

Abstract
Cardiovascular disease (CVD) risk factors, and particularly decreased high density lipoprotein cholesterol (HDL-C) dyslipidemia are prevalent in Assam, India. This study was undertaken to investigate whether Apolipoprotein A-I (APOA1) gene polymorphisms (G-75A and C+83T) were associated with i) the risk for decreased HDL-C, and ii) other CVD risk factors, viz. serum lipids, atherogenic indices, obesity, and blood pressure (BP). A total of 649 subjects were screened, from which 200 eligible individuals, classified as case group with decreased HDL-C levels (100 subjects) and control group with normal HDL-C levels (100 subjects) were enrolled and genotyped using polymersase chain reaction-restriction fragment length polymorphism (PCR-RFLP) and DNA sequencing. Lipid fractions [HDL-C, total cholesterol (TC), low density lipoprotein cholesterol (LDL-C), very low density lipoprotein cholesterol (VLDL-C), triglycerides (TG)] and atherogenic indices [Castelli’s Risk Indices-I and -II (CRI-I and -II), non-HDL-C fraction, atherogenic index of plasma (AIP), atherogenic coefficient (AC)] were estimated. The G-75A and C+83T loci were not associated with decreased HDL-C risk. This was confirmed across different genetic models (dominant, recessive, additive and allelic). Association was also absent with BP and obesity. However, the G-75A locus was associated with LDL-C, whereas the C+83T locus was associated with TG and VLDL-C. Furthermore, these sites had effects on atherogenic indices. The rare A allele at the G-75A locus was associated with adverse CRI-I, CRI-II, non-HDL-C and AC values, while the major C allele at the C+83T locus was associated with adverse AIP values. Thus, the pro-atherogenic G-75A polymorphism and the anti-atherogenic C+83T polymorphism represent important genetic loci that modulate CVD risk factors in subjects from Assam.
Keywords: Atherogenic indices, Cardiovascular disease (CVD) risk factors, C+83T, G-75A, Lipid profile, Northeast India
Introduction
Cardiovascular diseases (CVDs) are the leading cause of mortality and morbidity worldwide. In 2012, an estimated 17.5 million people died of CVDs, representing about 31.0% of all global deaths [1]. Atherosclerosis, which refers to the progressive hardening of arteries and narrowing of lumen due to fibro-fatty changes in the vessel wall, is the major pathophysiological process underlying CVDs [2]. The risk factors for atherosclerosis and CVDs are many, such as altered serum lipid levels (also called dyslipidemia), smoking, alcohol, obesity, male gender, advancing age, ethnicity and genetic predisposition, diabetes and hypertension [1-3]. The apolipoprotein A1 gene (APOA1), which is a part of the APOA1-CIII-AIV gene cluster on chromosome 11, is a major site controlling the expression of lipids and lipoproteins [4-7]. Thus, allelic variants of APOA1 that influence CVD risk are of interest. The role of two single nucleotide polymorphisms (SNPs), located in the regulatory regions of APOA1, namely G-75A and C+83T, is being increasingly appreciated in this regard. These SNPs have been implicated in various clinical conditions [8-11], but most notably with reference to CVDs and their risk factors [12-18]. The G-75A site lies in the promoter region, while the C+83T site lies in the first intron of APOA1 [19,20]. These polymorphic sites are also cleavage sites for the restriction endonuclease, MspI and thus are amenable to detection by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) [15,16,21]. These SNPs are thought to influence several quantitative traits that are risk factors for CVD, such as: serum lipids [viz. total cholesterol (TC), high density lipoprotein cholesterol (HDL-C), low density lipoprotein cholesterol (LDL-C), very low density lipoprotein cholesterol (VLDL-C) and triglycerides (TG)], indices of obesity [viz. waist circumference (WC) and body mass index (BMI)], and blood pressure (BP) [12-19].
Recently, the G-75A and C+83T polymorphisms were described in the native population of Assam, where significant differences were noted in their distribution as compared to populations of neighboring regions [21]. Assam is the most populated state in northeast India, an ethnically distinct region that is often regarded as the ethnological transition zone between the Indian subcontinent and east/southeast Asia. Decreased level of HDL-C is a major risk factor for CVD [1-3] and a common form of dyslipidemia in Assam [22,23]. Moreover, APOA1 codes for the major part of the proteome of HDL particles [19]. Assam also has a sizeable burden of many other conditions (high BP, diabetes, schizophrenia, increased TG, etc.) where APOA1 is implicated [23-26]. Thus, it is of interest to investigate whether the G-75A and C+83T polymorphisms have a role to play in relation to these conditions in the population of Assam.
On this basis, we decided to conduct a pilot study to explore if G-75A and C+83T SNPs of APOA1 were associated with cardiovascular risk factors in the population of Assam. Our primary objective was to determine whether G-75A and C+83T polymorphisms were associated with decreased HDL-C. Our secondary objective was to assess if these polymorphisms were associated with other CVD risk factors, viz. lipid fractions (TC, LDL-C, VLDL-C, TG); obesity indices (WC and BMI); and BP (systolic and diastolic). Additionally, we studied the associations of these SNPs with several atherogenic indices [also known as lipid ratios, namely: Castelli’s Risk Index-I (CRI-I), Castelli’s Risk Index-II (CRI-II), atherogenic index of plasma (AIP), atherogenic coefficient (AC), non-HDL cholesterol fraction (non-HDL-C)]. Atherogenic indices reflect the propensity of the serum lipids to induce atherosclerotic changes. They are increasingly recognized as valuable predictors of CVD risk [27-29], often better than the traditional lipid measures, but genetic factors influencing them have been scarcely investigated.
Materials and methods
Study Subjects
Individuals undergoing health check-ups at the Guwahati Medical College and Hospital, Assam, India, were screened clinically and with the help of available investigation reports. Information on past medical history, drug history, dietary habits (vegetarian or non-vegetarian), and current smoking and alcohol use status was also obtained. We screened 649 individuals, from which 200 subjects (100 cases and 100 controls) of either sex, who were native to Assam, were finally selected for a case-control study on the basis of the following eligibility criteria.
Subjects enrolled in the case group had decreased HDL-C dyslipidemia (defined as HDL-C levels <40.0 mg/dL) [30]. Individuals with confounding conditions known to decrease the levels of HDL-C, such as diabetes mellitus/hyperglycemia, liver diseases, thyroid disorders and other endocrinal disorders, acute infections and inflammatory conditions, using medications affecting serum lipids (such as β-blockers, statins, oral contraceptive pills, steroids, hormone replacement therapies, etc.) and pregnancy were excluded.
In the control group, healthy subjects with normal values of HDL-C (≥40.0 mg/dL) were included. The other serum lipid fractions were also within the normal range (i.e., total cholesterol <200.0 mg/dL, TG <150.0 mg/dL, LDL-C <130.0 mg/dL, VLDL-C ≤30.0 mg/dL) [30].
