

Abstract
Restoration of learning and memory deficits following traumatic brain injury (TBI) is attributed, in part, to enhanced neural stem/progenitor cell (NSPCs) function. Recent findings suggest gap junction (GJ)-associated connexin 43 (Cx43) plays a key role in the cell cycle regulation and function of NSPCs and is modulated following TBI. Here, we demonstrate that Cx43 is up-regulated in the dentate gyrus following TBI and is expressed on vimentin-positive cells in the subgranular zone. To test the role of Cx43 on NSPCs, we exposed primary cultures to the α-connexin Carboxyl Terminal (αCT1) peptide which selectively modulates GJ-associated Cx43. Treatment with αCT1 substantially reduced proliferation and increased caspase 3/7 expression on NSPCs in a dose-dependent manner. αCT1 exposure also reduced overall expression of Cx43 and phospho (p)-Serine368. These findings demonstrate that Cx43 positively regulates adult NPSCs; the modulation of which may influence changes in the dentate gyrus following TBI.
Keywords: Cx43, traumatic brain injury, neural stem progenitor cell, gap junction, and apoptosis
Introduction
Traumatic brain injury (TBI) affects over 1.7 million Americans each year and is a leading cause of long-term disability [46, 54, 55, 62], including physical, cognitive, and emotional dysfunction ranging from headaches to memory loss and depression [31, 43, 49, 62]. While the yearly incidence of TBI is more than triple that of most cancers [10], no effective therapies exist to prevent neural tissue damage and/or fully restore neurological function. Impairment in learning and memory is a major hallmark of the neurocognitive sequela of TBI [1, 21, 68] and is partly attributed to hippocampal neuronal and neural progenitor cell loss [4, 44, 68]. Neural stem/progenitor cells (NSPCs) in the dentate gyrus (DG) play a key role in hippocampal function [19, 35, 71]. Although NSPCs are initially vulnerable in this neurogenic region following TBI, subsequent promotion of neurogenesis could aid in replacing locally damaged neurons and enhancing neuronal circuit repair and restoration of neurocognitive decline [22, 27, 28, 56, 69, 70]. A comprehensive understanding of the cellular and molecular underpinnings controlling TBI-induced neurogenesis will promote future strategies targeting this adaptive mechanism after brain trauma.
Analysis of the neurogenic response in the human brain following trauma is severely limited due to lack of human tissue availability [13]. Therefore, rodent models of reproducible injury have been widely used to address the cellular and molecular changes occurring in the DG following trauma [25, 36, 40]. One such model, the moderate controlled cortical impact (CCI) injury results in a focal cortical contusion and selective cell loss in the hippocampus [5, 25, 64]. Recent studies using a transgenic approach to ablate Nestin-expressing NSPCs directly linked TBI-induced neurogenesis to cognitive and motor functional recovery [4, 11, 70]. These findings emphasize the critical role of endogenous neurogenesis and underscore the importance of expanding our knowledge regarding the nuances regulating the complex milieu in the neurogenic niches of the brain following injury.
Gap junctional (GJ) communication plays a key role in proliferation and migration within the neurogenic zone during brain development by differentially coupling radial-glial (RG) and dividing neural progenitor cells in the ventricular zone [8, 60, 61]. Gap junctions are formed by connexin hemichannels and are specialized sites of cell-cell contact that allow the passage of ions, intracellular metabolites and messenger molecules (less than 2kDa) as well as provide adhesion sites between adjacent cells [9, 15, 23, 45]. Clusters of neural progenitor and RG cells, but not migrating neuroblasts, exist within the developing ventricular zone [17] and are differentially coupled by GJs depending on the phase of the cell cycle and the discrete stage of neurogenesis [3]. The modulation of gap junction-associated Cx43 during cell division is regulated, in part, at the level of phosphorylation [30, 52, 53]. Phosphorylation of serine368 is important for cell division and peaks during mitosis as gap junction communication ceases. Interestingly, Cx43 also displays channel-independent roles in regulating migration and adhesion [12]. These findings suggest gap-junction associated Cx43 plays an important role in embryonic neurogenesis; however, no studies have addressed the expression and function of Cx43 in the adult neurogenic niches of the brain under homeostasis and following TBI.
To date it remains unclear whether modulation of GJ-associated Cx43 occurs in the clustered neural stem-progenitor cell niche of the SGZ of the DG and the effects of modulating Cx43 in these highly coupled cells. To address this question we evaluated the expression of Cx43 on vimentin-positive NSPCs in the SGZ in sham and CCI-injured mice at 4-days using confocal imaging microscopy. To further address the role of GJ-associated Cx43 in NSPC activities we exposed primary NSPC cultures to a mimetic peptide of Cx43, called α-connexin Carboxyl Terminal 1 (αCT1), [18, 51], and found increased Caspase 3/7 activation and reduced BrdU incorporation in a dose-dependent manner. Treatment with αCT1 reduced total levels of Cx43 expression on NSPCs at 8 and 24 hours post-treatment. Our findings suggest that Cx43 plays a positive role in regulating NSPC survival and proliferation, the modulation of which could influence key neurogenic changes following TBI.
Materials and Methods
Animals
All mice were generated and housed in an AAALAC approved, virus/specific antigen-free facility with a 12 h light-dark cycle; food and water ad libitum. CD1 mice were purchased from Charles Rivers and bred until desired numbers were generated for experimentation. All experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were conducted under the approval of the Virginia Tech Institutional Animal Care and Use Committee (IACUC; #15-063) and the Virginia Maryland Regional College of Veterinary Medicine.
CCI Injury
Male CD1 mice at 8–12 weeks of age were anesthetized with ketamine and xylazine intraperitoneal (i.p.) injection and positioned in a stereotaxic frame [2, 63, 64]. Body temperature was monitored with a rectal probe and maintained at 37°C with a controlled heating pad (homeothermic blanket system; Harvard Apparatus). A 5mm craniotomy was made using a portable drill over the right parietal-temporal cortex (−2.5 mm A/P and 2.0 mm lateral from bregma). Injury was induced by moderate CCI using the eCCI- 6.3 device (Custom Design & Fabrication; 3mm impounder) at a velocity of 3.5 m/s, depth of 1.0 mm, and 150 ms impact duration [2, 63]. Sham controls received anesthesia and craniotomy only. Following injury, the incision was closed using Vetbond tissue adhesive (3M, St. Paul, MN, USA) and the animals were placed into a heated cage to maintain body temperature for 1 h post-injury followed by daily monitoring for 4 days or until the day of euthanization.
