarization contribute to myeloid-dependent fibrosis
We examined the number of total leukocytes in the young B6J heart versus aged hearts from B6J, MCP-1KO and B6N mice (Supplemental Fig. 1A depicts the gating strategy). A flow cytometry analysis showed a 50% increase in accumulated CD45+ leukocytes in the aged B6J heart but not in other aged hearts (Fig. 1A). There are two sources of macrophages in the heart, residents that express lower levels of CD45 (CD45lo) that is characteristic of resident macrophages in other organs [51] and those derived from infiltrating monocytes (CD45hi) [15]. Superimposed histograms showed a population of cells expressing higher levels of CD45 (CD45hi) that predominated in isolates from aged B6J hearts but not in the others (Fig. 1A, upper panel). The number of CD45hi in aged B6J increased three-fold when compared with three other groups, suggesting ongoing infiltration of CD45+ leukocytes into aged B6J hearts (Fig 1A lower panel, Supplemental Figure 1). Immunofluorescence staining of heart sections from three aged models confirmed our flow cytometry data (Fig. 1B), where we saw evenly distributed CD45+ cells in the aged B6J hearts and very few positive cells in aged MCP-1KO or B6N. (In contrast to the flow cytometry data, the immunofluorescence staining of mouse heart sections only detects the CD45hi population.) We found less collagen deposition in MCP-1KO hearts and very modest collagen accumulation in B6N hearts (Fig. 1B). Similarly, the aging human heart follows what we have observed in the aged B6J heart, namely that we observed increased numbers of CD45+ cells and augmented accumulation of collagen in older healthy people compared to the young human heart (Fig. 1C).
Fig. 1.
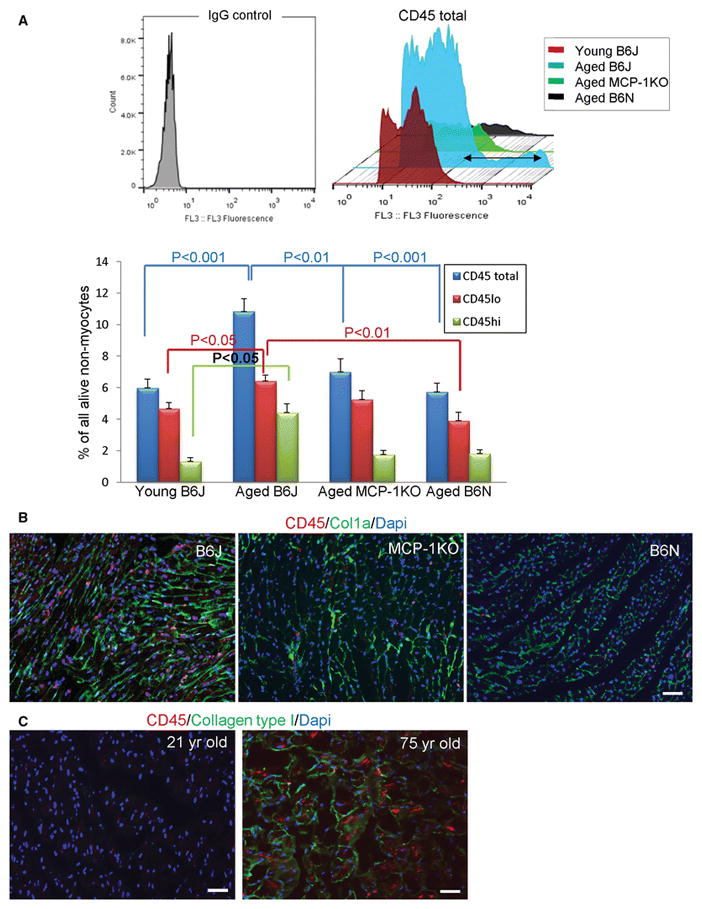
The increased number of CD45+ leukocytes in the aging B6J hearts. A, Flow cytometry analysis of calcein+ (viable) non-myocytes isolated from young (3 month-old, B6J), and aged (21–25 month-old B6J, MCP-1KO, B6N) mice was performed using CD45-PE antibody. Upper panel shows superimposed CD45+ alive cells from representative histograms depicting all four experimental groups. The black double arrow illustrates the expansion of CD45hi cell population that is abundant in cells isolated from aged B6J hearts. The lower panel portrays flow cytometry based quantification of CD45+ cells, cells that have low (CD45lo) or high (CD45hi) expression of CD45 in four experimental groups. Results are represented as mean ± SEM. N= 8, 12, 7, 7 for young B6J and aged B6J, MCP-1KO, and B6N respectively. B, Immunofluorescence staining of aged mouse paraffin heart sections or C, human frozen heart sections showing CD45 positive cells (red) within collagen type I deposition (green). Nuclei are stained with Dapi (blue). For mouse hearts, 21–24 month-old mice of an indicated genetic strain were used, N= 3, 2, 3 for biological repeats and N=3 for experimental repeats, scale bar=50 μm. For human heart N=1 per young (21 year-old) and aged (75 year-old), N=3 for experimental repeats, scale bar= 50 μm.
We have further characterized how CD45+ cells contribute to fibrosis by analyzing the number of M2a polarized macrophages/myeloid fibroblasts (Col1a+CD301+) in aging hearts gated on CD45lo or CD45hi populations (see Supplemental Figure 1 for the gating strategy). Aged B6N hearts had a reduced number of myeloid fibroblasts when compared with B6J and MCP-1KO hearts (Fig. 2A). In the aged B6J and MCP-1KO hearts we observed more than 5 and 2.5 fold higher levels of myeloid fibroblasts that derived from infiltrating monocytes (Col1a+CD301+CD45hi) than in young hearts (Fig. 2A). Myeloid fibroblasts that originated from resident macrophages (Col1a+CD301+CD45lo) were at similar levels in B6J and MCP-1KO hearts, with a trend towards reduced numbers in B6N hearts (Fig. 2A). Not only was the number of myeloid fibroblasts greatest in aged B6J hearts, but the level of collagen expression portrayed as mean fluorescence intensity (MFI) was almost two fold higher in aged B6J than in other hearts (Fig. 2B upper and lower panels).
Fig. 2.

The augmented participation from infiltrating monocytes into myeloid-dependent fibrosis in the aging B6J heart. A, Analysis of the contribution of resident (CD45lo) and infiltrating (CD45hi) leukocytes to the M2a macrophage/myeloid fibroblast pool by flow cytometry. B, An increased collagen expression by the M2a macrophages/myeloid fibroblasts in the aging B6J hearts. Upper panel shows representative images of M2a macrophage/myeloid fibroblasts (Col1a+CD301+CD45+) and the quantification of mean fluorescence intensity (MFI) of collagen type I in Col1+CD301+CD45+ M2a macrophage/myeloid fibroblasts. Results are represented as mean ± SEM. N= 6, 11, 6, 4 for young B6J and aged B6J, MCP-1KO, and B6N respectively.