The study was conducted as per the guidelines of the Declaration of Helsinki. It was approved by the Institutional Ethics Committee, Guwahati Medical College and Hospital, Assam, India. All subjects voluntarily provided written informed consent to participate prior to enrollment in the study. Some of the participants were selected from our previous studies [21,22].
Physical Measurements
The physical measurements performed in the subjects were: WC, BMI and BP. Waist circumference and BMI were used as indices of central and general obesity, respectively. They were measured according to World Health Organization guidelines [31]. Systolic and diastolic BP was measured as per Joint National Committee (JNC VII) guidelines [32].
Lipid Fractions and Atherogenic Indices
Measurements of TC, TG and HDL-C in fasting blood samples were done photometrically by homogenous enzymatic methods using dry chemistry reagent slides (Ortho-Clinical Diagnostics Inc., Rochester, NY, USA) in VITROS 5600 integrated system autoanalyzer (Ortho-Clinical Diagnostics Inc.). For quality control, these assays were validated using third-party control materials (Christian Medical College, Vellore, India and Bio-Rad Laboratories, Hercules, CA, USA). The LDL-C and VLDL-C were estimated indirectly using Friedewald’s formula [33]. Atherogenic indices were determined as follows: CRI-I = TC/HDL-C, CRI-II = LDL-C/HDL-C, AIP = log10 (TG/HDL-C), AC = (TC – HDL-C)/HDL-C and non-HDL-C = TC – HDL-C [34-36].
DNA Extraction and Genotyping
Genomic DNA extraction (by a rapid salting-out method) and genotyping for the G-75A and C+83T loci (by a PCR-RFLP method) were done from peripheral blood samples using the protocol described previously [21]. In brief, a 435 bp region at the 5’-end of the APOA1 gene that included the ‘-75’ and ‘+83’ loci was amplified using forward (5’-AGG GAC AGA GCT GAT CCT TGA ACT CTT AAG-3’) and reverse (5’-TTA GGG GAC ACC TAC CCG TCA GGA AGA GCA-3’) primers in 25 μL reaction mixture, followed by restriction digestion of the PCR product by MspI. The genotypes were inferred from the sizes of the restriction fragments analyzed in 12.0% polyacrylamide gel, as mentioned elsewhere [21]. Accordingly, for the G-75A site, the 66 and 114 bp fragments indicated a GG genotype; the 180 bp fragment indicated an AA genotype, and the 66, 114 and 180 bp fragments indicated a GA genotype. Likewise, for the C+83T site, the 46 and 209 bp fragments denoted a CC genotype, the 255 bp fragment denoted a TT genotype, and the 46, 209 and 255 bp fragments denoted a CT genotype.
We performed a quality-check by repeating the PCR-RFLP technique randomly in 20.0% of the samples ensuring that no differences were noted in the genotypes. Additional confirmation by direct sequencing (at SciGenom Pvt. Ltd., Cochin, Kerala, India) was done in 5.0% of the samples. All the genotypes detected for the two loci in the study were represented.
Statistical Analyses
Descriptive statistics were performed in Excel spreadsheets (Microsoft Office 2003, Redmont, WA, USA). For continuous data, means with standard deviation (SD) were expressed. The categorical data were summarized as counts and percentages. The allelic and genotype frequencies for the two SNPs were calculated using POPGENE 2.0 software (Molecular Biology and Biotechnology Centre, University of Alberta, Edmonton, AB, Canada). Conformity to Hardy Weinberg equilibrium (HWE) was assessed by the goodness-of-fit χ2 and G-square tests. Diplotype frequencies were also determined.
The statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS Inc., Chicago, IL, USA) version 11.5 software. The continuous data were verified for normality by the Kolmogorov-Smirnov test. The comparisons of allelic, genotype and diplotype frequencies between the case and the control groups were performed by the χ2 test (with Yate’s continuity correction, if required). The association of G-75A and C+83T poly-morphisms with decreased HDL-C was tested under different genetic models (dominant, recessive, additive and allelic). The crude association was first determined by calculating the unadjusted odds ratios (OR) with 95% confidence intervals (CI). Further, adjusted OR with 95% CI was calculated by multiple logistic regressions after adjusting for covariates such as gender, age, smoking, alcohol use, WC, BMI, TC and TG. The associations of the SNPs with other CVD risk factors (viz. concentrations of other lipid fractions, atherogenic indices, WC, BMI, systolic and diastolic BP) were examined by comparing their values (adjusted for covariates) across the detected genotypes using the analysis of covariance (ANCOVA) test. Comparisons were also performed across diplotypes. To quantify the significant relationships detected between the SNPs and CVD risk factors, the results were complemented by calculating the effect sizes (Cohen’s f) in G*POWER software (G*POWER 3.1.9.2; Düsseldorf, Germany). It provided an estimate of the magnitude of association that the SNPs had with the CVD risk factors. The effect sizes were interpreted as small f = 0.1), medium (f = 0.25) and large f = 0.4) effects as recommended by Cohen [37]. Further, power calculations were done to determine whether the statistical analysis and the results obtained therein were adequately powered. All calculations were two-tailed and a P value of less than 0.05 was considered as statistically significant.
Results
The baseline characteristics of the study subjects are presented in Table 1. The individuals in the case and the control groups were comparable (p >0.05) in terms of age and sex composition, and smoking and alcohol use status. Concentration of HDL-C was significantly lower in the case group than in the control group, as expected. Furthermore, the case group individuals had significantly higher (p <0.05) measures of obesity (BMI and WC), systolic and diastolic BP, lipid profile parameters (TC, TG, LDL-C and VLDL-C) and atherogenic indices in comparison to the control group individuals. None of the subjects in the study were vegetarians. During genotyping, we detected GG, GA and AA genotypes for the G-75A site, and CC and CT genotypes for the C+83T site (Figure 1).
Table 1.