Tissue sectioning and Immunostaining
Brains were dissected from the skull and embedded into tissue freezing medium (Optical Cutting Temperature (OCT) Compound, Fisher Healthcare) before immediately snap frozen. Five serial, coronal sections (30μm in thickness) were collected spaced 300μm apart using a cryostat (Thermo Fisher Cryostar NX50, Waltham, MA, USA). To label for neural stem/progenitor cells undergoing apoptosis, slides were fixed in 10% formalin (Fisher Chemicals, Pittsburgh, PA, USA) for 10 minutes, washed 3 times with 1X PBS, and permeabilized in 2:1 Ethanol:Acetic Acid for 10 minutes, washed 3 times in 1X PBS then incubated with 0.4% Triton X-100 for 5 minutes and washed with 1X PBS. Slides were incubated in primary antibody using block overnight at 4°C at 1:500 anti-Cx43 (Santa Cruz Biotechnology) and anti-vimentin (Santa Cruz Biotechnology, Dallas, TX, USA). Sections were washed with 1X PBS then incubated with anti-rabbit Alexa Fluor 594-conjugated and Alexa Fluor 488-conjugated anti-rabbit or Alexa Fluor 488-conjugated anti-goat (Molecular Probes, Carlsbad, CA) in block for 1h at RT.
Peptide Sequences
The inhibitor peptide, αCT1, includes the Cx43 C-terminal amino acids 374RPRPDDLEI382 that encompass the ZO-1-binding sequence. A control peptide was generated by reversing the Cx43 amino acid sequence (IELDDPRPR), and is termed “Reverse” throughout the manuscript. Both peptides contain a 16-amino acid antennapedia internalization vector (RQPKIWFPNRRKPWKK) linked to the N terminus of the Cx43 (or reversed Cx43) sequence; peptides were N-terminally biotinylated. These peptides were dissolved and used as previously described [24].
Neural stem cells assays
Neural stem/progenitor cells were isolated from the SVZ of adult CD1 mice (2 to 4 months old) and grown as monolayer cultures as previously described [65]. Once stem cells reached 70–80% confluency, they were passaged and 20,000 cells were plated in a 96-well plate using monolayer culture media (DMEM/F12 (Hyclone, Logan, Utah) containing N2 supplement (Life Technologies Corporation, Grand Island, NY), 35μg/mL bovine pituitary extract (Sigma-Aldrich, St. Louis, MO, USA), 1X pen-strep glutamine (100X stock, Life Technologies Co., Grand Island, NY), 5% fetal bovine serum, and 20ng/mL each epidermal growth factor (EGF, Chemicon International, Temecula, CA, USA) and basic fibroblast growth factor (EGF, Chemicon International, Temecula, CA, USA) in the presence of 150μM, 75μM and 37.5μM Reverse or αCT1 peptide. After 8 or 24 hours, cells were fixed for 10 minutes using 10% buffered formalin. For immunostaining, wells were washed 3 times in PBS before blocking in 2% fish gelatin with 0.1% Triton X-100 (2% fish gelatin/0.1% Triton-X100) for 1h at RT before primary incubation in 1:500 (rabbit anti-Cx43 polyclonal, Santa Cruz, Dallas, TX, USA) and 1:100 (mouse anti-Nestin, Santa Cruz Biotechnology, Dallas, TX, USA) for 1–2 hours at room temperature. Wells were washed with 1X PBS then incubated in anti-mouse Alexa-Flour 488-conjugated secondary antibody (1:250) or anti-rabbit Alexa-flour 594-conjugated secondary antibody (1:500) (Invitrogen) along with DAPI (1:2000, Cell Signaling, Danvers, MA, USA) for 1h at RT.
Caspase 3/7 analysis
To assess cellular apoptosis in vitro, NSPCs were plated in a 96-well plate and treated with peptides as described above. After 8h or 24h, CellEvent Caspase-3/7 Green Detection Reagent (ThermoFisher Inc, Waltham, MA, USA) was added to each well at a final concentration of 4μM and incubated for 30 min at 37°C. To quantify Caspase 3/7 expression, each well was imaged at 4X and overall expression was measured using mean intensity in ImageJ. To analyze mean intensity in Image J, we opened the image, selected “Analyze > Measure” and recorded the “Mean” value for mean intensity.
BrdU analysis
To assess proliferation we performed BrdU incorporation, which identifies cells in the s-phase of the cell cycle, after NSPCs were plated in a 96-well plate and treated as described above. After 8h or 24h of αCT1 treatment, 10 μM BrdU was added for 20 min before cells were fixed with 10% formalin and washed 3 times with 1X PBS. Cells were incubated in 1N HCL for 1h at 37°C to denature the DNA and the pH was neutralized in 0.1M-borate buffer at pH 8.5 and washed in 1X PBS. Cells were blocked in 2% fish gelatin/0.1% Triton-X100 for 1h before incubating with rat anti-BrdU antibody 1:2000 (LifeTechnologies, Eugene, OR, USA) in block overnight at 4°C. The following day, each well was washed in 1X PBS then incubated in anti-rat Alexa-Flour 594-conjugated secondary antibody (1:500) (Invitrogen, Inc) were added with DAPI (1:2000, Cell Signaling Inc., Dallas, TX, USA) for 1h at RT. After washing in 1X PBS, the ratio of BrdU-positive cells to total nuclei was determined by imaging each well 2 times at 10X and using the Cell Counter on ImageJ (NIH) to quantify total cell numbers. Each experiment was repeated 4–5 times with 4–5 wells per treatment.
