

Abstract
The phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR signaling pathway plays an essential role in regulation of normal cell growth, metabolism, and survival. Somatic activating mutations in the PI3K/AKT/mTOR pathway are amongst the most common mutations identified in cancer, and have been shown to cause a spectrum of overgrowth syndromes including PIK3CA-Related Overgrowth Spectrum, Proteus syndrome, and brain overgrowth conditions. Clinical findings in these disorders may be isolated or multiple, including sporadic or mosaic overgrowth (adipose, skeletal, muscle, brain, vascular, or lymphatic), and skin abnormalities (including epidermal nevi, hyper- and hypopigmented lesions), and have the potential risk of tumorigenesis. Key negative regulators of the PI3K-AKT signaling pathway include PTEN and TSC1/TSC2 and germline loss-of function mutations of these genes are established to cause PTEN Hamartoma Tumor Syndrome and Tuberous Sclerosis Complex. Mosaic forms of these conditions lead to increased activation of PI3K and mTOR at affected sites and there is phenotypic overlap between these conditions. All are associated with significant morbidity with limited options for treatment other than symptomatic therapies and surgeries. As dysregulation of the PI3K/AKT/mTOR pathway has been implicated in cancer, several small molecule inhibitors targeting different components of the PI3K/AKT/mTOR signaling pathway are under clinical investigation. The development of these therapies brings closer the prospect of targeting treatment for somatic PI3K/AKT/mTOR-related overgrowth syndromes. This review describes the clinical findings, gene function and pathogenesis of these mosaic overgrowth syndromes, and presents existing and future treatment strategies to reduce or prevent associated complications of these disorders.
Keywords: Somatic mosaicism, segmental overgrowth, therapy, PI3K/AKT/mTOR pathway
INTRODUCTION
The PI3K/AKT/mTOR (Phosphoinositide-3-kinase/Protein Kinase B/mechanistic target of rapamycin) signaling pathway plays a major role in cellular processes in the normal cell such as proliferation, growth, angiogenesis, cell survival, and metabolism. The components of this pathway interact with many other pathways, including Ras/MAPK (Ras family of small GTPase proteins/mitogen-activated protein kinases) pathway. Overactivation of the PI3K/AKT/mTOR pathway results in significant dysregulation of normal cellular functions, which in turn leads to competitive growth metabolic advantage, and angiogenesis [Porta et al., 2014]. Pathogenic somatic mutations and gains or losses in each these genes are linked with many different solid and hematological tumors or cancers [Vivanco and Sawyers, 2002; Yuan and Cantley, 2008; Weigelt and Downward, 2012; Porta et al., 2014]. Mutations in genes that activate the PI3K/AKT/mTOR pathway have now been identified in multiple mosaic overgrowth disorders [Lindhurst et al., 2011; Lindhurst et al., 2012, Lee et al, 2012, Riviere, et al, 2012; Kurek et al., 2012; Shirley et al., 2013; Keppler-Noreuil et al., 2014; Mirzaa et al., 2012; Mirzaa et al., 2013].
Phosphoinositide 3-kinases (PI3Ks) are lipid kinases that phosphorylate the 3-hydroxyl group of phosphoinositides. PI3K converts phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-triphosphate (PIP3), which interacts with AKT at the cellular membrane where it is phosphorylated and activated. An important negative regulator of PI3K is the tumor suppressor phosphatase and tensin homolog (PTEN), which dephosphorylates PIP3 [Weigelt et al., 2012]. AKT serves a range of cellular functions with effects on metabolism and cell survival, and through indirect activation of mTOR controls cell proliferation, protein translation, and autophagy. mTOR is a serine/threonine kinase that functions as a downstream effector of the PI3K/AKT signaling pathway. mTOR exists as two multi-protein complexes, mTORC1 and mTORC2, which differ in composition, sensitivity to sirolimus (rapamycin) and activity. mTORC2 lies upstream of and activates AKT, whereas mTORC1 lies downstream from AKT and is activated via AKT-mediated inhibition of tuberous sclerosis complex 1 and 2 (TSC1 and2) and proline-rich Akt substrate 40 (AKT1S1) [PRAS40]. The PI3K/AKT/mTOR signaling pathway is highly complex with multiple levels of feedback inhibition, and further exhibits significant cross-talk with the RAS/MAPK pathway [Figure 1].
Figure 1.
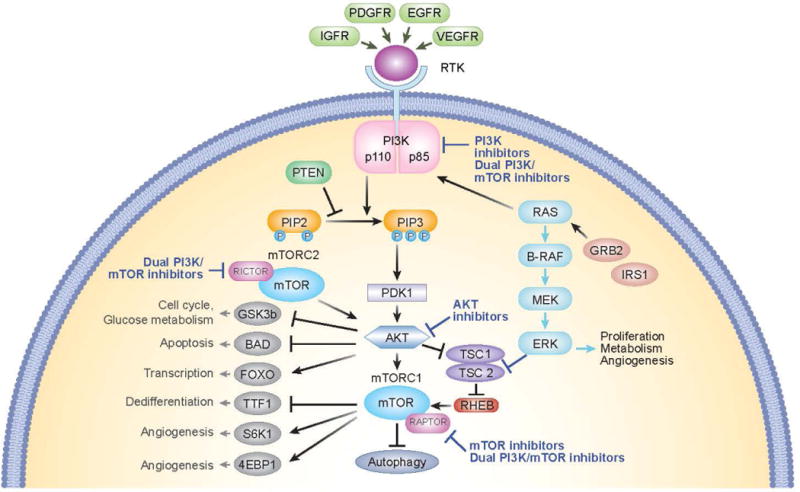
Diagram of the PI3K/AKT/mTOR pathway and interaction with PTEN and TSC1/TSC2. Potential sites of action of inhibitors are shown. PI3K converts PIP2 into PIP3, which interacts with AKT at the cellular membrane where it is phosphorylated and activated. PTEN is a negative regulator of PI3K, which dephosphorylates PIP3. AKT serves a range of cellular functions with effects on metabolism and cell survival, and through indirect activation of mTOR controls cell proliferation, protein translation, and autophagy. mTOR exists as two multi-protein complexes, mTORC1 and mTORC2. mTORC2 lies upstream of and activates AKT, whereas mTORC1 lies downstream from AKT and is activated via AKT-mediated inhibition of tuberous sclerosis complex 1 and 2 (TSC1 and2) and proline-rich Akt substrate 40 (PRAS40). The PI3K/AKT/mTOR signaling pathway exhibits cross-talk with the RAS/MAPK pathway.
Abbreviations: BAD, Bcl-2 associated agonist of cell death; EGFR. Epidermal growth factor receptor; FOXO, forkhead box protein O1; IGFR, insulin-like growth factor receptor; GSK3b,glycogen synthase kinase-3; mTOR, mechanistic target of rapamycin; mTORC, mTOR complex; PDGFR, platelet-derived growth factor receptor; PDK1, protein serine/threonine kinase-3′-phosphoinositide-dependent kinase 1; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-triphosphate; PTEN, phosphatase and tensin homolog; RTK, receptor tyrosine kinase; TSC, tuberous sclerosis complex; TTF1, Thyroid transcription factor 1; SGK1, serum and glucocorticoid regulated kinase 1; VEGFR, vascular endothelial growth factor receptor; 4EBP1, 4E binding protein 1.
There have been multiple phenotypes and genotypes described in association with mutations in the PI3K/AKT/mTOR growth pathway leading to a spectrum of segmental overgrowth syndromes [Lindhurst et al., 2011; Lindhurst et al., 2012, Lee et al, 2012, Riviere, et al, 2012; Kurek et al., 2012; Shirley et al., 2013; Keppler-Noreuil et al., 2014; Mirzaa et al., 2012; Mirzaa et al., 2013]. These syndromes include Proteus syndrome and other AKT-related disorders, PIK3CA-Related Overgrowth Spectrum, and mTOR-related disorders. Tuberous sclerosis complex (TSC), caused by loss-of-function mutations in either TSC1 or TSC2, and PTEN Hamartoma Tumor Syndrome (PHTS) are most frequently associated with germline mutations, but mosaic forms also appear as isolated (simplex or sporadic) occurrences [Mester & Eng, 2013; Tyburczy et al., 2015a, 2015b; Seibert et al., 2011; Cornu et al., 2013] [Table I]. The genes involved in these syndromes include gain-of-function mutations in PIK3CA, PIK3R2, AKT1, AKT2, AKT3, and mTOR, and loss of function mutations or deletions in PTEN, TSC1 and TSC2, and result in a spectrum of abnormal growth ranging from an isolated small skin, vascular, fibroadipose lesion with minimal or no overgrowth to extensive involvement of multiple tissues with striking extremity enlargement and tumor susceptibility [Laplante et al., 2012; Dazert et al., 2011; Inoki et al., 2002; Huang and Manning, 2009]. The common molecular basis of these disorders leads to significant clinical overlap, and raises the interesting biological question about the relative contributions of underlying genotype, timing of the mutation, and of the precise cell of origin of the founder mutation during development [Keppler-Noreuil et al., 2014].
Table I.
Somatic Overgrowth Disorders Associated with PI3K/AKT/mTOR Pathway
| Component of PI3K/AKT/mTOR pathway | Clinical Overgrowth Disorders |
|---|---|
| PI3K |
PIK3CA-Related Overgrowth Spectrum
|
| AKT |
|
| mTOR |
|
| PTEN | PHTS
|
| TSC1/2 | Tuberous Sclerosis Complex |
The genetic etiology for these disorders is a postzygotic mutation during embryogenesis in a population of somatic cells derived from the original zygote resulting in two or more genetically distinct cell lineages. Mosaicism for mutations in autosomal dominant single genes i.e. somatic heterozygous mutations, in either somatic or germline cells explains the variable clinical findings in these disorders. Whether mosaicism for a mutation involves only somatic tissue, the germline, or both depends on whether the mutation occurred before or after the separation of germline cells from somatic cells during embryogenesis. Mosaic mutations in somatic cells are typically sporadic and not inherited from a parent. The affected individual has a low risk of having an affected offspring or child.
The segmental mosaic forms of autosomal dominant genodermatoses have been described as type 1 or type 2 [Figure 2] [Happle et al., 1997; Paller, 2004]. In type 1 segmental mosaicism, the disease manifestations are limited to certain body regions whereas in type 2 they are more severe in certain body regions [Figure 2]. Type 1 mosaicism occurs due to a somatic mutation on a normal genetic background, whereas type 2 mosaicism is due to a second-hit mutation on a heterozygous germline background [Happle, 2014]. It is not known whether those with type 1 segmental mosaicism have somatic mosaicism alone or possibly somatic plus gonadal mosaicism, but all with type 2 mosaicism are expected to be heterozygous in the gonads. It is the type 2 segmental mosaic forms of PTEN Hamartoma Tumor Syndrome (PHTS) and TSC that may overlap the most with the overgrowth syndromes due to mutations that activate the PI3K/AKT/mTOR pathway. In type 2 segmental mosaicism, there is inactivation of both alleles of the tumor suppressor gene within the affected segment, i.e. somatic mosaicism on a background of a germline mutation causing a so-called “double-hit”, which has a similar functional consequence to an activating mutation in one allele of an oncogene along this pathway.
Figure 2.
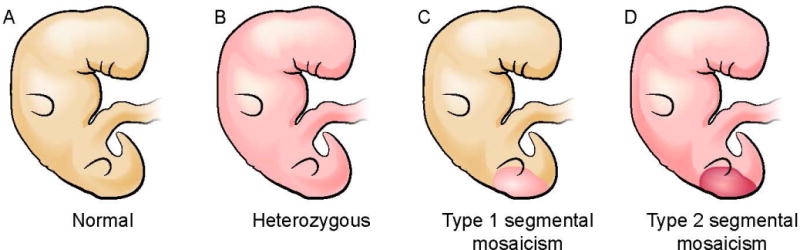
Type 1 and Type 2 segmental mosaicism with an autosomal dominant gene mutation. A. Normal embryo; B. Embryo with a germline heterozygous autosomal dominant mutation indicated with pink color throughout the body; C. Type 1 segmental mosaicism: an embryo with a post-zygotic (somatic) heterozygous mutation in a population of cells affecting a portion of developing tissues, shown in pink confined to a posterior region; D. Type 2 segmental mosaicism: an embryo like B with a germline mutation (pink shading), who in addition has a new post-zygotic mutation in a portion of cells (dark pink in posterior region), inactivating the other allele, also called loss of heterozygosity.
