

Abstract
Cytokine-induced endoplasmic reticulum (ER) stress is one of the molecular mechanisms underlying pancreatic β-cell demise in type 1 diabetes. Thrombospondin 1 (THBS1) was recently shown to promote β-cell survival during lipotoxic stress. Here we show that ER-localized THBS1 is cytoprotective to rat, mouse, and human β-cells exposed to cytokines or thapsigargin-induced ER stress. THBS1 confers cytoprotection by maintaining expression of mesencephalic astrocyte-derived neutrotrophic factor (MANF) in β-cells and thereby prevents the BH3-only protein BIM (BCL2-interacting mediator of cell death)-dependent triggering of the mitochondrial pathway of apoptosis. Prolonged exposure of β-cells to cytokines or thapsigargin leads to THBS1 and MANF degradation and loss of this prosurvival mechanism. Approaches that sustain intracellular THBS1 and MANF expression in β-cells should be explored as a cytoprotective strategy in type 1 diabetes.
Keywords: β-cell, cytokine, endoplasmic reticulum stress (ER stress), inflammation, interferon, IL-1, islet, mitochondrial apoptosis, thrombospondin, type 1 diabetes
Introduction
Type 1 and type 2 diabetes mellitus (T1D3 and T2D, respectively) are characterized by failure of the pancreatic β-cells. In T1D, this is caused by autoimmune aggression against β-cells that leads to progressive β-cell dysfunction and death. The pathogenesis of T2D is characterized by different degrees of β-cell failure/loss relative to variable degrees of insulin resistance (1). Apoptosis seems to be the main form of β-cell death in both forms of the disease. In T1D, invading immune cells trigger β-cell death by cell-to-cell contact and local production of proinflammatory cytokines, such as IL-1β and IFN-γ (2). The recent observation that gene expression in laser-captured islets from recent-onset T1D patients (3) is remarkably similar to gene expression in human islets exposed in vitro to IL-1β and IFN-γ (4) supports the idea that these cytokines (or other cytokines that elicit similar signal transduction) play a role in the human disease. In the context of T2D, the metabolic stress of chronic exposure to elevated levels of saturated free fatty acids, such as palmitate, and glucose contribute to β-cell dysfunction and apoptosis (5, 6).
It is of high interest to identify approaches that prevent both immune-mediated and metabolic β-cell demise; such an approach would be very useful in the prevention or early treatment of T1D and T2D. This task is made difficult, however, by the fact that proinflammatory cytokines (4) and palmitate (7) induce different gene networks and lead to pancreatic β-cell apoptosis by different mechanisms (1). One cellular stress response that is, however, present in β-cells in both forms of diabetes is endoplasmic reticulum (ER) stress (8, 9). Pharmacological modulation of the ER stress response might therefore hold promise for β-cell therapy (10, 11).
The multimeric Ca2+-binding glycoprotein thrombospondin 1 (THBS1) protects cardiomyocytes against ER stress via activation of ATF6 and downstream chaperones (12). We have shown recently that THBS1 protects human and rodent β-cells from palmitate-induced apoptosis (13). Different from cardiomyocytes, however, this takes place through activation of the ER stress transducer protein kinase R-like endoplasmic reticulum kinase (PERK) and the downstream transcription factor NRF2, increasing the β-cell capacity to withstand oxidative stress induced by saturated fatty acids (13).
Here we tested whether THBS1 is equally beneficial to rodent and human β-cells exposed to cytokines or chemically induced ER stress. THBS1 was clearly protective, but this was mediated by a different mechanism compared with protection against lipotoxic β-cell demise (13) or cardiomyopathy (12); namely, through induction of the mesencephalic astrocyte-derived neutrotrophic factor (MANF). This raises the intriguing possibility that the multifunctional protein THBS1 changes roles and/or partner affinities in a cell- or stress-specific manner, as suggested recently for other complex biological systems (14). Furthermore, these findings indicate that THBS1-inducing agents may represent a novel strategy for β-cell protection in both T1D and T2D.
Results
The cross-talk between THBS1 and ER stress- and proinflammatory cytokine–induced β-cell apoptosis
Knockdown of THBS1 in rat INS-1E cells by two independent siRNAs did not affect basal expression of cleaved caspase 9 and 3 and apoptosis (Fig. 1, A and B), but it augmented caspase cleavage and cell death following exposure to the chemical ER stressor thapsigargin (an inhibitor of the SERCA2 pump that depletes ER Ca2+) and the proinflammatory cytokines IL-1β and IFN-γ, which cause ER stress, at least in part, via inhibition of SERCA2 (15). The cleavage of both caspase 9 and 3 indicates that THBS1 knockdown favors activation of the intrinsic pathway of apoptosis by thapsigargin and cytokines (16). These findings were confirmed in human islets silenced for THBS1 (Fig. 1, C and D). Apoptosis was also detected by immunostaining for cleaved caspase 3 in THBS1-depleted human β-cells exposed to thapsigargin (Fig. 1E and supplemental Fig. S1) or cytokines (supplemental Fig. S1). Islets isolated from THBS1 knock-out mice were also significantly sensitized to thapsigargin and cytokines (Fig. 1F); in both cases, lack of THBS1 sensitized the cells to thapsigargin- and cytokine-induced apoptosis. In mirror experiments, THBS1 was overexpressed in INS-1E cells (Fig. 1G) and human islet cells (Fig. 1I) using an adenoviral vector. THBS1 overexpression partially reduced thapsigargin- and cytokine-induced caspase 9 and 3 cleavage (Fig. 1G) and apoptosis in rat β-cells (Fig. 1H) and human islet cells (Fig. 1J).
Figure 1.

THBS1 modulates ER stress- and cytokine-induced β-cell death. A and B, cleaved caspase 9 and 3 and THBS1 protein expression (A) and apoptosis (B) in INS-1E cells transfected with negative (N, control) or two THBS1 (T1 and T2) siRNAs and exposed to thapsigargin (THA) or IL-1β and IFN-γ (IL+IFN) for 16 h (n = 3–4). C–E, THBS1 mRNA expression (C), apoptosis (D), and immunostaining (E) for cleaved caspase 3 and insulin in dispersed human islet cells transfected with negative or THBS1 siRNA and exposed to thapsigargin or IL+IFN for 24 h (n = 3–7). F, cell death in wild-type (THBS1+/+) or THBS1−/− mouse islets exposed to thapsigargin or IL+IFN for 48 h (n = 4). G and H, cleaved caspase 9 and 3 and THBS1 protein expression (G) and apoptosis (H) in INS-1E cells infected with luciferase (L, control) or THBS1 (T) adenovirus (ad) and exposed to thapsigargin or IL+IFN for 16 h (n = 4). I and J, THBS1 mRNA expression (I) and apoptosis (J) in dispersed human islet cells infected with luciferase or THBS1 adenovirus and exposed to thapsigargin or IL+IFN for 24 h (n = 3–4). *, p < 0.05 versus control (CTL); #, p < 0.05 versus cytokine- or thapsigargin-treated cells transfected with negative siRNA or infected with luciferase-expressing adenovirus.
