

Abstract
GPR40 mediates free fatty acid–induced insulin secretion in beta cells. We investigated the safety, pharmacokinetics, and glucose response of MK‐8666, a partial GPR40 agonist, after once‐daily multiple dosing in type 2 diabetes patients. This double‐blind, multisite, parallel‐group study randomized 63 patients (placebo, n = 18; 50 mg, n = 9; 150 mg, n = 18; 500 mg, n = 18) for 14‐day treatment. The results showed no serious adverse effects or treatment‐related hypoglycemia. One patient (150‐mg group) showed mild‐to‐moderate transaminitis at the end of dosing. Median MK‐8666 Tmax was 2.0–2.5 h and mean apparent terminal half‐life was 22–32 h. On Day 15, MK‐8666 reduced fasting plasma glucose by 54.1 mg/dL (500 mg), 36.0 mg/dL (150 mg), and 30.8 mg/dL (50 mg) more than placebo, consistent with translational pharmacokinetic/pharmacodynamic model predictions. Maximal efficacy for longer‐term assessment is projected at 500 mg based on exposure–response analysis. In conclusion, MK‐8666 was generally well tolerated with robust glucose‐lowering efficacy.
Stdy Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ GPR40 agonists stimulate insulin secretion in a glucose‐dependent manner, and thus carry a low risk of hypoglycemia. MK‐8666, a partial GPR40 agonist, was generally well tolerated in healthy volunteers with no serious side effects following single and once‐daily dosing up to 10 days.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ This study aimed to characterize the safety, tolerability, and glucose response of MK‐8666 in patients with type 2 diabetes. Predictive accuracy of a diabetes translational PK/PD model was assessed. Use of prior knowledge coupled with PK/PD modeling and simulation provided a means of extrapolation to support potential design of a longer‐term phase IIb trial.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
✓ MK‐8666 was generally well tolerated after 2 weeks of treatment, with glycemic efficacy at 150 mg and maximal efficacy at 500 mg observed. The translational PK/PD modeling analysis adequately predicted clinical glucose response for MK‐8666.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓ The findings in this study further demonstrate the potential clinical efficacy of GPR40 agonists. By highlighting the predictive accuracy of a translational PK/PD model with clinical data and application of modeling and simulation to inform phase IIb study design, this may contribute to broader use of such quantitative approaches in early drug development.
Type 2 diabetes involves multiple metabolic defects that contribute to hyperglycemia, including β‐cell dysfunction and consequent decreased insulin secretion; abnormalities in the incretin axis; insulin resistance; increased lipolysis, lipogenesis, and plasma free fatty acid concentration; and increased glucose reabsorption, glucagon secretion, and hepatic glucose production.1 These metabolic defects offer multiple targets for drug development, with many of the drugs given concomitantly.1 Some classes of drugs, however, such as the insulin secretagogues (sulfonylureas and glinides), contribute to hypoglycemia by stimulating insulin secretion in a nonglucose‐dependent manner.2 A need exists for new drugs that work by other mechanisms and carry a low risk of hypoglycemia.
G‐protein–coupled receptor 40 (GPR40) is highly expressed in pancreatic β cells; its activation by fatty acids amplifies insulin secretion, but only when glucose levels are elevated.3, 4, 5 GPR40 agonists, like the incretin mimetics, stimulate insulin secretion in a glucose‐dependent manner, and thus carry a low risk of hypoglycemia.6, 7 GPR40 agonists represent a novel mechanism of action that may be complementary to other currently used therapies. A GPR40 agonist (TAK‐875) has been clinically validated in patients with type 2 diabetes, showing significant glucose‐lowering efficacy with a low risk of hypoglycemia,8, 9, 10 although its development program was halted because of liver toxicity.11
MK‐8666 is a potent and selective partial agonist for GPR40 that binds orthostatically. Based on preclinical pharmacology studies, MK‐8666 was shown to be selective for GPR40 relative to GPR119, GPR43, GPR41, or GPR120, and demonstrated weak pharmacological activity to other G‐protein‐coupled receptors (GPCRs). In healthy volunteers, MK‐8666 was shown to be generally well tolerated, with no serious side effects following single‐ and multiple‐dose daily administration up to 10 days and at doses exceeding those required for efficacy (Merck internal data). The present phase Ib, randomized, placebo‐controlled, multiple‐dose clinical study assessed the safety and tolerability, effects on indices of glycemic control including fasting plasma glucose (FPG) and 24‐h weighted mean glucose (WMG), and pharmacokinetics (PK) of MK‐8666 in patients with type 2 diabetes. As a companion to this study, the results from a translational PK/pharmacodynamic (PD) model are presented to assess the overall predictability of glucose response for a novel GPR40 agonist.12 This model describes the integrated glucose and insulin response profiles as previously published by Jauslin et al.,13 expanded to include GPR40 agonism in human and rodent models. Demonstrating predictability of clinical response established a quantitative translational tool for clinical and preclinical decision‐making. The glucose data obtained in this trial were further utilized in an exposure–response analysis to predict longer‐term glycemic response in support of phase II dose selection. The exposure–response model described the relationship between PK and FPG for MK‐8666, and was used to predict 12‐week HbA1c dose response14 by leveraging a previously published PK‐glucose‐HbA1c model for TAK‐875.15