Baseline characteristics of the study subjects. (Values are expressed as mean ± SD or n (%). Categorical variables were compared by the χ2 test. Continuous variables were compared by unpaired t-test.)
| Study Subjects (n = 200) | |||
|---|---|---|---|
| Variables | Case Group (n = 100) | Control Group (n = 100) | p Value |
| Age (years) | 43.12 ± 11.64 | 42.95 ± 11.60 | 0.92 |
| Sex | |||
| males | 66 (66.0%) | 61 (61.0%) | 0.56 |
| females | 34 (34.0%) | 39 (39.0%) | |
| Smokers | |||
| yes | 35 (35.0%) | 25 (25.0%) | 0.16 |
| no | 65 (65.0%) | 75 (75.0%) | |
| Alcohol use | |||
| yes | 20 (20.0%) | 32 (32.0%) | 0.08 |
| no | 80 (80.0%) | 68 (68.0%) | |
| Body mass index (kg/m2) | 24.79 ± 3.08 | 22.07 ± 3.18 | <0.05 |
| Waist circumference (cm) | 87.14 ± 5.89 | 80.72 ± 6.06 | <0.05 |
| Systolic blood pressure (mmHg) | 133.58 ± 16.44 | 117.86 ± 8.53 | <0.05 |
| Diastolic blood pressure (mmHg) | 84.32 ± 8.84 | 77.20 ± 5.92 | <0.05 |
| HDL-C (mg/dL) | 30.87 ± 5.19 | 54.61 ± 12.17 | <0.05 |
| Total cholesterol (mg/dL) | 175.54 ± 35.98 | 160.79 ± 25.70 | <0.05 |
| Triglycerides (mg/dL) | 145.95 ± 29.39 | 95.35 ± 23.55 | <0.05 |
| LDL-C (mg/dL) | 116.41 ± 28.19 | 87.37 ± 22.96 | <0.05 |
| VLDL-C (mg/dL) | 29.12 ± 7.62 | 19.03 ± 5.90 | <0.05 |
| Non-HDL-C (mg/dL) | 144.67 ± 32.47 | 106.18 ± 24.83 | <0.05 |
| Castelli’s risk index I | 5.71 ± 0.87 | 3.04 ± 0.65 | <0.05 |
| Castelli’s risk index II | 3.78 ± 0.79 | 1.68 ± 0.56 | <0.05 |
| Atherogenic index of plasma | 0.66 ± 0.13 | 0.26 ± 0.14 | <0.05 |
| Atherogenic coefficient | 4.71 ± 0.87 | 2.04 ± 0.65 | <0.05 |
HDL-C: high density lipoprotein cholesterol; LDL-C: low density lipoprotein cholesterol; VLDL-C; very low density lipoprotein cholesterol; non-HDL-C: non-high density lipoprotein cholesterol.
Figure 1.
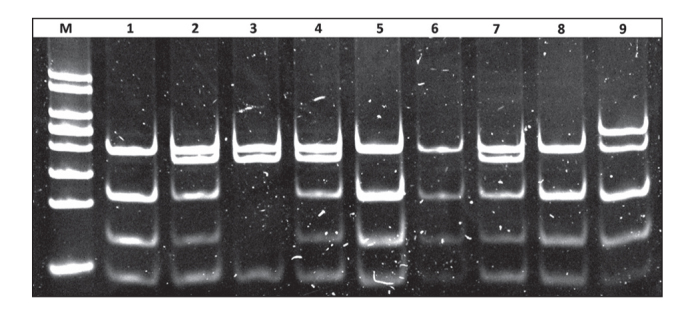
Polyacrylamide gel (12.0%) picture showing restriction fragments of various sizes, representing the different genotypes obtained for the G-75A/C+83T polymorphic sites. Lane M: 50 bp DNA lane marker (Thermo Fischer Scientific, Waltham, MA, USA); lanes 1, 5, 6 and 8: GG/CC (46, 66, 114 and 209 bp fragments); lanes 2, 4 and 7: GA/CC (46, 66, 114, 18 and 209 bp fragments; lane 3: AA/CC (46, 180 and 209 bp fragments); lane 9: GG/CT (46, 66, 114, 209 and 255 bp fragments). No TT genotype for the C+83T site was detected in the study.
Distribution of G-75A and C+83T Single Nucleotide Polymorphisms
The genotype, allelic and diplotype frequencies for G-75A and C+83T polymorphic loci are summarized in Table 2. Both the polymorphisms were in Hardy-Weinberg equilibrium. For the G-75A site, the GG genotype was most common, followed by GA and AA. The frequencies of GG, GA and AA genotypes were comparable between the case and the control groups (χ2 = 1.692, df= 2, p = 0.43). The allelic frequencies in the two groups were also comparable (minor A allele frequency = 0.205 in cases and 0.235 in controls, χ2 = 0.364, df = 1, p = 0.55). For the C+83T site, the homozygous TT genotype was not detected in either the cases or the controls. However, the frequency of CC and CT genotypes were comparable in the two groups (χ2 = 0.047, df = 1, p = 0.83). Similarly, the minor T allele frequency (0.055 in cases and 0.065 in controls) was also comparable (χ2 = 0.044, df= 1, p = 0.83). Five diplotypes were detected for the two polymorphic sites (Table 2). The diplotype frequencies did not vary significantly between the case and the control groups (χ2 = 2.041, df = 4, p = 0.73).
Table 2.
The distribution of G-75A and C+83T polymorphisms in the case and control groups. [Values are expressed as n (%).]
| Genotype Frequency | Allelic Frequency | |||||
|---|---|---|---|---|---|---|
| SNPs | Group | GG | GA | AA | G | A |
| G-75A | Case | 62 (62.0%) | 35 (35.0%) | 3 (3.0%) | 159 (79.5%) | 41 (20.5%) |
| Control | 60 (60.0%) | 33 (33.0%) | 7 (7.0%) | 153 (76.5%) | 47 (23.5%) | |
| χ2 = 1.692 (df = 2; p = 0.43 | χ2 = 0.364 (df = 1; p = 0.55) | |||||
| CC | CT | TT | C | T | ||
| C+83T | Case | 89 (89.0%) | 11 (11.0%) | – | 189 (94.5%) | 11 (5.5%) |
| Control | 87 (87.0%) | 13 (13.0%) | – | 187 (93.5%) | 13 (6.5%) | |
| χ2 = 0.047 (df = 1; p = 0.83) | χ2 = 0.044 (df = 1; p = 0.83) | |||||
| Diplotype Frequency | ||||||
| GG/CC | GA/CC | AA/CC | GG/CT | GA/CT | ||
| Combined | Case | 54 (54.0%) | 32 (32.0%) | 3 (3.0%) | 8 (8.0%) | 3 (3.0%) |
| Control | 50 (50.0%) | 30 (30.0%) | 7 (7.0%) | 10 (10.0%) | 3 (3.0%) | |
| χ2 = 2.041 (df = 4; p = 0.73) | ||||||
SNPs: single nucleotide polymorphisms; df: degree of freedom.
Association of G-75A and C+83T Single Nucleotide Polymorphisms with Decreased High Density Lipoprotein Cholesterol
Neither the genotypic variants nor the allelic variants of the G-75A or the C+83T polymorphism detected in the study were associated with HDL-C status (Table 3).
Table 3.