Because the above findings indicate that THBS1 protects human, rat, and mouse β-cells from cytokine- and ER stress–induced cell death, we evaluated whether these stresses affect THBS1 expression. Exposure of human islets to two different ER stressors, brefeldin A (which blocks transfer of cargo from the ER to the Golgi) and thapsigargin, decreased THBS1 mRNA expression by nearly 80% (Fig. 2A). A similar inhibition was observed in IL-1β and IFN-γ-exposed human islets (Fig. 2A), and these findings were confirmed at the protein level for thapsigargin and cytokines (Fig. 2B). Thapsigargin, which rapidly induces severe ER stress in INS-1E cells (15), already reduced THBS1 protein expression by 2 h, with a progressive decrease up to 24 h (Fig. 2C). Cytokines, which induce ER stress more slowly in INS-1E cells (by 6–8 h) (15), inhibited THBS1 expression by 8 h, with a nadir at 24 h. These observations suggest that chemical or cytokine-mediated ER stress progressively decreases THBS1 expression in β-cells, sensitizing these cells to a pro-apoptotic outcome. In addition to transcriptional inhibition (Fig. 2A), proteasomal degradation contributes to THBS1 depletion, as proteasome inhibition by MG132 prevented the loss of THBS1 (Fig. 2E).
Figure 2.

ER stress and proinflammatory cytokines decrease THBS1 expression. A and B, THBS1 mRNA (A) and protein (B) expression in human islets exposed to THA, brefeldin A (BRE), or IL-1β and IFN-γ (IL+IFN) for 48 h (n = 3). C and D, time course of THBS1 protein expression in INS-1E cells treated with thapsigargin or IL+IFN (n = 3). E, THBS1 protein expression in INS-1E cells treated with IL+IFN and MG132 (n = 3). *, p < 0.05 versus control (CTL).
Intracellular THBS1 protects β-cells against cytokines and chemical ER stress
THBS1 may reside in the ER and/or be secreted into the extracellular milieu (13, 17, 18). We thus investigated whether intra- or extracellular THBS1 mediates cytoprotection against cytokines or thapsigargin (Fig. 1). Conditioned medium from INS-1E cells overexpressing THBS1 (Fig. 3A) or exogenously added recombinant THBS1 (Fig. 3B), shown previously by us to transduce signals in β-cells (13), failed to protect INS-1E cells against thapsigargin- or cytokine-induced apoptosis. On the other hand, an adenovirus for THBS1 containing the ER retention sequence KDEL (13) was cytoprotective to the same extent as the control THBS1-FLAG adenovirus (Fig. 3, C and D).
Figure 3.

Intra- but not extracellular THBS1 improves β-cell survival. A, apoptosis in INS-1E cells exposed for 16 h to THA or IL-1β and IFN-γ (IL+IFN) in conditioned medium from INS-1E cells infected with luciferase (Luc) or THBS1 adenovirus for 48 h (n = 3). B, apoptosis in INS-1E cells exposed to recombinant THBS1 protein and thapsigargin or IL+IFN for 16 h (n = 3). C and D, THBS1 protein expression (C) and apoptosis (D) in INS-1E cells infected with luciferase (L), THBS1-FLAG (F), or THBS1-KDEL (K) adenovirus and exposed to thapsigargin or IL+IFN for 16 h (n = 4). E and F, THBS1 mRNA expression (E) and apoptosis (F) in dispersed wild-type (THBS1+/+) or THBS1−/− mouse islet cells infected with luciferase or THBS1-KDEL adenovirus and exposed to thapsigargin or IL+IFN for 48 h (n = 4). *, p < 0.05 versus untreated cells; #, p < 0.05 versus thapsigargin- or cytokine-treated cells infected with luciferase adenovirus or exposed to ad Luc medium; §, p < 0.05 as indicated.
Islet cells from THBS1−/− mice have increased susceptibility to cell death induced by ER stressors or cytokines (Fig. 1F), and we next evaluated whether ER-retained THBS1-KDEL could rescue this phenotype. The THBS1-KDEL adenovirus markedly induced THBS1 mRNA expression under both control or stressed conditions (Fig. 3E). As in human islet cells (Fig. 1J), THBS1+/+ mouse islet cells were protected against thapsigargin or cytokines (Fig. 3F). Importantly, overexpression of ER-retained THBS1 in islet cells that lack endogenous THBS1 (THBS1−/−) abrogated the marked susceptibility to thapsigargin or cytokines (Fig. 3F), indicating that the ER-retained form of THBS1 is necessary and sufficient for the cytoprotective effects.
THBS1 cytoprotection is not related to modulation of ATF6 or oxidative stress
It has been shown that THBS1 protects cardiomyocytes against ER stress by activating the transcription factor ATF6 and, thereby, up-regulating protective chaperones such as binding immunoglobulin protein (BiP) (12). This does not, however, explain β-cell protection by THBS1. THBS1 silencing did not impair ATF6 activity, as assessed using a luciferase reporter, or BiP expression (supplemental Fig. S2, A and B). In keeping with previous findings (12, 13), THBS1 knockdown reduced signaling in the PERK pathway (supplemental Fig. S2A). Conversely, THBS1 overexpression did not induce expression of BiP protein or mRNA, nor did it induce the ATF6 target GRP94 (supplemental Fig. S2, C and D).
Our recent findings indicate that THBS1 protects β-cells from lipotoxicity via the PERK-NRF2 pathway and consequent up-regulation of antioxidant defenses (13). THBS1 loss or gain of function did not, however, alter oxidative stress (supplemental Fig. S3, A and B). Cytokines, but not thapsigargin, increased oxidation of 2′,7′-dichlorofluorescein diacetate (DCF), and this was not altered by modulation of THBS1 expression (supplemental Fig. S3, A and B). In line with these findings, neither THBS1 silencing nor overexpression modified expression of the antioxidant genes GSTm1, catalase, and SOD2 (supplemental Fig. S3, C and D). THBS1 knockdown did not affect the activity of an antioxidant response element luciferase reporter (supplemental Fig. S3E), which we found previously to be activated by palmitate via up-regulation of NRF2 (13). As a whole, these observations indicate that THBS1 protects β-cells against palmitate or cytokines/ER stressors by different mechanisms.