METHODS
Patients
Key inclusion criteria for enrolled patients were age 18–65 years (inclusive), body mass index (BMI) 18–40.0 kg/m2 (inclusive), and treatment with diet/exercise alone (treatment‐naïve), or with no more than two oral antihyperglycemic agents (AHAs) (excluding thiazolidenediones) at the time of the prestudy (screening) visit. Patients who used injectable AHAs or thiazolidenediones previously must have discontinued these medications >6 months prior to screening. Patients taking a beta‐blocker or bupropion were excluded. Patients who were on mono or dual oral AHA therapy were washed off these medications beginning at least 2 weeks prior to administration of the initial dose of MK‐8666. Screening HbA1c had to be ≥7.0% (53 mmol/mol) and ≤10.5% (91 mmol/mol) for treatment‐naïve patients, and ≥6.5% (48 mmol/mol) and ≤10.0% (86 mmol/mol) for those on either the mono or dual oral AHA therapy. Patients had to be willing to follow a standard weight‐maintaining diet throughout the study and be nonsmokers and/or had not used nicotine or nicotine‐containing products for at least ∼3 months.
Study design
This phase Ib, multicenter, double‐blind, randomized, placebo‐controlled, four‐arm, parallel‐group study initiated October 14, 2013 and completed April 26, 2014 (ClinicalTrials.gov Identifier: NCT01971554, protocol 003). It was conducted in accordance with the ICH Harmonized Tripartite Guidelines for Good Clinical Practice, the ethical principles laid down in the Declaration of Helsinki, and applicable local regulations. All patients provided written informed consent.
Sixty‐three type 2 diabetes patients were randomized in a 2:2:1:2 ratio into one of four treatment arms: 500 mg (n = 18), 150 mg (n = 18), 50 mg (n = 9), and placebo (n = 18) and received medication once daily for 14 consecutive days. Randomization was done via a computer‐generated allocation schedule. Both patients and investigators were blinded to treatment; matching placebo capsules were used. Patients were domiciled for the entire duration of the treatment phase.
Study endpoints
Primary endpoints were measures of safety/tolerability and changes in FPG after 14 days of dosing with MK‐8666. Change from baseline to day 14 was of primary interest; differences from placebo are reported. Secondary endpoints were standard key PK parameters and changes in 24‐h WMG concentrations from baseline after 14 days. The relationship between MK‐8666 PK and parameters of glucose homeostasis (FPG, 24‐h WMG) during multiple timepoints in the 14‐day treatment period was assessed as an exploratory objective.
Study assessments
Blood for FPG was collected on Day –1 (preplanned dose), predose on Days 1, 3, 7, and 14, and 24 h postdose Day 14 (Day 15). On Day –1 (baseline) and Day 14, patients underwent an 18‐point glucose assessment during both fasting and postmeal times over a 24‐h period for measuring 24‐h WMG. Blood for MK‐8666 plasma concentration analysis was collected on Day 1 at predose, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 16, and 24 h postdose; and on Day 14 at predose, 0.5, 1, 2, 2.5, 3, 4, 6, 8, 12, 16, 24, 48, and 72 h postdose. MK‐8666 concentrations were assessed using a validated LCMS method.
Safety and tolerability were evaluated by clinical assessment of adverse experiences and other safety measurements conducted at prespecified timepoints (physical examinations, vital signs, 12‐lead electrocardiogram, laboratory safety tests (serum chemistry, hematology, and urinalysis), and frequent glucometer‐based glucose monitoring).
Pharmacokinetic analysis
Plasma concentration and nominal sampling time data collected pre‐ and postdose on Days 1 and 14 were used to conduct a noncompartmental PK analysis in WinNonlin v. 6.3 (Certara, Princeton, NJ). Peak and time of peak concentrations (Cmax, Tmax) and trough concentrations (Ctrough) were identified from the plasma concentration–time curve. Plasma AUC0‐tau and terminal elimination phase (t½) were calculated using the linear up/log down method. The accumulation ratio for AUC0‐tau, Cmax, and Ctrough was calculated as a ratio of Day 14 / Day 1 values. A formal statistical assessment of dose proportionality was not conducted, given the relatively early stage of development of the compound. However, descriptive and graphical data assessments were made to explore changes in exposure relative to MK‐8666 dose.