Association of decreased high density lipoprotein cholesterol with G-75A and C+83T polymorphisms under different genetic models. [Values are expressed as n (%) or OR (95% CI).]
| SNPs | Model | Genotype/Allele | Case | Control | Crude OR (95% CI) | Adjusted ORa (95% CI) |
|---|---|---|---|---|---|---|
| G-75A | Additive: GG vs. GA vs. AA | GG GA AA |
62 (62.0%) 35 (35.0%) 3 (3.0%) |
60 (60.0%) 33 (33.0%) 7 (7.0%) |
reference 1.03 (0.57-1.86) 0.42 (0.10-1.68) |
reference 0.82 (0.36-1.88) 0.23 (0.04-1.24) |
| Dominant: GG vs. (GA + AA) | GG GA +AA |
62 (62.0%) 38 (38.0%) |
60 (60.0%) 40 (40.0%) |
reference 0.92 (0.52-1.63) |
reference 0.67 (0.30-1.47) |
|
| Recessive: (GG + GA) vs. AA | GG + GA AA |
97 (97.0%) 3 (3.0%) |
93 (93.0%) 7 (7.0%) |
reference 0.41 (0.08-1.62) |
reference 0.24 (0.05-1.29) |
|
| Allelic: G vs. A | G A |
159 (79.5%) 41 (20.5%) |
153 (76.5%) 47 (23.5%) |
reference 0.84 (0.52-1.35) |
reference 0.61 (0.32-1.17) |
|
| C+83 T | CC vs. CT | CC CT |
89 (89.0%) 11 (11.0%) |
87 (87.0%) 13 (13.0%) |
reference 0.83 (0.34-1.97) |
reference 1.66 (0.54-5.13) |
| Allelic: C vs.T | C T |
189 (94.5%) 11 (5.5%) |
187 (93.5%) 13 (6.5%) |
reference 0.84 (0.36-1.94) |
reference 1.58 (0.54-4.59) |
SNPs: single nucleotide polymorphisms; OR: odds ratio; 95% CI: 95% confidence interval. Association between C+83T SNP and decreased HDL-C was not assessed by separate genetic models as no TT homozygotes were detected.
Odds ratio was adjusted for gender, age, smoking, alcohol use, WC, BMI, TC and TG. In every model, for each OR value, the corresponding 95% CI extended across 1. None of the OR values were statistically significant (p >0.05).
Association of G-75A and C+83T Single Nucleotide Polymorphisms with Other Lipid Fractions
The G-75A SNP was associated with LDL-C levels (Table 4). Values of LDL-C were significantly (p <0.05) higher in the A allele-containing controls as compared to the GG homozygotes. No significant association (p >0.05) was observed with other components of lipid profile. The C+83T locus was associated with TG and VLDL-C levels (Table 5). The case subjects with the CC genotype had significantly (p <0.05) higher TG and VLDL-C values than their CT counterparts. These findings persisted in the overall sample.
Table 4.
Influence of G-75A polymorphism on cardiovascular risk factors. [Values are expressed as adjusted mean±standard error (SE). Adjusted means compared between genotypes by analysis of covariance (ANCOVA). Statistically significant (p <0.05) differences among genotypes are highlighted in bold. Furthermore, means in a genotype column bearing an ‘a’ or ‘b’ as a superscript are significantly different.]
| Phenotypic Variables | Case Group (n = 100) | Control Group (n = 100) | Study Subjects (n = 200) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Lipid Profile | |||||||||
| GG | GA | AA | GG | GA | AA | GG | GA | AA | |
| HDL-C# | 30.7±0.5 | 31.3±0.6 | 29.5±2.3 | 55.4±1.5 | 54.8±2.0 | 47.0±4.6 | 42.7±1.1 | 42.9±1.5 | 42.2±3.9 |
| TC§ | 179.0±4.7 | 172.9±6.3 | 134.9±21.9 | 156.5±3.3 | 166.1±4.5 | 172.9±10.1 | 168.2±2.9 | 169.2±3.9 | 161.3±10.5 |
| TG§ | 145.5±4.9 | 148.1±6.7 | 129.6±23.2 | 93.8±3.7 | 94.9±4.9 | 110.3±11.2 | 120.4±3.5 | 121.1±4.8 | 121.5±12.7 |
| LDL-C§ | 120.7±3.8 | 113.3±5.1 | 87.2±17.8 | 82.7a±2.9 | 92.2b±3.9 | 105.1b±8.9 | 102.4±2.7 | 101.6±3.6 | 97.9±9.6 |
| VLDL-C§ | 29.0±0.9 | 29.6±1.3 | 25.8±4.7 | 18.7±0.7 | 18.9±0.9 | 21.9±2.2 | 24.2±2.5 | 24.2±0.9 | 24.0±0.7 |
| Atherogenic Indices | |||||||||
| Non-HDL-C§ | 147.9±4.2 | 141.9±5.7 | 109.5±19.8 | 101.1a±3.2 | 111.3a, b±4.3 | 125.9b±9.7 | 125.5±3.1 | 126.1±4.1 | 120.2±10.9 |
| CRI-I§ | 5.8±0.1 | 5.6±0.2 | 5.4±0.5 | 2.9a±0.1 | 3.1a±0.1 | 3.7b±0.2 | 4.4±0.1 | 4.3±0.2 | 4.1±0.5 |
| CRI-II§ | 3.9±0.1 | 3.6±0.1 | 3.4±0.5 | 1.6a±0.1 | 1.8a, b±0.1 | 2.2b±0.2 | 2.8±0.1 | 2.7±0.1 | 2.5±0.4 |
| AIP§ | 0.66±0.02 | 0.67±0.02 | 0.69±0.08 | 0.24±0.02 | 0.26±0.02 | 0.36±0.05 | 0.46±0.02 | 0.46±0.03 | 0.48±0.07 |
| AC§ | 4.8±0.1 | 4.6±0.2 | 4.4±0.5 | 1.9a±0.1 | 2.1a±0.1 | 2.7b±0.2 | 3.4±0.1 | 3.3±0.2 | 3.1±0.5 |
| Obesity Indices | |||||||||
| BMI† | 24.5±0.4 | 25.1±0.5 | 26.3±1.9 | 21.9±0.4 | 22.3±0.6 | 21.9±1.3 | 23.3±0.3 | 23.7±0.4 | 23.1±1.1 |
| WC† | 86.7±0.7 | 88.3±0.9 | 83.7±3.4 | 80.6±0.8 | 80.8±1.1 | 81.2±2.4 | 83.7±0.6 | 84.6±0.8 | 81.7±2.2 |
| Blood Pressure | |||||||||
| Systolic BP‡ | 135.4±2.1 | 130.7±2.8 | 129.1±9.7 | 118.1±1.0 | 117.2±1.4 | 118.7±3.1 | 127.0±1.2 | 124.0±1.6 | 121.5±4.5 |
| Diastolic BP‡ | 85.1±1.1 | 83.7±1.6 | 77.2±5.4 | 77.1±0.7 | 77.1±0.9 | 78.3±2.2 | 120.4±3.5 | 80.3±0.9 | 78.4±2.4 |
HDL-C: high density lipoprotein cholesterol; TC: total cholesterol; TG: triglycerides; LDL-C: low density lipoprotein cholesterol; VLDL-C; very low density lipoprotein cholesterol; non-HDL-C: non-high density lipoprotein cholesterol; CRI-I: Castelli’s risk index I; CRI-II: Castelli’s risk index II; AIP: atherogenic index of plasma; AC: atherogenic coefficient ; BMI: body mass index; WC: waist circumference; Systolic BP: systolic blood pressure; Diastolic BP: diastolic blood pressure.