Genetic somatic alterations in the PI3K/AKT/mTOR pathway cause many different cancers. Inhibitors of PI3K, AKT, and mTOR, either alone or in combination, have been effective in pre-clinical and clinical trials, and they have been approved for the treatment of these cancers. These studies have brought the application of targeted drug trials in segmental overgrowth dramatically closer to fruition.
This review of the PI3K/AKT/mTOR signaling pathway will focus on the clinical features, gene function, pathogenesis, and therapeutic strategies for the somatic overgrowth disorders of this pathway, including PIK3CA-Related Overgrowth Spectrum (PROS), Proteus Syndrome (PS), mTOR-related disorders, as well as mosaic presentations of the negative regulators in the pathway, PTEN and TSC1/TSC2.
PIK3CA-RELATED OVERGROWTH SPECTRUM (PROS)
Clinical Phenotype
PROS is characterized by congenital or early childhood-onset overgrowth, sporadic occurrence, and mosaic distribution. Clinical diagnostic criteria are outlined in Table II [Keppler-Noreuil et al., 2015]. Other characteristic findings may include: polydactyly, cutaneous syndactyly, kidney and urinary tract abnormalities (agenesis, hydronephrosis, duplicated ureter, cysts), and abnormalities of the ovaries (cysts) and testes (hydroceles). Clinical presentation is highly variable varied with different clinical entities described [Table I] and may be dependent upon the timing of the mutation during embryogenesis, the tissue type(s) affected, and potentially the PIK3CA genotype [Mirzaa et al., 2013; Keppler-Noreuil et al, 2014].
TABLE II.
Clinical Diagnostic Criteria for PROS
| Required: | Presence of somatic PIK3CA mutation | |
| Congenital or Early Childhood Onset | ||
| Overgrowth: Sporadic and Mosaic | ||
| Features as described in either A or B | ||
| A. | Two or more features (Spectrum) | |
| Overgrowth: Adipose, Muscle, Skeletal, Nerve | ||
| Vascular Malformations: Capillary, Venous, Arteriovenous, Lymphatic | ||
| Epidermal Nevus | ||
| B. | Isolated features | |
| Macrodactyly | ||
| Overgrown splayed feet/hands with overgrown limbs | ||
| Overgrowth of truncal adipose tissue | ||
| Large lymphatic malformation | ||
| Hemimegalencephaly or Dysplastic megalencephaly or Focal cortical dysplasia | ||
| Epidermal Nevus | ||
Adapted from Keppler-Noreuil et al., 2015
Segmental overgrowth is often congenital in onset, but it is usually noted by one year of age with progressive overgrowth of tissues persisting in some cases into adulthood [Figure 3]. Affected patients may suffer from severe functional impairment, hemorrhage, thromboembolism, epilepsy, and developmental delay, depending on the distribution and extent of disease [Lindhurst et al., 2012; Mirzaa et al., 2012; Lee et al., 2012; Kurek et al., 2012]. Currently, the only established management is serial debulking surgery, amputation, or vascular interventional techniques [Alomari et al., 2011].
Figure 3.
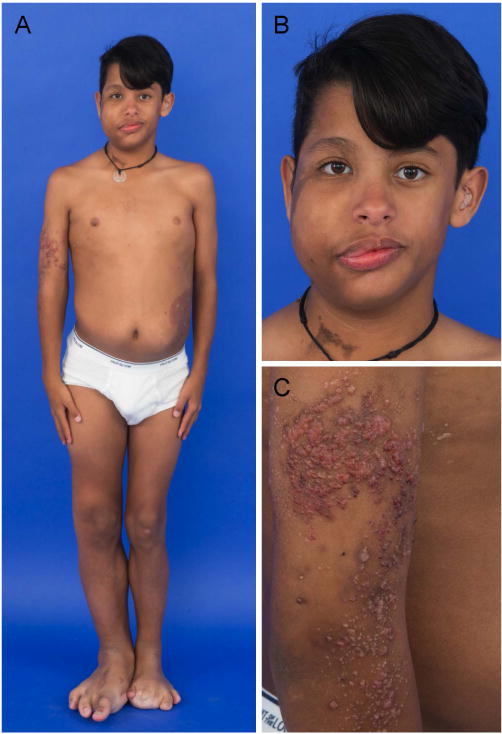
14 year old male with PROS. A. Frontal whole body view shows right facial, leg and bilateral feet hemihyperplasia (right more than left) after toe and metatarsal amputations, and left abdominal fibroadipose hyperplasia. B. Facial view shows right facial hemihyperplasia and epidermal nevus involving his right cheek and neck. C. Lymphovascular lesion involving his right arm and trunk.
The risk for tumorigenesis and development of malignancies is a theoretical concern because PIK3CA is somatically mutated or overexpressed in many cancers including colorectal, ovarian, breast, and hepatocellular carcinomas, and in glioblastomas [Vivanco and Sawyers, 2002; Campbell et al., 2004; Lee et al., 2005; Levine et al., 2005; Li et al., 2005; Velho et al., 2005; Li et al., 2005; Velho et al., 2005; Yuan and Cantley, 2008]. The mutations in PIK3CA in cancers and a large proportion of patients with PROS, especially those with the phenotypes of CLOVES, fibroadipose hyperplasia, and isolated macrodactyly, are most commonly located at hotspots within the helical and kinase domains and result in gain of function mutations, which have been implicated in oncogenicity [Samuels et al., 2004; Kang et al., 2005]. Four patients with PROS have been reported to have developed Wilms tumor or nephroblastomatosis; two of the four on whom there is clinical information were diagnosed at ages nine months and two years [Gripp et al., 2016]. However, further longitudinal studies with consistent surveillance are needed to accurately assess the potential risk.
PIK3CA Gene Function/Pathogenesis
PIK3CA is expressed in all cells and encodes the p110 alpha catalytic subunit of phosphoinositide 3-kinase (PI3K), which lies downstream from growth-associated receptors including the insulin and insulin-like growth factor 1 (IGF-1) receptors. PI3K is subdivided into three different classes according to substrate preference and composition; Class I PI3Ks are well characterized and comprise a regulatory and a catalytic subunit and phosphorylate PIP2 to PIP3, whereas class II and III PI3Ks possess a catalytic subunit only and phosphorylate phosphatidylinositol PI to phosphatidylinositol 4- phosphate (PIP). Class IA PI3Ks are predominantly involved in carcinogenesis and comprise a p85 regulatory subunit and a p110 catalytic subunit. There are four isoforms of p110 (class IA; p110 α, β, and δ and class IB; p110γ), of which α (PI3KCA) and β (PI3KCB) have been most closely associated with oncogenesis [Yuan and Cantley, 2008]. These isoforms, as well as class IB(γ), have roles in inflammatory and autoimmune diseases, thrombosis, and malignancy [Foster et al., 2012]. PIK3CA comprises five domains: adaptor binding (ABD), ras binding (RBD), C2, helical, and kinase domains. Mutations are found in all domains in PROS, with the exception of the RBD, and like those associated with cancer, are most commonly identified in the kinase and helical domains [Gripp et al., 2016]. These PIK3CA mutations promote a gain of function of PI3K activity leading to increased AKT and mTOR signaling [Vanhaesebroeck et al., 2012] with resultant abnormal growth in the affected tissue.
PROTEUS SYNDROME (PS) – AKT1 Somatic Mutation
Clinical Phenotype
PS is a rare segmental overgrowth disorder, which is progressive and associated with high morbidity and mortality. It is caused by a somatic gain of function mutation, c.49G→A, p.Glu17Lys) in the oncogene AKT1, encoding the AKT1 kinase [Lindhurst et al., 2011]. This activating mutation in AKT1 stimulates the AKT/PI3K pathway, resulting in increased cell proliferation and decreased apoptosis, and ultimately to the mosaic multisystem / multi-tissue overgrowth and tumor susceptibility manifested in PS.
Clinical diagnostic criteria for PS are summarized in TABLE III [Biesecker and Sapp, 2012]. A majority of the patients are born with few or no manifestations, but develop symptoms between 6 months and 2 years, with progressive, severe, and distorting overgrowth [Figure 4]. The overgrowth of PS can affect any organ and leads to many complications including orthopedic abnormalities, restrictive lung disease associated with severe scoliosis, cardiac, renal, pulmonary, gastrointestinal, and other parenchymal manifestations which can be fatal. Patients with PS appear to have a cancer predisposition with a heterogeneous array of tumors reported (ovarian and testicular cystadenoma, parotid adenoma, meningioma, hamartoma, mesothelioma, cysts of the liver, pancreas and epididymis), most of which are benign. However, a number of patients have had malignant tumors including mesothelioma, breast cancer, and papillary thyroid cancer. The most common life threatening manifestation is deep vein thrombosis and pulmonary embolism [Biesecker and Sapp, 2012]. The role of AKT1 in the predisposition to thrombosis in PS may be multifaceted; the underlying pathogenesis of this risk is being studied at the National Institutes of Health (NIH). One major risk factor is the presence of underlying vascular malformations compounded by surgery and immobility.
Table III.
Proteus syndrome diagnostic criteria.
| GENERAL CRITERIA | |
|---|---|
| Mosaic distribution | |
| Progressive course | |
| Sporadic occurrence | |
| SPECIFIC CRITERIA | |
| Category A | |
| Cerebriform connective tissue nevus | |
| Category B | |
| Linear epidermal nevus | |
| Asymmetric, disproportionate overgrowth of two of: Limbs, skull, external auditory canal, vertebrae, or viscera | |
| Specific tumors in the first decade of life: Bilateral ovarian cystadenomas Monomorphic parotid adenomas | |
| Category C | |
| Dysregulated adipose tissue | |
| Vascular malformations of one or more: Capillary, venous, and/or lymphatic | |
| Lung bullae | |
| Facial phenotype: Long face, dolichocephaly, down-slanted palpebral fissures, low nasal bridge, wide or anteverted nares, open mouth at rest |
Diagnosis requires all three general criteria plus one criterion from A, two from B, or three from C [Adapted from Biesecker and Sapp, 2012]
Figure 4.
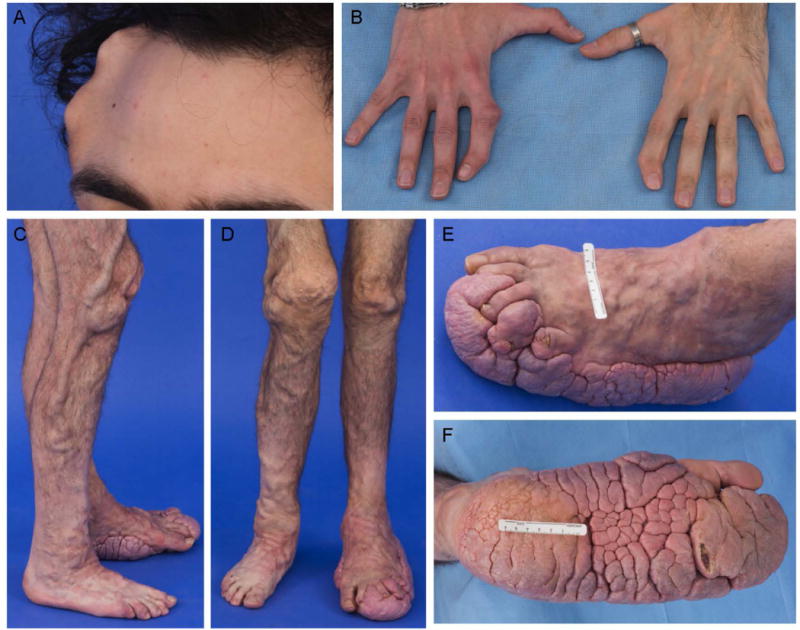
30 year old male with Proteus syndrome. A. Cranial hyperostosis involving his right frontoparietal bones. B. Bony overgrowth with ulnar deviation of his bilateral 2nd fingers. C. Lateral view of his right leg shows bony hemihyperplasia and vascular malformations – venous varicosities. D. Frontal view of his legs shows venous varicosities, bony overgrowth of his right leg. E. Dorsolateral view of left foot, and F. Plantar view of left foot with cerebriform connective tissue nevus (CCTN).
The only known treatments for patients with PS are symptomatic, primarily surgical. Symptomatic orthopedic surgical treatment for the skeletal overgrowth at the NIH Clinical Center has been employed for many years [Tosi et al., 2011]. Surgical treatment can be undertaken for other types of overgrowth including the soft tissue overgrowth, although this is far from satisfactory, and most patients are severely deformed and disfigured. A primary treatment for PS would clearly be beneficial to prevent or reduce the morbidity associated with this disease.