Statistical analyses
The per‐protocol population was used for the primary FPG, secondary 24‐h WMG, and secondary PK analysis. Any patients or data values excluded from analysis were to be identified, along with their reason for exclusion. At the end of the study, all patients who were compliant with the study procedure as aforementioned and had available data were included in the analysis dataset. The all‐subjects‐as‐treated population was used for the evaluation of safety and tolerability and comprised all patients who received at least one dose of MK‐8666.
The sample size for the study was chosen so that the study had ∼80% probability to establish that the high dose of MK‐8666 had at least a 50% posterior probability of reducing FPG at least 34 mg/dL more than placebo. With this criterion, and assuming a true between‐patient standard deviation of 24 mg/dL for each group, and a true reduction in FPG of 40 mg/dL more than placebo, the study planned to enroll 18 patients in the highest dose group of MK‐8666 and 18 patients in the placebo group.
For the analysis of the primary hypothesis to assess the effect of MK‐8666 on the reduction in FPG from baseline at Day 15, a constrained longitudinal data analysis (cLDA) model proposed by Liang and Zeger16 was fit to the FPG data from the MK‐8666 dose groups and placebo. This model assumes a common mean across treatment groups at baseline and a different mean for each treatment at each of the postbaseline timepoints. In this model, the response vector consisted of baseline and the values observed at each postbaseline timepoint, i.e., FPG at baseline, Day 3, Day 7, Day 14, and Day 15. Time was treated as a categorical variable so that no restriction was imposed on the trajectory of the means over time. An unstructured covariance matrix was used to model the correlation among repeated measurements. The hypothesis for the primary efficacy objective is that after 14 days of once‐daily treatment with MK‐8666, at a dose that is safe and well tolerated, the FPG reduction from baseline is ≥34 mg/dL more than placebo. To evaluate the primary hypothesis, the posterior probability, PostPr (δ ≥34 | data), where δ is the true mean placebo‐corrected reduction from baseline in FPG at Day 15, was computed using the estimated mean and standard error from the cLDA model and using an informative normal prior with mean of 35 mg/dL and standard deviation of 19 mg/dL (from published TAK‐875 data). The primary hypothesis would be supported if for the highest safe and well‐tolerated dose the PostPr (δ ≥34 | data) ≥0.50.
Summary statistics were generated for laboratory and vital sign parameters as deemed clinically appropriate. Statistical analysis was performed using SAS v. 9.3 (Cary, NC).
Pharmacokinetic/pharmacodynamic analyses
A translational PK/PD model12 developed to predict clinical response for novel GPR40 agonists was used to project the dose–response in this study. Briefly, this model is based on an integrated glucose–insulin dynamic model,13 extended to include response to GPR40 agonism for MK‐8666 and other GPR40 partial agonists. The model was developed using PK/PD (plasma drug, glucose, and insulin concentrations) data from prior clinical studies with GPR40 agonists including TAK‐875 and from in‐house Goto‐Kakizaki rat studies for a number of GPR40 agonist molecules. The model incorporated compound‐ and species‐specific GPR40 potency estimated from in vitro inositol monophosphate (IP1) assay data. A semimechanistic representation of the glucose–insulin feedback system was utilized, which captures the impact of insulin changes on glucose and vice versa. The majority of system parameters in this model, for instance, glucose volume of distribution and insulin‐independent clearance, were allometrically scaled between species rather than individually estimated, to better enable forward and backward translation. An in vitro–in vivo correlation relating IP1 in vitro potency in rat and human cells to model‐estimated in vivo insulin secretion was developed within the model to enable predictions within and across species. Further details describing the model, including a model schematic, table of model parameters used in simulations, and key equations that differ from the model published by Jauslin and colleagues15 are shown in Supplemental Figure S1 and Supplemental Table S1.
To enable prediction of glucose response in patients in a 2‐week study, a simple two‐compartment population PK model for MK‐8666 was developed using healthy subject PK data from two earlier phase I studies (unpublished data). Assuming similar PK in patients and healthy subjects, that two‐compartment PK model was incorporated into the translational PK/PD model to simulate 200 trials of 1–2,000‐mg doses administered for 2 weeks in 18 patients per dose. The resulting mean (90% confidence interval, CI) dose–response for 2‐week FPG change from baseline difference from placebo was calculated using a linear model, and compared graphically to the primary study results.
Following completion of the study, a post‐hoc population PK/PD model for MK‐8666 in patients was developed, incorporating PK data from the prior healthy volunteer studies in addition to the PK and FPG data in this trial.14 The PK of MK‐8666 was characterized by a two‐compartment model with dose‐dependent central volume of distribution and first‐order absorption rate constant. An indirect response model with stimulation of glucose elimination well described the PK‐FPG relationship. The next link to HbA1c was based on a previously published PK/PD model for the GPR40 agonist TAK‐875,15 where the rate of change of HbA1c is modeled as a difference of rates of HbA1c production and HbA1c degradation, with FPG influencing the production rate (see Supplemental Figure S2 and Supplemental Table S2). These two models were linked together to enable simulation of HbA1c following administration of MK‐8666 in a 12‐week phase IIb study setting.