THBS1 protects β-cells against cytokines and ER stress via MANF induction
We next explored alternative mechanisms underlying the THBS1 cytoprotection and examined the expression of MANF, shown previously to be up-regulated by THBS (12). MANF is an ER stress–regulated protein that is crucial for postnatal mouse β-cell survival (19). Thapsigargin and cytokines inhibited MANF protein expression, and this was aggravated by THBS1 knockdown (Fig. 4A). Palmitate, on the other hand, did not reduce MANF expression (Fig. 4A). A time course analysis in INS-1E cells exposed to cytokines showed that MANF expression already decreased at 4 h, and more so in THBS1-depleted cells (Fig. 4B). On the other hand, adenoviral THBS1 overexpression induced and preserved MANF expression for up to 8 h of cytokine exposure (Fig. 4C). MANF depletion by cytokines was not associated with a change in its subcellular localization, which remained cytoplasmic (Fig. 4D). As for THBS1, proteasome inhibition by MG132 prevented the cytokine-induced loss of MANF protein (Fig. 4E).
Figure 4.

THBS1 partially prevents ER stress- or proinflammatory cytokine–induced MANF degradation. A, MANF protein expression in INS-1E cells transfected with negative (N, control) or THBS1 (T1 and T2) siRNAs and exposed to palmitate (PAL), THA, or IL-1β and IFN-γ (IL+IFN) for 16 h (n = 3). B and C, time course of THBS1 and MANF protein expression in INS-1E cells transfected with negative or THBS1 siRNA (B) or infected with luciferase (L) or THBS1 adenovirus (T) (C) and exposed to IL+IFN (n = 2–3). D, MANF immunofluorescence in 8-h IL+IFN-exposed INS-1E cells (n = 3). E, MANF protein expression in INS-1E cells exposed to IL+IFN and MG132 (n = 3). F and G, MANF protein expression (F) and apoptosis (G) in INS-1E cells transfected with negative or MANF (M1 and M2) siRNAs and exposed to thapsigargin or IL+IFN for 16 h (n = 3). H, apoptosis in EndoC-βH1 cells transfected with negative or MANF siRNAs and exposed to thapsigargin or IL+IFN for 24 h (n = 4–5). I, immunostaining for cleaved caspase 3 and insulin in dispersed human islet cells transfected with negative or MANF siRNA and exposed to thapsigargin for 24 h (n = 2). These and other images are also presented in supplemental Fig. S4. *, p < 0.05 versus control (CTL) #, p < 0.05 versus thapsigargin- or cytokine-treated cells transfected or not with negative siRNA.
We next examined the role of MANF directly using two independent siRNAs. Efficient MANF knockdown (Fig. 4F) did not affect basal cell survival but sensitized INS-1E cells to both thapsigargin- and cytokine-induced apoptosis (Fig. 4G). A similar sensitization was seen in human insulin-producing EndoC-βH1 cells (Fig. 4H). Apoptosis was further confirmed by immunostaining for cleaved caspase 3 in MANF-depleted thapsigargin- or cytokine-treated EndoC-βH1 cells (supplemental Fig. S4). MANF silencing also induced caspase 3 cleavage in insulin-stained primary human β-cells exposed to thapsigargin (Fig. 4I and supplemental Fig. S5) or cytokines (supplemental Fig. S5).
We next examined whether cytoprotection could be achieved by exogenously added MANF protein. Recombinant MANF protein failed to protect clonal rat INS-1E cells against thapsigargin or cytokines (Fig. 5A). In clonal human EndoC-βH1 cells, however, partial protection was observed from both stresses (Fig. 5B). Comparable cytoprotection was seen in dispersed mouse islet cells (Fig. 5C). Apoptosis was further assessed by TUNEL assay, confirming that recombinant MANF decreases thapsigargin-induced apoptosis in insulin-positive mouse β-cells (Fig. 5D). Taken together, these data show significant protection conferred by exogenous MANF in mouse and human but not rat β-cells, pointing to a species difference.
Figure 5.

Extracellular MANF improves human and mouse but not rat β-cell survival. A, apoptosis in INS-1E cells treated with recombinant MANF protein and thapsigargin or IL-1β and IFN-γ (IL+IFN) for 16 h (n = 6). CTL, control. B, apoptosis in EndoC-βH1 cells treated with recombinant MANF protein and thapsigargin or IL+IFN for 16 h (n = 8). C, apoptosis in dispersed mouse islet cells treated with recombinant MANF protein and thapsigargin or IL+IFN for 48 h (n = 4). D, β-cell death assessed by TUNEL staining in mouse islets treated with recombinant MANF protein and thapsigargin for 16 h (n = 3–8). *, p < 0.05 versus control cells; #, p < 0.05 for rMANF versus vehicle-treated cells.
To evaluate whether MANF is indeed an important mediator of the protective effects of THBS1, we adenovirally overexpressed THBS1 and knocked down MANF in parallel (Fig. 6A). Inhibition of MANF abrogated the beneficial effects of THBS1 overexpression in INS-1E cells (Fig. 6B). Similarly, efficient THBS1 overexpression and MANF silencing in human islet cells (Fig. 6C) abolished the cytoprotection conferred by THBS1 (Fig. 6D). In mirror experiments, lentiviral MANF overexpression concurrent to THBS1 silencing (Fig. 6E) completely prevented sensitization to cytokine-induced apoptosis (Fig. 6F). These data show that MANF is necessary and sufficient for THBS1-mediated β-cell protection against cytokines or ER stress.
Figure 6.

MANF mediates the protective effects of THBS1 overexpression. A and B, MANF and THBS1 protein expression (A) and apoptosis (B) in INS-1E cells transfected with negative (Neg) or MANF siRNA, infected with luciferase (L) or THBS1 (T) adenovirus, and exposed to THA or IL-1β + IFN-γ (IL+IFN) for 16 h (n = 3). C and D, MANF and THBS1 mRNA expression (C) and apoptosis (D) in dispersed human islet cells transfected with negative or MANF siRNA, infected with luciferase or THBS1 adenovirus, and exposed to IL+IFN for 24 h (n = 3). E and F, MANF and THBS1 protein expression (E) and apoptosis (F) in INS-1E cells transfected with negative or THBS1 siRNA, infected with GFP (G, control) or MANF (M) lentivirus, and exposed to IL+IFN for 16 h (n = 4). *, p < 0.05 versus untreated cells; #, p < 0.05 versus thapsigargin- or cytokine-treated cells transfected with negative siRNA and infected with luciferase adenovirus or GFP-expressing lentivirus. §, p < 0.05 as indicated.