AKT1 Gene Function/Pathogenesis
AKT (Protein kinase B, PKB) is a serine/threonine kinase that belongs to the protein kinase A, G, and C families (PKA, PKC, PKG) (AGC) group, which affects a wide range of biological functions including cell proliferation and growth, metabolism, protein synthesis, migration, angiogenesis, and anti-apoptotic ability [Vivanco and Sawyers, 2002]. AKT is composed of three isoforms, including AKT1, AKT2, and AKT3 [Hubbard et al., 2014]. A specific somatic activating mutation in AKT1 (c.49G>A, p.Glu17Lys) was found to be causative of PS. This mutation is present in varying levels across many tissues and confirms the mosaicism hypothesis proposed by Happle [Lindhurst et al., 2011]. It has also been identified in sporadic tumors, including certain meningiomas [Keppler-Noreuil et al., 2016].
AKT2 AND AKT3 SOMATIC MUTATIONS
Clinical Phenotypes
Somatic mutations in codon 17 of AKT2 and AKT3 have both been reported and have distinct phenotypes from PS. Hussain et al. [2012] described an activating AKT2 mutation causing severe insulin-independent hypoglycemia, mild asymmetric overgrowth, and progressive obesity (OMIM# 240900). Activating AKT3 mutations have been associated with brain overgrowth, and Megalencephaly-Polymicrogyria-Polydactyly-Hydrocephalus (MPPH2) (OMIM# 615937) [Poduri et al., 2012; Riviere et al., 2012]. There is genetic heterogeneity for MPPH: MPPH1 (OMIM# 603387), which is caused by a heterozygous germline mutation in PIK3R2, MPPH2 (OMIM# 615937) caused by both somatic and germline mutations in AKT3, and MPPH3 (OMIM# 615938) caused by both somatic and germline mutations in CCND2 gene. There is considerable phenotypic similarity between this disorder and Megalencephaly-Capillary Malformation-Polymicrogyria syndrome (MCAP) (OMIM# 602501), which is caused by both germline and somatic heterozygous mutations in PIK3CA [Riviere et al., 2012; Lee et al., 2012; Mirzaa et al., 2016].
Pathogenesis
AKT2 is highly expressed in insulin-responsive tissues including skeletal muscle, liver, and fat, and is more closely implicated in metabolism than AKT3 [Whiteman et al., 2002]. AKT3 is predominantly expressed in the brain and the heart, and has been shown to regulate growth. Various mechanisms contribute to activation of the AKT pathway in human tumors, including dysregulation of those pathways that are upstream of PTEN and PI3K, and autocrine or paracrine stimulation of the receptor kinases. Somatic mutation and overexpression of AKT1 and AKT2 have been found in many human cancers, including breast, endometrial, lung, bile duct, ovarian, renal cell, melanoma, and colon cancers [Carpten et al., 2007; Yu et al., 2015].
mTOR-RELATED DISORDERS
Clinical Phenotypes
Mutations in a number of components of the PI3K-AKT-mTOR pathway lead to activation of mTOR and to hemimegalencephaly or focal cortical dysplasia type II [D’Gama et al., 2015]. Focal cortical dysplasia type II primarily presents in children in two subtypes that respond differently to clinical management based on histological findings: type IIA with dysmorphic neurons in the cortex, and type IIb with additional balloon cells. Initial histological analysis of abnormally enlarged or cytomegalic neurons in focal cortical dysplasia brain samples identified increased expression of markers of mTOR activation [Ljungberg et al., 2006]. Later studies identified gain of function mutations in components of the PI3K-AKT-mTOR pathway in the brains of individuals with hemimegalencephaly (Lee et al., 2012). Somatic mutations in mTOR itself were eventually identified to be sufficient to cause focal cortical dysplasia type IIb [Lim et al., 2015; Nakashima et al., 2015; Leventer et al., 2015], demonstrating that selective gain of function activation of mTOR accounts for this abnormal brain phenotype. In all of these cases, the identified variants were somatic and present in a mosaic distribution within the brain [Nakashima et al., 2015].
Most of the cases associated with mTOR mosaic gain of function mutations have significant intellectual disability, autism, and pigmentary skin changes including hypomelanosis of Ito [Figure 5]. A few case reports have described syndromic presentations associated with germline mTOR mutations. One family with a germline Glu1799Lysgain of function mutation manifested a phenotype that overlaps with PROS, including megalencephaly, polymicrogyria, intellectual disability, and skin pigmentary changes [Baynam et al., 2015]. An additional family with a germline Glu1799Lys gain of function mutation in mTOR presented with megalencephaly, autism, and intellectual disability [Mroske et al., 2015]. While there is some similarity of these phenotypes to those present in PROS, there is currently insufficient evidence to suggest that germline mTOR mutations are associated with a similar phenotype to PROS, which is typically associated with much more segmental involvement. In addition, given the complexity and cross-talk in the PI3K/AKT/mTOR pathway, it is as yet unclear whether gain of function mutations in mTOR are sufficient to cause the overgrowth phenotype observed in the syndromes associated with mutations in upstream components of the pathway (PI3K, PTEN, AKT).
Figure 5.
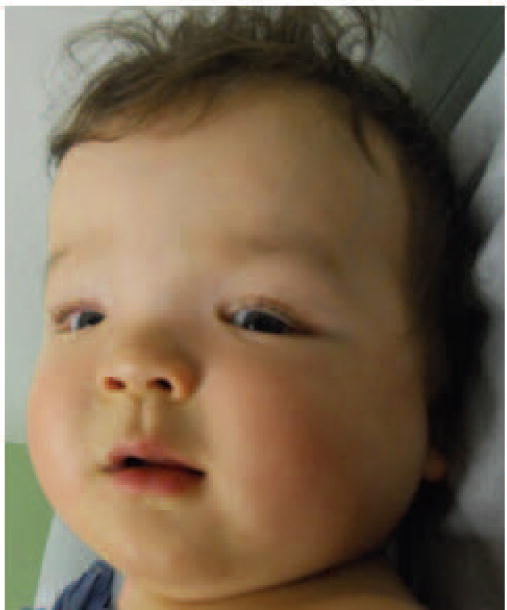
9 month old male infant with a mosaic c.5390C>T (p.Thr1977Ile) mutation in mTOR associated with megalencephaly, developmental delay, dysmorphic features, mosaic pigmentary skin changes, and polymicrogyria
In addition, syndromic germline variants in components of the GATOR1 complex, a negative regulator of mTOR, have been described associated with familial forms of focal cortical dysplasia. Mutations in components of the GATOR1 complex reduce its GTPase activating protein (GAP) activity, which is required for the inhibition of small GTPases that normally promote mTOR activation. Specifically, mutations in the GATOR complex components DEPDC5 and NPRL3 cause familial focal cortical dysplasia type IIa (OMIM# 607341) [Scerri et al., 2015; Sim et al., 2016]. Analysis of brain tissue samples has demonstrated activation of mTOR in these cases. In addition, mTOR negatively regulates autophagy, so it is not surprising that abnormalities in autophagy are present in these cells [Yasin et al., 2013]. These cases have established the beginnings of a syndromic presentation to germline mTOR gain of function mutations. These findings have also led to the suggestion of treating epilepsy and focal cortical dysplasia seizure disorders with mTOR inhibitors [Wong, 2010; Wong, 2013].
mTOR Gene Function/ Pathogenesis
mTOR integrates nutrient availability and environmental stress with cell proliferation and growth, as well as regulates autophagy, cell death and cell differentiation. As a nutrient-sensitive kinase, it responds to amino acids and glucose to promote cellular metabolism, and it mediates cellular adaptations to environmental stressors, including hypoxia.
mTOR function is carried out by two distinct complexes, as described earlier. The mTORC1 complex is made up of mTOR, RAPTOR, mLST8, and AKT1S1 (PRAS40). It is extremely sensitive to rapamycin and thus represents the target of the first-generation mTOR inhibitors. It activates ribosomal protein S6 kinase beta-1 (S6K1, RPS6KB1), which phosphorylates S6 to promote protein synthesis, and inactivates the repressor of protein synthesis eukaryotic translation initiation factor 4E binding protein 1 (4EBP1, EIF4EBP1), leading to protein translation and cell growth [Crino, 2011]. The mTORC2 complex is composed of mTOR, RICTOR, MAPKAP1, and mLST8; it is less sensitive to rapamycin. It is known to activate AKT, thereby promoting cell proliferation and survival.
mTOR activation causes increased production of multiple proteins, several of which have been implicated in the pathogenesis of multiple tumors, e.g., cyclin D1, which allows for progression of cells through the cell cycle, and hypoxia inducible factor (HIF), which drives the expression of pro-angiogenic growth factors such as vascular endothelial growth factor (VEGF) [Porta et al., 2014] [Figure 1]. The location of mTOR somatic mutations identified in cancer can vary across all of the protein sequence, including the kinase and FAT domains [Hardt et al., 2011]. Similarly, mutations in mTOR associated with pediatric disorders have been identified in the FAT and kinase domains, as well as outside both of these domains (Mirzaa et al, 2016). These mutations have different extents of activation of downstream targets of mTOR. They may differentially activate mTORC1 versus mTORC2 [Grabiner et al., 2014], which has implications for the therapeutic agents used.
PTEN HAMARTOMA TUMOR SYNDROME (PHTS)
Clinical Phenotypes
Haploinsufficiency due to loss of function mutations of PTEN, or dominant negative mutations in the protein, is the proposed mechanism of several autosomal dominant disorders, including macrocephaly-autism, Cowden syndrome (OMIM# 158350) and Bannayan-Riley-Ruvalcaba syndrome (OMIM# 153480) [Eng et al., 2016]. Macrocephaly-autism syndrome refers to a subset of individuals within the autism spectrum with extreme macrocephaly or head circumference more than two standard deviations above the mean. The association with mutations in PTEN was initially described in a family with Cowden syndrome [Goffin et al., 2001] and later confirmed in 10–20% of cases with autism and macrocephaly [Butler et al., 2005; Klein et al., 2013]. Cowden syndrome presents primarily with macrocephaly, skin findings that include trichilemmomas, and predisposition to lipomas and hamartomatous tumors of the gastrointestinal tract, skin, and thyroid gland. In addition, some individuals present with developmental delay, intellectual disability, or Lhermitte-Duclos disease, a dysplastic cerebellar gangliocytoma hamartomatous tumor associated with mutations in PTEN. Many of the phenotypic features of Cowden syndrome overlap with Bannayan-Riley-Ruvalcaba syndrome, which presents with intellectual disability, macrocephaly, macrosomia, dysmorphic features, and predisposition to hamartomas, primarily in childhood. Therefore, the phenotypic findings associated with mutations in PTEN are part of an overlapping spectrum, leading to the establishment of PHTS as being inclusive of all of its associated dominant conditions [Figure 6].
Figure 6.
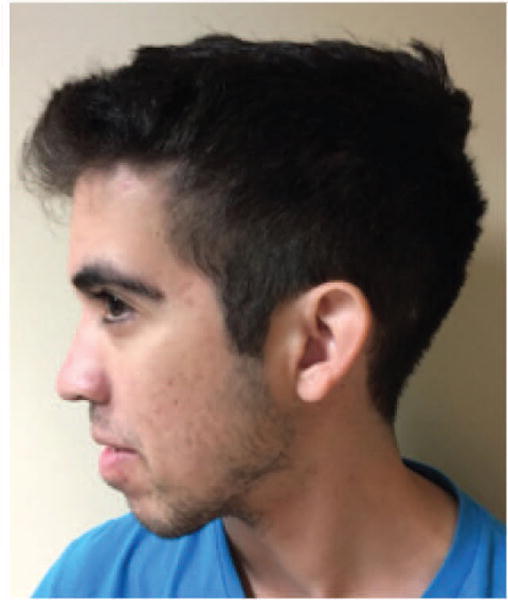
20 year old male with PTEN Hamartoma Tumor Syndrome, who has a heterozygous p.Pro38His variant in PTEN associated with extreme macrocephaly, dysmorphic features, pigmentary skin macules, developmental delay, and intellectual disability.
A small number of rare disorders are caused by type 2 segmental mosaicism with regional homozygous loss of PTEN function. One example is SOLAMEN syndrome, in which mosaic compound heterozygous mutations in PTEN cause segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (“SOLAMEN”) [Caux et al., 2007].