Means adjusted taking age, gender, smoking, alcohol use, TC, TG, WC and BMI as covariates.
Means adjusted taking age, gender, smoking, alcohol use, WC and BMI as covariates.
Means adjusted taking age, gender, smoking, alcohol use, TC, HDL-C and TG as covariates.
Means adjusted taking age, gender, smoking, alcohol use, TC, HDL-C, TG, BMI and WC as covariates.
Table 5.
Influence of C+83T polymorphism on cardiovascular risk factors. [Values are expressed as adjusted mean±SE. Adjusted means compared between genotypes by analysis of covariance (ANCOVA). Statistically significant (p <0.05) differences among genotypes are highlighted in bold. Furthermore, means in a genotype column bearing an ‘a’ or ‘b’ as a superscript are significantly different.]
| Phenotypic Variables | Case Group (n = 100) | Control Group (n = 100) | Study Subjects (n = 200) | |||
|---|---|---|---|---|---|---|
| Lipid Profile | ||||||
| CC | CT | CC | CT | CC | CT | |
| HDL-C# | 30.8 ± 0.4 | 31.1 ± 1.1 | 54.7 ± 1.2 | 53.7 ± 3.1 | 43.1 ± 0.9 | 39.9 ± 2.5 |
| TC§ | 176.5 ± 3.9 | 167.8 ± 11.2 | 162.6 ± 2.7 | 148.4 ± 7.1 | 169.7 ± 2.4 | 157.2 ± 6.5 |
| TG§ | 148.8a ± 123.2 | 123.2b ± 11.4 | 96.6 ± 3.0 | 87.1 ± 7.9 | 122.7a± 2.9 | 105.3b ± 7.9 |
| LDL-C§ | 116.9 ± 3.2 | 112.4 ± 9.1 | 88.7 ± 2.5 | 78.6 ± 6.4 | 102.9 ± 2.2 | 94.8 ± 5.9 |
| VLDL-C§ | 29.7a ± 0.8 | 24.7b ± 2.3 | 19.3 ± 0.6 | 17.3 ± 1.6 | 24.5a ± 0.6 | 21.1b ± 1.6 |
| Atherogenic Indices | ||||||
| Non-HDL-C§ | 145.6 ± 3.5 | 137.5 ± 10.1 | 107.7 ± 2.7 | 95.9 ± 6.9 | 126.7 ± 2.5 | 116.2 ± 6.8 |
| CRI-I§ | 5.7 ± 0.1 | 5.6 ± 0.3 | 3.1 ± 0.1 | 2.9 ± 0.2 | 4.4 ± 0.1 | 4.2 ± 0.3 |
| CRI-II§ | 3.8 ± 0.1 | 3.8 ± 0.2 | 1.7 ± 0.1 | 1.5 ± 0.2 | 2.7 ± 0.1 | 2.7 ± 0.2 |
| AIP§ | 0.67a ± 0.01 | 0.59b ± 0.04 | 0.27 ± 0.01 | 0.23 ± 0.04 | 0.47a ± 0.01 | 0.41b ± 0.03 |
| AC§ | 4.7 ± 0.1 | 4.6 ± 0.3 | 2.1 ± 0.1 | 1.8 ± 0.2 | 3.4 ± 0.1 | 3.2 ± 0.2 |
| Obesity Indices | ||||||
| BMI† | 24.9 ± 0.3 | 23.5 ± 0.9 | 22.1 ± 0.3 | 22.1 ± 0.9 | 23.5 ± 0.3 | 22.8 ± 0.7 |
| WC† | 87.3 ± 0.6 | 85.5 ± 1.7 | 80.6 ± 0.6 | 81.4 ± 1.7 | 84.0 ± 0.5 | 83.3 ± 1.4 |
| Blood Pressure | ||||||
| Systolic BP‡ | 133.6 ± 1.7 | 133.7 ± 4.9 | 117.7 ± 0.8 | 119.1 ± 2.2 | 125.6 ± 1.0 | 126.3 ± 28 |
| Diastolic BP‡ | 83.8 ± 0.9 | 88.3 ± 2.7 | 77.0 ± 0.6 | 77.9 ± 1.5 | 80.4 ± 0.6 | 82.9 ± 1.6 |
HDL-C: high density lipoprotein cholesterol; TC: total cholesterol; TG: triglycerides; LDL-C: low density lipoprotein cholesterol; VLDL-C; very low density lipoprotein cholesterol; non-HDL-C: non-high density lipoprotein cholesterol; CRI-I: Castelli’s risk index I; CRI-II: Castelli’s risk index II; AIP: atherogenic index of plasma; AC: atherogenic coefficient; BMI: body mass index; WC: waist circumference; Systolic BP: systolic blood pressure; Diastolic BP: diastolic blood pressure.
Means adjusted taking age, gender, smoking, alcohol use, TC, TG, WC and BMI as covariates.
Means adjusted taking age, gender, smoking, alcohol use, WC and BMI as covariates.
Means adjusted taking age, gender, smoking, alcohol use, TC, HDL-C and TG as covariates.
Means adjusted taking age, gender, smoking, alcohol use, TC, HDL-C, TG, BMI and WC as covariates.
Association of G-75A and C+83T Single Nucleotide Polymorphisms with Atherogenic Indices
The G-75A polymorphism was associated with several atherogenic indices (Table 4). The AA homozygotes had significantly (p <0.05) elevated non-HDL-C and CRI-II values, whereas the G allele carriers had significantly (p < 0.05) diminished CRI-I and AC values. On the other hand, the C+83T polymorphism was associated with the AIP values (Table 5). The CC subjects had significantly (p <0.05) raised AIP than their CT counterparts.
Association of G-75A and C+83T Single Nucleotide Polymorphisms with Obesity Indices and Blood Pressure
The G-75A and C+83T loci were not associated with obesity and BP in the subjects under the present study. Values of WC, BMI and systolic and diastolic BP were comparable (p >0.05) across the different genotypes detected for the two polymorphic sites (Tables 4 and 5).
Association of Diplotypes with the Cardiovascular Disease Risk Factors
The CVD risk factors did not differ significantly across the five diplotypes obtained in the current study (data not shown).