Confocal imaging and Cx43 quantification
In order to analyze images taken on Zeiss LSM 880 confocal laser scanning microscope, three series coronal sections per brain were imaged using z-stack at 20X and 40 X magnifications. To analyze z-stacks as one image rather than several images individually, we used Fiji by ImageJ (NIH) [47] to project the z-stack images into a 2D plane. We analyzed mean intensity expression on each 2D plane by placing a triangle shaped contour around the dentate gyrus, then using the Fiji by ImageJ program to calculate the “Mean” value for mean intensity by selecting “Analyze” > “Measure”. This recorded the mean intensity for Cx43 expression inside the contour of the dentate gyrus.
Western blot analysis
Protein of NSC cultures or hippocampal brain was extracted by lysing cells in RIPA buffer (1% NP-40, 1% sodium-deoxycholate, 0.1% SDS, 0.15 M NaCl, 2 mM EDTA, and 0.01M sodium phosphate) in the presence of complete protease inhibitor cocktail (Roche, Indianapolis, IN, USA) and phosphatase inhibitor cocktail 2 (Sigma, St. Louis, MO, USA). Supernatant was collected by centrifuging at 13000 g for 20 min at 4°C and the Lowry assay was used for determination of protein concentration (Pierce, Rockford, IL). Cell lysates (50 μg) were resolved on 10% SDS-PAGE gels and blotted onto PVDF membranes, blocked with 5% bovine serum albumin (BSA) in TBST buffer (20 mM Tris, 137 mM NaCl and 0.1% tween) then incubated in block overnight at 4°C with primary antibody against Cx43 (rabbit polyclonal, Santa Cruz, Dallas, TX, USA), p-Cx43 (s368, Cell Signaling, Danvers, MA, USA), or β-actin (mouse, 1:5000 Cell Signaling, Danvers, MA). Secondary antibodies (donkey anti-rabbit IRDye 800CW and donkey anti-mouse IRdye 680; Licor) were applied at 1:5000 to the membrane for 3 hours at room temperature before scanning the membranes with the Odyssey Fc imaging system (Licor, Lincoln, NE). Blots were quantified by densitometry using acquisition into Adobe Photo Shop (Apple, Cupertino, CA, USA). The level of protein expression was normalized according to β-actin control levels. Samples were run in duplicate.
Statistical analysis
Data was graphed using GraphPad Prism, version 4 (GraphPad Software, Inc., San Diego, CA). Student’s two-tailed t test was used for comparison of two experimental groups. Multiple comparisons were done by using one-way ANOVA where appropriate followed by post hoc Tukey test for multiple pairwise examinations. Differences were identified as significant if P < 0.05. Mean values were reported together with the standard error of mean (SEM).
Results
Connexin43 expression in the hippocampus and dentate gyrus following controlled cortical impact
Our preliminary findings demonstrate that Cx43 is highly expressed in the neurogenic compartments of the adult brain, specifically the subventricular zone and the subgranular zone (SGZ) of the dentate gyrus (DG) (supplementary Fig. 1). Cx43 expression has been shown to be increased after TBI, although it is unclear whether these changes occur in the hippocampus, an area vulnerable to brain trauma. To examine this we isolated protein from the hippocampus 4 days following sham or controlled cortical impact (CCI) injury and evaluated levels of Cx43 and pS368 using Western blot analysis. Using antibodies against Cx43 and pS368, we found no statistically significant difference in the expression of Cx43 and reduced p-S368 (Fig. 1a and 1b) (P>0.05 for both). To evaluate more cell-specific changes, we used serial coronal tissue sections of sham and CCI-injured brains and assessed Cx43 expression in the DG on vimentin-positive cells using immunohistochemistry. Vimentin is a type III intermediate filament protein present on neural stem cells and is upregulated in the DG following CCI injury (Fig. 1d) compared to sham (Fig. 1c). This upregulation coincides with an increase in Cx43 immunostaining in the SGZ and hilus of the DG following CCI injury (Fig. 1d, 1e and 1f) compared to sham (Fig. 1c, 1e and 1f). Vimentin is also upregulated on reactive astrocytes and is associated with astroglial proliferation. Therefore, Cx43 expression is present on both neural stem cells and reactive astrocytes in the DG after CCI injury.
Fig. 1. Cx43 expression in the hippocampus 4 days post-CCI injury.

(a) Western blot analysis showing total Cx43 and phospho (p) serine 368 protein levels in the whole hippocampus at 4 days post-sham or CCI injury. (b) Quantified data showing the fluorescence intensity of Cx43 relative to β-actin and p-S368 relative to total Cx43. (c) Immunohistochemistry for vimentin (red) and Cx43 (green) expression in the dentate gyrus (DG) of sham injury mice at 4 days post-sham compared to CCI injury (d). Increased expression by immunofluorescence is seen in the DG following CCI injury (e). (f) The mean fluorescence intensity of Cx43 expression in the region of the subgranular layer of the DG was significantly increased after CCI injury. *P<0.01 compared to sham injury.
Modulation of Cx43 on primary adult NSPCs using αCT1
Modulation of Cx43 using αCT1 has been shown to increase Cx43 gap junctional activity and impair proliferation and survival of breast cancer cells [20]. To investigate the role of gap junction-associated Cx43 on adult NSPC behavior, we exposed primary murine NSPCs [2, 11, 63] to the Cx43 mimetic peptide αCT1. Using BrdU incorporation, we found a significant reduction in proliferation in a dose dependent manner. At 24 hours post-treatment, the lowest concentration (37.5 μM) showed a significant reduction in proliferation (p<0.05) between cells treated with αCT1 (11.37 ± 1.923% positive BrdU cells) compared to Reverse control peptide (19.06 ± 1.957 % BrdU-positive cells). At 75μM there was also a significant difference in proliferation (p<0.001) between αCT1 (5.482 ± 1.356% BrdU-positive cells) and Reverse 16.88 ± 1.765% positive BrdU cells) treated cells. At the highest concentration (150μM), αCT1 (2.397 ± 0.440% BrdU-positive cells) inhibits proliferation (p<0.01) in comparison with the Reverse control peptide (7.567 ± 1.683% BrdU-positive cells) however, at this concentration the reverse also appears to have some suppressive effect on proliferation. To determine whether this effect was occurring earlier than 24 hours we examined BrdU incorporation at 8 hours post-αCT1 or -Reverse control treatment. We found no significant changes in the percent of BrdU-positive cells between 150μM (F=13.65, p=0.7057), 75μM (F=1.389, p=0.7831), or 37.5μM (F=8.740, p=0.1013) αCT1 and Reverse. These results demonstrate that modulation of Cx43 using the selective inhibitor αCT1 negatively affect NSPC proliferation.