We next evaluated the effector mechanisms downstream of THBS1-MANF that regulate β-cell apoptosis. Previous data indicate that the BH3-only proteins PUMA (p53-upregulated modulator of apoptosis, also known as BBC3) (20), death protein 5 (DP5) (21), and BCL2-interacting mediator of cell death (BIM) (22) are critical mediators of cytokine-induced apoptosis, whereas the Bcl-2 family proteins Bcl-2 and Bcl-XL antagonize these effects (16). Knockdown or overexpression of THBS1 did not modify the expression of DP5, PUMA, BIM, Bcl-2, and Bcl-XL in INS-1E cells (supplemental Fig. S6, A–E). BH3-only proteins may be a necessary component for cytokine-induced apoptosis, even under conditions where their expression is not up-regulated (16, 23). To test whether this was the case here, we silenced THBS1 in parallel with BIM, DP5, PUMA, or BCL-2-associated death promoter (BAD) (Fig. 7A). Efficient knockdown was achieved for BIM (by 52%; Fig. 7, B and D), DP5 (silencing of mRNA by 63%), PUMA (by 74%), and BAD (by 89%). BIM silencing, but not DP5, PUMA, or BAD knockdown, partially prevented cytokine-induced apoptosis in THBS1-depleted β-cells (Fig. 7A). In a similar experimental approach, BIM also mediated apoptosis induced by thapsigargin (Fig. 7, B and C). Because THBS1 cytoprotection requires MANF expression (see above), we examined whether BIM also mediates cell death induced by MANF deficiency. BIM knockdown fully abrogated sensitization to cytokine-induced apoptosis in MANF-depleted cells (Fig. 7, D and E). These observations indicate that BIM is the downstream mediator of β-cell apoptosis following inhibition of the THBS1–MANF pathway.
Figure 7.

BIM is the mediator of THBS1/MANF deficiency–mediated β-cell apoptosis. A, apoptosis in INS-1E cells transfected with negative or THBS1 siRNA in combination with BIM, DP5, PUMA, or BAD siRNAs and exposed to IL-1β + IFN-γ (IL+IFN) for 16 h (n = 3). B and C, THBS1 and BIM protein expression (B) and apoptosis (C) in INS-1E cells transfected with negative (N), THBS1 (T), and/or BIM (B) siRNAs and exposed to THA or IL+IFN for 16 h (n = 3). CTL, control. D and E, BIM and MANF protein expression (D) and apoptosis (E) apoptosis in INS-1E cells transfected with negative, MANF (M), and/or BIM siRNAs and exposed to IL+IFN for 16 h (n = 2–3). *, p < 0.05 versus untreated cells; #, p < 0.05 versus thapsigargin- or cytokine-treated cells transfected with negative siRNA. §, p < 0.05 as indicated.
Discussion
The local release of proinflammatory mediators by infiltrating immune cells contributes to pancreatic β-cell demise in T1D. Cytokine-induced ER stress is one of the molecular mechanisms activating the mitochondrial pathway of apoptosis in β-cells (9, 10, 24). This stress response also plays a role in β-cell failure in T2D and in monogenic forms of diabetes (25–27). Here we identified a novel β-cell protective role of the ER-resident proteins THBS1 and MANF in cytokine- and chemical ER stress–induced apoptosis. Partial cytoprotection was observed across species (rat, mouse and human). THBS1 maintains the expression levels of the anti-apoptotic protein MANF and thereby prevents BIM-dependent mitochondrial apoptosis. Prolonged exposure of β-cells to cytokines or thapsigargin leads to proteasomal THBS1 and MANF degradation and loss of this prosurvival mechanism (Fig. 8).
Figure 8.
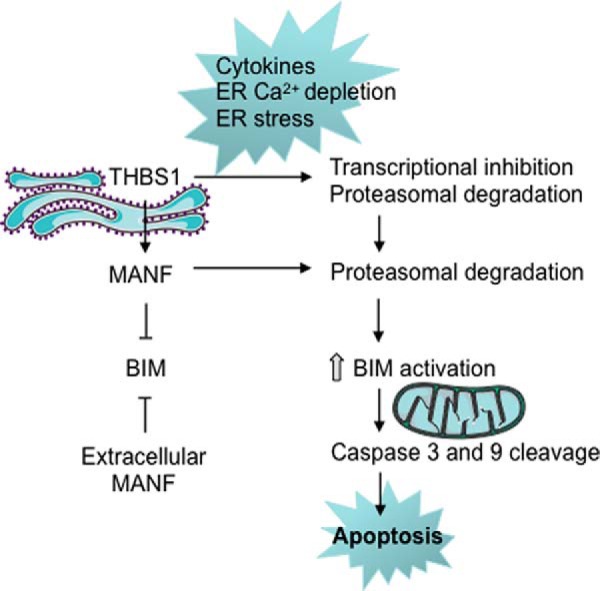
Schematic of the main findings. Cytokines, ER Ca2+ depletion, and ER stress lead to loss of THBS1 and MANF protein in rat, mouse, and human β-cells, triggering BIM-dependent β-cell apoptosis. The images of mitochondria and the ER are from Servier Medical Art.
THBS1 and MANF are both present in the ER, secreted via the constitutive secretory pathway, and regulated under conditions of ER stress. THBS1 is glycosylated and contains disulfide bridges. Under conditions of Ca2+ depletion and ER stress, the protein misfolds and is retained in the ER or targeted for degradation. MANF expression is induced by ER stress. In β-cell ER stress induced by misfolding insulin, MANF mRNA was induced, but protein levels were not examined (28). MANF is retained in the ER through different mechanisms: it has a C-terminal sequence that binds the KDEL receptor, and it binds BiP and reticulon proteins; under ER stress conditions, its secretion is enhanced (29).
THBS1 has a wide interactome. In cardiomyocytes, THBS interacts with ATF6 and promotes its ER-to-Golgi translocation, activation, and function, thereby eliciting an adaptive ER stress response (12). Under lipotoxic conditions, THBS1 promotes PERK phosphorylation and NRF2 activation, thereby equipping β-cells with an antioxidative stress defense (13). Extracellularly, THBS1 inhibits angiogenesis by interacting with a range of factors and receptors. In keeping with this biological effect, Carlsson and co-workers (30–32) showed that THBS1 knock-out mice have large hypervascularized islets. These THBS1−/− islets are dysfunctional, however, with reduced glucose oxidation and insulin biosynthesis and release and, at the whole-body level, impaired glucose tolerance. In db/db mice, THBS1 disruption leads to cardiomyopathy and heart failure (33). In man, a SNP in the THBS1 coding region reduces Ca2+ binding to the protein and impairs THBS1 folding and secretion; this has been associated with premature coronary artery disease (34, 35). Taken together, we suggest that maintenance of THBS1 expression is required to prevent or delay β-cell failure in diabetes and diabetes complications.