PTEN gene function/ Pathogenesis
PTEN encodes for a phosphatase that regulates PI3K signaling. PTEN dephosphorylates PIP3 to regulate and decrease pathway activation. In addition to its role in PI3K/AKT signaling, PTEN has roles in the nucleus on chromatin function and transcription that are less well characterized [Chen et al., 2014]. Furthermore, it is not clear that these roles are dependent on its lipid phosphatase domain [Song et al., 2011].
Inactivating heterozygous mutations in PTEN are associated with excessive PI3K activity. The location of the mutation and ultimate effect on protein function determine phenotype [Rodriguez-Escudero et al., 2011]; however, no unifying model has established clear genotype-phenotype correlations.
Many of the mutations identified in PTEN hamartoma syndrome cases are present in somatic cancers, many clustering at hot spots within the protein but most present at any position throughout the protein. A subgroup of mutations that preserve residual protein function are associated with milder clinical presentations, particularly with autism [Leslie and Longy, 2016]. A subset of variants in the PTEN gene, particularly those that alter residues outside of the phosphatase domain and are associated with cancer, may act in a dominant negative fashion [Papa et al., 2014]. While mouse and in vitro human cell models have demonstrated the efficacy of mTOR inhibitors in reversing part of the phenotypic consequences of PTEN haploinsufficiency, mostly on its canonical effects on cell growth and number [Krymskaya and Goncharova, 2009; Tilot et al., 2015], their efficacy on the non-canonical “moonlighting” functions of PTEN, such as chromatin and transcriptional activity, remains unclear. This has implications for therapeutic outcomes, since inhibiting the effects from excessive PI3K pathway activity may not be sufficient or may not target other aspects of PTEN function that extend beyond the PI3K-AKT-mTOR signaling pathway, including its functions in the nucleus. The dominant negative effect of a subset of PTEN mutations may not be as amenable to therapy as homozygous loss may cause excessive activation of PI3K-AKT-mTOR signaling that would require high doses of mTOR inhibitor. This is important in the setting of therapy, since the extent of pathway activation may determine the effectiveness of a particular dose of drug.
TUBEROUS SCLEROSIS COMPLEX (TSC)
Clinical Phenotype
TSC is ordinarily viewed as a non-mosaic, congenital disease with an autosomal dominant pattern of inheritance due to a germline mutation [DiMario et al., 2015]. The clinical features of individuals with this typical, non-mosaic form of TSC, as well as the less common mosaic forms of TSC, are almost entirely due to the development of tumors in multiple organs. One of the most common manifestations of TSC, seizures, is likely to be caused by cortical tubers. Other tumors include subependymal giant cell astrocytomas, cardiac rhabdomyomas, renal angiomyolipomas, lymphangioleiomyomatosis, and skin tumors such as facial angiofibromas. Diagnosis is made based on clinical criteria or by demonstrating a pathogenic mutation in either the TSC1 or TSC2 gene [Northrup et al., 2013].
TSC Gene Function/ Pathogenesis
The tuberous sclerosis complex comprises TSC1 and TSC2, and it binds to and inhibits Ras homolog enriched in brain (RHEB), [van Slegtenhorst et al., 1998; Aspuria and Tamanoi, 2004]. When the TSC complex is inhibited by AKT, RHEB activates mTOR [Long et al., 2005].
Mutations in either of the TSC genes result in tuberous sclerosis (OMIM# 191100 and 613254) [van Slegtenhorst et al., 1997]. In the non-mosaic condition, the individual has a germline heterozygous inactivating mutation in either the TSC1 or TSC2 tumor suppressor gene, which predisposes the person to the development of multiple characteristic tumors. The tumors occur through somatic “second-hit” mutations that inactivate the wild type allele, resulting in loss of function of the TSC1-TSC2 complex and constitutively active signaling through mTORC1 [Crino et al., 2010]. In organs with multiple tumors studied to date, however, it has been shown that tumors arise through independent second-hit mutations [Tyburczy et al., 2014; Tyburczy et al., 2015a].
Clinical Findings and Pathogenesis of Mosaic Forms of TSC
Mosaicism in TSC may be relatively silent clinically, remaining unnoticed and undiagnosed, for example, until a mosaic parent has an affected child [Verhoef et al., 1999]. The parent may have gonadal and somatic mosaicism, manifesting mild disease, or the parent may have only gonadal mosaicism, with no clinical evidence of TSC.
Patients with disseminated mosaicism may be clinically indistinguishable from non-mosaic forms of TSC except that all such cases will be sporadic. They are expected to have somatic or somatic plus gonadal mosaicism. A clinical scenario in which the possibility of disseminated mosaicism should be entertained is the patient who is diagnosed with TSC but has no mutations identified in DNA obtained from blood using standard methods of genetic testing. Many of these individuals have been shown to be mosaic, with low TSC1 or TSC2 mutant allele frequencies detectable in blood and/or saliva using next generation sequencing [Tyburczy et al., 2015b]. It has also been postulated that the absence of subependymal nodules in patients with tubers is consistent with mosaicism occurring in the neuroectoderm [Boronat et al., 2014].
Type 1 segmental mosaicism may manifest as unilateral facial angiofibromas [Trauner et al., 2003]. Those who may have type 2 mosaicism include those who have TSC plus features similar to the overgrowth syndromes discussed earlier. For example, there are individuals with TSC who also have unilateral or bilateral overgrowth of digits, hand, or an extremity [Soeiro E Sá et al., 2016] [Figure 7]. Other individuals with TSC have been reported to manifest vascular lesions analogous to those observed in the overgrowth syndrome, including port wine stains [Ben-Amitai et al., 2011], Klippel-Trenaunay syndrome [Troost et al., 1975; Assefa & Alemie, 2010], or Sturge-Weber syndrome [Chatterjee et al, 2015]. In these cases, which are suggestive for type 2 segmental mosaicism in TSC, mosaicism is presumed and remains to be proven.
Figure 7.
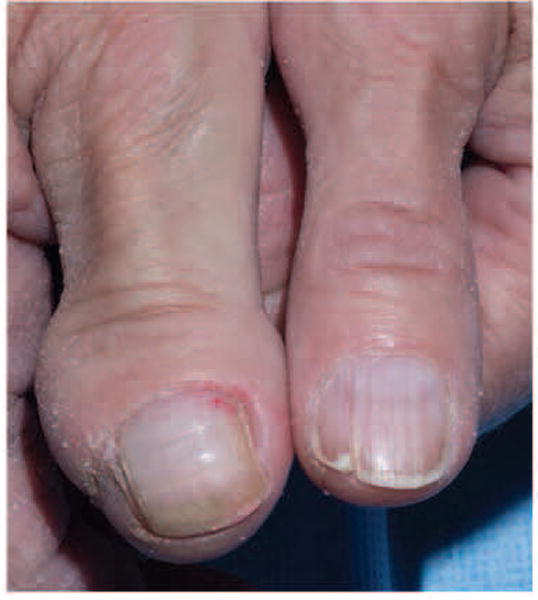
A female with TSC diagnosed based on renal angiomyolipomas, angiofibromas, and hypomelanotic macules, has congenital overgrowth of the right thumb and forearm that may represent type 2 segmental mosaicism.
THERAPEUTIC STRATEGIES – PI3K/AKT/mTOR PATHWAY INHIBITORS
As somatic genetic alterations in the PI3K/AKT/mTOR pathway cause cancer [Table IV], considerable research and development efforts have been deployed to generate inhibitors of PI3K, AKT and mTOR. Several of these compounds are in clinical trials for cancer and may be re-purposed as targeted therapies for segmental overgrowth conditions.
Table IV.
Reported Alterations in the PI3K/AKT/mTOR pathway in cancer
| Cancer Type | Type of Alteration |
|---|---|
| Glioblastoma | PTEN mutation/deficiency |
| Ovarian | PTEN mutations |
| Increased AKT1 activity | |
| AKT2 amplification and overexpression | |
| PI3K p110α amplification | |
| PI3K p85α mutation | |
| Breast | Increased AKT1 activity |
| AKT2 amplification and overexpression | |
| RSK amplification and overexpression | |
| Loss of heterozygosity at PTEN locus | |
| PI3K and AKT2 overactivation | |
| Endometrial | PTEN mutation |
| Hepatocellular carcinoma | PTEN mutation |
| Melanoma | PTEN mutation |
| Lung | PTEN inactivation |
| Renal cell carcinoma | PTEN mutations |
| Thyroid | PTEN mutations |
| AKT overexpression and overactivation | |
| Digestive Tract | PTEN mutation |
| PI3K p85α mutation | |
| Lymphoid |
PTEN mutations P85-EPPH fusion |
Adapted from Vivanco and Sawyers, 2002
PI3K inhibitors
PI3K inhibitors are a class of compounds under development for the treatment of cancer and respiratory disease (asthma and chronic obstructive pulmonary disease), and they may inhibit all PI3Ks or selectively target different isoforms [TABLE V]. Direct inhibition of PI3K is the most attractive prospect for treatment of PROS and PHTS. For PROS in particular, selective inhibition of p110α (PIK3CA) may directly reverse the prevailing molecular defect, whilst minimizing the risk of off-target effects from inhibition of different isoforms. Principal side effects associated with these compounds include hyperglycemia and gastrointestinal side-effects in selective p110α inhibitors, with concomitant immunosuppression in pan-PI3K inhibitors because of blockade of isoforms critical for immune function including p110δ and p110γ [Akinleye et al., 2013]. PI3K p110α inhibitors also block adipocyte insulin-dependent glucose uptake and regulation. There are not yet any clinical trials to evaluate these drugs in PROS, but pre-clinical studies in both PROS and cancers bearing PIK3CA mutations have yielded promising results [Gustin et al., 2008; Laconte et al., 2015].
Table V.
Class I isoform specific PI3K inhibitors in clinical trials*
| Inhibitor Name | International Nonproprietary Name | Primary Target | Development phase |
|---|---|---|---|
| BAY-80-6946 | Copanlisib | Class I PI3Ks | Phase II lymphoma |
| NVP-BKM120 | Buparlisib | Class I PI3Ks | Phase III combination breast cancer |
| NVP-BYL719 | Alpelisib | p110α | Phase II: Combination head and neck cancer |
| GS-9820 (CAL-120) |
p110δ | Phase Ib lymphoid malignancies | |
| GS-1101 | Idealisib | p110δ | Phase III – CLL Asthma/COPD; pre-clinical studies |
| GDC-0032 | Taselisib | p110α, p110δ | Phase II breast cancer |
| AZD-8186 | p110β, p110δ | Phase I : prostate cancer | |
| INK-1197 (IPI-145) |
Duvelisib | p110δ, p110γ | Phase III – CLL Phase II-Asthma, Pre-clinical studies COPD |
| GDC-0941 | Pictilisib | p110α p110β p110δ |
Phase II Combination breast cancer |
| PX-866 | Sonolisib | p110α p110δ, p110γ |
Phase II combination non-small cell lung cancer, ovarian cancer |
Data retrieved from http://Clinicaltrials.gov (May 2016)
AKT inhibitors
AKT inhibitors naturally form the focus as potential treatments for PS and AKT-related conditions. A degree of selectivity for the different isoforms of AKT can be achieved, but to a lesser extent than for PI3K isoforms in view of the higher degree of conservation across AKT isoforms. There are several AKT inhibitors in clinical trials for cancer [Table VI]. A Phase I dose finding trial of ARQ 092, a pan-AKT inhibitor is underway in children and adults with Proteus syndrome [ClinicalTrials.gov: NCT02594215]. The most common side effects associated with ARQ 092 and AKT inhibitors are hyperglycemia, gastrointestinal and myelosuppression [Alexander, 2011]. AKT regulates hepatic glycogenolysis and glucose uptake through its substrate glycogen synthase kinase-3 (GSK-3). Pre-clinical studies found that the mechanism for hyperglycemia is related to peripheral insulin resistance, increased gluconeogenesis and/or induction of glycogenolysis in the liver as well as inhibition of peripheral glucose uptake [Lamming et al., 2012]
Table VI.