To test whether αCT1 reduces proliferation due to changes in cell survival, we assessed Caspase 3/7 activation, which is indicative of cells undergoing apoptosis. At 24 hours post-Reverse or – αCT1 peptide treatment we found a significant increase in Caspase 3/7 activation (p<0.001) at 150μM αCT1 (2.427 ± 0.2943 mean intensity) (Fig. 3f and 3g) treated cells compared to 150μM Reverse (0.9813 ± 0.05619 mean intensity) (Fig. 3c and 3g), and in 75μM αCT1 (1.244 ± 0.08346 mean intensity) (Fig. 3e and 3g) compared to 75μM Reverse (0.7774 ± 0.02735 mean intensity) (Fig. 3b and 3g) treated cells. We did not see any significant changes (F=2.160, p=0.1293) between 37.5μM αCT1 (0.8428 ± 0.04075 mean intensity) (Fig. 3d and 3g) and 37.5μM Reverse (0.08158 ± 0.05118 mean intensity) (Fig. 3a and 3g) treatments. Under phase contract microscopy, cells treated with 150μM αCT1 were visibly unhealthy showing numerous pyknotic cells when compared to 37.5μM (supplementary Fig. 2). In addition, cells were fixed and immunostained for antibodies against Nestin and Cx43 at 24 hours after Reverse (Fig. 4a and 4b) or αCT1 (Fig. 4c and 4d) peptide exposure. Using fluorescence image analysis we found reduced Cx43 aggregates on the cell surface of Nestin-positive, αCT1 treated NSPCs compared to Reverse. Nestin expression also revealed significant morphological changes such as reduced extensions or processes in the αCT1 treatment groups compared to Reverse controls. Together, these results show that modulation of Cx43 suppresses the growth and survival of adult NSPCs.
Fig. 3. Modulation of Cx43 induces apoptosis in NSPCs.

Compared to Reverse control peptide (a–c), αCT1 dose dependently increased caspase 3/7 activation following 24 hour incubation (d–f). (g) Quantified data showing a significant increase in caspase 3/7 mean intensity following 150 μm αCT1 exposure compared to Reverse and an increase trend at 75 μm αCT1. ***P<0.001 compared to Reverse.
Fig. 4. αCT1 reduces Cx43 expression and induces changes in NSPC morphology.
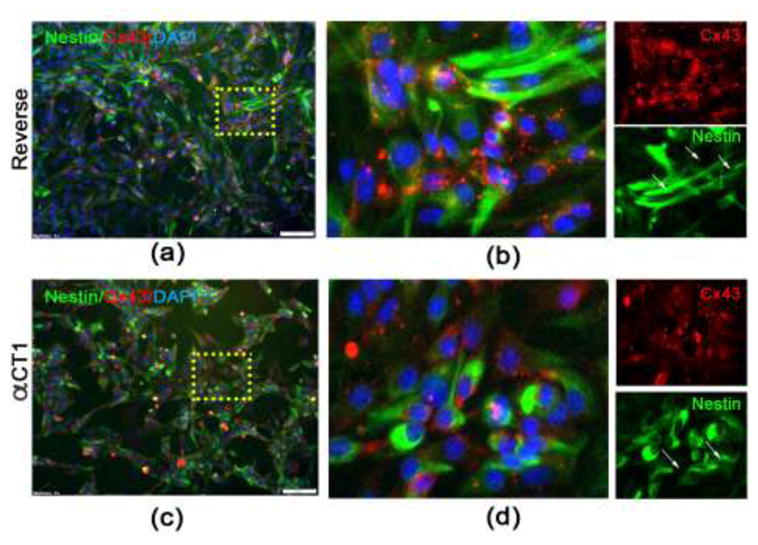
(a–b) Immunocytochemistry using antibodies against Cx43 (red) and Nestin (green) on monolayer cultures of NSPCS treated with 150 μm Reverse control peptide for 24 hours. (c–d) Significant morphological changes are seen following 150 μm αCT1 exposure, demonstrated by nestin staining showing NSPCs are more rounded with fewer protrusions. The density and aggregate number and size of Cx43 is also visibly reduced compared to Reverse control peptide.
To more quantitatively assess the effects of αCT1 treatment on Cx43 expression, we collected protein from αCT1 or Reverse treated cells at 24 hours and performed Western blot analysis (Fig. 5). We found a significant decrease in Cx43 expression in cells treated with 150μM αCT1 (0.325 ± 0.005 relative to β-actin) compared to 150μM Reverse peptide (2.24 ± 0.01 relative to β-actin) (Fig. 5a and 5b) and an overall decrease in the total levels of phosphorylated serine 368 (pS368) (Fig. 5a). However, relative to the total Cx43 levels, pS368 was significantly increased in αCT1 (2.25 ± 0.02 relative to Cx43) compared to Reverse (0.55 ± 0.02 relative to Cx43) treated NSPCs. Moreover, we observed a trend towards reduced expression of p-Akt (1.01 ± 0.03 Reverse vs 0.74 ± 0.01 αCT1 relative to total Akt) and Bcl-2 (0.31 ± 0.001 Reverse vs 0.17 ± 0.02 αCT1 relative to β-actin), although the results did not reach statistical significance. We also analyzed Cx43 and pS368 expression by Western blot at 8 hours post-treatment and found a significant reduction in both pS368 and total Cx43 expression and no change in p-AKT or bcl-2 (data not shown). This correlates with previous reports showing overexpression of Cx43 stimulated proliferation of neural progenitor cells, however, chemical-induced uncoupling of gap junctions reduced proliferation and increased cell death [7]. Our findings suggest that αCT1 negatively affects NSPC survival and proliferation by reducing total cellular Cx43.