As described by others, the spleen is an intermediate destination for monocytes between bone marrow and an inflamed organ [44]. Enhanced monocyte migration from the spleen to the heart has been observed in a mouse myocardial infarction model [44]. Since we have identified the increased presence of M2a polarized macrophages/myeloid fibroblasts in the aged B6J heart, we elected to analyze spleens isolated from young and aged B6J mice. H&E staining revealed a reduced number of cells in the sub-capsular red pulp in the aged spleens (Fig. 3A) in agreement with the notion that monocytes migrated to the inflamed heart in response to an elevated expression of MCP-1, as we have reported previously [11]. Next, we analyzed the migration of splenic monocytes in response to MCP-1. We found that monocytes isolated from aged B6J spleens had augmented chemotaxis towards MCP-1 in an in vitro assay (Fig. 3B), indicating the possibility that this may contribute to their increased presence in the heart.
Fig. 3.
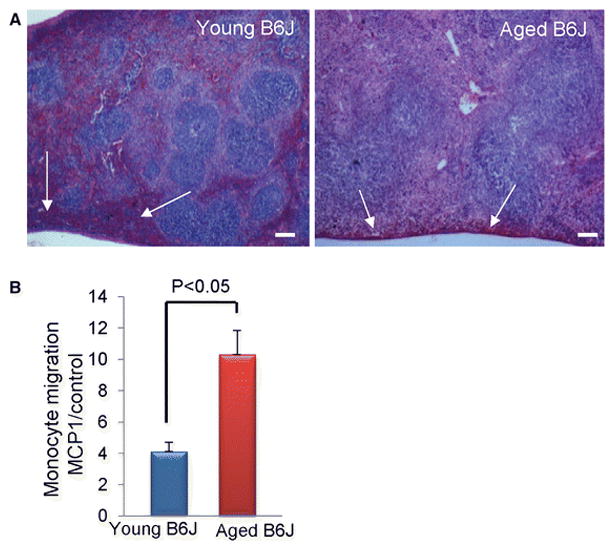
Aging affects splenic monocyte migration. A, The sub-capsular red pulp and the marginal zone in the old (spleens) are depleted as visualized by hematoxylin and eosin staining of spleen sections (white arrows). B, MCP-1 dependent migration of splenic monocytes isolated from aged mice is increased. Results are represented as mean ± SEM, * denotes P<0.05. N=3 per age group. Scale bar= 100 μm. Young denotes 3 month-old and aged denotes 30 month-old.
2. The link between increased oxidative stress and an inflammatory phenotype in the aging mesenchymal fibroblasts
Mesenchymal fibroblasts derived from the aging B6J heart change their phenotype into a pro-inflammatory state and secrete higher levels of various cytokines (MCP-1 and IL-6 among them) than fibroblasts derived from young B6J hearts [11]. We further demonstrated in the in vitro assay that although MCP-1 is necessary for transendothelial migration of leukocytes, IL-6 greatly facilitated the macrophage polarization into an M2a phenotype [11]. These discoveries encouraged us to postulate that the inflammatory state of mesenchymal fibroblasts may also be driving the leukocyte infiltration [49] and subsequent fibrosis.
Since leukocyte infiltration in aged B6N hearts is very modest when compared with aged B6J hearts, fibroblast conditioned medium was assayed using a protein array. Mesenchymal fibroblasts derived from B6J hearts secrete higher levels of MCP-1 (155%) and IL-6 (570%) (Fig. 4A, and Table 1) than B6N. In contrast to these two pro-inflammatory cytokines, the anti-inflammatory cytokine IL-10 was upregulated in B6N fibroblast culture medium by 60%, (p<0.053) but the detected amount of IL-10 was low (see representative membranes, green frames, Fig. 4A, and Table 1).
Fig. 4.
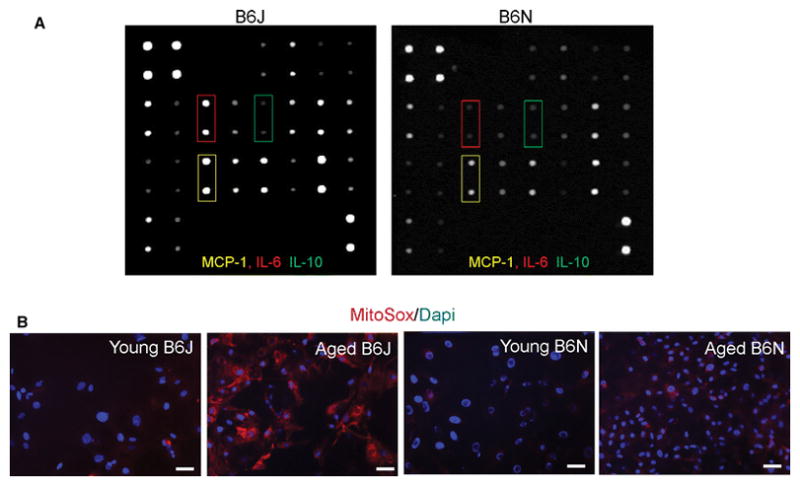
Mesenchymal fibroblasts derived from B6N hearts are protected from age-related ROS production and inflammation. A, Conditioned medium from mesenchymal fibroblast cultures containing 100 μg of protein was incubated with cytokine antibody array membranes. Panels depict representative membranes. B, Quiescent mesenchymal fibroblasts were stained with MitoSox (red) to determine ROS production. Scale bar = 20 μm. N= 3 per each strain and age group, young denotes 3 month-old and aged denotes 21–25 month-old mice.
Table 1. Cytokine antibody array membranes.
Fibroblast conditioned medium was analyzed by protein array. Densitometry data was acquired by analyzing digitalized membrane images using ImageJ software. Results are represented as mean ± SEM. N= 3 for fibroblasts derived from 21–25 month-old mice of each strain.