Dual PI3K/mTOR, mTOR and AKT inhibitors in clinical trials*
| Inhibitor Name | Primary Target | Development phase [Akinleye et al., 2013] |
|---|---|---|
| BEZ235 | PI3K/mTOR | Phase I/II solid tumors |
| BAY80-6949 | Class I PI3K | Phase I, Neoplasms |
| BGT226 | PI3K/mTOR | Phase I and II, solid tumors; breast |
| XL765 | PI3K/mTOR | Phase I and II, Advanced breast, gliomas, glioblastomas multiforme |
| SF1126 | PI3K/mTOR | Phase I, Advanced solid tumors |
| GSK2126458 | PI3K/mTOR | Phase I, Advanced solid tumors |
| PF-04691502 | PI3K/mTOR | Phase I and II, Advanced solid cancers; breast, endometrial |
| GDC-0980 | PI3K/mTOR | Phase I, (Advanced) solid cancers; non-Hodgkin’s lymphoma, bresast, prostate, endometrial, renal cell |
| PKI-587 | PI3K/mTOR | Phase I and II, Advanced solid cancers; endometrial |
| Sirolimus | mTOR | Phase I, II, III, Advanced solid cancers; breast, liver, rectum, NSCLC, leukemias, lymphomas, head and neck, pancreatic, ovarian, fallopian tube, glioblastomas, fibromatosis |
| OSI-O27 | mTOR/catalytic site | Phase I, solid tumors/lymphoma |
| AZD8055 | mTOR/catalytic site | Phase I/II, solid tumors/lymphoma |
| INK128 | mTOR/catalytic site | Phase I, solid tumors/multiple myeloma |
| Everolimus | mTORC1 | Approved, Renal cell carcinoma, neuroendocrine tumors/subependymal giant cell astrocytoma |
| Temsirolimus | mTORC1 | Approved, Renal cell carcinoma |
| Ridaforolimus | mTORC1 | Phase III, Metastatic soft-tissue and bone sarcomas |
| GSK2141795 | AKT | Phase I, Open-Label solid tumor malignancies (endometrial) or lymphomas; in combination with Trametinib for metastic Triple-Negative Breast Cancer; and metastic uveal melanoma |
| GSK2110183 | AKT | Phase I with Bortezomib, Dexamethasone for multiple myeloma |
| GOG229O | AKT | Endometrial cancer |
| Perifosine | AKT | Phase III Colorectal cancer/multiple myeloma; in combination with MEK inhibitor for multiple myeloma or solid tumor cancers |
| MK2206 | AKT1/2 | Phase I/II solid tumors; pancreatic/colon/breast/lung, lymphomas |
| AZD5363 | AKT1/ PIK3CA or PTEN | Phase I Advanced breast, gynecological cancers or other solid cancers |
| ARQ 092 | panAKT | Phase 1, Advance solid tumors and recurrent malignant lymphoma |
Data retrieved from http://Clinicaltrials.gov (May 2016)
As AKT transduces signals from PI3K downstream, AKT inhibitors could potentially be used for the treatment of PROS. There are currently no scheduled trials with AKT inhibitors in PROS.
mTOR inhibitors
Allosteric inhibitors of mTOR include sirolimus (rapamycin), everolimus, ridaforolimus, temsirolimus and are licensed therapies for a range of conditions from transplant suppression to cancer therapy [Table VII] [Lamming et al., 2013]. Allosteric inhibitors of mTOR act by binding to FKBP12 (FK binding protein 12 kDa), an immunophilin and form a complex termed FKBP12-rapamycin. This complex in turn binds to the FKBP12-rapamycin binding domain (FRB) of mTORC1. After binding to the FRB domain of the mTOR protein, the FKBP12-rapamycin complex potently inhibits the activity of mTORC1 by blocking the binding of mTORC1 to its substrates. In particular, the growth inhibitory effects of allosteric mTOR inhibitors are achieved through inhibition of the cell cycle and blockade of the progression from G1 to S phase of the cell cycle. Inhibition of mTORC2 is more variable and tissue specific; the mechanism is thought to be depletion of mTOR availability through formation of the FKBP12-rapamycin complex, which prevents assembly of mTORC2 complexes [Laplante and Sabatini, 2012].
Table VII.
PTEN Hamartoma Tumor Syndrome: Effects of Sirolimus Therapy
| PTEN mutation | Drug | Dose | Age start | Duration | Serum levels | Outcome | Reference |
|---|---|---|---|---|---|---|---|
|
De novo c.507delC |
Sirolimus | 0.1 mg/kg per day, divided into two doses | 16 months | 17 months | 5–10 ng/ml | -reduction soft tissue masses -reduction lymph node size -improved oral intake -development progression -no effect on lipomas |
[Marsh et al., 2008] |
| Deletion exons 2–9 | Sirolimus | 0.1 mg/kg per day | 46 months | 19 months | 5–10 ng/ml | -no effect on subcutaneous fat -reduced abdominal lipomatosis -decreased intestinal mucosa lymphatic tissue hyperplasia |
[Schmid et al., 2014] |
| c.913_914insT | Sirolimus | 0.8 mg/m2/dose, twice daily | 78 months | 12 months | 10–15 ng/ml | -decreased size vascular lesion | [Iacobas et al., 2011] |
As discussed, inhibitors of the PI3K/AKT/mTOR pathway have a common side effect of hyperglycemia. mTOR is important for insulin production and secretion in response to nutrients and also promotes beta-cell growth and proliferation in the pancreas and subsequently increases insulin secretion and reduces blood glucose levels [Laplante and Sabatini, 2012]. In particular, mTORC1 has a crucial role in end organ insulin uptake and its chronic activation, as shown in preclinical models, is associated with insulin resistance through ribosomal protein S6 kinase beta-1 (RPS6SKB1). Finally, liver mTORC2 is essential for insulin-induced AKT signaling regulation and its deletion or inactivation leads to dysregulated hepatic gluconeogenesis and glycogenolysis. Taken together, the above mechanisms provide a comprehensive explanation for the observation that inhibition of PI3K–AKT–mTOR pathway can lead to disrupted insulin function, impaired insulin secretion and development of insulin resistance [Lamming et al., 2012].
Use in PROS
As mTOR is a downstream effector of PI3K, blockade of mTOR is a rational strategy for indirect inhibition of PI3K signaling in PROS. Given the availability and established safety profile, sirolimus has been an attractive off-label candidate to trial as a treatment. A clinical trial investigating sirolimus in PROS (EudraCT 2014-000484-41, ClincalTrials.gov: NCT02428296) is currently underway with expected completion in 2017.
Use in PS
It is unclear if mTOR inhibitors could be beneficial therapies in PS, and there are currently no trials underway to assess this at present.
Use in mTOR-Related Disorders
Animal studies have demonstrated the efficacy of sirolimus therapy during a specific developmental window (E14.5-E18.5 in mouse embryos) in preventing cortical malformations associated with excessive mTOR activation, while postnatal therapy is not effective [Baek et al., 2015]. Similar effects are observed with mouse models of PTEN loss in which sirolimus therapy is effective in preventing but not correcting cortical malformations [Getz et al., 2016]. Despite this, effectiveness of sirolimus in reducing seizures in TSC cases has led to the proposal of using mTOR inhibitors as an adjunct treatment in epilepsy [Citraro et al., 2016]. Studies of the phosphoproteome have demonstrated that mTORC1 and mTORC2 and TSC1:TSC2 heterodimer can modulate several hundred potential candidate proteins, many widely expressed in the nervous systems, and this provides more targets for therapeutic manipulation in epilepsy, autism, and brain tumors, like glioblastoma [Crino, 2011]. Questions remain about the best approach to therapy, including duration (short term versus long term, pulsed versus continuous) and the findings from TSC are illuminating the best approaches.
Use in PHTS
Three individuals with PTEN mutations treated with mTOR inhibitors exist in the literature [Table VII]. Sirolimus has been the preferred agent of choice, mostly because of it available use as an immunosuppressant in organ transplantation. Two of the PHTS cases were severe and showed improvement in the size of lipomatous and vascular lesions during drug treatment. However, these changes reversed in all the cases upon discontinuation of the drug, suggesting that long-term drug administration may be necessary to achieve optimal outcomes.
Use in TSC
Until the discovery of the molecular pathogenesis of TSC, the only effective treatments for TSC included surgical approaches to remove, ablate, or embolize tumors [Flum et al., 2015; Jóźwiak et al., 2015], and antiepileptics to control seizures [Saxena & Sampson, 2015]. Tumors were insensitive to chemotherapy or radiation [Ergün et al., 1998]. Shortly after the discovery that loss of function of the TSC1-TSC2 complex caused hyperactivation of mTORC1, clinical trials were initiated for the use of mTOR inhibitors to treat TSC tumors. These studies resulted in approval by the US Food and Drug Administration of everolimus for the treatment of TSC-associated subependymal giant cell astrocytomas (SEGAs) and angiomyolipomas (AMLs), reviewed in [Franz, 2013], and the inclusion of mTOR inhibitors as one of the recommended therapies for TSC [Krueger et al., 2013]. Investigations are ongoing to determine the potential benefit of mTOR inhibitors for treating epilepsy in TSC [Curatolo et al., 2015].
There are several limitations to the use of mTOR inhibitors. They shrink, but do not eradicate, SEGAs and AMLs, and regrowth is observed after discontinuing treatment [Franz, 2013]. A variety of adverse effects have been reported with the use of mTOR inhibitors, including stomatitis, pneumonitis, hyperlipidemia or hypertriglyceridemia, hyperglycemia, infections, bone marrow suppression, and irregular menses [Sadowski et al., 2016]. These are generally mild but necessitate continuous monitoring of patients.
The allosteric mTOR inhibitors do not block activation of mTORC2 as avidly as mTORC1, and this effect is variable across different cell types [Sabatini, 2006]. Therefore, a second generation of mTOR inhibitors known as the “kinase inhibitors”, which more robustly inhibit both mTOR complexes, have been designed [Markman et al., 2010]. Clinical development of these compounds is less advanced than for allosteric inhibitors, or indeed PI3K and AKT inhibitors, but they remain potential candidates for clinical trials in PROS, TSC, PHTS and mTOR-related conditions in the future.
Dual inhibitors
Proteins in the PI3K-AKT-mTOR signaling pathway are highly conserved, and consequently, inhibitors targeting these proteins may unintentionally act upon multiple different components of the signaling pathway, with the potential for undesirable effects. However, this perceived drawback of therapy may turn out to be an advantage; blockade at two different sites within the pathway may be synergistic or help to overcome resistance to therapy [Elkabets et al., 2013]. Of greatest relevance to disorders of the PI3K-AKT signaling pathway are dual PI3K-mTOR inhibitors, which are currently in phase I trials for cancer [Maira et al., 2008].
Combination therapies
Drugs that can cross the blood-brain barrier effectively would be ideal to addressing the brain-associated phenotypes. Compounds like phenformin, which targets AMPK to inhibit mTOR activity and crosses the blood brain barrier, drugs that alter autophagy, or interventions that target cellular metabolism such as the ketogenic diet [Guerrini et al., 2015], may be attractive for combinatorial therapy. A clinical trial (ClinicalTrials.gov: NCT01687179) was conducted to examine the effects on lymphangioleiomyomatosis of combined sirolimus and autophagy inhibition using hydroxychloroquine [Taveira-DaSilva and Moss, 2014; Medvetz et al. 2015]. There is also evidence that simvastatin inhibits growth of TSC2-deficient cells [Atochina-Vasserman et al., 2013], so a combined trial of sirolimus with simvastatin is underway [Taveira-DaSilva and Moss, 2014]. In addition, other signaling pathways are dysregulated in association with specific phenotypes and genetic alterations [Patil et al., 2016], suggesting that more than one pathway may need to be targeted.
Other Drug Therapies
Metformin
Metformin, an established treatment for insulin resistant diabetes with a favorable safety profile, has been demonstrated to exhibit inhibitory activity over mTOR signaling [Ben Sahre et al., 2011]. Although metformin reduces cancer cell proliferation ex vivo and tumor growth in mouse models, this effect is achieved with supra-physiological doses of metformin, which would be toxic to humans. Metformin is therefore unlikely to be an effective monotherapy for disorders of the PI3K/AKT/mTOR signaling pathway, but its effect on mTOR signaling should be borne in mind as a low toxicity component of future combination therapy regimes in all of these conditions.
Aspirin
Aspirin, a well-established inhibitor of cyclo-oxygenase (COX 1 & 2) with anti-platelet and anti-inflammatory properties, and has been associated with a more favorable outcome in cancer patients with colorectal tumors bearing PIK3CA mutations [Liao et al., 2012]. Aspirin and other COX2 inhibitors are thought to inhibit AKT via a poorly characterized mechanism, and at doses within the therapeutic range for the drug [Wang et al., 2013]. These findings have led to the repurposing of aspirin and COX2 inhibitors as possible cancer therapies, and these compounds could therefore be evaluated as future treatments for AKT-related disorders, PHTS, and especially PROS, where dual effects on AKT activation and platelet aggregation could be advantageous in this group of patients with a heightened risk of thrombosis.