RESULTS
Patient characteristics
Overall, 63 patients were enrolled and treated. One patient in the MK‐8666 50‐mg group was lost to follow‐up. The mean age (range) of all patients was 55.0 (30–65) years; approximately half were white and half were African American. Demographic and baseline characteristics between groups were similar (Supplemental Table S3). For none of the results below were there any significant differences based on sex or ethnicity. Of the 63 subjects recruited into the study, 59 had at least one AHA and four subjects were treatment‐naïve. As subjects were washed off their prior AHA treatment 2 weeks prior to randomization, stratification based on prior treatment was not considered necessary.
Efficacy
Primary endpoint
The mean reduction from baseline in FPG of MK‐8666 at Day 15 was 54.1 mg/dL, 36.0 mg/dL, and 30.8 mg/dL, for patients taking 500 mg, 150 mg, and 50 mg, respectively, more than placebo; the differences were statistically significant for all dose groups at the nominal one‐sided 0.05 level. Figure 1 shows the mean FPG concentrations over time by treatment group. Table 1 shows the least squares means for the change from baseline in FPG at each timepoint for each treatment group and the mean differences from placebo in the change from baseline in FPG for each treatment group at each timepoint. The posterior probabilities for the MK‐8666 500‐mg and 150‐mg groups each vs. placebo were 0.97 and 0.59, respectively, meeting the primary hypothesis.
Figure 1.
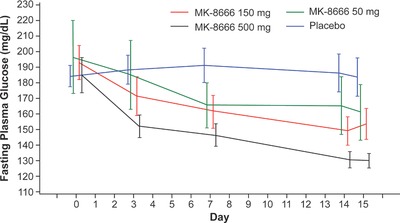
Mean fasting plasma glucose concentrations over time by treatment group (mean ± standard error).
Table 1.
Least squares means change from baseline in fasting plasma glucose (mg/dL) and changes from baseline in weighted mean glucose at 2 weeks
| Endpoint | Timepoint | Treatment | n | Least squaresMean change from baseline | Treatment comparison | Mean difference | 90% CI |
|---|---|---|---|---|---|---|---|
| Fasting plasma glucose | Day 3 | MK‐8666 500 mg | 18 | 33.3 | MK‐8666 500 mg vs. placebo | 37.2 | (27.4, 46.9) |
| MK‐8666 150 mg | 18 | 21.0 | MK‐8666 150 mg vs. placebo | 24.9 | (15.2, 34.7) | ||
| MK‐8666 50 mg | 9 | 10.5 | MK‐8666 50 mg vs. placebo | 14.4 | (2.5, 26.4) | ||
| Placebo | 18 | −3.9 | — | ||||
| Day 7 | MK‐8666 500 mg | 18 | 39.4 | MK‐8666 500 mg vs. placebo | 45.1 | (32.1, 58.1) | |
| MK‐8666 150 mg | 18 | 30.2 | MK‐8666 150 mg vs. placebo | 36.0 | (23.0, 49.0) | ||
| MK‐8666 50 mg | 9 | 28.9 | MK‐8666 50 mg vs. placebo | 34.7 | (18.8, 50.6) | ||
| Placebo | 18 | −5.7 | — | ||||
| Day 14 | MK‐8666 500 mg | 18 | 55.6 | MK‐8666 500 mg vs. placebo | 56.2 | (40.2, 72.2) | |
| MK‐8666 150 mg | 18 | 42.3 | MK‐8666 150 mg vs. placebo | 42.9 | (26.9, 58.9) | ||
| MK‐8666 50 mg | 9 | 28.5 | MK‐8666 50 mg vs. placebo | 29.1 | (9.5, 48.7) | ||
| Placebo | 18 | −0.6 | — | ||||
| Day 15 | MK‐8666 500 mg | 18 | 56.0 | MK‐8666 500 mg vs. placebo | 54.1 | (38.1, 70.0) | |
| MK‐8666 150 mg | 18 | 37.9 | MK‐8666 150 mg vs. placebo | 36.0 | (20.0, 51.9) | ||
| MK‐8666 50 mg | 9 | 32.8 | MK‐8666 50 mg vs. placebo | 30.8 | (11.3, 50.3) | ||
| Placebo | 18 | 2.0 | — | ||||
| 24‐H weighted mean glucose (mg/dL) | Day 14 | MK‐8666 500 mg | 18 | 45.4 | MK‐8666 500 mg vs. placebo | 48.8 | (35.6, 62.0) |
| MK‐8666 150 mg | 18 | 27.3 | MK‐8666 150 mg vs. placebo | 30.6 | (17.4, 43.8) | ||
| MK‐8666 50 mg | 9 | 19.0 | MK‐8666 50 mg vs. placebo | 22.3 | (6.2, 38.4) | ||
| Placebo | 17a | −3.3 | — |
WMG, weighted mean glucose.