| Cytokine | 6BJ [arbitrary units] | 6BN [arbitrary units] | 6BJ/6BN ratio | ttest |
|---|---|---|---|---|
| GCSF | 0.07 ± 0.004 | 0.09 ± 0.009 | 0.74 | 0.074 |
| GM-CSF | 0.13 ± 0.008 | 0.15 ± 0.013 | 0.85 | 0.196 |
| IFNγ | 0.05 ±.0.004 | 0.05 ± 0.006 | 1.05 | 0.754 |
| IL-2 | 0.04 ±.0.004 | 0.06 ± 0.006 | 0.70 | 0.052 |
| IL-3 | 0.04 ± 0.003 | 0.08 ± 0.015 | 0.54 | 0.069 |
| IL-4 | 0.27 ± 0.013 | 0.29 ± 0.018 | 0.94 | 0.443 |
| IL-5 | 0.06 ± 0.004 | 0.06 ± 0.004 | 1.11 | 0.356 |
| IL-6 | 0.47 ± 0.044 | 0.08 ± 0.002 | 5.70 | <0.001 |
| IL-9 | 0.10 ± 0.007 | 0.10 ± 0.008 | 0.97 | 0.787 |
| IL-10 | 0.05 ± 0.005 | 0.08 ± 0.013 | 0.57 | 0.053 |
| IL-12 p40p70 | 0.13 ± 0.007 | 0.12 ± 0.010 | 1.09 | 0.417 |
| IL-12 p70 | 0.28 ± 0.018 | 0.34 ± 0.050 | 0.83 | 0.313 |
| IL-13 | 0.13 ± 0.009 | 0.15 ± 0.017 | 0.85 | 0.312 |
| IL-17 | 0.07 ± 0.007 | 0.04 ± 0.031 | 1.52 | 0.026 |
| MCP-1 | 0.50 ± 0.046 | 0.33 ± 0.038 | 1.55 | 0.019 |
| MCP-5 | 0.18 ± 0.010 | 0.18 ± 0.020 | 0.98 | 0.851 |
| Rantes | 0.31 ± 0.042 | 0.39 ± 0.061 | 0.81 | 0.341 |
| SCF | 0.04 ± 0.008 | 0.04 ± 0.004 | 0.98 | 0.916 |
| sTNFRI | 0.64 ± 0.039 | 0.58 ± 0.039 | 1.11 | 0.270 |
| Thrombopoietin | 0.19 ± 0.011 | 0.18 ± 0.011 | 1.02 | 0.836 |
| TNFα | 0.08 ± 0.006 | 0.09 ± 0.011 | 0.97 | 0.834 |
| VEGF | 0.08 ± 0.023 | 0.09 ± 0.019 | 0.94 | 0.872 |
Nnt is an enzyme that transports hydrogen to catalyze reduction of NADP+ into NADPH, which indirectly decreases ROS levels. Because Nnt is present in the aged B6N mesenchymal fibroblasts, as opposed to B6J, wherein partial gene deletion makes the Nnt protein unstable and nonfunctional [26], we hypothesized that ROS production in these cells would be lower than in cells derived from aged B6J hearts. High superoxide anion levels were detected in mesenchymal fibroblasts derived from aged B6J when compared to cells isolated from young B6J hearts [9] using MitoSox. MitoSox is a fluorogenic dye which, when oxidized by superoxide anion, produces red fluorescence, as shown in Fig. 4B. We found mesenchymal fibroblasts derived from aged B6N hearts were protected from aging dependent increased superoxide anion production as opposed to mesenchymal fibroblasts derived from aged B6J hearts (Fig. 4B). The production of reactive oxygen species (ROS) has been shown to contribute to transcriptional activation of several cytokines [40] [28], which may explain the reduced secretion/production of various cytokines in mesenchymal B6N fibroblasts (Fig. 4A).
3. Mesenchymal fibroblast-dependent fibrosis
We have shown previously [8] in B6J hearts that with age progression the number of mesenchymal fibroblast precursors (CD44+CD45−) increases gradually. Flow cytometry analysis of non-myocytes isolated from young (B6J) and aged (B6J, MCP-1KO, B6N) hearts demonstrated the increased number of fibroblast precursors in aged B6J and MCP-1KO hearts by 661% and 470% respectively when compared with young B6J hearts (Fig. 5A, see Supplemental Figure 2 for the gating strategy). By contrast, in the aged B6N heart, fibroblast precursor numbers were not elevated (Fig. 5A). Immunofluorescence of heart sections confirms the flow cytometry data (Fig. 5B). We also examined heart sections from healthy young and elderly human donors that revealed an elevated presence of CD44+ cells in the aging myocardium (Fig. 5C).
Fig. 5.
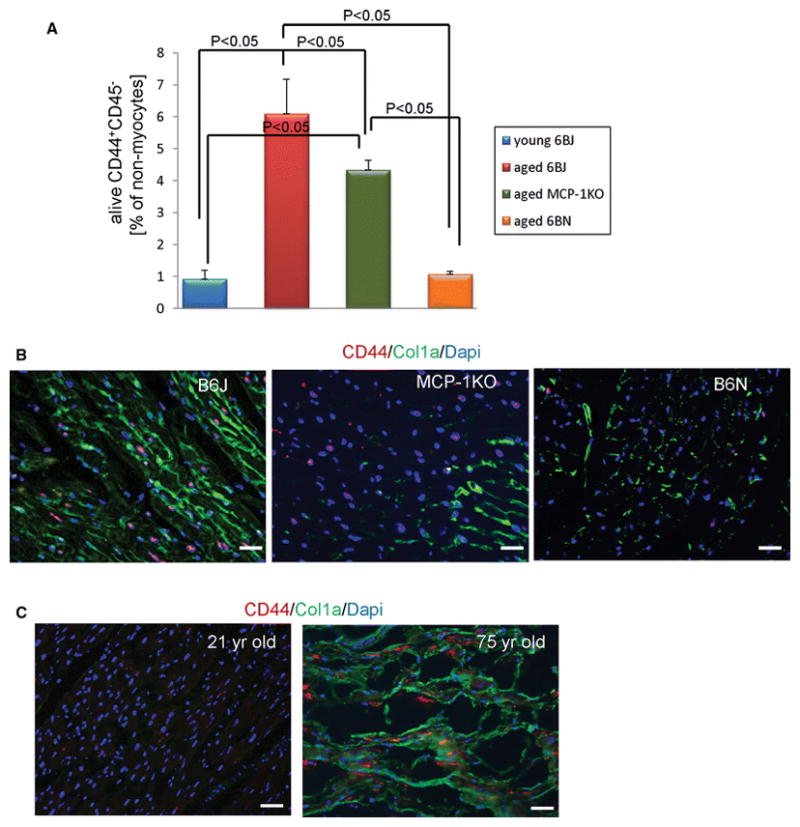
An increased number of mesenchymal fibroblast precursors in the aging heart. A, Quantification of flow cytometry analysis of calcein+ non-myocytes that are stained with CD44 and CD45 antibodies to distinguish CD44+CD45− mesenchymal fibroblasts from other non-myocytes. Results are represented as mean ± SEM. N= 4, 4, 8, 8 for young B6J and aged B6J, MCP-1KO, and B6N respectively. B, Immunofluorescence staining of aged mouse paraffin heart sections or C, human frozen heart sections showing CD44 positive cells (red) within collagen type I deposition (green) and Dapi stained nuclei (blue). 21–24 month-old mice of an indicated genetic strain were used, and young denotes 3 month-old. N= 3, 2, 3, scale bar=50 μm. Human heart sections obtained from 21 and 75 year-old donors were used, scale bar= 50 μm.