OBSTACLES TO INHIBITOR THERAPIES OF THE PI3K/AKT/mTOR PATHWAY
It will be important to incorporate and utilize the experience from oncology in using PI3K/AKT/mTOR inhibitors to treat cancer when considering clinical trials that may use these drugs for targeted treatment of segmental overgrowth disorders [Porta et al., 2014; Li and Wang, 2014; Yu et al., 2015]. In 2014, there were more than 50 drugs inhibiting the PI3K/AKT/mTOR pathway in development, with 60% of them being used in clinical trials [Li and Wang, 2014]. Potential problems may include toxic side effects and drug resistance. Not only will these drug inhibitors target cells with abnormal activity, they would also affect metabolism in healthy cells [Pallet et al., 2013; Porta et al., 2014; Petrulea et al., 2015]. As discussed earlier, dysregulation of glucose metabolism resulting in hyperglycemia is common.
Similar to treatment of cancers and the need to maintain remission in malignancy, patients with somatic activation of PI3K and AKT in PROS and PS respectively, may need life-long therapy with the inhibitor. This raises concern not only for the more common adverse effects of these inhibitors, including rash, mucositis, asthenia, but also unknown effects because of short-term evaluation in clinical trials to date [Holmes et al., 2011; Petulea et al., 2015].
In addition to reported and potential toxicities, there are also regulatory feedback and drug resistance issues that may decrease therapeutic effect of these drugs [Owonikoko and Khuri, 2013; Porta et al., 2014]. Multiple mechanisms have been suggested, including secondary target mutations, activation of alternative, parallel signaling pathways and amplification of downstream alterations in the same pathway [Hubbard et al., 2014; Porta et al., 2014]. With the somatic overgrowth syndromes, which are caused by a single post-zygotic mutation rather than multiple mutations seen in cancers, the first mechanism is unlikely to play a role. Resistance to mTOR inhibitors has been attributed to lack of feedback inhibition exerted by mTORC1/S6K, which leads to upregulation of the Receptor Tyrosine Kinases (RTKs) (Insulin Receptor, and PDGFRs), enhanced IRS1 signaling and increased Ras-MAPK/ Ras-PI3K signaling [Sarbossov et al., 2006; Pallet et al., 2013].
CONCLUSIONS
Elucidation of the genetic basis, clinical characterization, and ongoing understanding of the natural history of segmental overgrowth syndromes of the PI3K/AKT/mTOR signaling pathway provide the basis for developing targeted therapeutic strategies. This holds promise for treating progressive complications, although it is less likely that developmentally patterned defects e.g. polydactyly and hemimegalencephaly, will be amenable to therapy. Key issues for future clinical trials and treatment strategies with single or multiple agents will relate to management of drug toxicities and resistance, and to balance these risks against the clinical benefits achieved. Furthermore, existing and future clinical trials in somatic overgrowth syndromes may well provide useful information for treatment of cancer.
Acknowledgments
The authors are grateful to the patients and their families who have participated in this research review. KKN is supported by intramural funding of the National Human Genome Research Institute. VERP is supported by the UK National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre. JAMA is supported by the March of Dimes Birth Defects Foundation (6-FY12-324), NINDS (U54-NS092090), NIH/NICHD (P50-HD-055784), and NIH/NCATS (UL1TR000124). The ideas and opinions in this paper are those of the authors only and do not necessarily represent a position or policy of the National Institutes of Health or any other institutional organization with which any of the authors are affiliated.
Biographies
Kim M. Keppler-Noreuil, MD is Clinician Associate Investigator in the National Human Genome Research Institute, National Institutes of Health, and former Professor of Pediatrics /Medical Genetics at the University of Iowa. She is a clinical geneticist/ pediatrician with research interests in clinical delineation of multiple malformation syndromes, including somatic overgrowth disorders, and epidemiological studies of birth defects, inherited and chromosomal disorders.
Victoria E.R. Parker, PhD MBBS MRCP is an Associate Medical Director at MedImmune Ltd and holds an Honorary Consultant Endocrinologist position at Cambridge University Hospitals NHS Trust. She has a special interest in human diseases caused by dysregulation of the PI3K-AKT-mTOR signalling pathway with a particular focus on overgrowth conditions and diabetes mellitus.
Thomas N. Darling, MD, PhD, is professor and chair of Dermatology at the Uniformed Services University of the Health Sciences. He is a dermatologist who studies the skin manifestations of tuberous sclerosis complex and overgrowth syndromes.
Julian A. Martinez-Agosto, MD, PhD is Associate Professor in the Department of Human Genetics and Division of Medical Genetics, Department of Pediatrics at the David Geffen School of Medicine at UCLA. He is a medical geneticist and developmental biologist who studies genetic syndromes associated with abnormal growth that lead to intellectual disability, birth defects, autism, and cancer predisposition. His interests focus on novel regulators of growth signalling pathways in progenitor and stem cell maintenance.
Footnotes
The authors have no conflicts of interest.
References
- Akinleye A, Avvaru P, Furqan M, Song Y, Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol. 2013;6:88. doi: 10.1186/1756-8722-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander W. Inhibiting the akt pathway in cancer treatment: three leading candidates. Pharm Ther. 2011;36:225–227. [PMC free article] [PubMed] [Google Scholar]
- Alomari AI, Burrows PE, Lee EY, Hedequist DJ, Mulliken JB, Fishman SJ. CLOVES syndrome with thoracic and central phlebectasia:Increased risk of pulmonary embolism. J Thorac Cardiovasc Surg. 2010;140:459–466. doi: 10.1016/j.jtcvs.2010.04.023. [DOI] [PubMed] [Google Scholar]
- Arch EM, Goodman BK, Van Wesep RA, Liaw D, Clarke K, Parsons R, McKusick VA, Geraghty MT. Deletion of PTEN in a patient with Bannayan-Riley-Ruvalcaba syndrome suggests allelism with Cowden disease. Am J Med Genet A. 1997;71:489–493. [PubMed] [Google Scholar]
- Assefa G, Alemie B. Tuberous sclerosis complex (TSC) and Klippel-Trenaunay-Weber (KTW) syndromes association of two complete phakomatoses in a single individual. Ethiop Med J. 2010;48:315–320. [PubMed] [Google Scholar]
- Aspuria PJ, Tamanoi F. The Rheb family of GTP-binding proteins. J Cell Signal. 2004;16:1105–1112. doi: 10.1016/j.cellsig.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Atochina-Vasserman EN, Goncharov DA, Volgina AV, Milavec M, James ML, Krymskaya VP, Statins in lymphangioleiomyomatosis Simvastatin and atorvastatin induce differential effects on tuberous sclerosis complex 2-null cell growth and signaling. Am J Respir Cell Mol Biol. 2013;49:704–709. doi: 10.1165/rcmb.2013-0203RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek ST, Copeland B, Yun E-J, Kwon S-K, Guemez-Gamboa A, Schaffe AE, Kim S, Kang HC, Song S, Mathern GW, Gleeson JG. An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat Med. 2015;21:1445–1454. doi: 10.1038/nm.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baynam G, Overkov A, Davis M, Mina K, Schofield L, Allcock R, Laing N, Cook M, Dawkins H, Goldblatt J. A germline MTOR mutation in Aboriginal Australian siblings with intellectual disability, dysmorphism, macrocephaly, and small thoraces. Am J Med Genet A. 2015;167:1659–1667. doi: 10.1002/ajmg.a.37070. [DOI] [PubMed] [Google Scholar]
- Ben-Amitai D, Halachmi S, Lapidoth M. Are port wines stains a feature of tuberous sclerosis? J Eur Acad Dermatol Venereol. 2011;25:804–807. doi: 10.1111/j.1468-3083.2010.03866.x. [DOI] [PubMed] [Google Scholar]
- Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Augerger P, Tanti JF, Giorgetti-Peraldi S, Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- Biesecker LG, Sapp JC. Proteus Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet] Seattle (WA): University of Washington Seattle; 2012. Aug 9, pp. 1993–2016. Available from: http://www.ncbi.nlm.nih.gov/books/NBK99495/ [PubMed] [Google Scholar]
- Boronat S, Caruso P, Thiele EA. Absence of subependymal nodules in patients with tubers suggests possible neuroectodermal mosaicism in tuberous sclerosis complex. Dev Med Child Neurol. 2014;56:1207–1211. doi: 10.1111/dmcn.12523. [DOI] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou X-P, Talebizaded Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarsk R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Caux F, Plauchu H, Chibon F, Faivre L, Fain O, Vabres P, Bonnet F, Selma ZB, Laroche L, Gerard M, Longy M. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J Hum Genet. 2007;15:767–773. doi: 10.1038/sj.ejhg.5201823. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Mukherjee SB, Mendiratta V, Aneja S. Sturge Weber-Like Gyral Calcification Seen in Tuberous Sclerosis Complex 1. J Child Neurol. 2015;30:1070–1074. doi: 10.1177/0883073814542947. [DOI] [PubMed] [Google Scholar]
- Chen ZH, Zhu M, Yang J, Liang H, He J, He S, Wang P, Kang X, McNutt MA, Yin Y, Shen WH. PTEN interacts with histone H1 and controls chromatin condensation. Cell Rep. 2014;8:2003–2014. doi: 10.1016/j.celrep.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citraro R, Leo A, Constanti A, Russo E, De Sarro G. mTOR pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharm Res. 2016;107:333–343. doi: 10.1016/j.phrs.2016.03.039. [DOI] [PubMed] [Google Scholar]
- Crino PB. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med. 2011;17:734–742. doi: 10.1016/j.molmed.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013;23:53–62. doi: 10.1016/j.gde.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Crino PB, Aronica E, Baltuch G, Nathanson KL. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurol. 2010;74:1716–1723. doi: 10.1212/WNL.0b013e3181e04325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015;14:733–745. doi: 10.1016/S1474-4422(15)00069-1. [DOI] [PubMed] [Google Scholar]
- Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23:744–755. doi: 10.1016/j.ceb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- D’Gama AM, Geng Y, Couto JA, Martin B, Boyle, LaCoursiere CM, Hossain A, Hatem NE, Barry BJ, Kwiatkowski DJ, Vinters HV, Barkovich AJ, Shendure J, Mathern GW, Walsh CA, Poduri A. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol. 2015;77:720–725. doi: 10.1002/ana.24357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMario FJ, Jr, Sahin M, Ebrahimi-Fakhari D. Tuberous sclerosis complex. Pediatr Clin North Am. 2015;62:633–648. doi: 10.1016/j.pcl.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH, Serra V, Rodrik-Outmezguine V, Hazra S, Singh S, Kim P, Quadt C, Liu M, Huang A, Rosen N, Engelman JA, Scaltriti M, Baselga J. mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 2013;5:196ra99. doi: 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C. PTEN Hamartoma Tumor Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 2001. Nov 29, pp. 1993–2016. [Updated 2016 Jun2] Available from: http://www.ncbi.nlm.nih.gov/books/NBK1488/ [Google Scholar]
- Foster JG, Blunt MD, Carter E, Ward SG. Inhibition of PI3K Signaling Spurs New Therapeutic Opportunities in Inflammatory/Autoimmune Diseases and Hematological Malignancies. Pharmacol Rev. 2012;64:1027–1054. doi: 10.1124/pr.110.004051. [DOI] [PubMed] [Google Scholar]
- Ergün R, Okten AI, Yaman M, Gezici AR, Taşkin Y. Subependymal giant-cell astrocytoma associated with tuberous sclerosis: case report. Neurosurg Rev. 1998;21:185–188. doi: 10.1007/BF02389330. [DOI] [PubMed] [Google Scholar]