MANF expression is also required for β-cell survival. Antiapoptotic effects have been reported for intracellular and extracellular MANF in different cell types. The C-terminal domain of MANF is structurally similar to the BAX (BCL-2-associated protein X) inhibitory protein Ku70, and it was suggested to prevent neuronal apoptosis by inactivating BAX (36), but experimental evidence for this could not be found in a subsequent study (37). MANF knock-out mice develop severe diabetes because of progressive postnatal reduction of β-cell mass, caused by decreased proliferation and increased apoptosis (19). Deficient MANF induction underlies immune-independent β-cell failure and diabetes susceptibility of non-obese diabetic (NOD) mice when the protein load in the islets of these mice is increased by expression of hen egg lysozyme under the Ins2 promoter (38). Whether MANF expression is lost in β-cells in T1D models under less artificial conditions has not been reported. Serum MANF levels were found to be increased in children with T1D around the time of diabetes onset (39), but the reason for this is unclear. As MANF levels in long-standing T1D patients are similar to non-diabetic controls, and considering its ubiquitous expression, it seems plausible that most circulating MANF does not originate from β-cells (39). A homozygous MANF mutation (IVS1 + 1G>T) was recently reported as a likely pathogenic candidate in a 22-year-old woman with diabetes, hypothyroidism, primary hypogonadism, short stature, microcephaly, deafness, myopia, and alopecia (40). This case report awaits confirmation, but it may suggest a role for MANF loss of function in human diabetes.
Based on the evidence for chronic ER stress in pancreatic β-cells in T1D (9, 24), attempts have been made to protect β-cells from ER stress. In the NOD and RIP-LCMV-GP (rat insulin promoter-lymphocytic choriomeningitis virus-glycoprotein) mouse models of T1D, promoting a functional ER stress response by chemical chaperones confers survival potential to pancreatic β-cells (10). Peroxisome proliferator-activated receptor (PPAR)-γ activation by pioglitazone administration to NOD mice improved β-cell function and survival by promoting an adaptive ER stress response (41). Based on our findings, we suggest that reducing ER Ca2+ depletion and ER stress may be β-cell–protective through the preservation of THBS1 and MANF expression. Under conditions of severe ER stress, approaches that sustain intracellular THBS1 and MANF expression in β-cells should be explored as a cytoprotective strategy in T1D.
Experimental procedures
Culture of mouse islet cells, human islets, and INS-1E cells
Animals were used according to the Belgian Regulations for Animal Care with approval of the Université Libre de Bruxelles Ethical Committee. Islets were isolated from 10- to 13-week-old THBS1 knock-out (30) or wild-type mice. Human islets (from 20 donors; age, 64 ± 3 years; body mass index, 26 ± 1 kg/m2; 12 males and 8 females; cause of death cerebral hemorrhage (six), cardiovascular disease (eight), trauma (five) or unknown (one)) were isolated by collagenase digestion and density gradient purification. The islets were cultured, dispersed, and transfected as described previously (42). The percentage of β-cells, examined by insulin immunofluorescence, was 46% ± 3%. The rat insulin-producing INS-1E cell line (a kind gift from Prof. C. Wollheim, Centre Médical Universitaire, Geneva, Switzerland) was cultured in RPMI 1640 (with 2 mm GlutaMAX-I) containing 5% FBS (43) and used at passages 60–72. Human EndoC-βH1 cells (44) were kindly provided by Prof. Raphael Scharfmann (Université Paris-Descartes, Institut Cochin, Paris, France) and cultured as described previously (45).
Treatments
Mouse islets and INS-1E cells were exposed to a combination of IL-1β (10 units/ml, R&D Systems, Abingdon, UK) and recombinant mouse or rat IFN-γ (1000 or 100 units/ml, respectively; R&D Systems) or 1 μm thapsigargin in medium with 1% or 5% FBS, respectively. Recombinant human IL-1β (50 units/ml, R&D Systems) and human IFN-γ (1000 units/ml; Peprotech, London, UK), 1 μm thapsigargin, or 10 ng/ml brefeldin A (Sigma) were added in medium without serum for human islets (4, 7). MG132 (Sigma) was dissolved in DMSO and added at 2 μm concentration 2 h before and during cytokine exposure. INS-1E cells were exposed to 0.5 mm palmitate in the presence of 0.75% fatty acid-free BSA and 1% FBS, as described previously (13, 46). Lyophilized THBS1 (purified from human platelets; Calbiochem, San Diego, CA) was diluted in bidistilled water and used at 2 μg/ml. Recombinant MANF was used at 100 ng/ml. For the TUNEL assay (see below), mouse islets were cultured in medium with 10% FBS.
RNAi
Proteins were knocked down using previously validated siRNAs against THBS1, DP5, PUMA, BIM, and BAD (13, 20, 42, 47, 48). MANF siRNAs (rat MANF-1 s164483, rat MANF-2 s164484, and human MANF s15436) were from Life Technologies). The negative control of 21-nt duplex RNA (Qiagen, Hilden, Germany) does not affect β-cell function, gene expression, and viability (49). Lipid–RNA complexes were formed in Opti-MEM (1 μl of Lipofectamine RNAi-MAX, Invitrogen, to 150 nm siRNA) and added to the cells at a final concentration of 30 nm siRNA (50). Transfected cells were cultured for 2 days before treatment.
Real-time PCR
Poly(A)+ RNA was isolated from cells using oligo(dT) 25-coated polystyrene Dynabeads (DYNAL, Oslo, Norway) and reverse-transcribed to cDNA with the GeneAmp RNA PCR kit (PerkinElmer Life Sciences). Real-time PCR amplification was done using IQ SYBR Green Supermix (Bio-Rad) on a MyiQ2 instrument. PCR product concentration was calculated as copies per microliter using a standard curve, and values were corrected for the reference gene GAPDH. β-Actin, the expression of which is not modified by cytokine exposure (4), was used as the reference gene for mouse or human samples. The specific primer sequences are described in supplemental Table S1. Gene expression values are shown as -fold change of control, except in the mouse islet experiments.
Assessment of β-cell apoptosis
Apoptotic cells were counted by fluorescence microscopy after staining with the DNA-binding dyes Hoechst 33342 (10 μg/ml) and propidium iodide (5 μg/ml) (51). Caspase 3 and 9 cleavage confirmed apoptosis (see below). TUNEL staining was assessed by Click-iT TUNEL Alexa Fluor imaging assay (Thermo Fisher Scientific), performed according to the instructions of the manufacturer, was followed by insulin staining. Slides were mounted with Vectashield mounting medium containing DAPI (Vector Laboratories), and the number of apoptotic β-cells was quantified.
Western blotting
Western blot analyses were performed using 20 μg of whole-cell extract protein (51). Laemmli buffer was added, and the samples were boiled for 5 min. Protein expression and phosphorylation were analyzed using the specific primary rabbit/mouse antibodies described in supplemental Table S2. After incubation with secondary horseradish peroxidase–labeled anti-rabbit or anti-mouse antibodies (1:10,000, Jackson ImmunoResearch Laboratories, Baltimore Pike, PA), protein-specific signals were detected using chemiluminescence Supersignal (Pierce) and quantified using Scion Image (Scion Corp., Frederick, MD).