Furthermore, we observed that within the CD44+CD45− population, more cells derived from aged B6J hearts were expressing collagen type I (68.5%) than those from all three other groups (~20%) as demonstrated in Fig. 6A, B. These data suggest that in the aging B6J hearts an increased number of CD44+CD45− mesenchymal fibroblast progenitors (6% of total alive non-myocyte, Fig. 5A) and an increased number of collagen expressing cells within that population (68%, Fig. 6B) augment the pool of activated fibroblasts that participate in collagen synthesis. Fig. 6C shows the total number of mesenchymal fibroblasts as a percent of non-myocytes in the heart (Supplemental Figure 2 shows computation pertinent to Fig. 6C). Consistent with the flow cytometry data there was an increased number of cells that are actively producing collagen (procollagen type 1a+) in the aged B6J heart (Fig. 6D), an extension of data presented in Fig. 2B and 5B that show collagen deposition. The magnified field shows specifically CD44+procollagen type 1a+ double positive cells (Fig. 6D, lower panel).
Fig. 6.
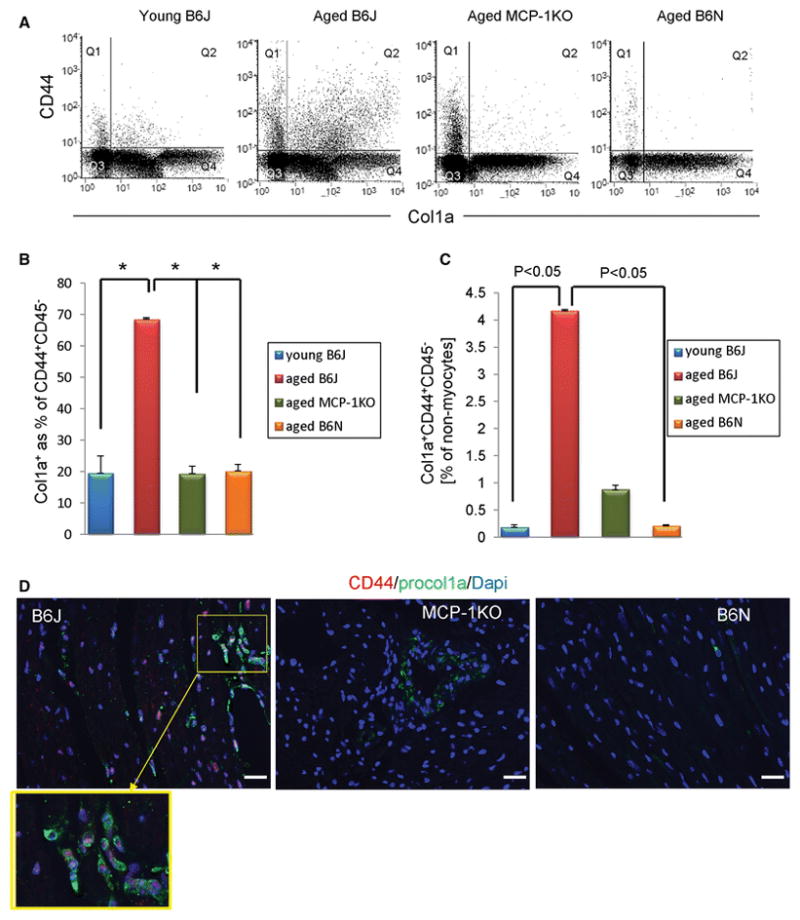
Augmented differentiation of mesenchymal fibroblast precursors into fibroblasts in B6J heart. A, Representative histograms of flow cytometry analysis of mesenchymal fibroblasts gated on the CD45− cell population. B, Percentage of collagen producing cells within the CD44+CD45− pool. C, Quantification of total mesenchymal fibroblasts in hearts. Results are represented as mean ± SEM. N= 3, 3, 6, 3 for young B6J and aged B6J, MCP1KO, and B6N respectively. D, Immunofluorescence staining of aged mouse paraffin heart sections showing CD44 positive cells (red) within procollagen type I deposition (green) and Dapi stained nuclei (blue). Yellow frame depicts enlarged view of double positive cells. 21–24 month-old mice of an indicated genetic strain were used, and young denotes 3 month-old. N= 3, 2, 3, scale bar=50 μm. Human heart sections obtained from 21 and 75 year-old donors were used, scale bar= 50 μm.
4. Heart function and fibrosis in the aging heart
We previously published an extensive comparison of heart function in young vs aged B6J mice [6, 33]. In the current study we investigated cardiac fibrosis in aged mice of three different genetic strains: B6J, MCP-1KO (that are backcrossed to the B6J background) and B6N. We compared the systolic and diastolic function of aged B6J with either aged B6N or aged MCP-1KO mice. The data are shown in Supplemental Table 1 and systolic function was similar among the groups. The aged MCP-1KO mice had slightly reduced cardiac outputs, and Peak Velocity and fractional shortening (FS) were preserved (Supplemental Table 1). The Left Ventricular Internal Dimension in diastole (LVID;d) and calculated Volume of the aged B6N were significantly larger than the other two groups, yet Peak Velocity and FS were not different from the other two old groups consistent with preserved systolic function. While we did not perform invasive studies in these aged mice the heart the heart rates were similar for the B6N and B6J, which allowed us to compare the E/A ratios as a marker of diastolic function. The E/A was higher for the B6N than B6J (Fig. 7A, Supplemental Table 1), suggesting better preserved diastolic function. However, to insure that this was not “pseudo-normalization” (a manifestation of elevated pressures and a very stiff ventricle), we used the spring model suggested by Kovacs [27]. As shown in Supplemental Table 1, the Peak E filling velocities were similar, but the stiffness coefficient k was almost 30% lower in the B6N compared to the B6J, consistent with better diastolic function in the B6N (Fig. 7B, Supplemental Table 2). Similarly, the normalized left atrial size was not larger for the B6N than the B6J, supporting the absence of high filling pressures in the B6N (Supplemental Table 1). The MCP-1KO mice data were between the B6N and the B6J for most variables. These data suggest that decreased interstitial fibrosis as seen in the old B6N is associated with better ventricular filling and less ventricular stiffness.
Fig. 7.
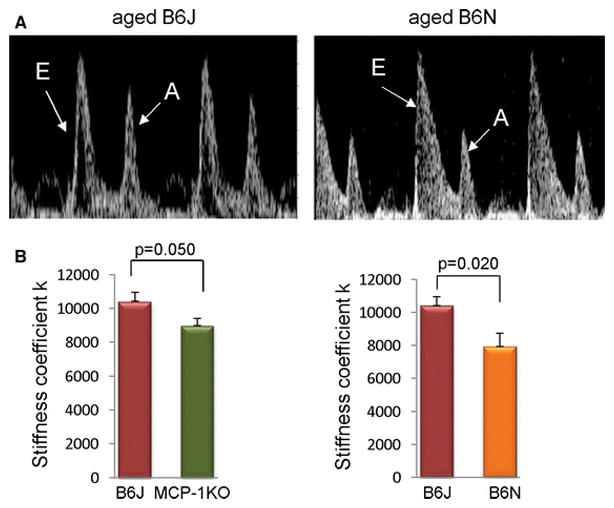
The spring stiffness coefficient of mitral inflow for aged B6J, MCP-1KO and B6N hearts. A, Representative images from aged B6J and B6N hearts showing E/A ratio for aged B6J and B6N hearts. N=10 per each group. B, Kovacs analysis of Doppler mitral early flow predicts left ventricular diastolic stiffness. N= 7, 4, 5 for aged B6J, aged MCP-1KO, and aged B6N respectively. 21–24 month-old mice were used. Data are presented as mean ± SEM.