Least squares means and confidence intervals from a constrained longitudinal data analysis model.
aData from one patient at Day 14 in the placebo group is missing, as the patient had to leave the study site for personal reasons.
Secondary endpoint
MK‐8666 significantly reduced 24‐h WMG in all dose groups relative to placebo. The mean reduction from baseline on Day 14 was 48.8 mg/dL, 30.6 mg/dL, and 22.3 mg/dL more than placebo, for patients taking MK‐8666 500 mg, 150 mg, and 50 mg, respectively. Table 1 shows the changes from baseline in 24‐h mean WMG after 2 weeks for each treatment group.
Pharmacokinetics/pharmacodynamics
Figure 2 shows the mean plasma concentration profiles for MK‐8666 for all dose groups for 1 day and 14 days. Table 2 shows values for various PK parameters for MK‐8666 following administration of daily oral doses for 1 day or 14 days. MK‐8666 was absorbed with a median Tmax of 2.0–2.5 h and demonstrated a biphasic decline. The mean apparent t½ was 22–32 h. Based on a comparison of AUC0‐24hr on Day 1 and Day 14, the MK‐8666 exposures following repeated daily dosing of 150 mg and 500 mg accumulate roughly 1.2–1.8× relative to a single dose. Descriptive and graphical analyses show trends of nonlinearity with slightly greater than proportional increases in exposure between 50 mg and 150 mg, and dose‐proportional increases beyond that.
Figure 2.
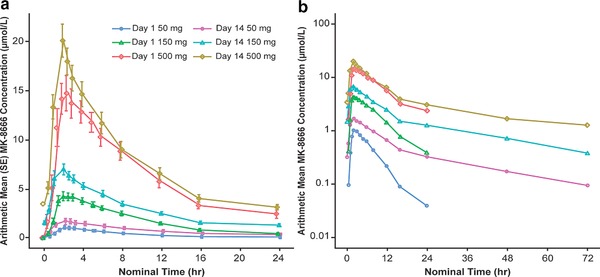
Mean plasma concentration profiles for MK‐8666 following administration of daily oral doses of 50 mg, 150 mg, and 500 mg for 1 day or 14 days to fasted adults with type 2 diabetes (mean ± SE). (a) Linear scale. (b) Semilogarithmic scale.
Table 2.
Pharmacokinetic parameter values for MK‐8666 following administration of daily oral doses for 1 day or 14 days to patients with type 2 diabetes
| Day | Dose (mg) | Tmax a (hr) | Cmax (μM)b | C24hr (μM)b | AUC0‐24hr b (μM•hr) | t1/2 b (hr) |
|---|---|---|---|---|---|---|
| 1 | 50 | 2.0 | 0.906 | 0.0314 | 6.11 | |
| (1.5‐12.0) | (102) | (89) | (88) | |||
| 14 | 50 | 2.0 | 1.50 | 0.264 | 15.3 | 21.6c |
| (1.0‐12.0) | (93) | (89) | (88) | (63) | ||
| Day 14/Day 1 d | 1.7 | 8.4 | 2.5 | |||
| 1 | 150 | 2.25 | 4.49 | 0.260 | 37.2 | |
| (1.5‐4.0) | (45) | (134) | (44) | |||
| 14 | 150 | 2.0 | 7.16 | 1.14 | 66.9 | 23.0 |
| (1.0‐6.0) | (37) | (54) | (32) | (50) | ||
| Day 14/Day 1 d | 1.6 | 4.4 | 1.8 | |||
| 1 | 500 | 2.5 | 17.1 | 1.86 | 143 | |
| (1.5‐8.0) | (40) | (84) | (33) | |||
| 14 | 500 | 2.0 | 19.9 | 2.86 | 172 | 31.6 |
| (0.5‐3.0) | (32) | (45) | (37) | (40) | ||
| Day 14/Day 1 d | 1.2 | 1.5 | 1.2 |
Lower limit of quantification is 1.92 nM.
Noncompartmental analysis based on nominal times (18 for the 500‐mg group, 18 for the 150‐mg group, 9 for the 50‐mg group).
aMedian (min–max). bGeometric mean (Geometric CV%). c n = 7. dGeometric mean ratio.
The predictions from the previously developed translational PK/PD model are shown overlaid with the observed study results in Figure 3. The resulting simulations closely capture both the mean and patient variability in the observed FPG dose–response data in this trial.
Figure 3.