Effect Size and Power
The G-75A locus produced medium-to-large sized effects (0.25 < Cohen’s f <0.4) on the CVD risk factors (non-HDL-C, CRI-I, CRI-II, AC and LDL-C values) with which it was associated, while the effect size of the C+83T locus on CVD risk factors (TG, VLDL-C and AIP) was small-to-medium (0.1 <Cohen’s f <0.25). The power available for detecting these medium and large effect sizes was more than 80.0% on most occasions. For detecting a very minimal effect size (0.10), the power was less than 80.0%.
Discussion
We explored the associations of G-75A and C+83T SNPs in the study population with cardiovascular risk factors. Although the G-75A and C+83T polymorphic sites were reported to be associated with HDL-C levels in some studies [9,15-17], we observed no such association in our study. These polymorphisms did not confer greater risk of developing decreased HDL-C dyslipidemia in the current population. Lack of association between these SNPs and low HDL-C levels was also reported by other investigators [7,12,38]. We verified our findings in different genetic models (additive, dominant, recessive and allelic) and no relationship was found. It is possible that changes in HDL-C values across the genotypic variants of G-75A and C+83T loci reported in some of the earlier studies were actually modulated by confounding factors. This is substantiated by the fact that the effects of G-75A and C+83T SNPs on phenotypic characteristics have been found to be influenced by gender differences [13,39], smoking [39] and alcohol use [40]. Such biases have not been controlled for in many of the previous studies [9,16,17]. We took care to ensure that the effects of these SNPs were assessed after controlling for potential confounders in the data analysis. Moreover, many conditions that may spuriously alter lipid levels in the cases were excluded in the design stage itself.
Among the other serum lipids, we found that the minor A allele in the G-75A locus was associated with adverse LDL-C levels. The AA and GA genotypes had high LDL-C values. This was consistent with the findings of previous investigators [4,38]. However, few studies have found the A allele to produce beneficial effects on LDL-C [41]. These discordant results may be due to linkage disequilibrium between the G-75A SNP and some other regulatory elements in close proximity that actually influence the levels of LDL-C. For example, strong linkage was observed between the A allele (of the G-75A site) and X2 allele (of the XmnI RFLP site) in the 5’-flanking region of APOA1 [4]. The X2 allele of the XmnI polymorphism has been found to be associated with LDL-C levels [42]. Future studies using sequencing would be of help in verifying such possibilities.
On the other hand, we found that the C>T transition at C+83T locus favorably affected TG and VLDL-C concentrations. Our results were in agreement to those by Ma et al. [43] who also found that the CT heterozygotes had lower TG than the CC homozygotes. The association with VLDL-C levels were expected because TG and VLDL-C metabolism are intimately related, and their values are proportional to one another [33]. It was suggested that the C+83T locus may have a role in regulating TG levels in Oriental subjects [43]. The biological mechanism causing these effects is uncertain. It was hypothesized that APOA1 variants may affect the lipase-mediated breakdown of TG particles in the plasma, thus modulating their concentration [14].
The association of the two SNPs with several atherogenic indices was a novel and significant finding of the current study. Genetic factors affecting atherogenic indices have been investigated infrequently. We found that the minor A allele at the G-75A site was associated with adverse values of CRI-I, CRI-II, non-HDL-C and AC, whereas, the minor T allele at the C+83T site was associated with favorable values of AIP. These pro-atherogenic effects of the G-75A polymorphism and the anti-atherogenic effects of the C+83T polymorphism on the atherogenic indices may be important modulators of CVD risks that are often described in relation to these two SNPs. Recent data have shown that atherogenic indices (viz. CRI-I, CRI-II, AIP, non-HDL-C, AC) are valuable predictors of CVD risk [27-30]. The usefulness of the traditional serum lipid measures (viz. TC, HDL-C, LDL-C, VLDL-C and TG) is mainly limited to predicting risk for those at the lower and the higher end of the CVD risk spectrum. In contrast, the atherogenic indices, being composite measures, take into account several lipid fractions. Thus, they reflect the bi-directional cholesterol traffic (in and outward) through the arterial intima and are more robust in predicting CVDs than individual lipid fractions [28,29].
There are contradictory reports about the effects of G-75A and C+83T SNPs on indices of obesity and BP. These SNPs were not associated with WC, BMI, systolic and diastolic BP in the current population. In conformity to the present findings, no association of the G-75A locus was detected with obesity and BP by Ma et al. [43] and de Franca et al. [6]. In contrast, Kim and Hong [18] reported association of the G-75A locus with obesity indices. On the other hand, Coban et al. [13] detected significant associations of the G-75A locus with BP, but not with obesity (BMI ≥30.0 kg/m2). For the C+83T locus, de Franca et al. [6] found no significant variation in obesity measures across the genotypes. This was in agreement with our findings. But Ma et al. [43] found the C+83T site to be associated with BP and obesity measures in diabetic subjects. In view of the above findings, it seems that these effects, if at all present, are remarkable only in some populations and that they are of an indirect nature, perhaps as a result of interaction with environmental and other genetic factors [43].
A limitation of our study was its design with respect to the secondary objectives. As our primary objective was to investigate the association of the polymorphisms with the risk of developing decreased HDL-C, we therefore had to recruit the subjects as case and control groups on the basis of HDL-C status. Thus, we could not use contingency tables and risk estimates (e.g., OR) to study the secondary objectives (i.e., association of the polymorphisms with other CVD risk factors such as TC, LDL-C, TG, VLDL-C, obesity indices, BP and atherogenic indices). However, this was circumvented by studying the associations using ANCOVA, and backing up the results with effect size calculations. It would also have been desirable to replicate and validate our findings in additional independent samples from other parts of northeast India and the adjoining regions.
Conclusions
The results from our pilot study suggested that the G-75A and C+83T polymorphisms of APOA1 were important modulators of CVD risk in the population of Assam. In general, the A allele at G-75A was pro-atherogenic and the C allele at the C+83T locus was anti-atherogenic. The G-75A locus was associated with LDL-C levels, while the C+83T locus was associated with TG and VLDL-C levels. But, these sites were not associated with the HDL-C status. Our study supports the hypothesis that the two SNPs do not play a major role in influencing HDL-C levels. Associations with TC, obesity and BP were also absent. Notably, these two loci were significantly associated with several atherogenic indices that are strong predictors of CVDs. Although drawn on a sample of modest size, yet these remarkable associations (with the detected effect sizes and available statistical power) from our exploratory study strongly suggested that the effect of genetic factors on atherogenic indices may be important while characterizing CVD risk. In the current population, the G-75A polymorphism seemed to be particularly important in this regard. Similar studies in other populations and larger sample groups would help in clarifying such utility of atherogenic indices.