DISCUSSION
Age-associated diastolic dysfunction has multiple contributing factors, including modifications of myocardial calcium fluxes and the extent of interstitial fibrosis. We simplify this to active and passive processes, the fibrosis contributing to the passive factors. As the drivers of interstitial fibrosis seen in aging and in HFpEF become better understood, the pro-inflammatory environment of the aged animal, specifically the aged heart, seems to play a critical role in these processes. Here we looked at the aging of three models, two of which (by different mechanisms) had reduced myocardial inflammation (MCP-1KO and B6N) compared to the B6J mouse that with aging develops an inflammatory cardiac phenotype.
Cardiac recruitment of T cells, dendritic cells and monocytes has been shown to contribute to fibrosis and heart failure [20, 36, 54]. MCP-1 is a critical cytokine responsible for chemoattraction of leukocytes into inflamed organs [14]. In the ischemia/reperfusion injury model (young mice), MCP-1 is induced only around vessels [22], whereas in the aging heart (without injury), mesenchymal fibroblasts express high levels of this cytokine as well [6, 11]. In the injured young mouse heart [22] and in the uninjured aged heart, inflammation depends on MCP-1 expression; aged MCP-1KO hearts were protected from leukocytic infiltration (Fig. 2). In this report, we further follow leukocytes expressing markers of macrophages to determine the number of M2a polarized macrophages/myeloid fibroblasts. Several attempts have been made to characterize the cardiac macrophage population, either after acute injury [16] [24, 44] or during aging [38]. Some of these models use genetically labeled markers that distinguish the resident macrophage population, but that distinction in steady-state models may be more difficult, especially when characterizing resident and monocyte-derived macrophages separately on various genetic backgrounds. The distinction we applied between resident and newly infiltrated cells was based on the intensity of expression of the pan leukocyte marker CD45. This strategy has been successfully used before in distinguishing between both populations in injury models of the brain [51] and the heart [15], where CD45hi infiltrating cells give rise to monocyte-derived macrophages and CD45lo represent resident macrophages.
When we compared the number of M2a macrophages derived from infiltrating monocytes (CD45hi) we found a ~5 fold increase in this population in aged B6J hearts compared to young B6J, but surprisingly MCP-1KO (that did not display an augmented infiltrating CD45 population) were not completely protected from myeloid-dependent fibrosis; the levels of M2a macrophages/myeloid fibroblasts were ~2.5 fold higher than those observed in the young heart. To explain this apparent discrepancy we examined the production of IL-6 in mesenchymal fibroblasts derived from aged B6J and MCP-1KO hearts. Previously we have demonstrated that IL-6 greatly facilitated macrophage polarization into the M2a phenotype [11] but plays no role in macrophage infiltration as demonstrated by Müller et al. [35]. Indeed, the analysis of fibroblasts derived from aged MCP-1KO and aged B6J hearts demonstrated a 3 fold increased IL-6 mRNA expression (measured by qPCR [8]) in fibroblasts derived from the aged MCP-1KO heart, and although this difference did not reach statistical significance (2.95 ± 1.05 vs 1.00 ± 0.39, P=0.16), it suggests a potential mechanism by which aged MCP-1KO hearts still have a pro-inflammatory environment and partially develop myeloid-dependent fibrosis. An increased macrophage infiltration in the aged B6J mouse heart has been described by others [31, 48], and in these studies the ratio of M1 (classically activated pro-inflammatory macrophage) to M2 alternatively activated macrophage was increased. We have found that both M1 (data not published) and M2a macrophages had an increased presence in the aging heart when compared with young controls. This partial discrepancy may be explained by the fact that we used different markers and we analyzed the M2a population, which is only a subtype of the much broader class of M2 cells. There are several pathways leading to transcriptional regulation of MCP-1 [50] and IL-6 (for review see [2]). The IL-6↔MCP-1 axis, where one cytokine enhances transcription of the other, has been postulated in macrophages [45]. However, the mechanism may be different in cardiac mesenchymal fibroblasts. We have examined transcriptional regulation of IL-6 [11] and MCP-1 (unpublished data) in cardiac mesenchymal fibroblasts and found that both cytokines in the aged B6J fibroblasts are regulated by the Erk pathway, which is in agreement with other authors [40]. Although we did not study which transcription factor was involved in this regulation, it may be possible that in the aged MCP-1KO fibroblasts, a common transcription factor that governs both IL-6 and MCP-1 transcription is abundantly available for IL-6 gene expression due to an MCP-1 null mutation, and this may explain an elevated IL-6 synthesis in MCP-1KO fibroblasts.
ROS mediated cytokine expression has been shown in various models (for review see [34]). We have found that mesenchymal fibroblasts derived from B6N hearts produce markedly less ROS (Fig. 4B), MCP-1 and IL-6 than fibroblasts derived from aged B6J hearts (Fig. 4A), suggesting that Nnt may play a protective role in a slowly-progressing aging heart. This is in contrast to observations that, with injury, the young B6N heart responds more severely to stress than age-matching B6J hearts [4, 37]. Although Nnt is considered an antioxidative enzyme, in pathophysiological conditions such as pressure overload (and perhaps in others) Nnt reverses its action and instead of generating NADPH, it depletes it to support NADH and ATP production. This results in an increase in ROS levels [37]. While it is not known how injury will affect the aged B6N heart, Nnt transcript levels are reduced in the aging B6N mouse heart [29].
The number of mesenchymal fibroblasts increases with age in B6J hearts [8]. This may prematurely deplete the existing progenitor pool and negatively influence the healing after injury such as myocardial infarction. The impaired reparative fibrosis in the aging B6J heart has been reported by our group before [7]. Here, we have demonstrated that MCP-1KO hearts (that are backcrossed to the B6J background) also display an increased number of CD44+CD45− progenitors, but aged B6N hearts were protected from this dysregulation. Interestingly, in MCP-1KO hearts the total number of Col1a+CD44+CD45− mesenchymal fibroblasts was also modestly increased (Fig. 5C) suggesting that the cross-talk between leukocytes and fibroblast progenitors is altered due to the lower number of infiltrating leukocytes [52] or because of a lack of MCP-1. MCP-1 has been shown to stimulate collagen expression directly [19]. Therefore, even though the number of myeloid fibroblasts is noticeably lower than of mesenchymal fibroblasts (20:80 ratio), our results indicate that the mesenchymal progenitor may need an input from myeloid cells to become activated and start producing collagen. In agreement, aged B6N hearts, which did not display increased leukocyte infiltration, did not exhibit an increased number of mesenchymal fibroblasts.