Fig. 5. αCT1 reduces Cx43 protein expression.

(a–b) Using Western blot analysis we find a significant reduction in Cx43 expression at 24 hours post-αCT1 treatment compared to Reverse. Quantified expression of Cx43 is graphed relative to β-actin levels. Although overall pS368 is reduced, the expression relative to the total levels of Cx43 is increased following 150 μm αCT1 exposure compared to 150 μm Reverse. (c–d) Expression of p-Akt (relative to total Akt) and Bcl-2 (relative to β-actin) show a trend towards reduced levels in αCT1 compared to Reverse. ***P<0.001 compared to Reverse control peptide.
Discussion
The gap junction (GJ)-associated protein Cx43 has been implicated in the response to TBI [16, 41, 57, 58]. Previous studies suggest that radial glial-like stem cells in the adult dentate gyrus form Cx43 associated GJs, which is necessary to maintain adult neurogenesis [29, 33, 39]. Our findings demonstrate that Cx43 is up-regulated in the subgranular zone of the dentate gyrus, specifically on vimentin-positive cells and is expressed by both horizontal and radial glia (RG)-like neural stem cells in the adult DG [48]. To identify the role of Cx43 in adult NSPC function, we exposed cultured NSPCs to the α-connexin carboxyl-terminal peptide, αCT1, [18, 24] which resulted in an overall reduction in Cx43 expression at 8 and 24 hours which coincided with a dose-dependent reduction in proliferation and increase in apoptotic expression of caspase 3/7. These effects were both present at the highest dosage (150μM). While there was no significant difference in Caspase 3/7 activation at 37.5μM or 75μM, there is still a significant reduction in proliferation at these dosages. This specific effect on cell cycle arrest might be attributed to time difference in activation of Caspase 3/7 vs exiting the s-phase of the cell cycle at the lower dosages. Alternatively, a dose dependent effect of αCT1 on overall Cx43 expression could exist, making the cells more vulnerable to apoptosis higher doses. This dosage response could also regulate differential effects on cell signaling pathways important for survival and/or proliferation [14]. Moreover, it is unlikely, these effects influenced the production of doublecortin-positive neuroblasts in the NSPC cultures. The defined cell culture conditions used generally do not allow for the differentiation of doublecortin positive cells as the NSPCs are kept in a tight self-renewal state. It is also unlikely that after 8 or 24 hours of alpha-CT1 treatment this would change given that differentiation is a much slower event.
Although not significant, a trend toward reduced expression of anti-apoptotic protein Bcl-2 and p-Akt growth signaling could suggest that Cx43 may positively regulate NSPC survival and proliferation by mediating these downstream pathways. Additionally, the mTOR pathway, which regulates the trafficking of Cx43, could also be disrupted by αCT1 treatment [50]. Immunocytochemistry revealed that αCT1 reduced the density and size of Cx43 aggregates and caused a morphological change in cell shape. These findings correlate with previous studies showing high levels of Cx43 and GJ communication are required for maintaining NSPCs in a proliferative state [7, 33, 37] and suggest that up-regulation of Cx43 on NSPCs may represent a compensatory mechanism to induce proliferation and cellular replacement in the DG after TBI. The role of Cx43 on NSPC proliferation and survival appear to be regulated, in part, by epidermal growth factor receptor signaling (EGF/EGFR), which has a modulatory effect on the functional activity and expression of connexin proteins [34, 38]. However, the impact of EGF/EGFR signaling axis on GJ coupling and Cx43 expression appears to be cell-type dependent. For example, EGF mediates down-regulation of Cx43 on cortical rodent astrocytes [66] and inhibits Cx43-mediated GJ communication in HEK cells and liver epithelial cells [6, 32]. On the other hand, EGF/EGFR signaling enhances GJ communication and Cx43 expression in kidney epithelial cells and granulosa cells [26, 67], as well as embryonic-derived neural progenitor cells [7]. Although we did not test Cx43 expression in adult NSPCs following withdrawal of EGF, we did observe enhanced cell death and growth restriction when cells were exposed to αCT1in the absence of EGF and FGF (data not shown). Overall, these data suggest that adult NSPCs are highly vulnerable to changes in Cx43 expression, and high level of Cx43 and strong cell-cell coupling may be required to maintain them in a healthy, proliferative state.
It remains unclear whether αCT1 may also disrupt GJ-independent actions of Cx43 such as hemi-channel-related cell survival signaling cascades [59]. Cx43 hemichannels can transduce survival signals in response to extracellular cues via ERK anti-apoptotic pathways [42]. Likewise, αCT1 may be mediating translocation of Cx43 to the mitochondria of NSPCs, where Cx43 was previously shown in pancreatic cancer cells to interact with Bax and initiate the mitochondria apoptotic pathway [59, 72]. Future studies elucidating these and other pathways are currently underway. To conclude, we find that adult NSPCs are selectively vulnerable to changes in Cx43 expression and that selective inhibition of Cx43 using the αCT1 peptide resulted in the induction of apoptosis and reduced proliferation of NSPCs. It is possible these effects may be mediated by both GJ-dependent and independent mechanisms. Although up-regulation of Cx43 following TBI has been linked to neural damage, the functional consequence of Cx43 dysregulation in the brain may be cell-type specific. The preservation and maintenance of Cx43 expression on NSPCs in the neurogenic compartment may confer protection of selective progenitor cell populations and be necessary and sufficient to enhance the neurogenic response following TBI although future studies will elucidate these cell-type specific longitudinal changes in Cx43 expression.
Supplementary Material
Fig. 2. Proliferation of NSPCs is reduced following αCT1treatment.