Acknowledgments
KB received financial support in the form of MDMS Thesis Scholarship from the Department of Biotechnology, Ministry of Science and Technology, Government of India for this study. The authors are grateful to State Biotech Hub (Assam), College of Veterinary Science, Khanapara, Guwahati for offering laboratory facilities for performing genotyping. KB and MSP conceptualized and designed the study; PB and IH participated in the design; KB and DD collected the clinical data; KB and MSP performed the biochemical estimations; KB, PB and IH performed the genotyping; KB, PB and DD analyzed the data; KB and PB wrote the manuscript with inputs from MSP, IH and DD; all authors approved the final manuscript.
Footnotes
Declaration of Interest. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M. et al. Heart disease and stroke statistics – 2015 update: A report from the American Heart Association. Circulation. 2015;131(4):e29–e322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Remaley AT, Rifai N, Warnick GR. Burtis CA, Ashwood ER, Bruns DE. Tietz Textbook of Clinical Chemistry and Molecular Diagnostics. 5th. New Delhi India: Elsevier (Reed Elsevier India Pvt. Ltd.); 2012. Lipids, lipoproteins, apolipoproteins and cardiovascular risk factors; pp. 731–805. 1st Indian reprint. [Google Scholar]
- 3.Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97(18):1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 4.Dallinga-Thie GM, Bu XD, Trip MLS, Rotter JI, Lusis AJ, de Bruin TWA. Apolipoprotein A-I/C-III/A-IV gene cluster in familial combined hyperlipidemia: Effects on LDL-cholesterol and apolipoproteins B and C-III. J Lipid Res. 1996;37(1):136–147. [PubMed] [Google Scholar]
- 5.Wojciechowski AP, Farrall M, Cullen P, Wilson TME, Bayliss JD, Farren B. et al. Familial combined hyperlipidaemia linked to the apolipoprotein AI-CIII-AIV gene cluster on chromosome 11q23q-q24. Nature. 1991;349(6305):161–164. doi: 10.1038/349161a0. [DOI] [PubMed] [Google Scholar]
- 6.de Franca E, Alves JG, Hutz MH. APOA1/C3/A4 gene cluster variability and lipid levels in Brazilian children. Braz J Med Biol Res. 2005;38(4):535–541. doi: 10.1590/s0100-879x2005000400006. [DOI] [PubMed] [Google Scholar]
- 7.Ding Y, Zhu MA, Wang ZX, Zhu J, Feng JB, Li DS. Associations of polymorphisms in the apolipoprotein APOA1-C3-A5 gene cluster with acute coronary syndrome. J Biomed Biotechnol. 2012;2012:509420. doi: 10.1155/2012/509420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swanson CR, Li K, Unger TL, Gallagher MD, van Deerlin VM, Agarwal P. et al. Lower plasma apolipoprotein A1 levels are found in Parkinson’s disease and associate with apolipoprotein A1 genotype. Mov Disord. 2015;30(6):805–812. doi: 10.1002/mds.26022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smach MA, Edziri H, Charfeddine B, Ben Othman L, Lammouchi T, Ltaief A. et al. Polymorphism in apoA1 influences high-density lipoprotein cholesterol levels but is not a major risk factor of Alzheimer’s disease. Dement Geriatr Cogn Disord Extra. 2011;1(1):249–257. doi: 10.1159/000329910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu ZH, Xiao YM, Tang LY, Jiang L, Wang YJ, Zhang RC. et al. Apolipoprotein A1 -75 G/A and +83 C/T polymorphisms and renal cancer risk. Lipids Health Dis. 2015;14:143. doi: 10.1186/s12944-015-0132-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu MC, Lee KT, Hsiao WC, Wu CH, Sun HY, Lin IL. et al. The dyslipidemia-associated SNP on the APOA1/C3/A5 gene cluster predicts post-surgery poor outcome in Taiwanese breast cancer patients: A 10-year follow-up study. BMC Cancer. 2013;13:330. doi: 10.1186/1471-2407-13-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miroshnikova VV, Rodygina TI, Demina EP, Kurjanov PS, Urazgildeeva SA, Gurevich VS. et al. Association of apoprotein A-1 genetic variants with development of atherosclerosis in the population of St. Petersburg. Russian J Genet Appl Res. 2011;1:411–415. doi: 10.1134/S207905971105011X. [DOI] [Google Scholar]
- 13.Coban N, Onat A, Guclu-Geyik F, Komurcu-Bayrak E, Can G, Erginel-Unaltuna N. Gender-specific associations of the APOA1 -75G>A polymorphism with several metabolic syndrome components in Turkish adults. Clin Chim Acta. 2014;431:244–249. doi: 10.1016/j.cca.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 14.Souverein OW, Jukema JW, Boekholdt SM, Zwinderman AH, Tanck MWT. Polymorphisms in APOA1 and LPL genes are statistically independently associated with fasting TG in men with CAD. Eur J Hum Genet. 2005;13(4):445–451. doi: 10.1038/sj.ejhg.5201362. [DOI] [PubMed] [Google Scholar]
- 15.Wang XL, Badenhop R, Humphrey K, Wilcken DE. New MspI polymorphism at +83 bp of the human apolipoprotein A1 gene: Association with increased circulating high density lipoprotein cholesterol levels. Genet Epidemiol. 1996;13(1):1–10. doi: 10.1002/(SICI)1098-2272(1996)13:1<1::AID-GEPI1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 16.Dawar R, Gurtoo A, Singh R. Apolipoprotein A1 gene polymorphism (G-75A and C+83T) in patients with myocardial infarction: A pilot study in a North Indian population. Am J Clin Pathol. 2010;134(2):249–255. doi: 10.1309/AJCPKPTXQ3QN1IFG. [DOI] [PubMed] [Google Scholar]
- 17.Liao BH, Cheng KQ, Dong SH, Liu HD, Xu ZL. Effect of apolipoprotein A1 genetic polymorphisms on lipid profiles and the risk of coronary artery disease. Diagn Pathol. 2015;10:102. doi: 10.1186/s13000-015-0328-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim YR, Hong SH. The apolipoprotein A1 polymorphisms were associated with decreased risk for metabolic syndrome in Koreans. Genes Genome. 2015;37(10):875–882. doi: 10.1007/s13258-015-0318-x. [DOI] [Google Scholar]
- 19.Pagani F, Sidoli A, Giudichi GA, Barenghi L, Vergani C, Baralle FE. Human apolipoprotein A-I gene promoter polymorphism: Association with hyperalphalipoproteinemia. J Lipid Res. 1990;31(8):1371–1377. [PubMed] [Google Scholar]
- 20.Wang XL, Badenhop R, Humphrey K, Wilcken DEL. C to T and G to A transitions are responsible for loss of a MspI restriction site in the 59-region of the human apolipoprotein AI gene. Hum Genet. 1995;95(4):473–475. doi: 10.1007/BF00208984. [DOI] [PubMed] [Google Scholar]