Infection with recombinant adenoviruses
Cells were infected with adLUC (a luciferase-expressing control virus), adTHBS1 (expressing mouse THBS1), adTHBS1-KDEL (expressing mouse THBS1 fused to KDEL), or adTHBS1-FLAG (expressing mouse THBS1 fused to the FLAG peptide) (12, 13) and used at a multiplicity of infection of 1. To induce MANF overexpression, cells were infected with the CMV-MANF lentivirus or control CMV-GFP lentivirus at a multiplicity of infection of 5. After 3–4 h of infection, the medium was changed, and cells were treated or collected 48 h later.
DCF assay
Oxidative stress was measured by incubating cells with the fluorescent dye DCF (10 μm, Sigma) for 30 min at 37 °C. Fluorescence was quantified in a Victor 2 reader (PerkinElmer Life Sciences) and corrected for total protein quantified in cell lysates (13).
Promoter reporter assay
The UPRE luciferase reporter construct was kindly provided by Prof. Prywes (Columbia University, New York, NY). The pARE-TI-luciferase reporter containing a single copy of the 41-bp murine GST-Ya antioxidant response element and minimal TATA-Inr promoter was kindly provided by Prof. Fahl (University of Wisconsin, Madison, WI). Cells were transfected using Lipofectamine 2000 with 250 ng of reporter construct and pRL-CMV plasmid (50 ng, with Renilla used as an internal control for transfection efficiency) and treated after 48 h. Luciferase activity of cell lysates was expressed as relative luciferase/Renilla activity.
Immunofluorescence
INS-1E cells were fixed with 4% formalin for 10 min, permeabilized with 0.1% Triton for 5 min, blocked in PBS with 3% goat serum for 30 min, and incubated overnight with MANF antibody. The slides were stained with Hoechst 33342 and secondary antibodies conjugated with Alexa Fluor 568 (Life Technologies) and analyzed by inverted fluorescence microscopy (Zeiss Axiovert 200, Oberkochen, Germany) at ×20 magnification and 20 °C. Double immunofluorescence for insulin and cleaved caspase 3 was performed on dispersed human islet cells as described previously (52).
Statistical analysis
Data are presented as means ± S.E. of the indicated number (n) of independent experiments. Comparisons were performed by analysis of variance followed by paired t test with Bonferroni correction for multiple comparisons where needed. p < 0.05 was considered statistically significant.
Author contributions
D. A. C., D. L. E., and M. Cnop contributed to the original idea and experimental design of the study. D. A. C., M. Cito, F. A. G., C. C., T. D., and L. L. carried out the experiments and performed data analysis. M. L., A. D., M. B., and P. M. contributed materials and data interpretation. D. L. E. and M. Cnop wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Isabelle Millard, Anyishai Musuaya, Nathalie Pachera, and Michael Pangerl (Université Libre de Bruxelles Center for Diabetes Research) for expert technical assistance. We also thank Dr. Jeffery D. Molkentin (Department of Pediatrics, Cincinnati Children's Hospital Medical Center, Cincinnati, OH) for providing the THBS1 adenovirus and Prof. Mart Saarma (Institute of Biotechnology, University of Helsinki, Helsinki, Finland) for support with the MANF studies.
This was supported by the European Foundation for the Study of Diabetes/Lilly Program; the European Union Horizon 2020 Research and Innovation Program, Project T2DSystems, under Grant Agreement 667191; the Fonds National de la Recherche Scientifique; Actions de Recherche Concertées de la Communauté Française, Belgium; and the Innovative Medicines Initiative 2 Joint Undertaking, Project INNODIA, under Grant Agreement 115797, which receives support from the European Union Horizon 2020 Research and Innovation Program, EFPIA, JDRF, and the Leona M. and Harry B. Helmsley Charitable Trust. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5 and Tables S1 and S2.
- T1D
- type 1 diabetes
- T2D
- type 2 diabetes
- ER
- endoplasmic reticulum
- PERK
- protein kinase R-like endoplasmic reticulum kinase
- MANF
- mesencephalic astrocyte-derived neutrotrophic factor
- BiP
- binding immunoglobulin protein
- DCF
- 2′,7′-dichlorofluorescein diacetate
- THBS
- thrombospondin
- PUMA
- p53-upregulated modulator of apoptosis
- DP5
- death protein 5
- BIM
- BCL2-interacting mediator of cell death
- BAD
- BCL-2-associated death promoter
- BAX
- BCL-2-associated protein X
- NOD
- non-obese diabetic
- THA
- thapsigargin
- ad
- adenovirus
- Luc
- luciferase.
References
- 1. Cnop M., Welsh N., Jonas J. C., Jörns A., Lenzen S., and Eizirik D. L. (2005) Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54, S97–S107 [DOI] [PubMed] [Google Scholar]
- 2. Eizirik D. L., Colli M. L., and Ortis F. (2009) The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 5, 219–226 [DOI] [PubMed] [Google Scholar]
- 3. Lundberg M., Krogvold L., Kuric E., Dahl-Jørgensen K., and Skog O. (2016) Expression of interferon-stimulated genes in insulitic pancreatic islets of patients recently diagnosed with type 1 diabetes. Diabetes 65, 3104–3110 [DOI] [PubMed] [Google Scholar]
- 4. Eizirik D. L., Sammeth M., Bouckenooghe T., Bottu G., Sisino G., Igoillo-Esteve M., Ortis F., Santin I., Colli M. L., Barthson J., Bouwens L., Hughes L., Gregory L., Lunter G., Marselli L., et al. (2012) The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 8, e1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cnop M. (2008) Fatty acids and glucolipotoxicity in the pathogenesis of type 2 diabetes. Biochem. Soc. Trans. 36, 348–352 [DOI] [PubMed] [Google Scholar]
- 6. Poitout V., and Robertson R. P. (2008) Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocr. Rev. 29, 351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cnop M., Abdulkarim B., Bottu G., Cunha D. A., Igoillo-Esteve M., Masini M., Turatsinze J. V., Griebel T., Villate O., Santin I., Bugliani M., Ladriere L., Marselli L., McCarthy M. I., Marchetti P., et al. (2014) RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 63, 1978–1993 [DOI] [PubMed] [Google Scholar]
- 8. Marchetti P., Bugliani M., Lupi R., Marselli L., Masini M., Boggi U., Filipponi F., Weir G. C., Eizirik D. L., and Cnop M. (2007) The endoplasmic reticulum in pancreatic β cells of type 2 diabetes patients. Diabetologia 50, 2486–2494 [DOI] [PubMed] [Google Scholar]