Exposure of NSPCs to selective inhibitor αCT1 (d–f) reduces BrdU incorporation in a dose dependent manner following 24 hour incubation compared to the control Reverse control peptide (a–c). (g) Quantified data showing a significant reduction in the percentage of BrdU-positive cells at 37.5 and 75 μm αCT1 exposure compared to Reverse. Non-specific effects of Reverse at 150 μm were also seen. (h) Quantified data showing no changes in NSPC proliferation are evident at 8 hours of αCT1 treatment. *P<0.01; ***P<0.001 compared to Reverse control peptide.
Highlights.
Cx43 maintains proliferation and survival of adult neural stem progenitor cells
Up-regulation of Cx43 in the dentate gyrus after TBI may mediate neurogenesis
αCT1 exposure reduces Cx43 expression, proliferation and survival of adult NSPCs
Acknowledgments
The authors would like to thank VT-IMSD Programs for student support (K.M.G.). This work was supported by R01 NS096281 (MHT), diversity supplement NS096281 (MHT, KG), R15 NS081623 (MHT) and VT’s Institute of Critical Technology and Science. We recognize the Virginia Maryland College of Veterinary Medicine for student and financial support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or any other funding agency.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arciniegas DB, Held K, Wagner P. Cognitive Impairment Following Traumatic Brain Injury. Curr Treat Options Neurol. 2002;4:43–57. doi: 10.1007/s11940-002-0004-6. [DOI] [PubMed] [Google Scholar]
- 2.Baumann G, Travieso L, Liebl DJ, Theus MH. Pronounced hypoxia in the subventricular zone following traumatic brain injury and the neural stem/progenitor cell response. Exp Biol Med (Maywood) 2013;238:830–841. doi: 10.1177/1535370213494558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bittman K, Owens DF, Kriegstein AR, LoTurco JJ. Cell coupling and uncoupling in the ventricular zone of developing neocortex. J Neurosci. 1997;17:7037–7044. doi: 10.1523/JNEUROSCI.17-18-07037.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaiss CA, Yu TS, Zhang G, Chen J, Dimchev G, Parada LF, Powell CM, Kernie SG. Temporally specified genetic ablation of neurogenesis impairs cognitive recovery after traumatic brain injury. J Neurosci. 2011;31:4906–4916. doi: 10.1523/JNEUROSCI.5265-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brickler T, Gresham K, Meza A, Coutermarsh-Ott S, Williams TM, Rothschild DE, Allen IC, Theus MH. Nonessential Role for the NLRP1 Inflammasome Complex in a Murine Model of Traumatic Brain Injury. Mediators Inflamm. 2016;2016:6373506. doi: 10.1155/2016/6373506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cameron SJ, Malik S, Akaike M, Lerner-Marmarosh N, Yan C, Lee JD, Abe J, Yang J. Regulation of epidermal growth factor-induced connexin 43 gap junction communication by big mitogen-activated protein kinase 1/ERK5 but not ERK1/2 kinase activation. Journal of Biological Chemistry. 2003;278:18682–18688. doi: 10.1074/jbc.M213283200. [DOI] [PubMed] [Google Scholar]
- 7.Cheng A, Tang H, Cai J, Zhu M, Zhang X, Rao M, Mattson MP. Gap junctional communication is required to maintain mouse cortical neural progenitor cells in a proliferative state. Dev Biol. 2004;272:203–216. doi: 10.1016/j.ydbio.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 8.Cina C, Maass K, Theis M, Willecke K, Bechberger JF, Naus CC. Involvement of the cytoplasmic C-terminal domain of connexin43 in neuronal migration. J Neurosci. 2009;29:2009–2021. doi: 10.1523/JNEUROSCI.5025-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contreras JE, Sanchez HA, Veliz LP, Bukauskas FF, Bennett MV, Saez JC. Role of connexin-based gap junction channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res Brain Res Rev. 2004;47:290–303. doi: 10.1016/j.brainresrev.2004.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA Cancer J Clin. 2016;66:31–42. doi: 10.3322/caac.21320. [DOI] [PubMed] [Google Scholar]
- 11.Dixon KJ, Theus MH, Nelersa CM, Mier J, Travieso LG, Yu TS, Kernie SG, Liebl DJ. Endogenous neural stem/progenitor cells stabilize the cortical microenvironment after traumatic brain injury. J Neurotrauma. 2015;32:753–764. doi: 10.1089/neu.2014.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elias LA, Wang DD, Kriegstein AR. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- 13.Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 14.Eungdamrong NJ, Iyengar R. Modeling cell signaling networks. Biol Cell. 2004;96:355–362. doi: 10.1016/j.biolcel.2004.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans WH, De Vuyst E, Leybaert L. The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J. 2006;397:1–14. doi: 10.1042/BJ20060175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002;22:644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freitas AS, Xavier AL, Furtado CM, Hedin-Pereira C, Froes MM, Menezes JR. Dye coupling and connexin expression by cortical radial glia in the early postnatal subventricular zone. Dev Neurobiol. 2012;72:1482–1497. doi: 10.1002/dneu.22005. [DOI] [PubMed] [Google Scholar]
- 18.Ghatnekar GS, O’Quinn MP, Jourdan LJ, Gurjarpadhye AA, Draughn RL, Gourdie RG. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen Med. 2009;4:205–223. doi: 10.2217/17460751.4.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goncalves JT, Schafer ST, Gage FH. Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell. 2016;167:897–914. doi: 10.1016/j.cell.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 20.Grek CL, Rhett JM, Bruce JS, Abt MA, Ghatnekar GS, Yeh ES. Targeting connexin 43 with alpha-connexin carboxyl-terminal (ACT1) peptide enhances the activity of the targeted inhibitors, tamoxifen and lapatinib, in breast cancer: clinical implication for ACT1. BMC Cancer. 2015;15:296. doi: 10.1186/s12885-015-1229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamm RJ, Lyeth BG, Jenkins LW, O’Dell DM, Pike BR. Selective cognitive impairment following traumatic brain injury in rats. Behav Brain Res. 1993;59:169–173. doi: 10.1016/0166-4328(93)90164-l. [DOI] [PubMed] [Google Scholar]