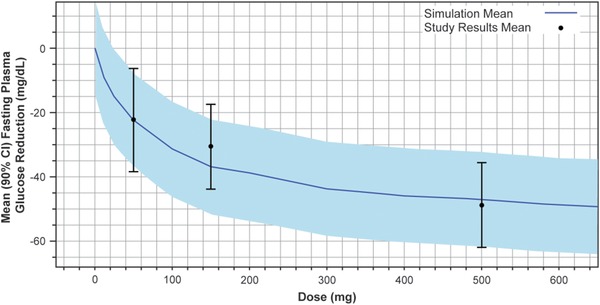
MK‐8666 mean simulated fasting plasma glucose lowering (placebo‐corrected reduction from baseline) vs. dose from translational PK/PD model, with observed mean (90% CI) study results.
The results of a further PK/PD modeling analysis of the plasma concentration data and FPG data obtained in this study and predicted into a 12‐week trial are shown in Figure 4. Based on this simulation, daily doses of 500 mg and 150 mg are predicted to achieve greater than 95% and ∼90% of maximum HbA1c response, respectively.14
Figure 4.
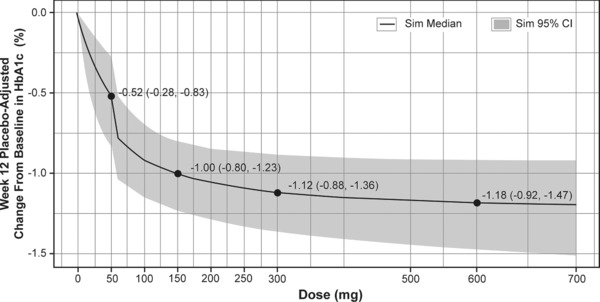
Pharmacokinetic/pharmacodynamic model 12‐week HbA1c dose–response predictions for MK‐8666.14 The number of subjects per dose was 200, and 1,000 simulations were run with uncertainty in population mean parameters and no variability. The shaded region is the confidence interval of the mean.
Safety and tolerability
A total of 65 adverse events (AEs) were reported by 27 out of 63 patients, of which 18 were considered drug‐related by the investigator. All AEs were mild to moderate in intensity. The most commonly reported AEs were pain (back/neck/extremity, 13 episodes), headache (10 episodes), constipation (9 episodes), nausea (6 episodes), diarrhea (5 episodes), and abdominal pain (4 episodes). No patient showed treatment‐related hypoglycemia. Supplemental Table S4 shows patients with AEs with incidence >0%.
A 53‐year‐old African‐American man in the 150‐mg group, who had normal liver safety tests at screening and prior to dosing with the exception of a modestly elevated gamma‐glutamyl transpeptidase (γ‐GT), developed alanine aminotransferase (ALT) and aspartate aminotransferase (AST) increases above baseline prior to dosing on Day 3, and continued to trend upward with continued dosing. The highest observed ALT/AST values were measured 24 h after the final dose of MK‐8666 (ALT 4–5× upper‐limit‐of‐normal (ULN) and AST 2–3× ULN), concurrent with a modest elevation in alkaline phosphatase (AP) ∼1.4× ULN. Two weeks after the completion of dosing, all liver safety tests had returned to normal or near‐normal concentrations with the exception of γ‐GT, which was trending back towards predosing baseline. Subsequent evaluation did not identify any specific cause for the liver safety test abnormalities, and it is possible that they were related to MK‐8666. There were no other notable individual liver safety test abnormalities. No relevant increases in mean ALT or AST levels in MK‐8666 vs. placebo‐treated patients were observed. There were no clinically significant abnormalities in urine chemistry panels, complete blood count, electrocardiogram, and physical examinations, including vital signs.
DISCUSSION
MK‐8666 was found to be generally well tolerated and effective relative to placebo at lowering FPG and WMG in a cohort of patients with type 2 diabetes. As expected from MK‐8666's mechanism of action, no patient showed treatment‐related hypoglycemia. MK‐8666 significantly reduced FPG in all dose groups; the proof‐of‐concept criterion of having a posterior probability of at least 0.50, of the placebo‐corrected reduction from baseline in FPG at Day 15 being at least 34 mg/dL, was met by the 500‐mg (posterior probability = 0.97) and 150‐mg (posterior probability = 0.59) dose groups. These clinical results were consistent with the predictions from the translational PK/PD model.12 This indicates that such a translational approach can be used to accurately predict human glucose response based on preclinical experimental results.
This study also provided insights into the PK of MK‐8666 in patients with type 2 diabetes. The observed half‐life of MK‐8666 (22–32 h) supported once‐daily dosing, which was seen as desirable. At steady‐state, the geometric mean half‐lives tended to show an increase at higher doses. Caution must be exercised in interpreting the physiological relevance of this finding, given the moderately high intersubject variability of each dose group and because in some subjects in the lowest dose group, concentrations towards the later timepoints were below quantifiable limits. Of note, a half‐life could not be calculated following a single dose given the duration of sampling (24 h) relative to the estimated half‐life (22–32 h). In addition, MK‐8666 demonstrated a lack of dose proportionality over the evaluated range. Greater than dose proportional increases were observed between 50 mg and 150 mg, with relatively proportional changes in exposure between 150 mg and 500 mg. The underlying reason for this lack of proportionality was not identified. MK‐8666 is mainly eliminated by metabolism, with a negligible fraction excreted unchanged in the urine (fe <0.1%). MK‐8666 oxidative metabolism is mediated by CYP3A, with preclinical evidence of UGT1A3 as another elimination pathway. Had the compound continued through development, there likely would have been efforts to understand this nonlinearity. However, as dose proportionality was observed in the likely clinically relevant range, this observation was considered of limited relevance for clinical dose decision‐making.