- 9. Marhfour I., Lopez X. M., Lefkaditis D., Salmon I., Allagnat F., Richardson S. J., Morgan N. G., and Eizirik D. L. (2012) Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 55, 2417–2420 [DOI] [PubMed] [Google Scholar]
- 10. Engin F., Yermalovich A., Nguyen T., Ngyuen T., Hummasti S., Fu W., Eizirik D. L., Mathis D., and Hotamisligil G. S. (2013) Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci. Transl. Med. 5, 211ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cunha D. A., Ladrière L., Ortis F., Igoillo-Esteve M., Gurzov E. N., Lupi R., Marchetti P., Eizirik D. L., and Cnop M. (2009) Glucagon-like peptide-1 agonists protect pancreatic β-cells from lipotoxic endoplasmic reticulum stress through upregulation of BiP and JunB. Diabetes 58, 2851–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lynch J. M., Maillet M., Vanhoutte D., Schloemer A., Sargent M. A., Blair N. S., Lynch K. A., Okada T., Aronow B. J., Osinska H., Prywes R., Lorenz J. N., Mori K., Lawler J., Robbins J., and Molkentin J. D. (2012) A thrombospondin-dependent pathway for a protective ER stress response. Cell 149, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cunha D. A., Cito M., Carlsson P. O., Vanderwinden J. M., Molkentin J. D., Bugliani M., Marchetti P., Eizirik D. L., and Cnop M. (2016) Thrombospondin 1 protects pancreatic β-cells from lipotoxicity via the PERK-NRF2 pathway. Cell Death Differ. 23, 1995–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu J. X., Thomas C. E., and Brunak S. (2016) Network biology concepts in complex disease comorbidities. Nat. Rev. Genet. 17, 615–629 [DOI] [PubMed] [Google Scholar]
- 15. Cardozo A. K., Ortis F., Storling J., Feng Y. M., Rasschaert J., Tonnesen M., Van Eylen F., Mandrup-Poulsen T., Herchuelz A., and Eizirik D. L. (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes 54, 452–461 [DOI] [PubMed] [Google Scholar]
- 16. Gurzov E. N., and Eizirik D. L. (2011) Bcl-2 proteins in diabetes: mitochondrial pathways of β-cell death and dysfunction. Trends Cell Biol. 21, 424–431 [DOI] [PubMed] [Google Scholar]
- 17. Bornstein P. (1995) Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J. Cell Biol. 130, 503–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Veliceasa D., Ivanovic M., Hoepfner F. T., Thumbikat P., Volpert O. V., and Smith N. D. (2007) Transient potential receptor channel 4 controls thrombospondin-1 secretion and angiogenesis in renal cell carcinoma. FEBS J. 274, 6365–6377 [DOI] [PubMed] [Google Scholar]
- 19. Lindahl M., Danilova T., Palm E., Lindholm P., Võikar V., Hakonen E., Ustinov J., Andressoo J. O., Harvey B. K., Otonkoski T., Rossi J., and Saarma M. (2014) MANF is indispensable for the proliferation and survival of pancreatic β cells. Cell Rep. 7, 366–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gurzov E. N., Germano C. M., Cunha D. A., Ortis F., Vanderwinden J. M., Marchetti P., Zhang L., and Eizirik D. L. (2010) p53 up-regulated modulator of apoptosis (PUMA) activation contributes to pancreatic β-cell apoptosis induced by proinflammatory cytokines and endoplasmic reticulum stress. J. Biol. Chem. 285, 19910–19920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gurzov E. N., Ortis F., Cunha D. A., Gosset G., Li M., Cardozo A. K., and Eizirik D. L. (2009) Signaling by IL-1β+IFN-γ and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic β-cell apoptosis. Cell Death Differ. 16, 1539–1550 [DOI] [PubMed] [Google Scholar]
- 22. Barthson J., Germano C. M., Moore F., Maida A., Drucker D. J., Marchetti P., Gysemans C., Mathieu C., Nuñez G., Jurisicova A., Eizirik D. L., and Gurzov E. N. (2011) Cytokines tumor necrosis factor-α and interferon-γ induce pancreatic β-cell apoptosis through STAT1-mediated Bim protein activation. J. Biol. Chem. 286, 39632–39643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marroquí L., Santin I., Dos Santos R. S., Marselli L., Marchetti P., and Eizirik D. L. (2014) BACH2, a candidate risk gene for type 1 diabetes, regulates apoptosis in pancreatic β-cells via JNK1 modulation and crosstalk with the candidate gene PTPN2. Diabetes 63, 2516–2527 [DOI] [PubMed] [Google Scholar]
- 24. Tersey S. A., Nishiki Y., Templin A. T., Cabrera S. M., Stull N. D., Colvin S. C., Evans-Molina C., Rickus J. L., Maier B., and Mirmira R. G. (2012) Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61, 818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cnop M., Foufelle F., and Velloso L. A. (2012) Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 18, 59–68 [DOI] [PubMed] [Google Scholar]
- 26. Abdulkarim B., Nicolino M., Igoillo-Esteve M., Daures M., Romero S., Philippi A., Senée V., Lopes M., Cunha D. A., Harding H. P., Derbois C., Bendelac N., Hattersley A. T., Eizirik D. L., Ron D., et al. (2015) A missense mutation in PPP1R15B causes a syndrome including diabetes, short stature, and microcephaly. Diabetes 64, 3951–3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Synofzik M., Haack T. B., Kopajtich R., Gorza M., Rapaport D., Greiner M., Schönfeld C., Freiberg C., Schorr S., Holl R. W., Gonzalez M. A., Fritsche A., Fallier-Becker P., Zimmermann R., Strom T. M., et al. (2014) Absence of BiP co-chaperone DNAJC3 causes diabetes mellitus and multisystemic neurodegeneration. Am. J. Hum. Genet. 95, 689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hartley T., Siva M., Lai E., Teodoro T., Zhang L., and Volchuk A. (2010) Endoplasmic reticulum stress response in an INS-1 pancreatic β-cell line with inducible expression of a folding-deficient proinsulin. BMC Cell Biol. 11, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lindahl M., Saarma M., and Lindholm P. (2017) Unconventional neurotrophic factors CDNF and MANF: Structure, physiological functions and therapeutic potential. Neurobiol. Dis. 97, 90–102 [DOI] [PubMed] [Google Scholar]