- 22.Han X, Tong J, Zhang J, Farahvar A, Wang E, Yang J, Samadani U, Smith DH, Huang JH. Imipramine treatment improves cognitive outcome associated with enhanced hippocampal neurogenesis after traumatic brain injury in mice. J Neurotrauma. 2011;28:995–1007. doi: 10.1089/neu.2010.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoang QV, Qian H, Ripps H. Functional analysis of hemichannels and gap-junctional channels formed by connexins 43 and 46. Mol Vis. 2010;16:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16:5686–5698. doi: 10.1091/mbc.E05-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson VE, Meaney DF, Cullen DK, Smith DH. Animal models of traumatic brain injury. Handb Clin Neurol. 2015;127:115–128. doi: 10.1016/B978-0-444-52892-6.00008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kennedy KL, Floyd AA, Clarkson AM, Lee VH. Epidermal growth factor regulation of connexin 43 in cultured granulosa cells from preantral rabbit follicles. Mol Reprod Dev. 2003;64:61–69. doi: 10.1002/mrd.10219. [DOI] [PubMed] [Google Scholar]
- 27.Kernie SG, Parent JM. Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiol Dis. 2010;37:267–274. doi: 10.1016/j.nbd.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleindienst A, McGinn MJ, Harvey HB, Colello RJ, Hamm RJ, Bullock MR. Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. J Neurotrauma. 2005;22:645–655. doi: 10.1089/neu.2005.22.645. [DOI] [PubMed] [Google Scholar]
- 29.Kunze A, Congreso MR, Hartmann C, Wallraff-Beck A, Huttmann K, Bedner P, Requardt R, Seifert G, Redecker C, Willecke K, Hofmann A, Pfeifer A, Theis M, Steinhauser C. Connexin expression by radial glia-like cells is required for neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A. 2009;106:11336–11341. doi: 10.1073/pnas.0813160106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laird DW, Puranam KL, Revel JP. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem J. 1991;273(Pt 1):67–72. doi: 10.1042/bj2730067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Leithe E, Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci. 2004;117:1211–1220. doi: 10.1242/jcs.00951. [DOI] [PubMed] [Google Scholar]
- 33.Lemcke H, Kuznetsov SA. Involvement of connexin43 in the EGF/EGFR signalling during self-renewal and differentiation of neural progenitor cells. Cell Signal. 2013;25:2676–2684. doi: 10.1016/j.cellsig.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 34.Lemcke H, Kuznetsov SA. Involvement of connexin43 in the EGF/EGFR signalling during self-renewal and differentiation of neural progenitor cells. Cellular Signalling. 2013;25:2676–2684. doi: 10.1016/j.cellsig.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 35.Leuner B, Gould E. Structural plasticity and hippocampal function. Annu Rev Psychol. 2010;61:111–140. C111–113. doi: 10.1146/annurev.psych.093008.100359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li SX, Wang BW, Liu D, He GL, Wang H, Duan YJ, Xing JJ, Zhou HY, Zhou YW. Advance in animal models of traumatic brain injury. Fa Yi Xue Za Zhi. 2011;27:286–289. 294. [PubMed] [Google Scholar]
- 37.Liebmann M, Stahr A, Guenther M, Witte OW, Frahm C. Astrocytic Cx43 and Cx30 differentially modulate adult neurogenesis in mice. Neurosci Lett. 2013;545:40–45. doi: 10.1016/j.neulet.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 38.Maldonado PE, Rose B, Loewenstein WR. Growth-Factors Modulate Junctional Cell-to-Cell Communication. J Membrane Biol. 1988;106:203–210. doi: 10.1007/BF01872158. [DOI] [PubMed] [Google Scholar]
- 39.Naus CC, Aftab Q, Sin WC. Common mechanisms linking connexin43 to neural progenitor cell migration and glioma invasion. Semin Cell Dev Biol. 2016;50:59–66. doi: 10.1016/j.semcdb.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 40.O’Connor WT, Smyth A, Gilchrist MD. Animal models of traumatic brain injury: a critical evaluation. Pharmacol Ther. 2011;130:106–113. doi: 10.1016/j.pharmthera.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Ohsumi A, Nawashiro H, Otani N, Ooigawa H, Toyooka T, Shima K. Temporal and spatial profile of phosphorylated connexin43 after traumatic brain injury in rats. J Neurotrauma. 2010;27:1255–1263. doi: 10.1089/neu.2009.1234. [DOI] [PubMed] [Google Scholar]
- 42.Plotkin LI, Manolagas SC, Bellido T. Transduction of cell survival signals by connexin-43 hemichannels. J Biol Chem. 2002;277:8648–8657. doi: 10.1074/jbc.M108625200. [DOI] [PubMed] [Google Scholar]
- 43.Rapoport MJ, McCullagh S, Shammi P, Feinstein A. Cognitive impairment associated with major depression following mild and moderate traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2005;17:61–65. doi: 10.1176/jnp.17.1.61. [DOI] [PubMed] [Google Scholar]
- 44.Rola R, Mizumatsu S, Otsuka S, Morhardt DR, Noble-Haeusslein LJ, Fishman K, Potts MB, Fike JR. Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Exp Neurol. 2006;202:189–199. doi: 10.1016/j.expneurol.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 45.Saez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV. Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta. 2005;1711:215–224. doi: 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santopietro J, Yeomans JA, Niemeier JP, White JK, Coughlin CM. Traumatic brain injury and behavioral health: the state of treatment and policy. N C Med J. 2015;76:96–100. doi: 10.18043/ncm.76.2.96. [DOI] [PubMed] [Google Scholar]
- 47.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seri B, Garcia-Verdugo JM, Collado-Morente L, McEwen BS, Alvarez-Buylla A. Cell types, lineage, and architecture of the germinal zone in the adult dentate gyrus. J Comp Neurol. 2004;478:359–378. doi: 10.1002/cne.20288. [DOI] [PubMed] [Google Scholar]