- 21.Bora K, Pathak MS, Borah P, Hussian MI, Das D. Single nucleotide polymorphisms of APOA1 gene and their relationship with serum apolipoprotein A-I concentrations in the native population of Assam. Meta Gene. 2016;7:20–27. doi: 10.1016/j.mgene.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bora K, Pathak MS, Borah P, Das D. Variation in lipid profile across different patterns of obesity – Observations from Guwahati Assam. J Clin Diagn Res. 2015;9(11):OC17–OC21. doi: 10.7860/JCDR/2015/15334.6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kotokey R, Hazarika S, De A. Lipid abnormalities in hypertensive urban population of Dibrugarh district of upper Assam. Indian Heart J. 2006;58(6):405–408. [PubMed] [Google Scholar]
- 24.Gururaj G, Girish N, Isaac MK. National Commission on Macroeconomics and Health (NCMH) Background Papers – Burden of disease in India. New Delhi India: NCMH, Ministry of Health & Family Welfare Government of India; 2005. Mental, neurological and substance abuse disorders: Strategies towards a systems approach; pp. 226–250. [Google Scholar]
- 25.National Cardiovascular Disease Database. Ministry of Health & Family Welfare Government of India and World Health Organisation. http://www.searo.who.int/india/topics/cardiovascular_diseases/NCD_Resources_National_CVD_database-Final_Report.pdf (Last accessed on June 21 2016.) [Google Scholar]
- 26.Shah SK, Saikia M, Snehlata C, Ramachandran A. High prevalence of type 2 diabetes in urban population of north eastern India. Int J Diabetes Dev Countries. 1999;19:144–147. [Google Scholar]
- 27.Ridkar PM, Rifai N, Cook NR, Bradwin G, Buring JE. Non-HDL cholesterol, apolipoproteins A-1 and B-100, standard lipid measures, lipid ratios, and CRP as risk factors for cardiovascular disease in women. JAMA. 2005;294(3):326–333. doi: 10.1001/jama.294.3.326. [DOI] [PubMed] [Google Scholar]
- 28.Pereira T. Kelishandi R. Dyslipidemia – From Prevention to Treatment. Rijeka. Croatia: InTech Europe; 2012. Dyslipidemia and cardiovascular risk: lipid ratios as risk factors for cardiovascular disease; pp. 279–302. [Google Scholar]
- 29.Kennel WB. Risk stratification of dyslipidemia: Insights from the Framingham Study. Curr Med Chem Cardiovasc Hematol Agents. 2005;3(3):187–193. doi: 10.2174/1568016054368250. [DOI] [PubMed] [Google Scholar]
- 30.National Cholesterol Education Program. Third and Final report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Circulation. 2002;106(25):3143–3421. [PubMed] [Google Scholar]
- 31.World Health Organisation Technical Report Series 854, Physical Status: The Use and Interpretation of An-thropometry. Geneva, Switzerland: World Health Organisation; 1995. World Health Organisation. Annex 2: Recom-mended measurement protocols and derivation of indices; pp. 424–438. [Google Scholar]
- 32.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr. et al. Seventh report of the Joint National Committee on prevention detection, evaluation, and treatment of high blood pressure. JAMA. 2003;289(19):2560–2572. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 33.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the low-density lipoprotein cholesterol concentration in plasma without the use of preparative centrifuge. Clin Chem. 1972;18(6):499–502. [PubMed] [Google Scholar]
- 34.Castelli WP, Abbott RD, McNamara PM. Summary estimates of cholesterol used to predict coronary heart disease. Circulation. 1983;67(4):730–734. doi: 10.1161/01.cir.67.4.730. [DOI] [PubMed] [Google Scholar]
- 35.Dobiasova M, Frohlich J. The plasma parameter log (TG/HDL-C) as an atherogenic index: Correlation with lipoprotein particle size and esterification rate in apoB-lipoprotein-depleted-plasma (FERHDL) Clin Biochem. 2001;34(7):583–588. doi: 10.1016/s0009-9120(01)00263-6. [DOI] [PubMed] [Google Scholar]
- 36.Nunes SOV, de Melo LGP, de Castro MRP, Barbossa S, Vargas HO, Berk M. et al. Atherogenic index of plasma and atherogenic coefficient are increased in major depression and bipolar disorder, especially when comorbid with tobacco use disorder. J Affect Disord. 2015;172:55–62. doi: 10.1016/j.jad.2014.09.038. [DOI] [PubMed] [Google Scholar]
- 37.Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd. New York NY, USA: Routledge Academic; 1969. [Google Scholar]
- 38.Al-Bustan SA, Al-Serri AE, Annice BG, Alnaqeeb MA, Ebrahim GA. Re-sequencing of the APOAI promoter region and the genetic association of the -75G>A polymorphism with increased cholesterol and low density lipoprotein levels among a sample of the Kuwaiti population. BMC Med Genet. 2013;14:90. doi: 10.1186/1471-2350-14-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saha N, Tay JS, Low PS, Humphries SE. Guanidine to adenine (G/A) substitution in the promoter region of the apolipoprotein AI gene is associated with elevated serum apolipoprotein AI levels in Chinese non-smokers. Genet Epidemiol. 1994;11(3):255–264. doi: 10.1002/gepi.1370110304. [DOI] [PubMed] [Google Scholar]
- 40.Qingwei Z, Mingpeng S, Ping S, Hua Z, Mengling Z. Interaction of alcohol and G to A substitution at the promoter region of apolipoprotein AI gene in determining plasma apolipoprotein AI levels in Yi and Han Chinese. Chinese Med J. 2000;113(5):471–474. [PubMed] [Google Scholar]
- 41.Li Y, Yin R, Zhou Y, Deng Y, Yang D, Pan S. et al. Associations of the apolipoprotein A-I gene polymorphism and serum lipid levels in the Guangxi Hei Yi Zhuang and Han populations. Int J Mol Med. 2008;21(6):753–764. [PubMed] [Google Scholar]
- 42.Kessling AM, Horsthempke B, Humphries SE. A study of DNA polymorphism around the human apolipoprotein AI gene in hyperlipidaemic and normal individuals. Clin Genet. 1985;28(4):296–306. doi: 10.1111/j.1399-0004.1985.tb00403.x. [DOI] [PubMed] [Google Scholar]
- 43.Ma YQ, Thomas GN, Tomlinson B. Association of two apolipoprotein A-I gene MspI polymorphisms with lipid and blood pressure levels. Int J Cardiol. 2005;102(2):309–314. doi: 10.1016/j.ijcard.2004.10.017. [DOI] [PubMed] [Google Scholar]