By utilizing a PK/PD modeling approach to further analyze the clinical trial data in this study, inferences to the anticipated performance of MK‐8666 in a larger and longer clinical trial can be made. This approach was used to assess the anticipated HbA1c dose–response relationship expected if a 12‐week phase IIb were to be conducted. The simulations showed that at 12 weeks doses of 500 mg and 150 mg would achieve greater than 95% and ∼90% of maximum HbA1c response, respectively, and define the plateau for maximal efficacy.14
As the clinical proof‐of‐concept study evaluated doses of 50 mg, 150 mg, and 500 mg, model‐informed analyses provided a more complete dose–response assessment, including evaluation of doses that were not included in the proof‐of‐concept study (Figures 3, 4), and allowed for robust planning of the phase IIb dose‐ranging study. A dose of 150 mg was considered to balance, providing a high predicted glucose response, while reducing the potential for exposure‐related AEs. In order to create a reasonable dose‐step between 150 mg and 500 mg (considered close to Emax), a dose of 300 mg was considered for inclusion. To ensure adequate exposure separation between doses, 600 mg was recommended as the highest dose that should be evaluated in the phase IIb study. Although the treatment period was short in this study, use of prior knowledge coupled with PK/PD modeling and simulation provided a means of extrapolation to support potential design of a longer‐term phase IIb trial.
MK‐8666 was generally well tolerated, although there appeared to be a greater incidence of certain AEs with higher doses. Gastrointestinal AEs including nausea, constipation, and loose stools appeared to be more common in the MK‐8666 500‐mg dose group than in other dose groups. Liver safety test abnormalities were found in a patient in the 150‐mg group. Abnormal hepatic function was also found in the phase III study of patients taking TAK‐875, although most of the patients in that study who experienced elevations of aminotransferases had confounding liver disease.10 Takeda announced termination of their TAK‐875 program in December 2013.11 It is possible the hepatic changes observed in the present study were related to MK‐8666, although it is not clear what mechanism could be causing the hepatic changes because, at least in rodents, GPR40 is not expressed in the liver.3, 5 Merck terminated the development program for MK‐8666 in August 2014 in anticipation of an unfavorable benefit/risk profile for the compound in the type 2 diabetes target population.
Overall, this study demonstrated that MK‐8666 was well tolerated after 2 weeks of treatment, where doses of 150 mg and higher were therapeutically efficacious, with maximum effect reaching at 500 mg.
Supporting information
Supplemental Table S1. Translational integrated glucose–insulin model parameters for proof‐of‐concept simulation
Supplemental Table S2. Summary of PK‐FPG model parameters for MK‐8666
Supplemental Table S3. Demographics and baseline characteristics, n (%)
Supplemental Table S4. Patients with adverse events (incidence >0%* in 1 or more treatment group)
SUPPLEMENTAL FIGURE TITLES AND LEGENDS
Supplemental Figure S1. Translational integrated glucose–insulin model for GPR40, with submodels for pharmacokinetics (black), in vitro–in vivo correlation of drug effect (orange), feedback control (red), glucose disposition (purple), and insulin disposition (green).
Supplemental Figure S2. PK‐FPG model was based on MK‐8666 proof‐of‐concept data .
Acknowledgments
Current address for MV Chakravarthy: Eli Lilly & Co., Lilly Corporate Center, Indianapolis, IN 46285, USA. Previously presented in abstract form at the American Diabetes Association 76th Scientific Sessions, June 10–14, 2016
Author Contributions
P.A.K., A.W.K., P.V., R.A.R., B.J.M., J.C., A.G.H.E., E.D., D.G.K., A.L.F., L.M., M.V.C., E.A.K., and D.T. wrote the article; B.J.M., J.C., A.G.H.E., and M.V.C. designed the research; P.A.K., A.W.K., P.V., B.J.M., J.C., A.G.H.E., E.D., D.G.K., A.L.F., L.M., M.V.C., E.A.K., and D.T. performed the research; P.A.K., A.W.K., R.A.R., and D.T. analyzed the data. Editorial and some medical writing assistance were provided by Steven Tresker of Cactus Communications. This assistance was funded by Merck & Co., Inc., Kenilworth, NJ, USA. The authors retained full control of the article content. Guarantor(s): A.W.K., R.A.R., P.V., D.A.T. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Funding for this research was provided by Merck & Co., Inc., Kenilworth, NJ, USA. The sponsor was involved in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Conflict of Interest
Alexander W. Krug, Radha A. Railkar, Bret J. Musser, Josee Cote, Eunkyung Kauh, Pavan Vaddady, Daniel A. Tatosian, and Prajakti A. Kothare are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA and may own stock/stock options in the company; Antwan G.H. Ederveen and Manu V. Chakravarthy are former employees. David G. Krefetz is an employee of PRA Health Sciences, which received payment to conduct the study. Emanuel DeNoia has an employment relationship with ICON plc. Almena L. Free has received personal fees from Pinnacle Research Group. Linda Morrow is an employee and shareholder of Profil Institute for Clinical Research, Chula Vista, CA.