In the B6J mouse, aging results in cardiac diastolic dysfunction that reflects altered calcium handling and increased interstitial fibrosis. In the B6N, the extent of the fibrosis is decreased and therefore allows us to dissect the additive factors of fibrosis and calcium handling. In the aged B6N we saw the E/A ratio was better, suggesting improved diastolic function (Fig. 7A, Supplemental Table 2). To test the possibility of this arising from a very stiff ventricle and increased filling pressures normalizing the velocities, we analyzed mitral filling by modeling it as a damped spring [27]. This gives a global stiffness parameter, k, which integrates geometry as well as tissue stiffness. If the aged B6N were stiffer, then the spring stiffness coefficient (k) for the aged B6N would be elevated. As shown in Fig. 7B, k was lower in the aged B6N, suggesting better diastolic function. For the aged MCP-1KO heart, the E/A ratio was not significantly higher than the aged B6J. The atrial size, corrected for the body size, may have been slightly less than that seen in the aged B6J (Supplemental Table 1). Overall, deletion of MCP-1 alone was not as effective as the B6N model in preventing the age-related impairment in diastolic function.
Conclusions
The purpose of this manuscript was to extend our previous work suggesting a role for reactive oxygen and inflammatory dysregulation in the production of interstitial fibrosis. Our initial studies in young mice describe the induction of the myeloid fibroblast in response to angiotensin treatment [22] or in an ischemia/reperfusion cardiomyopathy model [23]. In these acute models, the myeloid fibroblasts arose from blood monocytes and resulted in rapid onset of a fibrotic reaction. The reaction was dependent on the generation of MCP-1 and neither the infiltration nor the fibrosis occurred in mice with genetic deletion of MCP-1 (MCP-1 KO) [17, 22]. Chronic interstitial fibrosis associated with aging in the 6BJ mouse was also associated with progressive increases in MCP-1 production, generation of myeloid fibroblasts, and fibrosis (see schema Fig. 8). Since there was no acute inflammation-inducing intervention (as seen in the acute models), we investigated the source of the inflammatory phenotype and discovered that the aging mouse had a progressive defect in TGF-β signaling resulting from reduction in TGF-β receptor I expression [9, 10]. This defect originated in the mesenchymal stem cells and was passed to the mesenchymal fibroblast progeny. The restriction in TGF-β signaling resulted in an inflammatory phenotype as manifested by increased expression of many pro-inflammatory cytokines and chemokines. It has been demonstrated that mesenchymal stem cells have anti-inflammatory properties that are beneficial in reducing organ damage in various model of injury [30, 32], but in the aging heart mesenchymal stem cells seem to contribute to a pro-inflammatory milieu [9].
Fig. 8.
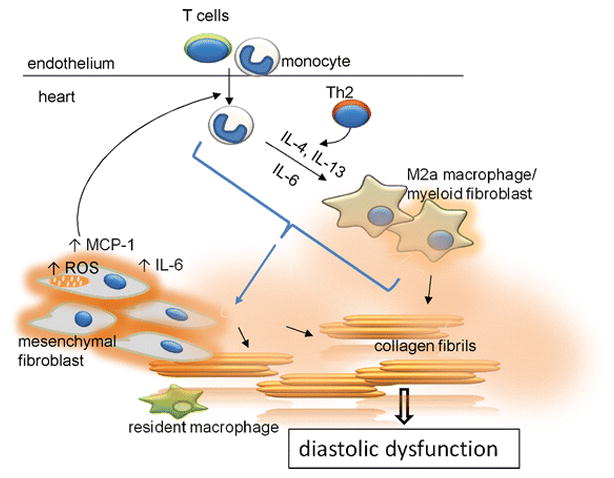
The two way interaction between infiltrating leukocytes and mesenchymal fibroblasts in the aging mouse heart. The dysregulated mesenchymal fibroblasts produce higher levels of ROS that promote expression of MCP-1 and IL-6. Secreted MCP-1 attracts leukocyte infiltration into the heart. Infiltrating monocytes polarize into alternatively activated M2a macrophages/myeloid fibroblasts in response to Th2 products (IL-4 and IL-13) and IL-6 (derived at least partially from dysregulated mesenchymal fibroblasts). Both types of fibroblasts (mesenchymal and myeloid) secrete collagen. Excessive collagen production by mesenchymal fibroblasts is controlled by the infiltrating leukocytes; without input from leukocytes/monocyte-derived macrophages, mesenchymal fibroblasts remain unactivated and do not overproduce collagen. Augmented collagen production contributes to fibrosis and diastolic dysfunction. Resident macrophages do not play an apparent role in this process.
The reduction of TGF-β signaling enhanced differentiation of mesenchymal stem cells to mesenchymal fibroblasts, and thereby increased the number of mesenchymal fibroblasts in the aged heart [8]. Thus, the dysregulated mesenchymal fibroblasts produced MCP-1, which augmented leukocyte uptake into the myocardium, and also IL-6, which was critical for generation of myeloid fibroblasts [11]. Our examinations demonstrated that this inflammatory state activated collagen synthesis via a Ras-Erk pathway despite the reduction of TGF-β signaling (usually associated with fibrosis). The inflammatory state was also associated with a marked increase in ROS in the inflammatory fibroblasts.
The current study was designed to further examine the interaction and role of two species of fibroblasts found to be associated with cardiac fibrosis in the aging mouse (Fig. 8). The experiments with MCP-1 deletion in B6J mice confirm a role for this chemokine in generating myeloid fibroblasts, which appear to arise mainly from the augmented infiltration of splenic monocytes (as opposed to resident macrophages). It is to be noted, however, that while genetic deletion of MCP-1 reduced the fibrotic reaction and blunted the ventricular stiffness, it did not eliminate the pathologic response. The second model studied took advantage of the fact that the B6J mouse is deficient in Nnt as a result of a spontaneous mutation. This enzyme causes reduction of oxidative stress and is also seen to be lower in hearts from patients suffering from heart failure when compared to healthy donors [41]. By contrast, the B6N strain, from which the B6J strain spontaneously mutated, contains a functional Nnt. We found that functional Nnt markedly decreased the elevated oxidative stress with age and markedly reduced the generation of both myeloid and mesenchymal fibroblasts as well as the interstitial fibrosis. This was accompanied by substantially better diastolic function when compared to the B6J mice. Thus, pathologic fibrosis in the aging animal is associated with and involves generation and activation of two fibroblast species, both related to oxidative stress and immunoinflammatory dysregulation.
Supplementary Material
Acknowledgments
This research was supported by NIH grant R01HL089792 (MLE), a Medallion Foundation grant (KAC) and the Hankamer Foundation.