- 30. Drott C. J., Olerud J., Emanuelsson H., Christoffersson G., and Carlsson P. O. (2012) Sustained β-cell dysfunction but normalized islet mass in aged thrombospondin-1 deficient mice. PLoS ONE 7, e47451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Olerud J., Johansson M., Lawler J., Welsh N., and Carlsson P. O. (2008) Improved vascular engraftment and graft function after inhibition of the angiostatic factor thrombospondin-1 in mouse pancreatic islets. Diabetes 57, 1870–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Olerud J., Mokhtari D., Johansson M., Christoffersson G., Lawler J., Welsh N., and Carlsson P. O. (2011) Thrombospondin-1: an islet endothelial cell signal of importance for β-cell function. Diabetes 60, 1946–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gonzalez-Quesada C., Cavalera M., Biernacka A., Kong P., Lee D. W., Saxena A., Frunza O., Dobaczewski M., Shinde A., and Frangogiannis N. G. (2013) Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ. Res. 113, 1331–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Topol E. J., McCarthy J., Gabriel S., Moliterno D. J., Rogers W. J., Newby L. K., Freedman M., Metivier J., Cannata R., O'Donnell C. J., Kottke-Marchant K., Murugesan G., Plow E. F., Stenina O., and Daley G. Q. (2001) Single nucleotide polymorphisms in multiple novel thrombospondin genes may be associated with familial premature myocardial infarction. Circulation 104, 2641–2644 [DOI] [PubMed] [Google Scholar]
- 35. Hannah B. L., Misenheimer T. M., Pranghofer M. M., and Mosher D. F. (2004) A polymorphism in thrombospondin-1 associated with familial premature coronary artery disease alters Ca2+ binding. J. Biol. Chem. 279, 51915–51922 [DOI] [PubMed] [Google Scholar]
- 36. Hellman M., Arumäe U., Yu L. Y., Lindholm P., Peränen J., Saarma M., and Permi P. (2011) Mesencephalic astrocyte-derived neurotrophic factor (MANF) has a unique mechanism to rescue apoptotic neurons. J. Biol. Chem. 286, 2675–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mätlik K., Yu L. Y., Eesmaa A., Hellman M., Lindholm P., Peränen J., Galli E., Anttila J., Saarma M., Permi P., Airavaara M., and Arumäe U. (2015) Role of two sequence motifs of mesencephalic astrocyte-derived neurotrophic factor in its survival-promoting activity. Cell Death Dis. 6, e2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dooley J., Tian L., Schonefeldt S., Delghingaro-Augusto V., Garcia-Perez J. E., Pasciuto E., Di Marino D., Carr E. J., Oskolkov N., Lyssenko V., Franckaert D., Lagou V., Overbergh L., Vandenbussche J., Allemeersch J., et al. (2016) Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nat. Genet. 48, 519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Galli E., Härkönen T., Sainio M. T., Ustav M., Toots U., Urtti A., Yliperttula M., Lindahl M., Knip M., Saarma M., and Lindholm P. (2016) Increased circulating concentrations of mesencephalic astrocyte-derived neurotrophic factor in children with type 1 diabetes. Sci. Rep. 6, 29058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yavarna T., Al-Dewik N., Al-Mureikhi M., Ali R., Al-Mesaifri F., Mahmoud L., Shahbeck N., Lakhani S., AlMulla M., Nawaz Z., Vitazka P., Alkuraya F. S., and Ben-Omran T. (2015) High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum. Genet. 134, 967–980 [DOI] [PubMed] [Google Scholar]
- 41. Maganti A. V., Tersey S. A., Syed F., Nelson J. B., Colvin S. C., Maier B., and Mirmira R. G. (2016) Peroxisome proliferator-activated receptor-γ activation augments the β-cell unfolded protein response and rescues early glycemic deterioration and β cell death in non-obese diabetic mice. J. Biol. Chem. 291, 22524–22533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Santin I., Moore F., Colli M. L., Gurzov E. N., Marselli L., Marchetti P., and Eizirik D. L. (2011) PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic β-cell apoptosis via regulation of the BH3-only protein Bim. Diabetes 60, 3279–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Asfari M., Janjic D., Meda P., Li G., Halban P. A., and Wollheim C. B. (1992) Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 130, 167–178 [DOI] [PubMed] [Google Scholar]
- 44. Ravassard P., Hazhouz Y., Pechberty S., Bricout-Neveu E., Armanet M., Czernichow P., and Scharfmann R. (2011) A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J. Clin. Invest. 121, 3589–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brozzi F., Nardelli T. R., Lopes M., Millard I., Barthson J., Igoillo-Esteve M., Grieco F. A., Villate O., Oliveira J. M., Casimir M., Bugliani M., Engin F., Hotamisligil G. S., Marchetti P., and Eizirik D. L. (2015) Cytokines induce endoplasmic reticulum stress in human, rat and mouse β cells via different mechanisms. Diabetologia 58, 2307–2316 [DOI] [PubMed] [Google Scholar]
- 46. Oliveira A. F., Cunha D. A., Ladriere L., Igoillo-Esteve M., Bugliani M., Marchetti P., and Cnop M. (2015) In vitro use of free fatty acids bound to albumin: a comparison of protocols. BioTechniques 58, 228–233 [DOI] [PubMed] [Google Scholar]
- 47. Cunha D. A., Igoillo-Esteve M., Gurzov E. N., Germano C. M., Naamane N., Marhfour I., Fukaya M., Vanderwinden J. M., Gysemans C., Mathieu C., Marselli L., Marchetti P., Harding H. P., Ron D., Eizirik D. L., and Cnop M. (2012) Death protein 5 and p53-upregulated modulator of apoptosis mediate the endoplasmic reticulum stress-mitochondrial dialog triggering lipotoxic rodent and human β-cell apoptosis. Diabetes 61, 2763–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cunha D. A., Gurzov E. N., Naamane N., Ortis F., Cardozo A. K., Bugliani M., Marchetti P., Eizirik D. L., and Cnop M. (2014) JunB protects β-cells from lipotoxicity via the XBP1-AKT pathway. Cell Death Differ. 21, 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moore F., Naamane N., Colli M. L., Bouckenooghe T., Ortis F., Gurzov E. N., Igoillo-Esteve M., Mathieu C., Bontempi G., Thykjaer T., Ørntoft T. F., and Eizirik D. L. (2011) STAT1 is a master regulator of pancreatic β-cell apoptosis and islet inflammation. J. Biol. Chem. 286, 929–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moore F., Cunha D. A., Mulder H., and Eizirik D. L. (2012) Use of RNA interference to investigate cytokine signal transduction in pancreatic β cells. Methods Mol. Biol. 820, 179–194 [DOI] [PubMed] [Google Scholar]
- 51. Cnop M., Ladriere L., Hekerman P., Ortis F., Cardozo A. K., Dogusan Z., Flamez D., Boyce M., Yuan J., and Eizirik D. L. (2007) Selective inhibition of eukaryotic translation initiation factor 2α dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic β-cell dysfunction and apoptosis. J. Biol. Chem. 282, 3989–3997 [DOI] [PubMed] [Google Scholar]
- 52. Marroqui L., Dos Santos R. S., Fløyel T., Grieco F. A., Santin I., Op de Beeck A., Marselli L., Marchetti P., Pociot F., and Eizirik D. L. (2015) TYK2, a candidate gene for type 1 diabetes, modulates apoptosis and the innate immune response in human pancreatic β-cells. Diabetes 64, 3808–3817 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.