- Flum AS, Hamoui N, Said MA, Yang XJ, Casalino DD, McGuire BB, Perry KT, Nadler RB. Update on the Diagnosis and Management of Renal Angiomyolipoma. J Urol. 2015;195:834–846. doi: 10.1016/j.juro.2015.07.126. [DOI] [PubMed] [Google Scholar]
- Franz DN. Everolimus in the treatment of subependymal giant cell astrocytomas, angiomyolipomas, and pulmonary and skin lesions associated with tuberous sclerosis complex. Biologics. 2013;7:211–221. doi: 10.2147/BTT.S25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz SA, DeSpenza T, Li M, Luikart BW. Rapamycin prevents, but does not reverse, aberrant migration in Pten knockout neurons. Neurobiol Dis. 2016;93:12–20. doi: 10.1016/j.nbd.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet A. 2001;105:521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH, Sabatini DM. A Diverse Array of Cancer-Associated MTOR Mutations Are Hyperactivating and Can Predict Rapamycin Sensitivity. Cancer Discov. 2014;4:554–563. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Baker L, Kandula V, Conard K, Scavina M, Napoli JA, Griffin GC, Thacker M, Knox RG, Clark GR, Parker VER, Semple R, Mirzaa G, Keppler-Noreuil KM. Nephroblastomatosis or Wilms tumor in a fourth patient with a somatic PIK3CA mutation. Am J Med Genet A. 2016;170:2559–2569. doi: 10.1002/ajmg.a.37758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini R, Duchowny M, Jayakar P, Krsek P, Kahane P, Tassi L, Melani F, Polster T, Andre VM, Cepeda C, Krueger DA, Cross JH, Spreafico R, Cosottini M, Gotman J, Chassoux F, Ryvlin P, Bartolomei F, Bernasconi A, Stefan H, Miller I, Devaux B, Najm I, Giordano F, Vonck K, Barba C, Blumcke I. Diagnostic methods and treatment options for focal cortical dysplasia. Epilepsia. 56:1669–1686. doi: 10.1111/epi.13200. [DOI] [PubMed] [Google Scholar]
- Gustin JP, Cosgrove DP, Park BH. The PIK3CA gene as a mutated target for cancer therapy. Curr Cancer Drug Targets. 2008;8:733–740. doi: 10.2174/156800908786733504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happle R. Mosaicism in Human Skin. Understanding Nevi, Nevoid Skin Disorders, and Cutaneous Neoplasia. Springer-Verlag; Berlin Heidelberg: 2014. Two Major Categories of Mosaicism; pp. 13–37. [Google Scholar]
- Hardt M, Chantaravisoot N, Tamanoi F. Activating mutations of TOR (target of rapamycin) Genes Cells. 2011;16:141–151. doi: 10.1111/j.1365-2443.2010.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes D. PI3K pathway inhibitors approach junction. Nat Rev. 2011;10:563–564. doi: 10.1038/nrd3527. [DOI] [PubMed] [Google Scholar]
- Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard PA, Moody CL, Murali R. Allosteric modulation of Ras and the PI3K/AKT/mTOR pathway: emerging therapeutic opportunities. Front Physiol. 2014;5:1–7. doi: 10.3389/fphys.2014.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain K, Challis B, Rocha N, Payne F, Minic M, Thompson A, Daly A, Scott C, Harris J, Smillie BJ, et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334:474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacoba I, Burrows PH, Adams DM, Sutton VR, Hollier LH, Chintagumpala MM. Oral rapamycin in the treatment of patients with hamartoma syndromes and PTEN mutation. Pediatr Blood Cancer. 2011;57:321–323. doi: 10.1002/pbc.23098. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signaling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Jóźwiak S, Mandera M, Młynarski W. Natural History and Current Treatment Options for Subependymal Giant Cell Astrocytoma in Tuberous Sclerosis Complex. Semin Pediatr Neurol. 2015;22:274–281. doi: 10.1016/j.spen.2015.10.003. [DOI] [PubMed] [Google Scholar]
- Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Parker VER, Blumhorst C, Darling T, Tosi LL, Huson SM, Whitehouse RW, Jakkula E, Grant I, Balasubramanian M, Chandler KE, Fraser JL, Gucev Z, Crow YJ, Brennan LM, Clark R, Sellars EA, Pena LDM, Krishnamurty V, Shuen A, Braverman N, Cunningham ML, Sutton VR, Tasic V, Graham JM, Geer J, Henderson A, Semple RK, Biesecker LG. Clinical Delineation and Natural History of the PIK3CA-Related Overgrowth Spectrum. Am J Med Genet A. 2014;164:1713–1733. doi: 10.1002/ajmg.a.36552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler-Noreuil KM, Rios JJ, Parker VE, Semple RK, Lindhurst MJ, Sapp JC, Alomari A, Ezaki M, Dobyns W, Biesecker LG. PIK3CA-related overgrowth spectrum (PROS): Diagnostic and testing eligibility criteria, differential diagnosis and evaluation. Am J Med Genet A. 2015;167:287–295. doi: 10.1002/ajmg.a.36836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler-Noreuil KM, Baker EH, Sapp JC, Lindhurst MJ, Biesecker LG. Somatic AKT1 mutations cause meningiomas Colocalizing with a characteristic pattern of cranial hyperostosis. Am J Med Genet A. 2016;170:2605–2610. doi: 10.1002/ajmg.a.37737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S, Sharifi-Hannauer P, Martinez-Agosto JA. Macrocephaly as a Clinical Indicator of Genetic Subtypes in Autism. Autism Res. 2013;6:51–56. doi: 10.1002/aur.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- Krueger DA, Northrup H. International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:255–265. doi: 10.1016/j.pediatrneurol.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krymskaya VP, Goncharova EA. PI3K/mTORC1 activation in hamartoma syndromes: Therapeutic prospects. Cell Cycle. 2009;8:403–413. doi: 10.4161/cc.8.3.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, Mulliken JB, Bowen ME, Yamamoto GL, Kozakewich HP, Warman ML. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90:1108–1115. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming D, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 2013;123:980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30–38. doi: 10.1016/j.semcdb.2016.01.030. [DOI] [PubMed] [Google Scholar]
- Leventer RJ, Scerri T, Marsh AP, Pope K, Gillies G, Maixner W, MacGregor D, Harvey AS, Delatycki MB, Amor DJ, Crino P, Bahlo M, Lockhart PJ. Hemispheric cortical dysplasia secondary to a mosaic somatic mutation in MTOR. Neurol. 2015;84:2029–32. doi: 10.1212/WNL.0000000000001594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Wang G. Computer-Aided Targeting of the PI3K/Akt/mTOR pathway: Toxicity Reduction and Therapeutic Opportunities. Int J Mol Sci. 2014;15:18856–18891. doi: 10.3390/ijms151018856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, Sun R, Nosho K, Meyerhardt JA, Giovannucci E, Fuchs CS, Chan AT, Ogino S. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;67:1596–1606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, Cho YW, Kim S, Kim HM, Kim JA, Kim J, Rhee H, Kang SG, Kim HD, Kim D, Kim DS, Lee JH. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21:395–400. doi: 10.1038/nm.3824. [DOI] [PubMed] [Google Scholar]
- Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, Blumhorst C, Brockmann K, Calder P, Cherman N, Deardorff MA, Everman DB, Golas G, Greenstein RM, Kato BM, Keppler-Noreuil KM, Kuznetsov SA, Miyamoto RT, Newman K, Ng D, O’Brien K, Rothenberg S, Schwartzentruber DJ, Singhal V, Tirabosco R, Upton J, Wientroub S, Zackai EH, Hoag K, Whitewood-Neal T, Robey PG, Schwartzberg PL, Darling TN, Tosi LL, Mullikin JC, Biesecker LG. A Mosaic Activating Mutation in AKT1 Associated with the Proteus Syndrome. N Engl J Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindhurst MJ, Parker VER, Payne F, Sapp JC, Rudge S, Harris J, Witdowski AM, Zhang Q, Groeneveld MP, Scott CE, Daly A, Huson SM, Tosi LL, Cunningham ML, Darling TN, Geer J, Gucev Z, Sutton VR, Tziotzios C, Dixon AK, Halliwell T, O’Rahilly SO, Savage DB, Wakelam MJO, Barroso I, Biesecker LG, Semple RK. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations inPIK3CA. Nat Genet. 2012;44:928–933. doi: 10.1038/ng.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungberg MC, Bhattacharjee MB, Lu Y, Armstrong DL, Yoshor D, Swann JW, Sheldon M, D’Arcangelo G. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol. 2006;60:420–429. doi: 10.1002/ana.20949. [DOI] [PubMed] [Google Scholar]
- Loconte DC, Grossi V, Bozzao C, Forte G, Bagnulo R, Stella A, Lestella P, Cutrone M, Benedicenti F, Susca FC, Petruno M, Varvara D, Germani A, Chessa L, Laforgia N, Tenconi R, Simone C, Resta N. Molecular and Functional Characterization of Three Different Postzygotic Mutations in PIK3CA-Related Overgrowth Spectrum (PROS) Patients: Effects on PI3K/AKT/mTOR Signaling and Sensitivity to PIK3 Inhibitors. PLoS One. 2015;10:1–12. doi: 10.1371/journal.pone.0123092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–13. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- Luks VL, Kamitaki N, Vivero MP, Uller W, Rab R, Bovee JV, Rialon KL, Guevara CJ, Alomari AI, Greene AK, Fishman SJ, Kozakewich HP, Maclellan RA, Mulliken JB, Rahbar R, Spencer SA, Trenor CC, 3rd, Upton J, Zurakowski D, Perkins JA, Kirsh A, Bennett JT, Dobyns WB, Kurek KC, Warman ML, McCarroll SA, Murillo R. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015;166:1048–1054. doi: 10.1016/j.jpeds.2014.12.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, Aimone P, Fretault N, Dharan B, Tavorath R, Hirawat S. PI3K inhibitors as new cancer therapetuics: implications for clinical trial design. Onco Targets Ther. 2015;9:203–210. doi: 10.2147/OTT.S89967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maira S-M, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, García-Echeverría C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget. 2010;1:530–543. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh DJ, Trahair TN, Martin JL, Chee WY, Walker J, Kirk EP, Baxter RC, Marshall GM. Rapamycin treatment for a child with germline PTEN mutation. Nat Clin Pract Oncol. 2008;5:357–361. doi: 10.1038/ncponc1112. [DOI] [PubMed] [Google Scholar]
- Medvetz D, Priolo C, Henske EP. Therapeutic targeting of cellular metabolism in cells with hyperactive mTORC1: a paradigm shift. Mol Cancer Res. 2015;13:3–8. doi: 10.1158/1541-7786.MCR-14-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester J, Eng C. When Overgrowth Bumps Into Cancer: The PTEN-Opathies. Am J Med Genet C, Semin Med Genet. 2013;163:114–121. doi: 10.1002/ajmg.c.31364. [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, Conway RL, Gripp KW, Lerman-Sagie T, Siegel DH, deVries LS, Lev D, Kramer N, Hopkins E, Graham JM, Jr, Dobyns WB. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158:269–291. doi: 10.1002/ajmg.a.34402. [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, Parry DA, Fry AE, Giamanco KA, Schwartzentruber J, Vanstone M, Logan CV, Roberts N, Johnson CA, Singh S, Kholmanskikh SS, Adams C, Hodge RD, Hevner RF, Bonthron DT, Braun KP, Faivre L, Rivière JB, St-Onge J, Gripp KW, Mancini GM, Pang K, Sweeney E, van Esch H, Verbeek N, Wieczorek D, Steinraths M, Majewski J, FORGE Canada Consortium. Boycott KM, Pilz DT, Ross ME, Dobyns WB, Sheridan EG. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat Genet. 2014;46:510515. doi: 10.1038/ng.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa GM, Timms AE, Conti V, Boyle EA, Girisha KM, Martin B, Kircher M, Olds C, Juusola J, Collin S, Park K, Carter M, Glass I, Krageloh-Mann I, Chitayat D, Parikh AS, Bradshaw R, Torti E, Braddock S, Burke L, Ghedia S, Stephan M, Stewart F, Prasad C, Napier M, Saitta S, Straussberg R, Gabbett M, O’Connor BC, Keegan CE, Yin LJ, Lai AHM, Martin N, McKinnon M, Addor MC, Boccuto L, Schwartz CE, Lanoei A, Conway RL, Devriendt K, Tatton-Brown K, Pierpont ME, Painter M, Worgan L, Reggin J, Hennekam R, Tsuchiya K, Pritchard CC, Aracena M, Gripp KW, Cordisco M, Esch HV, Garavelli L, Curry C, Goriely A, Kayserilli H, Shendure J, Graham J, Guerrini R, Dobyns WB. PIK3CA-associated developmental disorders exhibit distinct classes of mutations with variable expression and tissue distribution. 2016 doi: 10.1172/jci.insight.87623. http://dx.doi.org/10.1172/jci.insight.87623. [DOI] [PMC free article] [PubMed]