We would like to thank Dr. Jeffrey Crawford for conducting the monocyte chemoattractant assay. We thank Thuy Pham and Dorellyn Lee for excellent technical assistance.
Footnotes
CONFLICT OF INTEREST
On behalf of all authors, the corresponding author states that there is no conflict of interest.
ETHICS STATEMENT
The manuscript does not contain clinical studies or patient data.
BIBLIOGRAPHY
- 1.Arkblad EL, Tuck S, Pestov NB, Dmitriev RI, Kostina MB, Stenvall J, Tranberg M, Rydstrom J. A Caenorhabditis elegans mutant lacking functional nicotinamide nucleotide transhydrogenase displays increased sensitivity to oxidative stress. Free Radic Biol Med. 2005;38:1518–1525. doi: 10.1016/j.freeradbiomed.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 2.Brasier AR. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovascular Research. 2010;86:211–218. doi: 10.1093/cvr/cvq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. 2002;27:92–98. doi: 10.1007/s00059-002-2354-y. not available. [DOI] [PubMed] [Google Scholar]
- 4.Cardin S, Scott-Boyer MP, Praktiknjo S, Jeidane S, Picard S, Reudelhuber TL, Deschepper CF. Differences in cell-type-specific responses to angiotensin II explain cardiac remodeling differences in C57BL/6 mouse substrains. Hypertension. 2014;64:1040–1046. doi: 10.1161/HYPERTENSIONAHA.114.04067. [DOI] [PubMed] [Google Scholar]
- 5.Carlson S, Trial J, Soeller C, Entman ML. Cardiac mesenchymal stem cells contribute to scar formation after myocardial infarction. Cardiovasc Res. 2011;91:99–107. doi: 10.1093/cvr/cvr061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol. 2011;50:248–256. doi: 10.1016/j.yjmcc.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cieslik KA, Taffet GE, Crawford JR, Trial J, Mejia Osuna P, Entman ML. AICAR-dependent AMPK activation improves scar formation in the aged heart in a murine model of reperfused myocardial infarction. J Mol Cell Cardiol. 2013;63C:26–36. doi: 10.1016/j.yjmcc.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cieslik KA, Trial J, Carlson S, Taffet GE, Entman ML. Aberrant differentiation of fibroblast progenitors contributes to fibrosis in the aged murine heart: role of elevated circulating insulin levels. FASEB J. 2013;27:1761–1771. doi: 10.1096/fj.12-220145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cieslik KA, Trial J, Crawford JR, Taffet GE, Entman ML. Adverse fibrosis in the aging heart depends on signaling between myeloid and mesenchymal cells; role of inflammatory fibroblasts. J Mol Cell Cardiol. 2014;70:56–63. doi: 10.1016/j.yjmcc.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cieslik KA, Trial J, Entman ML. Defective myofibroblast formation from mesenchymal stem cells in the aging murine heart rescue by activation of the AMPK pathway. Am J Pathol. 2011;179:1792–1806. doi: 10.1016/j.ajpath.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cieslik KA, Trial J, Entman ML. Mesenchymal stem cell-derived inflammatory fibroblasts promote monocyte transition into myeloid fibroblasts via an IL-6-dependent mechanism in the aging mouse heart. FASEB J. 2015;29:3160–3170. doi: 10.1096/fj.14-268136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford JR, Pilling D, Gomer RH. Improved serum-free culture conditions for spleen-derived murine fibrocytes. J Immunol Methods. 2010;363:9–20. doi: 10.1016/j.jim.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duerrschmid C, Trial J, Wang Y, Entman ML, Haudek SB. Tumor Necrosis Factor: A mechanistic link between angiotensin-II-induced cardiac inflammation and fibrosis. Circ Heart Fail. 2015;8:352–361. doi: 10.1161/CIRCHEARTFAILURE.114.001893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, Kraemer D, Taffet G, Rollins BJ, Entman ML. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–592. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- 18.Gates PE, Tanaka H, Graves J, Seals DR. Left ventricular structure and diastolic function with human ageing. Relation to habitual exercise and arterial stiffness. Eur Heart J. 2003;24:2213–2220. doi: 10.1016/j.ehj.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 19.Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem. 1996;271:17779–17784. doi: 10.1074/jbc.271.30.17779. [DOI] [PubMed] [Google Scholar]
- 20.Glezeva N, Voon V, Watson C, Horgan S, McDonald K, Ledwidge M, Baugh J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: evidence of M2 macrophage activation in disease pathogenesis. J Card Fail. 2015;21:167–177. doi: 10.1016/j.cardfail.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Granillo A, Pena CA, Pham T, Pandit LM, Taffet GE. Murine echocardiography of left atrium, aorta, and pulmonary artery. J Vis Exp. 2017 doi: 10.3791/55214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, Taffet GE, Entman ML. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:499–507. doi: 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis NG, Entman ML. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci U S A. 2006;103:18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–1622. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoek JB, Rydstrom J. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem J. 1988;254:1–10. doi: 10.1042/bj2540001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang TT, Naeemuddin M, Elchuri S, Yamaguchi M, Kozy HM, Carlson EJ, Epstein CJ. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum Mol Genet. 2006;15:1187–1194. doi: 10.1093/hmg/ddl034. [DOI] [PubMed] [Google Scholar]
- 27.Kovacs SJ, Jr, Barzilai B, Perez JE. Evaluation of diastolic function with Doppler echocardiography: the PDF formalism. Am J Physiol. 1987;252:H178–187. doi: 10.1152/ajpheart.1987.252.1.H178. not available. [DOI] [PubMed] [Google Scholar]
- 28.Lakshminarayanan V, Lewallen M, Frangogiannis NG, Evans AJ, Wedin KE, Michael LH, Entman ML. Reactive oxygen intermediates induce monocyte chemotactic protein-1 in vascular endothelium after brief ischemia. Am J Pathol. 2001;159:1301–1311. doi: 10.1016/S0002-9440(10)62517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 30.Luger D, Lipinski MJ, Westman PC, Glover DK, Dimastromatteo J, Frias JC, Albelda MT, Sikora S, Kharazi A, Vertelov G, Waksman R, Epstein SE. Intravenously-Delivered Mesenchymal Stem Cells: Systemic Anti-Inflammatory Effects Improve Left Ventricular Dysfunction in Acute Myocardial Infarction and Ischemic Cardiomyopathy. Circ Res. 2017;117:310599. doi: 10.1161/CIRCRESAHA.117.310599. [pii] [DOI] [PubMed] [Google Scholar]