Previous Presentations
Previously presented in abstract form at the American Diabetes Association 76th Scientific Sessions, June 10–14, 2016.
List of Principal Investigators
David Krefetz, Emanuel DeNoia, Almena L. Free, Linda Morrow.
References
- 1. Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 58, 773−795 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Detournay, B., Halimi, S., Robert, J., Deschaseaux, C. & Dejager, S. Hypoglycemia hospitalization frequency in patients with type 2 diabetes mellitus: a comparison of dipeptidyl peptidase 4 inhibitors and insulin secretagogues using the French health insurance database. Vasc. Health Risk Manag. 11, 417−425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Briscoe, C.P. et al The orphan G protein‐coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 278, 11303−11311 (2003). [DOI] [PubMed] [Google Scholar]
- 4. Edfalk, S., Steneberg, P. & Edlund, H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57, 2280−2287 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Itoh, Y. et al Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422, 173−176 (2003). [DOI] [PubMed] [Google Scholar]
- 6. Burant, C.F. Activation of GPR40 as a therapeutic target for the treatment of type 2 diabetes. Diabetes Care 36, Suppl 2, S175−179 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mancini, A.D. & Poitout, V. GPR40 agonists for the treatment of type 2 diabetes: life after 'TAKing' a hit. Diabetes Obes. Metab. 17, 622−629 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Burant, C.F. et al TAK‐875 vs. placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet 379, 1403−1411 (2012). [DOI] [PubMed] [Google Scholar]
- 9. Kaku, K., Araki, T. & Yoshinaka, R. Randomized, double‐blind, dose‐ranging study of TAK‐875, a novel GPR40 agonist, in Japanese patients with inadequately controlled type 2 diabetes. Diabetes Care 36, 245−250 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaku, K., Enya, K., Nakaya, R., Ohira, T. & Matsuno, R. Efficacy and safety of fasiglifam (TAK‐875), a G protein‐coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: a randomized, double‐blind, placebo‐controlled, phase III trial. Diabetes Obes. Metab. 17, 675−681 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lead GPR40 agonist bites the dust. Nat. Rev. Drug Discov. 13, 91−91 (2014). [Google Scholar]
- 12. Tatosian, D. et al Application of translational pharmacokinetic and pharmacodynamic modeling in the development of GPR40 partial agonists [abstract]. Diabetes 65 ( Suppl. 1 ), A617 (2016). [Google Scholar]
- 13. Jauslin, P.M. et al An integrated glucose‐insulin model to describe oral glucose tolerance test data in type 2 diabetics. J. Clin. Pharmacol. 47, 1244–1255 (2007). [DOI] [PubMed] [Google Scholar]
- 14. Vaddady, P. et al Pharmacokinetic and Pharmacodynamic Modeling of GPR40 Agonist MK‐8666 Proof of Concept Data to Inform Clinical Decisions [abstract]. J .Pharmacokinet. Pharmacodyn. 43, S33 (2016). [Google Scholar]
- 15. Naik, H. et al Pharmacometric approaches to guide dose selection of the novel GPR40 agonist TAK‐875 in subjects with type 2 diabetes mellitus. CPT Pharmacometrics Syst. Pharmacol. 2, e22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liang, K.Y. & Zeger, S. Longitudinal data analysis of continuous and discrete responses for pre–post designs. Sankhyā Indian J. Stat, (Series B). 62, 134–148 (2000). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. Translational integrated glucose–insulin model parameters for proof‐of‐concept simulation
Supplemental Table S2. Summary of PK‐FPG model parameters for MK‐8666
Supplemental Table S3. Demographics and baseline characteristics, n (%)
Supplemental Table S4. Patients with adverse events (incidence >0%* in 1 or more treatment group)
SUPPLEMENTAL FIGURE TITLES AND LEGENDS
Supplemental Figure S1. Translational integrated glucose–insulin model for GPR40, with submodels for pharmacokinetics (black), in vitro–in vivo correlation of drug effect (orange), feedback control (red), glucose disposition (purple), and insulin disposition (green).
Supplemental Figure S2. PK‐FPG model was based on MK‐8666 proof‐of‐concept data .