- 49.Siopi E, Llufriu-Daben G, Fanucchi F, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M. Evaluation of late cognitive impairment and anxiety states following traumatic brain injury in mice: the effect of minocycline. Neurosci Lett. 2012;511:110–115. doi: 10.1016/j.neulet.2012.01.051. [DOI] [PubMed] [Google Scholar]
- 50.Smyth JW, Shaw RM. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013;5:611–618. doi: 10.1016/j.celrep.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soder BL, Propst JT, Brooks TM, Goodwin RL, Friedman HI, Yost MJ, Gourdie RG. The connexin43 carboxyl-terminal peptide ACT1 modulates the biological response to silicone implants. Plast Reconstr Surg. 2009;123:1440–1451. doi: 10.1097/PRS.0b013e3181a0741d. [DOI] [PubMed] [Google Scholar]
- 52.Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711:154–163. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 53.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spitz G, Schonberger M, Ponsford J. The relations among cognitive impairment, coping style, and emotional adjustment following traumatic brain injury. J Head Trauma Rehabil. 2013;28:116–125. doi: 10.1097/HTR.0b013e3182452f4f. [DOI] [PubMed] [Google Scholar]
- 55.Sun D. The potential of endogenous neurogenesis for brain repair and regeneration following traumatic brain injury. Neural Regen Res. 2014;9:688–692. doi: 10.4103/1673-5374.131567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun D, Bullock MR, McGinn MJ, Zhou Z, Altememi N, Hagood S, Hamm R, Colello RJ. Basic fibroblast growth factor-enhanced neurogenesis contributes to cognitive recovery in rats following traumatic brain injury. Exp Neurol. 2009;216:56–65. doi: 10.1016/j.expneurol.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun L, Gao J, Zhao M, Cui J, Li Y, Yang X, Jing X, Wu Z. A novel cognitive impairment mechanism that astrocytic p-connexin 43 promotes neuronic autophagy via activation of P2X7R and down-regulation of GLT-1 expression in the hippocampus following traumatic brain injury in rats. Behav Brain Res. 2015;291:315–324. doi: 10.1016/j.bbr.2015.05.049. [DOI] [PubMed] [Google Scholar]
- 58.Sun L, Gao J, Zhao M, Jing X, Cui Y, Xu X, Wang K, Zhang W, Cui J. The effects of BMSCs transplantation on autophagy by CX43 in the hippocampus following traumatic brain injury in rats. Neurol Sci. 2014;35:677–682. doi: 10.1007/s10072-013-1575-6. [DOI] [PubMed] [Google Scholar]
- 59.Sun Y, Zhao X, Yao Y, Qi X, Yuan Y, Hu Y. Connexin 43 interacts with Bax to regulate apoptosis of pancreatic cancer through a gap junction-independent pathway. Int J Oncol. 2012;41:941–948. doi: 10.3892/ijo.2012.1524. [DOI] [PubMed] [Google Scholar]
- 60.Sutor B. Gap junctions and their implications for neurogenesis and maturation of synaptic circuitry in the developing neocortex. Results Probl Cell Differ. 2002;39:53–73. doi: 10.1007/978-3-540-46006-0_3. [DOI] [PubMed] [Google Scholar]
- 61.Sutor B, Hagerty T. Involvement of gap junctions in the development of the neocortex. Biochim Biophys Acta. 2005;1719:59–68. doi: 10.1016/j.bbamem.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 62.Swanson TM, Isaacson BM, Cyborski CM, French LM, Tsao JW, Pasquina PF. Traumatic Brain Injury Incidence, Clinical Overview, and Policies in the US Military Health System Since 2000. Public Health Rep. 2017:33354916687748. doi: 10.1177/0033354916687748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Theus MH, Ricard J, Bethea JR, Liebl DJ. EphB3 limits the expansion of neural progenitor cells in the subventricular zone by regulating p53 during homeostasis and following traumatic brain injury. Stem Cells. 2010;28:1231–1242. doi: 10.1002/stem.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Theus MH, Ricard J, Glass SJ, Travieso LG, Liebl DJ. EphrinB3 blocks EphB3 dependence receptor functions to prevent cell death following traumatic brain injury. Cell Death Dis. 2014;5:e1207. doi: 10.1038/cddis.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Theus MH, Ricard J, Liebl DJ. Reproducible expansion and characterization of mouse neural stem/progenitor cells in adherent cultures derived from the adult subventricular zone. Curr Protoc Stem Cell Biol. 2012;Chapter 2(Unit 2D):8. doi: 10.1002/9780470151808.sc02d08s20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ueki T, Fujita M, Sato K, Asai K, Yamada K, Kato T. Epidermal growth factor down-regulates connexin-43 expression in cultured rat cortical astrocytes. Neuroscience Letters. 2001;313:53–56. doi: 10.1016/s0304-3940(01)02249-2. [DOI] [PubMed] [Google Scholar]
- 67.Vikhamar G, Rivedal E, Mollerup S, Sanner T. Role of cx43 phosphorylation and MAP kinase activation in EGF induced enhancement of cell communication in human kidney epithelial cells. Cell Adhes Commun. 1998;5:451–460. doi: 10.3109/15419069809005603. [DOI] [PubMed] [Google Scholar]
- 68.Whiting MD, Baranova AI, Hamm RJ. Cognitive Impairment following Traumatic Brain Injury. In: Levin ED, Buccafusco JJ, editors. Animal Models of Cognitive Impairment. Boca Raton FL: 2006. [PubMed] [Google Scholar]
- 69.Wu H, Lu D, Jiang H, Xiong Y, Qu C, Li B, Mahmood A, Zhou D, Chopp M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25:130–139. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- 70.Yu TS, Zhang G, Liebl DJ, Kernie SG. Traumatic brain injury-induced hippocampal neurogenesis requires activation of early nestin-expressing progenitors. J Neurosci. 2008;28:12901–12912. doi: 10.1523/JNEUROSCI.4629-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng W, ZhuGe Q, Zhong M, Chen G, Shao B, Wang H, Mao X, Xie L, Jin K. Neurogenesis in adult human brain after traumatic brain injury. J Neurotrauma. 2013;30:1872–1880. doi: 10.1089/neu.2010.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou JZ, Jiang JX. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions--an update. FEBS Lett. 2014;588:1186–1192. doi: 10.1016/j.febslet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.