- 31.Ma Y, Chiao YA, Clark R, Flynn ER, Yabluchanskiy A, Ghasemi O, Zouein F, Lindsey ML, Jin YF. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc Res. 2015;106:421–431. doi: 10.1093/cvr/cvv128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martire A, Bedada FB, Uchida S, Poling J, Kruger M, Warnecke H, Richter M, Kubin T, Herold S, Braun T. Mesenchymal stem cells attenuate inflammatory processes in the heart and lung via inhibition of TNF signaling. Basic Res Cardiol. 2016;111:54. doi: 10.1007/s00395-016-0573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medrano G, Hermosillo-Rodriguez J, Pham T, Granillo A, Hartley CJ, Reddy A, Osuna PM, Entman ML, Taffet GE. Left atrial volume and pulmonary artery diameter are noninvasive measures of age-related diastolic dysfunction in mice. J Gerontol A Biol Sci Med Sci. 2016;71:1141–1150. doi: 10.1093/gerona/glv143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller J, Gorressen S, Grandoch M, Feldmann K, Kretschmer I, Lehr S, Ding Z, Schmitt JP, Schrader J, Garbers C, Heusch G, Kelm M, Scheller J, Fischer JW. Interleukin-6-dependent phenotypic modulation of cardiac fibroblasts after acute myocardial infarction. Basic Res Cardiol. 2014;109:440. doi: 10.1007/s00395-014-0440-y. [DOI] [PubMed] [Google Scholar]
- 36.Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ Heart Fail. 2015;8:776–787. doi: 10.1161/CIRCHEARTFAILURE.115.002225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nickel AG, von Hardenberg A, Hohl M, Loffler JR, Kohlhaas M, Becker J, Reil JC, Kazakov A, Bonnekoh J, Stadelmaier M, Puhl SL, Wagner M, Bogeski I, Cortassa S, Kappl R, Pasieka B, Lafontaine M, Lancaster CR, Blacker TS, Hall AR, Duchen MR, Kastner L, Lipp P, Zeller T, Muller C, Knopp A, Laufs U, Bohm M, Hoth M, Maack C. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22:472–484. doi: 10.1016/j.cmet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Pinto AR, Godwin JW, Chandran A, Hersey L, Ilinykh A, Debuque R, Wang L, Rosenthal NA. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging (Albany NY) 2014;6:399–413. doi: 10.18632/aging.100669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rozenberg S, Tavernier B, Riou B, Swynghedauw B, Page CL, Boucher F, Leiris J, Besse S. Severe impairment of ventricular compliance accounts for advanced age-associated hemodynamic dysfunction in rats. Exp Gerontol. 2006;41:289–295. doi: 10.1016/j.exger.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, Koyasu S, Matsui H, Yamauchi-Takihara K, Harada M, Saito Y, Ogawa S. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res. 2001;89:661–669. doi: 10.1161/hh2001.098873. [DOI] [PubMed] [Google Scholar]
- 41.Sheeran FL, Rydstrom J, Shakhparonov MI, Pestov NB, Pepe S. Diminished NADPH transhydrogenase activity and mitochondrial redox regulation in human failing myocardium. Biochim Biophys Acta. 2010;1797:1138–1148. doi: 10.1016/j.bbabio.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 42.Simon MM, Greenaway S, White JK, Fuchs H, Gailus-Durner V, Wells S, Sorg T, Wong K, Bedu E, Cartwright EJ, Dacquin R, Djebali S, Estabel J, Graw J, Ingham NJ, Jackson IJ, Lengeling A, Mandillo S, Marvel J, Meziane H, Preitner F, Puk O, Roux M, Adams DJ, Atkins S, Ayadi A, Becker L, Blake A, Brooker D, Cater H, Champy MF, Combe R, Danecek P, di Fenza A, Gates H, Gerdin AK, Golini E, Hancock JM, Hans W, Holter SM, Hough T, Jurdic P, Keane TM, Morgan H, Muller W, Neff F, Nicholson G, Pasche B, Roberson LA, Rozman J, Sanderson M, Santos L, Selloum M, Shannon C, Southwell A, Tocchini-Valentini GP, Vancollie VE, Westerberg H, Wurst W, Zi M, Yalcin B, Ramirez-Solis R, Steel KP, Mallon AM, de Angelis MH, Herault Y, Brown SD. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013;14:R82. doi: 10.1186/gb-2013-14-7-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. 2001;280:C53–60. doi: 10.1152/ajpcell.2001.280.1.C53. not available. [DOI] [PubMed] [Google Scholar]
- 44.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A, 3rd, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toba H, Cannon PL, Yabluchanskiy A, Iyer RP, D’Armiento J, Lindsey ML. Transgenic overexpression of macrophage matrix metalloproteinase-9 exacerbates age-related cardiac hypertrophy, vessel rarefaction, inflammation, and fibrosis. Am J Physiol Heart Circ Physiol. 2017;312:H375–H383. doi: 10.1152/ajpheart.00633.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toba H, de Castro Bras LE, Baicu CF, Zile MR, Lindsey ML, Bradshaw AD. Increased ADAMTS1 mediates SPARC-dependent collagen deposition in the aging myocardium. Am J Physiol Endocrinol Metab. 2016;310:E1027–1035. doi: 10.1152/ajpendo.00040.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toba H, de Castro Bras LE, Baicu CF, Zile MR, Lindsey ML, Bradshaw AD. Secreted protein acidic and rich in cysteine facilitates age-related cardiac inflammation and macrophage M1 polarization. Am J Physiol Cell Physiol. 2015;308:C972–982. doi: 10.1152/ajpcell.00402.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trial J, Entman ML, Cieslik KA. Mesenchymal stem cell-derived inflammatory fibroblasts mediate interstitial fibrosis in the aging heart. J Mol Cell Cardiol. 2016;91:28–34. doi: 10.1016/j.yjmcc.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y, Okubo T. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. 1994;153:2052–2063. notavailable. [PubMed] [Google Scholar]
- 51.Umekawa T, Osman AM, Han W, Ikeda T, Blomgren K. Resident microglia, rather than blood-derived macrophages, contribute to the earlier and more pronounced inflammatory reaction in the immature compared with the adult hippocampus after hypoxia-ischemia. Glia. 2015;63:2220–2230. doi: 10.1002/glia.22887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. 2014;102:258–269. doi: 10.1093/cvr/cvu062. [DOI] [PubMed] [Google Scholar]
- 53.Vanoverschelde JJ, Essamri B, Vanbutsele R, d’Hondt A, Cosyns JR, Detry JR, Melin JA. Contribution of left ventricular diastolic function to exercise capacity in normal subjects. J Appl Physiol (1985) 1993;74:2225–2233. doi: 10.1152/jappl.1993.74.5.2225. notavailable. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Kwak D, Fassett J, Liu X, Yao W, Weng X, Xu X, Xu Y, Bache RJ, Mueller DL, Chen Y. Role of bone marrow-derived CD11c+ dendritic cells in systolic overload-induced left ventricular inflammation, fibrosis and hypertrophy. Basic Res Cardiol. 2017;112:25. doi: 10.1007/s00395-017-0615-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschope C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4:44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.