- Mirzaa GM, Campbell CD, Solovieff N, Goold CP, Jansen LA, Menon S, Timms AE, Conti V, Biag JD, Olds C, Boyle EA, Collins S, Ishak G, Poliachik SL, Girisha KM, Yeung KS, Chung BH, Rahikkala E, Gunter SA, McDaniel SS, Macmurdo CF, Bernstein JA, Martin B, Leary RJ, Mahan S, Liu S, Weaver M, Dorschner MO, Jhangiani S, Muzny DM, Boerwinkle E, Gibbs RA, Lupski JR, Shendure J, Saneto RP, Novotny EJ, Wilson CJ, Sellers WR, Morrissey MP, Hevner RF, Ojemann JG, Guerrini R, Murphy LO, Winckler W, Dobyns WB. Association of MTOR Mutations With Developmental Brain Disorders, Including Megalencephaly, Focal Cortical Dysplasia, and Pigmentary Mosaicism. JAMA Neurol. 2016;73:836–45. doi: 10.1001/jamaneurol.2016.0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroske C, Rasmussen K, Shinde DN, Huether R, Powis Z, Lu HM, Baxter RM, McPherson E, Tang S. Germline activating MTOR mutation arising through gonadal mosaicism in two brothers with megalencephaly and neurodevelopmental abnormalities. BMC Med Genet. 2015;16:1–11. doi: 10.1186/s12881-015-0240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M, Saitsu H, Takei N, Tohyama J, Kato M, Kitaura H, Shiina M, Shirozu H, Masuda H, Watanabe K, Ohba C, Tsurusaki Y, Miyake N, Zheng Y, Sato T, Takebayashi H, Ogata K, Kameyama S, Kakita A, Matsumoto N. Somatic Mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol. 2015;78:375–386. doi: 10.1002/ana.24444. [DOI] [PubMed] [Google Scholar]
- Northrup H, Krueger DA. International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243–254. doi: 10.1016/j.pediatrneurol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owonikoko TK, Khuri FR. Targeting the PI3K/AKT/mTOR Pathway: Biomarkers of Success and Tribulation. 2013 doi: 10.1200/EdBook_AM.2013.33.e395. http://meetinglibrary.asco.org/content/131-132:1-10. [DOI] [PMC free article] [PubMed]
- Paller AS. Piecing together the puzzle of cutaneous mosaicism. J Clin Invest. 2004;114:1407–1409. doi: 10.1172/JCI23580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf. 2013;12:177–186. doi: 10.1517/14740338.2013.752814. [DOI] [PubMed] [Google Scholar]
- Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, Lunardi A, Webster K, Ng C, Newton RH, Knoblauch N, Guarnerio J, Ito K, Turka LA, Beck AH, Pinton P, Bronson RT, Wei W, Pandolfi PP. Cancer-Associated PTEN Mutants Act in a Dominant-Negative Manner to Suppress PTEN Protein Function. Cell. 2014;157:595–610. doi: 10.1016/j.cell.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil VV, Guzman M, Carter AN, Rathore G, Yoshor D, Curry D, Wilfong A, Agadi S, Swann JW, Adesina AM, Bhattacharjee MB, Anderson AE. Activation of extracellular regulated kinase and mechanistic target of rapamycin pathway in focal cortical dysplasia. Neuropathology. 2016;36:146–156. doi: 10.1111/neup.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrulea MS, Plantinga TS, Smit JW, Georgescu CE, Netea-Maier RT. PI3K/Akt/mTOR: A promising therapeutic target for non-medullary thyroid carcinoma. Cancer Treat Rev. 2015;41:707–713. doi: 10.1016/j.ctrv.2015.06.005. [DOI] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BF, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, Walsh CA. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:1–11. doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios JJ, Paria N, Burns DK, Israel BA, Cornelia R, Wise CA, Ezaki M. Somatic gain-of-function mutations in PIK3CA in patients with macrodactyly. Hum Mol Genet. 2012;22:444–451. doi: 10.1093/hmg/dds440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, Cytrynbaum C, Drolet BA, Innes AM, Lauzon JL, Lin AE, Mancini GM, Meschino WS, Reggin JD, Saggar AK, Lerman-Sagie T, Uyanik G, Weksberg R, Zirn B, Beaulieu CL, Finding of Rare Disease Genes (FORGE) Canada Consortium. Majewski J, Bulman DE, O’Driscoll M, Shendure J, Graham JM, Jr, Boycott KM, Dobyns WB. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–40. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Escudero I, Oliver MD, Andres-Pons A, Molina M, Cid VJ, Pulido R. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet. 2011;20:4132–4142. doi: 10.1093/hmg/ddr337. [DOI] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sadowski K, Kotulska K, Jóźwiak S. Management of side effects of mTOR inhibitors in tuberous sclerosis patients. Pharmacol Rep. 2016;68:536–542. doi: 10.1016/j.pharep.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Saxena A, Sampson JR. Epilepsy in Tuberous Sclerosis: Phenotypes, Mechanisms, and Treatments. Semin Neurol. 2015;35:269–276. doi: 10.1055/s-0035-1552616. [DOI] [PubMed] [Google Scholar]
- Scerri T, Riseley JR, Gillies G, Pope K, Burgess R, Mandelstam SA, Dibbens L, Chow CW, Maixner W, Harvey AS, Amor DJ, Delatycki MB, Crino PB, Bahlo M, Lockhart PJ, Leventer RJ. Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC5. Ann Clin Transl Neurol. 2015;2:575–580. doi: 10.1002/acn3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid GL, Kassner F, Uhlig HH, Korner A, Kratzsch J, Handel N, Zepp F-P, Kowalzik F, Laner A, Starke S, Wilhelm FK, Schuster S, Viehweger A, Hirsch W, Kiess W, Garten A. Sirolimus treatment of severe PTEN hamartoma tumor syndrome: case report and in vitro studies. Pediatr Res. 2014;75:527–534. doi: 10.1038/pr.2013.246. [DOI] [PubMed] [Google Scholar]
- Seibert D, Hong CH, Takeuchi F, Olsen C, Hathaway O, Moss J, Darling TN. Recognition of tuberous sclerosis in adult women: delayed presentation with life-threatening consequences. Ann Intern Med. 2011;154:806–813. doi: 10.1059/0003-4819-154-12-201106210-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signaling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Surge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971–1979. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim JC, Scerri T, Fanjul-Fernández M, Riseley JR, Gillies G, Pope K, van Roozendaal H, Heng JI, Mandelstam SA, McGillivray G, MacGregor D, Kannan L, Maixner W, Harvey AS, Amor DJ, Delatycki MB, Crino PB, Bahlo M, Lockhart PJ, Leventer RJ. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann Neurol. 2016;79:132–137. doi: 10.1002/ana.24502. [DOI] [PubMed] [Google Scholar]
- Soeiro E, Sá M, Moldovan O, Sousa AB. Macrodactyly in tuberous sclerosis complex: Case report and review of the literature. Am J Med Genet A. 2016;170:1903–1907. doi: 10.1002/ajmg.a.37675. [DOI] [PubMed] [Google Scholar]
- Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, Pandolfi PP. Nuclear PTEN Regulates the APC-CDH1 Tumor-Suppressive Complex in a Phosphatase-Independent Manner. Cell. 2011;144:187–199. doi: 10.1016/j.cell.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveira-DaSilva AM, Moss J. Management of lymphangioleiomyomatosis. F1000Prime Rep. 2014;6:116. doi: 10.12703/P6-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilot AK, Frazier TW, Eng Charis. Balancing Proliferation and Connectivity in PTEN-associated Autism Spectrum Disorder. Neurotherapeutics. 2015;12:609–619. doi: 10.1007/s13311-015-0356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi LL, Sapp JC, Allen ES, O’Keefe RJ, Biesecker LG. Assessment and management of the orthopedic and other complications of Proteus syndrome. J Child Orthop. 2011;5:319–27. doi: 10.1007/s11832-011-0350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner MA, Ruben BS, Lynch PJ. Segmental tuberous sclerosis presenting as unilateral facial angiofibromas. J Am Acad Dermatol. 2003;49:S164–S166. doi: 10.1067/mjd.2003.146. [DOI] [PubMed] [Google Scholar]
- Troost BT, Savino PJ, Lozito JC. Tuberous sclerosis and Klippel-Trenaunay-Weber syndromes. Association of two complete phakomatoses in a single individual. J Neurol Neurosurg Psych. 1975;38:500–504. doi: 10.1136/jnnp.38.5.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyburczy ME, Wang JA, Li S, Thangapazham R, Chekaluk Y, Moss J, Kwiatkowski DJ, Darling TN. Sun exposure causes somatic second-hit mutations and angiofibroma development in tuberous sclerosis complex. Hum Mol Genet. 2014;23:2023–2029. doi: 10.1093/hmg/ddt597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyburczy ME, Jozwiak S, Malinowska IA, Chekaluk Y, Pugh TJ, Wu CL, Nussbaum RL, Seepo S, Dzik T, Kotulska K, Kwiatkowski DJ. A shower of second hit events as the cause of multifocal renal cell carcinoma in tuberous sclerosis complex. Hum Mol Genet. 2015a;24:1836–1842. doi: 10.1093/hmg/ddu597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyburczy ME, Dies KA, Glass J, Camposano S, Chekaluk Y, Thorner AR, Lin L, Krueger D, Franz DN, Thiele EA, Sahin M, Kwiatkowski DJ. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLoS Genet. 2015b;11:e1005637. doi: 10.1371/journal.pgen.1005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A, Halley D, Young J, Burley M, Jeremiah S, Woodward K, Nahmias J, Fox M, Ekong R, Osborne J, Wolfe J, Povey S, Snell RG, Cheadle JP, Jones AC, Tachataki M, Ravine D, Sampson JR, Reeve MP, Richardson P, Wilmer F, Munro C, Hawkins TL, Sepp T, Ali JB, Ward S, Green AJ, Yates JR, Kwiatkowska J, Henske EP, Short MP, Haines JH, Jozwiak S, Kwiatkowski DJ. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997:277805–277808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst M, Nellist M, Nagelkerken B, Cheadle J, Snell R, van den Ouweland A, Reuser A, Sampson J, Halley D, van der Sluijs P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet. 1998;7:1053–1057. doi: 10.1093/hmg/7.6.1053. [DOI] [PubMed] [Google Scholar]
- Verhoef S, Bakker L, Tempelaars AM, Hesseling-Janssen AL, Mazurczak T, Jozwiak S, Fois A, Bartalini G, Zonnenberg BA, van Essen AJ, Lindhout D, Halley DJ, van den Ouweland AM. High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet. 1999;64:1632–1637. doi: 10.1086/302412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The Phosphatidlinositol-3-kinase-AKT pathway in Human Cancer. Nat Rev. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wang L, Wu J, Zhang W, Zhi Y, Wu Y, Jiang R, Yang R. Effects of aspirin on the ERK and PI3K/Akt signaling pathways in rats with acute pulmonary embolism. Mol Med Rep. 2013;8:1465–1471. doi: 10.3892/mmr.2013.1676. [DOI] [PubMed] [Google Scholar]
- Weigelt B, Downward J. Genomic determinants of PI3K pathway inhibitor response in cancer. Front Oncol. 2012;2:1–16. doi: 10.3389/fonc.2012.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Michael. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:27–36. doi: 10.1111/j.1528-1167.2009.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M. Mammalian target of rapamycin (mTOR) activation in focal cortical dysplasia and related focal cortical malformations. Exp Neurol. 2013;244:22–26. doi: 10.1016/j.expneurol.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yasin SA, Ali AM, Tata M, Picker SR, Anderson GW, Latimer-Bowman E, Nicholson SL, Harkness W, Cross JH, Paine SM, Jacques TS. mTOR-dependent abnormalities in autophagy characterize human malformations of cortical development: evidence from focal cortical dysplasia and tuberous sclerosis. Acta Neuropathol. 2013;126:207–218. doi: 10.1007/s00401-013-1135-4. [DOI] [PubMed] [Google Scholar]
- Yu Y, Savage RE, Eathiraj S, Meade J, Wick MJ, Hall T, Abbadessa G, Schwartz B. Targeting AKT1-E17K and the PI3K/AKT Pathway with an Allosteric AKT Inhibitor, ARQ 092. PLoS One. 2015 Oct 15;10(10):e0140479. doi: 10.1371/journal.pone.0140479. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Huang SL. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin J Cancer. 2012;31:8–18. doi: 10.5732/cjc.011.10281. [DOI] [PMC free article] [PubMed] [Google Scholar]